| Adaptation et programmation d'un gestionnaire graphique de processus bioinformatiques
par Franck VALENTIN
Institut Pasteur
Traductions: Original: fr Source:
| |
Copyright (c) Franck VALENTIN
Permission is granted to copy, distribute and/or modify this document under the terms of the GNU Free Documentation License, Version 1.2
or any later version published by the Free Software Foundation; with no Invariant Sections, no Front-Cover Texts, and no Back-Cover Texts.
A copy of the license is included in the section entitled "GNU Free Documentation License".

MÉMOIRE
présenté en vue d'obtenir
Le Mastère Spécialisé en Bio-Informatique
Franck VALENTIN
Conception et programmation d'un gestionnaire graphique de processus
bioinformatiques d'analyse de séquences et application à l'identification
des résidus encodant la spécificité de la reconnaissance de l'interleukine-2
humaine par ses récepteurs.
Institut PASTEUR
Septembre 2004
Directeur de stage :
Thierry ROSE
Unité d'Immunogénétique Cellulaire
rose@pasteur.fr
Tél. : 33 01.45.68.85.99
Fax. : 33 01.45.68.86.39
2
Résumé
Les outils informatiques utilisés voire développés par les biologistes sont de natures très diverses, il
peut s'agir entre autres d'applications locales, de formulaires web, de scripts personnels ou de services
web.Pour répondre à leurs besoins il leur est souvent nécessaire d'enchaîner ces outils manuellement. Ce
travail devient fastidieux lorsqu'il s'agit de le répéter pour ne modifier que quelques paramètres par
exemple. L'utilisation d'un workflow permettant l'enchaînement et l'exécution de ces outils devrait
simplifier ce processus. De plus, une construction et une exécution graphiques permettent un meilleur
contrôle dans la conception et dans l'enchaînement et offre des qualités pédagogiques intéressantes.
Nous avons dans un premier temps défini clairement nos besoins puis fait un tour d'horizon des
solutions existantes. Nous avons retenu d'utiliser Ptolemy II, un workflow non dédié à la
bioinformatique auquel nous avons ajouté nos briques et celles d'autres projets.
Notre outil est initialement prévu pour analyser les bases de la reconnaissance des cytokines par
leurs récepteurs en général au cours de l'évolution et plus précisément la conservation des résidus de
l'interleukine-2 et de ses récepteurs. Nous montrerons donc son intérêt dans ce contexte et
l'illustrerons par quelques exemples simples. Enfin, nous conclurons en proposant des évolutions futures
et en exposant les problématiques de ce type de projet si ce n'est des applications de bioinformatique
en général.
Abstract
The IT tools used or even developed by biologists are diverse by nature. They can be, among other
things, local applications, web forms, personal scripts or web services.
To satisfy their needs, biologists often have to manual y enchain these tools. This work becomes
tedious when it is necessary to repeat it, for example, just to change a few parameters.
The use of a workflow that can link and execute these tools ought to improve this process.
Furthermore, a graphical construction and execution permit better control in the conception and in the
linking, as wel as offering interesting educational qualities.
First the requirements were clearly defined and then an overview of existing solutions was made.
Ptolemy II, a workflow not dedicated to bioinformatics, was chosen as a foundation to which custom-
made components and ones from other projects were added.
This new tool is primarily aimed at analysing the basis of cytokine recognition by their receptors in
general through evolution and more precisely residue conservation within interleukine-2 and its
receptor chains. Its usefulness in this context will be shown and illustrated through some simple
examples.
To conclude, future developments wil be suggested. Some problematical areas in this kind of
project, if not in bioinformatic applications in general, will also be shown.
Mots clés
Workflow, Ptolemy, Kepler/SPA, services web, java, interleukine-2
Remerciements
Je tient tout d'abord à remercier Thierry Rose qui m'a confié ce projet et qui s'est démené pour
que ce stage à l'institut Pasteur se déroule dans les meilleures conditions. Je le remercie de l'avoir
enrichi de son optimisme et de ses innombrables idées qui m'ont effrayées plus d'une fois !
Je suis reconnaissant envers Bernard CAUDRON, responsable du groupe Logiciels et Banques de
données qui m'a trouvé une place dans les locaux de son service.
Je remercie Catherine LETONDAL qui a accepté que cette place se situe dans son bureau. Je la
remercie aussi pour ses efforts de montrer que l'on peut aussi faire de l'informatique en pensant aux
utilisateurs et même les associer à la conception des outils qu'ils vont utiliser.
Je remercie Bertrand NERON qui m'a eu dans le dos pendant ces six mois, au seul sens propre
j'espère, et qui a rendu cette collocation vraiment agréable. Je ne désespère pas qu'il m'enseigne un
jour au Trocadéro à réduire le nombre de mes serpillières et autres pizzas.
Enfin, je ne peux oublier Catherine, Corinne, Daniel, Elodie, Eric, Gérard, Jérôme, Louis, Marc,
Marie-Christine, Nicolas et Olivier des groupes Logiciels et Banques de données et Systèmes et Réseau
qui ont largement contribué à l'ambiance conviviale de ce stage. Je me souviens encore des Magnums©
inopinés de cet été.
Glossaire
ACD
(Ajax Command Definitions) format de fichier utilisé par les programmes EMBOSS
pour décrire leurs paramètres.
Anticorps
Protéine synthétisée par les cellules lymphoïdes en réponse à l'introduction d'une
substance étrangère appelée antigène.
Antigène
Substance étrangère (microbe, toxine, matière organique, etc) capable d'induire,
lors de son introduction dans un organisme animal, la formation d'anticorps
spécifiques.
Applet
Programme en Java conçu pour être téléchargé via un réseau à chaque fois qu'on
veut l'exécuter, en général par un navigateur web.
BioMOBY
Projet pour la découverte, l'intégration et l'interopérabilité de services et de bases
de données biologiques.
BioPerl
Ensemble de librairies écrites en langage Perl et dédiées au domaine de la
bioinformatique.
Cladistique
Analyse cladistique : qui privilégie le degré de ressemblance phylogénique plutôt que
la ressemblance morphologique.
Cladogramme
Schéma exprimant des relations phylogénétiques à partir d'une analyse cladistique.
Cluster
Architecture de groupes d'ordinateurs reliés entre eux et pour lesquels les
échanges sont rapides. l'ensemble est considéré comme une seule et unique
machine.
CVS
(Concurrent Versions System) Outil de gestion de sources multi-utilisateurs
permettant de sauver et de récupérer les différentes versions de fichiers.
Cytokine
Substance peptidique ou protéique synthétisée par une cellule du système
immunitaire (lymphocyte, macrophage) et agissant sur d'autres cellules
immunitaires pour en réguler l'activité.
DAML-S
(DARPA Agent Markup Language Services) Langage de description sémantique de
services web. Il permet la description, la recherche, la sélection et l'exécution
d'un service web particulier mais aussi la composition de services entre eux.
DAML+OIL
Langage permettant de définir une ontologie pour un domaine particulier.
Dataflow
Application dans laquelle la modification de la valeur d'une variable entraîne
automatiquement la réévaluation des variables qui en dépendent (un tableur est un
exemple de dataflow). Les termes dataflow et workflow sont quelquefois assimilés.
DTD
(Document Type Definition) : Document permettant de définir la structure d'un
fichier XML.
EMBOSS
Ensemble de logiciels pour la biologie moléculaire.
FIFO
(First In First Out) File d'attente où les premières données entrées sont les
premières sorties.
Framework
Infrastructure logicielle qui facilite la conception des applications par l'utilisation
de bibliothèques ou de générateurs de programmes. En français "un cadre de
développement".
GRID computing
Architecture de groupes d'ordinateurs reliés entre eux par un réseau étendu pour
exécuter des tâches et pour lesquels les échanges peuvent être lents.
Hématopoïèse
Formation des cellules du sang dans la moelle rouge des os et dans le tissu
lymphoïde.
Homologue
L'homologie est la ressemblance héritée d'un même ancêtre commun. Deux
séquences sont dites homologues si elles ont un ancêtre commun.
Interleukine
(IL) cytokine sécrétée par les lymphocytes, qui active les leucocytes et déclenche
la sécrétion d'interférons.
Kinase
Enzyme capable de fixer un groupement phosphate en présence d'adénosine
triphosphate (ATP).
LSF
Nom d'une application permettant de répartir des processus sur plusieurs machines
hétérogènes.
MyGrid
Projet visant à développer une application de calcul distribué dédiée à la
bioinformatique. Le but est aussi de permettre la recherche de services web, et
l'exécution de workflows utilisant des ressources distribués sur un réseau étendu.
Monocyte
Globule blanc du sang qui passe dans les différents tissus, où il se transforme en
macrophage.
Ontologie
Une ontologie est un catalogue sémantique, dont les descriptions sont à la fois
concises, non ambiguës, et qui se doit d'être exploitable par un logiciel (description
formelle) comme par un opérateur humain (description littéraire) [src.
www.infobiogen.fr]
Orthologue
Gènes orthologues : gènes d'espèces différentes dont les séquences sont
homologues, la divergence faisant suite à une spéciation. S'il s'agit d'une évolution
après duplication au sein d'un individu on parlera de paralogue.
PBS
Nom d'une application permettant de répartir des processus sur plusieurs machines
proxy web
Ordinateur qui s'intercale entre deux réseaux et qui permet principalement
d'enregistrer dans un cache les pages web couramment utilisées pour ensuite les
délivrer sans qu'il soit nécessaire de se connecter sur le serveur initial.
RDF
(Resource Description Framework) Modèle et description de syntaxe en vue de
l'utilisation de méta données sur le web. Son objectif est de faciliter le traitement
automatisé des informations contenues sur le web en permettant leur description
ans ambiguïté.
service web
Application disponible sur le web et accessible par une interface standardisée. Elle
peut interagir avec d'autres services web indépendamment du système
d'exploitation et des langages de programmation utilisés.
SGE
(Sun GridEngine) Nom d'une application permettant de faire du calcul distribué.
SOAP
(Simple Object Access Protocol) Protocole de communication inter applicatif, au
dessus de HTTP, comportant un ensemble de règles pour structurer des messages
(XML) et invoquer un service web.
Soaplab
Ensemble de services Web permettant l'accès à des applications, essentiel ement
d'analyse de données. Intègre plus particulièrement la suite EMBOSS en proposant
une définition XML des fichiers ACD. Il intègre aussi un service d'enregistrement
et de recherche de services web.
Thread
Portion de programme pouvant s'exécuter en parallèle d'autres portions.
UDDI
(Universal Description Discovery and Integration) Norme permettant de créer et
de retrouver des services web. Un annuaire UDDI est un annuaire en ligne, basé
sur la norme UDDI, référençant un ensemble de services Web disponibles.
workflow
Application qui permet de séquencer des tâches suivant un modèle qui définit en
particulier comment ces tâches sont synchronisées. Voir aussi dataflow.
WSDL
(Web Service Description Language) Langage basé sur XML permettant la
description de l'interface d'un service web.
XML
(eXtensible Markup Languages)
XQuery
Langage pour interroger et manipuler les données d'un document XML.
XSLT
(eXtensible Style Language Transformation) langage permettant de transformer un
document XML en un nouveau document XML ayant une structure (et
éventuellement une DTD) différente.
Sommaire
Remerciements.................................................................................................................................................................. 4
Glossaire............................................................................................................................................................................. 5
1.Introduction.................................................................................................................................................................. 11
1.1 Encadrement et environnement........................................................................................................................ 11
1.2 Problématique du sujet...................................................................................................................................... 11
2.Nos besoins et les solutions existantes................................................................................................................. 12
2.1 Définition des besoins....................................................................................................................................... 12
2.2 Les solutions disponibles.................................................................................................................................. 12
2.2.1 Les solutions non graphiques................................................................................................................... 13
2.2.1.1 G-Pipe................................................................................................................................................... 13
2.2.1.2 Biopipe................................................................................................................................................. 13
2.2.2 Les solutions graphiques......................................................................................................................... 14
2.2.2.1 Taverna............................................................................................................................................... 14
2.2.2.2 ViPEr................................................................................................................................................... 16
2.2.2.3 PipeLine Pilot et VIBE.................................................................................................................... 16
2.2.2.4 Wildfire............................................................................................................................................. 18
2.2.2.5 Ptolemy II......................................................................................................................................... 18
2.2.2.6 Kepler................................................................................................................................................ 20
2.3 Choix de Ptolemy II avantages et inconvénients..................................................................................... 21
3.Fonctionnalités développées.................................................................................................................................... 23
3.1 Contraintes, outils et méthodologie.............................................................................................................. 23
3.2 Applications locales.......................................................................................................................................... 23
3.3 Checkpoint.......................................................................................................................................................... 25
3.4 Mobyle et le web............................................................................................................................................... 25
3.5 Services Web..................................................................................................................................................... 29
3.6 Architecture...................................................................................................................................................... 29
4.Application à la reconnaissance de l'IL-2.............................................................................................................. 31
4.1 Introduction : L'IL2 et ses récepteurs......................................................................................................... 31
4.2 Importance médicale de l'IL2, et du développement d'agonistes et antagonistes de l'IL2R.......... 33
4.3 Recherche en cours au laboratoire d'Immunogénétique Cellulaire (IGC).............................................33
4.4 Approche bioinformatique...............................................................................................................................34
4.4.1 Rechercher les séquences orthologues à chaque cytokine et chacune des chaînes de leurs
récepteurs............................................................................................................................................................ 35
4.4.2 Produire des alignements multiples afin de préparer des reconstructions des modèles
structuraux.......................................................................................................................................................... 36
4.4.3 Production des profils propres aux domaines encodés par ces famil es..................................... 38
4.4.4 Produire des arbres phylogénétiques.................................................................................................. 38
5.Conclusion - futur et réflexions............................................................................................................................. 39
5.1 Typage des données et services web............................................................................................................ 39
5.2 Exécutions réparties........................................................................................................................................ 40
5.3 Briques internes................................................................................................................................................ 40
5.4 Ergonomie et "utilisabilité"............................................................................................................................. 41
5.5 Intégration des autres projets..................................................................................................................... 43
Bibliographie................................................................................................................................................................... 44
Références Web............................................................................................................................................................ 46
Annexe A - Cahier des charges............................................................................................................................. 47
Annexe B - Diagramme statique............................................................................................................................ 55
Annexe C - Diagramme statique : MobyleApplication...................................................................................... 56
Annexe D - DTD de Mobyle................................................................................................................................... 57
Table des figures
Fig. 1 G-Pipe.......................................................................................................................................................... 13
Fig. 2 Taverna, exécution de BLAST.............................................................................................................. 15
Fig. 3 Taverna : Représentation minimale pour envoyer un mail............................................................... 15
Fig. 4 ViPEr. Visualisation d'une molécule..................................................................................................... 16
Fig. 5 PipeLine Pilot............................................................................................................................................. 17
Fig. 6 VIBE........................................................................................................................................................... 17
Fig. 7 Wildfire..................................................................................................................................................... 18
Fig. 8 Ptolemy II................................................................................................................................................. 20
Fig. 9 Kepler (SDM/SPA).................................................................................................................................. 21
Fig. 10 Configuration d'une brique "blastall"............................................................................................... 24
Fig. 11 Configuration d'une brique emacs...................................................................................................... 24
Fig. 12 Exécution d'un programme de visualisation à partir d'un"CheckPoint"....................................25
Fig. 13 MobyleApplication : Configuration.................................................................................................... 26
Fig. 14 MobyleApplication : Fenêtre de configuration et menus disponibles........................................27
Fig. 15 MobyleApplication : exécution........................................................................................................... 28
Fig. 16 Configuration et exécution d'un service web................................................................................. 29
Fig. 17 Quand et par quelles cellules est produite et sécrétée l'IL-2 ?................................................31
Fig. 18 Quels sont les récepteurs actifs de l'IL-2 ?.................................................................................. 31
Fig. 19 Comment agit l'IL-2 par la voie Jak-Stat ?.................................................................................... 32
Fig. 20 Quels sont les rôles de l'IL-2 ?........................................................................................................ 32
Fig. 21 Quels usages thérapeutiques pour l'IL-2 ?.....................................................................................33
Fig. 22 Modèles de la transition de conformation...................................................................................... 34
Fig. 23 Spécificité de p1-30 par rapport à l'IL-2 ?................................................................................... 34
Fig. 24 Blast en fonction de e pour l'IL-2.................................................................................................... 35
Fig. 25 Nombre de séquences en fonction des puissances de e...............................................................36
Fig. 26 cluscore et hmmsearch....................................................................................................................... 37
Fig. 27 Résultats de hmmsearch..................................................................................................................... 37
Fig. 28 Cosa. Niveau de conservation des résidus...................................................................................... 38
Fig. 29 Affichage complet et l'exécution correspondante....................................................................... 41
Fig. 30 Représentation des composants et exemple d'enchaînement repris de [r13]........................42
1.Introduction
1.1 Encadrement et environnement
Le projet s'est déroulé à l'institut PASTEUR de Paris et a été initié par Thierry ROSE de l'unité
d'Immunogénétique Cel ulaire. Cette unité développe trois grands axes de recherche :
· le rôle de l'interleukine-2 (IL-2) dans le contrôle de l'amplitude des réponses T et le maintien de
l'homéostasie;
· la caractérisation des voies de signalisation de l'IL-2 et de mimétiques de l'IL-2 humaine
développées à des fins thérapeutiques (cancers rénaux, mélanomes, infections VIH).
· l'impact de la dérégulation du système IL-2/RIL-2 sur l'immunodépression induite au cours de
l'infection par le VIH.
J'ai aussi été encadré par Catherine LETONDAL du Groupe Logiciels et Banques de données qui a
pour mission de fournir aux chercheurs de l'institut Pasteur les infrastructures, les logiciels et les
banques de données nécessaires à une recherche de pointe en biologie moléculaire, y compris la
génomique et la biologie structurale.
1.2 Problématique du sujet
Les outils informatiques utilisés voire développés par les biologistes sont de nature très diverses, il
peut s'agir entre autres d'applications locales, de serveurs web ou de scripts personnels. Pour répondre
à ces besoins il leur est souvent nécessaire d'enchaîner ces outils manuellement. Ce travail devient
fastidieux lorsqu'il s'agit de le répéter pour ne modifier que quelques paramètres par exemple.
L'intérêt de ce travail est donc de pouvoir utiliser des programmes de bioinformatique, de faciliter
leurs interactions et de rendre possible leur enchaînement tout en visualisant de manière graphique
l'architecture, le flux de données et l'exécution.
En outre cela permettra de valider des workflows réalisés par des experts et de les rendre
accessibles à des néophytes, ou aux experts de disposer d'un atelier de développement et
d'exploitation d'outils de bioinformatique de manière plus visuelle et plus souple autorisant des
approches classiques ou plus intuitives.
Enfin, une attention particulière doit être portée aux technologies émergentes comme les services
web et les formats d'échanges de données XML ainsi que le respect des ontologies.
Nous avons pour cela tout d'abord décrit nos besoins et vérifié qu'il n'existait pas de solution qui
pouvait satisfaire à notre cahier des charges. Celui-ci est important pour la compréhension de ce
document, il est détaillé en annexe A. Nous décrirons ensuite les composants que nous avons développés
et donnerons quelques exemples dans le cadre des études sur l'IL-2.
2.Nos besoins et les solutions existantes
2.1 Définition des besoins
La première partie du stage a consisté entre autres à préciser les fonctionnalités de l'application.
Les fonctions principales ainsi que les éléments graphiques ont été définis par Thierry ROSE. Ces
éléments ont évolué au cours de nos différentes réunions et des outils étudiés.
Afin de formaliser quelque peu les idées générées nous avons rédigé un cahier des charges de
l'application (cf. Annexe A). Ce document n'est pas à proprement parler un document de spécifications
fonctionnelles, certaines fonctions n'étant pour le moment que des résultats de discussions ou d'idées
générales à creuser, son rôle est d'une part de formaliser les fonctions retenues et d'autre part de
consigner les réflexions, analyses ou souhaits qui sont apparus au cours de ce stage. Comme nous le
verrons par la suite, il est aussi indispensable pour avoir une vision de l'avancement du projet.
Nous pouvons, sans rentrer dans les détails, retenir les besoins principaux suivants :
· Pouvoir glisser et déposer des applications/données/contrôles dans un environnement de travail.· Les relier entre elles pour définir le workflow.
· Exécuter des workflows séquentiels ou concurrents.· Suivre l'état de l'exécution du workflow.
· Pouvoir comparer des résultats entre plusieurs exécutions.· Permettre à l'utilisateur de rajouter simplement de nouvelles applications (intégration de briques
PISE [w11] par exemple).
· Définir des opérateurs de comparaison, des boucles.
· Pouvoir faire des exécutions pas à pas, ajouter des points d'arrêt, faire des reprises sur erreur.· Exécuter partiellement un workflow.
· Vérifier la compatibilité des données, les rendre compatibles par l'ajout d'adaptateurs.· Intégrer des services web.
· Permettre l'exécution en arrière plan sans interface graphique mais pouvoir ensuite visualiser à
chaque moment l'état du workflow et le modifier.
· Avoir des affichages différents en fonction des niveaux de représentation désirés.
L'idée maîtresse de cette application étant de permettre à l'utilisateur de concrétiser rapidement
un processus de pensée, de modifier rapidement le workflow et de vérifier la validité de ses
hypothèses.
La simplicité d'utilisation et la clarté du graphisme sont aussi des éléments majeurs dans la mesure
où cette application pourrait être utilisée comme outil pédagogique et les workflows résultants partagés
voire publiés.
2.2 Les solutions disponibles
Nous avons étudié les solutions existantes dans le domaine des workflows afin, soit d'utiliser une
application qui répondait majoritairement à nos besoins, soit d'adapter une solution existante à nos
propres desiderata et ainsi éviter de définir entièrement une architecture.
Nous nous sommes intéressés prioritairement aux applications à manipulation directe, c'est à dire
celles dont l'interface permettait de créer et exécuter un workflow graphiquement par glisser/déposer
de briques (traitements, données ou contrôles); nous utiliserons le terme "application graphique" pour la
suite. Nous donnerons cependant auparavant un aperçu des autres projets.
Les vocabulaire pour désigner les tâches à exécuter est différent d'une application à l'autre
(processeur, acteur, tâche, composant, etc.) nous utiliserons le terme générique de "brique" dans les
paragraphes suivants pour désigner une tâche qui agit sur les données qu'elle accepte en entrée.
2.2.1 Les solutions non graphiques
Bien qu'elles ne répondent pas à nos attentes en terme d'interactions, nous citerons ici quelques
applications. La liste n'est pas exhaustive mais mettent en évidence certaines problématiques qui se
rapprochent des nôtres.
2.2.1.1 G-Pipe
G-Pipe [w24] a été créé par Alexander Garcia et Samuel Thoraval. C'est un outil permettant de
créer des pipelines en chaînant des applications disponibles à partir d'un formulaire web créé par le
générateur d'interfaces Pise [w11]. Le pipeline est sauvé dans un fichier XML dont la DTD est une
évolution de la DTD de Pise.
Il met à profit des fonctions comme la conversion automatique des données entrées par l'utilisateur
et la détermination des applications qui peuvent traiter un type de résultat particulier.
Fig. 1 G-Pipe
Le menu en haut à gauche liste
les applications disponibles. Le
menu en bas à gauche permetde définir un ou des workflows
et de suivre leur exécution.
La partie droite affiche la pagede configuration d'une
application (telle qu'elle
apparaît pour Pise) ou desinformations sur les tâches
lancées.
2.2.1.2 Biopipe
Biopipe[r12] a été développé par plusieurs organismes de Singapour (Institute of Molecular and Cell
Biology; Genome Insitute of Singapore et Temasek Life Sciences Laboratory). Sa motivation est de
faciliter l'interaction entre des applications et des données hétérogènes en proposant un cadre de
développement (framework) écrit en Perl. Le workflow est défini par un fichier XML mais sauvé dans
une base de donnée pour être exécuté par un "PipelineManager". Cette base reflète également l'état de
l'exécution à tout moment et permet ainsi de la reprendre en cas d'arrêt inopiné dû à une erreur
matérielle ou logicielle.
Un effort a également été porté pour abstraire l'utilisateur des différents formats de fichiers (en
utilisant une représentation sous forme d'objets Bioperl) et des différentes interfaces des
programmes (par la définition d'interfaces communes (wrappers)). Enfin ce projet permet de répartir
les tâches sur un cluster par l'intermédiaire d'outils comme PBS [w21] ou LSF [w22] .
2.2.2 Les solutions graphiques
2.2.2.1 Taverna
Taverna [w6] n'est pas à proprement parler un workflow graphique à manipulation directe dans la
mesure où le workflow se construit à partir d'un menu en arbre et une représentation du résultat est
reconstruite automatiquement sous forme graphique. Il est le résultat d'une collaboration entre
l'European Bioinformatics Institute (EBI), IT Innovation, the Rosalind Franklin Centre for Genomic
Research (RFCGR), Newcastle Computer Science Faculty, Newcastle Centre for Life, Manchester
Computer Science Faculty et le Nottingham University Mixed Reality Lab.
Les briques ("processors") qui constituent les workflows sont choisies manuel ement mais il est prévu
à terme d'intégrer des mécanismes de découverte de service (UDDI, BioMOBY, etc.). Elles peuvent
être de 4 types :
· des services web
· des services Soaplab [r8], un ensemble de services web associé à des outils de recherche de service.· des scripts Talisman [w33], permettant de facilement interagir avec des bases de données, des
ressources diverses et d'autres workflows.
· des workflows Taverna.
Parmi les nombreuses fonctionnalités on peut noter :
· un mécanisme de synchronisation entre des applications qui n'échangent pas de données entre elles
(auquel cas la synchronisation se fait naturellement);
· la définition d'applications de substitution ou des reprises du workflow en cas d'erreur sur
l'exécution d'une application (paramétrables en fonction du nombre de tentatives voulues et de la
durée entre ces tentatives);
· l'itération du workflow sur une liste de données avec dans le cas de plusieurs entrées, une possibilité
de jointure sur ces entrées.
Ce workflow est basé sur le langage SCUFL (Simple Conceptual Unified Flow Language) représentable
par un fichier XML (Xscufl). Ce format a l'avantage de pouvoir être exécuté par les workflows deMyGRID [w4] (à l'exception de fonctionnalités de coordination entre plusieurs applications).
Les types de données ne sont pas encore pris en compte, les auteurs ont retenu de ne les considérer
que comme des types MIME éventuellement regroupées en listes.
Ce workflow est sous licence LGPL et codé en java. La version 0.1.beta.10 fut testée sur quelques
exemples fournis. La prise en main est relativement aisée, malheureusement l'ajout d'applications passe
forcément par la définition d'un fichier de description ce qui est handicapant pour construire
rapidement un workflow surtout lorsqu'il s'agit d'opérations simples. De plus l'édition et la
représentation sont relativement lourdes. L'exemple de la figure 3 montre par exemple un workflow
minimal pour envoyer un mail !
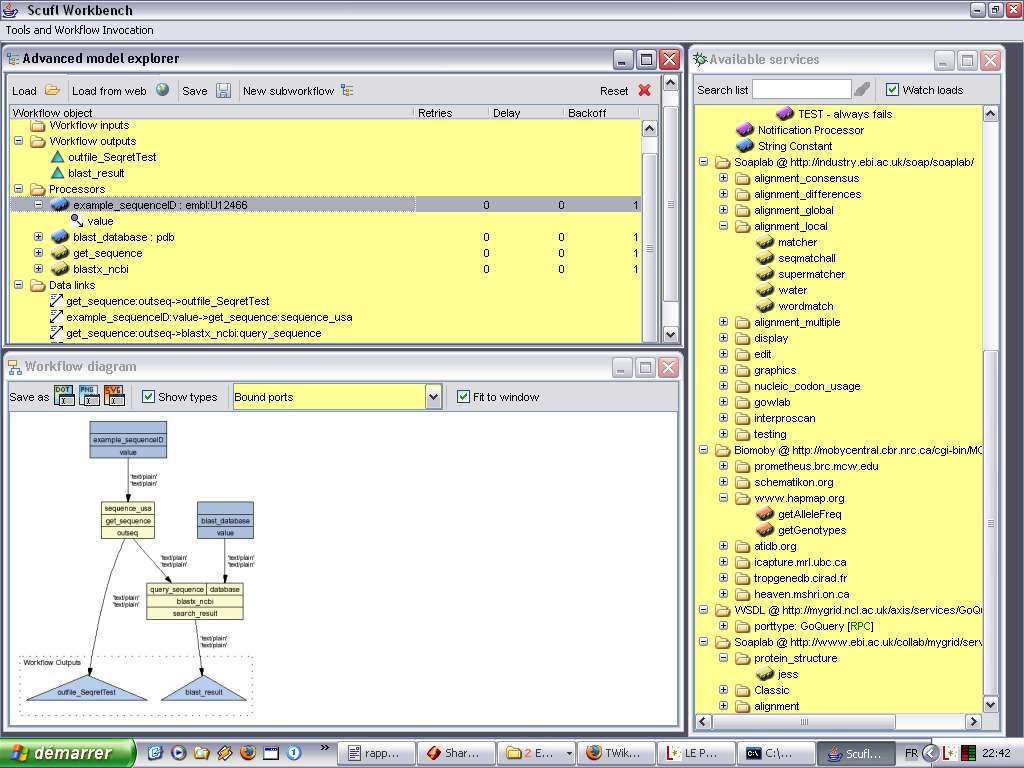
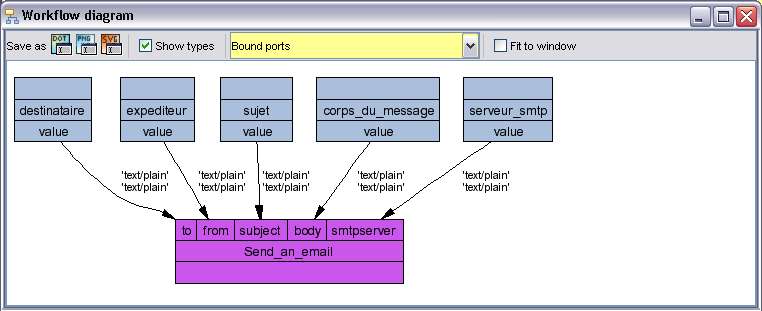
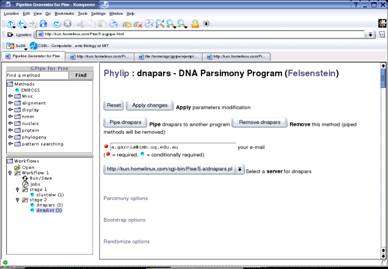
Fig. 2 Taverna, exécution de BLAST.
Ce workflow permet d'exécuter BLAST pour une séquence et une banque particulières. La fenêtre de droitereprésente les briques (processors) disponibles, retrouvées ici des environnements Soaplab et BioMOBY. La fenêtredu haut permet de créer le workflow à partir du choix des briques. L'affichage graphique est rafraîchi après chaquemodification. L'exécution (par le menu Tools and Workflow Invocation) permet de visualiser les tâches en cours et lesrésultats intermédiaires
Fig. 3 Taverna : Représentation minimale pour envoyer un mail.
2.2.2.2 ViPEr
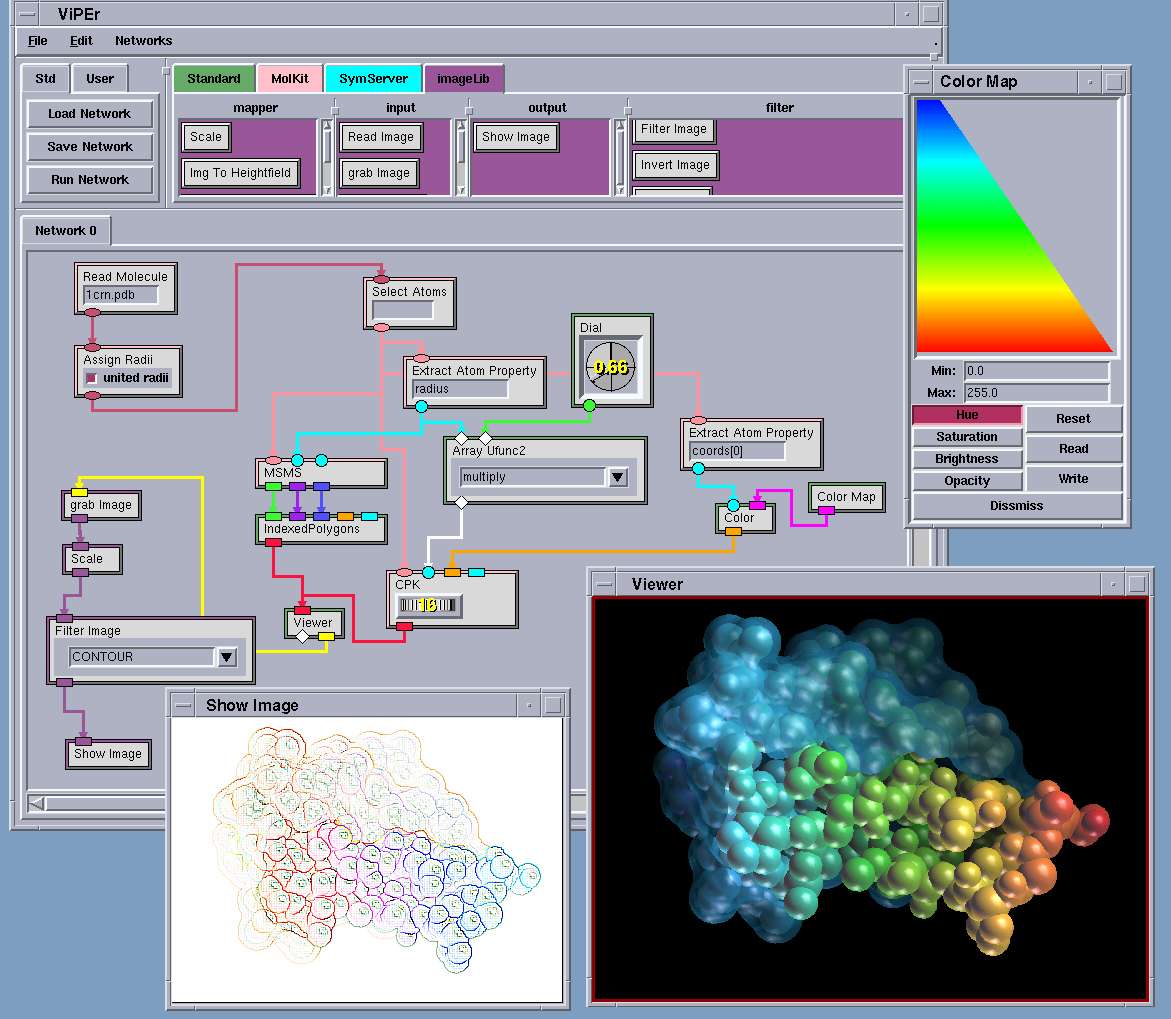
ViPEr (Scripps Research Institute, San Diego)[r9] est un environnement de programmation visuel
écrit en Python et Tkinter (librairie graphique). Il permet de définir graphiquement une série de
transformations appliquées à des données et de visualiser le ou les résultats, les tâches et modules de
visualisation disponibles sont essentiellement destinés à la biologie structurale. Il a la particularité de
ne pas se baser sur un mode 'exécution' mais de déclencher une brique dès qu'une donnée en entrée a
évolué et ainsi de propager les résultats et les exécutions, on a donc plus ici une application de dataflow.
De nouvelles briques écrites en Python peuvent être ajoutées aux librairies existantes.
Les workflows résultants sont sauvés sous la forme de code Python et peuvent ainsi être intégrés
dans d'autres applications.
Fig. 4 ViPEr. Visualisation d'une molécule.
2.2.2.3 PipeLine Pilot et VIBE
PipeLine Pilot de la société Scitegic (San Diego) [w7] et VIBE de la société Incogen (Wil iamsburg)
[w8] sont deux produits commerciaux que nous n'avons malheureusement pas pu tester car aucune
version de démonstration n'était facilement disponible. Pipeline Pilot a été conçu plus spécifiquement
pour l'industrie pharmaceutique alors que VIBE est proposé avec des applications d'analyse de
séquence.
Ils offrent tous deux la possibilité, entre autres, de construite des workflows graphiquement, de
les sauver sous un format XML, de suivre l'état de l'exécution et d'ajouter des composants
supplémentaires.
Nous avons relevé des fonctions intéressantes à intégrer dans notre application ou au moins à
prendre en compte comme fonctionnalités potentielles :
· Pipeline Pilot permet, après la définition d'un workflow, de l'exécuter à partir d'un formulaire web.
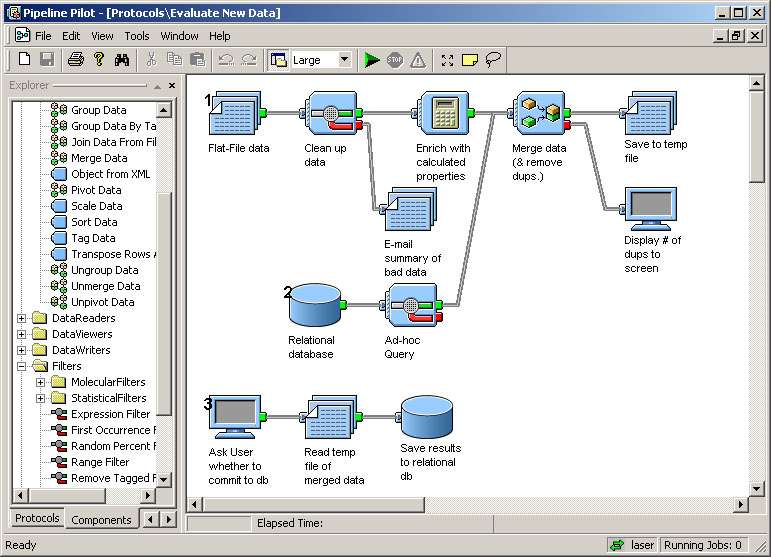
· Pipeline Pilot permet de définir des briques à partir d'un langage d'expressions.· VIBE propose une aide en ligne pour chaque composant et permet à l'utilisateur de rajouter ses
commentaires.
· VIBE propose plusieurs espaces de travail
· Avant une exécution, VIBE vérifie la validité et la compatibilité des données échangées ainsi que
l'initialisation des paramètres.
Fig. 5 PipeLine Pilot
Le panel de gauche liste les briques
disponibles par catégorie.
Fig. 6 VIBE
Les icônes représentent les
briques disponibles pour
construire le workflow. Le panelde droite permet de changer les
paramètres d'une brique,
d'accéder à son aide ou derajouter des commentaires.
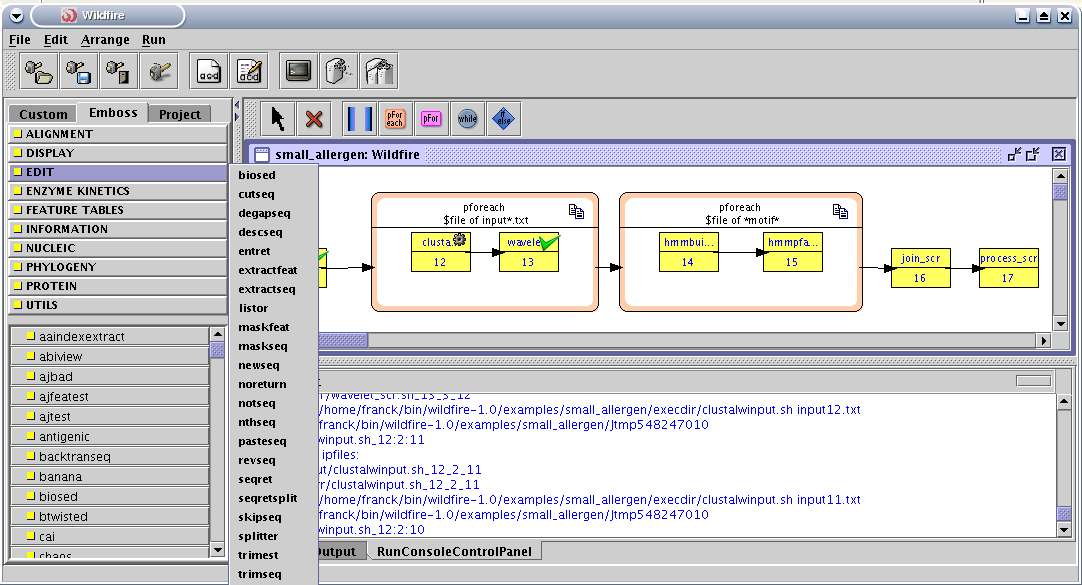
2.2.2.4 Wildfire
Wildfire [w18] est développé par l'A-STAR (Agency for Science Technology and Research à
Singapour). Il se différencie des autres projets par une définition des workflows plus proche de la
programmation procédurale (utilisation de boucles et de conditions). Il est fortement lié à la suite
EMBOSS dans la mesure où il propose comme briques de base les programmes de cette bibliothèque et
où la définition d'une nouvelle brique correspondant à un programme local se fait par l'intermédiaire de
la définition d'un fichier ACD. Une partie de l'application utilise d'ailleurs JEMBOSS, l'interface
graphique écrite en java pour les outils EMBOSS.
L'exécution concurrente des programmes s'appuie sur le langage GEL [r10], pour lequel a collaboré
l'auteur de Wildfire. Ce langage de script permet d'utiliser différentes technologies de distribution
de tâches comme SMP, les clusters (PBS, SGE, LSF) ou le "grid computing". L'approche retenue ici est
donc d'utiliser directement les applications telles quelles sans redéfinir d'interface web ou de service
web et de travailler sur des outils de répartition de charge, ce qui n'est pas étonnant si l'on considère
que c'est le domaine de recherche des auteurs.
L'inconvénient de cette plateforme est que les communications inter tâches se font exclusivement
par échange de fichiers.
Fig. 7 Wildfire
Le panel de gauche liste les applications EMBOSS disponibles. L'exécution peut être suivie sur l'affichage du workflowet par une console.
2.2.2.5 Ptolemy II
Ptolemy II [w19] est développé par une équipe du département EECS (Electrical Engineering and
Computer Sciences) de l'université Berkeley en Californie. Il est le résultat de précédents
environnements et recherches créés dès 1986 et notamment destinés à la conception de systèmes
embarqués. Cet environnement n'est pas spécifique à un domaine, il est principalement utilisé par ces
équipes pour implémenter et expérimenter divers techniques et travaux de recherche comme la
définition de modèles d'ordonnancement (la façon d'exécuter les tâches dans un workflow), la
génération de code à partir d'un modèle ou encore la conception objet.
Ainsi, aucune chance d'y trouver une brique de bioinformatique. Cependant, la librairie définie
dispense de l'écriture de briques quasi indispensables comme l'affichage des données sous plusieurs
formes, les fonctions sur les chaînes de caractères, les opérateurs de calcul ou encore la lecture de
fichiers. Étonnamment, c'est le seul environnement à offrir un tel panel. Ci-dessous nous listons
quelques-unes des fonctions offertes pour donner une idée de leur variété :
· Affichage de données sous la forme textuelle, en 2 dimensions et en diagrammes en barres.· Opération sur les tableaux.
· Conversion entre types de données.
· Lecture et écriture de données à partir de fichiers ou de datagrammes IP.· Opérations mathématiques complexes (différentiation, intégration, etc.), opérations sur les
matrices.
· Lecture, visualisation, transformation et filtres sur les images.
· Évaluation d'expressions issues du logiciel Matlab (a priori utile, voir par exemple [w20])· Possibilité d'intégrer des briques écrites en Python (utilisation de la librairie Jython qui
implémente en Java le langage Python).
En dehors de ces briques l'environnement offre des fonctions intéressantes comme :
· La possibilité de définir des paramètres utilisables dans l'environnement de travail et par
chaque brique.
· Un langage qui permet d'évaluer des expressions algébriques dont les termes peuvent être
des paramètres par exemple.
· La définition de types (entiers, flottants, chaînes de caractères, matrices) pour les
paramètres et les ports d'entrée et de sortie des briques.
· La possibilité de définir un workflow comme une nouvelle brique et de la sauvegarder dans
une librairie.
· La sauvegarde des workflows sous forme XML.
· La possibilité d'exécuter un workflow sans interface graphique.· La possibilité d'exécuter un workflow dans une applet java.
Enfin, il offre plusieurs modèles d'ordonnancement (models of computation) qui définissent la
manière dont interagissent les briques en fonction des données qu'el es reçoivent ou émettent, du
temps (continu ou discret) et de la manière de gérer la concurrence au sein du workflow. Un composant
appelé 'director' permet de définir le modèle pour un workflow donné.
Par exemple, dans le modèle PN (Process Network) les briques sont des tâches indépendantes dont
les ports d'entrées sont des canaux FIFO. La brique traite les données en entrée et envoie
éventuellement des résultats sur des ports de sortie. Elle se bloque dès que le canal est vide en
attendant de nouvelles données. Le modèle DE (Discret Event) quant à lui modélise un système pour
lequel la communication entre les briques est séquencée par des événements discrets au cours du
temps.
L'avantage ici n'est pas tant ce choix de modèles, la difficulté étant surtout de les comprendre et
d'en choisir un qui convienne, mais la possibilité d'en créer un qui nous permette de satisfaire notre
cahier des charges pour par exemple autoriser l'exécution partielle d'un workflow ou rajouter des
points d'arrêt.
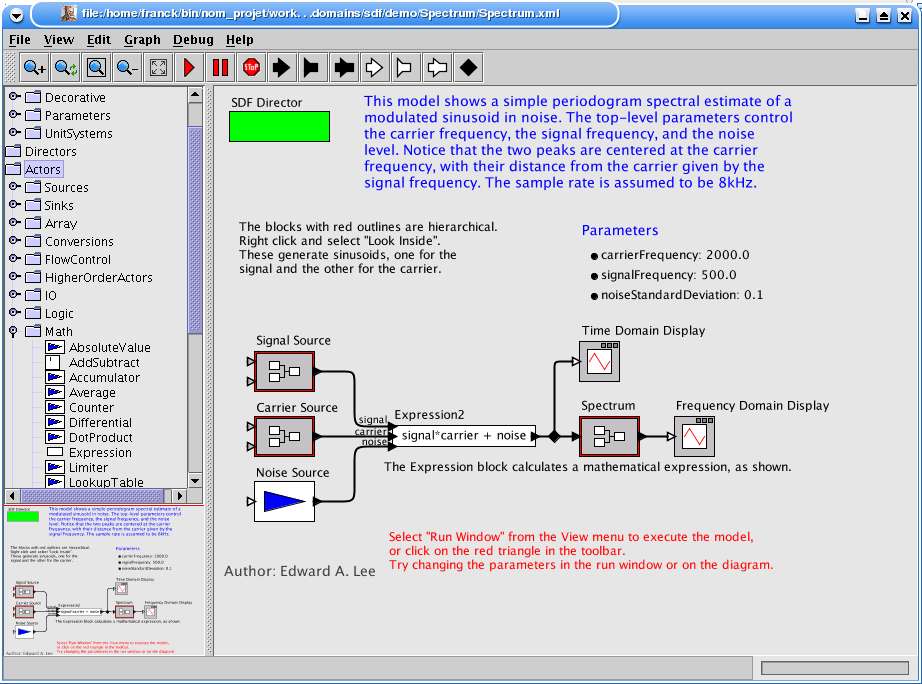
Fig. 8 Ptolemy II
Le panel de gauche liste les briques (acteurs) disponibles ainsi que d'autres types d'objets utilisables (paramètres,commentaires, directeurs, etc). Le panel en bas à gauche donne une vue d'ensemble du workflow. L'espace de travail àdroite donne un aperçu des possibilités : la définition de paramètres, l'ajout de commentaires, l'inclusion d'autresworkflows ou l'utilisation d'expressions algébriques.
2.2.2.6 Kepler
Le projet Kepler [w16] est issu de la collaboration de plusieurs organismes de disciplines
scientifiques différentes tels que SEEK (Science Environment for Ecological Knowledge), SDM
Center/SPA (Scientific Data Management Center/Scientific Process Automation), GEON
(Cyberinfrastructure for the Geosciences), ROADNet (Real-time Observatories, Applications, and Data
Management Network). Son but est de faciliter aux scientifiques de ces différentes disciplines la
création, l'exécution et le partage de workflows scientifiques.
Il utilise la plateforme Ptolemy II à laquelle il ajoute des fonctionnalités sous la forme de briques
supplémentaires [r7] qui peuvent se classer ainsi :
· les services web, soit sous forme générique, soit associés à des services particuliers (Blast,
ClustalW, etc.). Une brique particulière permet aussi de retrouver les services disponibles à
partir d'une url ou d'un annuaire UDDI.
· l'accès à des bases de données.
· les briques de transformation (XSLT, Xquery, Perl, etc.).
Nous n'avons malheureusement pu tester certaines des fonctions listées ci-dessus que tardivement.
En effet, le projet est actuellement en développement (version 1 alpha en juin 2004) et certaines
briques sont encore "ésotériques" faute de documentation.
La partie développée par SDM/SPA [w25] est spécifique à la bioinformatique, nous verrons dans les
chapitres suivants les développements que nous avons pu reprendre.
Fig. 9 Kepler (SDM/SPA)
Cet exemple repris de [w16] permet d'identifier les sites potentiels de fixation des facteurs de transcriptionpour une série de gènes. Les briques sont essentiellement des services web. Certaines briques sontspécifiques à ce workflow et permettent essentiellement une synchronisation entre les tâches. On peutremarquer l'utilisation de briques composites (constituées elles-mêmes de workflows).
2.3 Choix de Ptolemy II avantages et inconvénients
L'étude des différentes solutions nous a montré qu'aucune ne satisfaisait les fonctions principales
de notre cahier des charges. Il était raisonnablement impossible de créer une application de zéro qui
soit, même en partie, rapidement fonctionnelle.
Nous avons donc considéré Ptolemy II et Kepler qui étaient les deux candidats qui s'approchaient le
plus de nos desiderata. Bien que Kepler puisse sembler le plus approprié, il intègre en effet déjà des
briques bioinformatiques, nous avons choisi Ptolemy II pour les raisons suivantes :
· Kepler n'est pas spécifique à la bioinformatique, l'espace de travail regroupe différents domaines
qui nous sont inutiles et nous ne pouvions anticiper les axes de développements des prochaines
versions (modification de l'espace de travail, architecture). Ce choix nous a donné raison par la
suite car la dernière version ne prend pas encore en compte les dernières évolutions de Ptolemy
II.
· Les briques d'intérêt développées, essentiellement des services web, sont facilement
réutilisables seules.
· Ptolemy II est une application aboutie et pérenne. L'architecture est claire (relativement au
nombre de classes et de fonctionnalités présentes !), il existe une documentation détaillée et à
jour [r1][r2][r3] qui regroupe l'architecture logicielle (diagrammes statiques) et les
fonctionnalités.
Paradoxalement cette richesse est aussi un inconvénient. L'application est conséquente (plus de 1300
classes) et les concepts nombreux. Ainsi, un effort important doit être fait pour appréhender cet
environnement avant de développer de nouvelles applications.
3.Fonctionnalités développées
3.1 Contraintes, outils et méthodologie
Le choix des fonctions développées en priorité a été dicté par les projets de Perrine Barjou1 et de
Vldimir Dric2 qui nécessitient de disposer rpidement d'un environnement pour enchaîner des tâches.
Nous avons donc privilégié les premières briques nécessaires pour ces projets qui nous permettaient de
plus de valider notre approche. Les plus importantes de ces briques sont détaillées dans les paragraphes
suivants.
L'urgence dans les développements nous a amené à ne coder et déboguer que les fonctions
indispensables. Cependnt, afin d'avoir une vision sur l'état du projet nous avons associé à chaque
fonctionnalité un jeu de tests et à chaque test les bogues correspondants (non détaillés dans ce
document !).
Ptolemy II est entièrement écrit en Java. Les environnements et les outils utilisés furent les
suivants :
· Linux 2.6
· Java JDK 1.4.2
· Sources de Ptolemy II 3.0.2 puis mise à jour avec Ptolemy II 4.0.1· Environnement de développement Eclipse [w9].
· Création de diagrammes UML avec Poséidon [w10].
3.2 Applications locales
Les applications locales (commandes en ligne ou programmes avec une interface graphique) peuvent
être exécutées par l'acteur 'CmdLineApplication'. Cette brique permet de définir complètement et de
façon très souple la liste des paramètres d'une application.
Les spécifications sont détaillées en Annexe A, fonction P8 et ne sont pas reprises ici. Les exemples
simples suivants illustrent les possibilités de cette brique :
Exemple 1 : recherche de séquences (similaire à celle d'une requête dans une base de séquences
par la méthode BLAST)
"Je veux exécuter blastall sur un fichier présent sur le port d'entrée 'fic_fasta'. Le fichier de
sortie sera nommé 'blast.out' et sauvé dans un répertoire défini par le paramètre 'dir_resu'. Il sera
aussi envoyé vers le port de sortie 'fic_blast'. La valeur espérée (option -e) sera aussi déterminée par
un port d'entrée."
La ligne de paramètres de blastal sera alors :
-p blastp -i in{fic_fasta} -d nr -m8 -o out{fic_blast}={exp{dir_resu+"blast_out"}} -e in{e}
La figure 10 montre la fenêtre de configuration et la brique résultante. Les ports nouvellement
créés ont été connectés à des briques contenant des valeurs pour les ports d'entrée et à un afficheur
pour le port de sortie.
1 Analyse et prédiction de réseaux d'interaction de protéines.
2 Analyse et prédiction de structures de protéines.
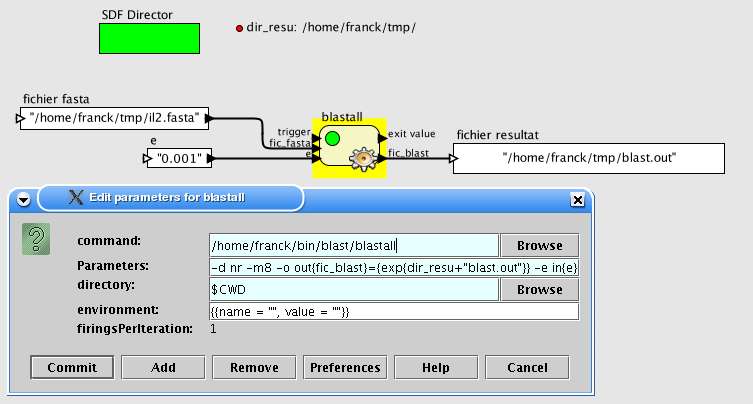
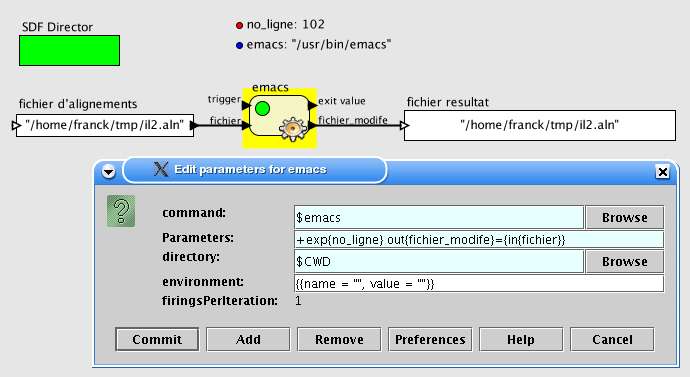
Fig. 10 Configuration d'une brique "blastall"
Exemple 2 : édition de séquences
"Je veux éditer une séquence dans emacs. Un port en entrée contient le fichier à éditer, lorsque
je ferme l'éditeur le fichier modifié est envoyé sur un port de sortie. A l'ouverture de l'éditeur je
veux accéder directement à un numéro de ligne défini par le paramètre 'no_ligne'".
La ligne de paramètres sera alors :
+exp{no_ligne} out{fichier_modifie}={in{fichier}}
La figure 11 montre la fenêtre de configuration et le résultat de l'exécution. Ici la commande est
elle-même définie par un paramètre de l'espace de travail ('$emacs') ce qui, dans le cas de workflows
plus importants, permet de plus facilement partager des workflows et de les adapter pour sa propre
configuration.
Fig. 11 Configuration d'une brique emacs
3.3 Checkpoint
Cette brique est une première implémentation de la fonctionnalité "Sauvegarde et visualisation de
données entre acteurs" (cf. Annexe A, fonction P16). Elle a été conçue pour visualiser le contenu des
fichiers échangés entre les acteurs et ainsi déboguer plus facilement nos premiers workflows. Bien que
cette fonctionnalité soit importante si ce n'est primordiale elle est souvent absente des workflows
étudiés.
Le "CheckPoint" de la figure 12 (brique ronde) est devenu vert lorsque des données lui ont été
présentées, l'utilisateur peut choisir de les visualiser à tout moment pendant ou après l'exécution du
workflow. Le programme de visualisation à utiliser est choisi lors de la configuration de cette brique et
peut être modifié à tout moment.
Fig. 12 Exécution d'un programme de visualisation à partir d'un"CheckPoint".
Les "CheckPoints" peuvent aussi être choisis pour toute application interactive si l'on souhaite que le
workflow ne s'arrête pas sur cette tâche lors de l'exécution.
3.4 Mobyle et le web
Un moyen efficace d'ajouter et de configurer de nouvelles applications est d'utiliser Mobyle [w34],
une évolution de Pise développée par Catherine LETONDAL et Bertrand NERON. PISE [w11] permet à
partir d'une définition en XML de l'interface du programme de générer l'interface de configuration et
d'exécution pour plusieurs types d'affichage (Web, SeqLab, Tcl/Tk et X11). L'interface Web est la plus
répandue, le formulaire HTML créé permet de lancer un programme CGI capable de lancer l'exécution
du programme et de récupérer les résultats.
Mobyle ajoute à la DTD de PISE des évolutions pour l'enchaînement des tâches et la prise en compte
de plusieurs types d'exécution (local, CGI, service web) par exemple (cf. DTD annexe D).
Nous avons donc défini un acteur (cf. annexe A, fonction P7) qui, à partir du formalisme utilisé par
Mobyle, permettrait de configurer et d'exécuter une application que se soit un programme local, un CGI
ou un service web.
La configuration se fait par l'intermédiaire d'un formulaire html, l'utilisateur peut, pour chaque
paramètre, spécifier s'il s'agit d'un port en entrée, d'une expression à évaluer au moment de
l'exécution (opération entre paramètres définis dans l'environnement de Ptolemy II) ou d'une valeur
classique.
Cette configuration est faite par l'intermédiaire d'une page web similaire à celle qui est générée par
Pise. Nous avons de plus étendu la fonction précédente à l'exécution de cgi accessibles par des pages
web et défini ainsi deux urls, la première correspond au fichier XML (url_XML), la seconde à la page
html (url_presentation) de configuration de l'acteur.
Nous avons alors les 3 cas suivants :
url_XML seule : la page html de configuration est générée à partir du fichier XML. A partir de
cette page l'utilisateur spécifie les ports d'entrée, les expressions et les valeurs. Les ports de sortie
sont aussi déterminés à partir du fichier XML.
url_html seule : la page html du site est affichée, l'utilisateur peut naviguer avec les liens
hypertextes pour trouver le formulaire qui correspond au service désiré. Ensuite il spécifie le type des
champs dans ce formulaire. A l'exécution de l'acteur, la requête est envoyée au serveur (de la même
manière que si l'utilisateur appuyait sur 'submit'). La page html retournée par le serveur est envoyée
sur un port de sortie.
url_XML et url_html : le comportement est le même que dans le premier cas à la différence que
l'affichage correspond à url_html. Ceci permet d'offrir une interface similaire à celle d'un site
d'origine (plus simplement qu'à partir de la définition du fichier XML). Cela suppose bien sûr que le
formulaire définit dans url_html soit compatible avec le fichier XML.
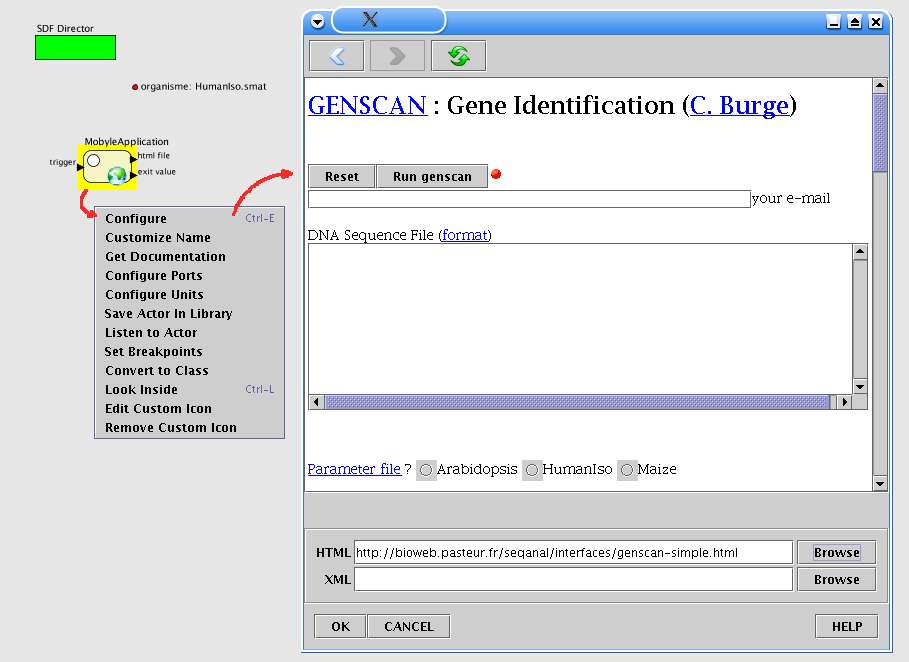
L'exemple ci-dessous il ustre une utilisation avec le logiciel GenScan disponible à partir d'une
interface web générée par Pise. La figure 13 montre l'interface de configuration après que l'utilisateur
ait choisi une url, la page html est alors présentée comme dans un navigateur classique.
Fig. 13 MobyleApplication : Configuration.
L'utilisateur saisie l'url d'une page et peut naviguer de la même manière qu'un navigateur classique.
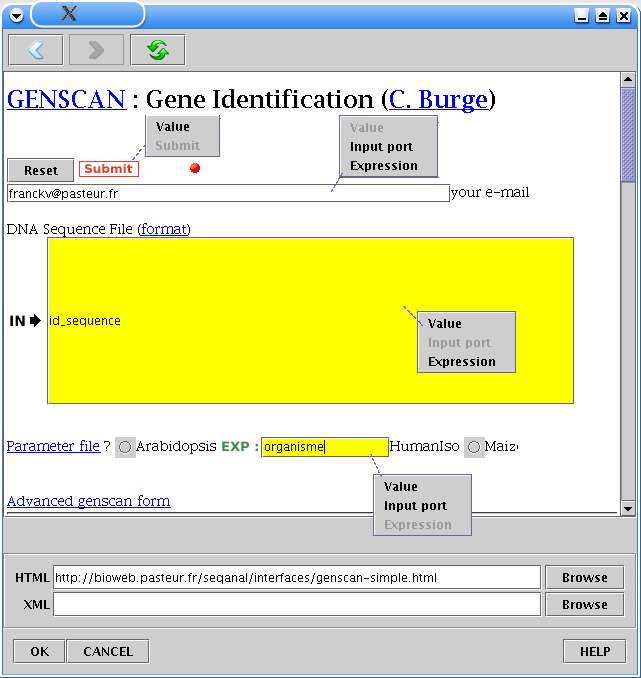
L'utilisateur spécifie ensuite le type de chaque champ. Sur la figure 14, le bouton "Run genscan" est
le bouton de soumission du formulaire, le champ mail reste une valeur classique, la séquence ADN
correspondra au contenu du port d'entrée 'id_sequence' et le fichier de paramètre (boutons radio
"parameter file") au contenu du paramètre 'organisme'.
Fig. 14 MobyleApplication : Fenêtre de configuration et menus disponibles.
Cet exemple montre les menus disponibles pour chaque champ du formulaire. La valeur griséecorrespond au type en cours. La taille d'un champ reste inchangée quel que soit le type pourconserver la mise en page initiale.
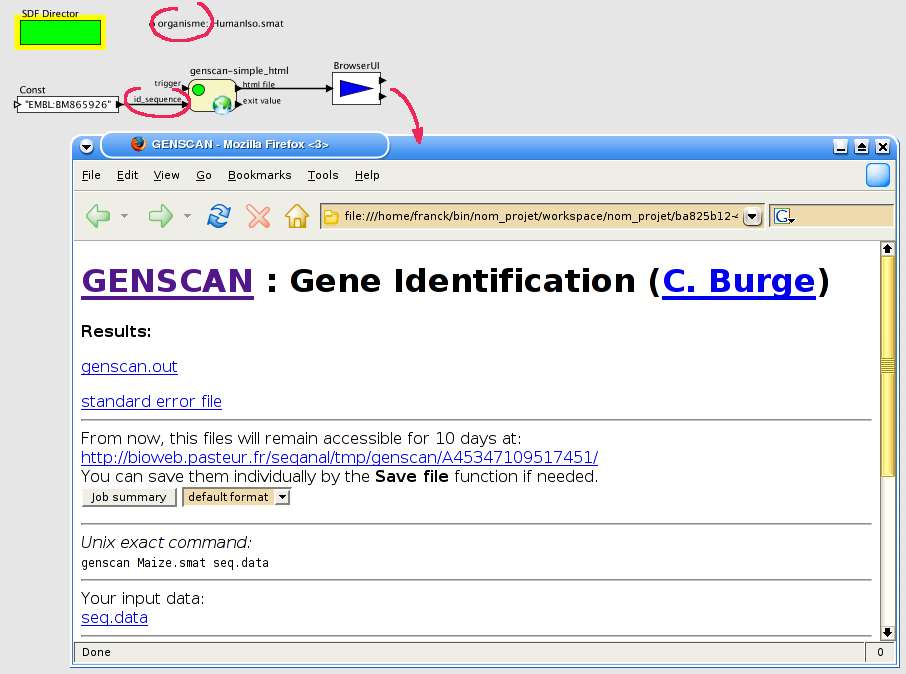
Après la validation de cette configuration, les ports sont ajoutés à l'acteur. La figure 15 montre le
résultat de l'exécution, le fichier html reçu après exécution de la brique est affiché dans un navigateur
externe (brique BrowserUI).
Fig. 15 MobyleApplication : exécution.
Les ports ont été ajoutés à la brique. Après l'exécution, le résultat est affiché dans unnavigateur externe.
Le prototype réalisé nous a permis en partie de valider la faisabilité de notre approche pour
l'intégration de MOBYLE, il reste cependant beaucoup de développements à réaliser comme
transformer un fichier XML pour générer l'interface html (a priori via une transformation XSLT) ainsi
que vérifier la validité des paramètres entrés par l'utilisateur, créer les ports de sortie et prendre en
compte le résultat en fonction du même fichier XML.
Concernant l'intégration de n'importe quel formulaire web, l'obstacle majeur tient dans les
bibliothèques java standards utilisées pour réaliser le navigateur web. Celles-ci ne supportent en effet
que la norme html 3.2 sans javascript et sans frame ce qui très insuffisant pour les nouveaux sites qui
utilisent à foison html 4.0, les frames, CSS1, CSS2, etc. Il faudrait alors se tourner vers des
composants comme xsmiles [w13] ou WebRendered [w14](plus abouti mais payant !)
Enfin, nous n'avons validé l'exécution que pour des formulaires web simples. Il reste encore
beaucoup de sites incompatibles (si ce n'est la majorité !). Un gros travail d'analyse et de conception
reste à faire car en plus de la nécessité de respecter les recommandations W3C pour la syntaxe des
formulaires [w12], de traiter les cas de connexion à travers un proxy et les sites accessibles par mot de
passe il faudra aussi prendre en compte les très nombreux formulaires hétéroclites du web.
Une dernière solution plus simple serait d'attendre que les sites web proposent une interface WSDL
pour leur cgi ! Ceci dit, le projet Gowlab [w15] permet déjà de définir dans un fichier au format ACD
(format utilisé pour la définition des programmes EMBOSS) les champs d'un formulaire web comme
l'url du cgi , le type de requête (POST, GET, etc.) et les différents paramètres. Ce fichier permet de
créer l'interface WSDL et de l'intégrer dans la liste des services web disponibles dans un
environnement Soaplab.
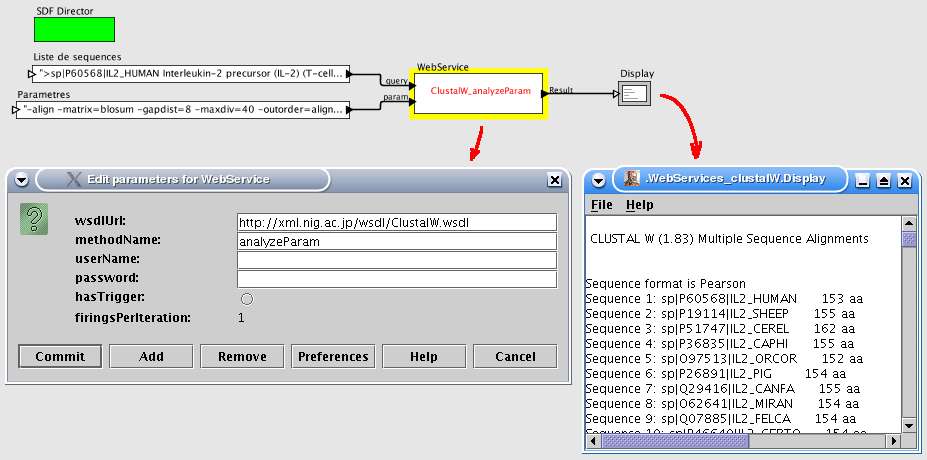
3.5 Services Web
Cette brique est reprise du projet Kepler. La figure 16 montre à la fois la configuration (choix de
l'url, nom de la méthode, utilisateur et mot de passe) et le résultat de la requête affiché dans un
éditeur classique.
Fig. 16 Configuration et exécution d'un service web.
Cet exemple montre la fenêtre de configuration ainsi que la fenêtre d'affichage du résultat (brique de Ptolemy II). Pource service, un port prend en compte la globalité des paramètres sous la forme d'une chaîne de caractères.
3.6 Architecture
Nous n'afficherons pas ici le code du projet (disponible à l'institut Pasteur) mais seulement les
diagrammes statiques UML (des scenarii ont également été créés pour la conception de la brique
'MobyleApplication').
Une partie de l'architecture logicielle est décrite par l'annexe B. Les attributs et les méthodes
privées et protégées ne sont pas représentés pour des raisons de lisibilité. Détail ons les classes
principales pour comprendre la structure générale :
· ExternalApplication : cette classe abstraite permet de définir les attributs et méthodes communes
de nos nouvelles briques (représentation à l'écran, nom de la brique, état de la tâche en cours, etc.).
· LocalApplication : classe permettant de lancer une application à partir de Java et de prendre en
compte les flux d'entrée et de sortie. Une chaîne de caractère représente l'ensemble des
paramètres de l'application.
· CmdLineApplication : classe dérivée de LocalApplication. La ligne de commande de l'application est
maintenant créée lors de l'exécution de la brique après analyse des paramètres entrés par
l'utilisateur. Ces paramètres sont traités par la classe CmdLineParameters.
· CmdLineParameters : liste (composition) d'objets de type CLPNode. CLPNode représente l'interface
pour les noeuds qui définissent un port en entrée (in{nom_du_port}), une expression (exp
{expression}), un port en sortie (out{nom_port}={contenu}) ou une valeur.
· CheckPoint : classe dérivée de LocalApplication (elle permet d'exécuter un application locale de
visualisation). Elle est associée à la classe checkPointControllerFactory qui permet de définir un
menu particulier pour cette brique.
L'annexe C décrit l'architecture utilisée pour la brique 'MobyleApplication'. La majeure partie du
travail a consisté à adapter les classes permettant de gérer et d'afficher les pages html (JeditorPane,
HTMLEditorKit, FormView. Voir aussi [w32]). Nous trouvons en particulier les classes :
· MobyleConfigurer : boite de dialogue qui affiche les pages html et qui gère l'historique.· ExtFormComponent : classe abstraite qui regroupe les méthodes et attributs communs aux classes
ExtJButton, ExtJComboBox, etc. Les instances de ces classes contiennent les widgets utilisés à la
place des widgets classiques d'une page html (boutons radio, cases à cocher, boutons, etc.). El es
permettent entre autres soit d'afficher le composant d'origine soit la zone de saisie d'un port ou
d'une expression.
· ExtFormComponentFactory : classe de création qui retourne une instance d'une sous-classe de
ExtFormComponent selon la valeur d'une balise html qui lui est fourni.
4.Application à la reconnaissance de l'IL-2
Ce gestionnaire graphique de processus bioinformatiques d'analyse de séquences a été conçu autour
d'une application commandée par l'utilisateur final, l'Unité d'Immunogénétique Cellulaire. L'application
est focalisée sur l'identification des résidus encodant la spécificité de la reconnaissance de
l'interleukine-2 humaine par ses récepteurs au cours de l'évolution par comparaison aux autres
systèmes cytokine-récepteur.
4.1 Introduction : L'IL2 et ses récepteurs
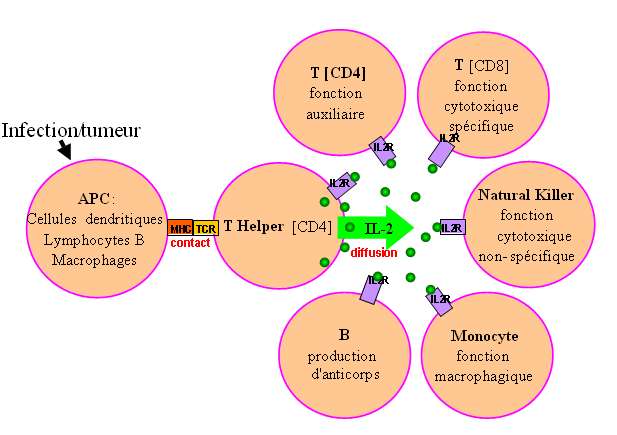
L'interleukine-2 (IL2) est une cytokine de 133 résidus sécrétée naturellement par les lymphocytes
CD4 lors d'une stimulation par les cellules présentant les antigènes (monocytes, cellules dendritiques,
lymphocytes B) au cours d'une infection ou en présence de cellules tumorales. L'IL2 est reconnue par
plusieurs types de récepteurs (IL2R) à la surface des lymphocytes (Fig. 17) et stimule leur
prolifération.
Fig. 17 Quand et par quelles cellules
est produite et sécrétée l'IL-2 ?
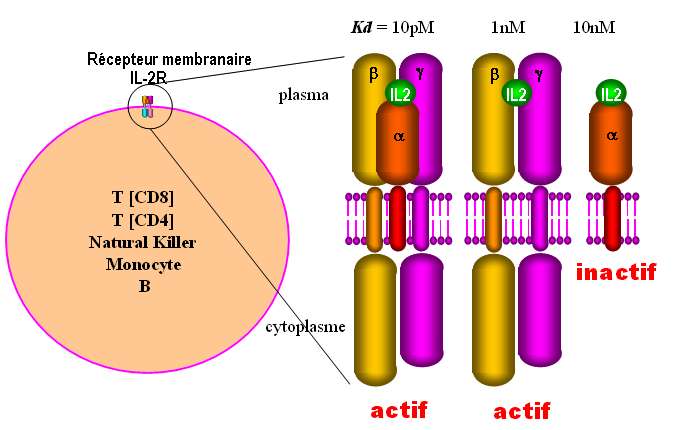
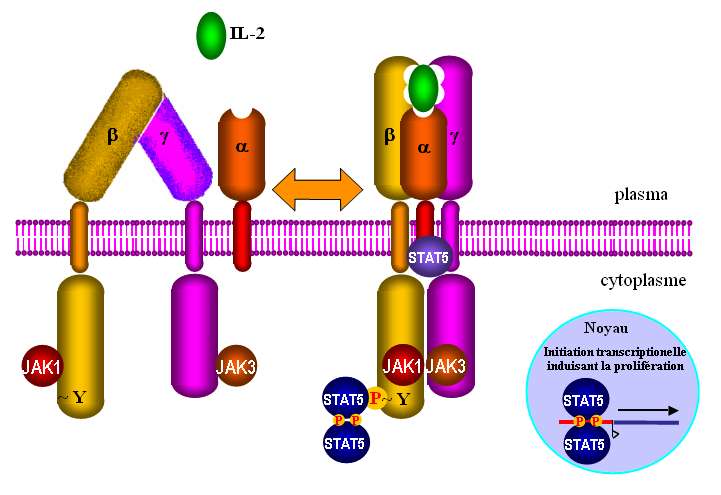
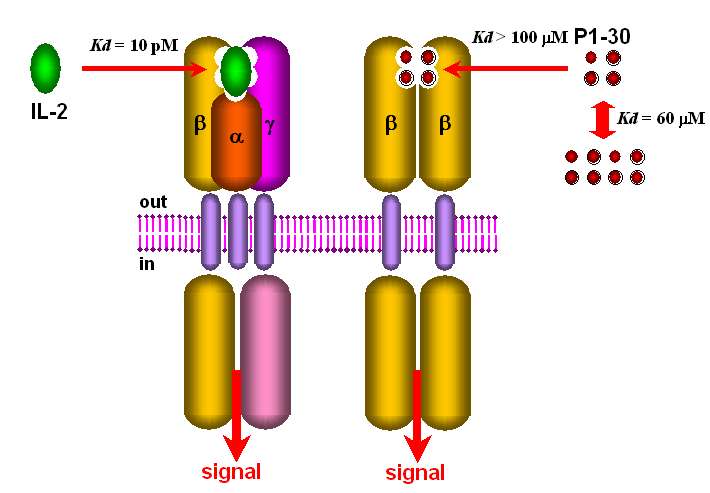
Les récepteurs de l'IL2 comprennent jusqu'à trois chaînes membranaires , et dont le contrôle de
l'expression et de la maturation varie d'un type de lymphocyte à l'autre. La proportion relative des
chaînes exprimées dicte la composition oligomériques des récepteurs. Ils se distinguent entre eux par
leurs affinités pour l'IL2: (10pM), (1nM) (Fig .18)
Fig. 18 Quels sont les récepteurs
actifs de l'IL-2 ?
Aucune structure des chaînes du récepteur n'a été résolue, pas même celles de ses domaines intra ou
extra-cytoplasmiques. Seules les structures cristallographiques et par RMN de l'IL2 ont été mises à
jour. Les bases moléculaires de la reconnaissance de l'IL2 par ses récepteurs et le mécanisme de
modification conformationnelle à l'origine de l'initiation transduction du signal ne sont pas documentés
expérimentalement. En particulier les hypothèses de pré-assemblage du récepteur avant la fixation de
la cytokine ou le rôle de l'assemblage induit par la cytokine sur la transduction du signal ne sont ni
démontrées ni invalidées.
Fig. 19 Comment agit l'IL-2 par la
voie Jak-Stat ?
Comme le décrit la figure 19, le domaine cytoplasmique de la chaîne IL2R fixe JaK1 (tyrosine kinase
Janus 1) et IL2R fixe JaK3. Le rapprochement des domaines cytoplasmiques met en contact JaK1 et
JaK3 qui forment un complexe actif qui catalyse la phosphorylation d'une tyrosine de l'extrémité C-
terminale de la chaîne IL2R. Cette tyrosine modifiée est reconnue par le complexe de facteurs de
transcription Stat5a-Stat5b qui s'active par auto-phosphorylation. Ces facteurs induisent l'expression
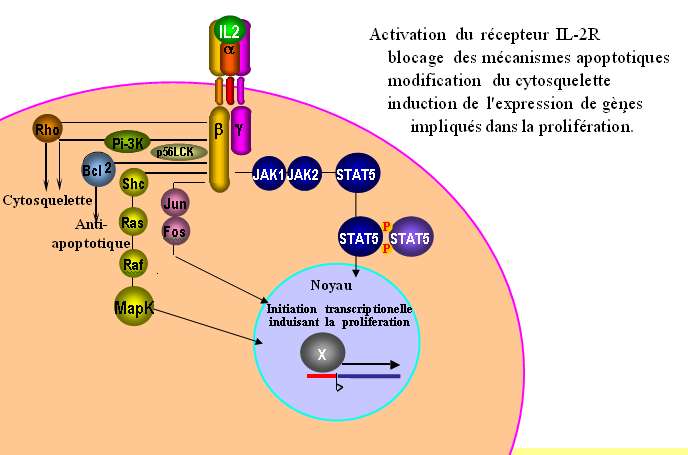
de plusieurs gènes, celui de l'IL2R par exemple. L'activation du récepteur IL2R induit aussi le blocage
des mécanismes apoptotiques via Bcl2, la modification du cytosquelette par la voie Rho et la
prolifération par l'activation de plusieurs gènes par la voie Ras/Raf/MapK (Fig. 20).
Fig. 20 Quels sont les rôles de l'IL-2 ?
4.2 Importance médicale de l'IL2, et du développement d'agonistes et
antagonistes de l'IL2R
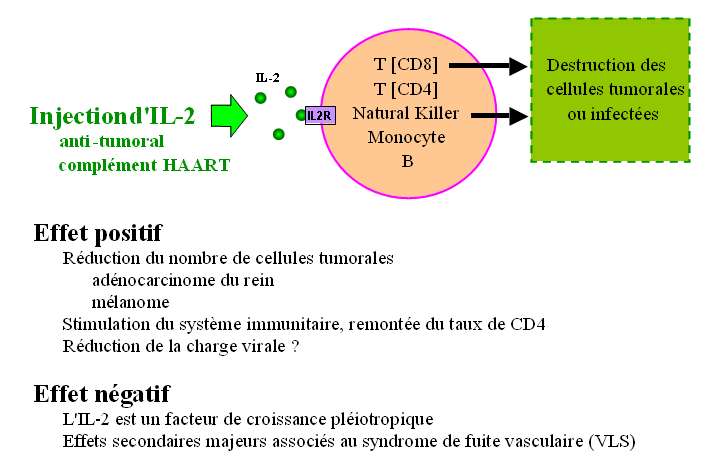
L'injection d'IL2 a été testée depuis 1980 chez des patients pour provoquer la prolifération de
lymphocytes et réduire le développement de tumeurs (Fig. 21). La thérapie a été agréée depuis une
dizaine d'années pour le traitement de carcinomes rénaux et de mélanomes. Plus récemment, l'IL2 est
utilisée pour reconstituer le taux de CD4 chez certains patients infectés par le virus VIH.
Malheureusement, l'IL2 induit aussi des effets secondaires indésirables. Le syndrome de fuite
vasculaire (VLS) responsable entre autres d'oedèmes pulmonaires en est un exemple. La gravité des
effets secondaires oblige à réduire les doses injectées ce qui diminue l'efficacité des traitements.
Aucun agoniste du récepteur ni aucune IL2 modifiée n'a permis jusqu'à présent d'améliorer
significativement l'index thérapeutique de l'IL2.
Fig. 21 Quels usages thérapeutiques
pour l'IL-2 ?
La recherche d'inhibiteurs du récepteur de l'IL2 offrirait des espoirs de thérapies contre certaines
maladies auto-immunes ou en complément de la cyclosporine prescrite dans le cas de greffes d'organes.
En effet, la cyclosporine est toxique pour le rein et le seul moyen actuellement d'en réduire les doses,
est l'injection d'anticorps anti-IL2R inhibiteur du récepteur IL2R.
4.3 Recherche en cours au laboratoire d'Immunogénétique Cellulaire (IGC)
L'Unité IGC est entre autre engagée dans deux projets conjoints l'un est l'identification du
mécanisme de transduction induit par l'IL2 sur les lymphocytes "Natural killers" et l'autre de
sélectionner ou concevoir des molécules susceptibles de stimuler cette transduction à la place de l'IL2,
toxique chez les patients en traitement.
Ce groupe a été le premier à démontrer la présence de récepteurs préassemblés de l'IL2 spécifiques
des NK [r15]. Ceux-ci sont aussi stimulables spécifiquement par des peptides mimant la structure de
l'IL2 entière [r16]. En absence de données cristallographiques Thierry Rose a reconstruit par
modélisation moléculaire comparative les structures tridimensionnel es du dimère de chaînes IL2R
libres et associés au tétramère de peptide p1-304 [r16] sur la base du complexe de l'érythropoïétine et
de son récepteur résolue par cristallographie. Ces simulations suggèrent un mécanisme de transduction
de signal à travers la membrane par la modification des interactions des deux chaînes pré-assemblées
(Fig. 22). Sous l'effet de la fixation du peptide, les extrémités C-terminales des deux domaines
extra-cytoplasmiques distantes de plus de 80Å, se rapprochent à moins de 30 Å. Par extension de ces
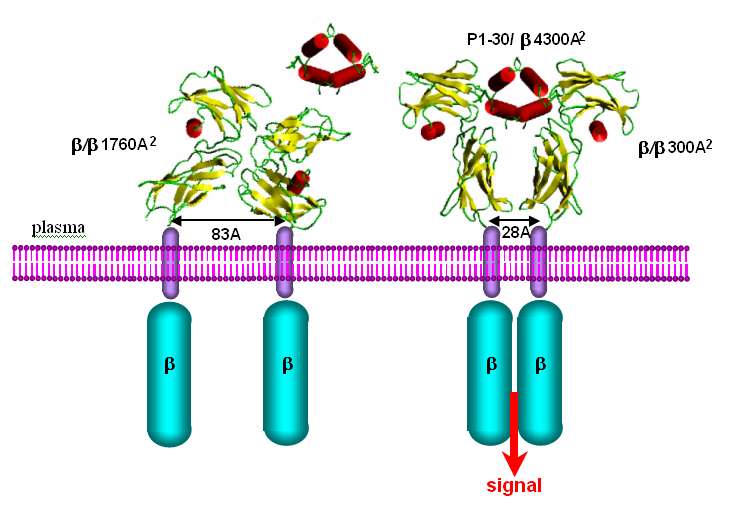
domaines, les hélices transmembranaires uniques sont elles aussi rapprochées de 80Å à 30Å. Ce
mouvement de plus de 50 Å serait responsable du rapprochement des domaines cytoplasmiques (Fig.
22). Chaque domaine cytoplasmique fixe une tyrosine kinase (JaK); le rapprochement des deux kinases
permettrait de phosphoryler une tyrosine de l'IL2R reconnue comme site de fixation du complexe de
facteur de transcription Stat5a-Stat5b.
Fig. 22 Modèles de la
transition de conformation
entre le récepteur libre et lié
au trétramère p1-30 ?
La symétrie du tétramère p1-304 serait responsable de l'association préférentielle au récepteur
symétrique plutôt que le récepteur asymétrique (Fig. 23). En effet, le peptide p1-30 a la même
séquence que l'hélice A de l'IL2 et l'interaction de A avec le récepteur IL2R, se ferait par
l'intermédiaire de la chaîne .
Fig. 23 Spécificité de p1-30 par
rapport à l'IL-2 ?
Ce mécanisme semble être généralisable à toutes les IL2 et mimétiques sur leurs récepteurs
naturels , et mais aussi pour tous les systèmes cytokine-récepteur de la famille des
hématopoiétines.
4.4 Approche bioinformatique
Les approches bioinformatiques que j'ai mises en application dans le cadre du stage ont pour objet de
· Rechercher les séquences orthologues à chaque cytokine et chacune des chaînes de leurs
récepteurs.
· Produire des alignements multiples afin de préparer des reconstructions des modèles structuraux.· Produire des profils propres aux domaines encodés par ces familles pour retrouver par des
méthodes types chaîne de Markov, de nouvelles séquences de systèmes homologues de familles
proches ou éloignées.
· Produire des arbres phylogénétiques au sein des systèmes cytokines-récepteurs et d'une sous-
famille à l'autre au sein des hématopoiétines.
4.4.1 Rechercher les séquences orthologues à chaque cytokine et chacune des chaînes de leurs
récepteurs
Nous avons sélectionné les séquences suivantes :
Cytokine : IL2 ; Récepteur : IL2R, IL2R, IL2R.
Cytokine : IL4 ; Récepteur : IL4R, IL2R.
Cytokine : IL7 ; Récepteur : IL7R, IL2R.
Cytokine : IL9 ; Récepteur : IL9R, IL2R.
Cytokine : IL13 ; Récepteur : IL13R, IL4R, IL2R.
Cytokine : IL15 ; Récepteur : IL15R, IL2R, IL2R.
Cytokine : IL21 ; Récepteur : IL21R, IL2R.
Les paramètres de recherche des séquences (matrice de substitution, espérance e, création et
extension de lacune) dans la base de séquences Nrprot (NCBI) par la méthode Blast (NCBI) ont été
optimisés par le workflow décrit dans la figure 24. Le niveau de détection de séquence n'est pas
amélioré dans les cas présents par l'utilisation de la méthode itérative PSI-Blast.
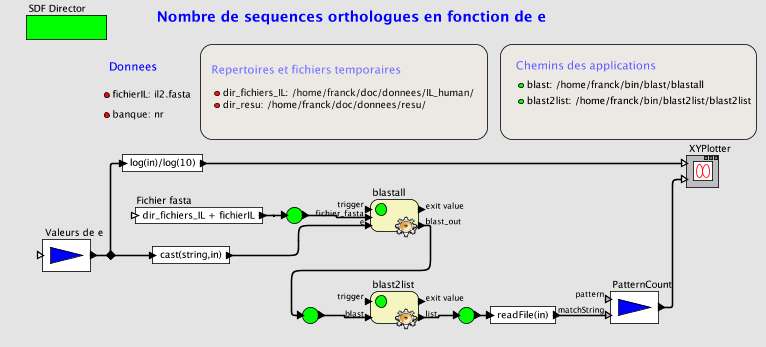
Fig. 24 Blast en fonction de e pour l'IL-2.
La brique 'valeurs de e' est une liste de 12 valeurs de 10-10 à 10. BLAST est exécuté pour chacune de ces valeurs. Lesidentifiants des séquences résultantes sont retrouvés par blast2list puis comptés. La courbe est affichée par unebrique de Ptolemy II.
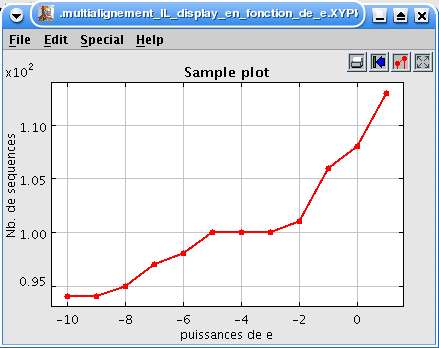
Fig. 25 Nombre de séquences
en fonction des puissances de
e.
4.4.2 Produire des alignements multiples afin de préparer des reconstructions des modèles
structuraux
Les fichiers résultats de recherche de séquences par Blast ont été filtrés par le programme
blast2list écrit par le groupe IGC et qui permet de sortir, filtrer et classer les séquences par score ou
organismes. Les listes ont été utilisées pour produire des fichiers où figurent les séquences complètes
au format fasta avec le programme fastacmd du package Blast (NCBI). Ce fichier est alors utilisé en
entrée par le programme ClustalW [w35] pour produire un fichier d'alignements multiples. Les
paramètres d'alignement ont aussi été testés (matrices de substitution, espérance e, création et
extension de lacune) en vue d'optimiser le taux d'identité par paire et le taux de similitude sur la
totalité du multialignement calculé par le programme cluscore comme décrit dans la figure 26.
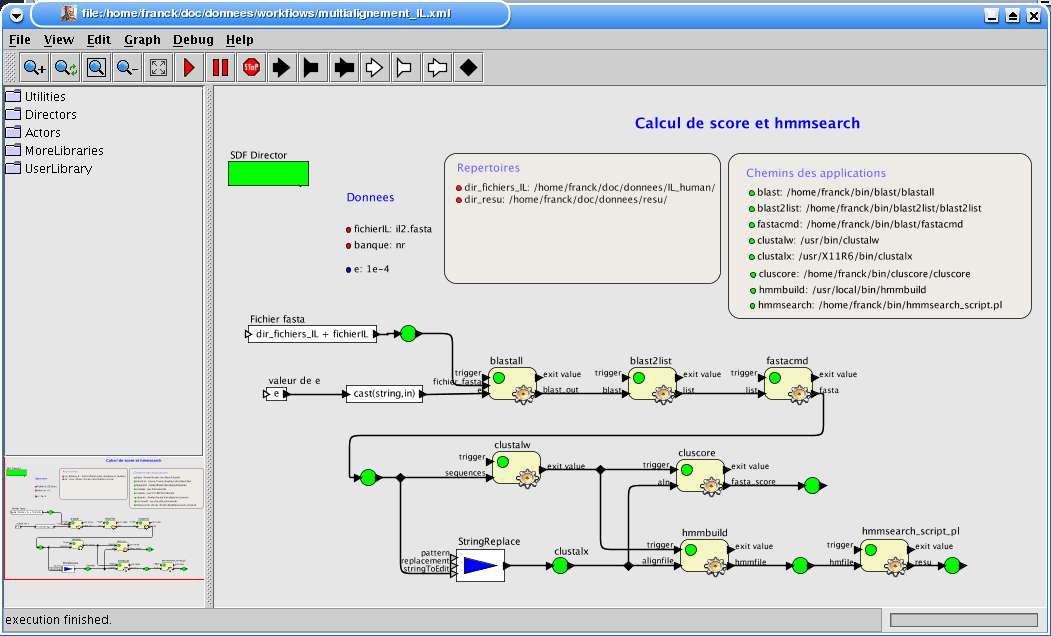
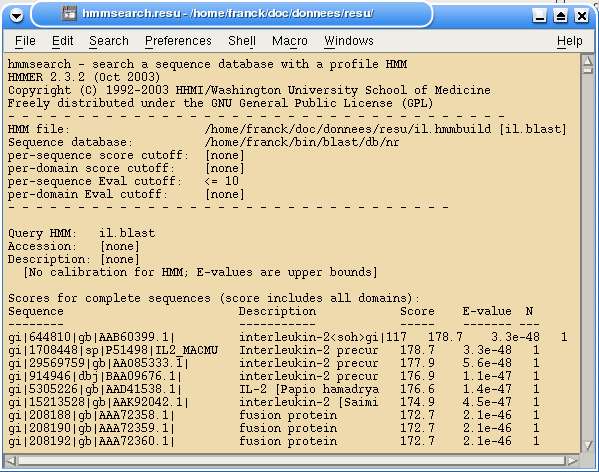
Fig. 26 cluscore et hmmsearch
Ce workflow montre à la fois l'utilisation de cluscore et la génération de profils par hmmsearch.
Fig. 27 Résultats de hmmsearch
Les fichiers aln produits par clustal ont été utilisés comme point de départ pour comparer plusieurs
logiciels interactifs d'alignement multiples Clustal X [r17], Jalview [w36], Indonesia [w37], T-coffee
[w38], 3D-coffee [w39]. L'ensemble de séquences Bali et l'évaluateur cluscore nous ont permis de
choisir les méthodes en fonction des cas. Nous avons écarté Indonesia, retenu ClustalX et Jalview dans
les cas généraux, T-coffee dans les cas plus difficiles et 3D-coffee lors de l'intégration de données
structurales, sur les structures secondaires non-sécables en particulier.
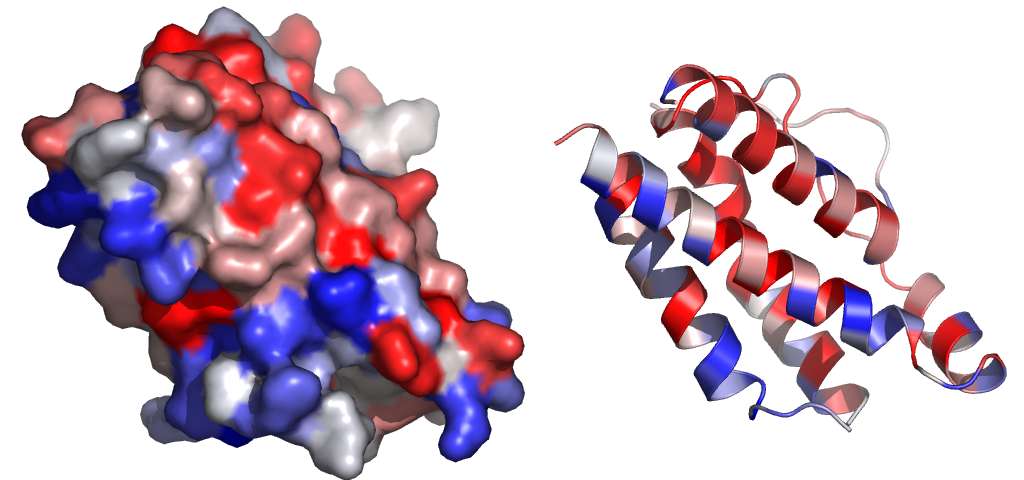
Les fichiers aln ont été utilisés pour évaluer le niveau de conservation de chaque résidu pour chaque
sous-famille au moyen du programme Cosa développé par le groupe IGC. Cosa produit une image d'un
modèle moléculaire tridimensionnel (Fig. 28) affichant le niveau de conservation des résidus à partir
d'un fichier au format clustal contenant parmi toutes les séquences au moins la séquence d'une protéine
de structure connue.
Fig. 28 Cosa. Niveau de conservation
des résidus.
Les zones en rouge sont fortement
conservées, les zones en bleu le sontfaiblement.
Ces fichiers d'alignements multiples au format clustal ont été utilisés pour reconstruire les modèles
moléculaires des cytokines et des différentes combinaisons de leurs récepteurs dans le cadre du projet
de Vldimir Dric.
De même, ces fichiers ont été utilisés par Perrine Barjou pour identifier les résidus impliqués dans
l'affinité et la sélectivité des reconnaissances cytokine-récepteur.
4.4.3 Production des profils propres aux domaines encodés par ces famil es
La production des profils propres aux domaines encodés par ces famil es permettent de retrouver
par des méthodes types chaîne de Markov, de nouvelles séquences de systèmes homologues de familles
proches ou éloignées
Les alignements multiples ont été utilisés pour générer avec le programme HMMER [w40] des profils
qui rendent compte des propriétés des acides aminés par position le long de la séquence.
Ces profils ont été utilisés pour rechercher des séquences distantes dans la database Nrprot avec le
programme HMMERsearch.
4.4.4 Produire des arbres phylogénétiques
ClustalW produits des arbres phylogénétiques par une méthode neighbor-jonction. Sans être
excellente cette méthode c'est avérée suffisante dans notre étude afin de produire les cladogrammes.
Perrine Barjou a exploré ces arbres afin de rechercher dans leur superposition en multicouche, une
couche par organisme, d'éventuelles corrélations entre l'évolution de la cytokine et de son récepteur.
5.Conclusion - futur et réflexions
5.1 Typage des données et services web
Un des aspects abordés dans le cahier des charges est le typage des données échangées entre les
applications qui composent un workflow. L'idée première était d'une part d'attirer l'attention de
l'utilisateur sur d'éventuelles incompatibilités (les connexions sont autorisées mais affichées
différemment) et d'autre part de transformer automatiquement ces données pour les rendre
compatibles par l'intermédiaire d'outils comme readseq [w1] ou squizz [w2] par exemple.
Le typage des données permet de plus de guider l'utilisateur en lui proposant les applications
disponibles pour un certain type de données.
Ptolemy II définit déjà des types pour les données échangées entre les acteurs (booléens, entiers,
flottants, chaînes de caractères, matrices, compositions...) [r2] et dispose d'un mécanisme automatique
de conversion et d'opérations polymorphiques entre ces types. Une solution serait donc d'étendre
cette hiérarchie aux types de données biologiques (séquence, séquence d'ADN, séquence protéique,
etc.), mais on se trouve alors confronté à leur définition voire à leur sémantique (on peut définir un type
'protéine' en voulant toutefois différencier une enzyme d'une cytokine) et on se rapproche au problème
connu de création d'ontologies.
La définition d'ontologies aussi bien pour les données que pour les services est un domaine en pleine
émergence mais ne couvre cependant pas tous les domaines. Gene Ontology [w3] par exemple décrit les
processus biologiques, les fonctions moléculaires et les composants cellulaires mais n'aborde pas la
structure des domaines protéiques ou les structures tridimensionnelles. Cependant les fonctionnalités
offertes ou prévues par des projets comme myGRID [w4] ou BioMOBY [w5] permettrait de profiter des
capacités offertes par les services web associés à ces ontologies. BioMOBY [r11] permet par exemple
de définir des tâches complexes associant la découverte, la distribution et le traitement de données
biologiques via des services disponibles sur le web (enregistrés en tant que tel sur un serveur 'MOBY
Central') [r4].
On pourrait ainsi imaginer des workflows composés de briques "services web" clientes découvertes
et proposées au fur et à mesure des données à analyser ou des workflows entiers à leur tour services
web.Cependant les objectifs de ces projets sont ambitieux et les notions utilisées pour les ontologies
(comme l'utilisation de langages d'ontologies DAML+OIL ou DAML-S, une ontologie permettant de
décrire les services web [r6]) et l'architecture sont non triviales à assimiler et donc encore moins à
implémenter !
On peut aussi noter l'approche de [r5] qui propose une ontologie dont les données sont définies par
un type structural (similaire aux types utilisés par les langages de programmation et qui détermine les
valeurs autorisées) et un type sémantique (type de haut niveau, conceptuel). Ils aspirent ainsi à
combiner des services structurellement différents mais qui sont sémantiquement compatibles. Cette
approche est intéressante dans la mesure où ils comptent utiliser et étendre Ptolemy II avec cette
démarche.
Enfin, grâce aux récents développements de Kepler, nous avons d'ores et déjà intégré des services
web à notre plateforme et par exemple testé les services offerts par l'Institut Nationale de Génétique
du Japon [w17]. L'utilisation d'acteurs de transformation (XSLT, Xquery) devrait par la suite
permettre de relier des services web syntaxiquement incompatibles [r7] .
5.2 Exécutions réparties
Il s'agit ici de répartir l'exécution d'un workflow sur des "clusters" (réseau d'ordinateurs pour
lesquels les échanges sont rapides) ou des grilles de calcul (grid computing) pour lesquels les noeuds
peuvent être hétérogènes, distants et les échanges de données lents. Chaque brique est alors exécutée
sur un noeud particulier. Ce domaine n'en est pas moins ambitieux, surtout si l'on considère plusieurs
aspects du problème liés au choix de la répartition en fonction de l'hétérogénéité des noeuds.
Reprenons par exemple les points abordés par [r10] :
· les noeuds peuvent être différents en terme de capacité de mémoire.
· les architectures peuvent être différentes (machines vectorielles ou scalaires par exemple), les
tâches peuvent s'exécuter plus ou moins efficacement selon ces architectures.
· Les tâches doivent être "proches" des données de taille importantes qu'el es utilisent car il peut
être prohibitif de les répliquer sur les noeuds à travers la grille de calcul.
· Certains logiciels possèdent des licences. Il serait coûteux de les installer sur tous les noeuds.
Sans entrer dans toutes ces considérations et sans prétendre proposer une solution et des
comparaisons exhaustives nous pourrions nous inspirer des projets tels que Biopipe, Wildfire ou Pise et
intégrer des outils de soumissions de tâches comme PBS [w21], LSF [w22] ou SGE[w23].
Une autre fonctionnalité à implémenter est la reprise sur erreur que ce soit pour les services web ou
les tâches reparties. Par exemple, des projets comme Biopipe ou Taverna permettent de retenter
l'exécution de chaque tâche un certain nombre de fois si celle-ci a échoué.
5.3 Briques internes
La possibilité d'utiliser des services web ou des acteurs génériques comme 'CmdLineApplication' ou
'MobyleApplication' pour les applications ou scripts locaux couvre a priori de nombreux domaines et
donc décharge du besoin de coder des briques spécifiques à Ptolemy II.
Cependant, il peut être avantageux de construire des briques dédiées (codées en java et intégrées
dans l'environnement) pour les raisons suivantes :
· Il est plus convivial pour l'utilisateur d'avoir une bibliothèque d'applications disponibles et
facilement paramétrables.
· Il est plus simple de partager des workflows sans avoir à se soucier du chemin des applications
locales.
· Dans le cas d'application locales ('CmdLineApplication'), l'échange de données se fait par fichiers.
L'intégration de composants permettrait de se dégager de cette contrainte en échangeant
directement les flux de données. La sauvegarde dans un fichier ne s'effectuant que lorsque c'est
nécessaire ou voulu (cf. P16).
· Enfin, l'ajout de briques de visualisation intégrées à l'environnement de Ptolemy II permettrait de
profiter d'un mode d'exécution qui permet d'exécuter un workflow sous une forme plus concise en
n'affichant que les entrées et sorties (textuel es ou graphiques). La figure 29 montre par exemple
un workflow entier et son exécution.
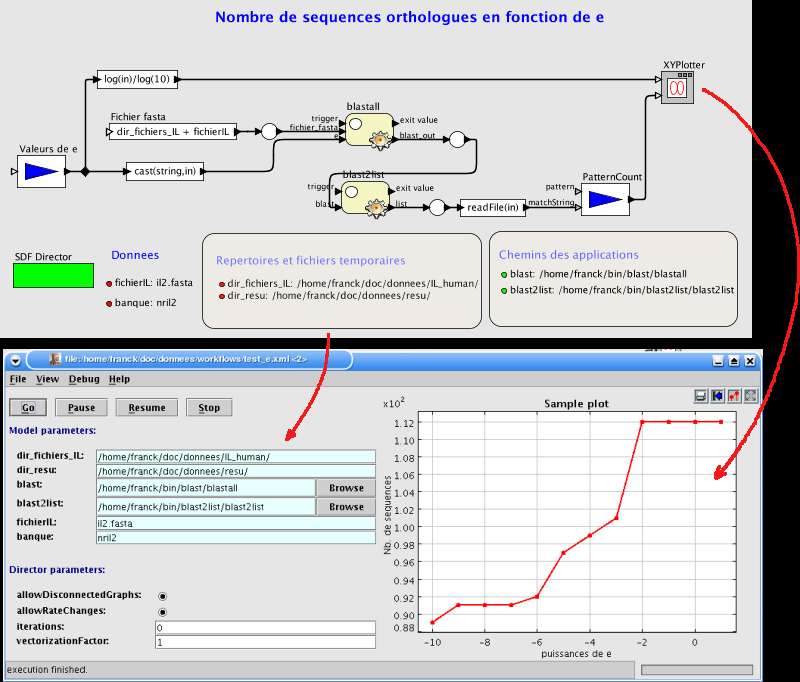
Fig. 29 Affichage complet et l'exécution correspondante.
Les briques d'affichage existantes sont, relativement à nos besoins, élémentaires mais on peut
imaginer de créer ou adapter d'autres projets de composants java de visualisation (ou autres) comme
par exemple FPV [w26] (structures protéiques), BugBrowser [w27] (séquences ADN), SstructView
[w28] (structures secondaires d'ARN), JaMBW [w29] (séquences), ATV [w30] (arbres phylogénétiques)
ou Cytoscape [w31] (interactions protéines-protéines).
5.4 Ergonomie et "utilisabilité"
Ce projet a été conçu autour d'un ensemble de critères définis par un utilisateur final le
laboratoire d'Immunogénétique cellulaire - pour enchaîner des tâches définies et répondre à des
questions précises.
Avant une diffusion plus large de ce projet et pour une utilisation plus aisée il conviendra d'adapter
l'espace de travail pour le rendre plus convivial comme par exemple élaguer les branches du menu listant
les acteurs (enlever les acteurs et les directeurs inutiles a priori), classer les applications et masquer
les menus et les fonctionnalités abscons.
Enfin, qu'en est-il de la robustesse de cet environnement et de sa généralisation aux tâches
courantes en bioinformatique ?"
[r13] permet de nous aiguiller sur ce point. Entre autres cette étude fait ressortir à partir de
consultations d'utilisateurs les composants principaux que devrait contenir une application de
bioinformatique, soit :
· Des collections d'objets (ordonnées ou non et avec redondance ou non).· Des filtres avec en entrée la collection d'objets à filtrer, une collection de restrictions (mots- clés,
identifiants de séquences, etc.) à appliquer sur ces objets et une collection qui détermine les champs
des objets sélectionnés à retourner.
· Des convertisseurs qui transforment chaque objet de la collection en entrée et retourne une
collection d'objets transformés.
· Des composants associant les fonctions de filtre et de convertisseur.
· Des branches conditionnelles ou non permettant les exécutions concurrentes.
La figure 30 donne une représentation graphique et un exemple de l'assemblage de ces composants.
Fig. 30 Représentation des composants et exemple d'enchaînement repris de [r13]
Le point délicat ici est l'intégration des collections dans nos workflows et plus précisément leur
traitement lors de l'exécution. Comment en effet combiner à la fois des traitements sur des objets
(ex. à partir d'un identifiant, rechercher dans GenBank la séquence correspondante) et des collections
d'objets (ex. à partir d'un tableau d'identifiants construire le tableau de séquences correspondantes).
Ce sujet est abordé par le projet Kepler/SPA dans [r14] qui propose d'améliorer un workflow
construit sous Ptolemy II permettant l'identification de promoteurs [w25].
Ce workflow est représenté par la figure 9 du chapitre 2.2.2.6, ses principaux inconvénients sont de
nécessiter des relations supplémentaires entre les briques pour contrôler le flux et donc d'avoir à
construire des briques spécifiques et difficilement réutilisables.
Deux solutions sont proposées, la première est d'intégrer des fonctions (briques) fcollection->liste et fliste-
>collection qui permettent respectivement les transformation d'une collection d'objets en une liste
d'objets (objets envoyés les uns à la suite des autres) et une liste en collection. La seconde solution
propose de traiter les tableaux comme des listes et d'insérer des marqueurs de début et de fin de
tableau.
A noter que dans tous les cas précédents le modèle d'ordonnancement choisi est PN (Process
Network), dans ce modèle les briques sont des tâches indépendantes dont les ports d'entrées sont des
canaux FIFO. La brique traite les données en entrée et envoie éventuellement des résultats sur des
ports de sortie. Elle se bloque dès que le canal est vide en attendant de nouvelles données. Il nous
faudra aussi adapter ce modèle pour implémenter l'exécution partielle d'un workflow [P4] (reprise sur
erreur ou choix par l'utilisateur d'un sous-ensemble par exemple).
5.5 Intégration des autres projets
Les paragraphes précédents nous ont montré que nous avions la possibilité de profiter de
développements extérieurs avec bien sûr principalement les projets Ptolemy II et Kepler/SPA qui sont
très actifs. Un des aspects à ne pas négliger est l'intégration de ces projets et la prise en compte de
leurs évolutions dans nos développements. Par exemple, un défaut du projet Kepler est d'être basé sur
une version antérieure de Ptolemy II (version 3) et donc de ne pas offrir les améliorations de la version
4. L'inclusion de leurs composants s'est fait par copier/coller avec quelques modifications dans le code,
ce qui devient ingérable pour l'intégration de bibliothèques et de mises à jour nombreuses.
Ceci passe évidemment par une gestion des sources (par CVS par exemple) mais aussi par des
développements qui ne bouleversent pas l'architecture logicielle en place. Par exemple, Ptolemy II
permet de personnaliser les différents menus et l'environnement de travail par l'ajout de nouvelles
classes, il ne serait pas judicieux de modifier celles existantes. Plus simplement, un nouveau directeur
PN sera défini plutôt que de modifier celui existant. Gageons cependant que nous aurons à contourner
ces voeux pieux.
L'utilisation d'un gestionnaire de sources nous permettra aussi de ne garder que les composants
utiles (est-il nécessaire par exemple de prendre en compte les systèmes à temps continu dans notre
environnement) est de réduire significativement la taille de l'application.
Bibliographie
[r1]
S. Bhattacharyya. C. Brooks, E. Cheong, J. Davis, M. Goel, B. Kienhuis, E. A. Lee, J. Liu, X. Liu, L.
Muliadi, S. Neuendorffer, J. Reekie, N. Smyth, J. Tsay, B. Vogel, W. Wil iams, Y. Xiong, Y. Zhao,
H. Zheng. PTOLEMY II. Heterogeneous Concurent Modeling and Design in Java. Volume 1:
Introduction to Ptolemy II. July 29, 2004.
[r2]
S. Bhattacharyya. C. Brooks, E. Cheong, J. Davis, M. Goel, B. Kienhuis, E. A. Lee, J. Liu, X. Liu, L.
Muliadi, S. Neuendorffer, J. Reekie, N. Smyth, J. Tsay, B. Vogel, W. Wil iams, Y. Xiong, Y. Zhao,
H. Zheng. PTOLEMY II. Heterogeneous Concurent Modeling and Design in Java. Volume 2:
Ptolemy II Software Architecture. June 24, 2004.
[r3]
S. Bhattacharyya. C. Brooks, E. Cheong, J. Davis, M. Goel, B. Kienhuis, E. A. Lee, J. Liu, X. Liu, L.
Muliadi, S. Neuendorffer, J. Reekie, N. Smyth, J. Tsay, B. Vogel, W. Wil iams, Y. Xiong, Y. Zhao,
H. Zheng. PTOLEMY II. Heterogeneous Concurent Modeling and Design in Java. Volume 3:
Ptolemy II Domains. June 24, 2004.
[r4]
M. D. Wilkinson and M. Links. BioMOBY: An Open Source Biological Web Services Proposal.
Briefing In bioinformatique, vol. 3 331-341, december 2002
[r5]
S. Bowers and B. Ludäscher. An Ontology-Driven Framework for Data Transformation in
Scientific Workflows. DILS 2004, LNBI 2994, pp 1-16, 2004
[r6]
Chris Wroe, Robert Stevens, Carole Goble, Angus Roberts and Mark Greenwood. A Suite of
DAML+OIL Ontologies To Describe Bioinformatics Web Services And Data. International
Journal Of Cooperative Information Systems Vol. 12 No.2 197-224
[r7]
Ilkay Altintas, Chad Berkley, Efrat Jaeger, Matthew Jones, Bertram Ludäscher, Steve Mock.
Kepler: An Extensible System for Design and Execution of Scientific Workflows. 2004
[r8]
Martin Senger, Peter Rice, Tom Oinn Soaplab - a unified Sesame door to analysis tools pages
509-513
[r9]
Sanner M.F., Stoffler D. and Olson A.J. (2002). ViPEr, a visual Programming Environment for
Python. In Proceedings of thre 10th International Python conference. 103-115. February 4-7,
2002. ISBN 1-930792-05-0
[r10] Chua Chung Lian, Francis TANG, Praveen Issac, Arun Krishnan. GEL: Grid Execution Language.
Bioinformatics Institute, Matrix#07-01-30 Biopolis St., Singapore. May 21, 2004
[r11] Wilkinson MD., Gessler D., Farmer A., Stein L. The BioMOBY project Explores Open-source,
Simple, Extensible Protocols for Enabling Biological Database Interoperability. Proceedings of
the Virtual Conference on Genomics and Bioinformatics. 2003
[r12] Shawn Hoon, Kiran Kumar Ratnapu, Jer-ming Chia, Balamurugan Kumarasamy, Xiao Juguang,
Michele Clamp, Arne Stabenau, Simon Potter, Laura Clarke, Elia Stupka. Biopipe: A flexible
Framework for Protocol_Based Bioinformatics Analysis. May 2003
[r13] Robert Stevens, Carole Goble, Patricia Baker and Andy Brass. A Classification of Tasks in
Bioinformatics. July 26, 2000
[r14] Bertrand Ludäscher, Ilkay Altintas. Technical Note: SciDAC-SPA-TN-2003-01. On Providing
Declarative Design and Programming Constructs for Scientific Workflows based on Process
Networks. August 2003
[r15] Eckenberg R, Rose T, Moreau JL, Weil R, Gesbert F, Dubois S, Tel o D, Bossus M, Gras H, Tartar
A, Bertoglio J, Chouaib S, Goldberg M, Jacques Y, Alzari PM, Theze J. The first alpha helix of
interleukin (IL)-2 folds as a homotetramer, acts as an agonist of the IL-2 receptor beta chain,
and induces lymphokine-activated killer cells. J Exp Med. 2000;191:529-40.
[r16] Rose T, Moreau JL, Eckenberg R, Thèze J. Structural Analysis and Modeling of a Synthetic
Interleukin-2 Mimetic and Its Interleukin-2R2 Receptor. Journal Biol Chem. 2003 278:22868-
76.
[r17] Thompson,J.D., Gibson,T.J., Plewniak,F., Jeanmougin,F. and Higgins,D.G. (1997) The ClustalX
windows interface: flexible strategies for multiple sequence alignment aided by quality analysis
tools. Nucleic Acids Research, 24:4876-4882.
Références Web
[w1]
readseq
http://bioweb.pasteur.fr/docs/man/doc/readseq2.1
[w2]
squizz
http://bioweb.pasteur.fr/seqanal/interfaces/squizz.html
[w3]
Gene Ontology (GO)
http://www.geneontology.org/
[w4]
MyGRID
http://www.mygrid.org.uk/
[w5]
BioMOBY
http://biomoby.org/
[w6]
Taverna
http://taverna.sourceforge.net/
[w7]
PipelinePilot
http://www.scitegic.com/products_services/pipeline_pilot.htm
[w8]
VIBE
http://www.incogen.com/index.php?type=Product¶m=VIBE
[w9]
Eclipse
http://www.eclipse.org
[w10] Poseidon
http://www.gentleware.com/
[w11] Pise
http://www.pasteur.fr/recherche/unites/sis/Pise/
[w12] Forms
http://www.w3.org/TR/REC-html40/interact/forms.html
[w13] X-smiles
http://www.xsmiles.org/
[w14] WebRendered
http://www.webrenderer.com/
[w15] Gowlab
http://industry.ebi.ac.uk/soaplab/Gowlab.html
[w16] Kepler
http://kepler.ecoinformatics.org/
[w17] XML central of DDBJ http://xml.nig.ac.jp/wsdl/index.jsp
[w18] Wildfire / GEL
http://web.bi .a-star.edu.sg/~francis/WildfireGEL/
[w19] Ptolemy II
http://ptolemy.eecs.berkeley.edu/ptolemyII/index.htm
[w20] Matlab
http://www.mathworks.com/access/helpdesk/help/toolbox/bioinfo/ug/a1
057170551.html
[w21] open PBS
http://www.openpbs.org/about.html
[w22] LSF
http://www.platform.com/products/LSF/
[w23] SGE
http://gridengine.sunsource.net/
[w24] G_Pipe
http://sirio.sanmartin.edu.co/Pise/gpipe.html
http://130.102.113.135/gpipe.htm
[w25] Kepler/SPA
http://kepler.ecoinformatics.org/spa.html
[w26] FPV
http://www.cs.ucsb.edu/~tcan/fpv/
[w27] BugBrowser
http://www.stdgen.lanl.gov/genomeMap/GenomeMapDocSTD.html
[w28] SstructView
http://www.smi.stanford.edu/projects/helix/sstructview/
[w29] JaMBW
http://hometown.aol.com/lucatoldo/myhomepage/JaMBW/
[w30] ATV
http://www.genetics.wustl.edu/eddy/atv/
[w31] Cytoscape
http://www.cytoscape.org/
[w32] HTMLEditorKit
examples.oreilly.com/jswing2/code/goodies/HtmlEdKit.pdf
[w33] Talisman
http://talisman.sourceforge.net/
[w34] Mobyle
http://www.pasteur.fr/recherche/unites/sis/Pise/pise-new.html
[w35] ClustalW
http://www.ebi.ac.uk/clustalw
[w36] Jalview
http://www.ebi.ac.uk/~michele/jalview
[w37] Indonesia
http://xray.bmc.uu.se/dennis/
[w38] T-Coffee
http://igs-server.cnrs-
mrs.fr/~cnotred/Projects_home_page/t_coffee_home_page.html
[w39] 3DCoffee
http://igs-server.cnrs-mrs.fr/Tcoffee/tcoffee_cgi/
[w40] HMMER
http://hmmer.wustl.edu
Annexe A - Cahier des charges
Fonctionnalités de la plate forme de construction des workflows
de programmes de manipulation et d'analyse de séquences
Cette page présente les fonctionnalités de l'application. El e sert de base de discussion et est
amenée à évoluer et à se détailler en fonction des nouveaux besoins et choix. Elle permet aussi de
suivre plus aisément l'avancement des développements et des tests.
Les fonctionnalités sont réparties en 2 groupes :
· Px : fonctionnalités du workflow proprement dit.
· Ax : fonctionnalités de la partie graphique (interaction et affichage)
· P?x/A?x : propositions de fonctionnalités à discuter (idées, fonctions optionnelles, etc.).
Définitions :
· TBD
(To Be Defined) : à voir.
· acteur
Bloc de traitement pouvant être une fonctionnalité interne à l'application (test,
affichage, boucle, comparateur,...) ou une application externe en local sur la machine ou
déportée (service web, ...)
· donnée
Donnée en entrée ou en sortie d'un acteur (ex. une séquence, une liste de
séquences). La donnée peut être l'information elle même (séquence FASTA, sortie XML d'un
service web, ...) ou une référence vers cette donnée (nom du fichier, URL).
ü
signifie que la fonctionnalité est ajoutée et disponible à l'état actuel du projet.
P1 ü
Liste des acteurs à écrire en priorité
Les fonctionnalités suivantes doivent être disponibles en priorité pour pouvoir tester
les autres projets (sous la forme d'une brique interne ou d'une application externe) :
P1.1
BLAST, PSI-BLAST, BLAST2P
P1.2
FastA
P1.3
ClustalW, ClustalX
P1.4
Smith-Waterman
P1.5
SignalP
P1.6
TMHMM
P1.7
ProSite
P1.8
HMMER, pfam
P1.9
TCoffee / 3DCoffe
P2
Chargement et sauvegarde d'un workflow
P2.1 ü
Le workflow créé par l'utilisateur doit pouvoir être sauvé sous la forme d'un fichier
XML.
P2.2 ü
Le fichier sauvé doit pouvoir être lu pour recréer un workflow ou définir un nouvel
acteur (éléments et enchaînements du workflow cachés dans une "boite noire").
P3
Moteur de workflow
Les fonctionnalités de traitement de l'application sont directement dépendantes des
possibilités du moteur de workflow (exécutions concurrentes, boucles, branchement, ...).
Ci-dessous sont listées les contrôles à implémenter (TBD liste à affiner et à mettre à
jour en fonction des besoins). Remarque : les fonctions de 'boucle' ne sont pas
présentes telles quelles dans Ptolemy II qui repose sur des itérations d'un workflow.
P3.1 ü
Exécutions séquentiel es.
P3.2 ü
Exécutions parallèles.
P3.3.1 ü
Boucle avec incrémentation d'un compteur.
P3.3.2
Boucle avec incrémentation des objets dans une liste de données ou une liste de listes
de données.
P3.3.3
Boucle qui permet de faire évoluer les paramètres des acteurs qui la composent et
dont la condition d'arrêt peut-être déterminée par ces paramètres.
P3.4 ü
Création d'une bifurcation logique. Définit l'acteur exécuté en fonction du résultat
vrai/faux d'une analyse des résultats de l'acteur précédent.
P?3.5
La synchronisation doit être possible entre des acteurs qui ne sont pas reliés entre
eux (qui ne convergent pas vers le même acteur ou qui n'échangent pas de données).
P4
Exécution partielles et reprise sur erreur.
Plusieurs options d'exécution du workflow doivent être disponibles :
P4.1
Exécution globale à partir du ou des points de départ.
P4.2
Exécution acteur par acteur avec attente de validation à chaque pause ou chaque tour
de boucle (mode pas à pas).
P4.3
Exécution d'une sous partie du workflow déterminée par une sélection d'une zone sur
l'interface graphique.
P4.4
Possibilité d'ajouter des points d'arrêt.
P?4.5
Pouvoir choisir les ressources où exécuter les processus. A priori cette fonction peut
être reléguée à une application extérieure (solution de cluster de type PBS par
exemple).
P?5
Benchmark
Un ordre de grandeur du temps d'exécution d'une partie ou de la globalité du
workflow doit pouvoir être fourni (TBD : possible ?)
P6
acteurs supplémentaires
P6.1
acteur STOP : arrêt avec reprise éventuelle (TBD : ce n'est pas trivial car cela
dépend du mode d'ordonnancement choisi dans Ptolemy II et donc serait à implémenter
pour chaque directeur).
P6.2 ü
acteur END : arrêt de l'ensemble du workflow
P6.3 ü
Arrêt du workflow à l'issue de l'exécution d'un acteur.
P6.4 ü
Envoi d'un mail (cet acteur est disponible dans Kepler mais est très basique). Les cas
d'utilisation sont à détailler.
P7
Ajout d'une application Mobyle
P7.1
Réutilisation du formalisme utilisé par Mobyle (prochaine évolution de Pise) : une
application externe doit pouvoir être définie par le fichier XML respectant la DTD de
Mobyle. Ce fichier décrit les paramètres d'entrée et de sortie et le type de l'application
(service web, application locale, CGI).
L'utilisateur peut, pour chaque paramètre d'entrée, spécifier s'il s'agit d'un port en
entrée, d'une expression à évaluer au moment de l'exécution (opérations entre
paramètres définis dans l'environnement de Ptolemy II) ou d'une valeur classique.
Cette configuration est faite par l'intermédiaire d'une page web similaire à celles qui
sont générées par Pise.
TBD : Un prototype est en cours pour valider la faisabilité. Il étend la fonction
précédente à l'exécution de cgi accessibles par des pages web. Il définit deux urls, la
première correspond au fichier XML (url_XML), la seconde à la page html
(url_presentation) de configuration de l'acteur.
Nous avons alors les 3 cas suivants :
· url_XML seule : la page html de configuration est générée à partir du fichier
XML. A partir de cette page l'utilisateur spécifie les ports d'entrée, les
expressions et les valeurs. Les ports de sortie sont aussi déterminés à partir
du fichier XML.
· url_html seule : la page html du site est affichée, l'utilisateur peut naviguer
avec les liens hypertexte pour trouver le formulaire qui correspond au service
désiré. Ensuite il spécifie le type des champs dans ce formulaire. A
l'exécution de l'acteur, la requête est envoyée au serveur (de la même
manière que si l'utilisateur appuyait sur 'submit'). La page html retournée par
le serveur est envoyée sur un port de sortie.
· url_XML et url_html : le comportement est le même que dans le premier cas
à la différence que l'affichage correspond à url_html. Ceci permet d'offrir
une interface similaire à celle d'un site d'origine (plus simplement qu'à partir
de la définition du fichier XML). Cela suppose bien sûr que le formulaire
définit dans url_html soit compatible avec le fichier XML. Les restrictions et
les différentes normes à prendre en compte sont à détailler à partir de
l'expérience du prototype !
P8
Ajout d'une application externe
P8.1 ü
L'acteur qui permet de définir une application externe a comme nom
'CmdLineApplication'. Il permet à l'utilisateur d'exécuter une application locale avec les
paramètres suivants :
Le chemin vers l'application.
La liste des paramètres de l'application. Le répertoire de travail.
Les variables d'environnement.
La liste des paramètres permet en fait de prendre en compte l'environnement
Ptolemy II comme par exemple les ports en entrée et en sortie et les paramètres de
l'espace de travail (workspace). Elle est de la forme suivante :
parameters_list :- (string | in_port | expression | out_port )*
in_port :- in{port_name}
expression :- exp{exprstring}
exprstring :- "Expression valide dans l'environnement de Ptolemy"out_port :- out{port_name}={out_port_value}
port_name :- "Nom de port valide dans l'environnement de Ptolemy"out_port_value :- (string | expression | in_port)+
out_port_string :- "chaîne de caractères quelconque qui ne contient pas les expressions définies pour in_portname ou expression."
string :- "chaîne de caractères quelconque non nulle qui ne contient pas les expressions définies pour in_portname, expression ou
out_port."
Après la saisie de ces paramètres, les ports d'entrée (définis par in{port_name}) et
de sortie (définis par out{port_name}) sont ajoutés ou supprimés conformément à ce qui
est rentré sur la ligne de commande. Lors de l'exécution :
· Les chaînes de type in{port_name} sont remplacées par le contenu des ports en
entrée.
· Les chaînes de type 'exp{expstring}' sont remplacées par le résultat de l'évaluation
de 'expstring' par Ptolemy.
· Les chaînes 'val_out' définies par out{port_out}={val_out} sont envoyées sur leur
port correspondant 'port_out'. Ces chaînes peuvent être le résultat d'une première
évaluation (cas où la valeur est de type 'parameter' ou port_in).
Deux ports supplémentaires sont automatiquement créés : 'trigger' : port en entrée
qui permet d'attendre un élément quelconque en entrée avant d'exécuter l'application.
Ce port est inactif s'il n'est pas relié. 'exit_value' : port en sortie qui contient la valeur
retournée par l'application après l'exécution.
Exemple : Programme cosa
nom de l'application : /usr/bin/cosa
paramètres : in{port_aln} exp{position} in{port_pdb}
out{sortie_pdb}={resu_pdb.pdb} out{sortie_txt}={exp{fichier_txt}} répertoire de travail : /home/user/tmp/
variables d'environnement : {{name = "", value = ""}}
Après l'entrée des paramètres les ports d'entrée 'port_aln', et 'port_pdb' sont
créés et le port de sortie 'sortie_pdb' est créé. 'position' et 'fichier_txt' sont des
paramètres définis dans l'espace de travail par l'utilisateur.
A l'exécution, les chaînes de type in{nom_port} sont remplacées par le contenu du
port 'nom_port', les expressions sont évaluées et les chaînes de type out{port_out}=
{val_out} sont remplacées par 'val_out' (éventuel ement évaluée).
La ligne de commande réel e exécutée est, avec position=4,
fichier_txt="~/tmp/resu.txt" et port_aln="fichier.aln", fichier_pdb="fichier.pdb" :
cosa fichier.aln 4 fichier.pdb resu_pdb.pdb ~/tmp/resu.txt
A la fin de l'exécution, les chaînes "resu_pdb.pdb" et "~/tmp/resu.txt" sont envoyées
respectivement sur les ports sortie_pdb et sortie_txt.
P?9
Ajout de types de données
Des types doivent pouvoir être définis pour les données échangées dans un workflow.
Ces types peuvent être primitifs (entiers, booléens,..), des types "biologiques" (séquence
d'ADN ou protéique, structures ,...), des collections de types (collections ordonnées ou
non, avec doublons ou non...) ou encore une référence vers une donnée (ex. nom de fichier
ou URN décrivant une séquence).
Cette fonctionnalité permet d'une part d'attirer l'attention de l'utilisateur sur
d'éventuelles incompatibilités (les connexions sont autorisées mais affichées
différemment) et d'autre part de transformer automatiquement ces données pour les
rendre compatibles par l'intermédiaire d'outils comme readseq ou squizz par exemple.
Le typage des données permet de plus de guider l'utilisateur en lui proposant les
applications disponibles pour un certain type de données.
Cette fonctionnalité est à approfondir :
· l'utilisateur peut-il rajouter ses types de données, définir leurs compatibilités
et les outils de transformations ?
· comment peut-on utiliser les ontologies déjà définies (cf. BioMOBY, myGRID,
GeneOntology, ...)
P10
Liens entre données et acteurs
P10.1
Le lien (entrée ou sortie) entre une donnée et le port d'entrée d'un acteur peut se
faire même s'ils ne sont pas compatibles.
P10.2
Si la donnée et l'acteur ne sont pas compatibles, le lien est représenté de manière
différente. L'utilisateur a la possibilité de demander à l'application d'ajouter un ou des
acteurs intermédiaires (wrappers) pour les rendre compatibles. Si plusieurs possibilités
existent une liste est proposée.
P10.3
Les acteurs intermédiaires ne font que de la traduction de données (adaptation de
formats par exemple).
P?11
Support des services web
A définir en fonction des solutions retenues pour les types de données. En plus du
service proprement dit, il serait intéressant d'utiliser des mécanismes de découverte de
ces services (à voir en fonction des solutions disponibles).
Remarque : Kepler implémente déjà des services web, voir comment les adapter à
notre plateforme.
P?12
Répartition de la charge d'exécution
L'application doit permettre une répartition de la charge si plusieurs ressources de
calcul sont disponibles. Cette fonctionnalité est à approfondir !
Nous pouvons a priori considérer au moins trois cas :
· les acteurs lancent des programmes locaux (création de processus). Un outil
externe comme PBS peut être utilisé.
· Les acteurs font partie intégrante de l'application (ils sont lancés sous forme
de threads).
· Les acteurs sont des services web ou font appel à des cgi.
P13
Comportement des acteurs
P13?.1
Sur erreur, il doit y avoir une possibilité d'avoir une alternative (soit arrêt du
workflow soit re-tentative avec un nombre définissable de tentatives et de durées entre
ces tentatives). Remarque : fonctionnalités implémentées dans Taverna.
P14
Exécution de plusieurs instances d'un modèle workflow, comparaison des résultats
P14.1
L'utilisateur doit pouvoir faire tourner plusieurs instances d'un modèle de workflow.
P14.2
Plusieurs modèles de workflow peuvent être ouvert et exécutés en même temps.
P14.3
Les résultats des différentes exécutions (issus d'un même modèle ou de modèles
différents) peuvent être comparés entre eux.
P15
Applications et données locales
P15.1 ü
L'utilisateur doit pouvoir choisir un ou des chemins par défaut pour l'exécution des
applications.
P15.2 ü
L'utilisateur doit pouvoir choisir un ou des chemins par défaut pour la localisation des
données locales.
P16
Sauvegarde et visualisation de données entre acteurs
Un acteur particulier permet de sauver sous la forme d'un fichier des données
échangées entre deux acteurs (il s'insère entre les deux acteurs), ceci permet de
pouvoir visualiser ces données et de définir des points de sauvegarde sur lesquels le
workflow pourra redémarrer (cas d'une exécution partielle ou d'une reprise sur erreur).
Il peut :
P16.1
Sauver les données qu'il reçoit en entrée et les fournir en sortie sans modification si
l'utilisateur n'y a apporté aucune modification. Remarque : il sera certainement
souhaitable d'empêcher la modification des données pour éviter des incohérences dans
le workflow (il faut s'assurer que c'est toujours la même donnée pour les différents
traitements !). En fonction du type de donnée on peut être amené à enregistrer
seulement la référence (cas d'un fichier ou d'une URN par exemple) ou à 'sérialiser' la
donnée (cas d'une séquence représentée par une chaîne de caractères par exemple) dans
un fichier ou une base de donnée puis la reconvertir dans son format d'origine en sortie
de l'acteur.
P16.2
Visualiser ces données en fonction de leur type en proposant plusieurs outils de
visualisations et/ou d'édition disponibles.
P16.3
Associer ces données à une instance particulière du workflow.
P16?.4
Sauver ces données sous un format compressé selon le choix de l'utilisateur.
P17 ü
Plateformes supportées
L'exécution sera indépendante de la nature du matériel et du système d'exploitation.
P18
Exécution en arrière plan
P18.1
Les workflows créés par l'utilisateur doivent pouvoir être lancés en arrière plan, leur
exécution ne sera alors pas interrompue par la fermeture du gestionnaire graphique.
P18.2
L'utilisateur pourra à partir du gestionnaire graphique rouvrir une instance à partir
de la liste des workflows lancés en arrière plan (issu de cette session graphique ou d'une
autre) par cet utilisateur.
P18.3
Le workflow ouvert affichera l'état en cours et sera modifiable comme un workflow
classique.
P19
Données
Les données en entrée (i.e. : qui ne sont pas issue d'une acteur) suivantes doivent être
définies en priorité, cette fonction est cependant dépendante du choix retenu pour P?9 :
P19.1
Une séquence de protéines, son identificateur ou une liste de séquences de protéines.
P19.2
Une structure de protéines, son identificateur ou une liste de séquences de protéines.
P19.3
Une ou des listes de paires de séquences de protéines formant un réseau continu ou
disjoint.
P19.4
Un arbre phylogénétique ou une liste d'arbres.
A1
Affichage des liens
Lors de la création d'un lien entre une donnée et un acteur, le trait est continu si la
compatibilité est confirmée et discontinu sinon.
A2
Zooms
A2.1
Zoom sur un acteur qui contient un workflow : les acteurs qui contiennent des
workflows peuvent être visualisés sous leur forme condensée (un acteur) ou éclatée
(éléments du workflow).
A2.2
Les éléments du workflow ont plusieurs représentations en fonction du zoom (avec
plus ou moins de détails). A confirmer cependant, il semble plus judicieux de garder la
même représentation quel que soit le zoom.
A2.3
Il doit être possible d'afficher un ensemble d'éléments sous la forme d'un seul
(différent de la transformation d'un workflow en acteur) par exemple après sélection
avec un menu (view as box) puis ensuite revenir à la vue éclatée.
A3
Représentation arborescente
A3.1
Les éléments du workflow (données, acteurs, liens) seront aussi représentés et
modifiables sous forme d'arbres.
A4
Facilités de visualisation
A4.1 ü
Un espace affichera une vue générale du workflow.
A?4.2
Une fenêtre "tip" affichera une aide sur l'acteur sélectionné dans le workflow ou
dans la liste des applications (voir le workflow VIBE).
A?4.3
Des commentaires peuvent être ajoutés par l'utilisateur à un acteur (voir le workflow
VIBE).
A4.4
Un espace affichera les événements liés au workflow et aux acteurs (état de
l'exécution, warnings, erreurs,.. )
A5
Recherche d'un acteur
Un menu doit permettre de rechercher les acteurs par le nom ou leur description.
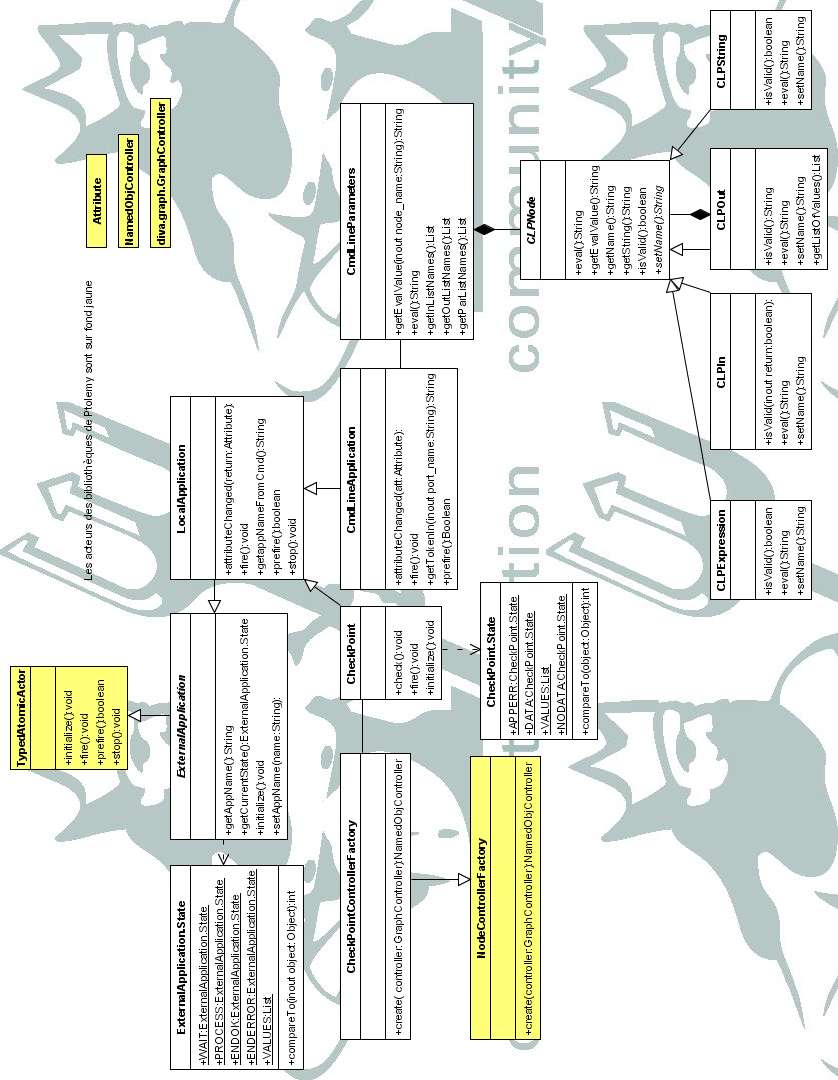
Annexe B - Diagramme statique
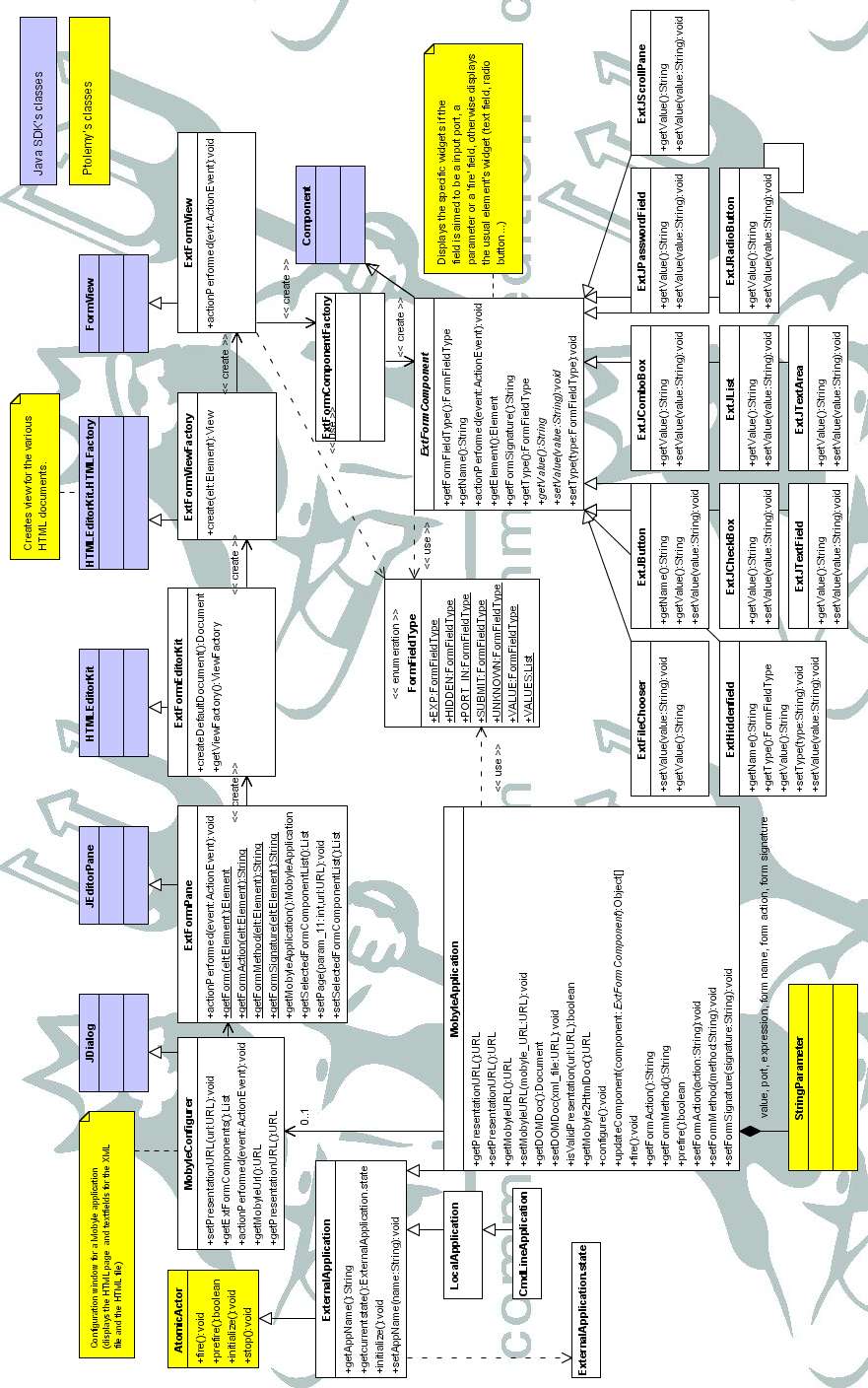
Annexe C - Diagramme statique : MobyleApplication
Annexe D - DTD de Mobyle
<!-- DTD for Mobyle descriptions -->
<!ENTITY % word "CDATA">
<!ENTITY % number "CDATA">
<!ENTITY % boolean "CDATA">
<!ELEMENT mobyle (program|pipeline)>
<!ELEMENT program (head, parameters)>
<!ELEMENT head (name, version?, doc, category*, command, layout?)>
<!ELEMENT name (#PCDATA)> <!-- the name of the program ex : blast2 -->
<!ELEMENT version (#PCDATA)> <!-- the version of the program --><!ELEMENT doc (title, description?, authors?, reference*, doclink*, help?)>
<!ELEMENT category (#PCDATA)>
<!-- to classified the programs in group
ex :DNA analysis, phylogeny ... -->
<!ELEMENT title (#PCDATA)>
<!ELEMENT description (text)+>
<!-- a short description of the program (1 line)
ex for melting it's: enthalpie, entropy and melting temperature -->
<!ELEMENT authors (#PCDATA)> <!-- the list of the authors -->
<!ELEMENT reference (#PCDATA)> <!-- the bibliographics references -->
<!ELEMENT doclink (#PCDATA)> <!-- a link to the documentation -->
<!ELEMENT help (text)+> <!-- a general help on the program -->
<!ELEMENT command (#PCDATA)>
<!ATTLIST command
name %word; #REQUIRED
type (local|GET|POST|POSTM|soap|xml-rpc) #IMPLIED
path %word; #IMPLIED
>
<!--
for local program:
name is the name of the command
type is local (by default)
path the path where is the program (by default the PATH variable)
for cgi:
name is the name of the script eg: myScript.cgi
type is the method to call the cgi (GET|POST|POSTM)
path is the url where is the script http://www.myDomain.org
for web service:
name is the method name
type is the protocol to call the ws soap|xml-rpc ...
path is the url of the wsdl
-->
<!ELEMENT parameters (parameter|paragraph|info)+>
<!ELEMENT paragraph (name,prompt,precond?,argpos?,format?,comment?,parameters, layout?)>
<!ELEMENT info (text)+> <!-- permit to insert text or code --><!ELEMENT layout (hbox|vbox)+> <!-- specified a layout to display a set of parameter -->
<!ELEMENT hbox (#PCDATA|(hbox|vbox)+)> <!-- to display the parameters horizontaly --><!ELEMENT vbox (#PCDATA|(hbox|vbox)+)> <!-- to display the parameters vertically -->
<!ELEMENT parameter (name,attributes,interface?)>
<!ATTLIST parameter
type (InFile|Sequence|Structure|OutFile|Results|Switch|Excl|List|Integer|Float|String)
#REQUIRED
ismandatory %boolean; #IMPLIED
iscommand %boolean; #IMPLIED
ishidden %boolean; #IMPLIED
isstandout %boolean; #IMPLIED
issimple %boolean; #IMPLIED
formfield %word; #IMPLIED
>
<!--
ismandatory : if true this parameter should be specified
iscommand : if true, this parameter specify the line of command to run the program used when
the command line is more complicated
isstandout : usualy the cgi redirect the output in a file programName.out.some programs have
already a text output and the cgi should not redirect the output.
issimple : specify if this parameter will be displayed only in the simple web form.
formfield : specify the look of this parameter in the interface
ex: in html :select, checkbox ...
-->
<!ELEMENT attributes (prompt|format|vdef|argpos|vlist|flist|comment|seqfmt|seqtype|ctrl|precond|
paramfile|filenames|scalemin|scalemax|scaleinc|separator|width|height|pipe|withpipe|example)+>
<!ELEMENT prompt (#PCDATA)>
<!ATTLIST prompt
lang %word; #REQUIRED >
<!-- suggestion xml:lang NMTOKEN iso639-1 -->
<!ELEMENT format (code)>
<!ELEMENT vdef ((value)+|(code)+)> <!-- the default value for the parameter -->
<!ELEMENT argpos (#PCDATA)>
<!--
in parameter: specify the position of this parameter on the commad line
in paragraph: specifiy the position on the command line for all parameters of this paragraph-->
<!ELEMENT vlist (value,label)+> <!-- a list of the available values for this parameter -->
<!ELEMENT label (#PCDATA)> <!-- the text associated to a value which will be display on
the form -->
<!ELEMENT flist (value,code)+>
<!-- it's used in conjonction with vlist, permit to associate code to a value of the vlist.
(flist it's an easy way to have a code associated whith each value in a list ) -->
<!ELEMENT comment (text)+> <!-- it's used to generate an help -->
<!ELEMENT seqfmt (value)+> <!-- the number of the sequence format in readseq 1. IG/Stanford
2. GenBank/GB 3. NBRF 4. EMBL etc ... -->
<!ELEMENT seqtype (#PCDATA)> <!-- the common values: dna, puredna, protein, pureprotein,
gapprotein, any, gapany -->
<!ELEMENT ctrl (message,code)+)> <!-- specified the possible values -->
<!ELEMENT message (#PCDATA)>
<!ELEMENT precond (code)+>
<!-- in parameter : the parameter is link to a condition
ex : this parameter could be used only if an other paramater has been specified -->
<!ELEMENT paramfile (#PCDATA)>
<!-- the name of the file when the parameters are specified in a file instead on command line -->
<!ELEMENT filenames (#PCDATA)>
<!-- the unix mask to retrieve the results files ex: *.aln ,*.dnd ... -->
<!ELEMENT scalemin (value|(code)+)>
<!ELEMENT scalemax (value|(code)+)>
<!ELEMENT scaleinc (#PCDATA)>
<!ELEMENT separator (#PCDATA)>
<!ELEMENT width (#PCDATA)> <!-- the width of an input or textarea in the html form --><!ELEMENT height (#PCDATA)> <!-- the height of an input or textarea in the html form -->
<!ELEMENT pipe (pipetype,(code)+)+>
<!ELEMENT withpipe (pipetype,(pipeparameter)+)+>
<!ELEMENT pipeparameter (#PCDATA)>
<!ELEMENT value (#PCDATA)>
<!ELEMENT code (#PCDATA)> <!-- a part of code in programmation language --><!ATTLIST code proglang %word; #REQUIRED>
<!-- proglang: the language in wich the code is written-->
<!ELEMENT name (#PCDATA)>
<!ELEMENT pipetype (#PCDATA)>
<!ELEMENT text (#PCDATA)>
<!ATTLIST text
proglang %word; #IMPLIED
lang %word; #IMPLIED
href %word; #IMPLIED
>
<!ELEMENT pipeline name, title?, (stage)+>
<!ELEMENT stage (arg*, pipe)>
<!ATTLIST stage
id %word; #REQUIRED
name %word; #REQUIRED
server %word; #REQUIRED
>
<!--
id : the identifier of the stage
name : a name of a program or a pipline (any xml file in pise)
server: the address where the service will be executed
-->
<!ELEMENT arg (#PCDATA)> <!-- a value for a parameter -->
<!ATTLIST arg name %word; #REQUIRED> <!-- name: the name of a parameter -->
<!ELEMENT pipe (in|out)+>
<!ELEMENT in (#PCDATA)> <!-- the previous stage id -->
<!ATTLIST in
id %word; #REQUIRED
pipetype %word; #REQUIRED
parameter %word; #REQUIRED
>
<!ELEMENT out (#PCDATA)> <!-- the next stage id -->
<!ATTLIST out
id %word; #REQUIRED
pipetype %word; #REQUIRED
parameter %word; #REQUIRED
>
<!ELEMENT interface (field+|layout?)>
<!ELEMENT field (name|prompt?|comment?) >
<!ATTLIST field formfield (upload|textarea|text)>
GNU Free Documentation License
Version 1.2, November 2002
Copyright (C) 2000,2001,2002 Free Software Foundation, Inc.
59 Temple Place, Suite 330, Boston, MA 02111-1307 USA Everyone is permitted to copy and distribute verbatim copies
of this license document, but changing it is not allowed.
0. PREAMBLE
The purpose of this License is to make a manual, textbook, or otherfunctional and useful document "free" in the sense of freedom: to
assure everyone the effective freedom to copy and redistribute it,with or without modifying it, either commercially or noncommercially.
Secondarily, this License preserves for the author and publisher a wayto get credit for their work, while not being considered responsible
for modifications made by others.
This License is a kind of "copyleft", which means that derivativeworks of the document must themselves be free in the same sense. It
complements the GNU General Public License, which is a copyleftlicense designed for free software.
We have designed this License in order to use it for manuals for freesoftware, because free software needs free documentation: a free
program should come with manuals providing the same freedoms that thesoftware does. But this License is not limited to software manuals;
it can be used for any textual work, regardless of subject matter orwhether it is published as a printed book. We recommend this License
principally for works whose purpose is instruction or reference.
1. APPLICABILITY AND DEFINITIONS
This License applies to any manual or other work, in any medium, thatcontains a notice placed by the copyright holder saying it can be
distributed under the terms of this License. Such a notice grants aworld-wide, royalty-free license, unlimited in duration, to use that
work under the conditions stated herein. The "Document", below,refers to any such manual or work. Any member of the public is a
licensee, and is addressed as "you". You accept the license if youcopy, modify or distribute the work in a way requiring permission
under copyright law.
A "Modified Version" of the Document means any work containing theDocument or a portion of it, either copied verbatim, or with
modifications and/or translated into another language.
A "Secondary Section" is a named appendix or a front-matter section ofthe Document that deals exclusively with the relationship of the
publishers or authors of the Document to the Document's overall subject(or to related matters) and contains nothing that could fall directly
within that overall subject. (Thus, if the Document is in part atextbook of mathematics, a Secondary Section may not explain any
mathematics.) The relationship could be a matter of historicalconnection with the subject or with related matters, or of legal,
commercial, philosophical, ethical or political position regardingthem.
The "Invariant Sections" are certain Secondary Sections whose titlesare designated, as being those of Invariant Sections, in the notice
that says that the Document is released under this License. If asection does not fit the above definition of Secondary then it is not
allowed to be designated as Invariant. The Document may contain zeroInvariant Sections. If the Document does not identify any Invariant
Sections then there are none.
The "Cover Texts" are certain short passages of text that are listed,as Front-Cover Texts or Back-Cover Texts, in the notice that says that
the Document is released under this License. A Front-Cover Text maybe at most 5 words, and a Back-Cover Text may be at most 25 words.
A "Transparent" copy of the Document means a machine-readable copy,represented in a format whose specification is available to the
general public, that is suitable for revising the documentstraightforwardly with generic text editors or (for images composed of
pixels) generic paint programs or (for drawings) some widely availabledrawing editor, and that is suitable for input to text formatters or
for automatic translation to a variety of formats suitable for inputto text formatters. A copy made in an otherwise Transparent file
format whose markup, or absence of markup, has been arranged to thwartor discourage subsequent modification by readers is not Transparent.
An image format is not Transparent if used for any substantial amountof text. A copy that is not "Transparent" is called "Opaque".
Examples of suitable formats for Transparent copies include plain
ASCII without markup, Texinfo input format, LaTeX input format, SGMLor XML using a publicly available DTD, and standard-conforming simple
HTML, PostScript or PDF designed for human modification. Examples oftransparent image formats include PNG, XCF and JPG. Opaque formats
include proprietary formats that can be read and edited only by
proprietary word processors, SGML or XML for which the DTD and/orprocessing tools are not generally available, and the
machine-generated HTML, PostScript or PDF produced by some wordprocessors for output purposes only.
The "Title Page" means, for a printed book, the title page itself,plus such following pages as are needed to hold, legibly, the material
this License requires to appear in the title page. For works informats which do not have any title page as such, "Title Page" means
the text near the most prominent appearance of the work's title,preceding the beginning of the body of the text.
A section "Entitled XYZ" means a named subunit of the Document whosetitle either is precisely XYZ or contains XYZ in parentheses following
text that translates XYZ in another language. (Here XYZ stands for aspecific section name mentioned below, such as "Acknowledgements",
"Dedications", "Endorsements", or "History".) To "Preserve the Title"of such a section when you modify the Document means that it remains a
section "Entitled XYZ" according to this definition.
The Document may include Warranty Disclaimers next to the notice which
states that this License applies to the Document. These WarrantyDisclaimers are considered to be included by reference in this
License, but only as regards disclaiming warranties: any otherimplication that these Warranty Disclaimers may have is void and has
no effect on the meaning of this License.
2. VERBATIM COPYING
You may copy and distribute the Document in any medium, eithercommercially or noncommercially, provided that this License, the
copyright notices, and the license notice saying this License appliesto the Document are reproduced in all copies, and that you add no other
conditions whatsoever to those of this License. You may not usetechnical measures to obstruct or control the reading or further
copying of the copies you make or distribute. However, you may acceptcompensation in exchange for copies. If you distribute a large enough
number of copies you must also follow the conditions in section 3.
You may also lend copies, under the same conditions stated above, and
you may publicly display copies.
3. COPYING IN QUANTITY
If you publish printed copies (or copies in media that commonly haveprinted covers) of the Document, numbering more than 100, and the
Document's license notice requires Cover Texts, you must enclose thecopies in covers that carry, clearly and legibly, all these Cover
Texts: Front-Cover Texts on the front cover, and Back-Cover Texts onthe back cover. Both covers must also clearly and legibly identify
you as the publisher of these copies. The front cover must presentthe full title with all words of the title equally prominent and
visible. You may add other material on the covers in addition.Copying with changes limited to the covers, as long as they preserve
the title of the Document and satisfy these conditions, can be treatedas verbatim copying in other respects.
If the required texts for either cover are too voluminous to fitlegibly, you should put the first ones listed (as many as fit
reasonably) on the actual cover, and continue the rest onto adjacentpages.
If you publish or distribute Opaque copies of the Document numberingmore than 100, you must either include a machine-readable Transparent
copy along with each Opaque copy, or state in or with each Opaque copya computer-network location from which the general network-using
public has access to download using public-standard network protocolsa complete Transparent copy of the Document, free of added material.
If you use the latter option, you must take reasonably prudent steps,when you begin distribution of Opaque copies in quantity, to ensure
that this Transparent copy will remain thus accessible at the statedlocation until at least one year after the last time you distribute an
Opaque copy (directly or through your agents or retailers) of thatedition to the public.
It is requested, but not required, that you contact the authors of theDocument well before redistributing any large number of copies, to give
them a chance to provide you with an updated version of the Document.
4. MODIFICATIONS
You may copy and distribute a Modified Version of the Document underthe conditions of sections 2 and 3 above, provided that you release
the Modified Version under precisely this License, with the ModifiedVersion filling the role of the Document, thus licensing distribution
and modification of the Modified Version to whoever possesses a copyof it. In addition, you must do these things in the Modified Version:
A. Use in the Title Page (and on the covers, if any) a title distinct from that of the Document, and from those of previous versions
(which should, if there were any, be listed in the History section of the Document). You may use the same title as a previous version
if the original publisher of that version gives permission.B. List on the Title Page, as authors, one or more persons or entities
responsible for authorship of the modifications in the Modified Version, together with at least five of the principal authors of the
Document (all of its principal authors, if it has fewer than five), unless they release you from this requirement.
C. State on the Title page the name of the publisher of the Modified Version, as the publisher.
D. Preserve all the copyright notices of the Document.E. Add an appropriate copyright notice for your modifications
adjacent to the other copyright notices.F. Include, immediately after the copyright notices, a license notice
giving the public permission to use the Modified Version under the terms of this License, in the form shown in the Addendum below.
G. Preserve in that license notice the full lists of Invariant Sections and required Cover Texts given in the Document's license notice.
H. Include an unaltered copy of this License.I. Preserve the section Entitled "History", Preserve its Title, and add to it an item stating at least the title, year, new authors, and
publisher of the Modified Version as given on the Title Page. If there is no section Entitled "History" in the Document, create one
stating the title, year, authors, and publisher of the Document as given on its Title Page, then add an item describing the Modified
Version as stated in the previous sentence.J. Preserve the network location, if any, given in the Document for
public access to a Transparent copy of the Document, and likewise the network locations given in the Document for previous versions
it was based on. These may be placed in the "History" section. You may omit a network location for a work that was published at
least four years before the Document itself, or if the original publisher of the version it refers to gives permission.
K. For any section Entitled "Acknowledgements" or "Dedications", Preserve the Title of the section, and preserve in the section all
the substance and tone of each of the contributor acknowledgements and/or dedications given therein.
L. Preserve all the Invariant Sections of the Document, unaltered in their text and in their titles. Section numbers
or the equivalent are not considered part of the section titles.M. Delete any section Entitled "Endorsements". Such a section
may not be included in the Modified Version.N. Do not retitle any existing section to be Entitled "Endorsements"
or to conflict in title with any Invariant Section.
O. Preserve any Warranty Disclaimers.
If the Modified Version includes new front-matter sections orappendices that qualify as Secondary Sections and contain no material
copied from the Document, you may at your option designate some or allof these sections as invariant. To do this, add their titles to the
list of Invariant Sections in the Modified Version's license notice.These titles must be distinct from any other section titles.
You may add a section Entitled "Endorsements", provided it containsnothing but endorsements of your Modified Version by various
parties--for example, statements of peer review or that the text hasbeen approved by an organization as the authoritative definition of a
standard.
You may add a passage of up to five words as a Front-Cover Text, and a
passage of up to 25 words as a Back-Cover Text, to the end of the listof Cover Texts in the Modified Version. Only one passage of
Front-Cover Text and one of Back-Cover Text may be added by (orthrough arrangements made by) any one entity. If the Document already
includes a cover text for the same cover, previously added by you orby arrangement made by the same entity you are acting on behalf of,
you may not add another; but you may replace the old one, on explicitpermission from the previous publisher that added the old one.
The author(s) and publisher(s) of the Document do not by this Licensegive permission to use their names for publicity for or to assert or
imply endorsement of any Modified Version.
5. COMBINING DOCUMENTS
You may combine the Document with other documents released under thisLicense, under the terms defined in section 4 above for modified
versions, provided that you include in the combination all of theInvariant Sections of all of the original documents, unmodified, and
list them all as Invariant Sections of your combined work in itslicense notice, and that you preserve all their Warranty Disclaimers.
The combined work need only contain one copy of this License, andmultiple identical Invariant Sections may be replaced with a single
copy. If there are multiple Invariant Sections with the same name butdifferent contents, make the title of each such section unique by
adding at the end of it, in parentheses, the name of the originalauthor or publisher of that section if known, or else a unique number.
Make the same adjustment to the section titles in the list of
Invariant Sections in the license notice of the combined work.
In the combination, you must combine any sections Entitled "History"
in the various original documents, forming one section Entitled"History"; likewise combine any sections Entitled "Acknowledgements",and any sections Entitled "Dedications". You must delete all sectionsEntitled "Endorsements".
6. COLLECTIONS OF DOCUMENTS
You may make a collection consisting of the Document and other documents
released under this License, and replace the individual copies of thisLicense in the various documents with a single copy that is included in
the collection, provided that you follow the rules of this License forverbatim copying of each of the documents in all other respects.
You may extract a single document from such a collection, and distributeit individually under this License, provided you insert a copy of this
License into the extracted document, and follow this License in allother respects regarding verbatim copying of that document.
7. AGGREGATION WITH INDEPENDENT WORKS
A compilation of the Document or its derivatives with other separate
and independent documents or works, in or on a volume of a storage ordistribution medium, is called an "aggregate" if the copyright
resulting from the compilation is not used to limit the legal rightsof the compilation's users beyond what the individual works permit.
When the Document is included in an aggregate, this License does notapply to the other works in the aggregate which are not themselves
derivative works of the Document.
If the Cover Text requirement of section 3 is applicable to these
copies of the Document, then if the Document is less than one half ofthe entire aggregate, the Document's Cover Texts may be placed on
covers that bracket the Document within the aggregate, or theelectronic equivalent of covers if the Document is in electronic form.
Otherwise they must appear on printed covers that bracket the wholeaggregate.
8. TRANSLATION
Translation is considered a kind of modification, so you may
distribute translations of the Document under the terms of section 4.Replacing Invariant Sections with translations requires special
permission from their copyright holders, but you may includetranslations of some or all Invariant Sections in addition to the
original versions of these Invariant Sections. You may include atranslation of this License, and all the license notices in the
Document, and any Warranty Disclaimers, provided that you also includethe original English version of this License and the original versions
of those notices and disclaimers. In case of a disagreement betweenthe translation and the original version of this License or a notice
or disclaimer, the original version will prevail.
If a section in the Document is Entitled "Acknowledgements",
"Dedications", or "History", the requirement (section 4) to Preserveits Title (section 1) will typically require changing the actual
title.
9. TERMINATION
You may not copy, modify, sublicense, or distribute the Document exceptas expressly provided for under this License. Any other attempt to
copy, modify, sublicense or distribute the Document is void, and willautomatically terminate your rights under this License. However,
parties who have received copies, or rights, from you under thisLicense will not have their licenses terminated so long as such
parties remain in full compliance.
10. FUTURE REVISIONS OF THIS LICENSE
The Free Software Foundation may publish new, revised versionsof the GNU Free Documentation License from time to time. Such new
versions will be similar in spirit to the present version, but maydiffer in detail to address new problems or concerns. See
http://www.gnu.org/copyleft/.
Each version of the License is given a distinguishing version number.
If the Document specifies that a particular numbered version of thisLicense "or any later version" applies to it, you have the option of
following the terms and conditions either of that specified version orof any later version that has been published (not as a draft) by the
Free Software Foundation. If the Document does not specify a versionnumber of this License, you may choose any version ever published (not
as a draft) by the Free Software Foundation.
|
|