CHAPITRE 3
COURBES ET SURFACES DE
POTENTIEL.
UNIVERSITE DE DOUALA /ECOLE
DOCTORALE DES SCIENCES FONDAMENTALES ET APPLIQUEES/ UFD PHYSIQUE ET SCIENCES DE
L'INGENIEUR / LABORATOIRE DE PHYSIQUE FONDAMENTALE.
3.1. INTRODUCTION
L'étude théorique d'un spectre
moléculaire nécessite, comme nous l'avons montré dans le
premier chapitre, la détermination précise des courbes
d'énergie potentiel des états électroniques
étudiés.
La fiabilité des résultats spectroscopiques
obtenus dépend de la qualité de ces courbes ou de ces surfaces
qui dépendent elles même essentiellement de la qualité de
la base d'OA et de la performance des méthodes de calcul
utilisées. Pour cela, nous les testons en essayant de reproduire
quelques résultats expérimentaux.
3.2. ETUDE DES FRAGMENTS NO, NO - ET N2O.
3.2.1. Bases utilisées
Les molécules NO et NO- sont diatomiques
hétéronucléaires. Elles appartiennent donc au groupe de
symétrie C8 v . Quant à la molécule N2O, elle
est linéaire et ne
possède pas de centre d'inversion. Elle appartient donc
également au même groupe de
symétrie . Comme les calculs
doivent être effectués dans une base finie, nous
C8 v
choisissons de les mener dans le groupe de symétrie qui
est un sous-groupe
C2 v
distingué de .
C8 v
Nous avons par la suite utilisé plusieurs bases
optimisées par Dunning . Il
[16]
s'agit notamment des bases aug-cc-avtz,
aug-cc-avqz et aug-cc-av5z auxquelles on a associé les
ensembles de fonctions spd, spdf, spdfg et spdfgh.
Pour mettre en évidence l'effet d'une base
donnée, nous avons effectué des calculs au niveau MCSCF avec des
bases différentes. Les résultats sont illustrés par les
courbes des figures 3.1, 3.2, 3.3, et 3.4.
UNIVERSITE DE DOUALA /ECOLE
DOCTORALE DES SCIENCES FONDAMENTALES ET APPLIQUEES/ UFD PHYSIQUE ET SCIENCES DE
L'INGENIEUR / LABORATOIRE DE PHYSIQUE FONDAMENTALE.
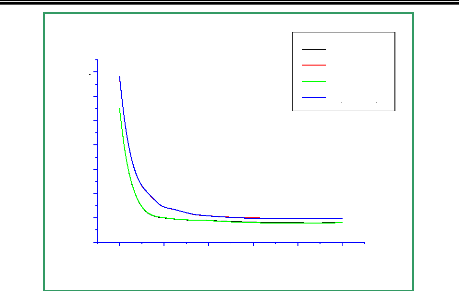
-128
-129
-130
1 2 3 4 5 6
Energie (u.a.)
-126
-127
-123
-124
-125
2Ó+ (avtz/spdf) 2Ä
(avtz/spd) 2Ó+ (avtz/spdf) 2Ä (avtz/spd)
Distance internucléaire (Bohr)
Figure 3.1 : Deux états
électroniques du NO ( 2 Ó+ ,1Ä )
calculés au niveaux MCSCF/avtz/spd et
MCSCF/avtz/spdf.
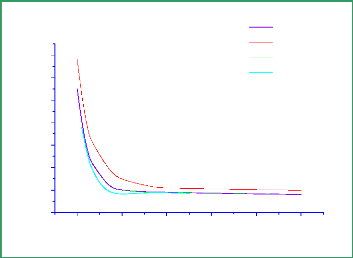
-123
-124
1 2 3 4 5 6
Distance internucléaire (Bohr)
-129
-130
-125
-126
-127
Energie (u. a.)
-128
2Ó+ (avtz/spd) 2Ä
(avtz/spd) 2Ó+ (avtz/spdf) 2Ó+
(avqz/spdf)
Figure 3.2 : Deux états
électroniques du NO ( 2 Ó+ ,1Ä )
calculés au niveaux MCSCF/avtz/spdf et
MCSCF/avqz/spdf.
UNIVERSITE DE DOUALA /ECOLE
DOCTORALE DES SCIENCES FONDAMENTALES ET APPLIQUEES/ UFD PHYSIQUE ET SCIENCES DE
L'INGENIEUR / LABORATOIRE DE PHYSIQUE FONDAMENTALE.
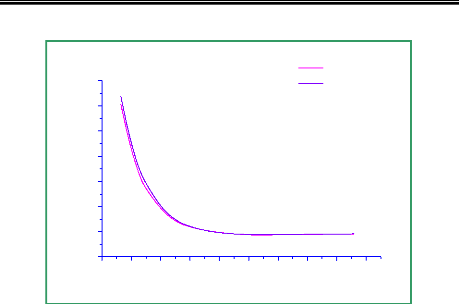
0,4 0,6 0,8 1,0 1,2 1,4 1,6 1,8 2,0 2,2
Distance internucléaire (Ang)
-123
-124
-125
Energie (u.a.)
-126
-127
-128
-129
-130
B2Ð avqz (spdf) B2Ð avtz (spdf)
Figure 3.3 : premier état excité
doublet 2Ð du NO calculé au niveau MCSCF/avtz/spdf et
MCSCF/avqz/spdf.
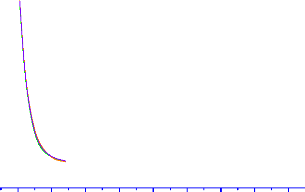
1Ó+ (av5z) 1Ó+ (avqz)
1Ó+ (avtz)






0,0 0,5 1,0 1,5 2,0 2,5 3,0 3,5 4,0 4,5
Distance internucléaire (Bohr)
-124
-125
-129
-130
-126
Energie (u. a.)
-127
-128
Figure 3.4 : Etat
1Ó+ du NO-
déterminé au niveau de calcul MCSCF/avtz, MCSCF/avqz et
MCSCF/av5z.
UNIVERSITE DE DOUALA /ECOLE
DOCTORALE DES SCIENCES FONDAMENTALES ET APPLIQUEES/ UFD PHYSIQUE ET SCIENCES DE
L'INGENIEUR / LABORATOIRE DE PHYSIQUE FONDAMENTALE.
Observations et
interprétations.
· La figure 3.1 montre que les effets de l'ajout des
fonctions f sur une triple zêta pour le monoxyde d'azote bien que
négligeables, sont plus marqués aux grandes distances. Ce qui est
très normal, puisque l'ajout de ces fonctions augmente la
flexibilité de la base.
· Les figures 3.2 et 3.3 montrent que pour un
état excité, les bases triple et quadruple zêta donnent les
mêmes résultats aux grandes distances, et des résultats
différents aux courtes distances. La différence étant
assez notable, le système NO est bien décrit au moins dans une
base quadruple zêta.
· La figure 3.4 quant à elle montre que dans le cas
de l'ion, il n'y a pratiquement pas de différence à travailler
dans une base triple zêta que dans une quadruple zêta. Une
différence notable aux très grandes distances n'apparaît
que lorsque les calculs sont effectués dans la base av5z. Compte tenu de
la croissance du coup informatique avec la base de calcul, si on
s'intéresse aux grandeurs énergétiques (énergie
totale, énergie d'ionisation et affinité électronique d'un
système moléculaire) ou aux grandeurs géométriques
(longueurs et angles de liaison, conformations), on peut sans erreurs notables
effectuer les calculs dans la base avqz. Il serait prudent d'effectuer les
calculs au moins dans une base av5z si on s'intéresse aux
propriétés électriques (moments dipolaires, multipolaires,
polarisabilités et
hyperpolarisabilités), magnétiques
(déplacement chimique et constantes de couplage de RMN) et
spectroscopiques.
3.2.2. Méthodes de calcul
utilisées.
Pour ces trois fragments du N 2 O 2 ou
duN 2 O- 2 , nous avons utilisé
successivement les méthodes HF, MCSCF, MRCI, MP4 et CCSD
(T) dans la base avqz.
UNIVERSITE DE DOUALA /ECOLE
DOCTORALE DES SCIENCES FONDAMENTALES ET APPLIQUEES/ UFD PHYSIQUE ET SCIENCES DE
L'INGENIEUR / LABORATOIRE DE PHYSIQUE FONDAMENTALE.
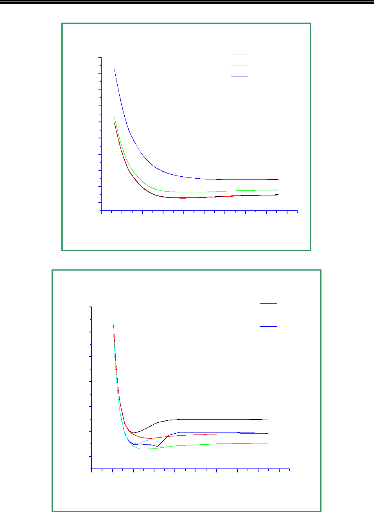
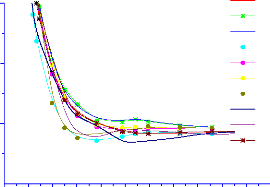
Figure 3.5 : Etat 2 II de la
molécule NO donnés par les méthodes HF, MCSCF et MRCI.
MRCI MRCI+Q MCSCF SCF-HF
-121
-122
-123
-124
-125
-126
-127
-128
-129
-130
Energie (u.a.)
0,4 0,6 0,8 1,0 1,2 1,4 1,6 1,8 2,0 2,2
Distance internucléaire (Ang)
-124
-125
-126
-127
-128
-129
Energie (u. a.)
R-HF MCSCF MRCI MD4 CCSD(T)
-130
0,0 0,5 1,0 1,5 2,0 2,5 3,0 3,5 4,0 4,5
Distance interatomique (Bohr)
Figure 3.6 : Etat 2 II de la
molécule NO donnés par les méthodes HF, MCSCF et MRCI, MP4
et CCSD. Observations et interprétations.
La figure 3.5 montre que la correction de Davidson
apportée à la méthode MRCI peut
être
négligée pour les états du NO .Les figures 3.5 et 3.6
montre qu'en terme de
UNIVERSITE DE DOUALA /ECOLE
DOCTORALE DES SCIENCES FONDAMENTALES ET APPLIQUEES/ UFD PHYSIQUE ET SCIENCES DE
L'INGENIEUR / LABORATOIRE DE PHYSIQUE FONDAMENTALE.
précision, on a l'ordre suivant : HF <<
MCSCF < CCSD(T) < MP4
< MRCI. Ce quiest bien en accord avec les
prévisions faites au chapitre 1.
3.2.3. Structure électronique du NO, NO- et
N2O.
Commençons par présenter quelques états
électroniques des systèmes étudiés.
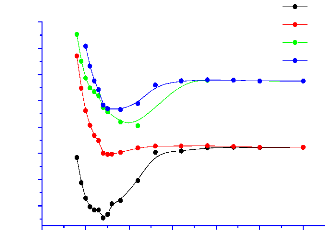
Energie (u.a.)
|
-127
-128
-129
|
|
|
X2Ó+
A2Ä
A2Ó+
B2Ó+
2Ä
X2Ð
A2Ð
B2Ð
C2Ð
D2Ð
|
|
|
-130
0,4 0,6 0,8 1,0 1,2 1,4 1,6 1,8 2,0 2,2
Distance internucléaire (Ang)
Figure 3.7 : les dix états
électroniques les plus bas du NO dans ses deux premières
symétries au niveau MCSCF/avqz.
UNIVERSITE DE DOUALA /ECOLE
DOCTORALE DES SCIENCES FONDAMENTALES ET APPLIQUEES/ UFD PHYSIQUE ET SCIENCES DE
L'INGENIEUR / LABORATOIRE DE PHYSIQUE FONDAMENTALE.
3Ä 3Ó+ 3Ä
3Ó+
X3Ð
3Ð 3Ð 3Ð
3Ó- 3Ó-
3Ä

0,4 0,6 0,8 1,0 1,2 1,4 1,6 1,8 2,0 2,2 2,4 2,6
Distance internucléaire (Ang)
-127
-130
-128
Energie (u.a.)
-129
Figure 3.8 : les onze états
électroniques les plus bas du NO- au niveau MCSCF/avqz.
-183,2
-183,3
1Ó+ 1Ä
1Ó+ 1Ó+
-183,4
Energie (u. a.)
-183,5
-183,6
-183,7
-183,8
-183,9
0,5 1,0 1,5 2,0 2,5 3,0 3,5
RN-N (Ang)
Figure 3.9 : les quatre états
électroniques les plus bas du N2O lorsqu'on tire sur la liaison N-N.
UNIVERSITE DE DOUALA /ECOLE
DOCTORALE DES SCIENCES FONDAMENTALES ET APPLIQUEES/ UFD PHYSIQUE ET SCIENCES DE
L'INGENIEUR / LABORATOIRE DE PHYSIQUE FONDAMENTALE.
|
-183,2
|
|
|
|
1Ó+ 1Ä
1Ó+
|
|
|
|
|
|
|
-183,3 -183,4 -183,5 -183,6 -183,7
|
|
|
|
|
|
|
|
|
|
|
-183,8
|
|
|
|
|
|
|
|
|
-183,9
|
|
|
|
|
|
0,5 1,0 1,5 2,0 2,5 3,0 3,5
RN-O (Ang)
Figure 3.10 : les trois états
électroniques les plus bas du N2O lorsqu'on tire sur la liaison N-O.
Observations et interprétations.
· Les figures 3.7 et 3.8 montrent respectivement que les
états fondamentaux du NO et du NO- sont
respectivement2 Ð et 3Ð. Il ressort des courbes
3.9 et 3.10 que le fondamental du N2O est l'état 1 .
Ó+
· Les états excités du N2O sont
très éloignés de leur fondamental, contrairement à
ceux du NO et du NO-. Ce qui signifie que les molécules NO et
NO- sont facilement excitables par rapport à la
molécule N2O.
· On observe également sur les figures 3.7 et 3.8
les croisements évités entre les états
2Ó d'une part et les états d'autre part.
2Ð
· Les énergies au seuil de dissociation des
systèmes NO et NO- sont respectivement -128. 569
eV et -129. 1746 eV.
· Les calculs montrent que le protoxyde d'azote N2O est
linéaire. Ce résultat était prévisible suivant la
règle de Walsh : La géométrie d'une molécule est
déterminée par le nombre d'électrons de valence. Les
molécules triatomiques ne contenant pas l'hydrogène et
possédant au plus 16 électrons de valence sont linéaires.
C'est bien le cas du N2O qui
UNIVERSITE DE DOUALA /ECOLE
DOCTORALE DES SCIENCES FONDAMENTALES ET APPLIQUEES/ UFD PHYSIQUE ET SCIENCES DE
L'INGENIEUR / LABORATOIRE DE PHYSIQUE FONDAMENTALE.
posséde 5 électrons de valence par atome d'azote
et 6 électrons de valence par atome d'oxygène.
3.3. ETUDE DE L'ETAT FONDAMENTAL DES SYSTEMES
N 2 O- 2 ET N 2 O2 Comme nous l'avons
mentionné à l'introduction générale, les
molécules N 2 O- 2 et N 2 O 2 appartiennent au
groupe ponctuel de symétrie . Nous présentons ci-dessous
C2 v
leurs structures géométriques. Par la suite, nous
déterminerons leurs modes de vibrations et leurs activités en
spectroscopie Raman et infrarouge.
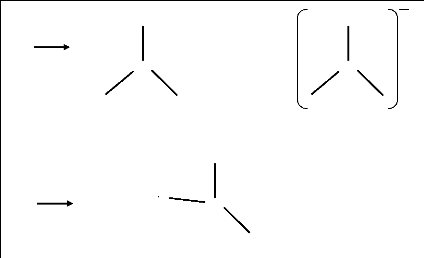
O
Cs
O
O
C2 v
N
O
N
N
O
O
N
N
N
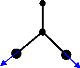
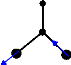
Figure 3.11 : Structures
géométriques du et du
N 2 O 2 N 2 O- 2 en symétrie C2v et
Cs.
3.3.1. Modes de vibration des molécules
N 2 O 2 et en symétries
et
N 2 O- 2 C2 v Cs
Chacune des molécules N 2 O 2 et
possède 4 atomes ; soit 6 modes
N 2 O- 2
normaux de vibrations indiquant ainsi que les vibrations de
chacun de ces deux systèmes sont décrites par 6
coordonnées internes ( r1 ,r2 ,r 3
,â 1, â 2, â 3) (voir figure
3.2) et 6 coordonnées de symétrie.
UNIVERSITE DE DOUALA /ECOLE
DOCTORALE DES SCIENCES FONDAMENTALES ET APPLIQUEES/ UFD PHYSIQUE ET SCIENCES DE
L'INGENIEUR / LABORATOIRE DE PHYSIQUE FONDAMENTALE.
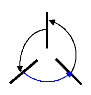
â 1
â3
r3
r2
O
O
â2
N
r1
N
Figure 3.12 : coordonnées internes du
ou .
N 2 O 2 N 2 O- 2
Par ailleurs, on montre que la représentation
irréductible qui engendre les vibrations de chacun de ces
systèmes est :
v = 3A 1 ? B 1?2B2 . (3.3)
Ainsi, celles engendrant les vibrations d'allongement et
angulaires sont données respectivement par :
r = 2A1 ? B 2 , (3.4)
= ? ? 2 . (3.5)
â A 1 B 1 B
Chacun de ces deux systèmes possède donc en
symétrie C 2v :
· Deux modes de vibration d'allongement complètement
symétriques suivant A 1.
· Un mode de vibration d'allongement complètement
antisymétrique suivant B2.
· Un mode de vibration angulaire complètement
symétrique suivant A1.
· Un mode de vibration angulaire complètement
antisymétrique suivant B2.
· Un mode de vibration angulaire antisymétrique
suivant B1.
Tous ces 6 modes de vibration sont actifs en spectroscopie
Infrarouge et Raman. Les fonctions d'onde de symétrie adaptées
à ces modes de vibration sont :
UNIVERSITE DE DOUALA /ECOLE
DOCTORALE DES SCIENCES FONDAMENTALES ET APPLIQUEES/ UFD PHYSIQUE ET SCIENCES DE
L'INGENIEUR / LABORATOIRE DE PHYSIQUE FONDAMENTALE.
Ø 1 (A 1 ) = r 1
|
|
Symmetric stretching
mode
|
|
1
Ø (A ) =
2 1 2
(r 2 r + 3 )
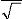
Symmetric stretching
mode
1
Ø (B ) =
3 2 2
(r 2 r - 3 )
asymmetric stretching
mode
1
Ø (A ) =
4 1 2
Ø 5 (B 1 ) = â 1
scisoring mode
Bending mode...
(1 â +â3)

1
Ø (B ) =
6 2 2
(1 â -â 3 )
Bending mode
En symétrie Cs pour le N 2
O2 , nous obtenons:
v = 5A ? A"' (3.6)
Ainsi, dans ce groupe, la molécule a :
· 5 modes de vibration suivant actifs en spectroscopie
infrarouge et Raman.
A '
UNIVERSITE DE DOUALA /ECOLE
DOCTORALE DES SCIENCES FONDAMENTALES ET APPLIQUEES/ UFD PHYSIQUE ET SCIENCES DE
L'INGENIEUR / LABORATOIRE DE PHYSIQUE FONDAMENTALE.
· 1 mode de vibration suivant actif en spectroscopie
infrarouge et Raman.
A "Contrairement aux calculs effectués par
Arnold et Neumark, où ils trouvent qu'en
symétrie C , toutes les vibrations ont lieu dans le plan
de la molécule, soit suivant .
A '
s
3.3.2. Géométries optimisées des
molécules N 2 O2 et .
N 2 O- 2
Nous présentons ci-dessous les géométries
optimisées sous Gaussian 03W[25].
méthode Energie RN-N RN-O1 RN-O2 < NNO1 <NNO2
UHF/6-31+G*
|
-258.493 764
|
1.2531
|
1.2541
|
1.2541
|
119.2542°
|
119.2542°
|
UMP2/6-31+G*
|
-259.165 960
|
1.2704
|
1.2528
|
1.2528
|
120.2575°
|
120.2575°
|
UMP2/6-311+G*
|
-259.274 329
|
1.2726
|
1.2402
|
1.2402
|
120.1256°
|
120.1256°
|
|
Tableau 3.1 : Résultats
ab-initio de l'anion N 2 O- 2 c
méthode
|
ù1
|
ù2
|
ù3
|
ù4
|
ù5
|
ù6
|
théorie
|
|
|
|
|
|
|
UMP2/6-31+G*
|
1411.3928
|
1243.0460
|
679.7760
|
997.7958
|
1539.0959
|
609.0024
|
UMP2/6-311+G* expérience
|
1421.1382
|
1222.9497
|
695.2410
|
1000.9260
|
1543.1580
|
623.7265
|
Ar matrix a
|
1356
|
1004
|
...
|
...
|
1199
|
...
|
Ne matrix b
|
1359
|
1008
|
...
|
...
|
1205.5
|
...
|
|
a Références [26] et
[27].
b Référence [26].
c Les énergies sont
données en u.a., les longueurs de liaison en angströms et les
fréquences vibrationnelles en cm-1
Tableau 3.2 : Fréquences
propres de vibration de l'anion N 2 O- 2 c
UNIVERSITE DE DOUALA /ECOLE
DOCTORALE DES SCIENCES FONDAMENTALES ET APPLIQUEES/ UFD PHYSIQUE ET SCIENCES DE
L'INGENIEUR / LABORATOIRE DE PHYSIQUE FONDAMENTALE.
Etat
|
paramètre
|
HF/6-31+G*
|
MP2/6-311+G*
|
Mode
|
HF/ 6-31+G*
|
MP2/ 6-311+G*
|
1
|
|
|
|
|
|
|
A1
|
RN-N
|
1.3021
|
1.2525
|
ù1
|
1534.5804
|
1391.6340
|
(A)
|
RN-O
|
1.1895
|
1.2299
|
ù2
|
1178.1499
|
1108.9664
|
|
<NNO1
|
114.3236°
|
114.0619°
|
ù3
|
691.8905
|
600.2176
|
|
<NNO2
|
114.3236°
|
114.0619°
|
ù4
|
875.5625
|
742.1835
|
|
Energie
|
-258.332 951
|
-259.123 993
|
ù5
|
1761.1334
|
1689.8163
|
|
|
-258.390 078e
|
|
ù6
|
175.8113i
|
800.7031i
|
1
A1
|
RN-N
|
1.127
|
1.1302
|
ù1
|
2196.3178
|
2318.5474
|
(B)
|
RN-O
|
1.3502
|
1.4735
|
ù2
|
1179.6093
|
1071.1945
|
|
<NNO1
|
146.8211°
|
147.4966°
|
ù3
|
895.7086
|
637.9780
|
|
NNO2
|
146.8211°
|
147.4966°
|
ù4
|
510.5793
|
520.0772
|
|
Energie
|
-258.293 542
|
-259.133 323
|
ù5
|
668.7958
|
983.2470
|
|
|
-258.332 402e
|
|
ù6
|
461.6837
|
51.6401
|
|
c Les énergies sont
données en u.a les longueurs de liaison en angströms et les
fréquences vibrationnelles en cm-1.
e Résultat HF dans la base
6-311+G*
c
Tableau 3.3 : Résultats ab-initio
des états singulets du en symétrie C
N 2 O 2 .
2v
|
Energie
|
RN-N
|
RN-O1
|
RN-O2
|
< NNO1
|
<NNO2
|
3
A1
|
|
|
|
|
|
|
UHF/6-31+G*
|
-258.426 981
|
1.225
|
1.270
|
1.270
|
127.5°
|
127.5°
|
MP2/6-31+G*
|
-258.998 122
|
1.182
|
1.294
|
1.294
|
129.6°
|
129.6°
|
3
|
|
|
|
|
|
|
B1
|
|
|
|
|
|
|
UHF/6-31+G*
|
-258.374 977
|
1.282
|
1.280
|
1.280
|
122.2°
|
122.2°
|
MP2/6-31+G*
|
-258.952 149
|
1.248
|
1.301
|
1.301
|
123.6°
|
123.6°
|
3
|
|
|
|
|
|
|
B2
|
|
|
|
|
|
|
UHF/6-31+G*
|
-258.341 847
|
1.144
|
1.388
|
1.388
|
127°
|
127°
|
MP2/6-31+G*
|
-258.953 932
|
1.188
|
1.311
|
1.311
|
116.6°
|
116.6°
|
3
|
|
|
|
|
|
|
A2
|
|
|
|
|
|
|
UHF/6-31+G*
|
-258.412 978
|
1.405
|
1.185
|
1.185
|
115.3°
|
115.3°
|
MP2/6-31+G*
|
-259.058 994
|
1.441
|
1.230
|
1.230
|
115.1°
|
115.1°
|
|
c Les énergies sont données en u.a.,
les longueurs de liaison en angströms .
c
Tableau 3.4 : Résultats ab-initio
des états triplets du en symétrie C
N 2 O 2 .
2v
UNIVERSITE DE DOUALA /ECOLE
DOCTORALE DES SCIENCES FONDAMENTALES ET APPLIQUEES/ UFD PHYSIQUE ET SCIENCES DE
L'INGENIEUR / LABORATOIRE DE PHYSIQUE FONDAMENTALE.
|
ù1
|
ù2
|
ù3
|
ù4
|
ù5
|
ù6
|
3
A1
|
|
|
|
|
|
|
UHF/6-31+G*
|
1543
|
1038
|
608
|
608
|
1288
|
576
|
MP2/6-31+G*
|
1972
|
1142
|
649
|
819
|
1776
|
585
|
3
|
|
|
|
|
|
|
B1
|
|
|
|
|
|
|
UHF/6-31+G*
|
1634
|
1032
|
657
|
437
|
923
|
545
|
MP2/6-31+G*
|
1467
|
1022
|
618
|
627
|
571
|
1180i
|
3
|
|
|
|
|
|
|
B2
|
|
|
|
|
|
|
UHF/6-31+G*
|
2037
|
869
|
495
|
618
|
977
|
561
|
MP2/6-31+G*
|
1646
|
959
|
611
|
541
|
1164
|
312
|
3
|
|
|
|
|
|
|
A2
|
|
|
|
|
|
|
UHF/6-31+G*
|
1576
|
1050
|
716
|
777
|
1868
|
565
|
MP2/6-31+G*
|
1431
|
902
|
625
|
638
|
1815
|
483
|
|
c Les fréquences vibrationnelles sont en
cm-1.
c
Tableau 3.5 : Résultats ab-initio
des fréquences de vibration des états triplets du en
symétrie C
N 2 O 2 .
2v
Observations et
interprétations.
L'analyse des tableaux 3.1, 3.2, 3.3, 3.4 et 3.5 montre que :
· Les résultats de calculs effectués dans la
base 6-31+G* sont en parfait accord avec ceux effectués par Arnold et
Neumark[1].
· L'optimisation de géométrie aux niveaux
HF/6-31+G* et MP2/6-311+G* prévoit que l'ion moléculaire N2O2 -
est plan. Il appartient au groupe de symétrie C2v et son fondamental est
l'état 2BB2 pour lequel la molécule a des longueurs et
des angles de liaison presque égaux. Ses angles de valence valent
environ 120° et Ses longueurs de liaison sont angström et
R N - O = 1.254
R N - N = 1.253 angström au niveau HF/6-31+G*. Au niveau
MP2/6-311+G*, ces longueurs valent respectivement 1.24 angström et 1.27
angström. Remarquons que la valeur calculée pour R N - N au niveau
HF/6-31+G* est caractéristique d'une double liaison ; par exemple ,
[28] R N - N = 1.252 Angström.
· Quant à la molécule neutre
N2O2, elle est également plane et possède deux
géométries d'équilibre (A et B) en symétrie
C2v et une en symétrie Cs. Son état
fondamental est l'état 1A1. Les calculs effectués au
niveau MP2/6-311+G*
UNIVERSITE DE DOUALA /ECOLE
DOCTORALE DES SCIENCES FONDAMENTALES ET APPLIQUEES/ UFD PHYSIQUE ET SCIENCES DE
L'INGENIEUR / LABORATOIRE DE PHYSIQUE FONDAMENTALE.
montrent que l'angle de valence <ONO vaut seulement
65° pour la structure B contre 132° pour la structure A. aussi, cet
angle est le paramètre géométrique qui varie le plus
lorsqu'un photodétachement sur l'anion conduit aux états triplets
de la molécule neutre. En plus, R N - N ne change pas beaucoup via un
photodétachement sur le N 2 O- 2 conduisant aux
états triplets
3 A 1 , B 1 , et B 2
3 3
|
.Ainsi,la liaison N-N dans ces états est
fondamentalement une
|
|
double liaison juste aussi comme dans l'anion.
Cependant, la longueur R N - N pour l'état triplet de la molécule
neutre mesure 0.17 angström de plus que
3 A2
dans l'anion. La valeur calculée (1.441 angström au
niveau MP2/6-31+G*) est
caractéristique d'une liaison simple N-N. par
exemple, angström
R N - N = 1.449
dans le [29]
N 2 H 4 .
· Par ailleurs, les énergies des molécules et
à leurs limites de
N 2 O- 2 N 2 O 2
dissociation sont respectivement -258.921eV et
-258.363 eV au niveau HF/avqz-spdf.
· En symétrie C2 V , La structure B de la
molécule neutre N 2 O2 est la plus stable.
· Lorsqu'on ne prend pas en compte la corrélation
statique, on peut classer les états triplets du comme suit : . Cet ordre
s'estompe
3 A 1 < A 2 < B 1 < B 2
3 3 3
N 2 O 2
lorsqu'on prend en compte ce type de corrélation (la
bonne description). Dans
ce cas, ces états sont classés comme
suit : au niveau MP2/ 6-
3 A 2 < A 1 < B 2 < B
3 3 3
1
31+G*. Notons que cet ordre pourrait bien changer en fonction du
niveau de calcul. En effet, les calculs effectués par Arnold et Neumark
[1] au niveau MP4/6-311+G* les classent comme suit : .
3 A 2 < A 1 < B 1 < B
3 3 3
2
· Les états triplets de la molécule N
2 O2 sont « en général » plus
hauts
énergétiquement que les états singulets
des trois géométries de cette molécule. Ce résultat
nous permettra certainement de simplifier l'étude de la
métastabilité de notre système.
UNIVERSITE DE DOUALA /ECOLE
DOCTORALE DES SCIENCES FONDAMENTALES ET APPLIQUEES/ UFD PHYSIQUE ET SCIENCES DE
L'INGENIEUR / LABORATOIRE DE PHYSIQUE FONDAMENTALE.
· L'optimisation de géométrie au niveau
HF ou MP2 surestime les fréquences des modes propres de vibrations de la
molécule et s'accorde raisonnablement avec les résultats
expérimentaux.
La molécule sous l'effet de ses vibrations, peut perdre
son état après un temps relativement court ou long. On dira ainsi
qu'elle présente des états métastables...
3.3.3. Stabilité ou métastabilité
d'un système moléculaire.
Un anion moléculaire est dit stable ou métastable
dans un état électronique donné lorsqu'il satisfait les
trois critères ci-dessous [30] :
1) L'affinité électronique de l'anion
relativement à cet état est positive. Par
définition, contrairement au potentiel d'ionisation, l'affinité
électronique X
A - d'un
anion traduit la difficulté à enlever un
électron d'une couche électronique de cet
X-
anion pour former la molécule neutre . C'est donc la
différence entre l'énergie de X
l'état fondamental de la molécule neutre (parent)
et celle de l'état fondamental de l'anion correspondant. Elle s'exprime
donc comme suit :

A - E (X) E (X )
-
= 0 - 0 . (3.7)
X
Ainsi, une affinité électronique positive
signifie physiquement qu'à partir des courbes de potentiel
électroniques, l'anion est en dessous de son neutre parent, plus
précisément dans la région d'équilibre de leurs
états fondamentaux respectifs.
Donnons une expression plus explicite de l'affinité
électronique. Pour cela, considérons la représentation
schématique ci-dessus.
UNIVERSITE DE DOUALA /ECOLE
DOCTORALE DES SCIENCES FONDAMENTALES ET APPLIQUEES/ UFD PHYSIQUE ET SCIENCES DE
L'INGENIEUR / LABORATOIRE DE PHYSIQUE FONDAMENTALE.
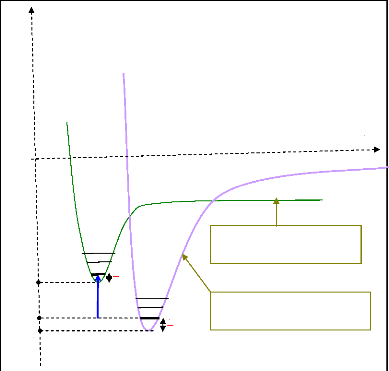
E
0
E-0
E0
A -
X
2 ù
1
0
2
1
ù0
-
Courbe de potentiel de la
molécule neutre.
Courbe de potentiel de
l'anion
R
Figure 3.13 : Illustration de
l'affinité électronique.
Ainsi, la relation de Chales permet d'écrire :
A + ù = E - + ù (3.8)
1 ( )
- 1
E 2 0
-
X - 2 0 0 0
Soit donc :
( 1 ) ( )
E E 1
-
A + (3.9)
X - 0 2 0
ù - 2 ù
-
= + 0 0
E0 et étant respectivement les énergies
électroniques des fonds des puits de
Å0 -
potentiel de la molécule neutre et de l'anion.
ù0 et 0
ù sont respectivement les fréquences
-
propres de vibration de la molécule neutre et de
l'anion en cm-1 , dans leurs états fondamentaux. 2 1 ù
0 et 1 2 ù- 0 sont appelées «
zero-point correction » respectivement de la molécule neutre et de
l'ion.
UNIVERSITE DE DOUALA /ECOLE
DOCTORALE DES SCIENCES FONDAMENTALES ET APPLIQUEES/ UFD PHYSIQUE ET SCIENCES DE
L'INGENIEUR / LABORATOIRE DE PHYSIQUE FONDAMENTALE.
2) La fonction d'onde associée à cet
état doit présenter de faibles interactions avec les états
du continuum électronique. Physiquement, ceci signifie que
le puit de potentiel électronique de l'anion doit être
suffisamment profond afin que la moindre excitation du système ne
l'amène hors de cet état.
3) Le couplage spin-orbite entre cet état
et un état de transition possible est pratiquement nul.
Physiquement, ceci se traduit par le non croisement de la courbe de potentiel
électronique de cet état et de celles des états du neutre
ayant une multiplicité de spin différente d'une unité de
celle de l'anion.
Pour étudier la stabilité ou la
métastabilité de l'anion N 2 O- 2 , nous avons effectué
des investigations sur cet anion et sur la molécule
neutre associée aux mêmes géométries. Dans la suite,
nous préciserons les bases et les méthodes de calcul
utilisées pour mener cette étude tout en présentant les
différents résultats obtenus.
3.3.4. Base et méthode de calcul
utilisée.
Michels et Montgomery[31], à partir des
investigations au niveau HF/6-31G* ont trouvé que l'état
fondamental du N 2 O 2 est l'état
1A1. En tenant compte de ce
résultat
Nous avons effectué des calculs dans les bases
aug-cc-pVTZ et aug-cc-pVQZ de Dunning au niveau HF vu nos limites dans les
outils informatiques. Les résultats sont présentés
à travers les courbes de potentiel ci-dessous.
3.3.5. Courbes de potentiel
électroniques.
Les calculs présentés ici ont été
faits pour les variations d'angles.
UNIVERSITE DE DOUALA /ECOLE
DOCTORALE DES SCIENCES FONDAMENTALES ET APPLIQUEES/ UFD PHYSIQUE ET SCIENCES DE
L'INGENIEUR / LABORATOIRE DE PHYSIQUE FONDAMENTALE.
112 113 114 115 116
Angle <NNO (°)


-258,1324
-258,1300
-258,1302
|
Energie (u.a)
|
-258,1304 -258,1306 -258,1308 -258,1310 -258,1312 -258,1314
-258,1316 -258,1318 -258,1320 -258,1322
|
|
|
Figure 3.14 : Etat fondamental
X1 du
A1 N 2 O2 pour des variations
d'angles /HF /avqz/spdf.
UNIVERSITE DE DOUALA /ECOLE
DOCTORALE DES SCIENCES FONDAMENTALES ET APPLIQUEES/ UFD PHYSIQUE ET SCIENCES DE
L'INGENIEUR / LABORATOIRE DE PHYSIQUE FONDAMENTALE.
-258,30

|
Energie (eV)
|
-258,32 -258,34 -258,36 -258,38 -258,40
|
|
|
90 95 100 105 110 115 120 125
Angle (dégrés)

-258,42
Figure 3.15 : Etat fondamental X
du
2 B2 N 2 O- 2 pour des variations d'angles
/HF/avqz/spdf
Après une étude minutieuse des configurations
dominantes des états représentés
sur les figures 3.14 et 3.15, il en ressort que l'état
X1 est le neutre parent associé à
A1
l'état . L'état est bien en dessous de son neutre
parent. Ainsi, l'ion
X B 2
2 X B 2
2 N 2 O- 2 a
une affinité électronique positive. En effet,
compte tenu des optimisations de
géométries
récapitulées dans les tableaux 3.1, 3.3 et de la formule (3.9),
l'affinité
électronique de la molécule N 2 O- 2 est :
A - = -259.133 323+0.012605 -259.274
329+0.014824 0.138787u.a. 0
( ) (
- ) = >
N 2 O2
Donc l'anion N 2 O- 2 existe bel et bien. Il est situé
à environ 3.8 eV en dessous du neutre.
UNIVERSITE DE DOUALA /ECOLE
DOCTORALE DES SCIENCES FONDAMENTALES ET APPLIQUEES/ UFD PHYSIQUE ET SCIENCES DE
L'INGENIEUR / LABORATOIRE DE PHYSIQUE FONDAMENTALE.
| 

