|

L'eau est l'élément le plus indispensable à
la vie humaine.
En effet, avec la révolution industrielle, l'eau n'a plus
l'unique intérêt vital mais aussi il présente un
intérêt économique et sanitaire.
L'eau est le seul composé qui peut se trouver dans les
trois états de la matière (solide, liquide, gazeux) aux
températures ordinaires ; A l'état solide ou glace, elle
constitue les calottes glaciers polaires, on la trouve également sous
forme de neige, de grêle et de grive dans les nuages. Elle est
présente à l'état liquide dans les nuages de pluie sous
formes de gouttelettes d'eau ; de plus elle recouvre environ les trois
quarts de la surface de la terre sous forme de marais, de lacs, de
rivières et d'océans. On la trouve aussi à l'état
gazeux ou vapeur d'eau dans le brouillard et les nuages.
Malgré son importance et la diversité de ses
ressources, l'eau présente une richesse en danger pour des raisons de
mal exploitation, de mauvaise distribution et surtout de problèmes de
pollution causés généralement par certaines industries.
Pour cela, la Tunisie a chargé
la SONEDE (Société
Nationale d'Exploitation et de Distribution des Eaux) de bien
veiller sur la bonne exploitation et distribution de cette richesse nationale
sur tout le territoire tunisien.
Les attributions de la SONEDE consiste
à :
ü La planification de l'approvisionnement en eau potable
dans tout le pays,
ü L'étude et la réalisation des installations
de captage, l'adduction et le traitement de
l'eau,
ü Gestion technique et financière des
réseaux.
La SONEDE dispose de 12 stations de
traitement de l'eau dont la station du complexe de GHDIR EL
GUOLLA est la plus importante. Le complexe couvre une surface de
5 hectares, situé au nord-Ouest de la capitale, sur la route reliant
Tunis à MJEZ EL BEB, à 9 km du centre ville, et
destiné à traiter les eaux distribuer à Tunis et au
banlieues, autrement il assure l'alimentation de 3.5 millions d'habitants
environ. Sa capacité est de 600.000 m3 par jour.
En dehors de ses attributions, la
SONEDE joue un rôle très important dans
l'encadrement des étudiants inscrits aux universités tunisiennes
dans plusieurs domaines technique et administratif en fournissant des stages
dans le but de participer à la formation des futurs professionnels.
Ce rapport de stage présente un résumé des
plus importants concepts et techniques appris le long d'un stage ouvrier d'un
mois du 01/07 jusqu'au 31/07/2008 effectué au sein de la
SONEDE au complexe de GHDIR EL
GUOLLA à Tunis dans le laboratoire
central concernant le contrôle de la
qualité bactériologique et physico-chimique de
l'eau.
![]()
Page
· Analyse des
chlorures......................................................3
· Mesure du
pH....................................................................3
· Analyse des résidus
secs................................................4
· Analyse des
sulfates........................................................5
· Analyse de la
dureté........................................................6
· Analyse de l'oxydabilité au KMnO4
................................8
· Mesure de la
turbidité......................................................9
· Mesure de
l'alcalinité.......................................................9
I/ Analyse des
chlorures :
I.1/ Principe :
L'analyse est basée sur le titrage
volumétrique des chlorures par le nitrate d'argent
Le chromate d'argent résulte de la réaction du
nitrate d'argent en excès avec le chromate de potassium quand tous les
chlorures disponibles ont été précipités par la
réaction plus favorisée entre l'argent et les ions chlorures
La fin de la réaction est indiquée par
l'apparition d'une légère teinte rouge brique
caractéristique du chromate d'argent ;
Ag+ + Cl-
? AgCl (précipité
blanc)
2Ag+ + CrO42-
? Ag2 CrO4 (Rouge
brique)
|
paramètre
|
plage
|
Limite de détection
|
Volume minimum reporté (VMR)
|
|
chlorure
|
1--2000mg/l
|
2 mg/l
|
6 mg/l
|
I.2/ Procédure de l'analyse des
chlorures :
· Préparer et vérifier le bon fonctionnement
de la burette automatique ;
· Chasser le vide dans le circuit de dosage en
rinçant la burette
· Remplir la burette de AgNO3 et s'assurer
qu'elle est réglée à zéro ;
· Prélever 100#177;0.15ml d'échantillon avec
pipette 100ml et l'introduire dans un Erlenmeyer 250ml. Ajuster le pH de
l'échantillon en ajoutant soit l'acide nitrique 0.1N, soit la solution
d'hydroxyde de sodium 0.1N selon le cas, et noter le volume sur la fiche
d'analyse.
· Essai à blanc :
prélever le même volume d'eau déionisée et ultra
pure
· Ajouter 3 gouttes du chromate de
potassium 10% m/v, ainsi la couleur devient jaune. Agiter.
· Tirer goutte à goutte et en agitant la solution du
nitrate d'argent 0.1 N jusqu'à apparition d'une teinte rouge brique
persistante marquant la fin du dosage des chlorures
Soit Vbl volume en ml du nitrate d'argent 0.1 N
utilisée pour le titrage du blanc et V net celui pour
l'échantillon d'eau
P : prise d'essaie de l'échantillon
V A : volume brut affiché sur la
burette
Vnet = VA -
Vbl : volume net pour l'échantillon
à analyser
La teneur en chlorure :
[Cl-] (mg/l) = 100/ (P * 35.5 *
Vnet)
II/ Mesure du pH :
II.1/ Principe :
Ø pH= - log [H+]
Elle spécifie la mesure du pH des eaux ayant une
température 0-60°C.
Le pH est le cologarithme des ions hydrogènes
mesurés à l'aide d'une électrode de verre associée
à une électrode de référence.
|
paramètre
|
plage
|
Limite de détection
|
Volume minimum reporté (VMR)
|
|
pH
|
3--10
|
0.1
|
3
|
La différence du potentiel existant entre
l'électrode de verre et une électrode de référence
est une fonction linéaire du pH.
III/ Analyse des résidus
secs :
III.1/ Principe :
C'est une analyse réservée à l'eau brute
pour production d'eau potable à distribution.
Elle se base sur la pesée des résidus secs d'un
volume d'eau après évaporation et séchage.
Les résidus secs permettent d'estimer la teneur en
matières dissoutes minérales ou organiques.
Un volume donné d'eau dans la capsule tarée est
évaporé lentement au bain marie et portée à sec
à l'étuve à température 105 #177; 5 °C
jusqu'à poids constant.
Ø Remarque :
À température 105 #177; 5°C, il peut y avoir
une perte plus ou moins négligeable de matières organiques.
|
paramètre
|
plage
|
Limite de détection
|
Volume minimum reporté (VMR)
|
|
RS
|
0--6000mg/l
|
26.3 mg/l
|
40 mg/l
|
III.2/ Réactifs
utilisés :
Ø Acide chlorhydrique concentré
· Pureté 36 - 38 %
· Densité 1.184 à 1.193
Ø Acide chlorhydrique dilué
· Chlorure de sodium en poudre
· Poids moléculaire 58.44
· Dureté 99% minimum
Ø Solution standard de CQA à partir de 12 g de Na
Cl/l
III.3/ Appareils et
équipement :
· Bain marie ;
· Etuve réglable 105 #177; 5 °C ;
· Dessiccateur ;
· Balance analytique ;
· Unité de production d'eau
déionisée ;
· Verreries nécessaires.
III.4/ Procédure de l'analyse des
résidus secs :
ü Préparation de l'échantillon
d'essai ;
ü Bien agiter l'échantillon pour
homogénéiser ;
ü Prélever 50ml #177; 0.1 ml de l'échantillon
avec une pipette jaugée 50ml ;
ü Introduire dans une capsule en porcelaine
tarée ;
ü Prélever un volume connu tel qu'il conduise
à résidus sec d'au moins 10mg et compris entre 100 et 200
mg ;
ü Faire évaporer progressivement eu bain marie dans
une capsule 50 ml d'eau ;
ü Porter la capsule à l'étuve durant 4h et
laisser refroidir environ 1 h au dessiccateur ;
ü Peser immédiatement et rapidement
P1 ;
Soit P2 le poids de la
capsule contenant le résidu :
RS = (P2 - P1) * 20000
mg/l
(Pour une prise d'essai 50ml #177; 0.1 ml)
IV/ Analyse des
sulfates :
IV.1/ Principe :
L'analyse des sulfates est basée la
méthode gravimétrique utilisant le
chlorure de baryum,
La méthode se base sur la précipitation des ions
sulfates SO42-
présents dans l'échantillon après leur
réaction avec le chlorure de Baryum :
Ba2+ + SO 42-
? BaSO4 2-
(précipité blanc)
La cohésion de ce précipité est
favorisée par une ébullition pendant 20 minutes suivies d'un
repos durant une nuit. Le précipité retenu par filtration sous
vide est séché et pesé.
Ø Remarque :
L'acidification et le chauffage de l'échantillon
permettent d'éviter une précipitation ultérieure de
carbonates.
|
paramètre
|
plage
|
Limite de détection
|
Volume minimum reporté (VMR)
|
|
sulfates
|
10--5000 mg/l
|
15.16mg/l
|
20 mg/l
|
IV.2/ Procédure de l'analyse des
sulfates :
Ø Prise d'essai :
· Agiter bien l'échantillon
· La prise d'essai doit être un volume compris entre
10 et 20 ml et ne doit pas contenir plus que 50 mg de
SO42-.
· Le choix de prise d'essai (V ml) est en fonction des
résidus secs.
· La prise d'essai est complétée à 20ml
de l'eau déionisée à l'aide d'une éprouvette.
|
RS (residues secs)
|
V (ml)
|
Tolérance (ml)
|
|
< 1000
|
200
|
#177; 0,4
|
|
1000<RS<2000
|
100
|
#177; 0,15
|
|
2000<RS <3000
|
50
|
#177; 0,1
|
|
3000<RS <5000
|
25
|
#177; 0,06
|
|
>5000
|
10
|
#177; 0,04
|
Ø Prétraitement :
· Prélever la prise d'essai choisi ;
· Introduire dans l'Erlenmeyer 500 ml ;
· Compléter par 200ml d'eau
déionisée ;
· Ajouter deux gouttes de méthylorange 1g/l à
l'aide du compte goutte ;
· Ajouter 2ml HCl 6N.
Ø Précipitation :
· Couvrir l'Erlenmeyer de verre ;
· Faire bouillir le contenu pendant 5 minutes ;
· Ajouter 10 ml #177; 0.04 ml de chlorure de
Baryum ;
· Laisser bouillir pendant 20 minutes à flamme
douce ;
· Laisser reposer jusqu'à demain à
l'étuve 50 #177; 10 °C.
Ø Filtration :
· Sécher un creuset en verre filtré à
105 #177; 5 °C pendant 4 h ;
· Laisser refroidir au moins 30 minutes ;
· Le peser avec une balance de résolution 0,1 mg,
soit P1 son poids à vide ;
· Filtrer le précipité ;
· Bien rincer l'Erlenmeyer à l'au
déionisée (4 fois) pour entraîner le
précipité adhérent au parois ;
· Placer le creuset au dessiccateur 105 #177; 5 °C et
laisser sécher 4 h ;
· Placer le creuset au dessiccateur et laisser sécher
au moins 30 minutes ;
· Peser le creuset et
précipité : P2
[SO 42-] =
(P2-P1) *411.6*1000 /V
V : prise d'essai en ml
|
Résultat (mg/l)
|
Arrondissement (mg/l)
|
|
<100
|
1
|
|
]100 ,,, 200]
|
5
|
|
>200
|
10
|
V/ Analyse de la
dureté :
V.1/ Principe :
La dureté hydrotimétrique correspond
à la somme des concentrations en cations bivalents et trivalents
métalliques et alcalino-terreux.
La dureté des eaux naturelles
est due en grande partie aux sels de calcium et de
magnésium.
La méthode repose sur la formation du
complexe comprenant le Ca et Mg.
Ø Dureté totale :
Les alcalino-terreux présents dans l'eau sont
amenés à forme complexe du type chélates par le sel
disodique de l'acide du type éthylène diamine
tétracétique à pH 10. la disparition des derniers traces
d'éléments libres à doser est décelée par le
virage du noir érichrome T du rouge foncé en présence des
ions calcium et magnésium au bleu.
En milieu convenablement tamponné pour empêcher la
précipitation de magnésium, la méthode permet de doser
l'ensemble des ions Ca 2+ et Mg 2+.
Ø Dureté
calcique :
Toutefois, comme le dosage se fait à pH (12-13), ceci
permet de se débarrasser du Mg à l'état d'hydroxyde
limitant ainsi la dureté à seule calcique.
Ø Dureté
magnésique :
La différence entre dureté calcique et totale.
|
paramètre
|
plage
|
Limite de détection
|
Volume minimum reporté (VMR)
|
|
Dureté
|
0--200 °F
|
1.07 °F
|
1.5 °F
|
|
Ca 2+
|
0--600 mg/l
|
1.87 mg/L
|
6mg/l
|
|
Mg 2+
|
0--250 mg/l
|
|
3mg/l
|
(1°F correspond à 10 mg de CaCO3/l,
à 4.008 mg Ca 2+/l et à 2.43 mg Mg2+/l.)
V.2/ Procédure de la mesure de la
dureté :
Ø Dureté totale:
ü Prélever 10ml #177; 0.04 ml d'eau à analyser
à l'aide d'une pipette jaugée 10 ml, ajouter 40ml d'eau
déionisée à l'aide d'une éprouvette graduée
50ml#177;0.6ml et homogénéiser ;
ü Ajouter 2 ml d'une solution tampon à l'aide d'une
pipette graduée de 10 ml #177; 0.1 ml et une pincée de noir
eriochrome T (NET) ;
ü Titrer à l'aide d'une solution d'EDTA
0.02 N à l'aide d'une burette automatique jusqu'au
virage de l'indicateur du rouge bordeau au
bleu ;
ü Noter V (ml) de la solution EDTA.
Ø Dureté calcique:
ü Prélever 10ml #177; 0.04 ml d'eau à analyser
à l'aide d'une pipette jaugée de 10 ml, ajouter 40ml d'eau
déionisée à l'aide d'une éprouvette graduée
de 50ml #177; 0.6 ml et homogénéiser ;
ü Ajouter 1ml d'agent masquant à l'aide d'une pipette
graduée de 5 #177; 0.05 ml, 3 gouttes d'acide ascorbique, 5 ml
d'hydroxyde de sodium 2 N à l'aide d'une pipette graduée 5 #177;
0.05 ml et une pincée de réactif de Patton et
Reeder ;
ü Titrer à l'aide d'une solution d'acide
éthylène diamine tétracétique jusqu'au virage de
l'indicateur du rouge bordeau ;
ü Noter V le volume de la solution EDTA.
Ø Dureté magnésique:
ü La différence entre dureté totale et
calcique.
VI/ Analyse de l'oxydabilité au KMnO4
:
VI.1/ Principe :
L'analyse est basée sur l'oxydabilité des
matières réductrices minérales ou organiques par le
KMNO4.
La procédure consiste à mesurer en
milieu acide et chaud la quantité d'O2 utilisée pour
la réduction du permanganate de potassium par la matière oxydable
contenu dans l'eau.
L'ajout d'une quantité d'oxalate de
sodium réducteur est opéré e sorte à
réagir avec le KMNO4 et laisser en
excès qui sera dosé par KMNO4 ;
2KMNO4 + 3 H2SO4
? 2Mn SO4 + K2SO4 +
3H2O + 5/2 O2
|
paramètre
|
plage
|
Limite de détection
|
Volume minimum reporté (VMR)
|
|
oxydabilité au KMNO4
|
0.10 mg O2/l
|
0.236
|
1
|
VI.2/ Réactifs
utilisés :
· Acide sulfurique concentré ;
· Solution de permanganate de potassium KMNO4
0.1N ;
· Solution mère KMNO4 0.1 N =
20mmol/l ;
· Solution fille de KMNO4 0.1 N = 2
mmol/l ;
· Oxalate de sodium en poudre ;
· Solution d'oxalate de sodium ;
· Eau déionisée exempte de matière
oxydable ;
· Solution fille et solution mère d'oxalate de
sodium ;
· Glucose anhydre ;
· Solution standard de CQA à 0.5 g/l à partir
du glucose ;
· Solution CQA à 5 mg O2 /l.
VI.3/ Procédure de l'analyse de
l'oxydabilité au KMnO4 :
· Préparer l'échantillon d'essai en laissant
décanter toute matière en suspension et faire filtrer
l'échantillon sur papier filtre.
· Transférer une prise d'essai de 25 #177; 0.06 ml
d'eau à analyser à l'aide d'une pipette jaugée de 25 ml de
classe B dans un tube à essai de 100ml
· Ajouter 5 #177; 0.05 ml d'acide sulfurique 2 mol/l
à l'aide d'une pipette graduée de 5 ml de classe B et
mélanger en agitant doucement.
· Placer le tube à essai dans le bain marie,
réglé à 100°C au temps t=0minute ;
· Après 5 minutes remettre le réglage du bain
marie à 95 °C ;
· Après 10 minutes, ajouter 5 ml #177; 0.03 ml de la
solution de permanganate de potassium 0.01 N à l'aide d'une pipette
jaugée de 5ml. Régler le bain marie à
100°C. ;
· Après 15 minutes remettre le bain marie à 95
°C ;
· Après 20 minutes, ajouter 5 #177; 0.03 ml de la
solution d'oxalate de sodium Na2 C2 O4
à 5mmol/l à l'aide d'une pipette jaugée de 5 ml et
attendre que la solution se décolore.
· Titrer pendant que la solution est encore chaude, avec la
solution de KMNO4 0.01 N jusqu'à une coloration rose pale
persistante pendant environ 30 secondes, noter le volume consommé.
Ø L'oxydabilité au KMNO4, exprimé
en milligrammes d'oxygène libérée par litre d'eau est
donnée par la formule suivante :
V *4 * 1.27
V : volume en millilitre de la solution de KMNO4
0.01N.
VII/ Mesure de la
turbidité :
VII.1/ Principe :
La mesure de la turbidité est basée sur les
méthodes quantitatives faisant appel au
turbidimètre optique par
mesurage de lumière diffusée et par atténuation de la
lumière incidente.
Elle s'applique à tout type d'eau brute destinée
à la production d'eau potable.
La turbidité est due à la
présence de matière en suspension finement
divisée.
L'appréciation de l'abondance de ces matières
mesure son degré de turbidité.
|
paramètre
|
plage
|
Limite de détection
|
Volume minimum reporté (VMR)
|
|
turbidité
|
0,1--500 NTU
|
0,024 NTU
|
0,1 NTU
|
Le résultat est affiché directement par le
turbidimètre.
VIII/ Mesure de
l'alcalinité :
VIII.1/ Principe :
L'alcalinité est en fonction des concentrations
hydrogénocarbonates, carbonates et hydroxyde.
La méthode repose sur l'acidification d'un
échantillon jusqu'à atteindre des valeurs de pH bien
déterminés.
|
paramètre
|
plage
|
Limite de détection
|
Volume minimum reporté (VMR)
|
|
alcalinité
|
4-50°F
|
0.11°F
|
0.5°F
|
La méthode basée sur le titrage d'un certain volume
d'eau par un acide minéral dilué à des valeurs de pH 8.3
et 4.5.
VIII.2/
Réactions :
La détermination du titre
alcalimétrique TA permet d'évaluer les teneurs en
hydroxydes alcalins et alcalino-terreux et carbonates.
Le titre alcalimétrique complet
TAC permet de connaître la teneur en hydroxydes, carbonates
et hydrogénocarbonates alcalins et alcalino-terreux.
La décoloration de la phénophtaléine se
produit dés que le pH est inférieur à 8,3
c'est-à-dire lorsque l'anhydride carbonique à
l'état libre commence à apparaître dans la
solution :
Ca (OH)2+ H2SO4
? Ca SO4 +
2H2O
2Ca CO3+ H2SO4
? Ca (HCO3)2 + Ca
SO4
À ce niveau (pH<8,3), tout ajout d'acide engendre la
réaction :
Ca (HCO3)2 +
H2SO4 ? Ca SO4 + 2
CO2 + 2 H2O
Le virage du méthylorange se
produit dés que le pH est inférieur à
4.5 c'est à dire dés qu'un excès fort
commence à apparaître.
Dans la méthode suivie, les points de pH 8.3,
4.5, 4.2 sont détectés par le
potentiomètre.
VIII.3/ Procédure de
mesure :
ü Vérifier le bon fonctionnement de la
burette automatique ;
ü Prélever 50 #177; 0.1ml d'eau à analyser
à l'aide d'une pipette jaugée de 50 ml de classe B ;
ü Introduire cette quantité dans un bécher de
100ml ;
ü Utiliser l'agitateur magnétique pour
homogénéiser ;
ü Verser goutte à goutte, l'acide
sulfurique 0.02 N, à l'aide d'une burette automatique
jusqu'à pH 4.5, soit
V1 le volume d'acide
utilisé ;
ü Continuer le titrage jusqu'à pH
4.2, soit V2 le volume total
d'acide utilisé.
VIII.4/ Exploitation des
résultats :
· Si le pH< 8.3 :
TAC (en d° français) :
(2V1-V2)*2
HCO3- en mg/l:
(2V1-V2) *2*12.2
· Si le pH > 8.3 : TA
existe
TA (en degré
français) : 2V
§ Si TA (°F) < TAC/2 °F :
[OH-] = 0 mg/l
[CO32-] = 2TA *6 en mg/l
[HCO3-] = (TAC - 2TA) *12.2 en mg/l
§ Si TA (°F) > TAC/2 °F :
[OH-] = (2TA -TAC)*3.4 en mg/l
[CO32-] =2*(TAC - 2TA) *6 en mg/l
[HCO3-] = 0 mg/l
§ Si TA (°F)= TAC/2 °F :
[OH-] = 0 mg/l
[CO32-] =2TA *6 en mg/l
[HCO3-] = 0 mg/l
§ Si TA =TAC :
[OH-] = TAC *3.4
[CO32-] =0 en mg/l
[HCO3-] = 0 mg/l
V : quantité d'acide verse jusqu'à pH 8,3
V 1: quantité d'acide verse jusqu'à
pH 4,5
V 2: quantité d'acide verse jusqu'à
pH 4,2.
![]()
Page
· Procédure du dosage des
fluorures............................................12
· Procédure du dosage des
nitrites...............................................13
· Procédure du dosage des
nitrates..............................................13
· Procédure du dosage de sodium
et de potassium par spectrométrie de
flamme..........................14
I/ Procédure du dosage des
fluorures:
I.1/ Principe :
Pour la détermination de la concentration des fluorures
dans un échantillon, on se base sur la méthode au
réactif SPADNS qui est axée sur la réaction
du fluorure avec la solution de laque rouge du
fluorure. Le fluore se combine avec une partie du zirconium pour former un
complexe zircorium-fluorure incolore
déclenchant une diminution de l'intensité de couleur inversement
proportionnelle à la concentration du fluorure et susceptible d'un
dosage colorimétrique.
|
paramètre
|
plage
|
Limite de détection
|
Volume minimum reporté (VMR)
|
|
fluorure
|
0--2 mg/l
|
0.078 mg/l
|
0.1 mg/l
|
I.2/ Réactifs
utilisés :
· Réactif SPADNS
· Solution CQA de fluorure à 1.7 mg/l et 0.85 mg/l
I.3/ Appareils et
équipement :
· Spectrophotomètre modèle DR
2010
· Pipettes jaugées (25ml ; 5ml)
I.4/ Procédure
d'analyse :
ü Effectuer un autotest du spectromètre ;
ü Entrer le numéro du programme
mémorisé pour le fluorure (190) ;
ü Presser ENTER, l'affichage
indique REGLER nm à 580
ü Tourner le bouton de réglage de la longueur d'onde
jusqu'à ce que l'affichage indique 580nm
ü Afficher correctement la longueur d'onde
580nm en faisant défiler les longueurs d'ondes
des valeurs les plus hautes vers les plus basses
ü Presser ENTER, l'affichage
indique mg/l F-
ü Remplir le flacon, comme blanc, avec 25 ml d'eau
déionisée à l'aide d'une pipette jaugée 25ml
ü Dans une série de flacons colorimétriques
numérotés, introduire 25ml d'échantillon à l'aide
d'une pipette jaugée de 25 ml
ü Ajouter 5ml de réactif de SPADNS à chaque
flacon à l'aide d'une pipette jaugée de 5ml et agiter pour
mélanger
ü Une coloration rouge se développe,
l'intensité de la couleur rouge est inversement proportionnelle à
la concentration du fluorure
ü Presser SHIFT TIMER, une
période de 1 minute commence
ü Lorsque le minuteur sonne et que l'affichage indique mg/l
F- placer le blanc dans le compartiment de mesure et fermer le capot
ü Presser ZERO, l'affichage
indique attendre, puis 0 mg/l F-
ü Placer immédiatement l'échantillon à
analyser dans le compartiment de masure et fermer le capot
ü Presser READ, l'affichage
indique attendre puis le résultat en mg/l F- s'affiche.
II/ Procédure du dosage des
nitrites :
II.1/ Principe :
Le dosage des nitrites est basé sur la
méthode au réactif de ZOMBELLI.
En effet, L'acide sulfanilique en milieu chlorhydrique en
présence d'ions ammonium et de phénol, forme avec les ions
NO2- un complexe coloré jaune dont
l'intensité est proportionnelle à la concentration en nitrites et
susceptible d'un dosage colorimétrique.
La coloration ainsi obtenue est stable pendant 24h.
|
Paramètre
|
Plage
|
Limite de détection
|
Volume minimum reporté (VMR)
|
|
Nitrite
|
0--2 mg/l
|
0.057mg/l
|
0.01mg/l
|
II.2/ Réactifs utilisés
:
· Ammoniaque pure
· Acide chlorhydrique
· Acide sulfanique
· Chlorure d'ammonium
· Réactif de Zombelli
II.3/ Appareils et équipement
:
· Spectromètre UV visible (modèle UV-160 PC
SHIMADRZU) assisté par un ordinateur
· Verreries nécessaires.
II.4/ Procédure
d'analyse :
ü Prélever 50ml de blanc exempt de nitrite à
l'aide d'une pipette et l'introduire dans un Erlenmeyer d 100ml ;
ü Prélever 50 ml d'eau destinée à
l'analyse à l'aide d'une pipette de 50 ml de la solution de
CQA prélevée à l'aide d'une
pipette et l'introduire dans un Erlenmeyer de 100ml ;
ü Ajouter 2ml de réactif de
Zambelli à l'aide d'une pipette graduée dans chaque
Erlenmeyer et laisser au repos pendant 10 minutes ;
ü Ajouter 2 ml d'ammoniaque pure à l'aide d'une
burette automatique ;
ü Effectuer la lecture au spectromètre à la
longueur d'onde 435nm en utilisant la courbe
d'étalonnage préalablement tracée.
III/ Procédure du dosage des
nitrates :
III.1/ Principe :
Le dosage est effectué par la méthode
au salicylate de sodium.
Il s'agit d'un dosage spectrométrique suite à la
formation du complexe paranitrosalicylate
de couleur jaune à partir de la réaction des
nitrates avec le salicylate.
La couleur du paranitrosalicylate de
Na+ est la plus intense des colorations fournies par
les nitrates. Elle est stable pendant une heure environ.
|
paramètre
|
plage
|
Limite de détection
|
Volume minimum reporté (VMR)
|
|
nitrate
|
0--50mg/l
|
0.447mg/l
|
0.50mg/l
|
III.2/ Réactifs utilisés
:
· Solution de Salicylate de sodium 1%
· Acide sulfurique concentré
· Hydroxyde de sodium
· Sel disodique de l'éthylénediamine
tétracétique de Na+ (EDTA)
· Des solutions étalons
· Acide acétique glacial
III.3/ Procédure
d'analyse :
ü Préparer le spectromètre UV 1061
DC ;
ü Prendre une capsule de diamètre 60-90
mm pour la préparation à blanc exempt de nitrate
préparé en solution de la même façon qu'à la
préparation des solutions des échantillons ;
ü Prélever, dans chaque capsule, 1ml d'eau claire
à analyser à l'aide d'une pipette graduée de 1 ml en
intercalant aléatoirement une solution de CQA
prélevée à l'aide d'une pipette graduée de 1ml
ü Diluer 10 fois la prise d'essai de 1 ml en y ajoutant 9 ml
d'eau déionisée ultra pure dans chaque capsule à l'aide de
la pipette graduée 10ml
ü Ajouter 0.5 ml d'azoture de sodium à l'aide d'une
pipette graduée. Mélanger et attendre
ü Poursuivre le dosage, en utilisant la pipette
graduée de 25ml
ü Effectuer la lecture au spectromètre, à la
longueur d'onde de 415 mm, en utilisant la courbe d'étalonnage,
préalablement préparée en prennent soin de rincer la
cellule de mesure par l'échantillon à passer et essuyer les
parois appropriées par un papier doux et absorbant avant chaque
mesure.
IV/ Procédure du dosage de sodium et de
potassium par spectrométrie de flamme :
IV.1/ Principe :
Pour pouvoir doser le sodium et le potassium on peut faire appel
à la méthode de spectrométrie de
flamme qui consiste à pulvériser une solution par
une flamme de façon à ce que l'eau de la solution
s'évapore et que les sels présents sont dissociés à
l'état atomique ; Ainsi les atomes les atomes de potassium ou de
sodium sont excités par une flamme d'énergie thermique.
|
Paramètre
|
longueur d'onde caractéristique
|
plage
|
Limite de détection
|
volume minimum reporté (VMR)
|
|
sodium
|
589nm
|
0--750mg/l
|
2.4mg/l
|
15mg/l
|
|
potassium
|
766,5nm
|
0--6 mg/l
|
0.07mg/l
|
0.75 mg/l
|
IV.2/ Réactifs
utilisés:
· Acide nitrique concentré
· Solution étalon de sodium
· Solution étalon de potassium
· Chlorure de potassium
IV.3/ Appareils et
équipement :
· Spectromètre de flamme
· Verreries nécessaires.
IV.4/ Procédure
d'analyse :
ü Effectuer les préparations préliminaires de
la série des échantillons et la solution de
CQA correspondante ;
ü Sélectionner l'élément à doser
à l'aide du sélecteur de filtre sur la position requise selon
l'intérêt ;
ü Etalonnage du Spectromètre de flamme ;
ü Calibrer l'appareil et établir la courbe
d'étalonnage ;
ü Analyser la série des échantillons ;
ü Relever les valeurs indiquées par l'appareil,
déterminer le degré de dilution et préparer une
deuxième série diluée des mêmes
échantillons ;
|
domaine de lecture
|
domaine de concentration (mg/l)
|
degré de dilution
|
volume de prise d'essai (ml)
|
volume d'eau déionidée ajouté (ml)
|
|
[0,,,100]
|
[0,,,30]
|
1
|
0
|
0
|
|
]100,,,150]
|
]30,,,60]
|
2
|
5
|
5
|
|
]150,,,200]
|
]60,,,90]
|
3
|
5
|
10
|
|
]200,,,240]
|
]90,,,120]
|
4
|
2,5
|
7
|
|
]240,,,270]
|
]120,,,150]
|
5
|
2
|
8
|
|
]270,,,320]
|
]150,,,210]
|
7
|
1
|
6
|
|
]320,,,390]
|
]210,,,300]
|
10
|
1
|
9
|
|
]390,,,480]
|
]300,,,450]
|
15
|
0,5
|
7
|
Ø Remarque :
Seules les concentrations appartenantes à la gamme
étalon [10......25mg/l] sont considérées.
ü Fixer le degré de dilution pour se situer dans la
gamme de lecture optimale [0......30] ;
ü Préparer les échantillons pour pré
analyse ;
ü Etalonner et recalibrer le spectrophotomètre
à flamme ;
ü Lire les nouvelles valeurs des concentrations.
![]()
Page
·
Introduction....................................................................................17
· Analyse par absorption
atomique...............................................17
· Analyse par absorption atomique : Mode
flamme....................................................................................17
· Analyse par absorption atomique avec
génération
d'hydrures..........................................................18
· Analyse par absorption atomique : Mode
four......................................................................................20
I/ Introduction :
Les impuretés en suspension et dissoutes
dans l'eau naturelle la rendent impropre pour de nombreux usages. Les
matières organiques et minérales indésirables en
suspension sont éliminées par plusieurs méthodes.
|
Substances
|
Dosages
|
Limites
|
|
Les substances indésirables
|
Fer
|
Méthode colorimétrique Méthode
d'absorption atomique
|
un niveau guide de 0.05mg/l et une concentration maximale
admissible de 0.3mg/l
|
|
Manganèse
|
Méthode d'absorption atomique
|
concentration maximale admissible de 0.1
|
|
Cuivre
|
Méthode colorimétrique Méthode par
absorption atomique
|
une teneur limite de 1mg/ l
|
|
Zinc
|
Méthode par absorption atomique
|
un niveau guide de 0.05 mg/l et une CMA de 0.2mg/l
|
|
Les substances toxiques
|
Arsenic
|
Méthode par absorption atomique
|
une CMA de 0.05mg/l
|
|
Mercure
|
Méthode d'absorption atomique
|
une CMA de 1mg/l
|
|
Cadmium
|
Méthode par absorption atomique
|
une teneur limite de 5mg/ l
|
|
Plomb
|
Méthode par absorption atomique
|
une CMA de 0.05mg/l
|
II/ Analyse par absorption
atomique :
II.1/ Procédure d'analyse du : fer,
cuivre, manganèse, zinc, argent, aluminium, silicium et chrome, par
absorption atomique : mode flamme :
Principe :
Un élément (cation) dispersé à
l'état atomique dans une flamme possède
la propriété d'absorber tout rayonnement de même
fréquence, il en résulte une absorption du
rayonnement spécifique émis par une lampe
appropriée à l'élément considéré par
la relation :
log (I0/I) = KLC
I0 : intensité de la radiation incidenté
I : intensité de la radiation après
traversée de la flamme
L : Langueur de chemin optique
K : coefficient
A partir de l'intensité du faisceau incident et du
faisceau transmis il est possible de déduire la concentration en
élément recherché.
Procédure
d'analyse :
ü Préparer de l'échantillon : Pour Fe,
Cu, Zn, Mn, Cr, Al et Ag 250 ml d'échantillon +0.5 ml de
H2SO4 de façon a avoir pH< 2 pour si on a
échantillon non acidifié ;
ü Calibrer de spectrophotomètre à absorption
atomique :
Ø
« Solaar .M » permet de
réaliser automatiquement les opérations suivantes :
· Placer la lampe de l'élément à
rechercher dans le compartiment approprié en face de la fente
réceptrice du faisceau lumineux ;
· Mettre la lampe sous tensions ;
· Choisir la méthode de l'élément
à doser en vérifiant les paramètres de celle-ci (langueur,
débit de C2H2, hauteur de brûleur, nombre de mesure) ;
· Lancer la méthode ;
· Allumer la flamme ;
· A la demande du logicielle, introduire une
5ème concentrée de l'élément à
analyser et faire un réglage de la flamme de manière à
assurer le maximum d'absorbance ;
· A la demande de logicielle, introduire une solution
concentrée de l'élément à analyser et faire un
réglage de la flamme de manière à assurer le maximum
d'absorbance ;
· à la demande de logicielle introduire un blanc
(eau deionisèe acidifier pour Fer, Cui, Zen, MN, Crs, Al et Agi et eau
deionisèe non acidifié pour Si) pour faire un réglage
optique ;
· Le logicielle demande d'introduire successivement les 3
solutions ;
· L'appareil tracera la courbe d'étalonnage et
affichera le coefficient de corrélation ;
ü Faire passer successivement les
échantillons ;
ü Faire un rinçage intermédiaire avec l'eau
ultra pure acidifiée pour éliminer les traces de
l'échantillon précédent ;
ü A la fin de l'analyse de tous les échantillons
préalablement enregistrés, le logicielle
affiche « analyse
terminée » ;
ü Les résultats sont directement affichés sur
le microordinateur exprimé en mg/l ou ug/l de l'élément
considères
II.2/ Analyse par absorption atomique avec
génération d'hydrure (Mercure et
Arsenic) :
Cette méthode concerne les éléments
suivants ; Mercure (Hg) et Arsenic (As) ; cette méthode est
axée sur la libération d'atomes par réaction avec un
réducteur approprié dans des conditions optimales et leur
transport par un gaz inerte. Ces atomes ont la propriété
d'absorber tout rayon de même fréquence.
|
paramètre
|
Plage (ug/l)
|
|
Hg
|
0--4
|
|
As
|
0--400
|
Méthode avec le
Mercure :
Ø Principe :
En raison de propriétés unique de mercure, il est
possible d'effectuer une analyse de la vapeur de l'élément
à T° ambiante, l'accessoire VP90permet par l'utilisation d'un
réducteur approprié Na BH4 (où Sncl2), la réduction
de la mercure en une vapeur élémentaire
Hg2+ +2BH4-
? H9+B2H2 +
H2
Après la réaction de réduction permettant la
transformation de mercure sous forme élémentaire, le
mélange est entraîné par un gaz inerte : l'argon, le
mélange traverse un séparateur gaz /liquide, qui en retire la
vapeur d'eau et le mercure arrive à une température de
piégeage T fixée sur le bulleur,
en face d'un faisceau lumineux émis par la lampe de mercure.
Les atomes ainsi pièges ont la propriétés
d'absorbes tout rayonnement de même fréquence il résulte
une absorption du rayonnement spécifique.
Cette absorption est liée à la concentration de
l'élément considérer par la relation :
Log (I0/I)=l0LC
Ø
Procédure :
ü Préparer les blancs, les étalons et les
échantillons :
|
Blanc
|
Etalon
|
Echantillons
|
|
1
|
2.5
|
5
|
|
HCl 37%
|
10 ml
|
10
|
10
|
10
|
2.5ml
|
3ème étalon 5ug de
As
|
KMnO45%
|
0.5
|
0.5
|
0.5
|
0.5
|
1.125ml
|
|
Hydroxylamonium chlorure 5%
|
2.5
|
2.5
|
2.5
|
2.5
|
0.625 ml
|
|
Volume final (ml)
|
100
|
100
|
100
|
100
|
25ml
|
ü Faire passer successivement les
échantillons ;
ü Faire un rinçage intermédiaire avec la
solution HCl 10%.
Méthode avec
l'Arsenic:
Ø Principe :
La procédure est similaire à celle de mercure
excepté le fait que cette fois on a besoin d'une source
d'énergie, la cellule « T » sera donc
chauffée par le bulleur (uniquement par une flamme
air/acétylène)
As3+ + 6BH-4 +
3H?AsH3 + 3B2H6 +
3H2
AsH3 chauffage As + 3/2
H2
Ø
Procédure :
ü Préparer les blancs, les étalons et les
échantillons :
|
Blanc
|
Etalon
|
Echantillons
|
|
1
|
2.5
|
5
|
|
HCl 37% (ml)
|
10
|
10
|
10
|
10
|
2,5
|
|
KI 10% (ml)
|
1
|
1
|
1
|
1
|
0,25
|
|
Acide ascorbique 10% (ml)
|
1
|
1
|
1
|
1
|
0,25
|
|
Volume final (ml)
|
100
|
100
|
100
|
100
|
25
|
ü Faire passer successivement les
échantillons :
ü Faire un rinçage intermédiaire avec HC
10%.
II.3/ Analyse par absorption atomique : Mode
four :
Principe :
|
paramètre
|
Plage de mesure (ug/l)
|
|
Plomb
|
0--100
|
Il s'agit d'un four graphite chauffé par effet joule,
Après introduction de l'échantillon liquide dans
le four, le chauffage qui conduit à
l'atomisation se fait en atmosphère inerte
(indispensable pour éviter une oxydation rapide de
l'élément chauffé et assure l'élimination des
vapeurs s'émanant de la matrice et formé avant l'atomisation)
selon trois étapes programmés en temps et en
température :
ü Injection
ü Séchage
ü Atomisation
Ces atomes ainsi générés ont la
propriété d'absorber tout rayonnement de même
fréquence. Il en résulte une absorption
du rayonnement spécifique émis par une lampe appropriée
à l'élément recherché. Cette
absorption est lié à la concentration de l'élément
considéré par la relation :
Log (I0/I) = KLC
I0= intensité de la radiation
incidents
I= intensité de la radiation
après traversé de la flamme
L= longueur du chemin optique
K= coefficient de corrélation
A partir de l'intensité du faisceau transmis, il est
possible de déduire la concentration de l'élément
d'intérêt.
![]()
Page
· Modalités et techniques de
prélèvement...........................22
· Les bactéries de
l'eau.........................................................23
· Le choix des indicateurs de
pollution................................24
· Norme Tunisiennes des eaux de
boisson........................24
· Analyse bactériologique de l'eau :
...................................24
· Recherche des
coliformes..........................................24
· Recherche des Streptocoques
fécaux........................27
· Recherche des anaérobies
Sulfito-réducteurs............27
· Recherche des germes
totaux....................................28
I/ Modalités et techniques de
prélèvement :
La plupart des échantillons seront prélevés
aux robinets, des réservoirs, des maisons, des lieux publics...
Il faut veiller à ce que les échantillons ne
subissent aucune contamination accidentelle au cours de
prélèvement.
Ø Techniques de
prélèvement :
1. Nettoyer le robinet :
· Retirer du robinet tout
accessoire qui risque de provoquer des contaminations ;
· Nettoyer soigneusement l'orifice du robinet à
l'aide d'un tissu propre.
2. Ouvrir le robinet :
· Laisser l'eau du robinet couler pendant au moins 2 minutes
pour être sûre que l'eau stagnante aura été
chassée de la tuyauterie avant le prélèvement.
3. Stériliser le
robinet :
· Passer le robinet à la flamme

4. Ouvrir un flacon stérilisé
prés du robinet.
5. Remplir le flacon d'eau :
· Tout en tenant la capsule dirigée vers le bas (de
façon à empêcher l'entrée de poussières
susceptibles d'être porteuses de germes) ;
· Placer immédiatement le flacon sous le jet d'eau et
le remplir.

6. Refermer le flacon :
· Ne pas remplir complètement de façon qu'on
puisse facilement secouer le flacon avant le prélèvement d'une
prise d'essai ;
7. Mettre une étiquette sur le flacon pour
éviter les erreurs ;
8. Placer le flacon dans la glacière et le
conserver au frais dans l'obscurité de préférence à
4°C.
Ø Mesures de
précautions :
· L'agent effectuant le prélèvement doit laver
et désinfecter ses mains avant chaque prélèvement.
· Toutes les précautions doivent être prises
pour éviter la contamination du flacon et celle de
l'échantillon.
· Les doigts de l'agent effectuant le
prélèvement ne doivent venir en contact ni avec
l'intérieur du goulot ni avec le bouchon.
II/ Les bactéries de
l'eau :
L'eau à épurer contient, une masse
bactérienne connue résultante de la croissance des
bactéries autochtones aux dépens de la matière organique,
et aussi, un nombre élevé des bactéries fécales
Nous allons axer sur les germes recherchés au
laboratoire central de la SONEDE. On distingue :
ü -Les bactéries
lactose-positives : Ce sont des bactéries pouvant former
des colonies en aérobiose à (36°C) et à (44°C)
sur un milieu de culture lactose sélectif et différentiel avec
production d'acide dans les 21h ; la présence de bactéries
coliformes peut bien que ce ne soit pas la preuve d'une contamination d'origine
fécale indique une défaillance du traitement ou de la
distribution de l'eau.
L'identification des souches isolées permet quelquefois
d'apporter une indication quant à leur origine.
ü -Les bactéries coliformes :
sont des bactéries lactose -positives qui sont oxydases
négatives.
ü -Les E.coli : sont des
bactéries coliformes qui produisent également
de l'indole à partir du tryptophane dans les (21#177;3)
h à (44#177;0,5) °C.
Les coliformes sont intéressants car un très grand
nombre d'entre eux vivent en abondance dans les matières fécales
des animaux à sang chaud et de ce fait, constituent des indicateurs
fécaux.
Par ailleurs, leur résistance aux agents antiseptiques,
et notamment au chlore et à ses dérivés, et voisine de la
résistance des bactéries pathogènes vis-à-vis des
quelles ce type de traitement est insaturé ; ils constituent donc
des indicateurs d'efficacité du traitement.
ü -Streptocoques fécaux : Sous
la dénomination générale de
« streptocoques fécaux », il faut entendre
l'ensemble des streptocoques possédant la substance antigénique
caractéristique du groupe D de Lance Field, c'est-à-dire
essentiellement : Streptococcus bovis, indicateur
d'efficacité de traitement de désinfection.
Streptococcus suis, Enterococcus faecalis, Enterococcus
faecium, Enterococcus durans, Enterococcus hiare, et Streptococcus
enquinus : Ces streptocoques du groupe D sont
généralement pris globalement en compte comme des témoins
de pollution fécale car tous ont un habitat fécal.
Les dénombrements des streptocoques fécaux
présumés sont rarement effectués indépendamment des
dénombrements de coliformes et coliformes fécaux
présumés. Les méthodes sont analogues pour ces deux types
d'indicateurs et seuls les milieux différents.
III/ Le choix des indicateurs de
pollution :
Le choix des indicateurs de pollution doit correspondre à
certain nombre de critères :
· Les indicateurs de pollution doivent être toujours
présents au cas ou des micro-organismes pathogènes sont
présents ;
· Ils doivent être présents en nombre plus
grand que celui des pathogènes ;
· Ils doivent avoir le même comportement que les
pathogènes dans l'environnement naturel et au cours des
procédés de traitement de l'eau ;
· Ils doivent être mis en évidence,
dénombrés et identifiés.
IV/ Normes Tunisiennes des eaux
de boisson :
|
Coliformes totaux par 100ml
|
Coliformes fécaux par 100ml
|
Streptocoques fécaux par 100ml
|
Anaérobies Sulfito-réducteurs par 100
ml
|
Conclusion
|
|
0
|
0
|
Absence
|
Absence
|
Eau propre
|
|
1 à 2
|
0
|
Absence
|
Absence
|
Eau satisfaisante
|
|
0
|
0
|
Absence
|
Présence
|
Eau à surveiller
|
|
0
|
0
|
Présence
|
Absence
|
Eau à surveiller
|
|
3 à 10
|
0
|
Absence
|
Absence
|
Eau douteuse
|
|
0
|
0
|
Présence
|
Présence
|
Eau à surveiller
|
|
>10
|
0
|
Absence
|
Absence
|
Eau impropre
|
|
0
|
1
|
Absence
|
Absence
|
Eau impropre
|
|
Présence
|
0
|
Présence
|
Absence
|
Eau impropre
|
Tableau : Normes tunisiennes de la qualité
bactériologique des eaux de boisson
(NT09.04 de SONEDE)
V/ Analyse bactériologique de
l'eau :
V.1/ Recherche des
coliformes :
Cette méthode s'applique aux eaux claires destinées
à la consommation humaine ne contenant pas de matières en
suspension pouvant colmater la membrane filtrante.
Pour les eaux turbides, il est nécessaire de
procéder à une dilution avec de l'eau distillée
stérile.
Procédure d'analyses des
coliformes fécaux :
Ø Préparation du milieu de
culture :
ü Préparation de la Gélose
lactosée :
· Mettre 54 #177; 1 g du milieu déshydraté
(gélose lactosée de base) dans 1 litre d'eau distillée.
· Attendre 5 min puis mélanger jusqu'à
obtention d'une suspension homogène,
· Chauffer lentement en agitant fréquemment puis
porter à l'ébullition jusqu'à complète
dissolution.
· Si nécessaire, ajuster le pH qu'après
stérilisation, il soit de 7.2 #177; 0.1 à 25°C.
· Répartir le milieu par quantité de 100 ml,
dans des flacons de 100ml, stériliser à l'autoclave à 121
#177; 3 °C pendant 15 min.
ü Préparation du TTC :
· Suspendre 56 #177; 0.1 g du milieu dans une litre d'eau
distillée.
· Chauffer en agitant fréquemment jusqu'à
ébullition.
· Distribuer dans des récipients adéquats et
stériliser à 121 #177; 3 °C pendant 15min.
ü Préparation de la Gélose
lactosée au TTC et Tergitol 7 :
· Ajouter pour chaque 0.9 litre de la préparation de
Gélose lactosée 45ml de la préparation de TTC.
· Agiter le flacon contenant le mélange, le milieu
sera coulé dans des boites de pétri stériles qui seront
par la suite utilisées pour la recherche des bactéries.
ü Conservation du milieu :
· Effectuer un contrôle de stérilité et
d'activité sur chaque lot de fabrication.
· Stocker les milieux prêts à l'emploi dans un
réfrigérateur afin de prolonger la durée de conservation
(ne pas conserver plus d'une semaine).
Ø Mode opératoire :
ü Allumer le bec bunsen.
ü Essuyer la paillasse et laver les mains avec de
l'alcool.
ü Placer l'entonnoir
ü Flamber la plaquette poreuse du support à l'aide du
bec benzène.

ü Mettre la membrane sur la grille
ü Agiter soigneusement le flacon (250ml)
d'eau à analyser.

ü Flamber le culot du flacon et verser 100 ml d'eau à
analyser.

ü Démonter l'appareil de filtration, et au moyen de
la pince, tenir la membrane par le bord et la placer soigneusement sur la boite
de pétri contenant le milieu Chapman au TTC en s'assurant qu'aucune
bulle d'air n'est emprisonnée.

ü Retourner la boite de pétri et incuber à
(44#177; 0,5) °C pendant (21#177; 3) heures.

ü Examiner les membranes et considérer comme
bactéries coliformes toute colonie typique dans la membrane ayant une
coloration jaune. 
ü Effectuer les tests de
confirmation :
· Repiquer de préférence toutes les colonies
typiques ou un nombre représentatif (au moins 10), respectivement, sur
la gélose non sélective (gélose au soja) et dans un
bouillon au tryptophane.
· Incuber la gélose non sélective à
(36#177;2) °C pendant (21#177;3) h et effectuer l'essai à l'oxydase
comme suit :

· Verser sur un papier-filtre 2 à 3 gouttes du
réactif à l'oxydase préparé extemporanément.
· A l'aide d'une tige en verre ou d'une anse de platine,
étaler une partie de la culture sur le papier filtre
préparé.
 
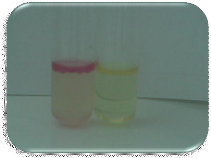
· La réaction est considérée positive
si, dans les 30 secondes apparaît une coloration bleu/violette
foncée.

· Après avoir incubé le milieu contenant le
bouillon au tryptophane à 44°C pendant 21°C, contrôler
la production de l'indole en ajoutant 0.2 ml à 0.3 ml de réactif
de Kovacs.
· L'apparition d'une coloration rouge à la surface du
bouillon confirme la production d'indole et par conséquent la
présence de l'E.coli.
V.2/ Recherche des Streptocoques
fécaux :
Principe :
La recherche des streptocoques fécaux est
quantitative.
Elle consiste à ensemencer un tube à essai
contenant le milieu de Roth avec 10 ml d'eau à
analyser et incuber à 37°C pendant 48h ; Ainsi la
présence d'un trouble dans le tube indique une présence des
streptocoques fécaux.
Procédure d'analyse des Streptocoques
fécaux :
Ø Préparation du milieu de
culture :
ü Le milieu de Roth est le
milieu sélectif pour la recherche des
streptocoques fécaux :
· Verser 69.4g de poudre dans 1 litre d'eau distillé.
Mélanger et stériliser à l'autoclave à 120°C
pendant 15 min à pH 7,5.
Ø Mode opératoire :
ü Ensemencer 10ml d'eau à analyser dans 10 ml de
milieu de Roth, homogénéiser et incuber à 37 °C
pendant 48h.
ü Examiner les tubes, si le milieu est trouble donc il y a
présence de streptocoques fécaux.
V.3/ Recherche des anaérobies
Sulfito-réducteurs :
Principe :
Ils sont caractérisés par la résistance de
leurs spores et par un équipement enzymatique.
Ils réduisent plus ou moins activement les sulfites et
les sulfures.
Procédure d'analyse des anaérobies
Sulfito-réducteurs :
Ø Préparation du milieu de
culture :
ü Le milieu sélectif pour cette
recherche est le «viande et
foie» :
· Mettre 46.5ml de poudre dans 1 litre d'eau
distillée.
· Attendre 5 min puis mélanger jusqu'à
obtention d'une suspension homogène.
· Porter lentement à l'ébullition.
· Ajuster, si nécessaire, le pH à 7.2.
· Stériliser à l'autoclave à 115°C
pendant 20 min.
Ø Mode opératoire :
ü Chauffer 10ml d'eau à analyser dans un bain marie
80°C pendant 10 min, afin de détruire les formes
végétatives.
ü Refroidir le tube à essai contenant l'eau à
analyser dans une bassine d'eau mise à la température ambiante
pour provoquer un «choc thermique» qui
induit le blocage de toutes réactions.
ü Verser l'échantillon préparé dans le
milieu de « viande foie ».
ü Incuber à 37 °C pendant 48h, l'apparition dans
le tube d'une coloration indique la présence des anaérobies
Sulfito-réducteurs.
V.4/ Recherche des
germes totaux :
Principe :
L'ensemble des germes totaux englobe les microorganismes
pathogènes d'une part et les microorganismes d'altération d'autre
part.
Ils sont capables de se développer à une
température entre 22 et 37 °C dans une
gélose nutritive.
Procédure d'analyse des germes
totaux :
Ø Préparation du milieu de
culture :
ü Mettre en suspension 23g du milieu
déshydraté dans un litre d'au distillée.
ü Mélanger et chauffer jusqu'à
ébullition pendant 2 minutes jusqu'à dissolution de produit.
ü Distribuer et stériliser à 120°C.
Ø Mode opératoire :
ü La recherche est réalisée par
incorporation en milieu
gélosé :
· Prélever 1 ml de l'échantillon à
analyser et le déposer dans une boite de pétri ;
· Mettre de la gélose nutritive tout en agitant
doucement à mouvement circulaire pour assurer un mélange
homogène de l'eau et de la gélose, sans former des bulles et sans
mouiller les bords de la boite.
· Incuber pendant 72h.
ü On identifie des colonies de différentes couleurs,
jaune clair, jaune d'or et leurs épaisseurs varient entre 4 et 6 mm de
diamètre.
![]()
D'une manière générale, ce stage a
été intéressant et bénéfique.
En effet, ce stage non seulement m'a permis de me familiariser
avec mes connaissances mais encore de les améliorer et d'acquérir
d'autres concepts et techniques plus utiles et plus efficaces à
l'échelle industrielle.
Ainsi, durant ce stage j'ai pris un aperçu sur les
différentes technologies et les méthodes d'analyses
utilisées dans le laboratoire central de la SONEDE et surtout
l'équipement de haute technologie adaptés à la nature des
analyses effectuées et leurs modes d'emploi (Spectrophotomètre
d'absorption atomique, Spectrophotomètre portatif modèle DR2010,
Spectrophotomètre à flamme, Spectrophotomètre UV visible,
Turbidimètre...).
Au cours de ce stage, j'ai eu une formation
générale en répartissant la durée globale dans les
quatre sections de laboratoires disponibles (SLACH (1(*)), SLBP (2(*)), SAA (3(*)), SLAB (4(*))).
De plus, j'ai pris un aperçu à l'organisation
administrative d'un laboratoire ainsi qu'à la vie socioprofessionnelle
et de ses exigences.
En dehors des aperçus techniques et administratifs, j'ai
pris conscience du rôle l'entreprise dans l'encadrement des
étudiants ainsi qu'à leur formation.
 
* (1 ) : Section de
Laboratoire d'Analyse Chimique
* (2 ) : Section de
Laboratoire de Biologie-Pollution
* (3 ) : Section
d'Absorption Atomique
* (4 ) : Section de
Laboratoire d'Analyse Bactériologique.
| 

