NAFTI
Yahia.
biohay2006@yahoo.fr
République Algérienne Démocratique
et Populaire
Ministère de l'Enseignement Supérieur et de la
Recherche Scientifique
Centre Universitaire ZIANE ACHOUR de
DJELFA
Institut des Sciences de la Nature et de la Vie
Département
de Biologie

Mémoire De Fin D'Étude En Vue De
l'Obtention Du Diplôme D'Ingénieur D'État
En
Biologie
Option : Contrôle de la Qualité et
Analyses

Thème
Contribution à l'étude de la
cinétique de libération d'un
principe actif: oxacilline sodique
encapsulé
en vue de déterminer les conditions de
conservation
Présenté par: Date de soutenance
:
' Mr.CHENOUF Ahmed 14/06/2008
' Mr.NAFTI Yahia
Devant le Jury:
' Mr. LAOUN khalil Maître Assistant, Chargé de cours
C.U.D Président
' Mr. DJEMOUI Amar Maître Assistant, Chargé de cours
C.U.D Examinateur
' Mr. RAHMANI Salah eddine Maître Assistant, Chargé
de cours C.U.D Examinateur
] Melle. BOUCHERIT N Maître Assistante, C.U.M
Promotrice
' Mr. BEN BRIDA Abdelghani Chef département de
développement SAIDAL Co-promoteur

äJÜ
ÜÜÜãIÚí äÜÜÜí
ÐáI íJÊÜÜÜÓí
áå áÜÜÜÞ *
ÑßÐÊÜ ÜÜí
ÇãÜ ÜÜäÅ ä~ãÜ
ÜÜÜ~Úí Ç áÇ
ä ÜÜÜÜí Ðá~ æ
* ÈÇÈÜ Üá
á~IjÜáæ
ãíÙÚáÇ
ááåÇ ÞÏÕ
~~ÒáÇ ÉÑæÓ ä
9~íUÇ
Tout d'abord je rends un grand hommage à la
mémoire de mon père, et je prie Dieu
le tout puissant de
l'accepter dans son vaste paradis
Je dédie ce mémoire à:
Ma chère maman, merci de votre amour et de toujours
croire en moi, j'espère
toujours rester fidèle aux valeurs
morales que vous m'avez apprises.
Je dédie ce mémoire ainsi
qu'a mes grands-parents trop tôt disparus.
Mon chèr
frère Attia et à mes précieuses soeurs.
Mes beaux frères : Chenouf Belgacem et Taleb Omar.
Mes proches les êtres chèrs comme on dit ... pour
moi, se serait plutôt mes
êtres-chèrs ... je vous aime
tous très fort...
Sans oublier mes amis avec qui je passe les bons moments en
particulier :
A.Abdelkader, B.Abdelrahman, B.Ahmed, B.Benaallia,
B.Boulerbah, B.Essadek,
B.Khaled, B.Radouan, B.R.Anouar, B.Naiel, B.Taher,
B.Tayeb, F.Abdelhafid,
F.Benyatou, G.Ameur,
G.Makhlouf, H.Abedelhadi, K.Ameur, L.Mohamed, M.Ali,
M.Ahmed, M.Attia,
M.Bachir, M.Elhadj, M.Elmagrbi, M.Lakhdar,
M.Moustafa, M. Toufik, R.Elembi,
R.Mostafa, S.Ben Saad, S.Mohamed l'ing,
Y.hssina, Z.Moussa Z.Khalil,, et à tous mes amis .

Une dédicace encapsulée à M. Djamel Eddine,
T Belkacem, H.Mouloud et son
équipe qui s'appelle la frappe, H.Salem et H.Mohamed,
H.Belel
Mon binôme
Tous mes amis du centre universitaire Zian Achour de Djelfa en
particulier toute
la promotion de 2006 de Biologie CQA.
Toute la Promotion de Biologie de l'université de Laghouat
2005 en particulier
B.Ryad, A. Saide, B.Mohamed, S.Nouredin,
B.Ali
Chenouf Ahmed
Dernière touche au manuscript, la Dédicace n'en
est pas moins importante.
Tout d'abord, je rends un grand hommage à
la mémoire de mon père, et je prie
Dieu le tout puissant de
l'accepter dans son vaste paradis.
Je dédie ce mémoire
à :
Ma mère qui m'a soutenu et encouragé durant la
période de mes études et à qui je
souhaite une longue
et heureuse vie.
Mes très chères soeurs : Djamila, Imane, et
Hanane.
Mes très chères nièces : Djihane, Roumaissa et
Rihab Nahla, ainsi qu'à leur père
Nafti Belkacem.
Mr.
Kacemi Djamal et toute sa famille.
Mon binôme.
Tous mes amis qui
m'ont encouragé.
Tous mes amis de Biologie option CQA promotion
2006.
Remerciements
Tout d'abord nous remercions le tout « PUISSANT ALLAH
» de nous montrer la
voie, guider et donner le courage de surmonter tous les
problèmes.
Et voilà, l'heure est maintenant aux remerciements
des personnes qui ont contribués
à l'élaboration de ce travail. La tiche n'est pas
simple mais il faut bien se
lancer...
Au terme de ce travail, il nous tient particulièrement
à coeur de remercier notre promotrice, M elle Boucherit N,
pour toute l'attention qu'elle nous a accordée.
Pour l'encadrement, les conseils, la disponibilité et la
patience, ainsi que notre
co-promoteur Mr. Ben brida A.
Nous adressons encore notre remerciement aux membres de
jury d'avoir
accepté de juger ce travail.
Nous remercions le médecin Dr. Zerguine Z pour son
soutien par ses références
bibliographiques concernant les médicaments et les
antibiotiques.
Un remerciement spécial à Mr. Kacemi M qui nous
aide avec plaisir.
Nos remerciements sont adressés à tous les
travailleurs de C.U.D sans exception
en particulier les personnels de la
bibliothéque, centre de calcul et le laboratoire
Nous remercions encore M elle Tahvchi F ,Benlahrech M
et Mr. Chenouf A
pour leur soutien dans les semaines qui ont
précédées la soutenance.
Liste des abréviations
· 6APA : Acide 6 amino
pénicilline
· 30 S : Sous unité 30
· 50 S : Sous unité 50
· ã : Tension interfaciale
· ADN : Acide
désoxyribonucléique
· ARN : Acide ribonucléique
· ARNpolymérase : Acide
ribonucléique polymérase
· ATB : Antibiotique
· BH+ : Base faible
· CMI : Concentration Minimale
Inhibitrice
· DCI : Dénomination Commune
Internationale
· DE 50 : Dose efficace 50
· DL50 : Dose létale 50
· ECH: Echantillon
· ex: Exemple
· fig: Figure
· HA : Acide
· HPLC : Chromatographie Liquide à
Haute Performance
· I: Intermédiaire
· IM : Intramusculaire
· IR : Intrarachidienne
· IV : Intraveineuse
· KF: Karl-Fisher
· L1 : Gouttelette de matière
active
· L2 : Phase continue
· L3 : Gouttelette de coacervat
· M : Concentration de
médicament
· m : Masse du principe actif
· Méti-R* :
Résistante à la méticilline
· Méti-S : Sensible à la
méticilline
· MRe : Complexe
médicament#177;récepteur.
· NCR : National Cash Registrer
Corporation
· OMS : Organisation Mondiale de la
Santé
· PA: Principe Actif
· pH : SRtectieIIG'hyGURJqce
· pKa : Constante de dissociation
· PLP : Pénicillines lient les
protéines
· ppm : Partie par million
· QSP : Quantité Suffisante Pour
· R : Résistante
· Re : Concentration du
récepteur.
· S1 : Surface du gouttelette de
matière active
· S2 : Surface de la phase continue
· S3 : Surface de gouttelette de
coacervat
· S: Sensible
· SBA: Sur Base Anhydre.
· STD : Standard
· T1/2 : Temps de demi-vie d'un
médicament
· T : Température
· Tf : Température de fusion
· t: Temps
· USA: United States of America
· USP : United States Pharmacopeia
· UV : Ultra -Violet
· Vd : Volume de distribution
Unités :
· 0C :
Degré Celsius
· g : Gramme
· h : Heure
· Kg : Kilogramme
· pg: Microgramme
· pl : Microlitre
· pm : Micromètre
· mg : Milligramme
· mg/l: Milligramme par litre
· ml : Millilitre
· mm: Millimètre
· mn : Minute
· nm : Nanomètre
· ng/ml : Nanogramme par millilitre
· N : Normal
· Cm : Centimètre
Liste des tableaux
Tableau (01) : Diversité des
antibiotiques de type beta-lactames: principaux cycles et ATB
représentatifs 20
Tableau (02) : Echelle exprimant la
solubilité d'une substance . 47
Tableau (03) : Résultats du
contrôle physicochimique de l'oxacilline sodique 60
Tableau (04) : Résultats du
contrôle physicochimique de l'acide alginique 63
Tableau (05) : Résultats d'absorbances
des solutions d'oxacilline sodique à différentes
concentrations 65
Liste des figures
Figure (01) : Illustration des
dénominations portées sur une boite d'un médicament ..
4
Figure (02) : Différentes formes
galéniques des médicaments 5
Figure (03) : Distribution d'un
médicament entre les deux formes : ionisée et non ionisée
7
Figure (04) : Schéma
général de la distribution d'un médicament 8
Figure (05) : Phases de transformations des
médicaments 9
Figure (06) : Antibiotiques et leurs sites
d'action 15
Figure (07) : Schéma simplifié de
l'enveloppe d'une bactérie GRAM- 21
Figure (08) : Structure chimique de la
pénicilline 22
Figure (09) : Action des pénicillinases
sur les pénicillines 23
Figure (10) : Action des acylases sur les
pénicillines 23
Figure (11) : Structure chimique de l'oxacilline
sodique 24
Figure (12) : Types de microparticule 27
Figure (13) : Comportement d'un coacervat (3)
vis-à-vis une phase liquide non miscible (1) 29
Figure (14) : Schéma de principe du
procédé de microencapsulation par coacervation complexe 30
Figure (15) : Schéma de principe du
procédé de microencapsulation par évaporation de solvant.
31
Figure (16) : Schéma de principe du
procédé d'encapsulation par gélification thermique
(hot
melt).......................................................................................................
. 32
Figure (17) : Etape de formation d'un film d'enrobage
par spraycoating sur des particules
solides........... . 33
Figure (18) : Schéma de principe du
procédé de gélification de gouttes 34
Figure (19) : Congélation de gouttes:
schéma de principe 35
Figure (20) : Mécanisme de la
polycondensation interfaciale 35
Figure (21) : Microréacteurs et
systèmes à libération déclenchée et
prolongée 37
Figure (22) : Processus de gonflement d'un
hydrogel .. 38
Figure (23) : Représentation
schématique d'une paroi matricielle . 39
Figure (24) : Profils de libération
obtenus à partir de différents types de microparticules 41
Figure (25) : Représentation
schématique de polymères linéaires 42
Figure (26) : Unités monosaccharidiques
constituant l'alginate 43
Figure (27) : Structure chimique de l'alginate .
44
Figure (28) : Etapes de préparation des
sphères 58
Figure (29) : Chromatogramme du STD (oxacilline
sodique) obtenu par analyse par HPLC 61
Figure (30) : Chromatogramme d' ECH (oxacilline
sodique) obtenu par analyse par HPLC 62
Figure (31) : Courbe d'étalonnage
d'oxacilline sodique 65
Figure (32) : Sphères de l'oxacilline
sodique 66
Figure (33) : Influence de la T sur la
libération du PA dans l'eau distillé à pH1,2 67
Figure (34) : Influence de la T sur la
libération du PA dans l'eau distillé à pH=3 67
Figure (35) : Influence de la T sur la
libération du PA dans l'eau distillé à pH=7 68
Figure (36) : Influence de la T sur la
libération de PA dans l'eau physiologique à pH=1,2.........
69
Figure (37) : Influence de la T sur la
libération de PA dans l'eau physiologique à pH=3 69
Figure (38) : Influence de la T sur la
libération de PA dans l'eau physiologique à pH=7 69
Figure (39) : Influence de la T sur la
libération de PA dans l'eau distillé à pH1,2........ .
70
Figure (40) : Influence de la T sur la
libération de PA dans l'eau distillé à pH3 . 70
Figure (41) : Influence de la T sur la
libération de PA dans l'eau distillé à pH7 . 71
Figure (42) : Influence de la T sur la
libération de PA dans l'eau physiologique à pH1,2........ 71
Figure (43) : Influence de la T sur la
libération de PA dans l'eau physiologique à pH=3 72
Figure (44) : Influence de la T sur la
libération de PA dans l'eau physiologique à pH=7 72
Sommaire
Liste des abréviations Liste des tableaux
Liste des figures
Introduction générale
Partie I : Etude bibliographique
|
Chapitre I: les médicaments
|
I.1. Généralités sur les médicaments
3
I.1.1. Définition d'un médicament 3
I.1.2. Composition d'un médicament 3
I.1.3. Origines des médicaments 3
I.1.4. Dénominations des médicaments 4
I.1.5. Fonctions du médicament 4
I.1.6. Classification des médicaments 5
I.1.7. Différentes formes pharmaceutiques
(galéniques) des médicaments ............................... 5
I.1.8. Voies d'administration des médicaments 5
I.2. Pharmacocinétique d'un médicament 6
I.2.2. Devenir du médicament dans l'organisme
I.3. Pharmacodynamique d'un médicament
I.3.2. Mécanisme d'action des médicaments
|
Chapitre II: les antibiotiques
|
II.1. G
II.1.3. Classement des
II.1.4. Les grandes fam
II.2.2. Activité des
II.2.3. Résistance
II.3. Antibiothérapie
II.3.1 Voies d'adm
II.3.3. Pharmacocinétique de
II.4. Antibiotiques Bêta-lactam
II.4.2. Mécanisme d'action des bêta-lactam
II.4.3. Résistance aux bêta-lactamines 21
II.4.4. Pharmacocinétique 21
II.5. Les pénicillines 22
II.5.1. Définition et structure chimique 22
II.5.2. Classification des Pénicillines 22
II.5.3. Propriétés physico-chimiques 22
II.6. Oxacilline sodique 24
II.6.1. Définition 24
II.6.2. Spectre d'activité antibactérienne 24
II.6.3. Pharmacocinétique 24
II.6.4. Conditions particulières de conservation 25
II.6.5. Formes galéniques de l'oxacilline 25
Chapitre III: la microencapsulation
III.1. Historique 26
III.2. Définition 26
III.3. Types de microparticules 26
III.4. Intérêt de microencapsulation 27
III.5. Procédés de microencapsulation 27
III.5.1. Procédés physico-chimiques 28
III.5.2. Procédés mécaniques 32
III.5.3. Procédés chimiques 35
III.6. Libération contrôlée (1P1)
PA 36
III.6.1. Définition 36
III.6.2. Mécanismes de la libération
Contrôlée 37
III.6.3. 3DIP ql1H01)flP11)2a1)lMEKIEpIalIR1) (IP1) 3$ 40
III.6.4. Profils de libération obtenus à partir de
différents types de microparticules 40
III.7. Les polymères 41
III.7.1. Définition 41
III.7.2. Fonctionnalité 41
III.7.3. Classification des polymères 42
III.7.4. Polymères utilisés dans la
microencapsulation 43
III.7.5. Acide alginique 43
Partie II: Etude expérimentale
Chapitre I : Matériels et
méthodes
I.1. Matériels utilisés 45
I.2. Méthodes 46
I.2.A. Contrôle physicochimique de la matière
première 47
I.2.A.1. Caractérisation du PA [oxacilline sodique] 47
I.2.A.2. Caractérisation de l'excipient [acide alginique]
52
I.2.B. Etude des paramètres influençant la
libération du PA [oxacilline sodique] encapsulé 56
I.2.B.1. Détermination de la courbe d'étalonnage
56
I.2.B.2. Préparation des sphères d'oxacilline
sodique 57
I.2.B.3. Détermination du rendement d'encapsulation 58
I.2.B.4. Étude de la stabilité des sphères
58
Chapitre II : Résultats et discussions
II.A. Résultats de contrôle physicochimique de la
matière première 60
II.A.1. Caractérisation physico-chimique du PA [oxacilline
sodique] 60
II.A.2. Caractérisation physico-chimique de l'excipient
[acide alginique] 63
II.B. Résultats de l'étude des
paramètres influençant la libération du PA [oxacilline
sodique]
encapsulé 65
II.B.1. Détermination de la courbe d'étalonnage
65
II.B.2. Préparation des sphères de l'oxacilline
sodique 65
II.B.3. Détermination du rendement d'encapsulation 66
II.B.4. Etude de la libération du PA encapsulé
66
Discussion générale 73
Conclusion générale 75
Références bibliographiques
Annexes
. Ù~6áÇ Øæ~Ô
ÐíÐ~Ê Ðå~ á-È~ã
íÏì4 ä~~~Ó4~ßæ :
Êá~Ú ÉÏIã Ñ~AÊ
Êß)Í ÊÓÇÑÏ í
Ê4~1~ã ÕÎ3ã
ã.Ìßí ñÏìØ
òõåõÓÈ~Sæ
:ÎäÈx ÍÏÈí
ñìÒ_Ñ
ÏÈÌõÌÍ ÒõÖ4Ñ
ðäÅ Îå4 òí Ðåó
ãð-äÇ ÇÐt
ááÇ4 òí
ßßiÒåä
ÎåËÈÞ
æìóÏìÙäÇ
ÏÈõõÌä ßðä
ÏÈÆóÒÌäÇ
ÍÏÐ,Òí ÊäÇìÞ
ò
|
- aziazz Edz Egzza
|
Îõõ9Ñ
ÊaÍ
|
-
|
|
- ÒÌÒ~Ñ
ÍÒõ~áÇ öÐh
|
ËõÍ
|
Îåð.Ò.ðäÇ
ÎõäæáÇ
ÏÇìðäÇ
ÎÌÞÇÒí
|
Ð,Ë ÇÐa
|
óìõÍ
|
ÎiËÈ~í -
ÈåõåÚ ãÙ.ðäÇ
ÌLÈÒõäÇ Ê.Í
:
Îõ4Èõðõ~ìóÒõ~äÇ
ãíÇì~äÇ Ö.Ë
ÒõËUÑ ÎÓÇÑÏ
íÒ~ Îå4 òíæ ,
EÎõßóÒíá Ç
ÎõóìóÈ~äÇ
ÕìÙõäÇ ò
ÎËìå~ðäÇ
ÒõóÈ2ðåä (
ÎÌØÑ æ ÎÈ~
ÏÈÌõÌ~äÇ ãß.
ßäÐSæ ÑÒ~ÒäÇ
ØÓæ æ ÍÑÇÒ~äÇ
Î~ÑÏ , 00 ~äÇ Èåõí
ÎäÈ..äÇ ÍÏÈðäÇ
ÑÒzÑ ðåÚ
ÎõäÈÒäÇ
Ì.ÈÒõäÇ ðåÚ
ÈõåÙzÑ ËõÍ
|
. ÎäÈ~~äÇ
ÍÏÈðä á Îõð~
ÒÌ~
|
ÑÒ~Ñ
|
ÎÙÍáÇí
ìÑ
|
q ,2 Eæ E7
EòõÒðõ~äÇ ÐõÚ :
00 ~äÇ
|
ÖÎó Èðõ
|
-
|
ÙLLN.11
Îð1áÇí 4.
4ÑÏ
.
|
q ÒÌÒ.Ñ
ÈðõõË ÑÒ-Òåä
Î~ÑÏ ò"Í ã~ðÑ :
EÍÑÇÒ-äÇ Î~ÑÏ
ÖÎó Èðõ
|
-
|
|
|
. Îõ~ÇæÐäÇ
ÎÛõÙäÇ öÐ~
ìóÐ~Òä ÎióÒØ
ò.Í ÈÌäÇ
ãßÔäÇ ÒÌÒ~ó
|
:EÏÈÌõÌ.äÇ
ãß- ÖÎó Èðõ
|
-
|
. Ò31ðäÇ
ÁÈðäÈË
ÎóÑÈ~í
ò+ìäìóÒõ~äÇ
ÁÈðäÇ ò ÒÌ6
ñìßó ÑÑÍÐäÇ
ñ ÙÍáÇó
|
:ÑÒ-ÒäÇ
ØÓæÉ Þå~Òó
Èðõ
|
-
|
|
,
|
ÈóìõÍ
ßß~Òåä ÎåËÈÞ
|
æìóÏìÙäÇ
ÏÈõõÌä
|
,
|
ã~Ìßí
|
, ñÏìØ
òõåõÓÈ~~æ
|
,
|
ÎäÈ. ÍÏÈí
|
,
|
ÏÈÌõÌÍ :
Ê.Í .~.áÇ
Ë4.1SáÇ
|
. ÑÒ-Ñ ,
Îõ1Èõðõ~ìóÒõ
ãíÇìÚ,ÎÌÞÇÒí
Résumé : Contribution à
l'étude de la cinétique de libération d'un principe actif:
oxacilline sodique encapsulé en vue de déterminer les conditions
de conservation.
Dans ce travail nous sommes interesses d'atteindre un double
objectif, le premier est d'encapsuler - selon le procede de gelification de
gouttes - un principe actif : oxacilline sodique dans des matrices polymeriques
biodegradables d'alginate de sodium, tout en contrôlant les
matières premières utilisees qui sont declarees - d'apr~s les
résultats obtenues - conformes aux normes de la pharmacopee americaine,
et le second est d'étudier l'effet de certains paramètres
physico-chimiques sur la liberation du principe actif tels que ; pH,
temperature , le milieu de dissolution et la forme des sphères
(sèche ou humide) dont on a atteint les resultats suivantes :
- Pour le pH : 7 et 1,2 correspondent à des valeurs dans
lesquelles la quantite liberee du principe actif est maximales ;
- Pour la temperature : 37 °C est la meilleure temperature
de liberation, tandis que la temperature 4°C est une temperature adequate
de conservation ;
- Pour la forme des sphères : la forme sèche
represente la meilleure forme pour presenter cette formulation ;
- Pour le milieu de dissolution : la libration est mieux
favorisée dans l'eau physiologique en comparant par l'eau distillee.
Mots clés : Encapsulation, principe
actif, oxacilline sodique, biodegradable, alginate de sodium, contrôle,
sphères, paramètres physico-chimiques, liberation.
Abstract : Contribution to the study of the kinetics
of the release of encapsulated active ingredient: sodic oxacillin in order to
determine the conditions of conservation.
In this work we are interested to attain a double objective;
the first is to encapsulate - using the process of prilling - an active
ingredient: oxacillin sodic in biodegradable polymeric matrix of sodium
alginate. This, after the control of the raw materials used which are declared
- according to the results obtained - in conformity with the standards of the
American pharmacopeia, and the second is to study the effect of certain
physicochemical parameters on the release of the active ingredient such as; pH,
temperature and the medium of dissolution and the spheres form (dry or wet).
Where we attained the following results:
- For the pH: 7 and 1,2 correspond to values in which the
released quantity of the active ingredient is maximum;
- For the temperature: 37 °C is the best temperature of
release, while the temperature 4°C is an adequate temperature of
conservation;
- For the form of the spheres: the dry form represents the best
way to represent this formulation;
- For the medium of dissolution: libration is favored in
physiological water comparing with the distilled water.
Key words: Encapsulation, active ingredient,
oxacillin sodic, biodegradable, sodium alginate, control, sphere,
physicochemical parameters, release .
Introduction générale
Introduction générale
L
a pharmacie galénique est la science et l'art de
préparer, de conserver, et de présenter les médicaments.
Actuellement, ce terme concerne la totalité des médicaments
contenant un principe actif qui nécessite une mise en
forme galénique pour son administration. Cette forme existait sous
plusieurs présentations mises sur le (s) marché (s) national
et/ou international (Lùllmann et al., 1998).
L'objectif de l'industrie pharmaceutique est
l'amélioration des propriétés des médicaments
produits ce qui reflète l'importance de ces derniers pour la
santé publique. Il est à noter que le développement et la
création de nouvelles préparations médicamenteuses,
dépendent de plusieurs paramètres y compris les
propriétés du principe actif, cette molécule doit - par
conséquent- conserver ses propriétés thérapeutiques
pendant la cascade: depuis la mise du point jusqu' à la cible
(Lùllmann et al., 1998).
Parmi les matières actives mises sous formes
galéniques en Algérie; les antibiotiques, qui sont des substances
particulières dans l'arsenal pharmaceutique par leur origine et leur
mode d'action. Le terme antibiotique désigne une substance d'origine
microbienne qui à très faible dose empêche la croissance
d'autres microorganismes ou les détruit, utilisée dans les
pathologies diverses du nouveau-né au vieillard.
La mise en point de nouvelles préparations
médicamenteuses nécessite un temps considérable et des
coûts importants que les pays en voie de développement ne peuvent
pas garantir. Le recours à la recherche de nouvelles préparations
faciles à mettre en oeuvre et similaires à celles
élaborées par les procédés appliqués par les
pays développés constitue un outil de choix permettant d'assurer
une amélioration notable à l'échelle de la recherche et du
développement locales. Il devient donc impératif, que la
formulation établie répond aux critères de la
qualité d'un médicament.
L'encapsulation est parmi les préparations
pharmaceutiques en cours d'élaboration par les grandes firmes
médicamenteuses, qui consiste à encapsuler selon un
procédé déterminé un principe actif dans une autre
matière inactive afin d'améliorer les propriétés de
conservation, de présentation et de biodisponibilité
(Richard et Benoit, 2000).
Dans le contexte où s'inscrit notre travail, nous avons
tenté d'atteindre l'objectif, qui consiste à appliquer le
procédé de gélification d'un polymère pour former
des microsphères comportant un principe actif "oxacilline sodique",
caractérisées par la suite par une cinétique de
libération qui dépend de plusieurs paramètres.
A cette fin nous proposerons de suivre le plan d'étude
suivant : > Une partie bibliographique devisée en trois chapitres
:
1°/- le premier chapitre, donne un aperçu sur les
médicaments, leurs composition, leur origine et leur comportement
dans l'organisme ;
2°/- le second chapitre, présente des notions de
bases sur les antibiotiques, leurs classifications, ainsi que les
principales familles existantes ;
3°/- le dernier chapitre, comporte une synthèse
bibliographique sur la microencapsulation, et les principaux
procédés couramment utilisés.
> Une partie expérimentale, consistant en premier lieu
à présenter les méthodes appliquées ainsi que le
matériel utilisé, et en deuxième lieu à montrer les
résultats établis à l'issu de ce travail.
Enfin une conclusion générale et des perspectives
achèveront l'étude.
Partie I
Etude bibliographique
Chapitre I
Les médicaments
I.1.Généralités sur les
médicaments :
I.1.1. Définition d'un médicament
:
Un médicament est défini d'une façon
très large comme une substance chimique qui affecte les processus de la
vie.
L'OMS donne une définition plus restrictive : «
Toute substance ou produit qui est utilisé pour modifier ou explorer les
systèmes physiologiques ou les états pathologiques pour le
bénéfice de celui qui reçoit la substance »
(Helali, 1994).
I.1.2. Composition d'un médicament :
Tout médicament est composé de deux
éléments :
> Principe actif : c'est la
molécule active détenant les propriétés curatives
ou préventives ; > Excipient : c'est une
substance inactive par elle-même, dont l'intérêt est de
faciliter l'utilisation du médicament, et notamment sa
libération.
Ainsi, dans un comprimé de 500 mg d'aspirine, on
trouvera 500 mg d'acide acétylsalicylique, qui est le PA, et une
quantité d'amidon (QSP), par exemple, qui constitue l'excipient
(Anonyme 1).
I.1.3. Origines des médicaments :
Les médicaments peuvent être obtenus de sources
très diverses :
> Origine végétale
:
C'est la source la plus ancienne, mais qui reste
d'actualité. Il est classique de distinguer parmi les produits
végétaux :
- Les alcaloïdes : tels que la quinine, strychnine morphine
;
- Les gommes : tels que les gommes pour suspension (arabique,
adragante) ;
- Les glycosides : ils contiennent des sucres dans leurs
structures chimiques, tels que la digitoxine.
> Origine animale :
- Extraits de sang humain tel que le fibrinogène ;
- Hormones polypeptidiques extractives tel que l'insuline ;
- Enzymes : tels que la trypsine,
chymotrypsine.et les kinases ;
Ils existent des excipients pharmaceutiques tels que la
lanoline.
> Origine synthétique :
La plupart des médicaments actuellement
commercialisés sont d'origine synthétique, obtenus par :
- Synthèse totale ;
- Hémi-synthèses : tels que certaines
pénicillines.
> Origine biogénétique
:
Les méthodes de génie génétiques
sont les dernières venues parmi les méthodes d'obtention des
médicaments : elles permettent de fabriquer par les cellules vivantes -
procaryotes ou eucaryotes - des substances naturelles polypeptidiques
présentant toutes les caractéristiques de leur modèle
humain.
La production de masse de ces protéines parfaitement
définies a permis d'obtenir de nouveaux médicaments :
- Hormones ;
- Facteurs de croissances (Moulin et Coqurel,
2002).

I.1.4. Dénominations des médicaments
:
Un même médicament peut avoir plusieurs noms
différents :
> Nom chimique : qui correspond
à la formule chimique du PA, ce nom n'apparaît pas sur le
conditionnement du médicament, exemple : 4-Thia-1-azabicyclo [3.2.0]
héptane-2- acide carboxylique, 3,
3-diméthyle-6-[[(5-méthyle-3-phényle-4-isoxazolyle)
carbonyle] - amino]-7- oxo-, sel monosodium, monohydraté, [2S-(2a, 5a,
6b)]- est le nom chimique de l'oxacilline sodique.
> Dénomination Commune Internationale
(DCI) : c'est le nom admis pour tous les pays, et il est
enregistré par l'OMS, exemple l'oxacilline sodique. La DCI est celle
qu'il faudra retenir de préférence, afin de pouvoir se retrouver
parmi les nombreuses marques du même médicament.
> Nom commercial, ou nom protégé
: c'est le nom sous lequel une firme pharmaceutique vend un
médicament donné. Etant donné qu'elle dépense un
certain budget pour la publicité autour de ce nom, ce nom sera
protégé par un brevet, dont la durée est variable suivant
les pays (de 10 à 99 ans), il y a par exemple près de 400 noms
différents protégés de composés contenant de
l'aspirine dans certains pays. Le nom commercial s'écrit avec un ®
(ex. OXACARE®). Comme illustré dans la figure ci-dessous
(Helali, 1994).
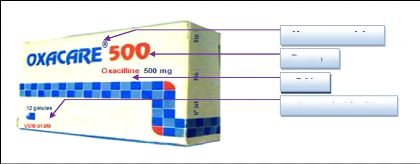
Dosage
Nom commercial
DCI
Mode d'utilisation
Figure (01) : Illustration des
dénominations portées sur une boite d'un
médicament.
I.1.5. Fonctions du médicament :
Un médicament peut exercer des fonctions diverses :
> Fonction thérapeutique :
c'est la plus habituelle, elle peut être :
* Préventive :
- Individuelle (vaccination, prévention individuelle du
paludisme, chimioprophylaxies diverses) ;
- Collective (chimioprophylaxies collectives de la
méningite, de la tuberculose). * Curative :
- Etiologique : le médicament s'attaque à la cause
de la maladie ;
- Substitutive : il apporte l'élément manquant
à l'organisme ;
- Symptomatique : il s'attaque seulement aux manifestations de la
maladie, sans pouvoir en traiter la cause.
> Fonction diagnostique :
Il peut agir d'opacifiant, de traceurs, d'agents
pharmacodynamiques divers, utilisés pour réaliser des
explorations fonctionnelles (Moulin et Coqurel, 2002).
I.1.6. Classification des médicaments
:
On peut définir les classes des médicaments de
différentes manières : classes selon leurs origines, leurs
compositions ou leurs structures chimiques, classes pharmacologiques selon
leurs actions sur l'organisme et classes thérapeutiques selon les
pathologies traitées (Anonyme 2).
I.1.7. Différentes formes pharmaceutiques
(galéniques) des médicaments :
La forme galénique1 d'un médicament
est sa présentation concrète : sirop, gélule, sachet,
comprimé, pilule, granulés, ampoule, flacon à perfusion,
seringue prête à l'emploi, ovule, collyre, aérosol,
pommade, crème, ces formes sont les formes les plus courantes, (fig.02)
(Anonyme 1).
Comprimé
Dragé
Solution injectable
Ampoule buvable
Gélule
Figure (02) : Différentes formes
galéniques des médicaments. (Anonyme 1)
I.1.8. Voies d'administration des médicaments
:
Il existe plusieurs voies d'administration, mais, selon la
voie utilisée, les PA n'ont pas le méme devenir dans l'organisme
et subissent des modifications métaboliques plus ou moins importantes,
ce qui peut altérer leur activité pharmacologique, surtout en ce
qui concerne le début, l'intensité et la durée de leur
action.
Les principales voies d'administration sont :
- Voie orale ;
- Voie parentérale ;
- Voies transmuqueuses : buccale, perlinguale, oculaire, nasale,
pulmonaire ...etc ; - Voie cutanée (Aiache et al.,
1995).
1 Le mot galénique référe au
CLAUDE GALIEN (v. 131-v. 201) : Médecin romain né à
Pergame, il commence dans cette ville ses études de philosophie et de
médecine. Sa thérapeutique est diététique et
médicamenteuse, et son étude des plantes médicinales
gardera le nom de « pharmacie galénique » (Anonyme
3).

I.2. Pharmacocinétique d'un médicament :
I.2.1. Définition :
C'est la partie de la pharmacologie qui étudie, en
fonction du temps, le devenir d'une substance après son introduction
dans un organisme vivant. Cette destinée comporte essentiellement quatre
phases : l'absorption ou résorption, la distribution, les
biotransformations ou métabolismes, et l'élimination, qui peuvent
être, en partie ou en totalité, fonction de la voie
d'administration et de la forme sous laquelle elle est administrée. Ces
facteurs conditionnent la quantité du produit pouvant atteindre les
cibles biologiques et produire une modification, à l'origine de l'effet
thérapeutique (Anonyme 3).
I.2.2. Devenir du médicament dans l'organisme
:
Après avoir pénétrer dans l'organisme, le
PA traverse un nombre variable de cellules pour aboutir dans la circulation
sanguine. Au niveau du tube digestif, et en particulier de l'intestin, la
résorption est facilitée par la très grande surface de
contact avec le contenu intestinal. La voie orale est donc la plus
utilisée, meme si elle n'est pas adaptée à toutes les
situations (Anonyme 3).
I.2.2.1. Absorption :
A) Les niveaux d'absorption :
L'absorption est le transfert d'un PA de son site
d'administration jusqu'à la circulation sanguine. Le taux et
l'efficacité d'absorption dépendent de la voie
d'administration (Champe et al., 2000).
> Absorption à partir des points
d'administration :
Elle est proportionnelle à la solubilité dans l'eau
du liquide extracellulaire. Les complexes peu solubles dans l'eau seront
absorbés lentement.
> Absorption au niveau de l'estomac
:
Elle peut avoir lieu à partir de la paroi stomacale
pour les petites molécules (alcool, eau) et pour les molécules
non ionisées au pH de l'estomac (ex. Aspirine). Mais l'absorption
stomacale est limitée dans le temps en cas de vidange rapide de
l'estomac. Cette motricité gastrique déterminera la vitesse de
délivrance à l'intestin, et sera ralentie par la présence
d'aliments.
> Absorption au niveau de l'intestin
:
La plus grande partie de l'absorption se fera au niveau de
l'intestin, qui présente une grande surface (Helali,
1994).
B) Mécanismes d'absorption :
> Transport passif :
Il peut se faire par :
- Diffusion simple le long du gradient de concentration pour les
substances liposolubles.
- Diffusion facilitée : dans ce cas la substance se
combine avec une molécule transporteuse de la membrane, qui agira sans
dépense d'énergie.
- Filtration sous un gradient de pression (cas de la filtration
glomérulaire).
> Transport actif :
Il est comparable à la diffusion facilitée, mais il
peut agir contre un gradient de concentration avec dépense
d'énergie. Le transport actif est sélectif et saturable
(Schmitt, 1980).

C) Facteurs influençant l'absorption
:
1. pH :
La plupart des médicaments sont des acides ou des bases
faibles (Champe et al., 2000). > Au
niveau de l'estomac :
Au niveau de l'estomac les acides faibles sont absorbés,
et les bases faibles sont excrétées.
> Au niveau de l'intestin :
Le pH intestinal étant alcalin, les pores aqueux rendent
possible l'absorption de molécules hydrosolubles (Schmitt,
1980).
La figure (03), illustre les formes ionisées du
médicament en fonction du pH du milieu d'absorption.
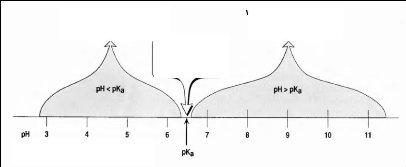
pK est le pH auquel 50 % de la
substance est
ionisée
Si : pH < pKa
HA et
BH+
prédominantes
Si : pH = pKa
HA = A-
BH+ = B
Si : pH > pKa
A- et
B
prédominantes
Figure (03) : Distribution d'un
médicament entre les deux formes : ionisée et non
ionisée
(Pour cette illustration : le médicament a un pKa =
6.5) (Champe et al, 2000).
2. Flux sanguin :
Le flux sanguin au niveau de l'intestin est plus grand qu'au
niveau de l'estomac, ce qui justifie l'importance d'absorption à ce
niveau.
3. Etendue de la surface absorbante
:
L'intestin présente une surface 1000 fois large que
l'estomac donc l'absorption intestinal est plus efficace.
4. Temps de contact avec la surface absorbante
:
Si le médicament se déplace rapidement tout le long
de tractus gastro-intestinal (cas d'une diarrhée sévère),
Il sera mal absorbé (Schmitt, 1980).
Il est à signaler que :
La biodisponibilité se
définit comme étant la fraction de la dose de médicament
administré qui atteint la circulation générale et la
vitesse à laquelle elle l'atteint (Lechat, 2007).
I.2.2.2. Distribution :
L'étape de distribution du PA, lorsqu'il atteint la
circulation générale, peut se diviser en deux phases :
A) Fixation d'un médicament aux
protéines plasmatiques (phase plasmatique) :
Les protéines plasmatiques ont un rôle très
important dans la distribution des

médicaments dont vont être transportés,
leurs fixations sur ces protéines se fait par des liaisons
réversibles parmi ces protéines l'albumine qui est la
plus importante sur le plan quantitatif (Touitou, 1995).
La molécule d'albumine est chargée, et
peut se lier de nombreuses substances. La partie liée aux
protéines par rapport à la quantité totale plasmatique est
variable : 90 % pour la pénicilline, moins de 10 % pour la
caféine (Helali, 1994).
La partie liée aux protéines n'a pas d'action
pharmacologique. Elle sert de réserve car le médicament se
défixe des protéines en fonction les besoins. C'est cette
fonction non liée aux protéines qui sera responsable de
l'activité thérapeutique (Touitou, 1995).
La liaison d'un médicament aux protéines
plasmatiques se caractérise par le pourcentage de médicament
fixé mais aussi par la force de la liaison (constante d'affinité
et le nombre de sites de fixation qui sont plus ou moins importants et pourront
être plus ou moins saturés).
B) Pénétration tissulaire (phase
tissulaire) :
La fraction libre du PA, véhiculé par le sang, va
pouvoir pénétrer dans les tissus de différents organes. La
pénétration tissulaire dépend de plusieurs facteurs :
- La taille de la molécule du PA ;
- L'hydrosolubilité et la liposolubilité du
médicament ;
- La fixation aux protéines plasmatiques et aux
protéines tissulaires : il existe un équilibre entre les formes
libres et les formes liées aux différentes protéines
plasmatiques et tissulaires ;
- La vascularisation : plus l'organe est vascularisé,
plus la pénétration sera importante et rapide. La liaison d'un
médicament sur ses récepteurs tissulaires et le plus souvent
réversible (Jolliet et al., 2000).
Les étapes de distribution sont résumées
dans la figure (04).
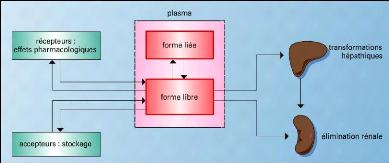
Figure (04) : Schéma
général de la distribution d'un médicament
(Anonyme 3).
Il est à signaler que :
Le volume de distribution : exprime
la quantité de médicament dans l'organisme divisée par la
concentration plasmatique du médicament. Ce volume est un volume
théorique qui serait atteint en cas de répartition
homogène de la molécule dans le volume, C'est à dire que
la concentration du médicament serait partout identique à celle
du plasma.
Vd =Quantité administrée /
Concentration plasmatique (Moulin et Coqurel, 2002).
I.2.2.3. Biotransformation (métabolisme)
:
Le terme métabolisme fait référencer
à la transformation par une réaction enzymatique d'un
médicament, en un ou plusieurs autres composés actifs ou inactifs
au plan pharmacologique.
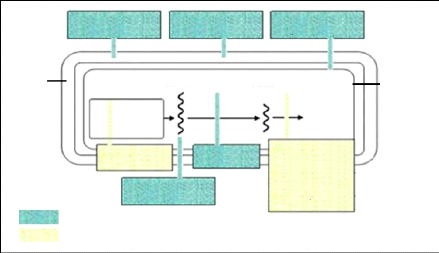
De nombreux tissus peuvent réaliser cette
transformation (peau, poumon, rein, intestin...). Néanmoins le principal
site de biotransformation se situe au niveau hépatique (Moulin
et Coqurel, 2002) (fig.05).
MEDICAMENT
PHASE 1
METABOLITES
PHASE 2



Métabolites libres
Métabolites conjugués
Médicament
conjugué
Médicament
inchangé



Figure (05) : Phases de transformations
des médicaments (Anonyme 2).
Les réactions de biotransformation sont divisées
en deux phases, phase 1 et phase 2, mais il faut signaler que
le passage d'une phase à autre et l'ordre dépend de la structure
du médicament lui-même et la capacité des enzymes
hépatiques (Anthony, 2002).
A) Réactions de la phase 1 :
Au cours de la phase 1, des réactions biochimiques
transforment la substance initiale en métabolites.
- Le métabolite formé peut être
pharmacologiquement actif. C'est un processus d'activation.
- Le métabolite formé peut être dangereux
pour l'organisme qui le fabrique. On parle de «métabolite
réactif », Il s'agit surtout de radicaux libres.
- Les métabolites formées peuvent être
inactifs (inactivation) ou moins actifs (désactivation) que la
molécule initiale.
Parmi les réactions de la phase 1 on trouve :
- Oxydation ;
- Réduction ;
- Hydrolyse (Anonyme 2).
B) Réactions de la phase 2 (conjugaison)
:
Les conjugaisons réalisent l'union des
médicaments ou de leurs métabolites avec un agent conjuguant
provenant du métabolisme physiologique. Le produit formé,
appelée conjugué, est inactif et facilement
éliminé. Le siège des conjugaisons est essentiellement
hépatique.
La glycurono-conjugaison est la conjugaison la plus
fréquente chez l'homme (Anonyme 2).
C) Facteurs influençant la biotransformation
:
- Les gènes : l'activité des enzymes du
métabolisme peut varier d'un individu à l'autre ; - L'age : sujet
âgé, enfant ;
- L'induction/inhibition : certains médicaments ont la
propriété de stimuler les systèmes enzymatiques
responsables du métabolisme ou de les inhiber ;
- L'insuffisance hépatique ;
- La grossesse (Jolliet et al., 2000).
I.2.2.4. Elimination (excrétion)
:
A) Voies d'élimination :
L'élimination correspond à la disparition du
médicament sous forme active. Cette élimination peut se faire
sous une forme inchangée dans l'urine et les selles ou bien après
une biotransformation en métabolites inactifs le plus souvent
(Saint-Maurice et al., 2004). L'excrétion d'un PA peut
s'effectuer par différentes voies :
> Excrétion par voie rénale
:
La voie rénale est la voie d'excrétion
principale, le rein joue son rôle physiologique d'excrétion. Le PA
passe de la circulation sanguine dans l'urine par deux processus : la
filtration glomérulaire, et la sécrétion tubulaire
(Jolliet et al., 2000).
> Excrétion par voie hépatique
:
Le foie participe à l'excrétion des
médicaments hors de l'organisme par le biais du système biliaire.
Après excrétion dans la bile, le médicament se retrouve
dans la lumière intestinale où il peut être
réabsorbé.
> Autres voies
d'excrétion:
Les autres voies (salivaires, pulmonaire...) sont usuellement
négligeables par rapport aux voies rénale et hépatique.
Néanmoins on soulignera l'importance de la voie lactée pouvant
donner des risques d'intoxications du nourrisson lors de l'allaitement
(Lechat, 2007).
Il est à signaler que :
La clairance : c'est la
capacité globale de l'organisme à éliminer une
molécule, elle correspond au volume de plasma totalement
épuré par unité de temps ; elle est ainsi habituellement
exprimée comme un débit en ml/min.
La clairance totale est égale à la somme des
clairances de chaque organe susceptible d'intervenir dans l'élimination
du médicament: clairance rénale, hépatique, intestinale,
pulmonaire ...etc (Lechat, 2007).
Le T (1/2) : c'est le temps
nécessaire pour que la concentration plasmatique d'un
médicament tombe par la moitie. Le T
(1/2) est déterminé par le volume de
distribution et la clairance ou bien l'élimination (Yang et al.,
2004).
I.3. Pharmacodynamique d'un médicament
:
I.3.1. Définition :
Le terme pharmacodynamique réfère à
l'action d'un médicament au niveau cellulaire, ce terme englobe la
fixation d'un médicament sur son récepteur, ou site du fixation,
et la relation entre la dose et la réponse physiologique
(Anthony, 2002).
I.3.2. Mécanismes d'action des médicaments
:
La plupart des médicaments agissent sur l'organisme grace
à leur affinité avec les
récepteurs de nos cellules. La nature des liaisons entre
le récepteur et le médicament conditionne fortement
l'activité thérapeutique (Anonyme 3).
I.3.2.1. Mécanisme général
:
> Action par fixation spécifique
:
Les médicaments agissent en général par
fixation sur des récepteurs, cette fixation est spécifique du
médicament et de son effet. Elle dépend étroitement de sa
structure et de ses propriétés chimiques.
> Sans fixation dans l'organisme
:
Ces médicaments agissent grâce à leurs
propriétés physiques (volume, pouvoir couvrant, etc.) ou en
modifiant celles du milieu extra cellulaire (pouvoir osmotique,
équilibre acidobasique, équilibre électrolytique, etc.).
Les structures chimiques peuvent être très différentes pour
un même effet.
> Action sur des organismes étrangers
:
Certains médicaments agissent sur des organismes
pathogènes (bactéries, virus, parasites, champignons). Les
mécanismes d'action sont semblables à ceux énumères
ci-dessus (Anonyme 2).
I.3.2.2. Mécanisme moléculaire
:
A) Définition d'un récepteur
:
Les récepteurs peuvent être définis comme
les éléments sensibles dans le système de communication
chimique qui coordonne les fonctions des différentes cellules de
l'organisme, les messagers chimiques étant des hormones, des
neurotransmetteurs ou des facteurs de croissance.
Les récepteurs sont classés en récepteurs
intra-cellulaires, principalement nucléaires et en récepteurs
membranaires (Jolliet et al., 2000).
B) Interaction entre un médicament et un
récepteur:
L'association chimique du PA avec le récepteur provoque
l'action thérapeutique, cette association se fait selon la
réaction réversible suivante :
1 2

M + Re MRe effet pharmacologique
Cette réaction est caractérisée par :
- Efficacité ;
- Effet pharmacologique (Helali, 1994).
C) Notion relation dose-effet :
La relation qui existe entre le logarithme de dose ou
concentration d'un médicament et la réponse biologique obtenue
après l'action de celui-ci, est une courbe sigmoïde approche la
réponse 0 % à faibles doses, puis la réponse maximale 100
% à hautes doses (Helali, 1994).
Il est à signaler que :
La DE 50 : est la dose (ou
concentration) de médicament produisant une réponse qui est
égale à la moitie (ou 50 %) de l'effet maximal obtenue chez
l'animal. La DE50 est utilisé pour la comparaison entre deux
médicaments: si deux médicaments ont la même
activité intrinsèque celui qui a la plus forte affinité
pour le récepteur a une représentation graphique
concentrationeffet déplacée vers la gauche et sa DE50 est plus
faible mais la hauteur des plateaux (l'effet maximum) est identique
(Helali , 1994).
La DL50 : est la dose (ou
concentration) de médicament qui tue 50 % des animaux au cours d'une
expérience. Les animaux les plus utilisés sont la souris, le
rat.
Les médicaments qui possèdent une DL50 plus
élevée, sont les médicaments qui offrent une
sécurité élevée et le contraire est juste
(Helali, 1994).
Chapitre II
Les antibiotiques
II.1. Généralités sur les
antibiotiques :
II.1.1. Découverte des antibiotiques :
Une découverte due au hasard
:
- ALEXANDER FLEMING (1881-1955), un médecin
bactériologiste, découvre les antibiotiques en 1928 ;
- Une découverte due au hasard : parti en vacances,
FLEMING laisse des cultures de la bactérie Staphylococcus
aureus dans son laboratoire. À son retour, il observe que certaines
de ces cultures ont été accidentellement contaminées par
une moisissure appelée Pénicillium (champignons), ce qui
a tué les bactéries, la pénicilline est donc le premier
antibiotique découvert.
Une découverte mal acceptée par le
monde scientifique :
- Malgré ses publications, FLEMING ne parvient pas
à intéresser les scientifiques qui pensent que toute substance
nocive pour les microbes l'est aussi pour l'homme ;
- Il faut attendre 1940 pour que la pénicilline soit
utilisée avec succès pour le traitement d'un malade atteint d'une
septicémie (infection généralisée de l'organisme)
causée par des staphylocoques ;
- En 1944, SELMAN ABRAHAM WAKSMAN (1878-1973) découvre un
puissant antibiotique, la streptomycine, actif, en particulier, sur le bacille
de la tuberculose.
Une découverte majeure :
- C'est une des principales découvertes dans l'histoire de
la médecine et de l'humanité ;
- La découverte, puis l'utilisation des antibiotiques ont
augmenté d'environ dix ans la durée de la vie humaine ;
- La plupart des espèces bactériennes infectieuses
à l'origine d'épidémies redoutables pour nos
ancêtres sont maintenant neutralisées par les antibiotiques
(Anonyme 4).
II.1.2. Définition des antibiotiques :
Par opposition au phénomène de symbiose, le mot
antibiotique dérive du terme "antibiose' crée en 1889
par VUILLEMIN pour désigner les phénomènes d'antagonisme
entre les micro- organismes vivants (Asselineau et Zalta,
1973).
En 1944 WAKSMAN définit les antibiotiques comme "toute
substance chimique produite par un micro-organisme, champignon ou
bactérie pouvant inhiber la croissance ou détruire d'autres
micro-organismes". Cette définition est aujourd'hui trop restrictive et
doit être abandonnée car des antibiotique peuvent être
obtenus par synthèse ou par hémi-synthèse. Un antibiotique
est donc actuellement défini comme une substance, d'origine biologique
ou synthétique agissant spécifiquement sur une étape
essentielle du métabolisme des bactéries (Leclere et al.,
1995).
II.1.3. Classement des antibiotiques :
Les antibiotiques sont classés d'après plusieurs
critères :
1. D'après leur spectre d'action
:
Les antibiotiques peuvent être à
(voir tableau 01 annexe I) :
- Spectre très large, ex. Tétracycline, ampicilline
;
- Spectre large, ex. Aminoside, rifamicine, fosfomycine ;
- Spectre moyen à prédominance sur les
GRAM+, ex. Pénicilline, macrolide, novobiocine ; - Spectre
étroit, ex.
o Pour les bacilles GRAM- : Quinolone, mecillinam ;
paroi cellulaire
bactérie
ATB bactéricide
ATB bactériostatique
pénicilline céphalosporine
Synthèse d'acide téra-
hydrofolique
sulfonamide triméthoprim
Inhibiteur de la gyrase nito-imadizole
ADN
bacitracine vancomycin
tétracycline
rifampicine aminoglycoside
chloramphénicol érythromycine
clindamycine
ARN
polymyxine
tyrothricine
protéine
membrane
cellulaire
o Pour les bactéries GRAM+ : Voncomycine,
fucidine ; o Pour les fongi : Amphotéricine,variotine (Maur,
1979).
2. D'après leur type d'action
:
Les antibiotiques et agents chimiothérapiques peuvent
être classés en :
- Bactériostatiques ex. Tétracycline,
chloramphénicol, macrolide ;
- Bactéricides ex. Pénicilline, cephalospirine,
aminoglycoside.
En général, les antibiotiques qui agissent sur
la paroi bactérienne ou sur la membrane cytoplasmique (protoplasmique),
sont bactéricides, et ceux qui agissent par inhibition de la
synthèse des protéines et /ou des acides nucléiques, sont
bactériostatiques (Maur, 1979).
3. D'après leur origine :
Les antibiotiques et agents chimiothérapiques sont
extraits de plusieurs sources : - Bactéries:
Lichenifirmis : Bacitracine ;
- Champignons : Pénicillium notatum :
Pénicilline ;
- Actinomycètes : Inyoensis streptomyces :
Sisomycine (Maur, 1979).
4. D'après leur point d'attaque
:
Les antibiotiques peuvent être classés de la
manière suivante : (fig.06)
- Antibiotiques qui inhibent la synthèse des mucopeptides
de la paroi bactérienne :
Le niveau de l'inhibition de la synthèse de la paroi
bactérienne s'exerce, soit :
o Sur la transpeptidase en fermant les ponts polyglycines du
mucopiptide pariétal comme : Pénicilline, céphalosporine,
novobiocine ;
o Soit sur le transfert et la polymérisation du
mucopiptide pariétale comme: Vancomycine, phosphonodipeptides
alaninomimétique ;
o Sur la synthèse de l'acide muramique comme :
Fosphomycine.
- Antibiotiques qui altèrent la membrane cellulaire
cytoplasmique bactérienne : Tyrothricine, polyènes antifongiques,
po1ymyxine-colistine.
o Les polymyxines se lient aux phospholipides des
bactéries GRAM- ;
o Les polyènes se lient aux stérols de la membrane
des fongi.
- Antibiotiques qui inhibent les mécanismes de
réplication et de transcription de l'ADN et l'ARN :
o Inhibition de l ' ARN-polymérase : Rifampicine,
quinolones ;
o Inhibition de la synthèse des protéines
bactériennes par action sur les ribosomes bactériens :
· Sur les sous-unités 30 S des ribosomes
oligosaccharides ;
· Sur les sous-unités 50 S des ribosomes:
Chloramphénicol, macrolides, lincomycine-clindamycine, fusidine ;
· Sur les deux : Tétracyclines.
- Antibiotiques qui agissent en tant qu'antimétabolites :
Sulfamides (antifolique), triméthoprime (antifolinique).
- Antibiotiques agissant par plusieurs mécanismes à
la fois : Oligosaccharides, novobiocine, synergistines, par exemple :
o Les oligosaccharides (streptomycine,....etc) disposent
à la fois d'une action d'inhibition des synthèses
protéiques (action ribosomique) et d'une action sur la membrane
protoplastique (Maur, 1979).
Figure (06) : Antibiotiques et leurs sites
d'action (Lùllmann et al., 1998).
5. D'après leur composition chimique
:
Les antibiotiques peuvent être classés de la
manière suivante :
- Dérivés d'un seul acide aminé :
Chloramphénicol, thiamphénicol, cyclosérine ;
- Dérivés de 2 acides aminés
condensés dans de nouveaux noyaux: Pénicillines,
céphalosporines, synergistines ;
- Peptides cycliques :
o Polypeptides surfactifs (peptolides): Polymyxinecolistine ;
o Polypeptides non surfactifs : Viomycine, capréomycine,
thyrothricine, saramycétine, amphomycine, bacitracine.
- Peptides à cycle lactonique : Synergistines
(streptogramine B) ;
- Aminosides (aminoglycosides) : Streptomycine,
néomycine, gentamicine, aminoglycosides semi-synthétiques
(amikacine, butirosine, kanendomycine) ;
- Macrolides (noyau lactonique): Erythromycine,
oléandomycine etc ;
- Complexes glyco-protéiques : Ristocétine,
vancornycine ;
- Dérivés de l'acétate : Noyau aromatique
naphtacène : Tétracyclines ;
- Stéroïdes : Fucidine ;
- Polyéne macrolides : Amphotéricine, nystatine,
pimaricine, trichomycine ;
- Structures non apparentées à d'autres
antibiotiques: Fosfomycine , novobiocine......etc (Maur,
1979).
6. D'après leur charge électrique
:
- Antibiotiques à caractère acide :
Pénicillines, céphalosporines, tétracyclines, novobiocine,
sulfamides, nitrofurantoine, quinolones ;
- Antibiotiques à caractère basique : Aminosides,
rifampicines, macrolides, lincomycineclindamycine, polymyxines ;
- Antibiotiques à caractère amphotère :
Tétracyclines (Maur, 1979).
7. D'après le caractère de la
résistance bactérienne :
- Résistance exclusivement chromosomique par mutations:
Quinolones, nitrofuranes, nitroimidazols, vancomicine, novobiocine.....etc ;
- Résistance extrachromosomique plasmidique
(multirésistance transférable) : Ce sont les antibiotiques
à large spectre d'action.
Certaines familles d'antibiotiques (bêta-lactamines,
aminosides, macrolides, triméthoprime) peuvent développer
à la fois des mutants résistants (résistance
chromosomique) et des résistances plasmidiques, ces dernières
étant toutefois prédominantes. A l'heure actuelle, les seuls
antibiotiques n'ayant pas développé de résistance
plasmidique transférable sont : Quinolones, nitrofuranes, fucidine,
polymyxine-colistine, nitroimidazols, novobiocine, rifampicine (Maur,
1979).
II.1.4. Les grandes familles d'antibiotiques
:
Les antibiotiques actuels sont groupés en plusieurs
familles et sous familles possédant un certain nombre de
caractères communs : Composition chimique ou origine apparentée,
spectre d'action similaire, mécanisme d'action identique, comportement
pharmacologique souvent similaire, résistance croisée, effets
secondaires rapprochés...etc.
Il existe plusieurs familles dont les principales sont :
Bêta-lactamines , macrolides et apparentés
(lincosanides) , tétracyclines,synergistines,antibiotiques à
structure d' acides aminés, polypeptides surfactifs , aminoglycosides.
(Voir tableau 02 annexe I) (Maur, 1979).
II.2. Propriétés générales
des antibiotiques :
II.2.1. Stabilité des antibiotiques :
La stabilité des antibiotiques dépends de la
nature de leur état physique (les pénicillines et les
tétracyclines sont plus stable en état solide qu'en solution), du
pH de la solution, de la température, de la présence des
réactifs, du temps de stockage et de leur durée d'action
(Dobreya, 1981).
II.2.2. Activité des antibiotiques :
Pour orienter le choix de l'efficacité
thérapeutique, il est nécessaire de connaître la
sensibilité du micro-organisme pathogène aux antibiotiques.
Certaines espèces bactériennes présentent des
résistances naturelles à un antibiotique, mais dans certain cas,
au sein d'une espèce a priori sensible à un antibiotique, des
résistances acquises par certaines souches. D'où certaines
méthodes de détermination de la sensibilité
bactérienne aux antibiotiques connues sous le nom
générique "antibiogramme" ont été
développées (Freney et al., 1994).
A. Principe d'un antibiogramme :
Le résultat pratique d'un antibiogramme est la
classification du microorganisme dans les catégories (S), (I) ou (R)
à chaque antibiotique (Freney et al., 1994).Cette
classification est établie à partir d'une grandeur de
référence CMI qui définie comme la plus faible
concentration inhibant toute croissance significative de la population
bactérienne. La confrontation de données bactériologiques,
pharmacologiques et cliniques permet la détermination de CMI
discriminantes (ou critiques) délimitant les classes de
sensibilité SIR. La comparaison de la CMI aux deux CMI discriminantes
prédéfinies pour chaque antibiotique permet alors la
classification du microorganisme dans une des trois catégories
(Laverdiere et Sabath, 1977).
B. Techniques de référence pour la
détermination de la CMI :
Il existe plusieurs méthodes pour déterminer la CMI
qui sont (voire annexe II) :
- Méthodes de dilution :
o En milieu liquide ;
o En milieu solide.
- Méthode de diffusion en gélose (méthode de
disque) (Duval et Soussy, 1980) ; - Méthode
turbidimétrique (Touitou, 1995).
II.2.3. Résistance aux antibiotiques :
II.2.3.1. Définitions :
Une souche bactérienne est dite résistante
à un antibiotique donné quand elle est capable de se
développer en présence d'une concentration en antibiotique
significativement plus élevée que celle habituellement active sur
les souches de cette espèce. En bactériologie médicale. La
définition de la résistance bactérienne à un
antibiotique prend également en considération la
pharmacocinétique de l'antibiotique : une souche est
considérée résistante à un antibiotique quand la
CMI de celui-ci est supérieure à la concentration sanguine
maximale d'antibiotique obtenue lors d'un traitement (Leclere et al.,
1995).
On distingue :
> Résistance naturelle :
c'est une insensibilité aux antibiotiques, existant naturellement chez
tous les membres d'un genre ou d'une espèce bactérienne. Elle
fait, partie du patrimoine génétique normal du germe.
(ex. Cas des streptocoques qui, en raison d'une
chaîne incomplète de transporteurs d'électrons. Elles
peuvent assurer le transport actif des aminoglycosides à travers la
membrane cytoplasmique et de ce fait, résistent à ces
antibiotiques) (Leclere et al., 1995).
> Résistance acquise : un
germe sensible, à un antibiotique donné peut au bout d'un certain
temps, devenir résistant à cet antibiotique, sous l'action des
facteurs génétiques situé soit au niveau des chromosomes
bactériens soit en dehors de ceux-ci.
o Résistance chromosomique due à des mutations ;
o Résistance extrachromosomique : la plus
fréquente, elle est liée à la production d'une enzyme
inhibitrice ;
o Résistance mixte : les staphylocoques et les
entérobactéries peuvent devenir résistants par
les deux mécanismes ;
o Résistance croisée : un germe devenu
résistant à un antibiotique peut de devenir résistant
à un autre antibiotique (Asselineau et Zalta, 1973).
II.2.3.2. Mécanismes de résistance
bactérienne aux antibiotiques :
La résistance bactérienne due aux effets des
antibiotiques, se manifeste par les mécanismes suivants :
- Production d'enzymes qui détruisent l'antibiotique ;
- Changement de la perméabilité aux antibiotiques
;
- Développement d'une altération structurelle du
récepteur à l'antibiotique ;
- Développement d'une autre voie métabolique qui
permet d'éviter l'inhibition provoquée par l'antibiotique ;
- Développement d'un changement enzymatique qui permet un
changement d'affinité à l'antibiotique de la part de la
bactérie (Helali, 1994).
II.3. Antibiothérapie :
II.3.1. Voies d'administration :
Comme tout autre agent thérapeutique les antibiotiques
peuvent être administré par :
- Voie intraveineuse (IV) ;
- Voie intramusculaire (IM) ; - Voie orale ;
- Voie intrarachidienne (IR) ;
- Voie cutanée et autres (Helali,
1994).
Les voies d'administration d'un antibiotique sont
conditionnées par plusieurs facteurs :
- La présentation disponible de l'antibiotique (forme
orale, injectable) ;
- L'urgence thérapeutique (Voie IV, IM) ;
- La nature de site infectieux ;
- L'état du réseau veineux du patient ;
- La possibilité d'administration orale ;
- Les thérapeutiques associes (ex. Anticoagulant et voie
IM) (Mouton et al., 2000).
II.3.2. Critères de choix d'un antibiotique
:
La prescription d'un antibiotique doit aboutir à
l'efficacité thérapeutique. Pour cela, une antibiothérapie
correcte repose sur la connaissance à la fois des données
bactériologiques du germe responsable de l'infection, par
détermination de la bactérie en cause et sa sensibilité,
de la pharmacocinétique de l'antibiotique prescrite et de la prise en
compte du terrain du ou des antibiotique (s) qu'il souhaite utiliser, l'effet
thérapeutique d'antibiotique choisi dépend de :
- Utilisation en monothérapie ou en association ;
- Dose prescrite ;
- Rythme et temps de traitement ;
- Voie d'administration (Bergogne et Dellamonica,
1995).
II.3.3. Pharmacocinétique des antibiotiques
:
Il ne suffit pas de préconiser l'antibiotique auquel la
bactérie est sensible pour garantir la guérison. Il faut
s'efforcer d'adapter le traitement, non seulement au profil de
sensibilité du germe incriminé et à la
sévérité de l'infection, mais aussi, à la nature du
foyer infectieux et aux capacités de biotransformation et
d'excrétion de l'organisme infecté (Duval et Soussy,
1980).
A) Absorption :
Dans l'organisme, l'ATB est absorbé pour atteindre le
milieu sanguin. La voie d'administration préférentielle est celle
qui aboutit à une absorption optimale.
Les antibiotiques administrés par voie orale doivent
résister aux sucs digestifs, à l'acidité gastrique et aux
bactériocines de la flore intestinale.
Parmi eux on distingue ceux qui traversent la muqueuse
intestinale et atteignent le compartiment plasmatique et ceux qui ne passent
pas la barrière intestinale, Alors que les premiers peuvent être
utilisés dans le traitement des infections générales
(pénicilline V, amoxicilline, pristinamycine), les seconds sont
réservés aux infections intestinales (sulfaguanidine)
(Duval et Soussy, 1980).
B) Biodiffusion ou diffusion :
À partir du sang, l'ATB passe dans les compartiments
interstitiels et cellulaires. La diffusion dans le compartiment interstitiel se
fait rapidement à cause de la grande perméabilité de la
membrane capillaire. Par contre, la pénétration à
l'intérieur de la cellule est un phénomène
beaucoup plus lent, fortement influencé par la
liposolubilité, le degré d'ionisation et l'affinité de
l'antibiotique pour les composants intracellulaires.
Ces facteurs permettent de distinguer les antibiotiques
à bonne diffusion tissulaire (macrolides, fluoroquinolones) de ceux
à diffusion moyenne (penicillines et cephalosporines) ou faible
(aminosides, polymyxines) (Duval et Soussy, 1980).
C) Transformation :
Les antibiotiques peuvent être ou non transformés
dans l'organisme :
- Certains antibiotiques ne sont pas modifiés dans
l'organisme, ils sont éliminés inchangés, sous forme
active par exemple : certaines céphalosporines (céfaloridine
...), les aminosides, les tétracyclines, les polymexines ... etc ;
- D'autres au contraire, subissent des transformations qui
peuvent aboutir à leur inactivation totale ou partielle
(dérivé antibactérienne nulle ou
inférieure) (Duval et Soussy, 1980).
D) Excretion :
Elle peut être :
- Rénale : ex. Pénicillines,
céphalosporines, aminosides, chloramphénicol en grande partie
inactivé ;
- Hépatique : ex. Ampicilline, dérivés et
analogues, rifamycines, macrolides ;
Il peut exister des excrétions par la salive, les
larmes..., ex. Macrolides (Duval et Soussy, 1980).
II.4. Antibiotiques Bêta-lactamine :
II.4.1. Définition et classification :
Les bêta-lactamines sont ainsi appelées parce que
leur molécule comporte un cycle betalactame. Cette famille comprend
comme l'indiqué le tableau 01 : les pénames, les
céphèmes, les carbapénèmes, les monobactames, et
les oxacéphèmes ainsi que les inhibiteurs des betalactamases
(clavame), qui n'ont pas d'activité antibactérienne
intrinsèque (Duval et Soussy, 1980 ; Touitou, 1995).
Tableau (01): Diversité des
antibiotiques de type beta-lactames: principaux cycles et ATB
représentatifs (Anonyme 5).
|
Noyau
|
|
Noyau
|
|
clavame
|
|
péname
|
|
7-oxo-4-oxa-
heptane-2-
1-azabicyclo [3.2.0] acide carboxylique
|
|
7-oxo-4-thia-1-azabicyclo [3.2.0]
heptane-2- acide
carboxylique
|
|
|
|
|
Noyau céphème
8-oxo-5-thia-1-azabicyclo [4.2.0]
octane -2- acide
carboxylique
|
8-oxo-5-oxa-1
octane-2-
Noyau oxacéphème
-azabicyclo [4.2.0]
acide carboxylique
|
|
2-oxoazéti
|
|
|
Noyau carbapénème
7-oxo-1-thia-1-azabicyclo [3.2.0]
heptane -2- acide
carboxylique
|
Noyau monobactame
dine-1- acide sulfinique
|
II.4.2. Mécanismes d'action des
bêta-lactamines :
L'activité des bêta-lactamines au niveau de la paroi
bactérienne s'opère en trois étapes:
> Pénétration par
l'intermédiaire des protéines transmembranaires ou porines
:
Le passage des bêta-lactamines au travers de la membrane
cellulaire externe s'effectue au
moyen d'un système de protéines transmembranaires
ou porines (fig.07) ;
> Attachement au récepteur
:
Les récepteurs aux bêta-lactamines sont les PLP
qui se trouvent à la partie interne sur la membrane cytoplasmique
(fig.07).La conséquence de la liaison d'une molécule de
bêtalactamines au PLP, est une inhibition de la réaction de
transpeptidation et un blocage de la synthèse du peptidoglycane ;
> Perturbation de la fonction bactérienne
:
Outre l'inhibition des enzymes de transpeptidation, d'autres
systèmes autolytiques interviennent par l'intermédiaire d'enzymes
lytiques (hydrolases), qui accélèrent l'éclatement de la
bactérie en milieu isotonique (Helali, 1994).
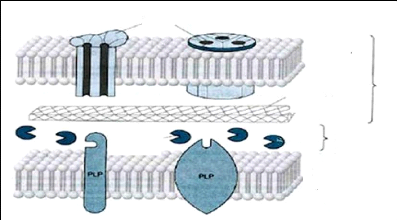
â- Lactamase
Porines
Peptidoglycane
Membrane cytoplasmique
Membrane externe
Espace periplasmique
Figure (07) : Schéma simplifié
de l'enveloppe d'une bactérie GRAM -.
La membrane externe à bi-couche lipidique, existe chez
les bactéries GRAM - mais pas chez les
GRAM + (Helali, 1994).
II.4.3. Résistance aux bêta-lactamines
:
Les bêta-lactamines peuvent perdre leurs efficacités
du fait de l'apparition d'une résistance des bactéries
(Gould, 1999).
Il y a trois mécanismes principaux :
- Par hydrolyse enzymatique du noyau bêtalactame par les
bêtalactamases: ces enzymes, produites par certaines bactéries,
hydrolysent et inactivent les bêta-lactamines par ouverture du cycle
bêtalactame, la synthèse de ces enzymes est codée soit par
les chromosomes, soit par les plasmides. La sécrétion peut
être constitutive ou induite ;
- Par modification des sites cibles (PLP): c'est le principal
mécanisme pour les staphylocoques résistants à la
méthicilline et pour les pneumocoques résistants
à la pénicilline ;
- Par réduction de la perméabilité des
membranes des bactéries GRAM -, par altération de certaines
porines, résultant en l'incapacité pour l'antibiotique à
pénétrer jusqu'à sa cible (Dukes et Aronson,
2000).
II.4.4. Pharmacocinétique :
Selon les molécules, l'administration se fait par voie
orale ou parentérale. Les bêtalactamines ont toutes en commun une
demi-vie d'élimination courte (30 mn pour oracilline, au maximum 8 h
pour la ceftriaxone). L'élimination est essentiellement rénale,
mais aussi biliaire pour certaines molécules. Il n'y a pas ou peu de
biotransformation. En cas d'insuffisance rénale sévère, il
est nécessaire d'adapter la dose pour certaines
spécialités (Dukes et Aronson, 2000).
II.5. Les pénicillines :
II.5.1. Définition et structure chimique
:
Antibiotiques d'origine biologique (extractive) ou de
semi-synthèse, faisant partie du groupe des béta-lactamines
(Maur, 1979).
Toutes les pénicillines contiennent le même
noyau-acide 6-APA et leur molécule présente un système
biocyclique condensé, constitué de deux cycles :
- Cycle (A) thialozidine : porteur d'un groupement
carboxylique
- Cycle (B) béta lactame : porteur d'un groupement
acylamino, dont le radical R. Varie dans les différentes
pénicillines (Dobreya, 1981).
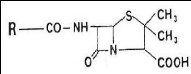
Figure (08) : Structure chimique de la
pénicilline (Dobreya, 1981).
Le radical R détermine le type des pénicillines,
ainsi que les propriétés antibiotiques de la molécule
(Dobreya, 1981). (voir tableau 03 annexe I).
II.5.2. Classification des Pénicillines
:
On peut classer les pénicillines actuelles dans
différentes catégories, selon les critères choisis qui
sont résumés dans le tableau 04 de l'annexe 1.
II.5.3. Propriétés physico-chimiques
:
II.5.3. 1. Caractères organoleptiques
:
Les pénicillines se présentent comme des poudres
blanches, cristallines, très solubles dans l'eau (à l'exception
de la pénicilline. v. Acide, de l'ampicilline et des sels et esters de
la pénicilline. G qui sont insolubles) (Afect,
1992).
II.5.3. 2.Caractère acide :
Le groupement -COOH en position 2 présente selon les
substituants R une valeur de pKa
comprise entre 2.5 et 2.75. Sa présence entraîne
pour les pénicillines les conséquences suivantes:
- Aptitude
de former des sels de sodium ou de potassium très solubles dans l'eau
;
- Aptitude de former des sels avec les amines ;
- Aptitude de former des esters qui seront des "prodrugs"
capables de libérer l'antibiotique in vivo (Afect,
1992).
II.5.3. 3. Stabilité :
La molécule de pénicilline est instable en solution
aqueuse, vis-à-vis des enzymes hydrolytiques : pénicillinases et
acylases.
- L'enzyme pénicillinase catalyse la coupure du cycle
bêta-lactame de la molécule des pénicillines en la
transformant en acide pénicillinique inactif (fig.09) (Afect,
1992).
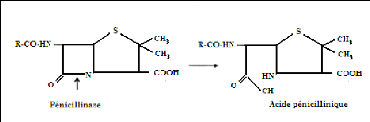
Figure (09) : Action des
pénicillinases sur les pénicillines (Afect,
1992).
- L'enzyme acylase provoque la rupture de la liaison amide,
elle provoque donc la désacylation du groupe amine, sur le carbone C6 et
l'obtention des noyaux moléculaires 6APA (fig.10).
(Afect, 1992).
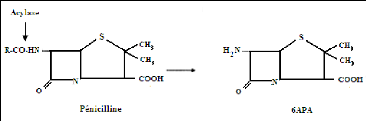
Figure (10) : Action des acylases sur les
pénicillines (Afect, 1992).
II.6. Oxacilline sodique :
II.6.1. Définition:
L'oxacilline est un ATB bactéricide de la famille des
bêta-lactamines, du groupe des pénicillines M
semi-synthétiques résistantes à la pénicillinase
(Anonyme 6).
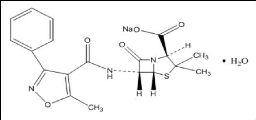
Figure (11) : Structure chimique de
l'oxacilline sodique (Anonyme 7).
II.6.2. Spectre d'activité antibactérienne
:
Les concentrations critiques séparent les souches
sensibles des souches de sensibilité intermédiaire et ces
dernières, des résistantes.
ex. Oxacilline (staphylocoques) : S = 2 mg/l et R > 2
mg/l.
Il est en principe active sur :
- Espèces sensibles :
o Aérobies à GRAM + : Staphylococcus
méti-S, Streptococcus pyogenes ; o Anaérobies :
Clostridium perfringens.
- Espèces résistantes : Staphylococcus
méti-R*.
La fréquence de résistance à la
méticilline est d'environ 30 à 50 % de l'ensemble des
staphylocoques et se rencontre surtout en milieu hospitalier
(Anonyme 6).
II.6.3. Pharmacocinétique :
Absorption :
L'administration de l'oxacilline peut se faire par la voie
orale et par la voie injectable. Par voie orale, la biodisponibilité est
de 41 % en raison de la biotransformation hépatique de l'oxacilline
(Anonyme 6).
Distribution :
- Par voie IM, une dose de 500 mg permet l'obtention d'un taux
sérique maximal de 11 ng/ml, après 30 mn ;
- Par voie IV lente, la même dose donne 43 ng/ml
après 5 mn ;
- La liaison aux proténes est d'environ 90 % ;
- La demi-vie est de l'ordre de 30 mn (par voie injectable) ;
- L'oxacilline diffuse rapidement dans la plupart des tissus de
l'organisme et notamment le liquide amniotique et le sang foetal
(Anonyme 6).
Biotransformation :
L'oxacilline est métabolisée à 45 % environ,
probablement dans le foie (Anonyme 6). Excretion
:
L'oxacilline s'élimine surtout par la voie urinaire
(Anonyme 6).
II.6.4. Conditions particulières de conservation
:
Après mise en solution, l'oxacilline est stable à
la température ambiante pendant 4 h dans les solutions glucosée
et salée isotoniques (Anonyme 6).
II.6.5. Formes galéniques de
l'oxacilline:
L'oxacilline existe sous plusieurs formes pharmaceutiques, on
cite : - Forme gélule ;
- Forme injectable ;
- Solution buvable (Anonyme 7).
Chapitre III
La microencapsulation
III.1.Historique :
Les premières publications sur la microencapsulation et
ses applications possibles dans le domaine pharmaceutique remontent à
1931.De 1931 à 1940, GREEN et son équipe à la NCR (USA)
ont établi un processus de microencapsulation basé sur
l'utilisation d'une enveloppe de gélatine (coacervation)
(Anonyme 8).
Depuis lors, l'industrie pharmaceutique a
développé plusieurs autres matériaux de revêtement
et beaucoup d'autres méthodes d'encapsulation.
Dans les 30 années, plusieurs brevets ont
été enregistrés au sujet de l'encapsulation des PA,
médicinaux et non médicinaux, comme des antibiotiques, vitamines,
et ainsi de suite.
D'autres industries ont été
intéressées, et le sont toujours, par cette technique :
l'industrie alimentaire, l'industrie photographique, l'industrie des engrais,
l'industrie de pesticides, etc.
En outre, l'industrie chimique avait développé
de nouveaux polymères avec des applications potentielles dans la
microencapsulation (Chang et Prakach, 2001).
III.2. Définition :
La microencapsulation regroupe l'ensemble des technologies qui
permettent la préparation de microparticules individualisées,
constituées d'un matériau enrobant contenant une matière
active.
- Les matériaux enrobants sont des polymères
d'origine naturelle ou synthétique, ou des
lipides.
- Les matières actives sont d'origines très
variées: principes actifs pharmaceutiques, actifs cosmétiques,
additifs alimentaires, produits phytosanitaires, essences parfumées,
microorganismes, cellules, ou encore catalyseurs de réaction chimique
...etc.
Les microparticules présentent une taille comprise
entre environ 1um et 1mm et contiennent typiquement entre 5 et 90 % (en masse)
de matière active (Richard et Benoit, 2000).
III.3. Types de microparticules :
Les procédés de microencapsulation permettent de
préparer des microparticules de deux types: (fig.12)
- Les microcapsules, la particule réservoir est
constituée d'un coeur de matière active liquide (plus ou moins
visqueux) ou solide, entourée d'une écorce solide continue de
matériau enrobant. Les microcapsules ne sont pas nécessairement
sphériques ;
- Les microsphères, un réseau
macromoléculaire ou lipidique continu formant une matrice dans laquelle
se trouve la matière active finement dispersée, à
l'état de molécules, de fines particules solides ou encore de
gouttelettes de solutions (Richard et Benoit, 2000).
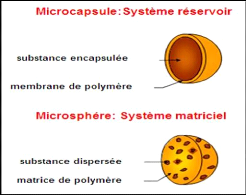
Figure (12) : Types de
microparticule (Anonyme 9).
Un certain nombre de facteurs physico-chimiques, permettent de
caractériser la membrane d'une microcapsule ou la matrice d'une
microsphère :
- Charge électrique de surface ;
- Mouillabilité ;
- Porosité ;
- Tortuosité des pores ;
- Degré de gonflement.
Le taux d'encapsulation (ou la teneur en matière
active) peut être très élevée dans les
microcapsules, de l'ordre de 85 à 90 % (rapport massique).
Comparés à ceux rencontrées dans les microsphères
qui sont plus faibles, de l'ordre de 20 à 35 % (Richard et
Benoit, 2000).
Le rendement d'encapsulation est le rapport entre la masse de PA
encapsulé et la masse de PA à encapsuler (Anonyme
9) :
× 100
Masse de PA à encapsuler
Rdt=
Masse de PA encapsulé
III.4. Intérêt de microencapsulation
:
Sur le plan industriel, la microencapsulation est mise en oeuvre
pour remplir les objectifs suivants :
- Assurer la protection, la compatibilité et la
stabilisation d'une matière active dans une formulation ;
- Réaliser une mise en forme adaptée (dosage plus
élevé dans de petits volumes) ; - Améliorer la
présentation d'un produit ;
- Masquer un goût ou une odeur ;
- Modifier et maîtriser le profil de libération
d'une matière active pour obtenir, par exemple, un effet prolongé
ou déclenché (Richard et Benoit, 2000).
III.5. Procédés de microencapsulation
:
Avant de procéder une préparation d'une
microencapsulation, il faut tenir compte : - La taille moyenne et la largeur de
distribution granulométrique;
- La teneur en matière active ou taux d'encapsulation ;
- La forme finale: dispersion de microparticules en phase aqueuse
ou en phase solvant, poudre sèche ;
- Les contraintes de stabilité au cours du stockage et au
cours de la mise en oeuvre ;
- La durée de conservation sans libération de
matière active, ainsi que le milieu dans lequel les particules seront
conservées ;
- Les conditions de libération et la cinétique
de libération. Si l'on souhaite une libération
déclenchée, il devra en particulier être
précisé quel est le paramètre de déclenchement:
pression ou cisaillement mécanique, variation de température,
variation de pH, dégradation enzymatique. Pour une libération
prolongée, la durée souhaitée de la période de
libération sera une des données du problème ;
- Les contraintes réglementaires liées au
domaine d'application et au mode d'administration qui sont prescrites dans les
réglementations nationales et internationales (Pharmacopée
Européenne ou USP par exemple, pour le domaine de la pharmacie).
Les choix du procédé et de la formulation
déterminent les caractéristiques finales de microparticules
(Richard et Benoit, 2000).
III.5.1. Procédés physico-chimiques
:
III.5.1.1. Procédé basé sur la
séparation de phase :
La coacervation est le phénomène de
désolvatation des macromolécules, conduisant à une
séparation de phases au sein d'une solution. A l'issue de la
coacervation, deux phases sont présentes dans le milieu :
- Le coacervat : Riche en polymère et pauvre en solvant
;
- Le surnageant : Pauvre en polymère et riche en
solvant.
Si, dans le même temps, une matière active est
dispersée dans ce milieu, sous forme de gouttelettes par exemple, le
coacervat formé pourra l'encapsuler si les conditions d'étalement
des phases en présence sont respectées.
La figure 13 représente trois situations possibles: Cas
où une encapsulation complète aura lieu, partielle, ou pas
d'encapsulation, (l'encapsulation est complète si le coacervat mouille
spontanément la surface de la matière active, c'est-à-dire
lorsque S3 > 0, S2 < 0 et S1
< 0) (Richard et Benoit, 2000).
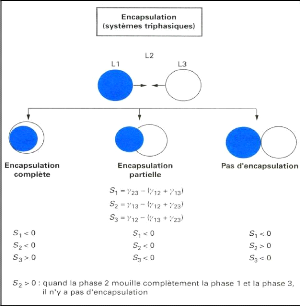
Figure (13) : Comportement d'un coacervat
(3) vis-à-vis une phase liquide
non miscible (1)
(Richard et Benoit, 2000).
III.5.1.2. Microencapsulation par coacervation
complexe :
La coacervation complexe est une désolvatation
simultanée de deux polyélectrolytes hydrosolubles portant des
charges opposées en provoquant par une modification de pH du milieu
aqueux. En effet, la structure du coacervat est complexe puisqu'elle comprend
deux polymères.
Le procédé de microencapsulation par
coacervation complexe se déroule de la façon suivante (fig.14)
:
- Dans un premier temps, le produit à encapsuler (sous
forme liquide ou solide) est dispersé dans une solution aqueuse
contenant les deux polymères (phase a).
- Dans un deuxième temps, la coacervation est induite
par un ajustement du pH de la solution, de façon que les charges
positives du premier polymère équilibrent les charges
négatives du second (phase b). L'attraction électrostatique des
deux polyélectrolytes provoque l'apparition d'un coacervat mixte.
- Dans un troisième temps, les gouttelettes de
coacervat formé viennent s'adsorber (phase c) à la surface de la
matière active à encapsuler et former un enrobage continu (phase
d). Finalement, cet enrobage est consolidé par réticulation
(phase e) des macromolécules constitutives du coacervat (Richard
et Benoit, 2000).
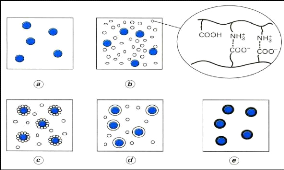
Figure (14) : Schéma de principe du
procédé de microencapsulation par
coacervation
complexe (Richard et Benoit, 2000).
Le polyélectrolyte chargé positivement qui est
généralement utilisé est la gélatine de haut point
isoélectrique (gélatine de type A telle que la gélatine de
peau de porc). Les polyanions les plus souvent utilisés sont la gomme
arabique, les alginates, les carraghénanes. La
carboxyméthylcellulose, les polyphosphates et d'autres.
Les particules obtenues sont des microcapsules. Leur taille
varie de quelques micromètres à quelques centaines de
micromètres. Les taux d'encapsulation peuvent être très
élevés, de l'ordre de 80%.
La microencapsulation par coacervation complexe est largement
utilisée dans de nombreux secteurs industriels. Dans le milieu
pharmaceutique, cette technique est également mise en oeuvre pour la
microencapsulation de paraffine liquide, d'huiles essentielles utilisées
en aromathérapie (Richard et Benoit, 2000).
III.5.1.3. Microencapsulation par coacervation simple
:
La coacervation simple se rapporte aux procédés
faisant intervenir la désolvatation d'un seul polymère par l'un
des facteurs suivants : abaissement de température, addition d'un
nonsolvant, addition d'électrolytes, addition d'un deuxième
polymère incompatible. Ce phénomène peut se
dérouler en milieu aqueux ou organique. Les étapes du
procédé sont en tous points identiques à celles
décrites pour la coacervation complexe.
Les particules obtenues sont généralement des
microcapsules. Toutefois, dans certains cas, le procédé par
coacervation simple permet d'obtenir des microsphères. C'est le cas
lorsque la proportion de substance active est faible par rapport au volume du
coacervat.
La taille des microparticules obtenues ainsi que la teneur en
matière active sont semblables à celles résultant du
procédé par coacervation complexe (Richard et Benoit,
2000).
III.5.1.4. Procédés
d'évaporation et d'extraction de solvant :
La méthode de microencapsulation par évaporation
de solvant repose sur l'évaporation de la phase interne d'une
émulsion sous agitation. Les étapes sont résumées
comme suit: Initialement, le matériau d'enrobage,
généralement un polymère hydrophobe, est dissous dans un
solvant organique volatil. La molécule active à encapsuler est
alors soit dissoute, soit dispersée dans la solution organique.
- La phase organique est émulsionnée sous agitation
dans une phase aqueuse, contenant un agent tensioactif.
- Une fois l'émulsion établie, le solvant
organique diffuse progressivement dans la phase continue sous agitation pour
s'évaporer, laissant le polymère précipiter sous forme de
microsphères (fig.15).
Ce procédé permet la fabrication de
microsphères de taille entre 0,5 et 200 um.
Le rendement de production peut aisément s'approcher de
100 % (Richard et Benoit, 2000).
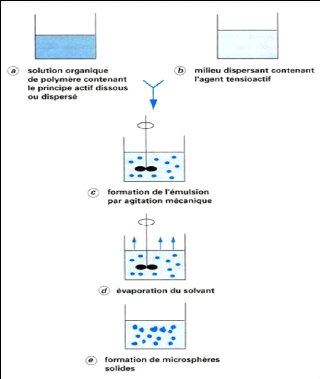
Figure (15) : Schéma de
principe du procédé de microencapsulation par évaporation
de solvant (Richard et Benoit, 2000).
III.5.1.5. Microencapsulation par gélification
thermique :
Ce procédé, encore appelé hot
melt, repose sur la fusion du matériau d'enrobage. La
matière active à encapsuler est dissoute ou dispersée dans
ce matériau fondu. L'ensemble est émulsionné dans une
phase dispersante, dont la température est maintenue supérieure
à la (Tf) de l'enrobage et pour laquelle la matière active n'a
aucune affinité: il s'agit d'eau distillée lorsque la substance
à encapsuler est lipophile, et d'huile de silicone, par exemple,
lorsqu'elle est hydrosoluble. La solidification des globules dispersés
est obtenue en refroidissant brutalement le milieu (fig.16) (Richard et
Benoit, 2000).
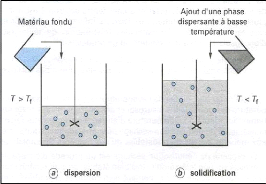
Figure (16) : Schéma de principe du
procédé d'encapsulation par gélification
thermique (hot
melt) (Richard et Benoit, 2000).
Comme de nombreuses substances actives sont thermolabiles, les
matériaux supports généralement utilisés dans ce
procédé de microencapsulation sont des lipides de bas point de
fusion. Les particules obtenues sont ici des microsphères d'une taille
pouvant aller généralement de 30 à 300 um. La teneur en
matière active est de l'ordre de 20 % (Richard et Benoit,
2000).
III.5.2. Procédés mécaniques
:
III.5.2.1. Procédé de
nébulisation/séchage :
Le procédé de nébulisation/séchage
est un procédé continu en une seule étape qui permet de
transformer une formulation liquide initiale en une forme microparticulaire
sèche. La formulation liquide initiale peut être constituée
:
- Soit d'une solution de matière active et de
matériau enrobant ;
- Soit d'une dispersion de particules solides de matière
active dans une solution ou une émulsion de matériau enrobant
;
- Soit encore d'une émulsion de matière active dans
une solution de matériau enrobant. Ce procédé comprend les
4 étapes séquentielles suivantes :
- Nébulisation de la formulation liquide initiale pour
former un aérosol ;
- Mise en contact de l'aérosol avec un flux d'air,
porté à une température contrôlée ; -
Séchage rapide de l'aérosol pour former des microparticules
solides ;
- Séparation de la poudre de microparticules et de l'air
contenant le solvant vaporisé.
Les microparticules obtenues par
nébulisation-séchage (Le plus souvent des microsphères)
sont d'une taille typiquement comprise entre environ 1 um et 50 um, pour un
séchage à cocourant. Le séchage à contre-courant
permet d'obtenir des microparticules de taille moyenne plus
élevée, comprise entre environ 50 et 200 um.
Le taux d'encapsulation est limité à environ 40 %
(en masse) (Richard et Benoit, 2000).
III.5.2.2. Procédé d'enrobage en lit
fluidisé :
Le procédé d'enrobage en lit fluidisé
s'applique exclusivement à des matières actives
constituées de particules solides (granulés, cristaux). Des
matières actives liquides peuvent néanmoins être
encapsulées après absorption par des supports particulaires
poreux. Le procédé
permet de réaliser un enrobage continu de particules qui
conduit donc à la production de microcapsules. Il comprend une
séquence cyclique en trois temps :
- Fluidisation de la poudre de particules ;
- Pulvérisation du matériau enrobant sur les
particules ;
- Séchage et filmification de l'enrobage.
Les formulations liquides qui sont pulvérisées sur
les particules en mouvement dans le lit fluidisé sont des solutions ou
des dispersions aqueuses ou organiques de polymères.
Quand les gouttelettes de la formulation liquide
pulvérisée rencontrent la surface des particules, la formation du
film d'enrobage s'effectue en plusieurs étapes successives (fig.17) :
- Contact microparticule-gouttelette ;
- Mouillage et étalement de la gouttelette sur la
particule ;
- Séchage par évaporation du solvant et
éventuellement pénétration dans la particule
(Richard et Benoit, 2000).
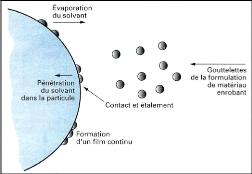
Figure (17) : Etape de formation d'un film
d'enrobage par spraycoating sur
des particules solides (Richard
et Benoit, 2000).
Les paramètres de procédé, qui doivent
être maîtrisés et ajustés pour obtenir un tel
enrobage, sont liés soit au séchage du film, soit à la
pulvérisation de la formulation liquide.
L'épaisseur des films d'enrobage déposés
sur des particules en lit fluidisé est généralement
comprise entre quelques micromètres et 20 um. Le film devant être
suffisamment épais pour masquer les défauts de surface.
Le taux d'encapsulation est généralement
élevé, compris entre 60 et 90 % (en masse) (Richard et
Benoit, 2000).
III.5.2.3. Frocédés de
gélification et congélation de gouttes (Frilling)
:
A- Gélification de gouttes :
La gélification de gouttes est basée sur la
formation d'une solution, dispersion ou émulsion de matière
active dans une solution aqueuse de polymères capables de former des
gels sous une action extérieure, physique ou chimique. Il s'agit, par
exemple, de l'alginate de sodium,
du chitosan ou de l'agarose. Dans le cas de l'alginate de
sodium, les gouttelettes, formées par extrusion à travers une
buse vibrante, sont réceptionnées dans une phase aqueuse
gélifiante contenant du chlorure de calcium. Elles se transforment
instantanément en des microparticules de gel sphérique (fig.18)
(Richard et Benoit, 2000).
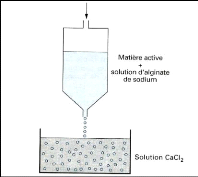
Figure (18) : Schéma de principe du
procédé de gélification
de gouttes
(Richard et Benoit, 2000).
Les microparticules peuvent ensuite être
transférées dans une solution de polylysine, pour former une
membrane rigide semi-perméable par complexation de l'alginate et du
polycation.
Lorsque le chitosane est utilisé, la phase aqueuse de
réception des microgouttelettes est une solution alcaline, qui
insolubilise le chitosane. Dans le cas de l'agar, c'est la variation de
température des gouttelettes de 80 0C à 30
0C qui induit la gélification. La solubilité de la
matière active dans le milieu de réception doit être la
plus faible possible pour minimiser les pertes par solubilisation
(Richard et Benoit, 2000).
B- Congélation de gouttes :
La congélation de gouttes (ou spray-congealing) fait
intervenir un matériau enrobant de type corps gras, glycéride ou
cire à point de fusion relativement bas, compris entre 50 0C
et 120 0C, elle consiste à préparer un fondu de ce
matériau enrobant dans lequel la matière active est
solubilisée ou dispersée soit sous forme particulaire solide,
soit sous forme de microgouttelettes d'émulsion inverse (eau dans
huile). Cette préparation fondue est maintenue à une
température supérieure à la température de fusion
du matériau enrobant et extrudée sous pression à travers
une buse vibrante. Sous l'effet de la vibration, pour une fréquence bien
choisie, le jet fondu est sectionné sous forme de gouttelettes
sphériques de taille uniforme (fig.19).Ces gouttelettes se refroidissent
dans le milieu de chute (air froid, azote ...) et se solidifient pour donner
des microparticules de type microsphère (Richard et Benoit,
2000).
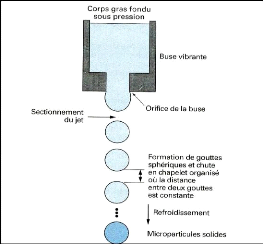
Figure (19) : Congélation de gouttes:
schéma de principe (Richard et Benoit, 2000).
Les microparticules obtenues par ces deux
procédés ont une distribution granulométrique très
étroite, dans une gamme de diamètres accessibles limitée
à des valeurs comprises entre seulement 200 um et 800 um.
Le taux d'encapsulation reste généralement faible
(entre 10 % et 30 % en poids) (Richard et Benoit, 2000).
III.5.3. Procédés chimiques :
III.5.3.1. Polycondensation interfaciale
:
La polycondensation interfaciale est un procédé
qui permet de préparer in situ une membrane polymère à la
surface de gouttelettes d'émulsion, grâce à une
réaction chimique entre deux monomères bien choisis, la
réaction se déroulant à l'interface entre la phase
dispersée et la phase dispersante. La méthode s'applique à
des solutions de matières actives, aussi bien organiques qu'aqueuses, ou
à des matières actives liquides. Le principe de ce
procédé est schématisé sur la figure 20
(Richard et Benoit, 2000).
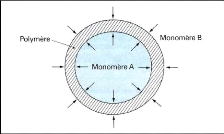
Figure (20) : Mécanisme de la
polycondensation interfaciale (Richard et Benoit,
2000).
La taille des microcapsules obtenues par polycondensation
interfaciale est comprise entre environ 0,5 um et 100 um selon la taille
initiale des gouttelettes d'émulsion (Richard et Benoit,
2000).
III.5.3.2. Autres procédés chimiques
:
Les autres procédés chimiques de
microencapsulation sont des procédés basés sur la
polymérisation radicalaire ou anionique en milieu dispersé. Il
s'agit des procédés de polymérisation en :
- Emulsion ;
- Microsuspension ;
- Dispersion ;
- Miniémulsion ;
- Microémulsion.
Les particules obtenues présentent des tailles
comprises entre quelques dizaines de nanomètres et quelques dizaines de
micromètres, et sont le plus souvent de type matriciel
(nanosphères ou microsphères).
Les taux d'encapsulation sont compris entre quelques pour cent et
50 % (en masse) (Richard et Benoit, 2000).
III.6. Libération contrôlée d'un PA
:
III.6.1. Définition :
Par définition, le rôle d'un système à
libération contrôlée est de délivrer la bonne
quantité d'un PA, au bon endroit et au bon moment.
Lorsqu'on considère les interactions matière
active/milieu extérieur, les microparticules peuvent être
classées en deux catégories: celles qui ne doivent pas
libérer leur contenu telles que les microréacteurs contenant des
enzymes ou des bactéries, et celles qui sont formulées de
façon à libérer la matière active
encapsulée. Dans ce dernier cas, il faut distinguer (fig.21) :
- Systèmes à libération
déclenchée ;
- Systèmes à libération prolongée
(Richard et Benoit, 2000).
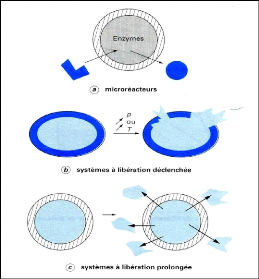
Figure (21) : Microréacteurs et
systèmes à libération déclenchée
et
prolongée (Richard et Benoit, 2000).
III.6.2. Mécanismes de la libération
contrôlée :
Les systèmes à libération
controlée possèdent des exigences particulières au (x)
matériau(x) impliquée (s), qui sont de nature polymérique
sous forme de matériau plein ou d'une membrane (Guery,
2006).
A) Systèmes à libération
déclenchée : sont généralement des
microcapsules formées d'une membrane de faible
perméabilité, qui vont libérer brutalement leur
contenu.
Les mécanismes de la libération connus dans ce cas
sont :
> Mécanismes de libération par
éclatement : sous l'effet d'une pression (mécanique
ou osmotique) ;
> Mécanismes de libération par
fusion : sous l'effet de la température (Guery,
2006).
B) Systèmes à libération
prolongée : sont majoritairement des microsphères
(Guery, 2006) Les mécanismes mis en jeu sont :
> Mécanismes de libération par
dégradation :
La plupart des polymères biodégradables se
dégradent par hydrolyse en composés de taille de plus en plus
faibles, biologiquement éliminables, dans certains
métabolique.
La dégradation peut s'effectuer selon une hydrolyse en
masse,il est uniforme dans toute la matrice polymère ou bien se produire
uniquement sur la surface du polymère.
> Mécanisme de libération par
diffusion uniquement :
La diffusion se produit quand un principe actif traverse le
polymère qui forme le système de libération .La diffusion
peut se produire à l'échelle macroscopique à travers les
pores dans la matrice ou à l'échelle moléculaire par le
passage entre les chaînes de polymères (lois de FICK).
> Mécanismes de libération par
gonflement suivi d'une diffusion :
Parmi les nombreux mécanismes de libération du PA,
nous souhaitons décrire plus particulièrement ce mécanisme
(le cas étudié).
La compréhension des mécanismes de gonflement
des polymères dans l'organisme est importante pour permettre de
concevoir la système particulier de libération
contrôlée et permet d'expliquer les comportements
cinétiques libération. Le PA est dissout ou dispersé au
sein d'une matrice polymèrque capable d'en sortir.
En prémier lieu, le polymère ne subit aucune
modification chimique, il n'est pas dégradé, l'eau diffuse
simplement à l'intérieur du réseau polymère, le
gonfle, ce qui permet aux médicaments piégés a
l'intérieur de se libérer.
Les systèmes de libération
contrôlées par gonflement sont initialement secs et quand ils sont
placés dans le corps, ils absorberont l'eau ou autres fluides du corps
et gonfleront .Ces systéme permettant la diffusion du PA à
travers: le réseau gonflé dans l'environnement externe .La plus
part des matières utilisées dans ces systéme sont les
hydrogels ( absorbant de l'eau ou autres fluides sans être dissoudre).
La capacité du gonflement de polymère se
manifeste quand le gonflement peut être déclenché par un
changement de l'environnement entourant le systéme de la
libération. Dépendant du polymère, le changement
environnement peut impliquer le pH , la température, ou la force
ionique, et le systéme peut se rétrécir ou gonfler sur un
changement de n'importe lequel de ces facteurs environnementaux.
La figure 22 illustre les changements de bases de la structure
de ce systéme sensibles. De nouveau, pour ce type de système, le
dégagement de drogue accompli seulement quand le polymère gonfle
(Xiaoling et Bhaskara, 2006)
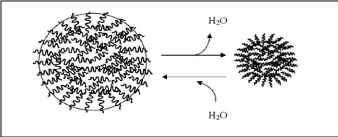
Figure (22) : Processus de gonflement d'un
hydrogel (Igor et Mattiasson, 2008)
À partir d'une microsphère, le
phénomène de diffusion de la molécule active à
travers une matrice sera décrit par une cinétique
obéissant à la loi d'HIGUCHI (Banker et Rhodes,
2002)
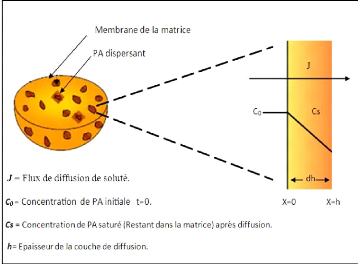
Figure (23) : Représentation
schématique d'une paroi matricielle (Banker et Rhodes,
2002)
= C0 ??h - ??????
d dh

Où :
dm : Variation de la masse de PA liberé par unité
de surface ;
dh : Epaisseur de la couche de diffusion ;
C0 : Concentration de PA initiale t=0 ;
Cs : Concentration de PA saturé (Restant dans la matrice)
après diffusion.
On a aussi à partir de théorie de diffusion :
Dm : Coefficient de diffusion dans la matrice ; t : Temps de
libération ;
À partir les deux équations précedentes et
après lintegration selon h, on trouve la formule suivante :
m = [Cs Dm (2 C0 -- Cs) t]1"2
On a toujours C0 >> Cs, donc
m = [2Cs Dm C0 t]1"2
Pour un systéme matriciel granulair et poreux :
m = [Ds Ca
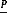
Où :
P : Porosité de la matrice ;
T : Tortuosité ;
Ca : Solubilité de PA dans le milieu de libération
; Ds : Coefficient de diffusion de PA dans le milieu.
On peut réduire cette équation pour obtenir la loi
de HIGUCHI :
m = k t1/2
La loi d'HIGUCHI montre que la quantité
libérée de matière active est directement proportionnelle
à la racine carrée du temps (Banker et Rhodes,
2002).
III.6.3. Param~tres influençant la
libération d'un PA :
Il est à noter que les paramètres
influençant la libération d'un PA encapsulé sont :
- Solubilité du PA dans le milieu de libération et
dans la paroi polymérique ;
- Taux d'encapsulation ;
- Interactions chimiques entre le PA et polymère ;
- - Caractéristiques morphologique de système de
libération (porosité, tortuosité, surface, forme) ;
- - Caractéristiques de polymère tel que poids
moléculaire (des études récentes montrent que les
polymeres de faibles poids moléculaires présentent une
porosité plus élevé que d'autre, donc une
libération plus élevée (Igor et Mattiasson,
2008).
III.6.4. Profils de libération obtenus à
partir de différents types de microparticules : La difusion du
PA selon les modes de la cinétique de libération obeissent aux
trajets de la figure 24 :
À partir des microcapsules, on peut obtenir des
cinétiques de libération d'ordre 0 ou d'ordre 1 (profils A et D),
en fonction de la solubilité dans l'eau de la matière active.
Les cinétiques d'ordre 0 peuvent titre modifiées
dans leur phase initiale: Soit la vitesse de libération est
exagérément marquée (effet de BURST) en raison de la
présence d'un excès de matière active dans la membrane
(profil B), soit un temps de latence qui précède la
libération, temps nécessaire par exemple pour que le principe
actif diffuse à travers la membrane, avant d'atteindre le milieu
extérieur (profil C).
À partir de microsphères, la cinétique
obéissant à la loi d'HIGUCHI (profil E) (Richard et
Benoit, 2000).
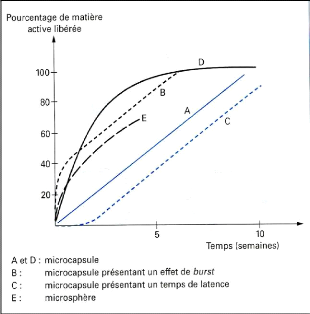
Figure (24) : Profils de libération
obtenus à partir de différents types
de microparticules
(Richard et Benoit, 2000).
Les microparticules à libération prolongée
sont très largement utilisées dans les domaines agroalimentaire
et pharmaceutique (Richard et Benoit, 2000).
III.7. Les polymères :
III.7.1. Définition :
Les polymères sont des matériaux composés
de macromolécules. Celles-ci sont constituées par la
répétition d'unités simples de faible masse
moléculaire, liées entre elles par des liaisons covalentes .Les
molécules qui s'enchaînent pour former le polymère sont
appelées monomères. Lorsqu'un polymère est obtenu à
partir de deux ou de plusieurs monomères différents, il est
appelé copolymère. Dans le cas contraire, on parle alors d
'homopolymère (Dubois, 2004).
III.7.2. Fonctionnalité :
Un monomère doit comporter au moins un groupe
fonctionnel capable de former des liaisons chimiques avec d'autres
molécules de monomère. Le nombre de groupes fonctionnels,
appelé nombre de fonctionnalité, à une influence
très importante sur la structure et les propriétés du
polymère. Un nombre de fonctionnalité unitaire entraîne la
synthèse de polymères linéaires; un nombre de
fonctionnalité supérieur permet la formation de polymères
réticulés (fig.25) (Dubois, 2004).
Figure (25) : Représentation
schématique de polymères
linéaires (a), ramifiés
(b) et réticulés (c) (Dubois, 2004).
III.7.3. Classification des polymères
:
Les polymères peuvent être classés selon
différents critères qui sont :
> Selon leur origine :
- Polymères naturels: sont issus des règnes
végétal ou animal, on peut cependant mentionner, la famille des
polysaccharides (cellulose, amidon), celle des protéines (laine,
soie...) ;
- Polymères artificiels : élaborés
chimiquement à partir d'un monomère naturel ;
- Polymères synthétiques : sont obtenus par
polymérisation de molécules monomères (les
monomères utilisés n'existent pas dans la nature).
> Selon la fonction géométrique
:
- Mono-dimensionnelle ;
- Bi-dimensionnelle ;
- Tridimensionnelle.
> Selon les propriétés
physico-chimiques :
- Polymères thermoplastiques (ou thermoplastes) : sont
constitués de macromolécules de taille limitée
linéaires ou ramifiées. Ils peuvent passer de l'état
rigide à l'état malléable par une faible
élévation de température. Ce processus est en
général réversible, ce qui confère à ces
polymères une certaine facilité de mise en oeuvre et de
recyclage. A ce jour, sont regroupés dans cette famille les
polymères dont la cohésion latérale est assurée par
des liaisons faibles (liaisons de type VAN DER WAALS, liaisons
hydrogènes) ;
- Elastomères, matériaux obtenus à partir
de polymères linéaires ayant des liaisons secondaires très
faibles. Ces matériaux sont ainsi considérés comme des
liquides très visqueux. L'introduction d'un certain nombre de liaisons
pontales entre les chaînes confère aux élastomères
une structure tridimensionnelle. Leur caractéristique principale est
leur grand déformabilité qui peut dépasser les 1000%. Ceci
est principalement dû à leur faible densité de
réticulation. Par ailleurs, le pontage rend les
élastomères difficilement recyclables ;
- Polymères thermodurcissables, qui sont des
polymères fortement réticulés. En effet, leur taux de
réticulation est de 10 à 100 fois plus élevé que
dans les élastomères. Ils forment un réseau
tridimensionnel et on peut considérer qu'ils ne sont constitués
que par une seule macromolécule d'une taille infiniment grande à
l'échelle atomique. Les fortes liaisons qui existent entre les
chaînes confèrent à ces polymères une
résistance mécanique et thermique nettement supérieure
à celle des thermoplastes. Les polymères thermodurcissables sont
insolubles, infusibles et non recyclables.
> Selon leur structure :
- Polymères amorphes, caractérisés par une
absence d'ordre des chaînes macromoléculaires. Celles-ci
s'enlacent les unes les autres, se replient sur elles mêmes. Ces
polymères sont habituellement imagés à un
plat de spaghetti cuit ;
- Polymères semi-cristallins,
caractérisés par la présence d'arrangements
réguliers des chaînes macromoléculaires reliés dans
une matrice amorphe, le taux de cristallinité caractérise
l'importance de structure cristalline (Dubois, 2004).
III.7.4. Polymères utilisés dans la
microencapsulation :
Dans la microencapsulation, variété de
polymères sont utilisés, on distingue :
Dans le cas de procédés physico-chimiques et
mécaniques, la formulation des microparticules met en oeuvre des
matériaux enrobant préformés, d'origine très
variées, qui sont choisis parmi des grandes classes.
(voir tableau 05 annexe I).
Dans le cas de procédés chimiques les
monomères utilisés sont habituellement:
- Monomères acryliques et méthacryliques de type
ester (acrylate de butyle, méthacrylate de méthyle ...) ou
porteurs de groupements fonctionnels tels que des acides carboxyliques (acide
acrylique ...), ou des fonctions hydroxyle (méthacrylate
d'hydroxyéthyle) ;
- Monomères vinyliques (acétate de vinyle ...) et
le styrène ;
- Monomères de type cyanoacrylate d'alkyle (cyanoacrylate
d'isohexyle).
Les deux premiers types de monomères réagissent
par un mécanisme radicalaire, alors que le troisième type
polymérise par voie ionique (voir tableau 06 annexe I)
(Richard et Benoit, 2000).
III.7.5. Acide alginique :
Acide de formule générale
(C6H10O7) n extrait des parois cellulaires
des Algues brunes. L'acide alginique est un biopolymère constitué
d'une succession de blocs formés de deux unités
monosaccharidiques : l'acide D-mannuronique (Ma) et son epimère en C5,
l'acide L-guluronique (Gu), liés en 1et 4 par des liaisons
glycosidiques, pour former une chaîne polymérique linéaire.
La proportion et la distribution de ces monomères déterminent en
grande patrie les propriétés de l'alginate. Les sels de l'acide
alginique sont nommés alginates, on distingue: les alginates de sodium,
de potassium et d'autres métaux alcalins sont parfaitement solubles dans
l'eau en donnant des solutions à haute viscosité, tandis que
l'acide alginique pur et son sel de calcium sont pratiquement insolubles dans
l'eau (Anonyme 10).
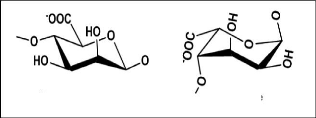
D-mannuronate L-guluronate
Figure (26) : Unités
monosaccharidiques constituant l'alginate (Onesippe,
2005).
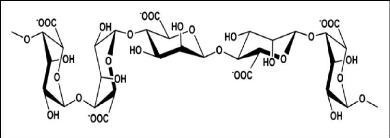
Figure (27) : Structure chimique de
l'alginate (Onesippe, 2005).
L'alginate a la
propriété de réticulation et ce dans les régions
d'acide guluronique quipeuvent être liées à une
région similaire dans une autre molécule d'alginate via le
Ca++ ou un
autre cation multivalent.
Du fait de leur absence de toxicité et de leur
capacité à former des gels, les alginates sont utilisés
dans de nombreux domaines (Anonyme 10).
Partie II
Etude expérimentale
Chapitre I
Matériels et méthodes
Objectif scientifique et protocole expérimental
:
L'étude expérimentale comporte deux
étapes, la première a été réalisée au
niveau du laboratoire de Contrôle de Qualité du complexe
ANTIBIOTICAL de MEDEA, dont l'objectif est d'analyser l'oxacilline sodique et
l'acide alginique, l'autre a été réalisée au niveau
du laboratoire de l'institut des science de la Nature et de la Vie du Centre
Universitaire ZIANE ACHOUR de Djelfa, dont l'objectif est d'étudier les
différents paramètres de conservation influençant la
libération du PA "oxacilline sodique" encapsulé.
Chaque échantillon analysé doit répondre aux
normes de la monographie qui précise un schéma d'analyse
spécifique. A cet égard, nous avons suivre les étapes
suivantes:
1/- Contrôle des échantillons (oxacilline sodique et
acide alginique) ;
2/- Préparation des microsphères de l'oxacilline
sodique ;
3/- Etude des différents paramètres
influençant la libération de PA encapsulé (pH,
température, force ionique, temps de libération et aspect des
microsphères "sèches ou humides").
I.1. Matériels utilisés :
A. Equipements :
- Agitateur magnétique ;
- Agitateur rotatif ;
- Appareil de Karl-Fischer (METROHM, E 452) ;
- Bain-marie ;
- Balance électrique de précision ;
- Barreau magnétique ; - Creuset en platine ;
- Dessiccateur ;
- Etuves thérmoréglées à 25 °C
et à 37 °C ;
- Four à moufle, T=600#177; 25°C ;
- HPLC (colonne C18 a phase inversé, appareil PERKIN
ELMER série 2 et série 200) ; - pH mètre (METROHM 632)
;
- Réfrigérateur réglé à 4
°C ;
- Spectrophotomètre UV-Visible. (BECKMAN DU 520).
B. Matières
premières:
- Acide alginique Lot N° 04BP075 ;
- Acide chlorhydrique (HCl) ;
- Chlorure de calcium (CaCl2) ;
- Eau distillée ;
- Eau physiologique ;
- Ethanol 96° (C2H5OH) ;
- Hydroxyde de sodium (NaOH) ;
- Méthanol (CH3OH) ;
- Oxacilline sodique: ECH Lot N° 07AP031 ;
STD Lot N° 06AP068.
C. Réactifs chimiques : voir
Annexe IV.
ii. Verreries et consommables : voir
Annexe IV.
I.2. Méthodes :
Le contrôle du PA et de l'excipient et même celle de
la préparation élaborée comporte les principales analyses
:
1. Analyse qualitative ou identification physicochimique,
chimique et/ou physique ;
2. Analyse quantitative ou essais de l'échantillon et/ou
de l'impureté associant.
Ces analyses sont basées sur les méthodes
d'identification suivantes :
- Soit la recherche des ions qui repose sur des réactions
chimiques spécifiques, ces réactions se divisent en trois groupes
:
1. Formation de gaz facile à identifier ;
2. Formation de précipites ;
3. Formation de composés colorés.
- Soit par recherche des constantes physiques : comme la
température de fusion, le pouvoir rotatoire, la densité relative,
les indices chimiques et la température d'ébullition, une fois
les constantes physiques sont mesurées, la recherche qualitative des
éléments fait appel à des essais importants. La
pharmacopée américaine1 décrit des
réactions d'identité des ions et des groupements fonctionnels,
cette recherche assure que l'échantillon n'est pas contaminé ;
- Soit par potentiomètrie: il s'agit d'une
méthode d'analyse électrochimique qui permet d'obtenir des
informations sur la composition d'un électrolyte, par mesure d'une
tension avec un courant d'intensité quasi nulle (par exemple : mesure de
pH) ;
- Soit par iodométrie : est l'une des méthodes
oxydométriques les plus souples, elle repose d'une part sur l'effet
oxydant de l'iode, d'autre part sur l'effet réducteur du ion iodure (ex
: KF). - Soit par chromatographie : c'est une technique de séparation et
de dosage ;
- Soit par spectrophotométrie: utilisée pour le
dosage, dans le but d'étudier la cinétique de la
libération du PA ;
- Soit par gravimétrie : ensemble de méthode de
dosage quantitative basé sur la mesure de la masse (sur la pesée)
(Anonyme 7).
1 La pharmacopée est un livre décrivant les
caractéristiques physiques, chimiques ou biologiques auxquelles doivent
répondre les formes pharmaceutiques et les matières
premières. La description des caractéristiques et de l'analyse
d'une matière première, est une monographie (Anonyme
7).
I.2.A. Contrôle physicochimique de la matière
première :
I.2.A.1. Caractérisation du PA [oxacilline sodique]
:
L'oxacilline sodique est parmi les substances décrites
par la pharmacopée américaine (USP), donc nous allons appliquer
les méthodes de référence de cette dernière, en
respectant ses normes.
Cette caractérisation comporte les analyses suivantes :
A/Caractère :
A.1/Aspect :
À examiner par une simple analyse visuelle de la
fluidité et de la limpidité des liquides, de
l'homogénéité des poudres, et par la vérification
de la couleur de l'odeur et de la saveur.
A.2/Solubilité :
Principe :
La solubilité est la quantité (exprimée en
(g) ou bien en (mole) par (litre) ou par (kg) de solvant) de composé que
l'on cherche à dissoudre dans un solvant donné.
Mode opératoire :
Préparer une série de tubes à essai
contenant chacun une masse de la substance à examiner qui correspond au
terme descriptif par apport au solvant.
Ce test est effectué dans l'eau, et dans l'alcool
(Méthanol).
Expression des résultats :
Les indications de solubilité figurant sous la rubrique
« caractères » sont exprimées en termes ayant la
signification suivante pour une température de 15 °C à 25
°C.
Tableau (02) : Echelle exprimant la
solubilité d'une substance (Anonyme 7).
|
Termes descriptifs
|
Volumes approximatifs de solvant en ml / g de substance
|
|
Très soluble
|
Inferieur à 1
|
|
Facilement soluble
|
de 1 à 10
|
|
Soluble
|
de 10 à 30
|
|
Assez soluble
|
de 30 à 100
|
|
Peu soluble
|
de 100 à 1000
|
|
Très peu soluble
|
de 1000 à 10000
|
|
Pratiquement insoluble
|
Plus de 10000
|
Le terme « partiellement soluble >> est
utilisé dans le cas d'un mélange dont seuls certains constituants
se dissolvent. Le terme « miscible >> est utilisé dans le cas
d'un liquide miscible en toutes proportions avec le solvant indiqué.
B/ Identification :
B.1/ Par HPLC :
Ce test est destiné d'une part à analyser
qualitativement l'ECH par identification et comparaison des temps de
rétention de la solution de STD et celle de l'ECH et d'autre part
à analyser quantitativement par détermination du titre en PA en
utilisant les aires de chaque pic de part et d'autre de l'ECH et du STD.
Principe :
En milieu hydro-organique, les solutés apolaire sont
exclus de la phase mobile et associent aux chaînes apolaires
greffées par effet hydrophobe.
Le solvant le moins polaire de la phase mobile s'adsorbe à
la phase stationnaire apolaire permettant le partage des solutés entre
la phase mobile et la phase liquide adsorbée.
Les temps de rétention des pics STD et ECH, doivent
êtes identiques.
Mode opératoire :
> Préparation de la solution STD
:
- Peser 109.4 mg de l'oxacilline sodique standard dans une fiole
jaugé de 200 ml. Puis ajuster jusqu'au trait de jauge avec de l'eau
distillée filtrée (double filtration) ;
- À l'aide d'un barreau magnétique agiter la
solution obtenue pendant 5 mn ;
- Prélever 10 ml de cette solution dans une fiole jauge de
50 ml, puis ajuster jusqu'au trait de jaugé avec l'eau distillée
filtrée (double filtration).
> Préparation de la solution ECH
:
- Peser 115.3 mg de l'oxacilline sodique échantillon dans
une fiole jaugé de 200 ml. Puis ajuster jusqu'au trait de jauge avec de
l'eau distillée filtrée (double filtration) ;
- À l'aide d'un barreau magnétique agiter la
solution obtenue pendant 5 mn ;
- Prélever 10 ml de cette solution dans une fiole jauge de
50 ml, puis ajuster jusqu'au trait de jaugé avec l'eau distillée
filtrée (double filtration).
> Préparation de la phase mobile
:
- Dissoudre 1.9 g de phosphate de potassium monobasique " KH2PO4"
dans 700 ml d'eau distillée ;
- Agiter la solution obtenue jusqu'à dissolution (environ
5 mn) ;
- Filtrer la solution obtenue (double filtration), et on obtient
alors la solution tampon ;
- Ajouter à la solution tampon (700 ml), un volume de 300
ml d'acétonitrile et un volume de 100 ml de méthanol.
> Conditions opératoires : -
Colonne type : C18 ;
- Détecteur : UV ;
- Longueur d'onde : 225 nm ; - Débit : 1 ml/min ;
- Pression : 80 bars ;
- Volume d'injection : 20 ul.
Procédé :
- Laisser circuler la phase mobile dans la colonne de l'appareil
pendant au moins 30 mn;
- Injecter séparément 20 ul pour la solution
standard et pour la solution échantillon dans le chromatographe au moyen
d'une micro seringue appropriée.
Expression des résultats :
Pour l'analyse quantitative, le temps de rétention de
l'ECH doit être identique au temps de rétention de STD.
B.2/ Identification de sodium :
Principe :
Ce test permet de détecter la présence du sodium
dans la molécule de l'ECH.
Mode opératoire :
Dissoudre 0,1 g de la substance à examiner dans 2 ml
d'eau distillé, Ajouter 2 ml de solution de carbonate de potassium
à 15 % et chauffer à ébullition. Il ne se forme aucun
précipité. Ajouter 4 ml de solution de pyroantimoniate de
potassium et chauffer à ébullition. Laisser refroidir dans l'eau
glacée et frotter si nécessaire la paroi du tube avec une
baguette de verre. Il se forme un précipité dense. Les
composés de sodium donnent une couleur jaune intense sous flamme.
C/Essais :
C.1/ Détermination du pH :
Principe :
Le pH est un nombre qui représente conventionnellement
la concentration en ions hydrogène ou hydronium d'une solution aqueuse.
La détermination potentiométrique du pH est effectuée par
mesure de la différence de potentiel entre 2 électrodes plongeant
dans la solution à examiner ; l'une de celles-ci est une
électrode sensible aux ions hydrogène (le plus souvent, une
électrode en verre) et l'autre une électrode de comparaison (de
référence).
Mode opératoire :
- Peser 900 mg de l'ECH dans 30ml d'eau distillée pour
avoir une concentration finale de 30 mg/ml, puis agiter jusqu'à
dissolution ;
- Rincer l'électrode du pH mètre avec de l'eau
distillée ;
- Calibrer l'appareil avec les solutions tampon de pH
différents ;
- Rincer l'électrode du pH mètre avec de l'eau
distillée ;
- Plonger l'électrode dans la solution à examiner
et effectuer la lecture dans les mêmes conditions que pour les solutions
tampons. Effectuer toutes les mesures à la même température
(20 °C à 25 °C) ;
- Lire la valeur du pH indiquée dans l'écran du pH
mètre ;
- Rincer l'électrode et plonger le, dans la solution de
garde (KCI).
Expression des résultats :
La valeur du pH doit être comprise entre: 4,5 et 7,5.
C.2/Teneur en eau :
La teneur en eau est déterminée par un appareil de
KARL-FISCHER qui est composé d'un dosimètre, d'une
électrode et d'un godet.
Principe :
Le principe établi par FISCHER pour la
détermination de la teneur en eau repose sur réaction ci-dessous,
et selon laquelle l'iode en présence d'eau réagit avec le dioxyde
de soufre :
I2 + SO 2 + 2H2O 2HI + H2SO4

L'eau dissolve l'iode et le dioxyde de soufre dans un solvant
non aqueux (le méthanol). Cette réaction d'oxydoréduction
s'effectue rapidement surtout si on associe une base à cette solution
(la pyridine).
I2 + SO2 + H2O + 3C5H5N 2C5H5N-HI + C5H5N-SO3
Donc le réactif de KARL-FISCHER contient l'iode, du
dioxyde de soufre et de pyridine dans le méthanol anhydre, ce
réactif est sous forme d'une solution prête à l'emploi.
On appelle Facteur de réactif KARL-FISCHER, le nombre de
mg d'eau titré par ml de
réactif.
F= mg (eau) /ml (réactif).
La substance utilisée pour la détermination du
facteur est dihydrate de sodium (Na2C2H4O62H2O).
Mode opératoire :
- Vérifier si la solution Karl-Fischer dans le godet n'est
pas saturée ;
- Neutraliser cette solution jusqu'à élimination
totale des traces d'eau, puis noter le volume initial (Vi) indiqué dans
le cadran du dosimètre ;
- Introduire la pesée « P » à
l'intérieur du godet, le dosage se fera automatiquement ;
- La fin du dosage automatique sera indiquée par
l'allumage de la lampe située dans l'appareil ; - Enfin, noter le volume
final (Vf) du dosage indiqué dans le cadran.
Expression des résultats :
La teneur en eau dans la prise de masse de l'ECH est
déterminée par la formule suivante :
|
(Vf - Vi) × F
Teneur en eau (%) = × 100
P
|
F : Facteur de la solution de dosage Karl-Fischer en mg/ml d'eau
(F = 3.2431 mg/ml) ; Vf : Volume final en ml ;
Vi : Volume initial en ml ;
P : Poids de l'échantillon en mg.
Limites d'acceptabilité: La valeur du Teneur en eau doit
être comprise entre: 3,5 à 5 %.
C.3. Dosage par HPLC :
Même principe et mode opératoire cité pour
l'identification par HPLC de l'oxacilline sodique.
Expression des résultats :
Pour l'analyse quantitative, le calcul se rapporte sur les
airs des pics au même temps de rétention dans le chromatogramme du
STD est ce lui de l'ECH et cela pour calculer le contenu en ug/mg de C
19 H 19 N 3 O 5 S dans
l'échantillon.
Le dosage par HPLC est évalué par la relation
suivante :
T =
Avec :
T : Titre ;
AIR STD : Air ou surface du pic du standard ;
AIR ECH : Air ou surface de pic de l'échantillon ;
P ECH : Pesée échantillon, (mg) ;
P STD : Pesée standard, (mg) ;
PUIS STD : Puissance du standard (ug/mg).
La limite d'acceptabilité : l'oxacilline sodique contient
un équivalent qui doit être supérieur à 815 ug et
inferieur à 950 d'oxacilline (C 19 H 19 N
3 O 5 S) par mg d'échantillon.
I.2.A.2. Caractérisation de l'excipient [acide
alginique] :
L'acide alginique est parmi les substances décrites par
la pharmacopée américaine (USP), donc nous allons appliquer les
méthodes de référence de cette dernière, en
respectant ses normes. Cette caractérisation comporte les analyses
suivantes :
A /Caractère
A.1 /Aspect :
À examiner par une simple analyse visuelle de la
fluidité et de la limpidité des liquides, de
l'homogénéité des poudres, et par la vérification
de la couleur de l'odeur et de la saveur.
A.2 /Solubilité :
Même principe et mode opératoire cités pour
déterminer la solubilité de l'oxacilline sodique.
B /Identification :
1/ - Dissoudre 1g d'acide alginique dans 150 ml de solution
d'hydroxyde de sodium (0.1N).
- À 5 ml de solution précédent, ajouter 1 ml
de solution de chlorure de calcium. Il se forme un précipité
volumineux et gélatineux.
2/ - Dissoudre 1g d'acide alginique dans 150 ml de solution
d'hydroxyde de sodium (0.1N).
- À 5 ml de solution précédent, ajouter 1 ml
d'acide sulfurique (4N). Il se forme un précipité lourd et
gélatineux.
3/ À 5 mg d'acide alginique, ajouter 5 ml d'eau
distillé, 1 ml d'une solution récemment préparée de
1,3-dihydroxynaphtalène à 10 g/l dans l'éthanol 96 % et 5
ml d'acide chlorhydrique. Faites bouillir doucement pendant 3 mn, refroidir
jusqu'à 15 0C, ajouter 5 ml d'eau distillé et agitez
avec 15 ml d'éther isopropylique. Effectuer un essai à blanc. La
couche supérieure du mélange obtenu avec la substance à
examiner présente une coloration rouge-bleu plus intense que celle
obtenue avec l'essai à blanc.
C. Essais
C.1/ Détermination du pH :
Principe :
Mêmes principe et mode opératoire cités pour
déterminer le pH de la solution d'oxacilline sodique. Ce test est
effectué pour une solution aqueuse de 3 mg/100 ml.
Expression des résultats :
La valeur du pH doit être comprise entre: 1,5 à
3,5.
C.2/ Arsenic :
Principe :
La détermination de la présence d'arsenic dans
l'ECH doit répondre aux limites d'acceptation.
Le principe est basé sur la transformation de l'arsenic
(As) de la prise d'essai en hydrogène arsénié qui
réagit avec le diethyldithiocarbamate d'argent pour former un complexe
de couleur rouge qui est comparée visuellement ou
spéctrophotométriquement avec la couleur de STD
préparé en utilisant la quantité d'arsenic de la limite
décrite dans la monographie. La couleur de l'ECH ne doit pas être
plus intense que le STD.
Mode opératoire :
Le mode de préparation des solutions STD et ECH ainsi le
montage utilisé sont donnés en annexe V.
Ces solutions sont traitées similairement comme suite :
Partie 1:
Dans la fiole, Ajouter 20 ml d'acide sulfurique (7N), 2 ml de
solution d'iodure de potassium, 0,5 ml de solution d'acide de chlorure stanneux
concentré, et 1 ml d'alcool isopropyle, et mélanger. Laisser
reposer à la température ambiante pendant 30 min.
Partie 2:
Mettre du coton dans un col en verre déjà
imbibé dans une solution saturée d'acétate de plomb, et
contenant un papier d'indication de la couleur.
- Transférer 3.0 ml de solution de
diéthyldithiocarbamate d'argent ;
- Ajouter 3.0 g de zinc granulaire au mélange dans la
fiole ;
Assembler aussitôt les 2 parties de l'appareil en
assurant la fermeture de l'ensemble; Laisser reposer à la
température ambiante permettant un dégagement régulier du
gaz et développement de couleur pendant 45 min ;
Déconnecter les 2 parties de l'appareil et
transférer la solution à une cellule d'absorption de 1cm.
Expression des résultats :
La couleur rouge produite par la préparation de l'ECH ne
doit pas être plus intense que celle de la préparation STD.
- Limite d'acceptabilité : < 3 ppm.
C.3/ Métaux lourds :
Principe :
Cet essai est fourni pour démontrer que la teneur des
impuretés métalliques qui précipitent à pH = 3,5
sous forme de sulfure colorés par l'action des ions sulfures ou des
réactifs capables de les engendrer (Thio acétamide
hydrolysé en milieu alcalin) ne dépasse pas la limite de
métaux lourds indiquée dans la monographie individuelle en termes
de pourcentage (en poids) de plomb dans l'ECH, ce pourcentage est
déterminé par comparaison visuelle concomitante. La couleur de la
solution l'ECH ne doit pas plus foncée que celle de la solution STD.
Les substances qui typiquement répondront à cet
essai sont : Pb, Cu, Ag, Hg, Co, Cd.
Mode opératoire :
La préparation des solutions STD et ECH est donnée
en annexe V.
À chacun des tubes contenant la solution STD et la
solution ECH, ajouter 2 ml de solution tampon d'acétate d'ammonuim de pH
3.5, puis ajouter 1,2 ml de solution de Thioacetamideglycérine,
diluer avec de l'eau distillé jusqu'à 50 ml,
mélanger, laisser reposer pendant 2 minutes, observer les tubes au
dessus d'une arrière plan blanche. La couleur de la solution à
examiner ne doit pas plus foncée que celle de la solution
témoin.
Expression des résultats :
La valeur de la teneur en métaux lourds pas plus de
0.004%.
C.4/Cendres totaux :
Principe :
Ce test a pour but de déterminer le pourcentage des
éléments minéraux présents dans la substance
à examiner, par chauffage au dessous de son point de
fusion.
Mode opératoire :
- Chauffer au rouge un creuset de platine pendant 30 min ;
- Laisser refroidir dans un dessiccateur, puis peser. Dans le
creuset, introduire 1.00g de la substance à examiner ;
- Distribuer uniformément la prise d'essai à
l'intérieur du creuset ;
- Desséchez pendant 1 h à 100-105 °C, puis
incinérer dans un four à moufle, à une température
de 600 #177; 25 °C. L'échantillon ne doit s'enflammer à
aucun moment de l'opération ;
- Continuer l'incinération jusqu'à masse constante
;
- Après chaque incinération, laisser refroidir
et peser le creuset au dessiccateur. Si les cendres contiennent encore des
particules noires, après une incinération prolongée,
reprenez-les à l'eau distillée chaude et filtrer sur un filtre
sans cendres. Incinérer à nouveau le résidu avec le
filtre. Réunir le filtrat et les cendres, évaporer prudemment
à siccité et incinérer jusqu'à masse constante ;
Expression de résultat :
La valeur du taux des cendres totaux est calculée par la
formule suivante:
X % = (Pcr-- Pcv) ×100 / PE
Avec:
Pcv: Poids du creuset de platine (vide) avant
l'incinération, (g) ;
Pcr: Poids du creuset de platine après
l'incinération (vide + résidu), (g) ;
PE: Prise d'essai, (g).
Limites d'acceptabilité: la valeur du taux des cendres
totaux n'est pas supérieure à 4 %.
C.5/ Perte à la dessiccation :
Principe :
La perte à la dessiccation est la perte de masse
exprimée en pourcentage m/m.
Cet essai permet de déterminer la proportion de tous les
produits volatils susceptibles d'être éliminés dans des
conditions spécifiques :
- Eau (le plus souvent) ;
- Eau « d'humidité de proportions variables »
;
- Eau d'hydratation ;
- Solvants organiques.
Mode opératoire :
Placez 0,1000 g d'acide alginique dans un flacon à
tare, desséché lui-même au préalable dans les
conditions précisées pour la substance à examiner. La
dessiccation de la substance se fait à l'étuve jusqu'à
masse constante pendant 4 h à (100 -105) °C.
Expression de résultat :
La valeur de la perte à la dessiccation est
calculée par la formule suivante:
X (%) = (VV - V A) x100/ PE
Avec:
VV: Poids du verre de montre (vide + échantillon) avant
dessiccation, (g);
VA: Poids du verre de montre (vide + échantillon)
après dessiccation, (g);
PE: Prise d'essai, (g).
Limites d'acceptabilité: la valeur de la perte à la
dessiccation ne doit pas être supérieure à 15,0 pour
cent.
D/ Dosage chimique (indice O'aciOe) :
Principe :
L'indice d'acide (IA) est le nombre en milligrammes qui
exprime la quantité d'hydroxyde de potassium nécessaire à
la neutralisation des acides libres présents dans 1 g de la substance
à analyser.
Mode opératoire :
Dissoudre 1 g d'acide alginique, exactement pesé, dans
un mélange de : 50 ml de l'eau distillé et de 30 ml de la
solution d'acétate de calcium (11 dans 250). Mélanger, laisser le
mélange se reposer pendant 1 h, ajouter la solution de
phénolphtaléine, et puis titrer l'acide acétique
libéré avec l'hydroxyde de potassium (1 N). Effectuer le dosage
du blanc (acide acétique seulement).
Expression de résultat :
L'indice d'acidité pris par la formule suivante :
T = 5.611 (A - B) /W
Avec :
5.611 : Equivalent gramme de la solution KOH 0.1 N ;
A et B : Volumes d'hydroxyde de potassium (0.1 N)
consommés dans le titrage de la préparation d'essai et du blanc,
respectivement, (ml) ;
W : Masse en g d'acide alginique pris.
Limites d'acceptabilité: L'indice d'acidité de
l'acide alginique, calculé sur la base sèche, n'est pas moins de
230.
I.2.B. Etude des paramètres influençant
la libération du PA [oxacilline sodique] encapsulé :
Cette partie mène à étudier la
libération de l'oxacilline sodique en fonction des paramètres de
milieu qui sont : le pH, la force ionique, la température, l'aspect des
microsphères "sèche ou humide".) , et le temps de
libération.
I.2.B.1. Détermination de la courbe
d'étalonnage :
Afin de caractériser la libération du PA
encapsulé, nous avons appliqué la spectrophotomètrie
UV-Visible, à cause de l'absorption de l'oxacilline sodique dans le
domaine de l'UV ; ceci nécessite la détermination de la courbe
d'étalonnage et de connaître la longueur d'onde pour l'absorption
maximale de l'oxacilline sodique.
Principe :
L'absorption moléculaire dans le spectre ultraviolet
(UV) et visible dépend de la structure électronique de la
molécule. L'absorption d'énergie est quantifiée et
résulte du passage des électrons d'orbitales de l'état
fondamental vers des orbitales d'un état excité d'énergie
supérieur.
Une expression plus adéquate de l'intensité
d'absorption est celle dérivée de la loi de Beer-Lambert, qui
établit la relation entre l'absorbance, l'épaisseur de
l'échantillon et la concentration des espèces absorbantes.
Cette relation s'écrit :
A= log (I0/I) = å bC
Avec :
A : Absorbance ou densité optique ;
I0 : Intensité de l'énergie d'irradiation arrivant
sur l'échantillon ;
I : Intensité de la radiation qui a traversée
l'échantillon ;
å : Constante caractéristique du
soluté (coefficient d'absorption moléculaire ou d'extinction
molaire : mol-1.l. cm-1) ;
b : Longueur du chemin optique à travers
l'échantillon (cm) ;
C: Concentration du soluté (mol.l-1).
Ou bien :
A= log (I0/I) = abC
Avec :
a: Absorptivité (g-1.l.
cm-1) ;
C: Concentration du soluté (g.l-1).
Mode opératoire :
- Préparer quatre solutions de l'oxacilline sodique de
concentrations suivantes : 0.01 mg/ml, 0.005 mg/ml, 0.00125 mg/ml, 0.000625
mg/ml ;
- Remplir la cuvette en quartz avec la solution ;
- Utiliser la même cuvette pour l'échantillon et
pour le blanc ;
- Mesurer l'absorbance de chaque solution ainsi pour le blanc, en
utilisant la longueur d'onde d'absorption maximale (ë max de
l'oxacilline sodique égale à 235 nm) (Anonyme 7)
;
-Tracer la courbe d'étalonnage
à partir de la moyenne des lectures obtenues avec les différentes
solutions ;
- Déterminer la concentration de l'oxacilline sodique dans
la solution à examiner à l'aide de la courbe obtenue.
I.2.B.2. Préparation des sphères de
l'oxacilline sodique :
Principe :
Le principe de la microencapsulation consiste à former
une mince paroi polymérique au tour des gouttelettes ou des particules
du produit en suspension de microcapsule dont la taille varie entre un et
plusieurs centaines de microns. Le polymère utilisé peut
être naturelle ou synthétique.
Le procédé utilisé pour la
préparation des microsphères est le procédé de
gélification des gouttes, le choix de ce procédé est
basé sur la simplicité et sur disponibilité du
matériel nécessaire à ce niveau.
Mode opératoire :
A/ Préparation de la solution d'acide alginique :
Mélanger 3 g d'acide alginique dans 25 ml d'eau
distillé, faire l'agitation à laide d'un barreau
magnétique.
B/ Préparation de la solution d'hydroxyde de sodium :
Dissoudre 1.66 g d'hydroxyde de sodium dans 25 ml d'eau
distillé.
C/ Préparation de la solution d'alginate de sodium et
d'oxacilline sodique :
- Mélanger les solutions préparées en (A) et
(B) sous agitation magnétique ;
- Mélanger 50 mg d'oxacilline sodique (PA) avec la
solution précédente (alginate de sodium), agiter jusqu'à
ce que la solution devienne homogène.
D/ Préparation de la solution du chlorure de calcium :
Dissoudre 0.325 g de chlorure de calcium (CaCl2) dans 25 ml d'eau
distillée.
E/ À l'aide d'une seringue goutter 2 ml de la solution
préparée en (C) dans une boitte de pétrie remplie
préalablement par la solution de chlorure de calcium.
F/ Laisser la préparation quelques minutes pour que les
sphères se solidifient, filtrer à l'aide des papiers filtres,
rincer les sphères par quelques ml d'eau distillé, pour
éliminer l'excès de CaCl2, mesurer l'absorbance de filtrat pour
calculer le rendement d'encapsulation.
Les étapes du procédé de formulation des
sphères sont schématisées dans la figure 28.
PA (oxacilline sodique) Alginate de sodium
Agitation
Seringue
Mélange d'alginate de sodium
et le PA
Aiguille de la seringue Gouttelettes de mélange
Solution de CaCl2
Sphères formés Boite de Pétri
Figure (28) : Etapes de préparation
des sphères.
I.2.B.3. Détermination du rendement d'encapsulation
:
Pour déterminer le rendement d'encapsulation, il faut
appliquer la relation suivante:
× 100
Masse de PA à encapsuler
Rdt=
Masse de PA encapsulé
I.2.B.4. Étude de la stabilité des
sphères :
Cette partie est consacrée à étudier la
stabilité des sphères mises dans des conditions de
températures différentes, dans des milieux différents (eau
physiologique, eau distillé) en vue de voir si les sphères
peuvent maintenir le PA dans ces conditions, c'est-à-dire la
détermination de la quantité libérée par les
sphères dans le temps, et ceci pour les cas suivants :
> Sphères humides se
mettent:
o Dans l'eau distillée à pH = 1.2 et T = (4, 25 et
37)°C. o Dans l'eau distillée à pH = 3 et T = (4,
25 et 37)°C.
o Dans l'eau distillée à pH = 7 et T = (4, 25 et
37)°C.
o Dans l'eau physiologique à pH = 1.2 et T = (4, 25 et
37)°C. o Dans l'eau physiologique à pH = 3 et T = (4, 25
et 37)°C.
o Dans l'eau physiologique à pH = 7 et T = (4, 25 et
37)°C.
> Sphères séchées se mettent
:
o Dans l'eau distillée à pH = 1.2 et T = (4, 25 et
37)°C. o Dans l'eau distillée à pH = 3 et T = (4,
25 et 37)°C.
o Dans l'eau distillée à pH = 7 et T = (4, 25 et
37)°C.
o Dans l'eau physiologique à pH = 1.2 et T = (4, 25 et
37)°C. o Dans l'eau physiologique à pH = 3 et T = (4, 25
et 37)°C.
o Dans l'eau physiologique à pH = 7 et T = (4, 25 et
37)°C.
N.B : Le séchage des
sphères se fait à l'étuve à 25 °C
pendent 48h.
Chapitre II
Résultats et discussions
II. A. Résultats de contrôle physicochimique
de la matière première : II.A.1. Caractérisation
physico-chimique du PA [oxacilline sodique] :
Les différents résultats établis pour la
caractérisation de l'oxacilline sodique sont figurés dans le
tableau ci-dessous associés aux spécifications de la monographie
et comparés aux normes.
Tableau (03) : Résultats du
contrôle physicochimique de l'oxacilline sodique :
|
Tests
|
Résultats
|
Normes
|
|
A/ Caractère :
|
|
|
|
- Aspect :
|
Poudre blanche, cristalline
|
-Poudre blanche, cristalline
|
|
avec une odeur
|
avec une odeur
|
|
caractéristique
|
caractéristique [acétate
d'éthyle].
|
|
- Solubilité :
|
|
|
|
Eau
|
1g/3ml
|
-Facilement soluble dans
l'eau.
|
|
Méthanol.
|
1g/5ml
|
-Facilement soluble dans le
|
|
|
Méthanol.
|
|
B/Identification :
|
|
|
|
- Par HPLC :
|
|
|
|
Temps de rétention
|
t (STD) = 3.312 mn
|
-Le temps de rétention du PA
|
|
t (ECH) = 3.337 mn
|
STD est identique à celui du l'ECH.
|
|
-Test de sodium
|
Positive
|
-Il se forme un précipité blanc et dense.
|
|
C/Essai :
|
|
|
|
- pH
|
4,92
|
-Entre 4,5 et 7,5.
|
|
- Teneur en eau (KF)
|
3,94 %
|
-Entre 3,5 et 5,0 %.
|
|
- Dosage
|
T= 927,18 ug /mg
|
-Entre 815 et 950 ug/mg.
|
Aspect :
L'analyse visuelle du PA portant sur les critères
d'odeur et de couleur a donné une poudre blanche, cristalline avec une
odeur caractéristique [acétate d'éthyle], cette odeur est
due au procédé d'obtention. La forme cristalline
détectée à l'aide du microscope optique polarisant permet
de détecter la couleur des cristaux. Ce résultat est conforme aux
normes exigées.
Solubilité :
Le résultat du test de solubilité porté
sur le tableau (03), montre que le PA est facilement soluble dans l'eau : par
ionisation du sodium et les groupements hydrophiles portés sur la
molécule d'oxacilline sodique. Sa solubilité dans le
méthanol est due essentiellement à la formation des ponts
hydrogène.
Identification par HPLC :
Les figures (29) et (30) schématisent respectivement
les chromatogrammes des solutions STD et ECH, qui permettent
simultanément d'identifier l'ECH et de déterminer son titre en
oxacilline.
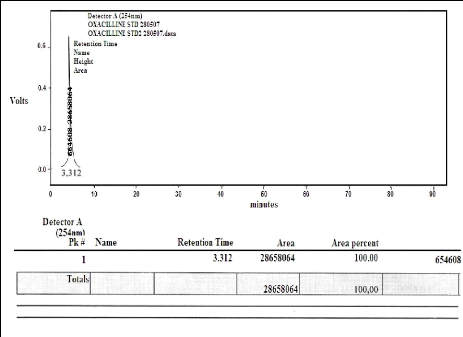
Figure (29) : Chromatogramme du STD
(oxacilline sodique) obtenu par analyse par HPLC.
[GROUPE
INDUSTRIEL SAIDAL Spa FILIALE ANTIBIOTICAL MEDEA]
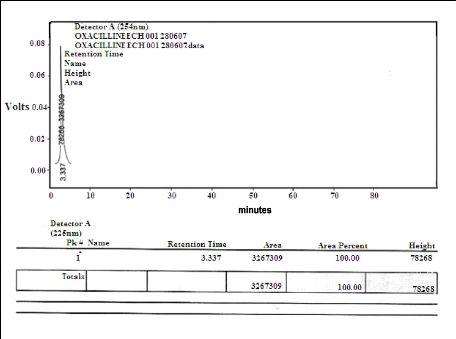
Figure (30) : Chromatogramme de l'ECH
(oxacilline sodique) obtenu par analyse par HPLC.
[GROUPE
INDUSTRIEL SAIDAL Spa FILIALE ANTIBIOTICAL MEDEA]
Le temps de rétention de l'ECH très proche de
celui du STD (en minute), ce qui permet de confirmer l'identification de la
molécule d'oxacilline, le faible écart peut être
interprété par les conditions de préparation et de
conditions opératoires ainsi que les propriétés de chacune
des substances ECH et STD.
Test de sodium :
Le résultat du test de sodium porté sur le tableau
(03) montre que la substance testée contient du sodium, parce qu'il
s'agit d'un sel sodique de l'oxacilline.
pH :
La mesure du pH de l'oxacilline sodique a
révélé une valeur de 4.92, situant confortablement dans
l'intervalle préconisé par les normes c'est-à-dire entre
4,5 à 7,5.
Teneur en eau (KF) :
La présence d'eau au sein d'une substance chimique peut
avoir pour conséquence d'en abaisser l'activité par diminution du
titre, et de modifier sa qualité pendant le stockage.
D'après le contrôle physicochimique de l'oxacilline
sodique ce dernier est déclaré conforme selon les normes de la
pharmacopée américaine.
II.A.2. Caractérisation physico-chimique de
l'excipient [acide alginique] :
Les différents résultats établis pour la
caractérisation de l'acide alginique sont figurés dans le tableau
(04) associés aux spécifications de la monographie et
comparés aux normes.
Tableau (04): Résultats du
contrôle physicochimique de l'acide alginique :
|
Tests
|
|
Résultats
|
|
Normes
|
|
A/ Caractère :
|
|
|
|
|
|
- Aspect :
|
|
Poudre amorphe,
inodore, insipide.
|
blanche,
|
- Poudre cristalline ou
amorphe, blanche ou brun jaunâtre, inodore, insipide.
|
|
- Solubilité :
|
|
|
|
|
|
Eau
|
|
Insoluble dans l'eau
|
|
- Insoluble dans l'eau
|
|
Alcool
|
|
1g/5000 ml
|
|
- Très peu soluble dans l'alcool.
|
|
B/Identification :
|
|
|
|
|
|
- A
|
|
Positive
|
|
- Il se forme un précipité volumineux et
gélatineux
|
|
- B
|
|
Positive
|
|
- Il se forme un précipité
lourd et gélatineux
|
|
- C
|
|
Positive
|
|
- Le mélange obtenu avec la
substance à examiner
présente une coloration
rouge-bleu plus intense que celle obtenu l'essai à
blanc
|
|
C/Essai :
|
|
|
|
|
|
- pH
|
|
2,22
|
|
1,5 à 3,5
|
|
- Arsenic
|
|
Conforme
|
|
= 3 ppm
|
|
- Métaux lourds
|
|
Conforme
|
|
= 0.004%
|
|
- Cendres totaux
|
|
1.21 %
|
|
= 4 %
|
|
- Perte à la dessiccation
|
|
9,58 %
|
|
= 15 %
|
|
- Dosage (indice d'acide
|
)
|
310,58 SBA
|
|
= 230 SBA
|
Aspect :
L'analyse visuelle de l'acide alginique portant sur les
critères d'odeur et de couleur a donné une poudre blanche,
amorphe, inodore et insipide. Ce résultat est conforme aux normes
exigées.
Solubilité :
Les résultats du test de solubilité
portés sur le tableau (04) montrent que l'acide alginique est insoluble
dans l'eau : par l'existence de longues chaînes hydrocarbonées
(polymère hydrophobe), la très faible solubilité dans
l'alcool est due essentiellement au caractère hydrophobe de la substance
et à la chaîne hydrocarbonée de l'alcool.
Identification :
Les tests d'identifications chimiques montrent que l'acide
alginique est conforme aux normes exigées.
pH :
La mesure du pH de l'acide alginique a
révélé une valeur de 2,22 : caractère acide se
situant confortablement dans l'intervalle préconisé par les
normes c'est-à-dire entre 1,5 à 3,5.
Arsenic :
Le résultat obtenu pour le taux de présence
d'arsenic dans l'acide alginique est conforme à la norme (=3 ppm).
Métaux lourds :
La présence d'un ou plusieurs métaux lourds au
sein d'une substance chimique peut avoir pour résultat d'abaisser
l'activité par diminution du titre, mais aussi d'augmenter la
toxicité lorsque l'impureté est particulièrement
nocive.
Le résultat obtenu dans l'essai de détermination
des métaux lourds est très faible ou négligeable par
rapport à la norme (= 0.004%).
Cendres totaux :
Le résultat du pourcentage des cendres totales obtenues
pour l'acide alginique a montré qu'il est conforme à la norme qui
prévoit une valeur de cendres de 1.21 %.
Perte à la dessiccation :
Le résultat de Perte à la dessiccation obtenue pour
l'acide alginique a montré qu'il est conforme à la norme qui
prévoit une valeur de cendres de 9,58 %.
Dosage (indice d'acide) :
310,58 SBA : ce résultat est conforme aux normes de la
pharmacopée américaine, qui prévoit une limite
supérieure à 230.
D'après le contrôle physicochimique de l'acide
alginique ce dernier est déclaré conforme selon les normes de la
pharmacopée américaine.
II.B. Résultats de l'étude des parametres
influençant la libération du PA
[oxacilline sodique] encapsulé :
II.B.1. Détermination de la courbe
d'étalonnage :
La variation de l'absorbance pour une longueur d'onde maximale
(ë = 235 nm), pour des solutions aqueuses d'oxacilline sodique à
différentes concentrations, sont résumés sur le tableau
suivant :
Tableau (05) : Résultats dVEIRLEECHI
des solutions d'oxacilline sodique à différentes concentrations
:
|
Concentration
en mg/ml
|
0
|
0,000625
|
0,00125
|
0,005
|
0,01
|
|
Absorbance
(A)
|
0
|
0,027
|
0,048
|
0,183
|
0,352
|
À partir de ces valeurs nous avons tracé la courbe
d'étalonnage :
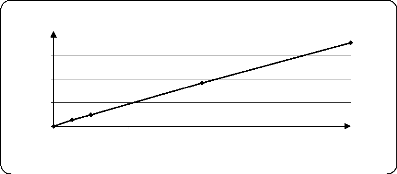
0 0,0025 0,005 0,0075 0,01
Concentration
mg/ml
Absorbance
0,4
0,3
0,2
0,1
0
Y= 35.04 X +0.003
R2 = 0.999
Figure (31) : Courbe d'étalonnage
d'oxacilline sodique.
La courbe d'étalonnage obtenue est une droite dont la
pente égale à 35.04 avec un coefficient de corrélation R=
0.999. Par la comparaison avec la loi de BEER-LAMBERT, on trouve :
La pente = a (absorptivité) = 35.04
g-1.l.cm-1
II.B.2. Préparation des sphères de
l'oxacilline sodique :

L'alginate de sodium résulte de la réaction de
neutralisation de l'acide alginique avec la soude selon l'équation
suivante :
Acide alginique + hydroxyde de sodium alginate de sodium + eau
> Aspect de la solution d'alginate de sodium
: - Solution visqueuse ;
- De couleur jaune.
> Aspect des sphères
résultantes :
Les sphères obtenues sont de couleur blanche, de forme
sphérique et d'une taille moyenne situant au voisinage : 2.4 mm
(fig.32).
Figure (32) : Sphères de
l'oxacilline sodique.
II.B.3. Détermination du rendement d'encapsulation
:
Le calcul du rendement d'encapsulation a
révélé une valeur de 31,25 %.
Rdt = 31,25 %.
II.B.4. Etude de la libération du PA
encapsulé :
L'étude de la cinétique et de la stabilité
des billes, nous a mené à respecter les deux conditions suivantes
:
En premier lieu nous avons tenté de comparer la
libération du PA à travers deux solutions aqueuses : eau
distillée et eau physiologique ;
En deuxième lieu, nous avons étudié
l'influence de deux paramètres : température et pH sur la
libération pour les premiers cas.
Les billes dont nous avons étudié la
libération du PA en fonction de différents paramètres sont
de deux types: billes séchées à l'étuve et billes
telles quelles sont ou humides.
À partir d'une microsphère, le
phénomène de diffusion de la molécule active à
travers une matrice sera décrit par une cinétique
obéissant à la loi d'HIGUCHI qui montre que la
quantité libérée de matière active est directement
proportionnelle à la racine carrée du temps.
m = k t1/2
Les résultats établis des tests
expérimentaux portés sur les billes préparées,
déterminantes la quantité libérée dans le temps,
sont récapitulés comme suit :
+ Cas des billes non séchées (humides)
:
> Libération de PA dans l'eau
distillée :
Les résultats des tests de libération du PA des
billes non séchées effectués dans l'eau distillée
à différentes températures pour des pH : 1,2, 3 et 7, sont
illustrés sur les figures 33, 34, 35 (voir annexe VI, tableau 7) :
N.B : La masse cumulative est la masse du PA
contenant dans le milieu de libération.
. pH = 1,2
Masse cumulative (mg)
0,6
0,5
0,4
0,3
0,2
0,1
0
1 2 22 23 24 46 47 48 49 70 71
t (h)
T= 37°c T= 25°c T= 4°c
Figure (33) : Influence de la
température sur la libération du PA
dans l'eau distillé
à pH=1,2.
. pH =3
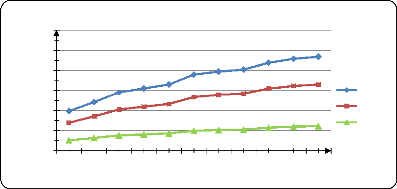
Masse cumulative (mg)
0,6
0,5
0,4
0,3
0,2
0,1
0
1 2 22 23 24 46 47 48 49 70 71
t(h)
T= 37°c T= 25°c T= 4°c
Figure (34) : Influence de la
température sur la libération du PA
dans l'eau distillé
à pH=3.
. pH =7
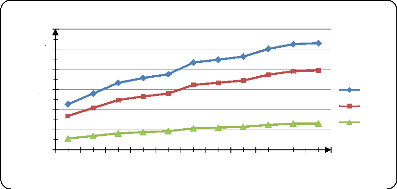
Masse cumulative (mg)
0,6
0,5
0,4
0,3
0,2
0,1
0
1 2 22 23 24 46 47 48 49 70 71
t(h)
T= 37°c T= 25°c T= 4°c
Figure (35) : Influence de la
température sur la libération du PA
dans l'eau distillé
à pH=7.
> Liberation de PA dans l'eau physiologique
:
Les résultats des tests de libération de PA des
billes non séchées effectués dans l'eau physiologique
à différentes températures pour des pH : 1,2, 3 et 7, sont
illustrés sur les figures 36, 37 et 38 (voir annexe VI, tableau 8):
. pH = 1,2
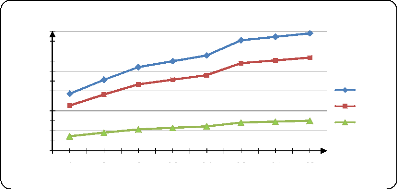
Masse cumulative (mg)
0,6
0,5
0,4
0,3
0,2
0,1
0
1 2 4 24 26 28 46 48
t(h)
T= 37°c T= 25°c T= 4°c
Figure (36) : Influence de la
température sur la libération de PA
dans l'eau physiologique
à pH=1,2.
. pH = 3
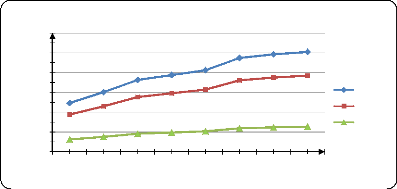
Masse cumulative (mg)
0,6
0,5
0,4
0,3
0,2
0,1
0
1 2 4 24 26 28 46 48
t(h)
T= 37°c T= 25°c T= 4°c
Figure (37) : Influence de la
température sur la libération de PA
daj l'eau physiologique
à pH=3.
. pH = 7
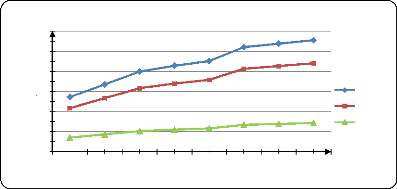
Masse cumulative (mg)
0,6
0,5
0,4
0,3
0,2
0,1
0
1 2 4 24 26 28 46 48
t(h)
T= 37°c T= 25°c T= 4°c
Figure (38) : Influence de la
température sur la libération de PA dans
l'eau physiologique
à pH=7.
+ Cas des billes séchées
:
> Libération de PA dans l'eau
distillée :
Les résultats des tests de libération de PA des
billes non séchées effectués dans l'eau distillée
à différentes températures pour des pH : 1,2, 3 et 7, sont
illustrés sur les figures, 39, 40, 41 (voir annexe VI, tableau 9) :
. pH = 1,2
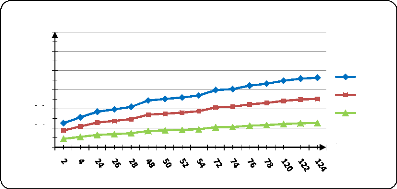
Masse cumulative(mg)
0,6
0,5
0,4
0,3
0,2
0,1
0
t(h)
T= 37°c T= 25°c T=
4°c
Figure (39) : Influence de
température sur la libération de PA
dans l'eau
distillée à pH=1,2.
. pH = 3
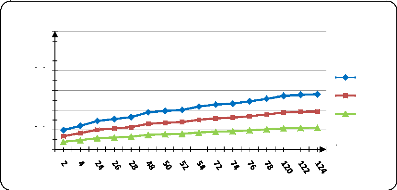
Masse cumulative(mg)
0,6
0,5
0,4
0,3
0,2
0,1
0
t(h)
T= 37°c T= 25°c T=
4°c
Figure (40) : Influence de
température sur la libération de PA
dans l'eau distillé
à pH3
. pH = 7
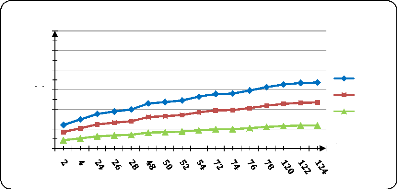
Masse cumulative(mg)
0,6
0,5
0,4
0,3
0,2
0,1
0
t(h)
T= 37°c T= 25°c T= 4°c
Figure (41) : Influence de
température sur la libération de PA
dans l'eau
distillée à pH=7.
> Libération de PA dans l'eau
physiologique :
Les résultats des tests de libération de PA des
billes non séchées effectués dans l'eau physiologique
à différentes températures pour des pH : 1.2, 3 et 7, sont
illustrés sur les figures 42, 43,44 (voir annexe VI, tableau 10) :
. pH = 1.2
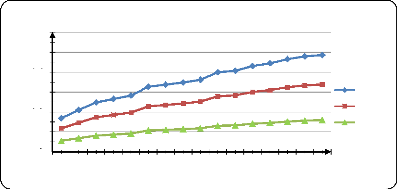
Masse cumulative(mg)
0,6
0,5
0,4
0,3
0,2
0,1
0
2 4 24 26 28 46 48 50 52 70 72 74 76 94 96 98
t(h)
T= 37°c T= 25°c T= 4°c
Figure (42) : Influence de la
température sur la libération de PA
dans l'eau physiologique
à pH=1,2.
. pH = 3
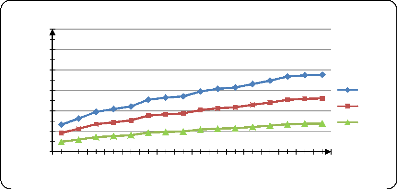
Masse cumulative(mg)
0,6
0,5
0,4
0,3
0,2
0,1
0
2 4 24 26 28 46 48 50 52 70 72 74 76 94 96 98
t(h)
T= 37°c T= 25°c T= 4°c
Figure (43) : Influence de la
température sur la libération de PA
dans l'eau physiologique
à pH=3.
. pH = 7
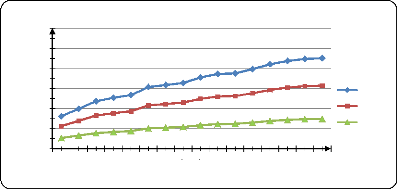
Masse cumulative(mg)
0,6
0,5
0,4
0,3
0,2
0,1
0
2 4 24 26 28 46 48 50 52 70 72 74 76 94 96 98
t(h)
T= 37°c T= 25°c T= 4°c
Figure (44) : Influence de la
température sur la libération de PA
dans l'eau physiologique
à pH=7.
Discussion générale
On peut organiser la discussion selon les points suivants:
A. Analyses et discussion des graphes :
1- Pour les billes humides :
Nous pouvons diviser les courbes cumulatives en 3 parties:
* La première partie, on remarque que la
libération est importante: de 1 h à 4h (cas de l'eau
physiologique) et de 1h à 22 h (cas de l'eau distillée), dans ce
cas la libération est favorisée par la différence de
concentration en oxacilline sodique entre le solvant de dissolution (l'eau
distillée) et le milieu interne (la matrice du polymère);
* Dans la deuxième partie qui est plus longue, de 4 h
à 28 h (cas de l'eau physiologique) et de 22h à 49 h (cas de
l'eau distillée) ,la libération est plus faible que la
première partie, ce qui implique l'abaissement de différence de
concentration en PA entre les deux milieux;
* Dans la troisième partie : à partir de 28 h
(cas de l'eau physiologique) et de 49 h (cas de l'eau distillée), la
quantité libérée est très faible (libération
presque constante due à la saturation de milieu), ce qui implique que la
quantité encapsulée est presque absente dans la bille dans les
deux cas (eau distillé et l'eau physiologique).
2- Pour les billes sèches: dans ce cas les allures sont
divisées en deux cas :
2-a/ Cas de la libération dans l'eau distillée,
le phénomène est très faible ce qui est traduit par
l'allure (presque une droite) et cela a été étudiée
dans un temps long: 1h à 124 h, pour une quantité totale
libérée comprise entre 0.11 mg et 0.36 mg, pour l'intervalle des
températures et des pH étudiées.
2-b/ Cas de la libération dans l'eau physiologique, les
allures sont discutées de la même manière que celles
décrites plus haut. Mais dans ce cas la quantité maximale
libérée est atteinte rapidement et plus faible que celle atteinte
par apport à celle des sphères non séchées.
Cependant, à la température de 4°C, la
libération est faible ce qui signifie que la température
adéquate de conservation (réfrigérateur) est cette valeur.
On remarque aussi, que la valeur de 37°C qui est la température
physiologique constitue une valeur optimale pour la libération du PA, ce
processus qui dure dans les conditions de pH (1.2, 3, 7) 71 h pour une
libération dans l'eau distillée et une valeur supérieure
pour une durée de 48 h dans l'eau physiologique.
Par ailleurs, la température 25°C (la
température de préparation), la quantité
libérée est en 2 h presque la moitié de celle de la
quantité maximale pour un pH = 7. Donc il faut tenir compte d'une part
les conditions de préparation et celles de la quantité suffisante
pour garantir l'effet thérapeutique.
B. Interprétation :
La libération du PA encapsulé dans la matrice
polymérique suit deux principales étapes: le gonflement: qui est
assuré par l'entré du solvant et la diffusion qui est régi
par la loi d'HIGUCHI, (les billes formulées sont de type
sphère et non microcapsule). Ce resultat est bien conforme aux allures
des courbes établies. Cependant l'effet des paramètres
physico-chimiques peut etre interprété de la manière
suivante:
1/ Influence de la temperature :
La température peut avoir un double effet, et cela d'une
part, sur la conformation des
chaines polymériques, cet effet se manifeste quand la
température est augmentée, les chaines polymériques
s'éloignent les unes des autres, se qui permet au PA de se
libérer.
Et d'autre part sur la modification du coefficient de
diffusion, qui est liée à la température par la relation
établie par STOCKES-EINSTEIN (Xiaoling et Bhaskara,
2006), où le coefficient de diffusion (D) augmente lorsque la
température augmente , ce qui traduit par l'augmentation de la masse de
PA diffusé.
2/ Influence de pH :
L'alginate est un polymère anionique, qui libère
un proton en milieu basique. Le gonflement dans ce cas est
accéléré par la force de répulsion qui apparait au
fur et à mesure que le pH augmente.
N.B : Pour la libération
à pH=1,2, la quantité libérée est inversement
proportionnelle au pH, ce qui peut être justifié par la haute
solubilité de PA dans un milieu acide (Igor et Mattiasson,
2008).
3/ Influence de la force ionique :
Le Na+ provient de milieu entre dans une
compétition avec Ca+2 de l'alginate de calcium, d'où
resulte,à nouveau, la formation de l'alginate de sodium qui est soluble
dans l'eau, avec le temps le PA piégé se libére
(Xiaoling et Bhaskara, 2006).
4/ Influence de séchage :
Le séchage des sphères a pour but de voir
l'effet de ce dernier sur les chaînes de polymère, avec le
séchage, les chaînes se contractent progressivement, donc elles
adoptent une conformation condensée, l'influence sur la
libération est exprimée par une diminution si cette
libération est comparée avec le cas normal (sphères
humide) (Rubinstein et Colby, 2003).
Enfin cette étude nous a permis de synthétiser les
constatations suivantes :
* Pour une formulation d'encapsulation il est
nécessaire de prendre en compte tous les facteurs dépendant la
formulation et les conditions de conservation;
* La température de stockage influe sur la
préparation médicamenteuse élaborée par
modification du temps ou de la cinétique de libération du PA. Ce
même paramètre permet de donner les informations sur le
comportement cinétique au sein de l'organisme humain;
* le pH, autre paramètre d'étude tres important
de mettre en oeuvre, permet de connaître d'une part la stabilité
du PA et son cinétique de libération dans l'organisme et de
choisir la voie d'administration qui peut être orale ou
parentérale;
* le solvant de conservation, constitue un autre paramètre
d'étude, qui permet le choix du liquide dont la libération est
favorisée.
Conclusion générale
Conclusion générale
Conclusion générale
Cette étude a été effectuée en trois
étapes:
- la première a été consacrée
à un contrôle de la matière première, principe actif
: oxaciline sodique et l'acide alginique.
- la deuxième consistait à la mise d'encapsulation
de l'oxaciline sodique;
- et la troisième, permet d'étudier les
paramètres influençant la cinétique de libération
du PA.
En effet, nous pouvons admettre que la formulation
préparée est basée sur le principe de gélification
de goute, dont le polymère utilisé est un polymère naturel
hydrophobe modifiée par la soude c'est-à-dire l'utilisation
d'alginate de sodium, et par la présence de CaCl2, afin de modifier la
rigidité de la sphère.
D'après cette étude, nous avons constaté
d'une part, que la mise en point d'une microencapsulation par un tel
procédé permet d'obtenir des billes dont le PA est
dispersé dans toute la masse (type sphère et non capsule), dont
le rendement d'encapsulation a atteint 31,25 %. D'autre part, l'analyse des
cinétiques de libération mène à dire que les
conditions de préparation telles que le pH et la température
peuvent influencer d'une manière notable la quantité et le temps
de libération.
En ce qui concerne le pH, les valeurs 7 et 1.2 correspondent
à des valeurs maximales de la quantité libérée ;
La cinétique de libération est fortement
influencée par la température, nous concluons que la valeur
optimale de libération est 37°C, mais la valeur 25°C peut
constitue une contrainte de préparation, tandis que la valeur 4°C
est une température adéquate de conservation.
Un dernier paramètre d'étude est le solvant de
dissolution, dont nous pouvons constater que l'eau physiologique est le
meilleur solvant comparé par apport à l'eau distillée.
Cette différence est remarquable dans le cas des billes
séchées, et cela en terme de temps de libération et en
terme de quantité libérée.
Enfin, bien que cette étude nous a permet de
réaliser une méthode d'encapsulation d'un antibiotique et de
savoir les conditions de libération d'un médicament, il est
recommandé de mettre en formulation d'autres procédés et
d'appliquer une caractérisation des microsphères afin
d'élucider le phénomène de libération par d'autres
méthodes plus précises.
Références bibliographiques
Références bibliographiques
· Anonyme 1, Encyclopédie
médicale pratique, CD-ROM., 1996.
· Anonyme 2,
http://
www.pharmacologie.ubordeaux2.fr/Enseignement/documents/poly_
Pharmacologie_générale.
· Anonyme 3, Encyclopédie
Universalis , DVD-ROM ., 2007., version 12.
· Anonyme 4, Encyclopédie
encarta, CD-ROM ., 2007.
· Anonyme 5,
http://
www.md.ucl.ac.be/infect/antiinfectieux/PLS/Beta-lactames/PLS-Betalactameshome.html
· Anonyme 6, Vidal, CD-ROM.,
2006.
· Anonyme 7, United States
Pharmacopeia 24 - NF 19, CD-ROM., 2001, version 4,2.
· Anonyme 8,
http://
www.swri.org/index.html
· Anonyme 9,
http://
www.materiatech-carma.net/html/pdf/clubmat29_catalyse.pdf
· Anonyme 10, Encyclopédie
Hachette multimedia, CD-ROM., 2006.
· Anonyme 11, Méthodes
spectrométriques d'analyse et de caractérisation, Edition
Ecole des Mines de Saint-tienne, centre SPIN," Génie des
Procédés" P 09-11 et 15-28.
· AFECT ., 1992, Traite de chimie
thérapeutique : Médicaments antibiotiques, Techniques et
documentation Lavoisier, Volume 2, p 989-990.
· AIACHE J.M ., AIACHE S et RENOUX R .,
1995, Initiation à la connaissance des
médicaments, Masson, Paris ,2e édition, p.24.
· ANTHONY K.P., 2002, Pharmacology
Secrets, Hanley & Belfus, Philadelphia, 1ere
édition, p 05.
· ASSELINEAU J., ZALTA J.P., 1973, Les
antibiotiques structure et exemples de mode d'action, Hermann, Paris, P
1-6.
· BANKER G.S., RHODES C.T., 2002,
Modern Pharmaceutics, Marcel Dekker, NewYork , 4eme édition, P 7-8
et 10-11.
· BERGOGNE B ., DELLAMONICA P., 1995,
Antibiothérapie en pratique clinique, Masson, Paris, P
245-246.
· CHAMPE C.P., HARVEY A.R et MYCEK J.M.,
2000, Pharmacology, Lippincott Williams & Wilkins,
Philadelphia. 2eme édition, P 04-06.
· DOBREYA N ., 1981, Manuel de
formation vrac, Cheminvest, Bulgarie, Tome1, P 2-3 et 23.
· DUBOIS F., 2004, Elaboration et
caractérisations électro-optique et diélectrique de
composites à cristaux liquides ferroélectriques dispersés
dans une matrice polymère, Docteur en physique
(Spécialité : Sciences des Matériaux), Université
du littoral-cote d'opale, Laboratoire de lemcel #177; upres-ea à Calais,
P23-25.
· DUKES M.N.G., ARONSON J.K., 2000,
Meyler's Side Effects of drugs, Elsevier, Paris, 14eme
édition, P 7-8.
· DUVAL J., SOUSSY C.J., 1980,
Abrégé d'antibiothérapie bases bactériologiques
pour l'utilisation des antibiotiques, Masson, Paris, 2eme
édition, P 33-36.
· FRENEY J., RENAUD F., HANSEN W et BOLLET C.,
1994, Manuel de bactériologie clinique, Elsevier,
Paris. p 431.
· GOULD I.M., 1999, A review of the
role of antibiotic policies in the control of antibiotic resistance.
Antimicrob Chemother, 43: P 459.
· GUERY J., 2006, Emulsions doubles
cristallisables : stabilité, encapsulation et relargage, Docteur en
physique (Spécialité : Sciences des Matériaux),
Université du Paris VI, Laboratoire de Liquides Ioniques et Interfaces
Chargées à Paris, P38.
· HELALI A., 1994, Pharmacologie
fondamentale et clinique à l'usage des étudiants en
médecine, ENAG, Alger, P 13-56 et 146-147.
· IGOR G., MATTIASSON B.O., 2008,
Smart polymers Applications in Biotechnology and Biomedicine , CRC Press,
New york, 2eme édition, p 148-150 et 166.
· JANTZEN G.M., ROBINSON J.R., 2002,
Sustained and contolled release drug delivery systems, in Modern
Pharmaceutics, P 498-499 et 501-502, Marcel Dekker, New-York, 4eme
édition.
· JOLLIET P., FONTAINE M., HERLIN B et CHAMBRAUD
E., 2000, Pharmacologie nouveaux cahiers de l'infirmiqre,
Masson, Paris, p16-17.
· LAVERDIERE M., SABATH L.D., 1977,
Historical survey of tests to determine bacterial susceptibility to
antimicrobial agents. The Mount Sinaï Journal of Medicine.
44: P73.
· LECHAT P, 2007, Pharmacologie, Edition
CHU-PS, paris. P 65,75 et 76.
· LECLERE H., GAILLARD J.L et SIMONET M.,
1995, Microbiologie générale. La bactérie et
le monde bactérien, Doin, Paris, P 505 et 507.
· LULLMANN H ., MOHR K., ZIEGLER A., 1998,
Atlas de poche de pharmacologie, Flammarion, Paris, P 265.
· MAUR N., 1979, VADE-MECUM des
antibiotiques et agents chimiotérapeutiques antiinfectieux, Maloine
S. A, Paris ,4eme édition, p 7-25 et 99-105.
· MOULIN M., COQUREL A., 2002,
Pharmacologie connaissance et pratique, Masson, Paris, P11 et 37.
· MOUTON Y., BINGEN E., DEBOSCKER Y., DUBREUIL L.,
2000, Antibiotique antiviraux antiinfectieux, John
libey eurotext , France, P 41.
· ONESIPPE C ., 2005, Etude des
systèmes polyeléctrolyte /tensioactif en phase aqueuse et a
l'interface liquide / gaz application a l'élaboration de micro -
capsules, Docteur en Chimie théorique, physique et analytique
(Spécialité Sciences des Matériaux), Université
Montpellier II, Laboratoire des Agrégats Moléculaires et
Matériaux Inorganiques (LAMMI) à Paris, p 5.
· RICHARD J., BENOIT J.P., 2000,
Microencapsulation, 7E-FLEqDE-UGE- l'UKTE-DrEGRF, j 2210, P1-18.
· RUBINSTEIN M., COLBY R.H., 2003,
polymer physics, Oxford university press ,USA, p 13.
· SAINT-MAURICE C., DAILLAND P., LANDAIS A., LOISEL
B., RAGUENEAU J.L., TANDONNET F., 2004, Cours I.A.D.E,
Pharmacologie, Lamarre, Paris, p 09.
· SCHMITT H ., 1980, Eléments
de pharmacologie, Flammarion, Paris, 7eme édition,
p.19.
· TOUITOU Y., 1995,
Pharmacologie, Masson, Paris, 7eme édition, P 24-25 et
296.
· XIAOLING L.I., BHASKARA R.J., 2006,
Design of Controlled Release Drug Delivery Systems, McGRAW-HILL, New York,
P 115-120 , 146-149, 206,281 et 419.
· YANG K.Y., GRAFF L et CAUGHEY A.B.,
2004, Pharmacology, Blackwell Publishers, Massachusetts, p
02.
Annexes
ANNEXE I
Le tableau 01 rassemble quelques données sur la
sensibilité aux antibiotiques les plus usuels d'un certain nombre
d'espèces bactériennes à gram positif ou négatif
(Asselineau et Zalta, 1973).
Tableau (01): CMI des antibiotiques en g/ml
de milieu (Asselineau et Zalta, 1973).
|
Bactérie à Gram positif
|
Bactérie à Gram négatif
|
|
Streptococcus
pyogènes
|
Pseudoeonas
Pyocyaneae
|
Salmonella sp.
|
Pénicilline
|
0.012
|
0.006
|
5-20
|
>32
|
Erythromycine
|
0.12
|
0.03
|
-
|
-
|
Novobiocine
|
0.12
|
2
|
-
|
-
|
bacitracine
|
2
|
0.25
|
-
|
-
|
polymyxine
|
-
|
-
|
0.12
|
0.12
|
|
Tableau (02) : Grandes familles des
antibiotiques (Maur, 1979).
Bêtalactaminès
|
Macrolides
et apparentés
(lincosanides)
|
Tétracyclines
|
Synergistines
|
Antibiotiques
à structure
d'acides
aminés
|
Pénicillines Céphalosporines Mecillinam-
Pivmécillinam Acide
clavulanique
|
Erythromycine Oléandomycine Spiramycine Kitasamycine
Lincornycine - clindamycine Rosamyclne Midecamycine
|
Tétracycline Chlorotétracycline
Déméthylchlortétracycline Oxytetracycline Methacycline
Doxycycline Minocycline
|
Viriginiarnycine Pristinamycine
|
Thiamphenicol Chloramphénicol
|
Polypeptides
surfactifs
|
Aminoglycosides
|
|
IIeme
sous-groupe
|
IIIeme
sous-groupe
|
IVeme
sous-groupe
|
Polymyxine- colisfine
1
|
Streptomycine Dihydrostrept- omycine
1
|
Neomycine Framycetine
|
Kanamycine Paromomycine Lividomycine Amicacine Bekenamycine
|
Gentamicine Tobramycine Sisomicine Ribostamycine Vistamycine
|
|
Tableau (03) : Principaux substituant des
chaînes latérales des pénicillines (Maur,
1979).
Nom générique
|
Groupe R
|
Benzylpénicilline (pénicilline G )
|
|
|
Phénoxyméthyl pénicilline
(pénicilline V)
|
|
|
Phénéthicilline
|
|
|
Propicilline
|
|
|
Oxacilline
|
|
|
Ancillin
|
|
|
|
|
|
Tableau (04) : Classement des
pénicillines selon différents critères (Maur,
1979).
a) Selon leur
origine
|
· Pénicillines (naturelles) obtenues par la
seule fermentation (pénicilline G et V);
· Pénicillines de semi-synthèse par
production « dirigée », grâce à des
précurseurs ajoutés au milieu de fermentation: toutes les
autres.
|
b) Selon leur
absorption ou leur
non
absorption
digestive
(administration
orale
ou
parentérale)
|
· Pénicilline d'administration exclusivement
parentérale : pénicilline G,
méthicilline,... etc.
· Pénicilline d'administration exclusivement orale :
-Phénoxyalkylpénicilline: pénicilline V,
clométocilline,
phénéthicilline...etc
- Azidocilline #177; pivampicildine - amoxycilline.
· Pénicilline d'administration orale et
parenterale:
- Isoxazolylpenicillines: Oxacilline, cloxacilline,
dicloxacilline,
flucloxacilline
-Ampicilline et certain de ses dérivés :
ampénicilline, hétacilline,
épicilline.
|
c) Selon qu'elles
sont résistantes ou
non
à la
dégradation par
les
pénicillinases
bactériennes
|
· Résistantes : Méthicilline, isoxazolyl
pénicillines, pyrazocilline;
· Non résistantes : Pénicillines G et V,
clométocilline, phénéthicilline, propicilline,
azidocilline, ampicilline et dérivés, carbénicilline et
dérivés.
|
|
|
d) Selon leur spectre d'action,
|
1) Pénicillines G, ayant le spectre moyen comprenant
surtout les germes GRAM+ et les cocci GRAM-,
mais a des dose élevées ayant encore une certaine activité
sur certains germes CRAM- (surtout par concentration, en milieu
urinaire) ;
2) Pénicillines orales non résistantes aux
pénicillinases bactériennes, de spectre moyen, mais ayant perdu
une bonne partie de leur action sur les germes GRAM- :
phénoxyalkylpénicillines, Clométocilline;
3) Pénicillines à large spectre d'action,
incluant, en plus les bactéries GRAM-, les bacilles
GRAM- et dont les représentants sont l'ampicilline et ses
dérivés et les céphalosporines ;
4) Pénicillines a très large spectre incluant en
plus le pseudomonas et les proteus indol+ :
sulfoxypenacillines, ureidopenicillines...etc.
5) Pénicillines "antistaphylocoiques",
résistant aux pénicillines
bactériennes, ayant perdu
complètement toute action sur les germes GRAM- et
orientées principalement vers les staphylocoques producteur de
pénicillinases, bien que leur spectre d'action inclue d'autre
cocci GRAM+, mais ayant sur ceux-ci une activité
inferieure a celle de la pénicilline G et V:Lafcilline,
ancilline....etc.
|
Tableau (05) : Principaux matériaux
enrobants utilisés dans les procédés physico-chimiques et
mécaniques de microencapsulation (Richard et Benoit,
2000).
Principaux matériaux
enrobants
Procédés de mise In oeXYEI
Exemples de domaines
d'application
Polymères d'origine naturelle
|
Gélatine
|
|
Alginate de sodium
|
|
Coacervation complexe Coacervation simple
|
|
Coacervation complexe Prilling
|
-Arômes
-Parfums
-Pharmacie
-Autocopiants
|
-Biomédical: encapsulation de cellules
-Arômes
-Cosmétique
-Parfums
-Phytosanitaire
|
|
Chitosane
|
|
Amidon
|
|
Coacervation complexe Prilling
Spray-drying
Spray-coating
|
|
Spray-drying
|
|
-Pharmacie
-Administration orale -Libération gastrique -Masquage
de goût
|
-Alimentaire : encapsulation d'arômes, d'huiles
essentielles ou aromatiques, de vitamines et d'épices
Polymères cellulosiques
|
Ethylcellulose (EC) Hydroxypropylcellulose (HPC)
|
Hydroxypropylméthylcellulose (H PMC)
|
Esters de cellulose entérosolubles
|
Phtalate d'hydroxypropylmé- thylcellulose
|
Coacervation simple
Spray-coating
Spray-drying
Évaporation - Extraction de solvant
|
|
-Pharmacie
-Masquage de goût -Administation orale -Libération
prolongée ou déclenchée (entérique)
|
Polymères synthétiques
|
Copolymères acryliques et méthacryliques
|
|
Spray-drying
Spray-coating
Évaporation - Extraction de solvant
|
|
-Pharmacie
-Administration orale -Libération gastrique
-Libération entérique -Libération prolongée
-Masquage de goût
|
|
Polyoléfines
|
(Co-) extrusion #177; sphéronisation
Évaporation de solvant
|
-Chimie
-Biomédical
-Phytosanitaire
-Libération prolongée ou déclenchée
par élévation de température
|
|
Copolymères (acrylo-) vinyliques
|
|
Homopolymères et copolymères d'acide
lactique et d'acide glycolique (PLA, PLGA)
|
Coacervation simple Spray-drying
Évaporation - Extraction de solvant
|
-Pharmacie
-Libération prolongée -Administration
parentérale de peptides
|
|
Lipides et cires minérales
|
|
Corps gras solides (esters, alcools, acides gras)
|
Prilling
Solidification d'émulsion (hot melt)
Spray-coating
Procédé de coacervation en milieu supercritique
|
-Cosmétique -Chimie
-Pharmacie
-Libération prolongée ou -
déclenchée
par élévation de température -Stabilisation
de matières actives
-Masquage de goût
|
|
Glycérides
|
|
Cires (d'abeilles, de Carnauba, de Candelilla ...)
|
|
Cires minérales
|
Tableau (06) : Monomères
utilisés dans les procédés chimiques de microencapsulation
et matériaux polymère formé (Richard
et Benoit, 2000).
|
Monomères
|
Matériau polymère
formé
|
Procédés de mise
Rn oeX\1R
|
Domaines
d'application
|
|
1 ,6-Hexaméthylènediamine /
Chlorure de
sébacoyle
|
Polyamide nylon 6-10
|
Polycondensation
interfaciale
|
Phytosanitaire
Biomédical
|
|
L-Lysine/Chlorure de téréphtaloyle
|
Polyamide
|
Polycondensation
interfaciale
|
Biomédical
|
|
Pipérazine/Chlorure de
téréphtaloyle
|
Poly(téréphtaloylpi-
pérazine)
|
Polycondensation
interfaciale
|
Biomédical
|
|
1 ,6-Hexaméthylènediami-
ne/Chlorure de
téréphtaloyle
|
Poly(téréphtalamide)
|
Polycondensation
interfaciale
|
Biomédical
|
|
2,2-bis (4-Hydroxyphényl)-
propane/Chlorure de
sébacoyle
|
Polyester
|
Polycondensation
interfaciale
|
Phytosanitaire
|
|
Méthylènediisocyanate
|
Polyurée
|
Polycondensation
interfaciale
|
Biomédical
|
|
1
,6-
Hexaméthylènediamine/Méthylène-
diisocyanate
|
Polyurée
|
Polycondensation
interfaciale
|
Vétérinaire
|
|
Cyanoacrylate de butyle
|
Poly(cyanoacrylate de
butyle)
|
Polymérisation
anionique
en émulsion
|
Pharmaceutique
|
|
Cyanoacrylate d'hexyle
|
Poly(cyanoacrylate
d'hexyle)
|
Polymérisation
anionique
en émulsion
|
Pharmaceutique
|
|
Méthacrylate de méthyle (MAM)
|
Poly(méthacrylate de
méthyle)
|
Polymérisation
radicalaire
en émulsion
|
Cosmétique
|
|
Acrylate de butyle (ABu)/MAM
|
Poly(acrylate de
butyle-co-méthacrylate
de
méthyle)
|
Polymérisation
radicalaire
en émulsion
|
Cosmétique
|
ANNEXE II
Technique de référence pour la
détermination de la CM
1. Méthodes de dilution :
> en milieu liquide :
On prépare une série d'une dizaine de tubes
à hémolyse dans lesquels on répartit une même
quantité de bouillon nutritif ensemencé (ce qui ne se traduit par
aucune opacité appréciable à l'oeil).
On distribue ensuite dans chaque tube (Fig.01), à
l'exception du premier qui servira de témoin, des quantités
croissantes de l'antibiotique étudié, réalisant une gamme
de concentrations en progression géométrique de raison 2
(Duval et Soussy, 1980).
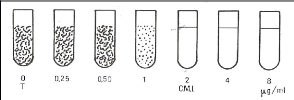
Figure (01) : Méthode de dilution
en milieu liquide (Duval et Soussy, 1980).
Les tubes sont placés à l'étuve à
37 °C pendant 24 h, après quoi on les observe macroscopiquement,
Dans un certain nombre de tubes, la culture s'est développée, un
trouble nettement visible en est la traduction. A partir d'une certaine
concentration, il n'y a plus de culture visible. Le premier tube dans lequel il
n'y a plus de culture visible indique la concentration minimum inhibitrice,
encore appelée taux inhibiteur ou taux de sensibilité de la
souche étudiée.
en milieu solide :
la méthode de dilution en gélose consiste a
incorporer l'antibiotique dans de la gélose coulée en boites de
pétri, réalisant comme précédemment une gamme de
concentrations croissantes. La souche est ensemencée en une strie
à la surface de chaque boite (fig.02) (Duval et Soussy,
1980).
Figure (02) : Méthode de dilution en
milieu solide (Duval et Soussy, 1980).
2. Méthode de diffusion en gélose
(méthode de disque) :
Elle consiste a déposer a la surface de la
gélose d'une boîte de Pétri des disques de papier buvard
imprégnés des différents antibiotiques testés.
Chaque antibiotique diffuse au sein de la gélose a partir du disque et y
détermine des concentrations inversement proportionnelles a la distance
du disque. Si. Avant de déposer les disques, on a ensemence
uniformément la surface de la gélose avec le germe à
étudier, les disques apparaissent. Après 24 heures
d'étuve, entourés d'une zone d'inhibition dont le diamètre
permet de mesurer la CMI, (il est évident que la culture s'arrête
là oil, dans la gélose, existe une concentration d'antibiotique
égale à la CMI) (fig.03) (Duval et Soussy,
1980).
Figure (03) : Méthode de diffusion en
gélose (Anonyme 10).
Pour mesurer cette dernière. Il suffit d'avoir au
préalable étalonné le système disquesmilieu de
culture avec un grand nombre de souches de CMI connues
déterminées par les méthodes précédentes :
On mesure le diamètre de la zone d'inhibition obtenue pour chacune de
ces souches; les chiffres obtenus permettent de tracer la droite de
régression ou courbe de concordance ». Donnant la correspondance
entre les CMI et les diamètres des zones d'inhibition pour les disques
et le milieu de culture considérés (fig.04). Pour
déterminer la CMI d'une souche x, il suffit de mesurer le
diamètre de sa zone d inhibition. Puis de reporter ce chiffre sur le
graphique: on en déduit la CMI (Duval et Soussy,
1980).
Diamètre de
la zone
01iQhiEitiRQ
CMI en ug/ml
Figure (04) : Exemple de courbe de
concordance ; relation diamètre des zones
d'inhibition et CMI
(Duval et Soussy, 1980).
3. Méthode turbidimétrique :
Consiste à mesurer l'opacité d'un tube où
se trouve un milieu renfermant des germes et l'antibiotique à tester.
Plus l'opacité est grande, plus l'activité de l'antibiotique est
faible (Touitou, 1995).
ANNEXE III
Spectrophotométrie d'absorption dans
l'ultraviolet et le visible
1. Principe :
La spectrométrie d'absorption moléculaire dans
le domaine ultraviolet (UV), de 185 à 380 nm environ, et visible (VIS),
de 380 à 800 nm environ, est une technique courante de contrôle et
d'analyse de composés chimiques. Elle s'applique à des
groupements d'atomes (ex : molécules, ions, polymères) qui
absorbent le rayonnement électromagnétique dans le domaine
UV-VIS.
L'absorption de la lumière UV-VIS par les
molécules se produit, comme pour les atomes, du fait de transitions
électroniques entre différents niveaux d'énergie. Un
électron à l'état fondamental absorbe des radiations d'une
énergie E suffisante pour l'élever à un niveau
d'énergie supérieur, l'état excité, (ce qui
détermine la longueur d'onde l, par la relation :
E = h c / l, oil « h » est la constante de Planck et
« c » la vitesse de la lumière). Le retour au plus bas niveau
d'énergie, l'état fondamental, se produit par perte
d'énergie sous forme de chaleur ou, occasionnellement, par
rémission de radiation (fluorescence ou phosphorescence).
Absorbance Absorbance
tat fondamentale
état fondamentale / xcitation
excitation
/ EE
Figure (05) : Spectre d'une
seule
transition (Anonyme 12).
Figure (06) : Spectre d'absorptio
Figure (06) : Spectre
dabsortion UV-VIS.
UV-VIS(Anonyme 12).
Absorbance Absorbance
tat excité état excité
tat fondamentale état fondamentale
1 / E
/ E
iure (07) : Spectre de plusieurs
Figure (07) : Spectre de sieurs transitions
[45] transitions (Anonyme
12).
Figure (08) : Spectr résultant de
Figure (08) : Spectre
résulta
plusieurs transitions (Anonyme 12).
de plusieurs transitions [45]
tat excité état excité
S'il n'y avait qu'un seul (fig.05), le spectre d'absorption
UV-VIS (fig.06) n'aurait qu'une seule raie à la longueur d'onde
correspondant à l'énergie nécessaire à la
transition. Dans ce cas idéal, la spectrométrie UV-VIS serait un
outil immédiat d'analyse qualitative : la longueur d'onde exacte
d'absorption serait parfaitement caractéristique de la molécule,
Cependant, de nombreux autres niveaux d'énergie (dus à des
vibrations, des rotations et des transitions moléculaires) se
superposent aux niveaux d'énergie électroniques, et plusieurs
transitions sont possibles (fig.07). Le spectre résultant prend alors la
forme d'une bande large sans caractéristique très marquée
(fig.08) (Anonyme 12).
Ce spectre permet à la fois l'identification (analyse
qualitative), mais dans certaines limites seulement et l'estimation
(analyse quantitative) d'un composé.
Les chromophores qui peuvent être détectés
par des spectromètres UV-VIS comprennent toujours des liaisons doubles
entre atomes de carbone (C=C) ou des liaisons doubles ou triples entre le
carbone et certains autres atomes (C=O, CN notamment).Ils peuvent être
conjugués à d'autres groupes fonctionnels pour former des
complexes chromophores. Un exemple typique est le benzène
(Anonyme 12).
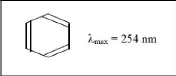
Figure (09) : molécule de
benzène (Anonyme 12).
Ainsi, la spectrométrie d'absorption moléculaire
UV-VIS permet d'étudier, à l'état gazeux, liquide ou
solide, des composés organiques principalement mais aussi des
composés inorganiques (Anonyme 12).
2. Analyse quantitative :
L'analyse quantitative par la spectrométrie UV-VIS est
très utilisée (beaucoup plus que l'analyse qualitative) car
l'absorption est plus ou moins importante selon le nombre de groupements
d'atomes placés sur le trajet de la lumière : des lois connues
relient cette absorption à ce nombre dans certaines conditions
opératoires. Ce sont les lois de LAMBERT et de BEER
(Anonyme 12).
· Loi de BEER- LAMBERT :
L'absorption de la lumière est directement proportionnelle
à la fois à la concentration du milieu absorbant et à
l'épaisseur de la cuve où se trouve le milieu.
Une combinaison de ces deux lois (la loi de BEER-LAMBERT) donne
la relation entre l'absorbance (A) et la transmittance (T) :
A = log (I0 / I) = log (100/ T) = å c x
Avec :
A = absorbance (sans unité).
I0= Intensité de la lumière incidente.
I = Intensité de la lumière transmise (I toujours
inférieure à I0).
T = Transmittance.
å coefficient d'absorption molaire ou d'extinction
(mol-1. dm3.cm-1).
c = concentration molaire (mol. dm-3).
x = longueur de la cuve (cm) ou trajet lumineux.
La relation entre transmittance et concentration n'est pas
linéaire (fig.10) mais la relation entre l'absorbance et la
concentration est linéaire (fig.11), ce qui est à la base de la
plupart des analyses quantitatives (Anonyme 12).
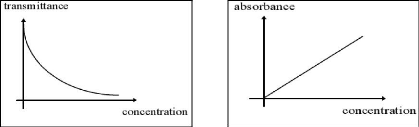
igure (10) : Relation entre
transmittance Figure (11) : Relation entre
l'absorbance.
et concentration (Anonyme 12). et
la concentration (Anonyme 12).
La simple relation linéaire entre l'absorbance et la
concentration et la facilité relative de mesure de la
lumière UV-VIS sont donc les raisons pour lesquelles la spectroscopie
UV-VIS est jà la base d'un grand nombre de méthodes d'analyse
quantitative.
N.B : A température ambiante, il
est donc hautement probable que toutes les molécules soient à
l'état électronique fondamental.
· Limites de la loi de
BEER-LAMBERT:
- Les mesures ne sont valables que dans une gamme de
concentrations très faible, et l'absorbance ne dépasse pas 3.
- Il est important de noter que å est une
fonction de la longueur d'onde et donc que la loi est seulement vraie en
lumière monochromatique, c'est une limite lié à
l'appareil (Anonyme 12).
3/Appareillage :
Les spectromètres classiques comprennent les
éléments suivants : (fig.12)
Une source, un porte-échantillons, un monochromateur, un
détecteur, un appareil de lecture (Anonyme 12).
Source Monochromateur Cellule Détecteur Processeur
Figure (12) : Principaux constituants d'un
spectrophotomètre (Anonyme 12).
> La source :
Pour la plupart des spectrophotomètres UV-visible, on
utilise principalement deux types de lampes afin de couvrir la totalité
du spectre. Ainsi, pour la partie UV du spectre, on emploie des lampes au
deutérium ou parfois au xénon à haute pression. Les lampes
au deutérium émettent en effet un rayonnement dont les longueurs
d'onde sont comprises approximativement entre 180 et 400 nm, et ont une
durée de vie d'environ 1000 h, En ce qui concerne la partie
visible du spectre, les plus utilisees sont les lampes
halogène au quartz à filaments de tungstène dont le
rayonnement, continu, est compris entre 350 et 1300 nm.
> Monochromateurs et polychromateurs
:
La fonction du monochromateur est de selectionner une
longueur d'onde parmi le spectre du rayon incident. Les plus simples sont
composes de filtres ne laissant passer qu'une seule longueur d'onde; il est
possible d'en utiliser plusieurs afin d'obtenir differentes longueurs d'onde.
Les monochromateurs les plus utilises sont composes en general d'une fente
d'entree, d'un dispositif de dispersion comme un prisme ou un reseau, et d'une
fente de sortie. L'echantillon et le detecteur, places juste derrière le
monochromateur.
> Les détecteurs :
Il existe deux types principaux de detecteurs: le detecteur
unicanal et le detecteur multicanal. En fait, le premier convient à un
monochromateur alors que le second est utilise avec un polychromateur.
> Le recueil du signal (lecture)
:
Le detecteur est relie grâce à un convertisseur,
à un microprocesseur, qui recueille toute la serie de mesures
(Anonyme 12).
4/ Méthodologie :
Le blanc utilisé doit contenir la solution à
laquelle l'élément étudié a été
retiré.
Dans un premier temps, on cherche la longueur d'onde
d'absorption maximale de cet élément, en traçant son
spectre d'absorption (A en fonction de ë) : ë max est
determinee.
Ensuite, à ë max (pour n'effectuer
qu'une seule mesure tout en évitant au mieux les lumières
parasites), on trace la courbe d'étalonnage de l'élément.
Pour cela, on prépare environ 5 échantillons à des
concentrations connues et on mesure à chaque fois l'absorbance A. Parmi
ces echantillons, on prepare un echantillon qui donne A = 0 (solvant seul sans
l'élément étudié) et un autre qui donne A
= 100% (élément seul). Cette courbe
d'étalonnage permet de calculer le coefficient d'extinction å
défini précédemment. Alors, les concentrations inconnues
de l'élément peuvent être determinees, à ë
max en reportant sur la courbe les absorbances mesurees (Anonyme
12).
5/ Qualité du spectre :
La qualité du spectre obtenu par utilisation d'un
instrument est une fonction de la monochromaticite du rayon incident. Celle-ci
depend de:
> La largeur des fentes du monochromateur :
Lorsque la largeur des fentes diminue, la forme de la courbe spectrale se
modifie jusqu'à ce qu'une image plus ou moins nette apparaisse.
Cependant, pour des fentes très étroites, une faible
quantité d'énergie atteint le detecteur, et du bruit peut alterer
la qualite spectrale.
> la quantité de lumière parasite :
Le spectre peut egalement être modifie par le rayon émergeant du
monochromateur qui possède une lumière assez différente de
celle que l'on devrait obtenir theoriquement. On appelle cette lumière
la lumière parasite, due aux reflexions involontaires dispersives dans
le monochromateur. Si l'instrument est réglé à une
longueur d'onde correspondant à un maximum d'absorption connu
(ë max) pour un echantillon, la lumière
parasite sera en général moins absorbée max par
l'échantillon que celle à d'autres longueurs d'onde
(Anonyme 12).
6/ Exactitude et précision des
résultats :
L'exactitude détermine la façon dont s'approche
la valeur spectroscopique mesurée (A ou ë) de la vraie valeur. La
précision des résultats servant à obtenir la valeur
moyenne (qui correspond à la meilleure estimation du résultat
réel que les expériences ont trouvé) est donnée par
leur dispersion (Anonyme 12).
"Vrai" Valeur Valeur moyenne, A

Exactitude
Dispersion
A
Figure (13) : conceptions d'exactitude et de
dispersion (Anonyme 12).
On trouve la valeur moyenne A pour l'ensemble des
résultats A1, A2, ..., A n grâce à la formule :
(Anonyme 12).
A = A1 + A2 + ...A n / n
ANNEXE IV

A- VERRERIES ET CONSOMMABLES
- Béchers en verre ; - Boites de Pétri ;
- Burette ;
- Cuvette en Quartz ;
- Entonnoir en verre et en plastique ;
- Eprouvette en verre graduée ;
- Fioles jaugées en verre ; - Papiers filtre ;
- Papier hygiénique ;
- Papier réactif de pH ; - Papier tournesol ;
- Poire ;
- Portoirs ;
- Pipettes en verre gradué ;
- Pipette pasteur ;
- Récipients en verre ; - Seringue à aguille ; -
Spatule ;
- Tubes à essai ;
B- REACTIFS CHIMIQUES UTILISES
- 1,3-dihydroxynaphtalène ; - Acétate d'LP P RniIXP
;
- Acétate de calcium ;
- Acétate de plomb ;
- Acide acétique ;
- Acide nitrique ;
- Acide sulfurique ;
- Chlorure stanneux ;
- Carbonate de potassium ; - Chlorure de calcium ;
- Diéthyldithiocarbamate d'argent ; - Éther
isopropylique ;
- Glycérine ;
- Hydroxyde de potassium ; - Isopropyl alcool ;
- Iodure de potassium ;
- Nitrate de plomb ;
- Peroxyde d'hydrogène à 30 % ; -
Phénolphtaléine ;
- Phosphate de potassium monobasique (KH2PO4) ;
- Pyroantimonate de potassium ;
;
- Thioacetamide ;
- Trioxyde d'arsenic
- Trioxyde sulfurique ;
- Zinc granulaire.
C- PREPARATION DES SOLUTION
Acide Chlorhydrique HCl :
Contient au minimum 36,5 pour cent m/m et au maximum 38,0 pour
cent m/m d' HCl.
Acide nitrique HNO3 (Mr 63,0) :
Contient au minimum 69,0 pour cent m/m et au maximum 71,0 pour
cent m/m de HNO3.
Acide sulfurique H2SO4 (Mr 98,1) :
Contient au minimum 95,0 pour cent m/m et au maximum 98,0 pour
cent m/m de H2SO4.
Carbonate de potassium K2CO3 (Mr 138.21)
:
Carbonate de potassium contient pas plus de 99.5 % et pas moins
de 100.5 % de K2CO3.
Ethanol 96° C2H5OH (Mr 46,07) :
Contient au minimum 92.3 % et au maximum 93.8 %, de poids, qui
correspondent au minimum 94.9 % et au maximum 96.0 % de volume à
15.56°C de C2H5OH.
Solution d'acide de chlorure stanneux concentré
:
Dissoudre 40 g du chlorure stanneux dans 100 ml d'acide
chlorhydrique. Entreposer dans des récipients en verre, et l'utiliser
pendant 3 mois.
Solution de Chlorure de Calcium CaCl2 :
Dissoudre 7.5 g de chlorure de calcium dans 100 ml d'eau
distillé
Solution de diéthyldithiocarbamate d'argent
:
Dissoudre 1 g de diéthyldithiocarbamate d'argent dans
200 ml de pyridine d'une bouteille fraîchement ouverte ou qui a
été récemment distillées. Entreposer dans des
récipients résistant à la lumière, et l'utiliser
pendant 30 jours.
F
Solution d'iodure de potassium :
Dissoudre 16.5 g d'iodure de potassium dans l'eau pour faire 100
ml. Entreposer dans des récipients résistant à la
lumière.
F
Solution de nitrate de plomb :
Dissoudre 159.8 mg du nitrate de plomb dans 100 ml de l'eau
auxquels a été ajouté 1 ml d'acide nitrique, puis diluer
avec de l'eau à 1000 ml. Préparer et conserver cette solution
dans des récipients en verre dépourvues des sels solubles de
plomb.
Solution de pyroantimoniate de potassium :
Dissoudre 2 g de pyroantimonate de potassium dans 95 ml d'eau
chaud. Refroidir rapidement, et ajouter une solution contenant 2.5 g
d'hydroxyde de potassium dans 50 ml d'eau distillé et 1 ml de solution
d'hydroxyde de sodium (8.5 dans 100). Laisser reposer pendant 24 h, filtrer, et
diluer avec l'eau distillé à 150 ml.
Solution de phénolphtaléine :
Dissoudre 1 g de phénolphtaléine dans 100 ml
d'éthanol 96°.
Solution Standard d'Arsenic :
Transférer 10.0 ml de solution de trioxyde d'arsenic
dans une fiole jaugé de 1000 ml, ajouter 10 ml d'acide sulfurique (2N),
puis ajuster jusqu'au trait de jauge avec de l'eau distillé
récemment bouillie et refroidie, et mélanger. Chaque ml de
solution standard d'arsenic contient l'équivalent de 1ug d'arsenic.
Conserver cette solution dans des récipients en verre, et l'utiliser
dans une période ne dépassant pas 3 jours.
Solution standard de plomb :
Au jour de l'utilisation, diluer 10.0 ml de solution de nitrate
de plomb avec de l'eau à 100 ml. Chaque 1 ml de solution standard de
plomb contient l'équivalent du 10 ug de plomb.
Solution tampon d'acétate d'ammonium de pH 3,5
:
Dissoudre 25,0 g d'acétate d'ammonium dans 25 ml d'eau
distillé. Et ajoutez 38,0 ml d'acide chlorhydrique (6N). Ajustez le pH,
si nécessaire, avec de l'acide chlorhydrique (6N) ou de hydroxyde
d'ammonium (6N) jusqu'au pH 3,5 et complétez à 100,0 ml avec de
l'eau, et agiter.
Solution de Thioacetamide :
Dissoudre 4 g de thioacetamide dans 100 ml d'eau.
Solution de thioacetamide-glycérine :
Mélanger 0.2 ml de solution de thioacétamide et 1
ml de glycérine, Chauffez dans un bain-marie pendant 20 secondes.
Utiliser le mélange immédiatement.
Solution de trioxyde d'arsenic(As2O3) :
Dissoudre 132.0 mg de trioxyde d'arsenic - exactement
pesé et séché à 105°C pendant 1 h dans 5 ml de
solution d'hydroxyde de sodium (1/5) dans une fiole jaugé de 1000 ml.
Neutraliser la solution avec de l'acide sulfurique (2 N), ajouter en plus 10 ml
d'acide sulfurique (2N), puis ajuster jusqu'au trait de jauge avec de l'eau
distillé récemment bouillie et refroidie, et mélanger
(Anonyme 7).
lE l
1. Préparation des solutions STD et ECH pour
le test d'arsenic :
> Préparation de solution STD
:
Pipeter 3.0 ml de solution standard d'arsenic dans une fiole
jaugé, ajouter 2 ml d'acide sulfurique, mélanger, et ajouter la
mrme quantité de peroxyde d'hydrogène à 30 %,
utilisé dans la préparation à examiner. Chauffer le
mélange sous flamme, refroidir, ajouter avec précaution 10 ml
d'eau distillé, réchauffer sous flamme. Répéter
cette procédure avec autre 10 ml d'eau pour éliminer toutes
traces de peroxyde d'hydrogène. Refroidir, et diluer avec l'eau
distillé jusqu'à 35 ml.
> Préparation de solution ECH
:
Utiliser une quantité, en g, de substance à
examiner comme prévu par la formule :
3.0 / L
Où: L est la limite d'arsenic en ppm.
Transférer dans une fiole conique (1g) de substance
à tester, Ajouter 5 ml d'acide sulfurique, lchauffer de
préférence sur une plaque chauffante à température
ne dépassant pas 120°C, jusqu'à ce que la carbonisation
commence. (Ajouter si nécessaire l'acide sulfurique pour humidifier
l'échantillon, le volume ajouté ne dépasse pas 10 ml).
Avec précaution, ajouter, goutte à goutte, le peroxyde
d'hydrogène à 30 %, laisser la réaction se reposer, et
encore faire chauffer entre les gouttes. Les premières gouttes sont
ajoutées lentement, avec agitation, pour éviter une
réaction rapide.
Arrester le chauffage si les écumes sont successivement
formées. Quand la réaction est éteinte, chauffer avec
précaution, faire l'agitation de temps en temps pour éviter la
fixation de l'échantillon sur les parois de fiole exposé à
la chaleur. Ajouter une quantité de solutionlde
peroxyde d'hydrogène à 30 % ljusqu'à ce
que lle mélange devient brun ou sombre. Continue la
digestion jusqu'à ce que la matière organique est
complètement détruite, augmenter la température
graduellement jusqu'à ce que les vapeurs de trioxyde sulfurique
lsont émit copieusement, et la solution dévient
incolore. Refroidir, ajouter avec précaution10 ml d'eau distillé,
mélanger, réchauffer jusqu'à l'évaporation,
Répéter cette procédure pour éliminer toutes traces
de peroxyde d'hydrogène. Refroidir, ajouter avec précaution10 ml
d'eau distillé, rincer les parois de la fiole conique avec quelques ml
d'eau distillé, et diluer avec l'eau distillé jusqu'à 35
ml (AEMEEf l7). l
171117fl714): Montage pour l'essai limite
de l'arsenic (EEEEMfl7).l
2. Préparation des solutions STD et ECH pour
le test des métaux lourds :
> Préparation de la solution STD
:
Dans un tube à essais de 50 ml, pipeter 4 ml de la
solution standard de plomb (40 ug de Pb), et diluer avec l'eau distillé
jusqu'à 25 ml. Ajuster avec l'acide acétique (1N) ou d'hydroxyde
d'ammonium (6N) jusqu'à un pH entre 3.0 et 4.0, en utilisant le papier
réactif de pH en tant qu'indicateur externe, dilué avec de l'eau
distillé jusqu'à 40 ml, et agiter.
> Préparation de la solution ECH
:
Utiliser une quantité, en g, de substance à
examiner comme prévu par la formule :
2.0/ (1000L)
Oil : L est la limite de métaux lourds, en pourcentage.
Transférer la quantité pesée (0.5 g) de
la substance dans un creuset de platine, ajouter suffisamment l'acide nitrique
pour mouiller la substance, et enflammer soigneusement à une basse
température jusqu'à ce que la carbonisation complète.
Ajouter à la masse carbonisée 2 ml d'acide nitrique et 5 gouttes
d'acide sulfurique, et les chauffer prudemment jusqu'à ce que les
vapeurs blanches ne soient plus émit. Enflammer, de
préférence dans un four à moufle, de 500°C à
600°C, jusqu'à ce que le carbone soit complètement
incinéré. Refroidir, ajouter 4 ml d'acide chlorhydrique (6 N),
les couvrir, digérer dans un bain de vapeur pendant 15 minutes,
découvrir, et laisser vaporiser lentement dans le même bain
à sec. Mouiller le résidu par 1 goutte d'acide chlorhydrique,
ajouter 10 ml d'eau distillé chaude, et digester pendant 2 minutes.
Ajouter l'hydroxyde d'ammonium (6 N) goutte-à-goutte, jusqu'à ce
que la solution soit toute alcaline ~ contrôler l'alcalinité par
le changement de couleur de papier tournesol #177; diluer avec de l'eau
distillé jusqu'à 25 ml, et ajuster avec l' acide acétique
(1N) jusqu'à un pH entre 3.0 et 4.0, en utilisant le papier
réactif de pH en tant qu'indicateur externe. Filtrer si
nécessaire, rincer le creuset et le filtre avec 10 ml de l'eau
distillé, combiner le filtrat et le rinçage dans un tube à
essais de 50 ml, dilué avec de l'eau distillé jusqu'à 40
ml, et mélanger (UUMUU UOD).O
Annexes
Annexe VI
Tableaux de masses cumulatives utilisés dans le
traçage des graphes
de libération du PA
Tableau(07) : Masse cumulative du PA dans
le milieu en mg (forme humide GDQWKILLWIP
pH =7
pH = 3
pH = 1,2
T= 4°c
T= 25°c
T= 37°c
T= 4°c
T= 25°c
T= 37°c
T= 4°c
T= 25°c
T= 37°c
0,05571508
0,16859576
0,22659271
0,05142857
0,13900135
0,19703441
0,058527
0,17710474
0,23802878
1
0,06875477
0,20805435
0,27962504
0,063
0,17027665
0,24136715
0,07286014
0,22047734
0,29632154
2
0,08179447
0,24751293
0,33265738
0,07585714
0,20502699
0,29062575
0,08599886
0,26023554
0,34975657
22
0,08772161
0,26544865
0,35676299
0,081
0,21892712
0,31032919
0,091971
0,27830746
0,37404522
23
0,09246332
0,27979723
0,37604747
0,08614286
0,23282725
0,33003263
0,09794314
0,29637937
0,39833387
24
0,10668844
0,32284295
0,43390093
0,099
0,26757759
0,37929124
0,11347071
0,34336634
0,46148436
46
0,11024472
0,33360438
0,44836429
0,10285714
0,27800269
0,39406882
0,117054
0,35420949
0,47605755
47
0,11380101
0,34436582
0,46282766
0,10542857
0,28495276
0,40392054
0,12063729
0,36505264
0,49063074
48
0,12328442
0,37306297
0,50139663
0,11442857
0,30927799
0,43840156
0,125415
0,37951017
0,51006166
49
0,12921156
0,39099869
0,52550224
0,11957143
0,32317813
0,458105
0,13855371
0,41926837
0,5634967
70
0,13039699
0,39458583
0,53032336
0,12214286
0,3301282
0,46795672
0,14094257
0,42649714
0,57321216
71
Temps
(h)
T= 4°c
T= 25°c
0,13780591
0,06964384
0,13336055
0,08594347
0,10224309
0,14225126
0,10965201
0,11557915
T= 37°c
0,26630956
0,33555005
0,31681655
0,39918885
0,21580258
0,27191125
0,33977427
0,42811558
0,41323898
0,52068111
0,44078825
0,55539319
0,42701361
0,53803715
0,35814045
0,45125697
0,10337143
0,0972
0,12342857
0,09102857
0,06171429
0,0756
0,12651429
0,1188
T= 4°c
T= 25°c
T= 37°c
0,22987348
0,30170894
0,29555161
0,38791149
0,38468623
0,50490067
0,18765182
0,24629301
0,27678643
0,36328219
0,37530363
0,49258602
0,36122975
0,47411404
0,31431679
0,41254079
pH =7
pH = 3
T= 4°c
T= 25°c
0,07315875
0,22669407
0,09107518
0,28221099
0,35558585
T= 37°c
Temps
(h)
0,28563453
1
Tableau(08) : Masse cumulative du PA dans
le milieu (forme humide dans l'eau physiologique).
0,46726738
0,58875689
0,35623354
0,44885426
0,3331015
0,41970788
0,43950892
0,55378124
0,45338815
0,57126906
0,10749857
0,11496375
0,37936559
0,47800065
pH = 1,2
24
2
4
0,12242893
26
0,14183839
0,1463175
0,15079661
48
28
46
0,06620499
0,06978364
0,08320357
0,09751816
0,09841282
0,10378079
0,11451674
0,11720072
0,06173168
0,08051958
0,08588755
0,09304485
0,11809538
0,11004343
0,04204911
0,0518904
T= 4°c
0,10313216
0,18492664
0,19381734
0,13869498
0,16003267
0,16536709
0,19559548
0,20626432
0,23471458
0,23293643
0,08357261
0,12269171
0,13158241
0,17070151
0,21871131
0,22760201
T= 25°c
pH =7
0,14787089
0,17591537
0,18866286
0,19886086
0,22945484
0,24475182
0,29574179
0,31358827
0,32633577
0,33398426
0,33653376
0,23710333
0,11982641
0,26514781
0,2804448
0,2778953
T= 37°c
0,03881402
0,04754717
0,05725067
0,08636119
0,09024259
0,09218329
0,09703504
0,10188679
0,10770889
0,06113208
0,06501348
0,07471698
0,11061995
0,07762803
0,07956873
0,1096496
T= 4°c
Tableau(09) : Masse cumulative du PA dans le
milieu (forme seche dans l'eau distille).
0,18838286
0,19177714
0,11370857
0,15783429
0,16122857
0,16971429
0,19347429
0,10013143
0,13577143
0,06788571
0,13916571
0,15104571
T= 25°c
0,08316
0,10692
0,13068
pH = 3
0,1782
0,12012462
0,15444594
0,18876726
0,20102487
0,23289467
0,24515229
0,27211904
0,27702208
0,14463985
0,16425203
0,19612183
0,21818553
0,22799163
0,09806091
0,27947361
0,2574099
T= 37°c
0,04417132
0,05498879
0,08563827
0,08834264
0,10456884
0,11268194
0,11628776
0,12530232
0,06941208
0,07391935
0,10637175
0,09465283
0,12710523
0,09104701
0,0649048
0,1216965
T= 4°c
0,21141386
0,22395536
0,24903836
0,25262164
0,12899829
0,17020607
0,20783057
0,24187179
0,18095593
0,23112193
0,10929021
0,14691471
0,0877905
0,1379565
0,1881225
pH = 1,2
0,175581
T= 25°c
0,12587402
0,18495774
0,21064632
0,24404147
0,25174804
0,26973004
0,29798747
0,30312519
0,32110719
0,33138262
0,34679577
0,19780203
0,15670031
0,25945461
0,36220891
0,3570712
T= 37°c
Temps
(h)
120
122
124
26
28
48
50
76
78
24
52
54
72
74
2
4
pH = 1,2
pH = 3
pH =7
T= 4°c
0,12301602
0,14761923
0,10735944
0,10064947
0,13755428
0,0771646
0,08722954
0,1465009
0,1218977
0,12972599
0,10400446
0,11630606
0,08275623
0,06486299
0,14314592
0,05256139
T= 25°c
0,31451753
0,26209794
0,22874002
0,29307315
0,21444377
0,25971523
0,16440689
0,18585127
0,27639419
0,31213482
0,2215919
0,24780169
0,17632043
0,1381971
0,3049867
0,1119873
T= 37°c
0,37579603
0,45095524
0,3723797
0,396294
0,32796744
0,44753891
0,26647355
0,2357266
0,42020829
0,30746948
0,35529806
0,43728993
0,31771846
0,198147
0,25280824
0,16056739
Tableau(10) : Masse cumulative du PA dans le
milieu (forme seche dans l'eau physiologique).
0,13827493
0,11522911
0,08126685
0,09339623
0,1212938
0,11280323
0,07156334
0,12735849
0,13706199
0,09946092
0,10795148
0,09703504
0,07641509
0,05943396
0,13463612
T= 4°c
0,04851752
0,26119029
0,21765857
0,22911429
0,176418
0,13517743
0,21307629
0,18787371
0,15350657
0,24057
0,25889914
0,18329143
0,20391171
0,144342
0,112266
0,25431686
T= 25°c
0,09164571
0,37728937
0,31440781
0,33095559
0,2548358
0,1952638
0,27138358
0,30778869
0,34750337
0,37397981
0,22174024
0,26476447
0,29455047
0,20850202
0,16216824
0,3673607
T= 37°c
0,13238223
T= 4°c
0,13296469
0,15888154
0,1566279
0,14085243
0,1453597
0,11380876
0,09239919
0,13071105
0,081131
0,10704784
0,15212062
0,06873598
0,08676509
0,11831604
0,1104283
0,05521415
0,338513
0,28329457
0,24248094
0,19686572
0,3337114
0,27849297
0,30970338
0,22807614
0,1728577
0,30010018
0,25208415
0,14644889
0,18486171
0,32410819
0,23527854
T= 25°c
T= 37°c
0,11763927
0,40618775
0,4784754
0,43028364
0,44405271
0,39930321
0,24784337
0,34766918
0,28226607
0,32701556
0,48535994
0,46470633
0,20997841
0,36143825
0,26505472
0,33734237
0,16867119
72
46
70
52
48
50
98
94
28
26
4
2
76
74
96
24
Temps
(h)
|
ANNEXE V
|
|
Appareils utilisés :
|
|
|
|
|
|
|
|
Montage de calcination
|
|
Dessiccateur
|
|
|
|
|

Appareil de KARL-FISHER

Spectrophotomètre UV-Visible


