|
THESE de DOCTORAT de l'Université Pierre et
Marie Curie
Paris VI
Présentée par
Sandra SUAREZ
Pour obtenir le grade de
DOCTEUR de l'UNIVERSITE PARIS VI
Spécialité : NEUROSCIENCES
LES TROUBLES COGNITIFS AU COURS DE L'INFECTION PAR LE
VIH-1
Soutenue le 30 Mars 2000, devant le jury composé
de:
Pr. Jean Louis VILDE Rapporteur
Dr. François BOLLER Rapporteur
Pr. Jean-François ALLILAIRE Examinateur
Dr. Jacques GASNAULT Examinateur
Pr. Bruno DUBOIS Codirecteur de thèse
Pr. Jean-Jacques HAUW Directeur de thèse
Je dédie ce travail aux patients, car c'est avant tout
pour eux et avec eux que tout s'est fait.
A mon directeur, Monsieur Jean-Jacques Hauw, pour son soutien,
ses enseignements, sa patience et pour l'estime dont il m'honore
A mon codirecteur, Monsieur Bruno Dubois, pour ses conseils et
le temps qu'il m'a consacré
Je remercie les membres du Jury,
Messieurs Jean-Louis Vildé et François Boller,
qui ont bien voulu assumer la lourde tâche de rapporteurs
Messieurs Jean-François Allilaire et Jacques Gasnault
qui me font l'honneur d'accepter de juger ce travail
A mes collègues et amis neurologues, le professeur
Catherine Lubetzki qui a dirigé une partie des travaux
présentés ici, merci aussi pour ses conseils et sa gentillesse. A
Thierry Dubard, et Enrique Turell. Et à Bruno Stankoff, précieux
compagnon de travail, compétent et plein d'humour
Au professeur François Bricaire, qui m'a honorée
de sa confiance et a tous ceux du pavillon d'infectiologie, pour leur accueil
chaleureux et pour avoir rendu ce travail possible
A Laurence Baril, qui a activement pris part à ce
travail et qui est à présent une amie
A tous les collègues des disciplines variées qui
ont participés à ces études, A. Coutellier, V. Calvez, L.
Lacomblez, A. Tourbah, M. Khellaf, C. Dufouil, C. Katlama
A mes collègues Leslie Conquy et Ouriel Rosenblum qui
ont initié ce travail avec moi
A mes collègues neuropsychologues, médecins et
orthophonistes du centre du langage et de neuropsychologie, pour m'avoir
accueillie parmi eux et m'avoir soutenue
A mes collègues de l'institut Pasteur, Catherine Vidal
et à mon amie Sylvie Granon, avec le voeux que notre future
collaboration soit fructueuse
Au Docteur Jacques Gasnault, qui a participé à
ce travail, merci pour sa grande compétence et pour l'amitié dont
il m'honore. Et à tout le petit monde de l'unité de suites et de
réadaptation, à Bicêtre, Géma, Pascale, Jean-Paul,
Gilles, Alioune, Annie, ma remplaçante Férial et tout le
personnel infirmier ainsi que les aides soignantes, qui rendent ce service si
attachant.
A ceux qui ont rempli ces quatre années de vie, le
personnel du laboratoire Escourolle :
Mon amie Karima Mokhtari, et aussi Marianne Candau, Susy
Clavier, Marie-Anne Colle, Benoît Delatour, Brigitte Ducteil,
Stéphane Haïk, Jean Hogenhuis, Mireille Juncosa, Nathalie Kipson,
Thierry Maisonobe, Christian Nze, Nicolas Privat, Christelle Py, Odile
Russaouen, Véronique Sazdovitch, Danielle Seilhean, Eléonore
Tang, Marcelle Techel, Catherine Zunz et tous ceux qui y vivent, ceux qui y
sont passés, Maria Rio, Yolande Arends, Frédéric Dessi,
Françoise Lazarini, Franck Letournel, Yves Grignon, Flore Colo... Merci,
pour les sourires, le café, les repas pleins d'humour, et pour les
petits tests des magazines féminins! Pour tous ces moments de joie qui
ont soutenu mon travail. Au professeur Duyckaerts qui rend le service moins
vide quelques dimanches
A ma famille
A mes amis
A Bertrand
Plan
Pages
LISTE DES ABREVIATIONS
14
AVANT PROPOS
17
I. INTRODUCTION
19
1. Historique
19
1.1. Apparition du VIH
19
1.2. Apparition des troubles cognitifs associés
à l'infection par le VIH-1
21
2. Epidémiologie
23
2.1. Infection par le VIH-1 Données mondiales
23
2.2. Infection par le VIH-1 en Europe et en France
25
2.3. Troubles cognitifs liés à l'infection
par le VIH-1
26
3. Données biologiques sur le VIH-1
27
3.1. Le rétrovirus, le cycle viral et le tropisme
cellulaire
28
3.1.1. Biologie du VIH-1
28
3.1.2. Structure du VIH-1
29
3.1.3. Cycle de réplication du VIH et mode d'action des
traitements.
30
A) Fixation du VIH à la cellule cible
30
B) Internalisation (fusion et pénétration)
31
C) Transcription
32
D) Intégration
33
E) Transcription du gène viral
33
F) Assemblage et maturation des protéines du virus
34
G) Bourgeonnement
35
3.2. Variabilité génétique du VIH
35
3.3. Physiopathologie de l'infection par le VIH-1
36
3.3.1. Evolution de l'infection par le VIH-1
37
3.3.2. La primo-infection
37
3.3.3. La phase asymptomatique
38
3.3.4. La phase de SIDA
40
3.4. Neuroinvasion et aspects neuropathologiques
41
3.4.1. Neuropathologie de l'infection par le VIH-1
41
3.4.2. Physiopathologie des troubles neurologiques induits par le
VIH1
42
A) Réplication du VIH-1 dans les différents types
de cellules du Système Nerveux Central
42
B) Infection du SNC et déclenchement du dysfonctionnement
cérébral
43
C) La neuropénétrance
43
D) Le neurotropisme
44
E) La neurovirulance
45
3.4.3. Facteurs génétiques
45
II. EVALUATION DES TROUBLES COGNITIFS LIES A L'INFECTION
PAR LE VIH-1
46
1. La démence du SIDA
46
1.1. Classification et critères du DSM-IV : Formes
sévères
48
1.2. Classification et critères de l'American
Association of Neurology: Formes sévères
49
1.3. Stades de Price et Worley
49
1.4. Les échelles de démence
50
1.4.1. L'échelle de démence du SIDA de
Power-McArthur
50
1.4.2. Les batteries neuropsychologiques de la démence
50
2. Evaluation des troubles cognitifs
modérés
51
2.1. Classification et critères du DSM-IV:
Troubles cognitifs modérés
51
2.2. Classification et critères de l'American
Association of Neurology: Formes légères
52
3. Données neuropsychologiques
53
3.1. Troubles cognitifs et controverses
54
3.1.1. Cohérence entre les populations testées
55
3.1.2. Paramètres biologiques et troubles cognitifs
56
3.2. Evolution des troubles
57
3.3. Le ralentissement psychomoteur
58
3.4. Les troubles des fonctions exécutives et
attentionnelles
59
3.5. Les troubles mnésiques
60
3.5.1. Cadre théorique des différents troubles
mnésiques
61
3.5.2. La mémoire de travail
62
3.5.3. Mémoire épisodique
64
3.5.4. Métamémoire
65
3.6. Les modèles animaux
66
3.6.1. Processus mnésiques
67
3.6.2. Processus attentionnels
68
3.6.3. Motricité fine
68
3.6.4. Conclusion
69
3.7. La dépression et les autres troubles
psychiatriques
69
3.7.1. Importance des troubles de l'humeur et des troubles
psychiatriques dans la pathologie VIH : Fréquence et gravité
70
3.7.2. Reflet de la plainte cognitive
71
3.7.3. Relations entre les troubles de l'humeur et les troubles
cognitifs
73
3.7.4. Relations entre les troubles de l'humeur et
l'évolution de la maladie
74
3.7.5. Interactions entre les troubles cognitifs et la
toxicomanie
74
4. Article 1 : Mise au point d'une batterie
neuropsychologique. « Similar subcortical pattern of cognitive
impairment in AIDS patients with and without dementia »
76
4.1. Introduction : Pourquoi une nouvelle
batterie ?
77
4.2. Matériel et méthodes
78
4.3. Examen Neuropsychologique
80
4.4. Résultats
82
4.4.1. Efficience cognitive globale
82
4.4.2. Ralentissement psychomoteur
83
4.4.3. Fonctions exécutives
84
4.4.4. Mémoire
86
4.4.5. Dépression
86
4.5. DISCUSSION GENERALE ET CONCLUSIONS
87
III. PARAMETRES NEUROBIOLOGIQUES ET TROUBLES COGNITIFS
92
1. Immunodépression et troubles cognitifs
92
2. Articles II et III : La charge virale et les
troubles cognitifs
93
2.1. Introduction : Définition et techniques
de mesure de la charge virale
93
2.2. Réplication virale et troubles cognitifs
94
2.3. Article 2 : Human immunodeficiency virus type 1
DNA and RNA load in brains of demented and nondemented patients with acquired
immunodeficiency syndrome
98
2.3.1. Objectifs de l'étude
98
2.3.2. Patients et méthodes
98
2.3.3. Résultats
99
2.3.4. Conclusions
99
2.4. Article 3 : Plasma and cerebrospinal fluid
human immunodeficiency virus type-1 (HIV-1) RNA levels in HIV-1-related
cognitive impairment.
100
2.4.1. Objectifs de l'étude
100
2.4.2. Patients et méthodes
100
2.4.3. Résultats
101
2.4.4. Conclusions
101
3. Article IV : Imagerie cérébrale et
troubles cognitifs
102
3.1. Introduction : Imagerie (IRM et Imagerie
cérébrale fonctionnelle) et troubles cognitifs.
102
3.1.1. IRM et troubles cognitifs dans l'infection par le VIH
102
3.1.2. Imagerie cérébrale fonctionnelle, potentiels
évoqués et troubles cognitifs
103
A) IRM fonctionnelle (IRMf)
103
B) Potentiels évoqués
103
3.2. Spectroscopie par résonance magnétique
(SRM)
104
3.2.1. Généralités sur la technique de SRM
104
3.2.2. La SRM dans la pathologie VIH
104
3.2.3. SRM et effets des traitements
106
3.3. Article 4: Clinical and Spectroscopic
improvement in HIV associated cognitive impairment: A longitudinal study
107
3.3.1. Objectifs de l'étude
107
3.3.2. Patients et méthodes
107
3.3.3. Résultats
108
3.3.4. Conclusions
109
IV. EFFETS DES TRAITEMENTS
110
1. Historique des traitements et des troubles cognitifs
110
1.1. Les Inhibiteurs de la transcriptase inverse
110
1.1.1. Apparition des analogues nucléosidiques,
monothérapies et bithérapies
110
1.1.2. Apparition récente des inhibiteurs non
nucléosidiques de la transcriptase inverse (NNRTI)
111
1.2. Effets des associations incluant une
antiprotéase ou un NNRTI sur les troubles cognitifs
112
2. Article V : Outcome of patients with HIV1-related
cognitive impairment on highly active antiretroviral therapy
116
2.1. Objectifs de l'étude
116
2.2. Patients et méthodes
116
2.3. Résultats
117
2.4. Conclusions
117
V. DEVENIR DES PATIENTS AYANT PRESENTE DES TROUBLES
COGNITIFS
118
1. Evolution de la mortalité et de la
morbidité
118
2. Séquelles cognitives
120
3. Qualité de vie
121
4. Article VI : Long term outcome of HIV1-infected
patients with neurological disability (en préparation)
123
4.1. Objectifs de l'étude
124
4.2. Patients et méthodes
124
4.3. Résultats
126
4.4. Conclusions
126
VI. CONCLUSION
128
VII. BIBLIOGRAPHIE
129
VIII. ANNEXES
130
LISTE DES ABREVIATIONS
AAN : American Academy of Neurology ; Association de
neurologues américains à l'origine, notamment, de critères
standardisés de définition des troubles cognitifs
modérés ou sévères liés à l'infection
par le VIH-1.
ADC : « AIDS Dementia Complex »,
syndrome démentiel associé au SIDA.
ADN : Acide désoxyribonucléique.
ARN : Acide ribonucléique.
AZT : Azidothymidine ou Zidovudine (Retrovir), analogue
nucléosidique inhibiteur de la transcriptase inverse.
CDC : « Center for Disease Control »
organisme surveillant l'incidence des maladies et la consommation de
médicaments aux USA.
CMV : Cytomégalo-Virus.
Combinaisons thérapeutiques : Associations
thérapeutiques incluant au moins une molécule antiprotéase
ou un inhibiteur non nucléosidique de la et deux
antirétroviraux.
Combivir : Association de AZT et 3TC, deux analogues
nucléosidiques inhibiteurs de la transcriptase inverse.
DDC : Zalcitabine (Hivid), analogue nucléosidique
inhibiteur de la transcriptase inverse.
DDI : Didanosine (Videx), analogue nucléosidique
inhibiteur de la transcriptase inverse.
DMP : (Sustiva) (Efavirenz), inhibiteur non
nucléosidique de la transcriptase inverse.
D4T : Stavudine (Zerit), analogue nucléosidique
inhibiteur de la transcriptase inverse.
EOF : (Saqui; Fortovase) inhibiteur de
protéase.
EU : Etats Unis d'Amérique.
FIV : Virus de l'immunodéficience
féline.
GEE : Abréviation anglo-saxonne de
« Generalized Estimating Equation », modèle
d'équations généralisées permettant d'analyser des
données pour des variables discrètes et continues.
HAART : Abréviation anglo-saxonne de
« Highly Active Antiretroviral Thérapy »,
« combinaison thérapeutiques » incluant deux
antirétroviraux et une molécule antiprotéase ou un
inhibiteur non nucléosidique de la transcriptase inverse.
HTLV-III : Nom donné au virus du SIDA par
l'équipe du professeur Robert Gallo.
Indinavir : (Crixivan), inhibiteur de protéase.
IRM : Imagerie par résonance magnétique.
IRMf : Imagerie par résonance magnétique
fonctionnelle.
LEMP : Leucoencéphalopathie multifocale
progressive.
LAV : (Lymphadenopathy Associated Virus) premier nom
donné au virus du SIDA, par l'équipe du professeur Luc
Montagnier.
LCR : Liquide céphalo-rachidien.
MTS : Mémoire de travail spatiale.
Nelfinavir : (Viracept), inhibiteur de
protéase.
NNRTI : Inhibiteur non nucléosidique de la
transcriptase inverse.
OMS : Organisation mondiale de la santé.
PI : Inhibiteur de protéase.
Ritonavir : (Norvir), inhibiteur de protéase.
Saqui : (Fortovase) (EOF), inhibiteur de
protéase.
SRM : Spectroscopie par résonance magnétique.
UE : Union européenne.
USA : United States of America.
Saquinavir : (Invirase), inhibiteur de
protéase.
SIDA : Syndrome de l'immunodéficience acquise.
SIV : Virus de l'immunodéficience simienne.
SNC : Système nerveux central.
SRM : Spectroscopie par résonance magnétique.
Sustiva : (Efavirenz) (DMP 266), inhibiteur non
nucléosidique de la transcriptase inverse.
PET : Tomographie par émission de positron.
VIH-1 : Virus de l'immunodéficience humaine, agent
étiologique du syndrome de l'immunodéficience acquise (SIDA) chez
l'homme.
Viramune : (Nevirapine), inhibiteur non
nucléosidique de la transcriptase inverse.
3TC : Lamivudine (Epivir), analogue nucléosidique
inhibiteur de la transcriptase inverse.
1592 : (Abacavir), analogue nucléosidique
inhibiteur de la transcriptase inverse.
AVANT PROPOS
L'infection par le VIH est l'une des épidémies
les plus dangereuses et les plus meurtrières du 20ème
siècle. 33,6 millions de personnes sont atteintes et plus de 95% des
personnes infectées n'ont pas accès à des traitements.
Les troubles cognitivo-moteurs modérés
apparaissent chez 20,7% des patients séropositifs pour le VIH et la
démence chez 24% d'entre eux, principalement aux stades tardifs de la
maladie. Dans les pays industrialisés, ces chiffres ont
été bouleversés par l'apparition des combinaisons
thérapeutiques qui ont été un progrès très
important dans le traitement de cette affection. Cependant, les effets
bénéfiques de l'utilisation des traitements sont à
relativiser. En effet, peu de patients en bénéficient et l'action
des molécules est moins efficace sur les pathologies du système
nerveux central, du fait de leur difficulté à traverser la
barrière hémato-encéphalique.
Les troubles cognitifs entraînés par le VIH sont
en général mal connus : leur diagnostic est rendu difficile
par les nombreuses autres causes d'altération neuropsychologiques qui
existent au cours de cette maladie ; leur origine et les mécanismes
de leur déclenchement restent mal élucidés.
Le but de ce travail a été tout d'abord
d'étudier et de décrire ces troubles cognitifs,
modérés comme sévères, en constituant et en suivant
longitudinalement une cohorte de 120 patients infectés par le VIH-1,
pendant plus de trois ans. Cela nous a permis de préciser la nature des
troubles et de décrire notamment les particularités de l'atteinte
mnésique.
Une deuxième partie de ce travail a consisté
à rechercher les paramètres neurobiologiques liés à
ces troubles cognitifs en étudiant les relations entre les
déficits neuropsychologiques et d'une part la charge virale dans le sang
et dans le liquide céphalo-rachidien, d'autre part les modifications
métaboliques observées par la technique de Spectroscopie par
Résonance Magnétique (SRM). Cela nous a permis de mettre en
évidence une relation non linéaire entre la quantité de
virus dans le cerveau et les troubles cognitifs. Nous avons aussi pu montrer,
par la SRM, des modifications en faveur de la présence de processus
inflammatoires dans le cerveau et d'une souffrance neuronale
prédominante dans les régions frontales.
Au cours de ce travail une révolution
thérapeutique est survenue : nous avons pu être des
observateurs privilégiés de l'action de ces nouvelles
associations thérapeutiques sur le système nerveux central et
avons constaté une amélioration rapide des troubles cognitifs.
Nous avons pu montrer qu'il existait une cinétique particulière
du ralentissement psychomoteur : celui-ci apparaît plus tardivement
et continue de s'améliorer plus longtemps que les autres domaines
cognitifs après l'introduction des traitements. La baisse de la charge
virale dans le sang et dans le liquide céphalo-rachidien s'accompagne de
la réversibilité de certaines anomalies métaboliques
reconnues par la SRM, notamment en ce qui concerne le N-acetyl aspartate
(composé reflétant le fonctionnement neuronal) dans le lobe
frontal.
Enfin nous avons analysé l'évolution sous
traitement des patients (en terme de mortalité et de handicap), en
élargissant, cette fois, notre étude à l'ensemble des
affections neurologiques du SIDA. Il existait certe une baisse de la
mortalité mais des difficultés modérés ou
sévères dans la vie quotidienne étaient présentes
chez plus de la moitié de ces patients.
Ce travail très pluridisciplinaire n'a
été possible que grâce à de multiples collaborations
réunies autour d'un seul but, la compréhension des troubles
cognitifs et du handicap qu'ils entraînent au cours de l'infection par le
VIH-1.
I.
INTRODUCTION
1.
Historique
1.1. Apparition du VIH
En 1959, au Congo Belge (aujourd'hui République
Démocratique du Congo), un Bantu mourut d'une maladie inconnue. Un
échantillon de plasma fut recueilli. Analysé des années
plus tard il permettra d'identifier le premier cas d'infection par le
VIH-11.
En 1981, le « Center for Disease
Control » (CDC) (organisme d'Atlanta surveillant l'incidence des
maladies et la consommation de médicaments aux USA) constate une
augmentation de consommation de Pentamidine (médicament utilisé
pour le traitement de la Pneumopathie à pneumocystis carinii
(pneumocystose)) chez de jeunes hommes homosexuels. Peu de temps après,
plusieurs dizaines de cas de sarcome de Kaposi (un cancer rare) sont
découverts, toujours chez des homosexuels, dont certains sont aussi
atteints de pneumocystose2. La conjonction de ces deux maladies
habituellement rarissimes est inquiétante et une enquête est
ouverte aux USA. Les données épidémiologiques font
soupçonner une nouvelle maladie.
En 1982, une nouvelle maladie est découverte chez un
nombre croissant d'homosexuels et d'hémophiles ; la transmission par
voie sexuelle et sanguine est démontrée. Les premiers cas chez
l'enfant sont décrits. Un déficit profond de l'immunité
cellulaire est constaté chez tous les malades. Les CDC publient la
description et les critères diagnostiques de cette nouvelle
entité clinique : le syndrome de l'immunodéficience acquise
(SIDA)3, 4.
En 1983, la croissance de la maladie aux USA est
exponentielle : 2000 cas sont diagnostiqués et 800 personnes sont
mortes. L'équipe du professeur Luc Montagnier à l'institut
Pasteur isole le virus du SIDA baptisé L.A.V. (Lymphadenopathy
Associated Virus)5. Elle est suivie par l'équipe du
professeur Robert Gallo qui découvre le virus H.T.L.V.-III en mai
19846. Une controverse s'installe sur l'antériorité et
la dénomination de la découverte du virus, qui sera
désigné sous le nom de Virus de l'Immunodéficience Humaine
(V.I.H.). Les Etats-Unis reconnaîtront l'antériorité de la
découverte du VIH par l'institut Pasteur en 1995.
En 1984, l'activité antirétrovirale de
l'Azidothymidine (AZT) est mise en évidence. C'est le premier traitement
disponible contre le VIH. En 1986, un deuxième rétrovirus, le
VIH-2 est isolé par l'équipe du professeur Montagnier, il semble
circonscrit à l'Afrique de l'Ouest7, 8.
De 1993 à 1996, la supériorité des
associations d'antiviraux sur la monothérapie est
démontrée. Les premiers inhibiteurs de protéase font leur
apparition et les « combinaisons thérapeutiques »,
des combinaisons thérapeutiques incluant au moins une
antiprotéase ou un inhibiteur non nucléosidique de le
transcriptase inverse et deux antirétroviraux, vont soulever d'immenses
espoirs thérapeutiques.
La question de l'origine du virus est posée dès
sa découverte. Très vite des similitudes avec le virus de
l'immunodéficience simienne (SIV) seront observées9.
Dans le début des années 90, l'origine probable du VIH2 chez le
singe Mangabey (Cercocebus atys) va être avancée10. En
1999, enfin, des analyses génétiques vont permettre de mettre en
évidence l'origine probable du VIH1 chez le chimpanzé
troglodyte11. Une deuxième étude viendra confirmer ces
travaux en montrant les similitudes du VIH1 avec le virus simien, chez trois
chimpanzés troglodytes originaires du Cameroun12.
1.2. Apparition des troubles cognitifs
associés à l'infection par le VIH-1
L'identification de détériorations
intellectuelles chez les patients présentant un syndrome
d'immunodéficience acquise s'est faite très rapidement
après la découverte de la maladie. Ces troubles seront d'abord
attribués au Cytomégalo-Virus (CMV)13, 14. D'abord
identifié sous le terme « d `encéphalopathie
subaiguë ou progressive » (subacute encephalitis, progressive
encephalopathy)15-17, ce syndrome clinique est ensuite
dénommé « syndrome démentiel associé au
SIDA » (AIDS dementia complex) (ADC) par l'équipe de
Navia et Price, à New York, EU18, 19. Très rapidement
les difficultés du diagnostic apparaissent, chez ces patients pour
lesquels les signes cliniques sont peu spécifiques et les autres causes
d'altération des fonctions supérieures très nombreuses
(germes opportunistes, lymphomes) et peuvent être à l'origine de
signes neurologiques intriqués surtout à la fin de
l `évolution de la maladie15, 18, 19. De plus, la
fréquence des manifestations psychiatriques rend difficile
l'évaluation de la démence chez ces patients souvent
déprimés ou confus20-24.
La présence de particules virales dans le
système nerveux central sera rapidement
révélée25 et du VIH va être isolé
à partir de liquide céphalo-rachidien et de tissu neuronal chez
des patients présentant un syndrome neurologique du
SIDA26-28. La mise en évidence du passage précoce de
la barrière hémato-encéphalique par le VIH-1
n'apparaîtra que plus tard29.
En 1988 Price et Brew définissent une échelle
évaluant le degré de détérioration de la
démence (Stades démentiels du SIDA)30.
Si la présence d'une démence ne fait plus de
doute, un débat va par contre persister sur l'origine et la
présence de troubles cognitifs modérés tôt ou tard
dans l'avancée de la maladie
En 1991, une nouvelle nomenclature, élaborée
conjointement par l'O.M.S. et par «l'American Academy of Neurology»
(A.A.N.), vise à standardiser le diagnostic clinique et à rendre
l'ensemble des recherches comparables31. Elle fournit une nouvelle
terminologie ainsi qu'un nouvel ensemble de critères diagnostiques (voir
tableau I). Les formes sévères sont regroupées soit sous
l'appellation de «HIV-1-associated dementia complex » soit sous
celle de « HIV-1-associated myelopathy » et sont
suffisantes pour poser le diagnostic de SIDA avéré.
Les formes légères font partie du
« HIV-1-associated minor cognitive/motor disorder » et ne
permettent pas de poser le diagnostic de SIDA avéré bien qu'elles
puissent être observées chez les sidéens.
Les appellations nouvelles et anciennes des formes
sévères et légères des troubles cognitivo-moteurs
associés au VIH-1 sont présentées dans le tableau I.
Tableau I :
|
Nouvelle nomenclature
(en vigueur après 1990)
Syndrome cognitivo-moteur associé au VIH-1
|
Ancienne nomenclature (jusqu'en 1990)
Syndrome démentiel du SIDA
|
|
Manifestations sévères
|
1) Syndrome démentiel associé au VIH-1
2) Myélopathie associée au VIH-1
|
1) encéphalite subaiguë, encéphalopathie
VIH, démence liée au SIDA
2) encéphalopathie VIH
|
|
Manifestations légères
|
Troubles cognitivo-moteurs associés au VIH-1 (voir
tableau II pour détails)
|
1) désordres neurocognitifs associés au VIH
2) anomalies neuro-comportementales associées au VIH
|
Pendant les 13 années qui suivent la découverte
du VIH, l'ADC va rester incurable et il faudra attendre l'apparition des
antiprotéases pour observer les premières rémissions chez
des patients.
2.
Epidémiologie
2.1. Infection par le VIH-1
Données mondiales(*)
En quelques années, l'infection par le VIH est devenue
un très important problème de santé publique. La
pandémie existe presque partout dans le monde et on estime qu`elle
deviendra la 9ème cause de décès par ordre de
fréquence en 2020 avec des situation particulièrement graves en
Afrique et en Asie32, 33.
Alors que débute le XXIème siècle,
l'Organisation Mondiale de la Santé (OMS) estime que 33,6 millions
personnes vivent avec le VIH/SIDA à fin 99. Parmi ces personnes, 32,4
millions seraient des adultes, 14,8 millions des femmes et 1,2 millions des
enfants de moins de quinze ans. La répartition des populations
infectées dans le monde est présentés dans la figure 1. Le
nombre de nouveaux cas d'infection en 1999 s'élèverait à
5,6 millions. Le nombre de personnes déjà
décédées du SIDA serait en fin 1999, de 16,3 millions,
dont 2.6 millions pour la seule année 1999 -un total mondial
supérieur à celui de n'importe laquelle des années
écoulées depuis le début de l'épidémie-
malgré les thérapies antirétrovirales qui, dans les pays
riches, ont ralenti le SIDA et les décès qui lui sont
associés.
Même si les programmes de prévention parvenaient
à ramener à zéro le nombre de nouvelles infections, les
décès parmi les personnes déjà infectées
continueraient d'augmenter pendant plusieurs années. Or la population
séropositive s'accroît toujours et l'OMS prévoit que le
chiffre annuel des décès dus au SIDA augmentera pendant de
nombreuses années avant d'atteindre son maximum.
Près de la moitié de toutes les personnes
infectées le sont avant l'âge de 25 ans et décèdent
avant leur 35ème année. A la fin de 1999, le SIDA a
laissé dans son sillage un total cumulé de 11,2 millions
d'orphelins du SIDA, c'est à dire d'enfants qui ont perdu leur
mère avant l'âge de 15 ans.
Dans les pays industrialisés, les comportements sexuels
sans risque sont en baisse parmi les homosexuels masculins. En Amérique
du Nord et en Europe, la baisse du nombre de décès imputables
à la thérapie antirétrovirale s'amenuise.
Les infections sont aussi en rapide augmentation en Europe
orientale et en Asie centrale. En Amérique centrale et dans les
Caraïbes sévit une des plus graves épidémies du VIH
hors de l'Afrique. En Afrique subsaharienne se trouvent près de 70% du
total des personnes infectées avec une espérance de vie moyenne
d'une dizaine d'années ce qui pourrait avoir pour conséquence
d'abaisser l'espérance de vie en Afrique australe de 59 ans
(années 90) à 45 ans vers 2005.
En résumé, l'écart entre pays riches et
pays pauvres s'est accru pour ce qui concerne taux d'infection par le VIH et de
décès dus au SIDA. Il risque de se creuser encore dans les
années à venir. On comptait plus de 15000 nouveau cas d'infection
à VIH par jour en 1999. Plus de 95% des personnes infectées
vivent dans les pays en voie de développement.
2.2. Infection par le VIH-1 en Europe et
en France(*)
Les données pour 1999 ne sont pas disponibles pour la
France et la Norvège. Ces deux pays sont donc exclus de l'analyse des
tendances récentes.
Au 30 juin 1999, un total de 202973 cas de SIDA a
été déclarés dans l'Union Européenne (UE).
Soixante pour cent d'entre eux sont décédés. La diminution
de l'incidence du SIDA observée depuis 1996 se poursuit en 1998: 10855
cas diagnostiqués en 1998, à comparer à 13352 en 1997:
-19%, (France et Norvège exclues). Elle s'est ralentie au premier
semestre 1999 (5012 cas).
Depuis 1996, l'incidence du SIDA a diminué dans tous
les pays de l'Union Européenne, sauf au Portugal, avec des
disproportions importantes: en 1998, elle variait de 3,1 cas par million
d'habitants en Finlande à près de 90 cas par million en Espagne
et au Portugal.
Dans la partie centrale et orientale de la Région
Europe de l'OMS, l'incidence du SIDA a augmenté globalement de 18% entre
1997 et 1998. Une augmentation considérable du nombre de cas de SIDA est
à prévoir dans plusieurs autres pays de l'Est, notamment en
Biélorussie et en Fédération de Russie.
Globalement, dans l'UE, la diminution du nombre de nouveaux
cas de SIDA se poursuit pour tous les groupes de transmission entre 1997 et
1998 (rapport homosexuel : -24%, utilisation de drogue par voie injectable
: -28%, rapport hétérosexuel : -10%. La transmission
hétérosexuelle est responsable d'une proportion croissante des
cas de SIDA (26% en 1998) et elle est devenue majoritaire parmi les nouveaux
cas de SIDA dans plusieurs pays dont la France, la Norvège et la
Suède.
La baisse de l'incidence du SIDA est due en grande partie
à une diminution du nombre de cas diagnostiqués comme infection
à VIH avant l'apparition des manifestations du SIDA, et ayant donc pu
bénéficier des associations d'antirétroviraux. Les
personnes ignorant leur séropositivité au moment de l'apparition
de la maladie constituent une proportion croissante des nouveaux cas de SIDA :
28% en 1996, 44% en 1998. Parallèlement à cette baisse
d'incidence du SIDA, on observe une diminution du nombre des
décès dans l'UE depuis 1996 avec 13984 décès
déclarés en 1996 et 6880 en 1998 (-32%). La prévalence des
personnes infectées par le VIH augmente.
En France, au 31 décembre 1998, sur 60,37 millions
d'habitants (incluant les DOM), 49421 cas de SIDA ont été
déclarés depuis le début de l'épidémie, dont
30190 personnes décédées. En 1998, sur un total de 1810
nouveaux cas de SIDA, les facteurs de risque étaient:
hétérosexuels (701), homosexuels/bisexuels (552), toxicomanes
(327), toxicomanes homo/bisexuels(12), transfusions (22), Transmission
maternelle (2), autre/indéterminé (194).
2.3. Troubles cognitifs liés
à l'infection par le VIH-1
La prévalence des troubles cognitifs
modérés liés au VIH varie selon le stade de la maladie.
Aux stades asymptomatiques (défini par les
critères biologiques), les données ont été
très hétérogènes. L'existence même de
troubles cognitifs à ce stade a pu être discutée, alors que
leur présence ne peut être remise en question aux stades
ultérieurs de la maladie. Le débat sur la présence ou non
de troubles cognitifs aux stades asymptomatiques visait à évaluer
les risques d'accidents dans des professions particulières comme par
exemple chez les aviateurs34.
L'incidence des troubles cognitifs modérés
augmente avec la baisse des lymphocytes CD4.
Selon l'AAN (1996) 35, les troubles
cognitivo-moteurs modérés représentent 20,7% des patients
séropositifs pour le VIH, contre 24% de démences, tous stades de
la maladie due au VIH-1 confondus. Ces chiffres ont été obtenus
avant 1995, date à laquelle les combinaisons thérapeutiques ont
été systématisées.
Selon le rapport n° 61 de l'OMS et de l'ONUSIDA, de
surveillance du VIH/SIDA pour la région Europe de l'OMS (France, Monaco,
Norvège: pas de données déclarées en 1999),
l'encéphalopathie due au VIH représente 5.2% des
pathologies indicatrices du SIDA diagnostiquées en 1999 chez les
adultes/adolescents (5.1% chez les hommes, 5.6% chez les femmes et 6.3% chez
les enfants).
En France, d'après le centre de données
épidémiologiques sur immunodéficience humaine, de
l'INSERM(*) (N° du 6-Mars 1999),
l'incidence de l'encéphalopathie VIH pour 1000 patients-année a
diminué de 14.4 au premier semestre 1996 à 4.5 au premier
semestre 1997 et à 4.1 au premier semestre 1998.
3.
Données biologiques sur le VIH-1
3.1. Le rétrovirus, le cycle viral
et le tropisme cellulaire
3.1.1. Biologie du VIH-1
Le virus de l'immunodéficience humaine (VIH-1) est
l'agent étiologique du syndrome de l'immunodéficience acquise
(SIDA).
Le VIH-1 appartient au groupe des lentivirus, famille de virus
à acide ribonucléique (ARN), appartenant à la classe des
rétrovirus. Les lentivirus sont à l'origine de maladies à
évolution lente et de syndromes d'immunodéficience. Ils ont
souvent un tropisme particulier pour le système nerveux
central36.
Les rétrovirus peuvent transcrire leur ARN
génomique monocaténaire en ADN bicaténaire grâce
à une enzyme, la transcriptase inverse. Les rétrovirus ne peuvent
pas se répliquer en l'absence d'une cellule hôte dont ils
utilisent le matériel de réplication. Le génome des
rétrovirus contient trois gènes majeurs, gag
(antigène de groupe), pol (polymérase) et env
(enveloppe). Ces gènes codent les précurseurs
polypeptidiques des protéines majeures et structurelles des
rétrovirus. Dans le cas du VIH, le gène env code les
deux glycoprotéines d'enveloppe, l'enveloppe la plus extérieure,
gp120 et la glycoprotéine transmenbranaire gp41, dérivée
d'une protéine précurseur, la gp160. Les composant codés
par le gène gag incluent les protéines de la capside du
nucléoïde (du core), p55, p40, p24 (antigènes de la
capside), p17 (protéine de la matrice), p7 (nucléocapside), p9,
p6 (protéines de l'ARN). Les protéine importantes codées
par pol sont les protéines des enzymes virales, p66 et p51
(pour la transcriptase inverse), p11 (pour la protéase) et p32 (pour
l'intégrase). Les gènes accessoires portés par le VIH-1
incluent tat, rev, nef, vif, vpr
et vpu. Le gène tat (transcripteur) produit une
protéine de régulation qui active la rétrotranscription et
la synchronisation de la production virale. Le gène rev
(régulateur de l'expression des virions) code une protéine de
régulation qui active la transcription de l'ARN viral, le gène
nef (negative regulatory factor) produit une protéine de
régulation agissant sur la réplication et l'infectiosité
du virus. Les gènes vif, vpr et vpu codent
des protéines jouant un rôle dans l'infectiosité
générale et les effets pathologiques des virus37-39
(figure 2: Diagramme schématique du génome du VIH-1).
Il existe trois groupes de rétrovirus, les
spumavirus, qui ne sont associés à aucune maladie
connue; les oncovirus, a l'origine de sarcomes ou de leucémies,
et les lentivirus, auxquels appartiennent les VIH-1 et VIH-2 ainsi que
des virus responsables de syndromes d'immunodéficience chez le singe
(SIV : simian immunodeficiency virus), les bovins (BIV : bovine
immunodeficiency virus), le chat (FIV : feline immunodeficiency virus) et
la souris (MIV : murine immunodeficiency virus).
3.1.2. Structure du VIH-1
Le virion du VIH-1 a l'apparence d'un petite sphère
d'environ 100 nanomètres de diamètre (figure 3 : Structure
du VIH-1). Le VIH-1 est composé :
- D'une enveloppe composée de deux parties
glycoprotéiques : l'enveloppe externe (gp120) et l'enveloppe
transmenbranaire (gp41). Ces deux glycoprotéines jouent un rôle
majeur dans la pénétration du virus dans une cellule (cf. plus
bas, cycle du VIH).
- D'une matrice, tapissant l'intérieur de la particule
virale.
- D'une membrane d'origine cellulaire enveloppant la matrice
(elle est acquise par le virus lors de l'exocytose).
- D'une capside virale, complexe d'intégration
englobant et protégeant le matériel génétique du
virus. C'est cette partie qui pénètre dans la cellule lors de son
infection.
- De deux brins d'ARN, à l'intérieur de la
capside. Ces deux ARN portent les informations génétiques du
virus nécessaire à sa reproduction.
- D'une transcriptase inverse, enzyme permettant au virus de
fabriquer à partir de son ARN une copie sous forme d'ADN. Cet ADN
s'intègre ensuite à l'ADN de la cellule dans son noyau.
- D'une protéase, enzyme permettant la maturation des
virus produits par la cellule infectée.
3.1.3. Cycle de réplication du VIH et mode d'action
des traitements.
Annexe 1: Les différents traitements contre le
VIH-1.
Le cycle de réplication du VIH peut être
décomposé en plusieurs étapes, de la fixation à la
cellule cible jusqu'à la libération de nouveaux virions par cette
dernière (figure 4: Cycle de réplication du VIH-1).
A) Fixation du VIH
à la cellule cible
Des glycoprotéines de l'enveloppe du VIH, la gp120,
sont attirées par des récepteurs cellulaires spécifiques
des cellules cibles, les CD4. Les CD4 sont des motifs protéiques
portés par les lymphocytes T4 (ou CD4+ ou auxiliaires), les lymphocytes
B, les lymphocytes tueurs, les cellules dendritiques (cellules de Langerhans,
cellules dendritiques), les cellules du système
hématopoïétique, les cellules endothéliales, certains
macrophages présents dans les ganglions lymphatiques, des cellules
souches de la moelle osseuse, des cellules épithéliales
gastro-intestinales, et certaines cellules souches du systèmes nerveux
(voir plus bas le paragraphe « neuropathologie de l'infection
par le VIH-1 »). Le VIH infecte principalement les cellules
présentant des CD4 Cependant, certaines cellules ne possédant pas
le récepteur CD4 (comme les fibroblastes) peuvent également
être infectées. D'autres récepteurs du VIH, le CXCR4
(fusines, Fc), et le CCR5, ont été identifiés. Il s'agit
de récepteurs aux chémokines que le VIH utilise en même
temps que la molécule CD4 pour pénétrer dans les cellules
cibles38, 40-42. Les fusines sont utilisées par les souches
de VIH-1 à tropisme lymphocytaire43, tandis que les CCR5 sont
utilisés par les souches à tropisme
macrophagique44-46.
D'autres récepteurs n'exprimant pas la molécule
CD4 mais interagissant avec la protéine gp120 ont aussi
été proposés, comme les lectines47 et le
galactosylcéramide (GalC)48.
Certaines cytokines (les chémokines) permettraient
de bloquer les récepteurs de la cellule et donc d'empêcher
l'infection des cellules.
Pour empêcher la fixation du VIH, les recherches
s'orientent vers l'élaboration de substances dérivées de
chémokines permettant de bloquer les récepteurs de la cellule ou
des substances destinées à bloquer le virus avant qu'il ne se
fixe à la cellule (CD4 solubles, anticorps anti-VIH)38,
49.
B) Internalisation
(fusion et pénétration)
La fixation de la glycoprotéine gp120 à la
molécule CD4 a entraîné un changement de sa conformation et
notamment a exposé des sites de clivages sensibles à des
protéases cellulaires. Leur clivage produit une deuxième
modification conformationnelle aboutissant à l'exposition de
l'extrémité hydrophobe N-terminale de la gp41, catalyseur de la
fusion des membranes virale et cellulaire (figure 5 : Récepteurs
membranaires du VIH-1 et de la cellule hôte).
Un second récepteur de la surface de la cellule cible
(le corécepteur) est nécessaire à la
pénétration du VIH. Ce corécepteur est une chimiokine,
molécule de surface incluant les récepteurs CXCR4 et CCR5. La
protéine d'enveloppe la plus interne, la gp41, achève la fixation
et permet la fusion des membranes virales et cellulaires. Les récepteurs
des chimiokines CXCR4 et CCR5 produisent un changement de conformation de la
protéine gp41 qui permet la fusion50. Le complexe
d'intégration (et son matériel infectieux) est alors
internalisé dans le cytoplasme de la cellule cible désormais
contaminée50.
Les différences existant dans les corécepteurs
(les chimiokines) présents à la surface d'une cellule permettent
de comprendre pourquoi différentes souches de VIH peuvent infecter des
cellules préférentiellement. Par exemple, certaines souches
« T-trophiques » vont interagir
préférentiellement avec les récepteurs de chimiokines
CXCR4 pour infecter les lymphocytes. D'autres,
« M-trophiques », vont interagir avec le corécepteur
CCR5 des chimiokines pour infecter les macrophages. La présence d'un
mutation du récepteur CCR5 peut expliquer certains cas de
résistance. En général, les mutations du VIH peuvent
augmenter l'habileté de ce dernier a infecter certaines souches
cellulaires51. Des molécules en cours de
développement, les inhibiteurs de fusion, tentent de bloquer ce
mécanisme.
C) Transcription
La nucléocapside du virus est détruite et l'ARN
viral est transcrit en ADN double brin linéaire grâce à une
enzyme, provenant de la capside virale, la transcriptase inverse.
La transcriptase inverse permet, à l'aide des
nucléosides contenus dans la cellule, de construire un brin d'ADN viral
à partir de l'ARN. L'ADN ainsi produit sera ensuite
intégré à l'ADN cellulaire52.
Plusieurs substances capables d'inhiber le processus de
rétrotranscription de la transcriptase inverse ont été
développées ces dernières années et sont
actuellement utilisées : les inhibiteurs de la transcriptase
inverse analogues de nucléosides, comme la zidovudine (AZT), la
didanosine (DDI), la zalcitabine (DDC), la lamividune (3TC) et la stavudine
(D4T); et les inhibiteurs de la transcriptase inverse non nucléosidique,
comme la delavirdine, le lovirid, la nevirapine, l'efavirenz ou
l'hydroxyurée (annexe 1).
D)
Intégration
L'ADN linéaire issu de la phase de transcription
inverse est transporté dans le noyau de la cellule. Cet ADN est
intégré à l'ADN cellulaire grâce à l'action
d'une enzyme du virus, l'intégrase. L'intégrase sectionne l'ADN
cellulaire et intègre l'ADN viral à l'ADN de la cellule
infectée52. Une fois intégré au génome
de la cellule hôte, l'ADN viral ne peut plus être détruit
à moins de détruire la cellule hôte elle-même.
E) Transcription
du gène viral
Une fois l'ADN intégré dans le patrimoine
génétique de la cellule le provirus se comporte comme un
gène de la cellule hôte et utilise la machinerie cellulaire. A ce
stade, le virus peut rester latent (et être transmis aux cellules filles
générées lors de la mitose; on parle alors de latence
virale) ou s'exprimer. S'il s'exprime, il s'opère alors une production
d'ARN messagers et de protéines nécessaires à la
production de nouveaux virus. L'ARN messager est décrypté par la
machinerie cellulaire de la cellule hôte, qui produit les
éléments (protéines de la capside, protéase,
matrice) permettant la synthèse de nouveaux virus.
La réplication du virus peut être modulée
par certaines cytokines, comme des interleukines (IL-6) ou des facteurs de
nécrose tumorale (TNF-, TNF-) qui activent les lymphocytes CD4 les
rendant plus actifs a produire des virions52, 53. D'autres cytokines
ont des effets inhibiteurs, comme les interférons54.
Cette fonction étant codée par les
gènes régulateurs du virus (tat, rev), les recherches s'orientent
vers des inhibiteurs de ces gènes viraux.
F) Assemblage et
maturation des protéines du virus
Les protéines formées précédemment
doivent subir l'action d'une enzyme avant leur assemblage. La maturation et
l'assemblage des protéines du virus s'effectuent grâce à
une enzyme nommée la protéase, elle-même issue de
l'étape de synthèse. La protéase permet d'ajuster la
structure des protéines en coupant les éléments superflus.
L'action de cette enzyme est indispensable à la création de virus
viables. L'assemblage des particules virales est ensuite amorcé par le
précurseur Pr55Gag55.
Les molécules de la famille des
antiprotéases inhibent l'action de cette enzyme rendant les virus
produits incapables d'infecter de nouvelles cellules. Plusieurs inhibiteurs de
protéase (antiprotéases) comme le saquinavir, le ritonavir,
l'indinavir, le nelfinavir, le saqui (EOF) sont actuellement utilisés et
ont une action incontestable contre la prolifération virale. D'autre
substances du même groupe devraient bientôt
apparaître.
G)
Bourgeonnement
C'est l'étape finale durant laquelle les virus
formés quittent la cellule. Les nouvelles particules virales produites
vont bourgeonner à la surface de la cellule infectée puis se
détacher, entourées d'un segment de la membrane cellulaire qui
leur procure une protection supplémentaire. La protéine p24
intervient dans l'encapsidation du complexe nucléoprotéique
(ARN-Gag) et au cours du bourgeonnement56. Les nouvelles particules
virales pourront alors infecter d'autres cellules.
Certaines recherches tentent d'empêcher ce processus
notamment par l'utilisation de substances de la famille des
interférons.
3.2.
Variabilité génétique du VIH
Figure 6 : Phylogenèse et répartition
géographique des isolats du VIH-1 et du virus simien.
Il existe une grande variabilité de sous-types du VIH.
L'émergence continue de nouveaux isolats est liée à la
mauvaise fidélité de retranscription de la transcriptase inverse
ajoutée au taux de renouvellement très élevé du
virus57. La mutation rapide des virions du VIH explique l'apparition
fréquente de résistances aux traitements.
La variabilité génétique du VIH provoque
aussi des différences dans les caractéristiques
phénotypiques des effets pathogènes du VIH. Trois grand groupes
peuvent être observés :
La variante (1) « n'induisant pas de
syncytium » (non-syncytium-inducing; NSI) avec une faible
capacité réplicative. (2) la variante NSI avec une haute
capacité réplicative et (3) la variante « induisant du
syncytium » (syncytium-inducing; SI), dérivant des variants
NSI, associé à un tropisme cellulaire pour les CD4+ et le
déclin rapide de ces cellules, une charge virale plasmatique plus
élevée et une progression plus rapide de la maladie. La
moitié environ des patients avec un SIDA ont une variante
SI58.
Les études phylogénétiques ont
identifié des clusters génétiques du gène
env du VIH-1, qui sont identifiés comme des sous-types. La
variabilité du gène env est élevée. La
séquence d'amino-acides V3 de ces variants génétiques
influence le phénotype du VIH et la réponse immune, faisant
varier les propriétés biologiques du VIH59. La
modification génétique au cours de l'infection peut aboutir
à un changement de tropisme cellulaire du VIH-1.
Les différents sous-types de VIH-1 qui ont surgit et
continueront à surgir dans l'évolution de
l'épidémie ont été identifiés avec certaines
répartitions géographiques, suivant les mouvements des
populations infectées par le virus au cours de l'histoire. La
variabilité biologique de ces sous-types permet pour une part
d'expliquer les différences dans la répartition de l'infection
dans les différents groupes à risque60. La
variabilité des sous-types du VIH-1 pourrait aussi perturber les
dépistages car le diagnostic, la sensibilité et la
spécificité des tests de laboratoire pourraient être
différents selon les isolats du virus61.
La figure 6 représente schématiquement la
phylogenèse des isolats du VIH-1. Les sous-types A à H
appartiennent au groupe le plus important. Le sous-type
« O », par contre est très différent et
apparaît génétiquement plus proche du virus simien (SIV) et
du VIH-262.
3.3.
Physiopathologie de l'infection par le VIH-1
L'histoire naturelle de l'infection par le VIH-1 est
dépendante de facteurs cliniques et infectieux. Depuis l'arrivée
des combinaisons actives, le pronostic de la maladie a énormément
évolué. Nous traiterons dans ce chapitre l'histoire naturelle de
l'infection par le VIH, sans tenir compte dans un premier temps de
l'interaction avec les traitements existants.
3.3.1. Evolution de l'infection par le
VIH-1
En règle générale, chez l'adulte, la
contamination est suivie d'une période de 8 à 10 ans pendant
laquelle le patient ne ressent pas ou très peu de symptômes, c'est
la primo-infection. Cependant, dans 10% des cas, la phase symptomatique (le
SIDA) peut se manifester en moins de deux ans suivant la contamination et
inversement 10% des patients n'auront pas évolué vers la phase de
SIDA plus de 10 ans après la contamination63. Il reste
néanmoins clair que la probabilité de l'apparition de la phase
SIDA et du décès du patient est directement reliée
à la durée depuis laquelle il est infecté. Même s'il
existe des patients dont la phase asymptomatique est très longue, aucune
donnée ne permet, à ce jour, de démontrer une
impossibilité du virus à évoluer vers le SIDA dans ces
cas64.
3.3.2. La primo-infection
La primo-infection est la phase suivant immédiatement
l'exposition à l'agent infectieux. Dans 50 à 90% des cas, elle
peut produire des symptômes physiques limités.
A partir de l'exposition au virus, la virémie change en
environ 4 à 11 jours. Les symptômes apparaissent environ 2
à 6 semaines après l'exposition. Les symptômes persistent
durant 1 à 2 semaines puis s'éteignent en 1 à 2 mois.
Les symptômes de la primo-infection sont
pseudo-mononucléosiques. Les manifestations les plus fréquentes
sont la fièvre, fatigue, arthralgies, myalgies, lymphadénopathie,
pharyngite, érythèmes diffus, rush du tronc, diarrhées,
nausées, vomissements, perte de poids, sueurs nocturnes,
ulcérations(mucocuteaneous) et céphalées. Plus rarement,
une méningoencéphalite peut être
observée65.
Durant cette phase aiguë de l'infection par le VIH-1, il
existe une réplication virale très active,
particulièrement dans les lymphocytes CD4. La virémie plasmatique
cellulaire est très élevée, elle se situe entre 1,000,000
copies/ml et 10,000,000 copies/ml. Le minimum observé est de 50,000
copies/ml. L'antigénie p24 est généralement
positive66.
Durant cette phase, le VIH-1 envahit l'organisme, y compris le
système nerveux central. Des altérations des cellules
monoclucléaires sanguines se produisent, traduites par un déclin
des lymphocytes CD4. Les personnes infectées sont alors très
contagieuses en conséquence de leur taux élevé de VIH dans
le sang et dans les sécrétions génitales66.
Trois semaines à trois mois après la
contamination, la virémie VIH redescend, et le taux de CD4 remonte
rapidement. Les mécanismes de la réponse immune semblent
impliqués dans cette remontée mais certains auteurs ont aussi pu
démontrer par un modèle mathématique que la
cinétique du virus entre les compartiments cellulaires et
extracellulaires pourrait aussi expliquer par elle-même la chute de la
virémie67.
3.3.3. La phase asymptomatique
Après la phase de primo-infection, le virus devient
cliniquement latent pendant une période variant en moyenne de 8 à
10 mais avec des extrémités pouvant aller de 18 mois à
plus de quinze ans (voir plus haut).
Pendant cette période, la réplication
détectable dans les munonucléaires périphériques
sanguins est faible voir indécelable. Le taux de lymphocytes T CD4+ du
sang périphérique décroît de manière lente et
progressive. Pas ou très peu de signes cliniques sont décrits
(lésions cutanées ou des muqueuses).
Malgré une virémie faible ou indécelable,
le virus continue cependant de se répliquer en particulier dans les
organes lymphoïdes. La réponse immune est active (plus de 99%,
environ 2 milliard, des virus produits seraient éliminés, puis
renouvelés chaque jour) mais insuffisante pour prévenir la
réplication virale continuelle dans les organes lymphoïdes et, en
conséquence, le système immunitaire s'épuise
progressivement.
Chez une minorité de personnes infectées par le
VIH une prolifération polyclonale des lymphocytes CD4 dirigés
contre l'infection VIH et contrôlant la virémie a pu être
démontrée. Cette réponse est médiée par les
cytokines (interferon gamma et beta chimiokines). Une réponse de ce type
peut apparaître avec les thérapies antirétrovirales.
La marque de l'émergence de la phase asymptomatique
vers la phase symptomatique est un déclin plus marqué des
lymphocytes CD4 et une augmentation de la virémie suite à la
disparition des cellules folliculaires dendritiques qui piégeaient le
virus dans les ganglions68.
Concernant la durée de la phase asymptomatique, il
existe des progresseurs typiques (de 8 à 10 ans de progression) des
progresseurs rapides (10% des personnes évoluant vers le SIDA en 2
à 3 ans) et des non progresseurs lents (10% des personnes n'ayant pas
évolué vers le SIDA plus de 10 ans après l'infection). Le
tabac et l'âge sont associés à une progression plus
rapide69.
On observe des troubles cognitivo-moteurs chez près de
30% des patients considérés comme asymptomatiques.
Les patients séropositifs pour le VIH peuvent
être catégorisés comme symptomatiques ou asymptomatiques,
suivant des critères biologiques (taux de lymphocytes associés
aux CD4), les pathologies opportunistes apparues ou les troubles cognitifs des
patients31. Il n'y a pas de corrélation entre ces
différents types de critères. Ceci rend l'analyse de la
littérature particulièrement difficile quant à la
signification du terme "asymptomatique", qui n'est parfois pas
spécifiée. Les précisions sur la signification du terme
« asymptomatique » sont d'autant plus nécessaires
que les molécules antirétrovirales provoquent une
réaugmentation du taux de CD4 alors que leur effet sur le cerveau et, a
fortiori, sur les déficits cognitifs, est encore largement inconnu. Ces
ambiguïtés ont conduit certains auteurs à s'orienter
dorénavant vers une catégorisation des patients discernant ceux
n'ayant jamais atteint le seuil fatidique d'un nombre de CD4 inférieur
à 200 cellules/ml et ceux ayant déjà, à un moment
quelconque de la maladie, dépassé cette limite. Cette nouvelle
définition rend plus complexe encore les comparaisons entre les diverses
séries de la littérature.
3.3.4. La phase de SIDA
Le développement des signes et symptômes
caractéristiques de la phase de SIDA déclaré (phase
symptomatique) est généralement parallèle à la
diminution du taux de lymphocytes CD4. En dessous de 200
CD4/millimètres3, la probabilité d'apparition des
symptômes augmente considérablement et le taux de mortalité
augmente.
La charge virale influence aussi l'apparition des
symptômes, environ 70% des personnes ayant une charge virale
élevée (>100 000 copies/ml) voient aussi apparaître des
symptômes cliniques.
Le «Center for Disease Control (CDC)» fournit et met
régulièrement à jour les critères de diagnostic
d'un SIDA avéré70. La classification
révisée de 1993 est basée sur trois catégories
cliniques, A, B et C, chacune subdivisée en trois catégories
selon le taux de lymphocytes CD4.
Une sérologie VIH positive associée à
démence du SIDA est considérée suffisant pour
diagnostiquer un SIDA déclaré.
3.4. Neuroinvasion
et aspects neuropathologiques
3.4.1. Neuropathologie de l'infection
par le VIH-1
Les lésions du système nerveux central ont
été très tôt décrites dans l'histoire de
l'infection par le VIH. Dès 1986, en effet, Navia et coll.18
décrivent, dans la substance blanche et les noyaux gris centraux, une
pâleur myélinique, des infiltrats lymphocytaires
périvasculaires et des macrophages pigmentés.
Cependant les signes cliniques (troubles cognitifs et
démence) ne reflètent pas toujours les lésions
neuropathologiques. En effet, les études clinico-pathologiques ont
montré que seules 50 % des démences du SIDA sont associées
à une encéphalite réplicative71. Pour tenter de
comprendre les relations existant entre les lésions observées et
les troubles cognitifs, les chercheurs ont tenté de décrire plus
en détail les lésions attribuées à la
neurovirulance du VIH-1 et leurs relation avec la réplication virale.
Quatre catégories de lésions ont ainsi été
décrites72 :
L'encéphalite à cellules géantes,
caractérisée par la présence de cellules géantes
multinucléées résultant d'une fusion (induite par le VIH)
de cellules microgliales-macrophagiques infectées. Elle semble
liée à la réplication intracérébrale du VIH.
Elle est souvent associée à des nodules micogliaux (amas de
cellules microgliales) qui ne sont cependant pas spécifiques d'une
infection le VIH.
La poliodystrophie, caractérisée par une gliose
astrocytaire (multiplication et surtout hypertrophie) de la substance grise,
associée à une activation microgliale et à une spongiose
plus ou moins marquée des couches superficielles du cortex.
La leucodystrophie (gliose astrocytaire et pâleur
myélinique) affectant la substance blanche profonde. La pâleur
myélinique semble due plus à un oedème myélinique
qu'à une démyélinisation authentique, qui peut cependant
être observée dans les cas très sévères. Elle
coexiste avec une augmentation de la densité des cellules
microgliales.
La myélopathie vacuolaire est une atteinte de la moelle
épinière avec des lésions de vacuolisation de la substance
blanche, ici encore associées à une infiltration
macrophagique.
3.4.2. Physiopathologie des troubles
neurologiques induits par le VIH1
A) Réplication du VIH-1 dans les différents
types de cellules du Système Nerveux Central
Le VIH-1 a été identifié dans plusieurs
types de cellules du SNC :
Les cellules immunes périphériques (monocytes
circulants, macrophages et lymphocytes), issues du sang circulant.
Les cellules microgliales (monocytes résidents), qui
ont été les premières identifiées comme
siège de la réplication du virus.
Les astrocytes, qui pourraient se comporter comme
réservoir de virus.
Les cellules endothéliales, qui joueraient un
rôle de réservoir et interviendraient dans le processus de
neuroinvasion
L'infection des oligodendrocytes serait possible mais semble
un phénomène rare.
L'infection des neurones est, en revanche, très
controversée, ce qui soulève de nombreuses questions sur le
mécanisme de dysfonctionnement neuronal.
B) Infection du SNC et
déclenchement du dysfonctionnement cérébral
Dans le cerveau infecté par le VIH, les lymphocytes,
les macrophages et les astrocytes sont activés. Ils expriment alors des
marqueurs antigéniques spécifiques sur leur membrane et
produisent des cytokines. Le degré d'activation macrophagique est
lié à la présence de la démence73. Le
virus se propage principalement dans les cellules du système
monocyte-macrophage circulantes et résidentes (microgliales) qui sont
activées. Ces dernières produisent des médiateurs
inflammatoires (cytokines et chimiokines) et des radicaux libres. La production
de cytokines pourrait notamment induire la sécrétion d'oxyde
nitrique (NO) par les astrocytes et d'anions superoxydes par la microglie. Ces
composés se combinent secondairement en peroxynitrate, toxique pour le
SNC. Ainsi l'inflammation dans le système nerveux central pourrait
être à l'origine de lésions neuronales 53, 74,
75.
L'infection du système nerveux central par le VIH-1 et
le déclenchement du dysfonctionnement cérébral sont
liés à trois propriétés du virus : 1) la
neuropénétrance, capacité de pénétration du
virus dans le système nerveux central; 2) le neurotropisme,
capacité du VIH-1 à infecter des cellules du système
nerveux : 3) la neurovirulance, capacité d'induire une maladie
neurologique 74, 76.
C) La
neuropénétrance
Le VIH envahit précocement le système nerveux
central (SNC) sans que la majorité des malades ne présente de
troubles neurologiques au début de la maladie. L'ADN proviral peut
être détecté précocement dans le cerveau des
patients séropositifs avec le plus souvent une faible charge virale
intracérébrale à ce stade77. Aux stades
précoces de l'infection par le virus (stades I et II), survient une
réaction inflammatoire du SNC, avec rupture de la barrière
hémato-encéphalique et lésions de la substance blanche qui
semblent être réversibles29, 78.
Différentes hypothèses sur les voies
d'entrée du virus ont été proposées : 1) Le
franchissement direct de la barrière
hémato-encéphalique79, 2) L'infection des cellules
endothéliales80, 3) Le passage à travers la
barrière hémato-encéphalique de cellules immunitaire
infectées et activées provenant du sang
périphérique81-83. La dernière hypothèse
est la plus fréquente. Cependant ces mécanismes pourraient
exister concomitamment.
Dans des conditions de culture in vitro, l'IL1- et le TNF-
(des cytokines) peuvent induire l'expression de l'ICAM-1 (intercellular
adhesion molecule-1) et de VCAM-1 (vascular cell adhesion molecule-1).
ICAM-1 va permettre l'ancrage des monocytes-macrophages sur les cellules
endothéliales et leur passage à travers la barrière
hémato-encéphalique. Ce mécanisme participe à
l'initiation et au maintient de la réaction inflammatoire du SNC84,
85. Il existe au stade de SIDA une activation de l'endothélium
avec un recrutement des monocytes-macrophages périvasculaires,
indépendamment du développement de troubles neurologiques ou
cognitifs86.
D) Le
neurotropisme
Les souches virales infectant le système nerveux
central ont un tropisme macrophagique. Cette faculté est liée
à des séquences de la boucle V3 de la protéine gp120. Il
n'existe par contre pas de preuves convaincantes de l'existence d'un tropisme
neuronal. Les chimiokines CCR3 et CCR5 permettent aux souches virales à
tropisme macrophagique de pénétrer dans les cellules microgliales
du SNC44. Certains groupes de neurones s'avèrent en outre
capables d'exprimer les récepteurs aux chimiokines87.
E) La
neurovirulance
La neurovirulance apparaît bien plus tardivement que la
neuroinvasion. Son apparition pourrait être due à
l'émergence de souches virales adaptées au système nerveux
et au recrutement accru de monocytes. Cependant, la réplication active
du virus n'est sans doute pas le seul facteur responsables de la
démence. La neurovirulance est aussi liée à des
mécanismes indirects mis en jeu par les réponses immunologiques
et notamment la production de cytokines88 (voir article 2).
3.4.3. Facteurs
génétiques
Le fait que seul un petit sous-groupe de patients soit atteint
d'affections neurologiques au cours de l'infection par le VIH suggère
une composante génétique s'exerçant par exemple sur les
récepteurs viraux et/ou la réponse immune75. Une
étude génétique récente rapporte d'ailleurs (sur un
petit nombre de patients) une susceptibilité accrue pour la
démence chez des patients porteurs de l'allèle E4 de
l'alipoprotéine E (APOE) (impliquée par ailleurs dans la maladie
d'Alzheimer et dans d'autres affections voisines)89. De même,
des délétions ou des mutations sur différents gènes
codant pour des corécepteurs viraux ou pour des protéines
contrôlant l'intensité de la réponse immune ou encore sur
les récepteurs au chimiokines, pourraient jouer un rôle dans
l'apparition des troubles cognitifs liés au VIH-1.
II. EVALUATION DES
TROUBLES COGNITIFS LIES A L'INFECTION PAR LE VIH-1
1. La démence du SIDA
La démence du SIDA, aussi appelée syndrome
démentiel cognitivo-moteur du VIH-1 ou encéphalopathie
VIH-190 affecte 15% des patients séropositifs78.
C'est une complication neurologique du SIDA, apparemment liée à
l'infection du système nerveux par le VIH, dont le mécanisme
exact n'est pas connu. Cette démence a été décrite
pour la première fois par Navia et coll.19 et introduite dans
la définition du SIDA en 1987 (Une encéphalopathie VIH-1
associée à une sérologie VIH-1 positive permet de poser le
diagnostic du SIDA).
La démence du SIDA est caractérisée par
des troubles mnésiques, des troubles des fonctions exécutives et
des fonctions attentionnelles ainsi que par des manifestations
comportementales. Ces troubles rappellent en première analyse ceux
décrits dans les démences sous-corticales.
Les données concernant la date d'apparition, le mode
d'installation et la nature de l'évolution dans les principales
descriptions de la littérature restent contradictoires.
Pour Navia et collaborateurs19, le processus
débute le plus souvent de manière insidieuse avec une progression
régulière (quelques mois) ou une stabilisation. Plus rarement, il
survient de façon aiguë ou subaiguë et progression rapide
(quelques semaines).
Pour Selnes et collaborateurs91, la démence
apparaît dans la grande majorité des cas de manière rapide.
Une fois le diagnostic posé la durée estimée de survie est
de 6 mois.
Description clinique:
Classiquement, le patient est atteint de troubles cognitifs
modérés évoluant de façon rapide, le mode
d'entrée dans la maladie pouvant être, plus rarement, brutal.
Rapidement, les altérations intellectuelles conduisent à un
état déficitaire plus important. L'inertie et le ralentissement
sont les éléments les plus marquants. Souvent réduit, le
langage est pourtant largement préservé. Le raisonnement est, en
revanche, nettement déficitaire, surtout si les tâches
imposées obligent à des opérations successives
reliées entre elles. Les réalisations motrices sur ordre sont
lentes et des consignes complexes ne sont pas exécutées ou
demeurent inachevées. L'impression générale est celle d'un
appauvrissement global de l'activité mentale et d'un déficit de
toutes les performances aux épreuves explorant les fonctions
cognitives.
En fin d'évolution, la majorité des patients
sont atteints d'un dysfonctionnement cognitif global et de troubles moteurs
majeurs (akinésie, tremblements, myoclonies). En outre, certains sont
affectés de signes frontaux à l'examen neurologique.
L'indifférence affective, l'apragmatisme et le mutisme ainsi que le
développement de troubles du contrôle sphinctérien vont
contribuer à l'établissement d'une situation d'abandon, de
passivité et de perte de contact avec l'entourage. A certains moments,
pourtant, de courtes phases de lucidité inattendue contrastent avec
l'aspect général de démence globale.
Actuellement, deux nomenclatures regroupant l'ensemble des
manifestations cognitives, motrices et comportementales permettent de poser le
diagnostic de "démence probable": le DSM-IV (Critères
diagnostiques et généraux de démence du
DSM-IV92 et la classification de l'American Academy of
Neurology31.
1.1.
Classification et critères du DSM-IV : Formes sévères
Critères généraux de démence
du DSM-IV92:
La démence est définie :
1- Par la présence d'un déficit mnésique
2- L'association du déficit mnésique à
au moins l'une des perturbations cognitives suivantes:
- Aphasie
- Apraxie
- Agnosie
- Déficit des fonctions exécutives
(Pensée abstraite et capacité de planifier, initier,
exécuter, contrôler et arrêter un comportement complexe).
3 - Le déficit doit être suffisamment
sévère pour causer des perturbations dans la vie sociale ou
professionnelle du patient.
4- Ce déficit doit être acquis et
évolutif.
Critères diagnostiques d'une démence du SIDA
(DSM-IV)92
« Le patient présente des pertes de
mémoire, des difficultés de concentration, des difficultés
à résoudre des problèmes, une apathie et un retrait
social. Occasionnellement, on peut observer un délire, des illusions ou
des hallucinations. L'examen physique montre souvent un tremblement, une
difficulté à exécuter des mouvements rapides et
répétés, des troubles de l'équilibre, une ataxie,
une hypertonie, une hyper-réflexie généralisée, des
signes d'un syndrome frontal et des mouvement oculaires saccadés ou une
perturbation des mouvements oculaires de poursuite ».
Ces critères reposent donc sur l'observation d'un
déficit mnésique associé à un déficit des
fonctions exécutives.
1.2.
Classification et critères de l'American Association of Neurology:
Formes sévères
La classification de L'American Academy of Neurology
(AAN)31 (annexe 2), est une nomenclature, élaborée
conjointement par l'OMS et par l'AAN, qui vise à standardiser le
diagnostic clinique et à rendre l'ensemble des recherches comparable.
Elle fournit une nouvelle terminologie ainsi qu'un nouvel ensemble de
critères diagnostiques.
Les formes sévères sont regroupées soit
sous l'appellation de "HIV-1-associated dementia complex" (annexe 2) soit sous
celle de "HIV-1-associated myelopathy". Elles sont suffisantes pour poser le
diagnostic de SIDA avéré.
On observe, par rapport au DSM-IV, qu'il n'est pas
indispensable d'observer un déficit mnésique pour poser un
diagnostic de démence.
Les formes légères font partie du
"HIV-1-associated minor cognitive / motor disorder". Elles ne permettent pas de
poser le diagnostic de SIDA avéré bien qu'elles puissent
être observées chez les patients sidéens.
1.3. Stades de
Price et Worley
Une échelle évaluant le degré de
détérioration de la démence (Stades démentiels du
SIDA) a été défini par Price et Worley93
(annexe 3).
Cette échelle évalue la gravité des
atteintes cognitives et motrices, ainsi que leur répercussion sur la vie
quotidienne. Elle comprend cinq stades ordonnés de 0 (normal) à 4
(stade final) et un sous-stade (0,5) dans lequel sont classés les
patients dont les symptômes sont équivoques. Le stade 1 signal
l'entrée dans la démence.
1.4. Les
échelles de démence
1.4.1. L'échelle de
démence du SIDA de Power-McArthur
Une équipe travaillant sur le neuro-SIDA à
Baltimore (EU) a développé en 1994-1995 une échelle rapide
évaluant la démence du SIDA, comportant un score allant de 0
(détérioration maximum) à 16 (pas de
déficit)94 (annexe 4). Cette échelle permet d'obtenir
une évaluation rapide des troubles mnésiques et du ralentissement
psychomoteur typiques de la démence du SIDA90.
1.4.2. Les batteries
neuropsychologiques de la démence
Un certain nombre de tests et de batteries neurologiques ou
neuropsychologiques ont été
développées95-113. La plupart des grandes batteries
américaines sont très longues (3-4 heures de passation) et
limitent souvent les résultats à un score global calculé
à partir des résultats aux différents tests (exemple, 0
point pour un score équivalent à celui de la moyenne de la
population, 1 point pour un score inférieur d'un écart-type
à celui de la population générale, 2 points pour un score
s'écartant de deux écart-types; ou encore 1 point pour un test
d'une catégorie cognitive anormal, 2 points si deux tests sont
anormaux). Pour revue voir dans le livre de Harrison et McArthur90.
2. Evaluation des troubles cognitifs
modérés
2.1.
Classification et critères du DSM-IV: Troubles cognitifs
modérés
Addendum du DSM-IV114.
Le désordre neurocognitif modéré est
défini comme correspondant à des perturbations cognitives
insuffisamment sévères pour entraîner un diagnostic de
démence, mais suffisantes pour avoir des implications dans la vie
courante. On note que les diagnostiques de "démence" et de
"désordre neurocognitif modéré" pourraient avoir des
pronostics différents et que la relation existant entre ces deux
désordres reste encore obscure, notamment en ce qui concerne
l'évolution des troubles dans le temps (Becker et al., 1994). Ce
désordre neurocognitif doit présenter au moins deux des
perturbations des fonctions cognitives suivantes, évoluant depuis au
moins deux semaines : 1- mémoire (apprentissage ou rappel) 2- fonctions
exécutives (planification, abstraction, etc.) 3- capacités de
concentration 4- capacités motrices 5- langage (compréhension,
production...). On doit pouvoir mettre en évidence une étiologie
d'une perturbation cognitive liée à un dysfonctionnement du SNC
grâce à un examen physique ou une technique de laboratoire (y
compris l'imagerie cérébrale). Un déclin ou une
anormalité aux tests neuropsychologiques doit être observable. Le
déficit cognitif doit être la cause d'une perturbation moyenne
dans les activités socioprofessionnelles et représenter un
déclin par rapport à un niveau antérieur. Enfin, la
perturbation cognitive ne doit pas correspondre à un désordre
cognitif ou psychiatrique connu.
2.2.
Classification et critères de l'American Association of Neurology:
Formes légères
l'AAN a proposé des critères de
définition d'un trouble cognitivo-moteur mineur du VIH-131
(annexe 5). Les déficits observés, contrairement au syndrome
démentiel, sont insuffisants pour poser un diagnostic de SIDA
déclaré. La continuité entre les troubles mineurs et la
démence n'a pas été prouvée. La différence
majeure entre le syndrome démentiel et les troubles cognitivo-moteurs
associés au VIH-1, tient au degré de handicap dans la vie de tous
les jours31. Ce degré de handicapé est
évalué cliniquement par des échelles de vie quotidienne et
doit être objectivement vérifiable par l'interrogation d'un
informateur clef. Les patients présentant des troubles cognitivo-moteurs
mineurs sont capables de réaliser la plupart des activités de la
vie quotidienne. Bien que la majorité des patients soient capables de
maintenir un travail usuel, les performances au travail, comme les
activités sociales, peuvent être moyennement perturbées.
Cependant, les patients ne sont pas dépendants d'autres personnes. Ils
peuvent se nourrir, maintenir leur hygiène personnelle, manipuler de
l'argent, faire des achats, utiliser des transports publiques ou conduire une
voiture. Les activités plus complexes de la vie quotidienne, comme le
souvenir d'un rendez-vous ou la prise des médicaments, peuvent
être occasionnellement perturbées.
En 1996, Marder et coll.35 ont rendu
opérationnelles les définitions de l'article de Janssen de
199131. Suivant ces nouveaux critères, les patients ont des
troubles cognitifs si leur score à l'un des tests proposés
dévie de 2 écart-types (ET), par rapport à la moyenne des
sujets témoins, ou bien de 1 ET si ce score est calculé sur deux
tests. Les tests proposés incluent six domaines cognitifs:
mémoire verbale, mémoire visuelle, construction, habiletés
psychomotrices, construction, habiletés motrices et fonctions frontales.
L'importance de la gène dans la vie de tous les jours est d'autre part
mesuré par des échelles de vie quotidienne.
La principale différence entre les troubles cognitifs
mineurs et majeurs (démence) concerne, comme il était possible de
le prévoir à la lecture de l'ensemble des critères actuels
de démence, le degré de détérioration des
activités de la vie quotidienne. Dans le cas du syndrome
démentiel associé au VIH-1, l'altération des
capacités de travail, professionnelles et privées, est
évidente alors que dans le cas du trouble cognitivo-moteur mineur,
seules les activités les plus exigeantes sont affectées. On
connaît mal les liens existant entre ces deux formes. En particulier, on
ne sait ni si elles correspondent à la même entité ni si
les patients qui ont une forme mineure évolueront vers une forme
sévère.
3. Données neuropsychologiques
3.1. Troubles
cognitifs et controverses
La présence de troubles cognitifs modérés
liés au VIH dépend du degré d'évolution de la
maladie. Les données de la littérature concernant les stades
asymptomatiques, comme défini par les critères biologiques (c'est
à dire nombre de CD4>200 cellules/ml), montrent une grande
hétérogénéité dans le statut cognitif des
patients, alors que la présence de troubles cognitifs au stade de SIDA
est rarement contestée. La question de la présence ou non de
troubles cognitifs aux stades asymptomatiques a une grande importance car elle
peut avoir des conséquences dans des professions particulières
comme par exemple les aviateurs militaires34. Selon l'article
récent de l'AAN (1996)35, et tous stades du VIH-1 confondus,
20,7% des patients séropositifs pour le VIH ont des troubles
cognitivo-moteurs modérés, contre 24% de démences. Ces
chiffres ont été obtenus avant 1995, date à laquelle les
combinaisons thérapeutiques ont été
systématisées en France. On voit que la proportion de patients
concernés par l'existence de troubles modérés
était, avant les antiprotéases, très importante
comparativement à la proportion des patients atteints de démence.
Ce premier constat appelle deux questions :
1) quelle va être l'évolution des
déficits chez les patients atteints de troubles modérés
avant l'introduction des combinaisons thérapeutiques?
2) chez les patients soumis à ces combinaisons, des
troubles modérés vont-ils émerger et, si oui, vont-ils
évoluer vers une démence?
Dans une revue antérieure à 1996 (c'est à
dire portant sur les études effectuées jusqu'en 1995 avant
l'avènement des antiprotéases), Sahakian et coll.108
relèvent les controverses majeures de la littérature. En effet,
certains auteurs n'ont mis en évidence aucun déficit cognitif aux
stades précoces de l'infection, et ce malgré le grand nombre de
patients testés115. Pour eux, et malgré l'usage de
tests de temps de réaction, censés être les plus sensibles
(voir plus loin;104, 116, les troubles cognitifs
n'apparaîtraient qu'au stade de SIDA102. D'autres auteurs, en
revanche, observent des troubles cognitifs chez près de 30% des patients
séropositifs au cours de la période asymptomatique, et ce sans
relation évidente avec des paramètres immunologiques et
virologiques113, 117. Enfin, il a été
suggéré que la présence de troubles cognitifs
précoces puisse prédire une évolution rapide vers un SIDA
et qu'elle était parallèle à une diminution
accélérée des CD4 ainsi qu'une augmentation de la
positivité à l'antigène p24118. On peut se
demander pourquoi de telles discordances sont observées alors que les
études portaient généralement sur des cohortes d'une
centaine de patients. Deux principales réponses peuvent être
suggérées : le manque de cohérence entre les
populations testées, notament la mauvaise adéquation des groupes
de contrôle et l'absence de relation directe entre les paramètres
biologiques et troubles cognitifs.
3.1.1. Cohérence entre les
populations testées
Les niveaux socioculturels des populations diffèrent
certainement d'une étude à l'autre et l'on connaît l'effet
de cette variable sur les tests neuropsychologiques classiquement
utilisés, notamment au cours des suivis prospectifs119.
Plusieurs auteurs ont souligné l'importance d'une "réserve
cognitive" dans l'apparition tardive des troubles cognitifs, les patients avec
un niveau éducatif plus bas étant plus sensibles au déclin
précoce des fonctions cognitives109, 120, 121. Par ailleurs,
l'un des principaux problèmes au cours de ces études a
consisté à apparier les patients séropositifs avec des
sujets témoins "compatibles" sur le plan de l'âge et du niveau
socioculturel. Selnes et collaborateurs122 ont ainsi posé
l'hypothèse d'une origine multifactorielle de ces troubles (troubles
psychiatriques, dépression, toxicomanie, carences, etc.) sans lien avec
l'infection par le VIH, expliquant que les troubles cognitifs directement
liés au virus, sont assez rares aux stades précoce de la
maladie.
3.1.2. Paramètres biologiques et
troubles cognitifs
Des études récentes d'imagerie
cérébrale et de neuropathologie post-mortem ne retrouvent pas de
relation entre ces marqueurs morphologiques et l'intensité des troubles
cognitifs. L'atrophie cérébrale, qui prédomine dans la
substance grise sous-corticale123, 124 est retrouvée
fréquemment chez les patients avec un SIDA déclaré et ne
présentant pas de démence125. Il a été
récemment montré que si l'atrophie cérébrale peut
être associée au stade CDC, aucune donnée univoque ne
permettait de la corréler au degré de troubles cognitifs126,
127. L'IRM anatomique des patients présentant des troubles
cognitifs modérés peut être anormale. Néanmoins, il
n'y a pas de corrélation entre l'intensité des anomalies
observées en IRM et la gravité de l'atteinte
cognitive128. D'un autre coté, chez des patients
décédés du SIDA et ayant développé une
démence, les marqueurs neuropathologiques (cellules géantes
multinucléées et la palleur myélinique) ne sont
observés que dans 40 à 50 % des cas71, 129. Cette
proportion est probablement encore moindre chez les patients ayant
présenté des troubles cognitifs modérés. Des
études prospectives130 et rétrospectives131
n'ont pas retrouvé de corrélations entre la perte neuronale
corticale et les troubles cognitifs.
De plus, la relation entre l'immunodépression (taux de
lymphocytes CD4) et l'apparition des troubles cognitifs est très
discutée (voir chapitre 2, titre I).
3.2. Evolution des
troubles
Certains auteurs suggèrent que la progression de la
maladie soit associée à une évolution rapide des troubles
neuropsychologiques110. D'autres, au contraire, constatent une
évolution extrêmement discrète des troubles cognitifs.
Ainsi, Dunbar et collaborateurs132 ont étudié
l'évolution des performances neuropsychologiques de patients ARC
(AIDS-Related- Complex, CDC groupes IVA et IVC2) évoluant (progresseurs)
ou n'évoluant pas (non-progresseurs) vers un SIDA déclaré.
Le suivi longitudinal de ces patients a montré, d'une part, que les deux
groupes de patients séropositifs, progresseurs et non progresseurs,
étaient plus ralentis et présentaient plus de troubles
attentionnels que des patients séronégatifs; d'autre part, les
progresseurs montraient une tendance (non statistiquement
vérifiée) à être plus ralentis que les non
progresseurs.
Les troubles cognitifs modérés correspondent au
ralentissement psychomoteur, aux déficits de mémoire verbale
épisodique118, 133, ainsi qu'aux déficits des
fonctions exécutives et attentionnelles108. Les fonctions
cognitivo-motrices les plus sensibles concernent la dextérité
manuelle et la coordination bi-manuelle, la fluence et la mémoire
verbale, les temps de réaction et les fonctions attentionnelles. Les
fonctions frontales sont plus atteintes chez les patients avec SIDA
déclaré que chez les séropositifs asymptomatiques, ce qui
pourrait suggérer que les déficits frontaux, comme les
déficits visuo-spatiaux, ne soient pas caractéristiques des
stades précoces de la maladie mais plutôt des stades
tardifs133. Enfin, chez les patients au stade de SIDA, sans troubles
cognitifs, un ralentissement moteur discret est un signe constamment
observé.
3.3. Le
ralentissement psychomoteur
L'importance capitale de la valeur prédictive du
ralentissement psychomoteur dans l'apparition d'une démence a
déjà été mentionnée plus haut. Certains
auteurs suggèrent que les tests de temps de réaction seraient les
plus sensibles, en faveur d'un déficit central de rapidité
motrice ou psychomotrice99, 134.
Des études de suivi longitudinal de cohortes de
patients séropositifs ont permis de démontrer que l'augmentation
des temps de réaction psychomoteurs est le signe précurseur le
plus significatif de l'évolution des troubles mineurs vers une
démence91. En effet, si les différentes études
peuvent aboutir des conclusions différentes concernant les performances
aux tests cognitifs, elles montrent une remarquable cohérence quant
à l'effet de l'infection par le virus sur le ralentissement moteur et/ou
cognitif dès les stades précoces de la maladie. Une étude
récente sur la contribution des différents tests
neuropsychologiques dans le tableau des troubles cognitifs montre que les
troubles moteurs, associés aux troubles mnésiques, expliquent
à eux deux la majorité des tableaux de
détérioration135.
L'importance de la valeur prédictive du ralentissement
moteur dans l'apparition d'une démence a été
récemment confirmée par le suivi prospectif de patients. Dans un
suivi longitudinal de 9 ans de patients séropositifs, Sacktor et
collaborateurs136 ont montré que les patients asymptomatiques
qui présentaient un ralentissement psychomoteur avaient plus de risque
d'évoluer vers un SIDA et/ou de développer une démence.
Baldeweg et collaborateurs95 remarquent que l'EEG topographique
montre que l'activation anormale de plusieurs aires motrices est un signe plus
sensible que les mesures des tests psychomoteurs comportementaux.
Les études menées après l'introduction
des combinaisons thérapeutiques antirétrovirales ont
montré l'influence de ces molécules dans l'augmentation de la
vitesse psychomotrice137, 138, cependant, y compris chez les
patients traités par des antiprotéases, le ralentissement
psychomoteur reste associé a une plus grande
mortalité139 (voir chapitre 3, titre II).
3.4. Les troubles
des fonctions exécutives et attentionnelles
Des troubles des fonctions exécutives peuvent
apparaître dès le stade asymptomatique de la maladie. Sahakian et
collaborateurs108 ont étudié les fonctions
exécutives de trois groupes de sujets : des patients séropositifs
asymptomatiques, des patients séropositifs pour le VIH-1 symptomatiques
et des témoins séronégatifs grâce à une
batterie de tests informatisés (CANTAB) permettant d'étudier la
mémoire visuospatiale, l'attention et les fonctions exécutives.
Ces auteurs ont ainsi montré que les patients symptomatiques comme
asymptomatiques présentaient des déficits aux tests des fonctions
exécutives et attentionnelles mais pas de troubles de la mémoire
visuelle. Les troubles exécutifs concernent la mémoire de travail
spatiale (MTS), les capacités de planification des actions (test de la
Tour de Londres-TOL) et le transfert des ressources attentionnelles (test dit
de EDID shift). Ces trois tests ont en particulier montré leur
sensibilité aux atteintes du cortex préfrontal et du circuit
striato-préfrontal monoaminergique (pour revue voir Robbins et coll.
1994140.
Dans une étude récente, Jasiukaitis et
Fein141 ont étudié d'une les effets de facilitation
par la répétition d'un stimulus (répétition ou
priming par le même stimulus), généralement
gérés par les fonctions visuoperceptives associées au
cortex extra-striatal et, d'autre part, le priming sémantique (ou
associé verbalement) correspondant à une fonction
attribuée aux régions frontales antérieures. Il ont pu
montrer, chez les patients séropositifs pour le VIH, des troubles du
priming sémantique avec un priming par le même stimulus intacte.
Ces résultats, consistants avec ceux de l'imagerie montrant des
lésions sous corticales, suggèrent un dysfonctionnement
sous-cortico-frontale dans la pathologie VIH. Ce déficit dans le priming
sémantique, avait déjà été
démontré dans une étude de Nielsen-Bohlman et
coll.142 datant de 1997, où des patients séropositifs
pour le VIH, avec des troubles cognitifs modérés, montraient
aussi un trouble de l'effet de priming sémantique(*) suggérant un déficit
d'activation des réseaux sémantiques automatiques.
Les déficits des patients sont en outre
corrélés au degré de difficulté des tâches et
à l'impossibilité qu'éprouvent les patients à
mettre en place une stratégie (notamment pour la MTS et la TOL). Ces
deux dernières observations suggèrent que les troubles
modérés des patients VIH soient liées à un
dysfonctionnement frontal, compte tenu de leur sensibilité aux facteurs
«complexité» et «activation de
stratégies».
Enfin, une étude récente de Llinkin et
coll.143 réalisée avec une version adaptée du
test de Stroop, montre un ralentissement psychomoteur chez les patients
séropositifs pour le VIH ainsi qu'un effet d'interférence. Ces
résultats suggèrent une déficience de l'inhibition chez
ces patients.
Tous ces résultats montrent que toute étude sur
les fonctions cognitives au cours de l'infection VIH se doit de tester de
façon la plus complète possible le fonctionnement frontal puisque
les tests classiquement employés en neuropsychologie, tels que la
fluence verbale, ne révèlent pas toujours de troubles aux stades
précoces de la maladie.
3.5. Les troubles
mnésiques
3.5.1. Cadre théorique des
différents troubles mnésiques
La psychologie cognitive a permis, au cours de la
dernière décennie, de mettre en évidence l'existence d'un
polymorphisme de la mémoire. Des formes variées de mémoire
se différentient par le mode de stockage, de codage et de rappel de
l'information, ainsi que par les structures cérébrales qu'elles
mettent en jeu144. Nous allons très brièvement en
donner un aperçu (pour revue, voir le livre de Tulving traduit par B.
Deweer145). La première grande distinction observée
est celle d'une différence de durée de conservation de
l'information à laquelle s'ajoute une différence de
capacité de stockage. On observe sur ce critère la mémoire
à court terme et la mémoire à long terme (modèle
d'Atkinson et Shiffrin), ces deux stocks de mémoire fonctionnant
probablement en parallèle. On distingue actuellement la mémoire
de travail qui stocke des informations qui ne sont valables que pour
l'activité en cours, est sensible aux interférences et est
régulièrement remise à zéro. On fait aussi la
distinction entre la nature et le type de traitement qu'impliquent le stockage
des informations reçues. Sur ce mode, on a pu différencier la
mémoire déclarative et la mémoire
procédurale146 et, dans la première, la mémoire
épisodique et la mémoire sémantique147.
Chez les patients séropositifs pour le VIH-1, la
plainte mnésique est fréquente et pose souvent la question d'une
dépression sous-jacente. Il est important de déterminer
précocement si un déficit mnésique réel existe afin
d'évaluer le handicap que cela représente pour le patient dans sa
vie courante. Nous ne développerons dans ce chapitre que les troubles
mnésiques ayant été reportés dans la
littérature comme liés à la pathologie VIH-1, c'est
à dire ceux concernant principalement la mémoire de travail, la
mémoire épisodique verbale et la métamémoire.
3.5.2. La mémoire de travail
La mémoire de travail a été
définie par Baddeley148 comme un ensemble de processus dans
lequel un centre de gestion alloue les ressources attentionnelles
nécessaires à la manipulation et au traitement des informations
dans deux stocks de mémoire tampon : la «boucle phonologique»
et le «calepin visuo-spatial». Cette mémoire active, aussi
appelée «mémoire
représentationnelle»149, met ainsi en jeu d'une part des
caractéristiques verbales et d'autre part, la manipulation mentale
d'items visuo-spatiaux.
L'infection VIH est associée à un déclin
de certaines fonctions exécutives et attentionnelles qui sont en
connexion étroite avec les capacités de mémoire de
travail. Stout et collaborateurs111 ont mis en évidence un
déficit des patients séropositifs symptomatiques (non
déments) dans des tests d'empan de lecture et d'empan chiffré
inversé. Les patients asymptomatiques montrent une tendance (non
significative) à être déficitaires dans ces tests,
suggérant ainsi que ce type d'atteinte de mémoire de travail
puisse débuter aux stades asymptomatiques. Ces résultats
suggèrent en outre que la progression de la maladie est sensible
à des tâches de mémoire de travail impliquant une
manipulation des informations temporairement stockées mais
préserve la mémoire à court terme «non active»,
évaluée dans des tâches impliquant un simple stockage des
informations - empan chiffré en ordre direct, par exemple.
Les résultats obtenus par Sahakian et
collaborateurs108 portent principalement sur la capacité
d'utilisation de la mémoire de travail spatiale. Le test consistait
à retrouver des objets virtuels (sur écran d'ordinateur)
cachés dans des boîtes que l'on peut visiter au cours d'essais
successifs. Le test est conçu de telle manière qu'on
évalue les performances en fonction de la difficulté de la
tâche en présentant au sujet un nombre plus ou moins grand de
boîtes (de 3 à 8) et qu'on estime, outre le temps
d'exécution, la stratégie développée par les
sujets. Les résultats à ce test montrent que les patients
symptomatiques et asymptomatiques ne diffèrent pas entre eux. Ces deux
populations sont nettement déficitaires par rapport aux sujets
séronégatifs, uniquement lorsque la difficulté de la
tâche est accrue (8 boîtes). De même, la stratégie
développée par les sujets, qui est corrélée
positivement au nombre de bonnes réponses, est significativement
réduite chez les deux groupes de patients par rapport aux
témoins. Ce résultat est retrouvé par ces mêmes
auteurs avec la tâche de la «Nouvelle Tour de Londres» qui met
en jeu la mémoire de travail et la capacité à planifier
des mouvements : cette tâche requière d'imaginer des
déplacements de billes de couleur de façon à copier un
modèle d'arrangement spatial de ces billes. La tâche sera d'autant
plus complexe en terme d'attention soutenue et de mémoire de travail que
le nombre de mouvements nécessaire est grand. Les patients, là
encore, ne sont déficitaires que lorsque le nombre de mouvements
à imaginer est important.
Ce profil (mauvais scores aux tests de mémoire de
travail spatiale étroitement corrélés à la
complexité et l'utilisation de stratégies
autosuggérées) est typique d'un déficit
préfrontal150 et suggère que les déficits de
mémoire de travail interviennent indépendamment du stade CDC sans
prédire l'évolution de la maladie. Par ailleurs, ce type de
déficit montre qu'il est possible d'observer un dysfonctionnement du
cortex préfrontal aux stades CDC II et III108.
Le déficit au test de fluence verbale,
caractéristique des fonctions frontales151, est
fréquent bien que non systématique chez les sujets
présentant des troubles cognitifs modérés. En effet, la
réalisation de ce test, qui met en jeu la manipulation en mémoire
de travail de mots répondant à une consigne (début du mot
par une lettre ou appartenance à une catégorie) n'est pas
toujours affectée108, 119. Il a été
récemment montré, grâce à l'imagerie
cérébrale par résonance magnétique fonctionnelle
(IRMf) que la réalisation de cette tâche requière
l'activation de structures frontales : le sillon frontal inférieur
gauche, le cingulaire antérieur et le sillon frontal
supérieur152. L'activation et l'intégrité de
ces structures frontales sont également nécessaires à la
réalisation des tâches de mémoire de travail
décrites plus haut mais il semble que ce soit l'hémisphère
droit qui soit recruté153 lors de l'exécution d'un
plan d'action (planification, organisation temporelle). On ignore cependant si
les déficits des patients peuvent être liés, non à
des lésions du cortex préfrontal droit (nous avons vu plus haut
qu'il n'y avait pas de corrélation à ce niveau) mais à des
dysfonctionnements (potentiellement réversibles) de ces structures.
3.5.3. Mémoire
épisodique
Plusieurs autres études portant sur la mémoire
verbale ont suggéré un profil mnésique
«sous-cortical» chez les patients séropositifs. En effet,
35,5% des patients séropositifs ont des troubles de l'acquisition et de
la rétention de mots alliés à une relative
préservation des capacités de reconnaissance154. Dans
une étude de la mémoire verbale antérograde de patients
séropositifs utilisant le «California verbal learning test»,
Peavy et collaborateurs155 ont montré que les patients
séropositifs symptomatiques présentaient par rapport à des
témoins séronégatifs, plus d'erreurs dans des mesures
d'acquisition et de rétention de la mémoire verbale. Ils
utilisaient moins souvent une stratégie d'organisation sémantique
des mots cibles pour le rappel. Le profil de troubles des patients
symptomatiques était similaire à celui de patients souffrant de
maladie de Huntington et différent de celui de patients avec maladie
d'Alzheimer. Il consiste en un rappel libre faible, avec des intrusions y
compris en rappel différé. Nos résultats112
montrent que des patients présentant un SIDA déclaré avec
troubles cognitifs modérés présentent plus de troubles du
rappel libre dans une épreuve d'apprentissage d'une liste de mots,
comparés à des séropositifs asymptomatiques. Le rappel
indicé (la performance de rappel après avoir donné un
indice sémantique des mots oubliés) est diminué, alors que
la reconnaissance est correcte. Une épreuve identique chez des patients
déments par rapport à ceux présentant des troubles
modérés montre une aggravation du rappel libre alors que la
sensibilité aux indices reste identique. Cette dissociation dans la
mémoire épisodique entre un rappel libre très
altéré et une relative préservation du rappel
indicé est caractéristique du profil mnésique
sous-cortical156. Les patients présentant ce profil ont
tendance à avoir un taux de CD4 plus bas et à être en phase
de SIDA déclaré, ce qui suggère que, contrairement aux
troubles de mémoire de travail, les troubles de mémoire
épisodique apparaissent plus tard dans le décours de la
maladie154.
3.5.4. Métamémoire
La relation entre la métacognition et les troubles
cognitifs chez des patients séropositifs, a été
explorée par plusieurs équipes et les résultats sont
contradictoires. Hinkin et collaborateurs157 se sont
intéressés plus spécifiquement à la relation entre
les troubles mnésiques, la dépression et la plainte
mnésique. Ils ont identifié d'une part, un sous-groupe de
patients surévaluant leurs troubles mnésiques et
significativement plus déprimés, et un sous-groupe de patients
qui, au contraire, sous évaluent leurs troubles cognitifs. Ces auteurs
émettent l'hypothèse que cette anosognosie des troubles pourrait
être liée à l'atteinte sous-corticale.
3.6. Les
modèles animaux
Des anomalies sont observées dans les potentiels
évoqués corticaux en réponses à des stimuli visuels
ou auditifs chez le singe et le chat infectés respectivement par le SIV
(simien immunodéficience virus) ou le FIV (félin
immunodéficience virus), comme c'est le cas chez l'homme158.
Le principal modèle animal utilisé à l'heure actuelle est
le macaque Rhésus, pour des raisons de facilité de
conditionnement à des tâches comportementales complexes. Un
modèle félin (FIV) a également été mis au
point159 mais l'étude du comportement chez le chat est
généralement plus difficile à mener que chez le singe et
très peu d'études existent.
L'inoculation du SIV à des macaques Rhésus
conduit à un syndrome d'immunodéficience proche du SIDA
humain160. Les singes Rhésus infectés par le SIV ont
une immunosuppression et développent des troubles neuropathologiques et
neuropsychologiques proches de ceux des humains161. La
différence la plus importante est cependant une progression plus rapide
de la maladie simienne162. Les études comportementales
conduites chez le singe ont un objectif double : 1) déterminer la nature
des déficits cognitifs et moteurs provoqués par l'infection chez
le singe et voir quelles structures cérébrales sont liées
aux fonctions perturbées et 2) mesurer l'efficacité des
thérapies sur ces déficits163. L'utilisation d'un
modèle animal des effets de l'infection par le virus sur les fonctions
cognitives et motrices permet en outre de connaître les performances
cognitives et habiletés motrices des sujets avant l'infection par le
virus, ce qui n'est pas le cas chez l'homme. Cette dernière
donnée est importante car, incontrôlable chez les sujets humains,
elle est source de variabilité interindividuelle et peut être une
des causes de contradiction entre les différentes études (voir
plus haut). En outre, les études conduites chez les modèles
animaux permettent de tester les troubles fonctionnels induits par le VIH-1 en
recherchant notamment les relations avec les modifications de taux de certains
marqueurs biologiques.
Comme chez les patients, l'infection par le SIV provoque des
déficits moteurs dans une très grande proportion des singes
infectés et des déficits cognitifs dans 50% des cas. Les
mêmes anomalies concernant la barrière
hémato-encéphalique, et le relargage de protéines virales
et de substances neurotoxiques sont trouvées158.
3.6.1. Processus mnésiques
Le nombre d'animaux utilisés dans ces études est
très restreint (de 6 à 15 en général) ce qui rend
difficile une présentation de résultats en terme statistiques.
Pour cette raison, nous mentionnerons simplement les tâches pour
lesquelles des déficits ont été répertoriés.
Les tests effectués chez le singe mettent en jeu
différents processus de mémoire visuelle : rétention et
apprentissage d'une discrimination visuelle, mémoire de travail reposant
sur ces discriminations. La reconnaissance visuelle simple, comme chez l'homme,
n'est qu'exceptionnellement affectée163. Les fonctions plus
généralement touchées concernent la discrimination
visuelle (rétention et apprentissage) et la mémoire de
travail.
Il s'agit généralement de tâches
d'appariement de stimuli (MTS); à chaque essai, on présente
à l'animal plusieurs stimuli qui apparaissent un par un sur un
écran tactile; après un délai variable pendant lequel
l'écran est vierge; chaque stimulus-cible, présenté
précédemment est affiché sur l'écran associé
à un nouveau stimulus. Pour obtenir la nourriture, l'animal doit toucher
les stimuli-cibles. Ce test met en jeu la reconnaissance visuelle des stimuli
présentés et la mémoire de travail. Murray et
collaborateurs163 ont ainsi montré que trois singes sur dix
présentaient des déficits dans ce test. Fox et
coll.158 ont mis au point une batterie de tests cognitivo-moteurs
pour le macaque. Dans cet ensemble de tests, la mémoire de travail est
évaluée par une procédure identique à celle
employée chez le sujet humain108. Contrairement à ce
qui est observé chez l'homme, ces auteurs ne mettent pas évidence
de déficits de mémoire de travail chez les macaques
infectés alors que l'exécution d'autres tâches (voir plus
loin) est affectée.
3.6.2. Processus attentionnels
Le test de transfert des ressources attentionnelles
(EDID-shift) utilisé chez le singe158, comme celui de la
mémoire de travail, a été adapté à partir du
CANTAB, mis au point chez l'homme108. Les animaux ont un
déficit de transfert de l'attention d'une dimension de la scène
visuelle à une autre dimension. En revanche, les capacités
d'adaptation à une nouvelle règle ou de discrimination visuelle
sont intactes. Des résultats similaires au même test ont
été obtenus chez les patients symptomatiques et
asymptomatiques108 et chez des primates humains et non humains ayant
des lésions du cortex préfrontal140. Dans le
modèle félin (FIV), ce sont essentiellement une augmentation du
taux d'activité et une distractibilité importante qui sont.
L'importance des troubles étant en relation directe avec la diminution
du taux de CD4 observés164, 165. Ces troubles chez le chat
seraient en lien avec des anomalies du lobe frontal, en particulier un
diminution du taux de N-acétyl-aspartate166 et une
réduction de la densité neuronale dans le cortex frontal167,
168.
3.6.3. Motricité fine
Chez le singe, les déficits moteurs sont plus
fréquents et interviennent plus tôt après l'inoculation que
les déficits cognitifs163.
En fait, ces déficits de motricité fine sont
quasiment systématique chez les animaux infectés.
L'habileté motrice chez le singe est généralement
testée grâce à un dispositif permettant d'évaluer la
dextérité des animaux à prélever, sur une table
rotative à vitesse variable, des morceaux de nourriture placés
dans de petites cupules ou un équivalent du test de Pegboard.
L'expérimentateur mesure la vitesse maximale de la table pour laquelle
l'animal parvient à prélever la moitié de la nourriture.
Les animaux infectés, entraînés préalablement dans
cette tâche, ne parviennent pas à prendre la nourriture à
la même vitesse que les animaux non infectés, ce qui
suggère, comme chez l'homme, un ralentissement moteur au cours de la
réalisation de tâche de dextérité fine. Les
perturbations à ce test sont corrigées par les traitements
à base de zidovudine161.
3.6.4. Conclusion
L'utilisation de modèles simiens de l'infection par le
SIV a globalement montré que les mêmes fonctions cognitives et
motrices étaient affectées que chez l'homme, notamment en ce qui
concerne les habiletés motrices fines et les fonctions attentionnelles.
De plus, comme c'est le cas chez les sujets humains, les dysfonctionnements
observés n'ont jamais pu être corrélés avec la
localisation ni l'extension des lésions inflammatoires du SNC. Ils ont
cependant pu être corrélés, dans certains cas, au taux
d'acide quinolinique dans le liquide céphalo-rachidien, et ce chez
l'homme comme chez l'animal. Il semble donc probable que les déficits
neuropsychologiques observés chez l'homme comme chez le singe,
infectés respectivement par le VIH et le SIV, résultent d'effets
indirects des virus.
3.7. La
dépression et les autres troubles psychiatriques
3.7.1. Importance des troubles de
l'humeur et des troubles psychiatriques dans la pathologie VIH :
Fréquence et gravité
Dans une revue de la littérature, Lyketsos et
Federman24 montrent qu'il existe à la fois une augmentation
du risque d'une infection VIH dans la population présentant des troubles
psychiatriques et une relativement haute fréquence de troubles
psychiatriques chez les patients séropositifs. Des troubles
psychiatriques ont été fréquemment décrits au cours
de l'infection par le VIH. Notamment, la dépression,
l'anxiété, les troubles de la personnalité, les
toxicomanies, l'alcoolisme et les troubles thymiques sur un versant maniaque.
Ces derniers représentent 8% des manifestations de la démence du
SIDA23 et ils semblent être secondaires à l'infection
par le VIH169.
Les états dépressifs sont connus pour perturber
les fonctions cognitives chez certains patients170. Il a
été clairement démontré, notamment par une
étude de l'organisation mondiale de la santé, qu'il existe une
augmentation de la prévalence de la dépression chez les patients
infectés par le VIH171. La dépression est un des
principaux diagnostics différentiels des troubles cognitifs
modérés de l'infection par le VIH. Cependant, les études
sur la relation existant entre la dépression et les résultats
neuropsychologiques des patients séropositifs tendent à montrer,
que si un patient peut évidemment présenter des troubles
liés à une dépression, l'existence et la nature des
troubles cognitifs du VIH est indépendante de la
dépression172, 173. Cependant cette question reste l'objet
d'un débat car certaines études trouvent au contraire plus de
troubles cognitifs chez les patients plus déprimés174.
Afin de déterminer, chez un patient séropositif
concomitamment dépressif, la nature des troubles cognitifs, des
traitements antidépresseurs «d'épreuve» sont
fréquemment proposés.
3.7.2. Reflet de la plainte
cognitive
Les questions sur la relation entre la plainte cognitive des
patients, leurs performances effectives et les troubles de l'humeur,
fréquents dans la pathologie VIH, ont été longtemps
débattues. Il est important en effet pour l'avenir du patient de savoir
si la plainte cognitive reflète ou non une atteinte des fonctions
supérieures et quelles sont ses relations avec une éventuelle
dépression sous-jacente. Dans la pratique clinique, on constate que ces
phénomènes sont extrêmement intriqués,
l'amélioration d'une symptomatologie anxio-dépressive peut
être concomitante d'une diminution, voire d'une disparition des troubles
des fonctions supérieures et la persistance des troubles peut faire
poser l'hypothèse d'une encéphalopathie au VIH
débutante175.
Plusieurs études ont suggéré qu'une part
des plaintes cognitives des patients était liée à des
troubles anxio-dépressifs plutôt qu'à des
dysfonctionnements cognitifs. Cependant les populations concernées par
ces études étaient souvent particulières. Dans une
première étude de Van Gorp et coll.176 par exemple, ce
sont essentiellement des patients asymptomatiques qui sont
étudiés, et les auteurs ne trouvent pas de relation entre la
plainte cognitive et les résultats neuropsychologiques, alors que la
dépression est associée aux plaintes cognitives. Une autre
étude de Wilkins et coll. incluait essentiellement des patients ayant
des troubles psychiatriques177. Les auteurs concluaient à une
relation entre les troubles psychiatriques (en particulier la
dépression) et la plainte cognitive, indépendamment de la
performance neuropsychologique. Dans cette étude, la plainte motrice
était cependant associée à des troubles moteurs. Une
troisième étude, portant sur 92 patients séropositifs pour
le VIH, ne trouve pas de relation entre la plainte et les performances
cognitive alors que la plainte est associée à une détresse
émotionnelle178.
D'autres études ont démontré que, au
contraire, il existe une relation étroite entre la plainte cognitive et
les troubles neuropsychologiques, en particulier les troubles de la
mémoire épisodique et le ralentissement moteur179.
Stern et coll.180 ont comparé 84 hommes
séronégatifs à 46 patients hospitalisés (et
séronégatifs) et à 78 patients séropositifs pour le
VIH-1 (49 asymptotiques et 29 symptomatiques). Ils ont montré non
seulement que les plaintes cognitives étaient plus fréquentes
chez les patients porteurs du VIH-1, mais aussi que dans ce même groupe
ces plaintes étaient liées aux résultats des tests
neuropsychologiques. De façon similaire, Mapou et coll.181
ont comparé 27 sujets contrôles séronégatifs
à 79 patients porteurs du VIH1 (asymptomatiques et symptomatiques) et
ont montré que les patients séropositifs qui se plaignaient de
difficultés cognitives avaient significativement plus de troubles aux
tests attentionnels, rapidité motrice et de mémoire que ceux qui
ne se plaignaient pas. Cependant, les patients ayant une plainte cognitive
avaient aussi plus de symptômes dépressifs et anxieux. Les auteurs
en concluent à l'indépendance des troubles neuropsychologiques et
affectifs.
Dans une autre étude, Beason-Hazen et
coll.182 ont examiné 133 patients asymptomatiques
séropositifs pour le VIH-1 et 80 témoins
séronégatifs. Ils ont trouvé une relation entre la plainte
cognitive et des anomalies aux tests de rapidité psychomotrice et de
temps de réaction. Ce lien persiste après correction des
données pour la dépression, ce qui conduit les auteurs a conclure
que la dépression n'intervient pas dans la relation entre la plainte et
les performances cognitives.
Dans la pratique, le clinicien dois donc être
particulièrement attentif, chez les patients séropositifs pour le
VIH-1 aux plaintes concernant la mémoire, l'attention ou un
ralentissement psychomoteur qui risquent d'être le reflet d'un trouble
cognitif, parfois très discret.
3.7.3. Relations entre les troubles de l'humeur et les
troubles cognitifs
La relation directe entre les déficits
neuropsychologiques et la présence ou l'absence de dépression a
intéressé de nombreux auteurs. Certain183 montrent que
si les symptômes de la dépression sont plus fréquents chez
les sujets séropositifs pour le VIH, il n'y a pas de relation directe,
par contre, entre la dépression et les troubles cognitifs. Dans une
étude plus récente, Castellon et coll.184 rapportent
que si la dépression n'est pas liée à un ralentissement
psychomoteur, il existe en revanche un lien entre la vitesse psychomotrice et
l'apathie, suggérant que des symptôme d'apathie
indépendants du syndrome dépressif puissent être un
indicateur important de l'atteinte du système nerveux central.
Enfin, Une étude chez 79 militaires atteints du VIH (et
27 contrôles séronégatifs) montre qu'il existe un lien
entre la plainte cognitive et le déficit neuropsychologique et entre la
plainte cognitive et les troubles de l'humeur (dépression et
anxiété)181. Par contre il n'existait pas de relation
entre le déficit neuropsychologique et les troubles de l'humeur, c'est
à dire que la plainte cognitive peut refléter soit un
déficit neuropsychologique effectif, soit un trouble de l'humeur, mais
que les effets de chacun sont indépendants.
La dépression n'est donc probablement pas à
l'origine des troubles cognitifs observés dans l'infection par le VIH-1.
Son caractère fluctuant, chez ces patients, alors que les troubles
cognitifs sont persistants explique probablement l'absence de
corrélation dans ces études s'intéressant à un
nombre important de patients. Au niveau individuel cependant il est possible
qu'à partir d'un certain seuil de dépression
sévère, celle-ci intervienne dans les troubles cognitifs. Des
résultats similaires, sur l'absence de lien entre la dépression
et les troubles cognitifs, suggérant l'existence d'un seuil de
sévérité de la dépression dans cette interaction
ont pu être trouvés notamment dans la maladie de
Parkinson185.
3.7.4. Relations entre les troubles de
l'humeur et l'évolution de la maladie
D'autres auteurs se sont interrogés sur le stade
d'apparition de l'anxiété et la dépression186,
187 au cours de l'infection par le VIH. Ils ont pu montrer que ces
symptômes sont indépendant du stade de l'infection VIH et sont
essentiellement liés à la présence de facteurs
psychologiques et psychosociaux. Par contre il semble qu'inversement, davantage
de stress et de difficultés psychosociales puissent
accélérer le cours de l'infection VIH188. Cela est
particulièrement important quand dans certaines couches de la population
américaine avec de lourds problèmes psychosociaux, des auteurs
ont montré que l'apparition d'une infection par le VIH chez les femmes
reste moins prédictif que les difficultés psychosociales
déjà existantes d'un avenir psychologique très
perturbé189, ce qui laisse présager d'un
développement de la pathologie VIH plus rapide et plus
sévère chez ces femmes.
Enfin il ne faut pas négliger l'influence des facteurs
psychosociaux et socio-démographiques sur la compliance aux traitements
antirétroviraux. La mauvaise compliance au traitement est en effet un
des facteurs les plus importants de l'échec thérapeutique et la
reprise de la maladie. Dans une étude espagnole, les auteurs montrent
notamment que les facteurs de mauvaise compliance aux traitements sont un
âge plus jeune, la toxicomanie, la dépression et un manque de
support social190.
3.7.5. Interactions entre les troubles
cognitifs et la toxicomanie
Bien qu'il soit reconnu depuis de nombreuses années que
la toxicomanie soit un facteur de risque important dans l'acquisition de
l'infection VIH191, l'importance du problème dans la
propagation de l'épidémie commence seulement a être
réalisée et prise en charge. Des nouvelles mesures visent en
effet à considérer l'effet des substances toxicomaniaques sur la
pathologie VIH. On s'interroge aussi sur l'influence du comportement des
patients toxicomaniaques dans la propagation de l'épidémie par
voie sexuelle, puisque les toxicomanes usant des drogues comme la cocaïne,
les methamphetamines, les morphiniques et l'alcool ont un taux plus
élevé de comportement sexuels a risque192.
Certains auteurs comme Selnes et coll. ou Concha et
coll.98, ont montré que dans le cas des toxicomaniaques (par
injection intraveineuse) il ne semble pas y avoir de différence au sein
de la population séropositive avec les autres groupes à risque en
ce qui concerne l'apparition et l'évolution des troubles
cognitifs193. Cependant ces résultats sont largement
contestés et il semble y avoir de nombreux arguments pour penser que, au
contraire, la toxicomanie peut influencer l'apparition et l'évolution
des troubles cognitifs.
Les drogues194 et le VIH peuvent, tous deux
affecter le système nerveux central. Plusieurs études ont pu
démontrer la fréquence plus élevée de
démence du SIDA dans la population toxicomane195. Il a
été suggéré en particulier que la toxicomanie
pourrait potentialiser le développement de la démence du SIDA via
un effet sur le système immunitaire196. Une étude
italienne a pu démontrer une augmentation de la fréquence des
troubles cognitifs chez les séropositifs toxicomanes197. Plus
alarmant encore, des études neuropathologiques dans une cohorte
Britannique ont montré que 56% des cerveaux de patients
séropositifs toxicomanes avaient des lésions d'encéphalite
du VIH (des cellules géantes multinucléées et un
antigène p24 positif) contre seulement 15 % des cerveaux des
séropositifs homosexuels non toxicomanes129, 198. Enfin des
patients séropositifs et toxicomanes avec un important ralentissement
psychomoteur ont une détérioration neurologique plus rapide avec
une activation des macrophages dans le SNC importante73.
Si des troubles cognitifs et des lésions
neuropathologiques sont donc plus fréquemment trouvés dans la
population séropositive toxicomane, on ignore encore largement, par
contre comment les drogues peuvent contribuer à ces troubles.
Un des problème de ces études tiens à la
nature même de la toxicomanie et à la difficulté de suivre
l'évolution de ces patients.
Des études expérimentales ont pourtant
montré que la cocaïne peut contribuer à léser la
barrière hémato-encéphalique rendant le cerveau plus
accessible à au virus199, 200. De plus, La cocaïne et
les méthamphétamines causent des troubles des
cathécholamines201 et la cocaïne ainsi que les
amphétamines agissent sur les neurones dopaminergiques202 (la
cocaïne étant un inhibiteur non sélectif des transporteurs
de dopamine et les amphétamines pouvant produire des
dégénérescences des extrémités
dopaminergiques) alors que les patients séropositifs pour le VIH ont des
taux de cathécholamines plus faible dans le liquide
céphalo-radichien et des signes cliniques comme, le
Parkinsonisme203 et les myoclonies204, qui
suggèrent des troubles dopaminergiques et noradrénergiques.
Enfin, Les opiacés (comme l'héroïne et la
morphine) inhibent directement les fonctions immunes205 et
pourraient aggraver l'infection VIH206-208.
La toxicomanie doit donc être considérée
comme un facteur de risque supplémentaire à l'apparition des
troubles cognitifs, dans le cas particulier des patients infectés par le
VIH.
4. Article 1 : Mise au point d'une batterie
neuropsychologique. « Similar subcortical pattern of cognitive
impairment in AIDS patients with and without dementia »
4.1.
Introduction : Pourquoi une nouvelle batterie ?
Le mécanisme des troubles cognitifs compliquant le
VIH-1 est mal connu. Des études de neuropathologie et d'imagerie
cérébrale récentes ne trouvent pas de relation entre les
marqueurs anatomiques et l'intensité des troubles cognitifs.
L'étude neuropathologique de cerveaux de patients
décédés d'une démence du SIDA a montré que
les cellules géantes multinucléées et la paleur
myélinique, qui sont les marqueurs neuropathologiques de
l'encéphalite VIH, sont observées seulement dans 40 à 50 %
des cas71, 129. De plus, des études
prospectives130 et rétrospectives131 n'ont pas
retrouvé de corrélations entre la perte neuronale corticale et
les troubles cognitifs. D'un autre côté, l'atrophie
cérébrale, qui prédomine dans la substance grise
sous-corticale123, 124 est aussi retrouvée fréquemment
chez les patients au stade de SIDA et ne présentant pas de
démence125. Une autre étude ne trouve pas de relation
entre l'atrophie sous corticale et les résultats
neuropsychologiques126. Un travail récent a
précisé ce problème de corrélations en
démontrant que s'il existe une relation entre l'atrophie
cérébrale et le stade clinique de la maladie, il n'en existe pas,
entre l'atrophie et les troubles cognitifs209.
Finalement, la relation entre l'atrophie et les troubles
cognitifs est peu claire. Cependant, la réponse à cette question
est importante car elle permettrait d'élucider le fait que la
démence soit liée directement à une perte neuronale
néocorticale, ou qu'elle pourrait résulter de mécanismes
indirects comme des atteintes neuronales, potentiellement réversibles
!
Les études neuropsychologiques qui ont
été réalisées sont peu nombreuses et n'ont pas
recherché spécifiquement un profil cortical ou sous cortical des
troubles. L'absence de corrélation avec un tableau de troubles cognitifs
«global» pourrait faire place à des relations
spécifiques avec des troubles cognitifs sous corticaux.
Le but de cette étude est d'évaluer les
fonctions cognitives de patients infectés par le VIH, avec et sans
démence, et d'élucider l'implication corticale ou sous corticale
dans ces troubles.
Nous utiliserons une batterie neuropsychologique,
élaborée à partir d'une revue de la littérature,
dans le but de sélectionner les tests les plus sensibles aux troubles
observés dans ce cadre spécifique de l'encéphalite du VIH.
Cette batterie neuropsychologique permet d'évaluer les six domaines
cognitifs spécifiques, proposés dans les critères du
groupe de travail sur le VIH de l'Académie de Neurologie
Américaine31 : l'attention/concentration, la
rapidité psychomotrice, l'abstraction/raisonnement, la
mémoire/apprentissage, les habiletés visuospatiales, ainsi que le
langage.
Il a été décidé d'élaborer
une batterie courte, afin de pouvoir examiner dans leur lit d'hôpital les
patients les plus immunodéprimés en diminuant le biais
attentionnel dû à leur fatigue générale.
4.2.
Matériel et méthodes
Quarante-cinq patients au stade de SIDA et 8 patients
séropositifs asymptomatiques (SP) ont été examinés.
Tous les patients étaient hospitalisés dans des service
d'Infectiologie ou de Médecine Interne. Les critères d'exclusion
comprenaient une histoire psychiatrique, une toxicomanie ou un alcoolisme
actif, une infection opportuniste du système nerveux central, une
infection oculaire opportuniste, un lymphome cérébral et une
incapacité des patients à répondre à des questions
très simples. Un examen neurologique était réalisé
à la demande du service ayant en charge le patient, ou à la
demande du neuropsychologue en cas de présence de troubles cognitifs.
Les patients étaient répartis en quatre groupes
suivant les critères du DSM-IV et le score à l'échelle de
Mattis (Mattis Dementia Rating Scale)210(annexe 6).
Le groupe de patients séropositifs asymptomatiques (SP)
ne présentait pas de troubles cognitifs (Mattis = 136).
Les trois autres groupes concernaient des patients au stade de
SIDA.
Les patients qui ne présentaient Pas de Troubles
Cognitifs (PTC) avaient une Mattis = 136 (n=25).
Les patients qui présentaient un trouble cognitif,
défini par un score de Mattis < 136 mais qui ne rencontraient pas les
critères de démence du SIDA du DSM-IV étaient
appelés «Troubles cognitifs modérés» (TCM)
(n=12).
Les patients qui rencontraient les critères de
démence du DSM-IV étaient appelés «Démence du
SIDA» (n=8), indépendamment de leur score à la Mattis.
Les facteurs de risque étaient attribués
à des comportements homosexuels (n=34), hétérosexuels
(n=6), toxicomaniaques par injection intraveineuse (n=8), une exposition
professionnelle (n=1) et des facteurs inconnus (n=4).
Les 45 patients sidéens avaient un âge moyen
(#177; SD) de 38,9 #177; 7,4 ans. Au moment de l'examen, cinq patients
n'étaient pas sous antirétroviraux, six étaient sous
monothérapie, 17 sous bithérapie et 17 sous trithérapie
(sous combinaisons thérapeutiques). Les CD4 moyens étaient de
107,8 #177; 135,8 cellules/mm 3, répartis de 3 à 553 (36
patients < 200 CD4 cellules/mm 3 et 9 patients > 200
cellules/mm 3). La durée moyenne de la maladie (#177; SD)
était de 8,38 #177; 0,45 années.
Les 8 patients asymptomatiques avaient un âge moyen de
37,4 #177; 6,5 ans. Au moment de l'examen, un patient ne recevait pas
d'antirétroviraux, 5 étaient sous bithérapie et 2 sous
trithérapie. Les CD4 moyens (#177; SD) étaient de 492,2 #177;
190,5 cellules /mm3. La durée moyenne de la maladie (#177; SD)
était de 6,12 #177; 1,02 années.
4.3. Examen
Neuropsychologique
Les patients passaient un examen neuropsychologique d'une
durée moyenne d'une heure.
L'efficience cognitive générale était
évaluée au moyen de deux échelles, le «Mini-Mental
State Examination» (MMSE)211 et l'échelle
d'évaluation de la démence de Mattis. Une composante
«langage» du MMSE a été considérée,
comprenant les épreuves de dénomination,
répétition, compréhension, lecture et écriture.
L'échelle de Mattis est divisée en cinq sous-tests :
l'attention, l'initiation, la construction, la conceptualisation et la
mémoire, son score global est compris entre zéro (performance
minimale) et 144 (performance maximale).
La rapidité psychomotrice et le contrôle
exécutif étaient évalués par le Trail Making Test,
partie A (TMT/A) et partie B (TMT/B)212. La différence entre
les deux scores (TMT/B-A) permet d'éliminer la composante
«rapidité motrice» du test commune à ces deux parties
pour estimer le ralentissement cognitif.
Le contrôle moteur fin et la dextérité
motrice étaient évalués avec le Purdue Pegboard Test (main
dominante (PP-md), main non dominante (PP-mnd) et deux mains (PP-2m))213,
214. Un patient avec une neuropathie périphérique des
membres supérieurs n'a pas pu passer ce test.
La mémoire épisodique était
évaluée avec le test de Grober et Buschke (G&B)215
qui permet de contrôler l'encodage et le rappel des informations par des
indices sémantiques. L'encodage était contrôlé en
demandant au patient de montrer et lire chacun des items à
mémoriser, présentés 4 par 4, en réponse à
leur catégorie sémantique. Tous les 16 items devaient être
rappelés immédiatement avec un rappel indicé avant le
début du test mnésique. Le rappel s'effectuait sous la forme de
trois séries de rappels libres et indicés, séparées
par 20 secondes d'interférence (compter en arrière), plus une
série de rappel libre et indicé différés, 20
minutes plus tard. Après chaque série de rappel libres, les
catégories sémantiques des mots oubliés étaient
proposées comme indice sémantique. Les réponses correctes
obtenues au rappel libre et au rappel indicé étaient
additionnées pour obtenir un score de rappel total. Après le
rappel différé, un test de reconnaissance des mots parmi des
distracteurs associés sémantiques ou neutres était
proposé. Afin de déterminer si de faibles performances des
patients au rappel libre étaient liées à un déficit
d'activation des stratégies de récupération en
mémoire d'informations stockées, ou à une
incapacité à fixer l'information, un index de sensibilité
aux indices (ISI) était calculé pour déterminer la
proportion de réponses correctes en réponse aux indices fournis.
Cet index était calculé par la formule : ((rappel total
global - rappel libre total) / (48 - rappel libre total)) X 100. Le nombre
total de réponses correctes dans les trois rappels libres était
appelé score de «rappel libre» et le nombre total de mots
rappelés spontanément ou après un indice appelé
«rappel total».
Ainsi cette batterie permettait d'explorer rapidement les
fonctions sous-cortico-frontales, potentiellement altérées dans
la pathologie VIH. A cet examen, nous avons ajouté un évaluation
clinique et qualitative de la dépression, par le test de MADRS
(Montgomery and Asberg Depression Rating Scale)216. Les patients
étaient considérés comme déprimés s'ils
obtenaient un score supérieur à 19.
4.4.
Résultats
4.4.1. Efficience cognitive globale
Les quatre groupes de patients (SP, PTC, TCM, Déments)
ne différaient pas en âge ou en nombre d'années
d'éducation. Il n'y avait pas de corrélations entre le niveau
cognitif global (MMSE ou Mattis) et l'âge ou le taux de CD4. Les scores
de MMSE et de Mattis étaient corrélés (r=0,662; p<
0,0001).
Les données neuropsychologiques sont montrées
dans l'annexe 7:
Les quatre groupes de patients étaient
significativement différents dans presque tous les tests
neuropsychologiques réalisés, à l'exception notable de la
composante langage du MMSE.
Les scores de tous les sous-tests de l'échelle de
Mattis étaient significativement différents dans les quatre
groupes de patients : attention (p=0,004), construction (p=0,0061),
conceptualisation (p<0,0001), mémoire (P<0,0001) et initiation
(p<0,0001). Les patients déments étaient significativement
plus déficitaires que les patients avec des TCM au score global de
l'échelle de Mattis (p=0,0022). La figure correspondant au
résultats des sous-tests de la Mattis est montrée dans l'article
1.
Comparés aux patients PTC, les patients avec des TCM
montraient une détérioration significative au MMSE (p=0,001)
(annexe 7).
4.4.2. Ralentissement psychomoteur
Les scores moyens des quatre groupes de patients au
ralentissement psychomoteur sont montrés, pour le test de Purdue
Pegboard (figure 7):
Si les quatre groupes de patients sont significativement
différents dans une analyse globale, la comparaison des groupes deux
à deux montre qu'il n'existe pas de différence significative
entre le groupe de patients avec des troubles cognitifs modérés
(TCM) et les patients sidéens sans troubles cognitif (PTC). Tous les
autres groupes de patients sont significativement différents deux
à deux. Ceci est valable quel que soit la modalité du test, c'est
à dire avec la main dominante, avec la main non dominante et avec les
deux mains.
La comparaison des résultats neuropsychologiques entre
les patients avec des TCM et les patients SP (annexe 7) montre un
ralentissement moteur significatif chez les patients avec des TCM, dans les
test de Purdue Pegboard (PP-md (p=0,002), PP-mnd (p=0,008), PP-2m (p=0,003) et
TMT/A (p=0,015). Comparés aux TCM, les patients déments
étaient significativement plus mauvais aux scores du Purdue Pegboard
(PP-md (p=0,023), PP-mnd (p=0,004), PP-2m (p=0,003)).
Figure 7. Scores moyens au test de Purdue Pegboard,
par groupe de patients

SP: Séropositif; PTC: Patient sans trouble
cognitif; TCM: Trouble cognitif modéré.
Les scores sont exprimés en moyenne #177;
ES.
4.4.3. Fonctions exécutives
Les scores moyens des quatre groupes de patients pour le test
TMT sont montrés dans la figure 8. Les quatre groupes de patients sont
globalement différents. Les analyses comparant les groupes deux à
deux montrent que:
Dans la composante TMT/A, la plus impliquée dans
l'évaluation de la rapidité motrice, il n'existe pas de
différence significative entre le groupe de patients avec des troubles
cognitifs modérés (TCM) et les patients sidéens sans
troubles cognitif (PTC). Tous les autres groupes de patients sont
significativement différents deux à deux.
Dans les composante TMT/B et surtout dans la composante
TMT/B-A, plus impliquées dans l'évaluation d'un ralentissement
« cognitif », il n'y a pas de différence entre les
groupes de patients PTC et SP et entre les patients Déments et TCM. La
différence significative apparaissant uniquement dans les comparaisons
entre un groupe de patients ne présentant pas de troubles cognitifs (SP
ou PTC) et un groupe de patients présentant des troubles cognitifs (TCM
ou Déments).
Comparés aux patients PTC, les patients avec des TCM
montraient une détérioration significative au sous-test attention
de la Mattis (p=0,0002), au sous-test conceptualisation de la Mattis
(p=0,0006), au sous-test initiation de la Mattis (p=0,002), au TMT/B (p=0,006)
et TMT/B-A (p=0,007) (annexe 7).
Figure 8: Scores moyens des différents groupes de
patients au Trail-Making test
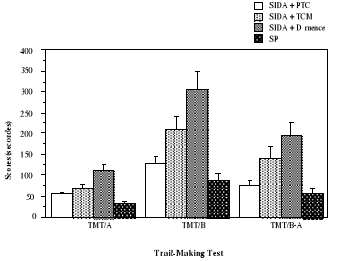
SP: Séropositif; PTC: Patient sans trouble
cognitif; TCM: Trouble cognitif modéré.
Les scores sont exprimés en moyenne #177;
ES.
4.4.4. Mémoire
Une analyse sur les mesures répétées du
G&B a été réalisée pour prendre en compte
l'effet de la répétition (de l'apprentissage) sur le rappel
(figure 4). Les quatre groupes de patients (groupes) étaient
significativement différents dans les scores de rappel libre
(p<0,0001). L'effet de la répétition des essais (essai1, 2, 3,
différé) (répétition) était aussi
significatif (p<0,0001) mais l'interaction
groupes*répétition n'était pas significative (la
figure sur les résultats du test de Grober et Buschke est donnée
dans l'article 1).
Le rappel total, au contraire, montrait un effet des
groupes (p<0,0001), un effet de répétition
(p<0,0001) et une interaction groupes*répétition. Les
patients déments faisaient significativement plus d'intrusions que les
autres patients (p<0,0001). Trois patients déments avaient un ISI
< 50% et quatre un ISI = 50%, ces deux groupes n'étaient pas
différents à leur score de Mattis ou de MMSE.
Comparés aux patients PTC, les patients avec des TCM
montraient une détérioration significative au sous-test
mémoire de la Mattis (p=0,008). Au test de Grober et Buschke, les
patients TCM présentaient un score plus bas de rappel libre (p=0,033),
un moins bon index de sensibilité aux indices (p=0,004) et un moins bon
score de rappel total (p=0,0006) que les patients PTC. Comparés aux TCM,
les patients déments étaient significativement plus mauvais au
sous-test mémoire de la Mattis (p=0,015)
Les patients déments avaient un score plus bas au
rappel libre du G&B (p=0,002) mais pas au score de rappel total.
4.4.5. Dépression
Les niveaux de dépression, mesurés par
l'échelle de MADRS, sont globalement différents dans les quatre
groupes. Cette différence est due au fait que les patients avec un TCM
étaient plus déprimés que les patients PTC (p=0,0029) et
séropositifs (p=0,0029) (figure 9) mais n'étaient pas
différents des déments.
Le score de MADRS n'était pas corrélé
avec l'efficience cognitive globale (Mattis, MMSE) ou avec le ralentissement
psychomoteur.
Figure 9: Scores des quatre groupes de patients à
l'échelle de dépression de MADRS
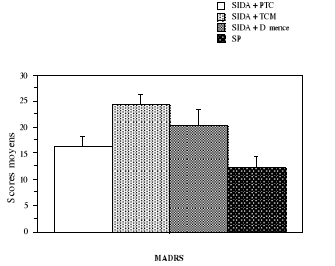
SP: Séropositif; PTC: Patient sans trouble
cognitif; TCM: Trouble cognitif modéré.
Les scores sont exprimés en moyenne #177;
ES.
4.5. DISCUSSION GENERALE ET
CONCLUSIONS
Les troubles cognitifs du VIH ont été
décrits par plusieurs auteurs au cours de ces dix dernières
années91, 102, 106, 110, 126, 217, 218. Bien qu'il soit
généralement reconnu que la démence du SIDA est une
démence sous corticale156, peu d'auteurs se sont
intéressés à préciser les caractéristiques
du tableau neuropsychologique.
Cette étude avait pour but de définir plus
précisément le patron sous-cortical des troubles cognitifs des
patients au stade de SIDA, avec ou sans démence. Nous avons, pour ce
faire, choisi une batterie de tests neuropsychologiques permettant d'identifier
des patients avec une atteinte cognitive modérée et
sévère. Cette batterie était suffisamment courte pour
être bien tolérée.
Nos résultats montrent que les troubles cognitifs
n'étaient pas liés à la dépression, puisque les
patients avec des troubles cognitifs modérés étaient plus
déprimés que les patients des autres groupes, sans aucune
relation avec le ralentissement psychomoteur ou les troubles mnésiques.
Une telle absence de lien entre la dépression et les troubles cognitifs
des patients sidéens a déjà été
trouvée172.
Nos résultats permettent d'analyser les trois
degrés de troubles cognitifs rencontrés chez ces patients
sidéens : NTC, TCM, Déments.
Le premier groupe (NTC) était différent des
séropositifs asymptomatiques uniquement en terme de ralentissement
psychomoteur. Ces résultats sont concordants avec des études
longitudinales réalisées chez les séropositifs
évoluant vers un SIDA déclaré, qui montraient qu'un
ralentissement psychomoteur est le seul facteur présymptomatique,
précurseur d'une évolution vers une démence.
Le second groupe (TCM) était caractérisé
par:
1) une aggravation significative du ralentissement
psychomoteur et des troubles du contrôle exécutif (ralentissement
des performances au TMT/B et TMT/B-A). Ce qui suggère que la
bradyphrénie soit un élément important et un trait
précoce des troubles cognitifs modérés observés
chez les patients avec un SIDA déclaré.
2) Une chute des performances aux sous-tests «initiation,
attention, conceptualisation et mémoire» de l'échelle de
Mattis;
3) une altération précoce du rappel libre et du
rappel indicé.
L'ensemble de ces éléments est en faveur d'un
dysfonctionnement frontal précoce.
Dans le groupe des déments, le ralentissement
psychomoteur était plus marqué, avec une composante importante de
bradykinésie (Purdue Pegboard, TMT/A), alors que la composante cognitive
était plus stable (TMT/B-A). L'importance du ralentissement psychomoteur
chez les patients sidéens (avec ou sans démence) a
déjà été soulignée106, 133, 218.
Dans les sous-tests de l'échelle de Mattis, le sous-test
«mémoire» était le seul qui était
significativement détérioré par rapport aux patients avec
des TCM. Dans le test de Grober et Buschke, le tableau mnésique des
patients déments, comparé à celui des patients avec des
troubles modérés, était caractérisé par une
aggravation significative des troubles du rappel libre, mais le rappel total
restait identique, confirmant, chez ces patients, la nature
sous-cortico-frontale des troubles.
Nos résultats confirment donc la distinction entre des
patients présentant des troubles modérés
caractérisés par une bradyphrénie, une inattention, une
baisse de la capacité de conceptualisation, ainsi qu'une baisse de
l'initiation et troubles mnésiques et des patients déments chez
qui la bradyphrénie et les troubles du rappel libre sont plus
sévères. Elle souligne cependant l'identité des troubles
dans les deux groupes. Pourtant, nous n'avons pas pu examiner les patients
déments incapables d'effectuer les épreuves et ne pouvons
affirmer que le profil neuropsychologique reste identique chez ces patients
présentant un stade avancé de démence.
Le profil neuropsychologique caractéristique de la
démence sous corticale a été défini par
Albert219 comme étant «une association d'une forme
particulière de trouble mnésique, d'un ralentissement
général des activités intellectuelles et d'une
altération de la personnalité, d'une altération de la
capacité à manipuler des connaissances acquises».
Dans cette étude, le tableau neuropsychologique des
patients avec des troubles cognitifs modérés était en
accord avec une perturbation sous corticale précoce des troubles
cognitifs. Comme attendu, les patients déments montraient un
déclin significatif dans toutes les mesures, à l'exception
notable de la composante langage du MMSE. Cependant, ces patients avec une
démence débutante, montraient un tableau neuropsychologique
caractérisé par la présence de bradyphrénie et
bradykinésie et de troubles mnésiques en l'absence d'aphasie,
apraxie, agnosie. Le profil mnésique, concernant à la fois le
rappel libre et l'index de sensibilité aux indices, montrait une baisse
de performance chez les patients avec des TCM et les patients déments,
comparés aux patients, au stade SIDA, sans troubles cognitifs.
Cependant, le déficit au rappel libre était plus
sévère chez les patients déments que chez les patients
avec des troubles cognitifs modérés, et il n'y avait pas de
différence en terme de réponse à l'aide d'un indice
sémantique. Ce type de troubles du rappel libre amélioré
par un indice sémantique était, globalement en accord avec le
tableau mnésique généralement observé dans les
démences sous corticales156, 220. Cependant, si cette
amélioration par un indice était supérieure à ce
qui a été observé chez les patients avec une maladie
d'Alzheimer, elle était moins bonne que chez des patients avec d'autres
démences sous corticales, comme la maladie de Huntington ou de
Parkinson156. La variabilité dans le rappel total
observée dans les performances des patients présentant une
démence du SIDA, nous a conduits à définir deux
sous-groupes, en fonction de l'index de sensibilité aux indices (ISI).
Premièrement, un sous-groupe de patients avec un ISI élevé
(=50), ce qui suggère que l'information encodée ait
été stockée, mais que les patients ont du mal à la
récupérer spontanément, un trait similaire à ce qui
est généralement le cas dans les démences sous corticales.
Et deuxièmement, un sous-groupe de patients avec un ISI faible (<50)
ce qui suggère que l'information encodée n'ait pas
été stockée et ne peut donc pas être
récupérée avec une aide indicée, un trait similaire
à ce qui est observé dans la maladie d'Alzheimer. Les deux
tableaux mnésiques sont indépendants de la
sévérité de la démence.
Ces résultats suggèrent donc une
hétérogénéité dans la démence du
SIDA. Cette hypothèse nécessite cependant d'autres
investigations.
Cette étude souligne la plus grande sensibilité
de l'échelle de Mattis aux troubles cognitifs du stade SIDA. En effet,
contrairement au MMSE, la Mattis permet de distinguer entre la démence
et les troubles cognitifs modérés et de préciser le niveau
de déficit. Les résultats du MMSE comme ceux de la Mattis
montrent qu'il n'y avait pas de différence entre les patients
asymptomatiques et ceux au stade de SIDA sans troubles cognitifs,
suggérant que l'immunodépression ne soit pas une condition
suffisante dans le développement des troubles cognitifs. Les scores du
MMSE chez les patients déments restaient relativement
élevés (25/30), reflétant une préservation globale
des fonctions explorées par ce test, alors que les scores de ces
patients à l'échelle de Mattis étaient plus nettement
perturbés (118/144).
Cette plus grande sensibilité de la Mattis dans les
troubles cognitifs de la démence était liée au fait que
cette échelle explore les troubles neuropsychologiques sous corticaux,
contrairement au MMSE qui est plus corrélé aux troubles
corticaux.
Enfin, cette étude propose une batterie
neuropsychologique courte, permettant un examen au lit du patient et de
caractériser le fonctionnement cognitif à la fois des patients
déments et de ceux présentant des troubles cognitifs modérés.
III. PARAMETRES
NEUROBIOLOGIQUES ET TROUBLES COGNITIFS
1. Immunodépression et troubles cognitifs
Il existe des sous-groupes de patients qui,
indépendamment de leur statut immunologique, présentent des
déficits cognitivo-moteurs discrets104, 108, 113, 118, 121, 133,
182, 221. Une relation directe entre le nombre de CD4 et l'existence de
troubles cognitifs ou moteurs est peu probable. En revanche, l'apparition des
troubles cognitifs est plus fréquente chez les patients plus
immunodéprimés222 et l'évolution vers les
stades plus avancés de la maladie est souvent associée à
une détérioration progressive des résultats aux tests
neuropsychologiques110, 112, 133. Ainsi, il y a plus de
différence dans les résultats de tests neuropsychologiques de
patients SIDA déclarés, comparés à des
témoins séronégatifs, qu'entre des patients
séropositifs asymptomatiques et des témoins
séronégatifs133. Cependant, ces données sont
à réévaluer en fonction de l'apparition des combinaisons
thérapeutiques antirétrovirales.
2. Articles II et
III : La charge virale et les troubles cognitifs
La
charge virale plasmatique est un marqueur prédictif de
l'évolution de la maladie. En revanche, la relation entre cette charge
virale et la survenue de troubles cognitifs est controversée222,
223. Seule une relation entre la charge virale (du plasma et du liquide
céphalo-rachidien) et le ralentissement psychomoteur
(évalué par le test de Purdue Pegboard) a été
trouvée224. La relation entre les paramètres
biologiques et les troubles cognitifs est donc peu claire. Cette absence de
corrélation entre les paramètres biologiques et les troubles
cognitifs pose la question de l'origine des troubles cognitifs associés
au VIH.
2.1.
Introduction : Définition et techniques de mesure de la charge
virale
La charge virale permet de mesurer l'évolutivité
de la maladie VIH en complément de la mesure des CD4 et de
l'appréciation des signes cliniques. L'expression des résultats
se fait en nombre de copies d'ARN/ml (100 à 10 Millions) ou en
logarithme du nombre de copies (2 à 7), dans un millimètre de
sang.
Depuis quelques années, la détection et la
quantification de l'ADN et de l'ARN viraux, dans le plasma et le liquide
céphalo-rachidien (LCR), est devenue plus précise, grâce
à l'amélioration des techniques de biologie
moléculaire.
Trois principaux « kits » commerciaux
permettant de détecter l'ARN plasmatiques sont utilisés
actuellement :
La technique « Quantiplex HIV RNA
(Chiron) », qui utilise une amplification du signal d'hybridation
moléculaire; la technique Amplicor HIV-1 monitor (Roche), basée
sur la technique de RT-PCR (polymerase chain reaction) et la technique NASBA QR
System (nucleic acid sequence-based amplification) (Organon Teknica), qui
utilise une amplification isotherme de l'ARN.
L'ADN proviral représente le génome du VIH
intégré dans la cellule infectée, alors que la charge
virale de l'ARN rétroviral évalue la réplication
virale.
Actuellement, la charge virale plasmatique est
régulièrement suivie chez les patients, ce qui permet de mieux
prendre en charge l'évolution de la maladie, les résistances
infectieuses qui pourraient apparaître et donc l'efficacité des
traitements.
2.2.
Réplication virale et troubles cognitifs
Les progrès de la biologie moléculaire ont rendu
possible le suivi régulier de la charge virale du plasma et du LCR. De
nombreux auteurs s'interrogent sur la valeur diagnostique et pronostique de la
charge virale dans l'atteinte cérébrale liée au VIH et
dans l'évaluation de la réponse aux traitements225.
Ils explorent, notamment, les liens existant entre la charge virale dans le
sang et dans le LCR et ceux qui existent entre la charge virale dans le LCR et
les troubles cognitifs. Pour certains, il n'y a pas de relation entre la charge
virale sanguine et dans le LCR226. Pour d'autres, plus nombreux, il
existe une corrélation entre la charge virale dans le plasma et celle du
LCR (voir article 3)227, 228, ce que nous avons pu
confirmer224. Une contamination plasmatique, explicable par les
altérations de la barrière hémato-encéphalique a
pourtant été suggérée. Que ces anomalies existent
aussi chez des patients asymptomatiques avec une barrière
hémato-encéphalique préservée, suggère
plutôt l'hypothèse d'une production locale de VIH-1228.
Cette idée est renforcée par les travaux de l'équipe de Di
Stephano229 qui montrent des virus à phénotypes
différents dans des échantillons de sang et de LCR, ce qui
suggére une évolution différente de l'infection dans ces
deux compartiments. Certaines études neuropathologiques renforcent les
arguments en faveur d'une production locale de virus associée à
la détérioration neurologique en montrant notamment une
antigénie p24 positive dans la substance blanche frontale (et dans la
substance grise dans un cas sur 2) chez des patients ayant une
encéphalite réplicative du VIH. Ces études montrent, de
même, une corrélation entre la charge virale
(déterminée par PCR quantitative) et l'encéphalite du
VIH198. Dans ce cas, la diminution de la charge virale dans le LCR
suivant l'administration des combinaisons antirétrovirales226, 230,
231 pourrait ne pas indiquer systématiquement un contrôle
effectif du VIH dans le système nerveux central.
En ce qui concerne les éléments apportés
par la détection et la quantification de l'ADN proviral dans le LCR, peu
d'éléments en faveur d'une éventuelle valeur diagnostique
ont été dégagés. En effet, la fréquence de
détection de l'ADN proviral dans le LCR des patients a été
estimée à 90%, indépendamment de l'existence de troubles
cognitifs232 et, de plus, la quantité d'ADN proviral dans le
LCR n'est pas plus élevée chez les patients présentant des
troubles neurologiques associés au SIDA que chez les patients sans
trouble cognitif233.
En revanche, la quantification de l'ARN pourrait
présenter plus d'intérêt. Un débat important sur la
valeur pronostique de cet outil a partagé les scientifiques.
Plusieurs observations ont été faites :
D'une part, certains patients asymptomatiques peuvent avoir
une charge virale élevée dans le LCR, sans troubles
cognitifs228, 234 et sans altération de la barrière
hémato-encéphalique228, ce qui suggère une
production locale de VIH-1. D'autre part, des résultats contradictoires
sur une éventuelle corrélation entre la charge virale dans le LCR
et les troubles cognitifs ont été trouvés.
Pour certains auteurs, il existe un lien entre la charge
virale dans le LCR et les troubles cognitifs et/ou neurologiques, surtout dans
les stades tardifs235. Dans l'étude de Di Stephano, la charge
virale dans le LCR était liée aux troubles
neurologiques229. Et Cinque et coll. ont pu montrer, par des
études neuropathologiques, qu'il existe une corrélation entre la
charge virale dans le LCR et l'encéphalopathie du VIH, ce qui
suggère une implication directe de la réplication virale à
l'origine des lésions236. Pour d'autres auteurs, ni la charge
virale du LCR, ni la charge virale dans le sang222 ne sont
directement liées à l'intensité des troubles
cognitifs227, 237. Nos travaux confirment ces résultats (voir
article 3). Cette idée est renforcée par les travaux
neuropathologiques relevant des discordances entre la quantification de l'ARN
du VIH-1 dans le cerveau et la démence : certains patients
déments ont une charge virale faible88 (Voir article 2).
Les différents résultats sur la
corrélation entre la charge virale et l'intensité des troubles
cognitifs montrent donc des résultats divergents. La plupart des auteurs
qui ne trouvent pas une corrélation directe entre les troubles
cognitifs et la charge virale montrent pourtant que la charge virale est en
moyenne plus élevée chez les patients présentant des
troubles cognitifs sévères (déments) que chez les patients
n'en ayant pas223, 224, 226, 235, 238. Ces résultats, qui
peuvent être rapprochés de ceux obtenus dans le tissu
cérébral (Voir article 2), suggèrent que des facteurs
indirects de neurotoxicité puissent intervenir dans la relation entre la
charge virale et les troubles cognitifs239 (voir chapitre I, 3.4).
En conclusion, la mise en évidence d'une charge virale
positive dans le LCR ne constitue pas un élément de diagnostique
précoce de l'encéphalopathie VIH. Cependant, l'apparition d'une
charge virale élevée dans le cerveau, quelque soit le stade de la
maladie, doit être surveillée car seules des études
longitudinales pourront démontrer que ces patients sont plus à
risque que d'autres de développer une atteinte cognitive
ultérieure225. Les premiers travaux longitudinaux sur l'effet
des thérapies antirétrovirales prouvent cependant deux points:
une réplication importante augmente le risque de complications
neurologiques ; d'autre part, les nouvelles combinaisons
thérapeutiques antirétrovirales semblent avoir
considérablement changé les données de la situation en
diminuant fortement l'incidence des affections neurologiques240.
2.3.
Article 2 : Human immunodeficiency virus type 1 DNA and RNA load in brains
of demented and nondemented patients with acquired immunodeficiency
syndrome
2.3.1. Objectifs de l'étude
Les examens post-mortem des patients infectés par le
VIH-1 trouvent des lésions neuropathologiques (cellules géantes
multinucléées et pâleur myélinique) dans 70 à
90% des cas241, 242. Ces marqueurs neuropathologiques de
l'encéphalite réplicative du VIH, ne sont pourtant
observés que dans 50% des cas de démence. Le lien entre la charge
virale dans le cerveau et les troubles cognitifs est, de même, encore mal
compris.
Cette étude a pour but de chercher le lien existant
entre la démence et la charge virale dans le cerveau.
2.3.2. Patients et méthodes
Les patients étaient classés en deux
groupes : déments et non déments. Seize patients ont
été évalués sur le plan cognitif par le MMSE. Les
patients ayant un score supérieur à 24 (n=9) étaient
considérés non déments. Deux patients ont
bénéficié d'une batterie plus complète et ont
été classés, respectivement, déments
modérés et sévères. Un patient était
asymptomatique et deux autres n'ont pas eu de test psychométrique. Au
total, sur 21 patients, 9 étaient considérés comme
déments, 9 non déments et trois n'étaient pas
classés.
Les quantités d'ADN proviral et d'ARN du VIH-1 ont
été extraites à partir d'un prélèvement de
circonvolution frontale moyenne par une technique de PCR quantitative
(réalisée par une biologiste, Françoise Lazarini) chez les
21 patients, tous décédés au stade de SIDA, sans
lésion focale cérébrale. Les prélèvements
ont été fixés puis inclus en paraffine et les cellules
positives pour la protéine gp41 du VIH-1 ont été
dénombrées.
2.3.3. Résultats
Dans 18 cas sur 21 (y compris les cas asymptomatiques), de
l'ADN ou de l'ARN viral a été détecté dans la
circonvolution frontale moyenne. Les charges virales en ADN et en ARN du VIH-1
étaient statistiquement liées (p=0,0051, r=0,441). Les charges
virales en ADN (p=0,0063, r=0,424) et en ARN (p=0,042, r=0,305) étaient
liées à la densité de la protéine gp41. Il
n'existait pas, en revanche, de corrélation entre la charge virale dans
le cerveau et la présence d'une démence (mais la charge virale
était en moyenne plus élevée chez les déments).
Enfin, 4 patients déments avaient de faibles charges virales.
2.3.4. Conclusions
Le petit nombre de patients ne permet pas de rejeter
l'hypothèse d'un lien entre les troubles cognitifs et la charge virale
du tissu nerveux dans certains cas, ce travail renforce l'hypothèse
selon laquelle la démence n'est pas la conséquence directe de la
réplication du virus dans le cerveau. Des mécanismes secondaires,
comme des facteurs immunologique et une production de cytokines pourraient
intervenir.
Article 3 : Plasma
and cerebrospinal fluid human immunodeficiency virus type-1 (HIV-1) RNA levels
in HIV-1-related cognitive impairment.
2.3.5. Objectifs de l'étude
Etudier les relations existantes entre la quantification de la
charge virale (ARN rétroviral) dans le sang et le LCR d'une part et les
troubles cognitifs sous-corticaux-frontaux caractéristiques de
l'encéphalite du VIH d'autre part.
2.3.6. Patients et méthodes
Entre avril 1996 et Mars 1998, 30 patients séropositifs
pour le VIH depuis 3 à 14 ans, âgés de 30 à 66 ans,
sans pathologie opportuniste du système nerveux central et sans tumeur
cérébral ont été inclus de manière
prospective. Tous les patients ont eu un examen neurologique, psychiatrique et
neuropsychologique. La batterie neuropsychologique utilisée comprenait
le MMSE, la Mattis-DRS, le Trail A et B, le Purdue Pegboard test, le Grober et
Buschke et un examen de la dépression avec la MADRS. Cette batterie vise
à mettre en évidence les troubles cognitifs
caractéristiques de l'infection par le VIH-1. Elle est décrite
dans l'article 1. Les ponctions lombaires ont été
réalisées sur indication médicale,
généralement pour l'évaluation d'une fièvre ou d'un
trouble des fonctions supérieures. L'écart entre le
prélèvement sanguin et du LCR et l'évaluation des troubles
cognitifs n'excédait pas un mois. Les charges virales dans le sang et le
LCR ont été évaluées par la technique de RT-PCR
(Roche amplicor) avec une limite de détection de 200 copies par ml.
2.3.7. Résultats
Cette étude a montré une forte relations entre
la charge virale dans le sang et celle du LCR (p<0.0001). Il n'y avait pas
de corrélation, en revanche, entre la charge virale dans le sang ou du
LCR et le statut cognitif global (mesuré par la Mattis DRS). Cependant,
quand les patients ont été catégorisés en trois
groupes selon leurs troubles cognitifs (pas de troubles, troubles
modérés ou dément), les déments avaient une charge
virale dans le LCR plus élevée, en moyenne, que les autres
groupes. Chez le sous-groupe de patients ayant un SIDA déclaré,
cette différence était significative entre les déments et
les patients sans troubles cognitifs (p=0.036). Enfin, il existait une
corrélation entre les résultats au Purdue Pegboard Test et la
charge virale dans le sang (p=0.041) et dans le LCR (p=0.039).
2.3.8. Conclusions
La détection d'un lien entre la charge virale sanguine
et la charge virale dans le LCR suggère un transport passif du VIH
à travers la barrière hémato-encéphalique ou une
réplication synchronisée du virus dans les deux compartiments. De
façon surprenante, si une corrélation avec les troubles cognitifs
globaux n'était pas trouvée, le ralentissement psychomoteur et la
charge virale dans le sang et le LCR étaient, par contre,
statistiquement liés. Enfin, le manque de corrélation directe
entre la charge virale et les troubles cognitifs dans leur ensemble ne permet
pas d'avoir un indice pronostique sur l'évolution neurologique
ultérieure des patients ayant une charge virale élevée
dans le LCR à un moment de la maladie. Ceux-ci seront-ils plus à
risque que d'autres de développer ultérieurement des troubles
cognitifs, ou bien la baisse importante de la charge virale grâce aux
nouveaux traitement les préservera t-elle de la survenue de ces
troubles ? Seules des études longitudinales peuvent maintenant
répondre à ces nouvelles questions.
3. Article IV : Imagerie cérébrale et
troubles cognitifs
3.1.
Introduction : Imagerie (IRM et Imagerie cérébrale
fonctionnelle) et troubles cognitifs.
3.1.1. IRM et troubles cognitifs dans
l'infection par le VIH
L'Imagerie par Résonance Magnétique (IRM) est
une technique permettant d'obtenir une image cérébrale en captant
une onde électromagnétique émise par les protons contenus
dans les tissus cérébraux(*).
Chez les patients séropositifs pour le VIH, l'Imagerie
par Résonance Magnétique (IRM) est généralement
normale jusqu'aux stades avancés de la maladie243. Aux stades
avancés de la maladie, peut survenir une atteinte
cérébrale avec atrophie corticale, diminution de volume des
noyaux gris centraux124 et atteinte de la substance blanche de type
réduction de volume et hyposignal diffus124. Cette
leucoencéphalopathie du VIH prédomine dans la substance blanche
périventriculaire et le centre semiovale.
Les résultats de l'IRM sont peu
spécifiques244 et peu corrélés aux
résultats anatomopathologiques245, ni aux données
cliniques. En particulier, l'atrophie cérébrale
prédominante dans les noyaux gris centraux est trouvée chez des
patients au stade SIDA non déments123, 246 et l'atrophie
sous-corticale n'est pas liée aux performances
neuropsychologiques126. Il semble qu'en fait, l'atrophie soit
globalement plus indicatrice du stade CDC que des troubles
cognitifs127.
3.1.2. Imagerie cérébrale
fonctionnelle, potentiels évoqués et troubles cognitifs
A) IRM
fonctionnelle (IRMf)
La technique d'IRMf, utilisant l'injection d'un agent de
contraste a été utilisée dans l'étude de la
démence du SIDA et a permis de montrer une diminution relative du volume
sanguin cérébral particulièrement dans la substance grise
profonde (et non dans la substance blanche) correspondant à une
aggravation du stade CDC247. Ces résultats sont concordant
avec les travaux en Tomographie par émission de Positron (PET) montrant
un hypométabolisme de ces régions248.
B) Potentiels
évoqués
Enfin, nous citerons comme technique d'exploration
fonctionnelle qui pourrait s'avérer utile cliniquement et sur le plan
thérapeutique, les technique de potentiels évoqués,
où une diminution d'amplitude et augmentation de la latence de l'onde
P300 est observée en réponse à des stimuli visuels, chez
des patients asymptomatiques, avant que des déficits cognitifs puissent
être mis en évidence par des tests
neuropsychologiques249. Ce profil d'altération étant
retrouvé dans les modèles animaux du VIH.
3.2. Spectroscopie
par résonance magnétique (SRM)
3.2.1. Généralités
sur la technique de SRM
La spectroscopie par résonance magnétique (SRM)
est une technique qui permettant de mesurer des concentrations de
métabolites cérébraux.
Le principe de la SRM consiste en la détection du
signal résultant de l'interaction entre les noyaux atomiques et le champ
magnétique créé. La technique la plus utilisée se
focalise sur les noyaux d'hydrogène. Chaque type de molécule a
des propriétés magnétiques différentes et donc le
signal émit par leurs protons peut être
distingué250.
3.2.2. La SRM dans la pathologie
VIH
La technique de SRM permet, dans la pathologie VIH, d'obtenir
des indicateurs sensibles de l'avancée de la pathologie
cérébrale251-253. Les composants neurochimiques
souvent étudiés dans l'atteinte du SNC par le VIH-1
sont :
Le myoinositol (myo) : Il comprend des
composés phosphorés de l'inositol qui joue le rôle de
second messager. Une augmentation de ce pic a été décrite
dans les processus de prolifération astrocytaire et pourrait, donc,
refléter la gliose d'un tissu.
La choline (Cho) : C'est un composé entrant
dans la composition des membranes cellulaires. Les astrocytes contiennent plus
de Cho que les neurones. Une augmentation du pic de choline a été
décrite dans les processus inflammatoires, la prolifération
microgliale (les glioses) et/ou la démyélinisation254.
La créatine (Cr) et la
phosphocréatine : Ces deux molécules
représentent le réservoir énergétique d'un tissu
où elles sont en équilibre permanent. La créatine,
très stable, est utilisée en standard interne.
Le N-Acetyl Aspartate (NAA) : C'est un
composé prédominant dans l'encéphale. Il a une
localisation intraneuronale. Il est synthétisé par les
mitochondries des neurones de la substance grise. Précurseur d'acides
aminés neurotransmetteurs, il reflète la densité et/ou le
fonctionnement neuronal.
La Lactate (LAC) : Reflète la glycolyse
anaérobie
Les lipides : Les pics de lipides sont
augmentés dans les processus de démyélinisation mais aussi
de remyélinisation.
L'intensité de chacun des pics indique la concentration
de la molécule.
La figure 10 représente les images de spectro-IRM
caractéristiques de la démence du SIDA. La région
analysée est la substance blanche frontale, mais des images similaires
peuvent être obtenues dans les noyaux gris centraux. Le spectre utilise
la créatine comme référence interne.
Il existe une augmentation du pic de choline et de
myoinositol, alors que le pic de NAA décroît. La
décroissance du signal du NAA reflète une souffrance ou une perte
neuronale255-259. L'hypothèse d'un lien entre la
réduction du signal de NAA et la souffrance et/ou perte neuronale est
renforcé par des études anatomopathologiques
post-mortem260-262. Cependant, ces changements peuvent
apparaître alors même que les examens cliniques et/ou
neuropsychologiques sont normaux259, 263-268, ce qui suggère
que ces modifications puissent précéder l'apparition des troubles
cognitifs259. L'augmentation du pic de choline indique probablement
une gliose astrocytaire. Cette augmentation a été trouvée
chez des patients avec une démence du SIDA255, 267. Les
études transversales269 et longitudinales251 en
SRM de patients déments montrent que, avec l'avancée de la
démence et sans traitement, la décroissance du pic de NAA
s'aggrave ainsi que l'augmentation du pic de Cho et myo. La diminution du taux
de NAA est de même, liée au taux de lymphocytes CD4269
et au degré d'encéphalopathie252, 269.
3.2.3. SRM et effets des
traitements
Dès 1995, l'influence de la zidovudine (AZT) sur les
modifications métabolites cérébraux avaient
été montrée, avec notamment une réaugmentation du
pic de NAA263, 270-272. Une étude de Salvan et coll.,
cependant ne montre pas d'effet sur le taux de Cho273, ce qui
indique que la souffrance cellulaire et la gliose pourraient persister, au
moins jusqu'à un certain point. Une étude récente de Chang
et coll. confirme ces tendances sous AZT et indique que les nouvelles
combinaisons thérapeutique (HAART) améliorent les troubles
métaboliques, en particulier le taux de myoinositol272. Dans
cette étude, la modification du taux de myoinositol dans la substance
blanche frontale est liée à l'augmentation du taux de lymphocytes
CD4 et à l'amélioration du stade de démence. Les travaux
réalisés par notre équipe sur une suivi longitudinal de 22
patients268, 274 (voir article 4) montrent une diminution du taux de
NAA dans la substance blanche frontale, qui s'améliore après 9
mois, alors que le taux de choline reste élevé. Ces
résultats indiquent une persistance de l'inflammation, parallèle
à l'amélioration de la souffrance neuronale. La spectroscopie
apparaît, par conséquent, un bon outil pour suivre
l'évolution des lésions, sous traitement.
3.3.
Article 4: Clinical and Spectroscopic improvement in HIV associated
cognitive impairment: A longitudinal study
3.3.1. Objectifs de l'étude
Analyser l'évolution des troubles neurologiques et
cognitifs du VIH-1 et les anomalies métaboliques
cérébrales chez les patients traités par combinaisons
thérapeutiques.
3.3.2. Patients et méthodes
Un total de 22 patients présentant un SIDA (11 sans
troubles cognitifs, 9 présentant des troubles cognitifs
modérés, et 2 déments) a été inclus dans
l `étude. Nous avons pu suivre 15 patients pendant 9 mois (6 sans
troubles cognitifs, 9 présentant des troubles cognitifs
modérés). Les 7 autres sont sortis de l'étude après
le premier, deuxième ou troisième examen. 19 patients recevaient
une combinaison thérapeutique antirétrovirale incluant au moins
une antiprotéase au début de l'étude. Parmi les trois
autres patients, deux ont reçus des combinaisons thérapeutiques
dans les premiers mois de l'étude et le troisième est
resté non traité.
Une batterie neuropsychologiques permettant d'identifier les
troubles sous-corticaux-frontaux caractéristiques de l'atteinte par le
VIH-1112 (voir article 1) ainsi qu'un examen neurologique
standardisé étaient réalisée à l'inclusion
puis tous les trois mois (soit 4 fois).
Les IRM et la SRM était réalisée sur 18
patients au début de l'étude. Onze patients ont pu être
suivi jusqu'à neuf mois (5 sans troubles cognitifs et 6 avec des
troubles cognitifs). La SRM était extraite de trois régions
spécifiques : dans la substance blanche frontale, dans la substance
blanche du centre semiovale et dans la substance grise
pariéto-occipitale médiane. Le taux de créatine
était utilisé comme référence interne.
L'atrophie était estimée sur la base de
l'élargissement des espaces sous-arachnoïdiens et des
ventricules.
La charge virale en ARN du VIH-1 était
quantifiée à l'aide du kit Roche Amplificator de RT-PCR de Roche
(voir chapitre sur la charge virale).
3.3.3. Résultats
Aucun patient ne s'est aggravé. Les patients ayant des
troubles cognitifs ont amélioré leurs performances (La Mattis DRS
dans ce groupe est passée d'une moyenne de 135 1.9 à 138.5
1.1). La charge virale sanguine a diminué dans les deux groupes de
patients.
Lors des examens initiaux, une atrophie
cérébrale était détectée chez 2/8 des
patients sans troubles cognitifs et 7/10 des patients avec troubles cognitifs.
Seuls les patients avec des troubles cognitifs avaient une diminution du pic de
NAA dans la substance blanche frontale (mais pas dans la zone
postérieure). Les taux de NAA dans la substance blanche frontale
étaient corrélés au sous-test
« mémoire » de la Mattis DRS. Le pic de choline
était augmenté dans tous les groupes et dans les trois
régions. Chez les patients avec des troubles cognitifs, le pic de
myoinositol dans la substance blanche postérieure, était
supérieur à la moyenne, alors qu'il était normal dans la
substance blanche frontale chez tous les patients. Les pics de NAA, Cho et
Myoinositol étaient normaux et similaires dans la substance grise
postérieure, dans les deux groupes cognitifs.
Neuf mois après le premier examen, le pic de NAA est
resté similaire chez les patients sans troubles cognitifs alors qu'il a
augmenté dès le troisième mois et jusqu'au neuvième
mois (s'est normalisé) dans le groupe avec des troubles cognitifs au
départ. Les taux de Cho, ont diminué dans la substance blanche
postérieure des patients avec des troubles cognitifs alors qu'ils sont
restés élevés dans les deux groupes dans la substance
blanche antérieure. Le pic de myoinositol a diminué
jusqu'à des valeurs normales dans la substance blanche
postérieure des patients avec des troubles cognitifs et est resté
normal dans la substance blanche frontale. Dans la substance grise
postérieure, les métabolites sont restés normaux.
3.3.4. Conclusions
Cette étude longitudinale explorait principalement des
patients avec des troubles cognitifs modérés. Les troubles
cognitifs des patients se sont améliorés sous traitement. Cette
amélioration est concomitante avec une augmentation (une normalisation)
du pic de NAA dans la substance blanche sous corticale. Cette
amélioration du marqueur neuronal parallèlement à
l'amélioration cognitive pourrait indiquer des phénomène
de souffrance neuronale réversibles (du moins jusqu'à un certain
stade) impliqués dans les troubles cognitifs du VIH.
IV. EFFETS DES
TRAITEMENTS
1. Historique des traitements et des troubles cognitifs
Cf. Annexe 1: classification des traitements.
1.1. Les Inhibiteurs de la transcriptase inverse
Les inhibiteurs de la transcriptase inverse sont
divisés en deux catégories: les analogues nucléosidiques
(zidovudine, didanosine, zalcitabine, lamivudine, stavudine, abacavir) et les
non nucléosidiques, plus récents (Nevirapine, Efavirenz,
Delavirdine) (annexe 1). Ils inhibent la transcriptase inverse, enzyme
permettant au virus de fabriquer à partir de son ARN un
équivalent sous forme d'ADN (cf. chapitre I, 3.1. « Structure
du VIH »).
1.1.1. Apparition des analogues nucléosidiques,
monothérapies et bithérapies
En 1984, l'activité antirétrovirale de
l'Azidothymidine (finalement dénommée zidovudine) (AZT) est mise
en évidence. C'est le premier traitement disponible contre le VIH, qui
s'avérera finalement être, avec la stavudine et
l'abacavir275 un des traitements passant le mieux la barrière
hémato-encéphalique (avec un bon coefficient de
pénétration)129, 218, 276, 277. La zidovudine sera
conseillée dans le traitement de la démence du
SIDA278.
De 1993 à 1996, de nouveaux antirétroviraux vont
apparaître et la supériorité des associations d'antiviraux
sur la monothérapie va être démontrée, les
monothérapies seront progressivement écartées, en
particulier car elles favorisent la sélection de souches
résistantes.
A partir de l'année 1995, et grâce à
l'apparition de nouveaux médicaments, les bithérapies vont
être progressivement instituées. Elles vont représenter,
dans un premier temps, un progrès considérable dans la survie des
patients, certains patients sont encore traités par ces associations. De
nombreuses études se sont intéressées à leur effet
sur le système nerveux central, montrant globalement un effet
bénéfique279.
Cependant, ces thérapies posent aujourd'hui le
problème de l'apparition de multiples résistances
croisées, les souches résistantes apparaissant grâce
à l'incapacité de ces combinaisons à inhiber totalement la
réplication virale et à leur mauvaise pénétration
dans les sites sanctuaires comme le système nerveux central.
1.1.2. Apparition récente des inhibiteurs non
nucléosidiques de la transcriptase inverse (NNRTI)
Les NNRTI (Non Nucleosidic Reverse Transcriptase Inhibitors),
sont apparus récemment. Les données sur la
pénétration des molécules de cette famille dans le
système nerveux central sont encore très parcellaires. Il semble
néanmoins que l'atévirdine ait des résultats favorables
dans le traitement des troubles cognitifs280.
Des associations de NNRTI et d'analogues nucléosidiques
sont proposées dans le traitement des affections du SNC liées au
VIH-1281.
1.2. Effets des
associations incluant une antiprotéase ou un NNRTI sur les troubles
cognitifs
Les inhibiteurs de protéase agissent sur la
protéase, enzyme permettant la maturation des virions produits par la
cellule infectée). Sans cette maturation, les virions produits ne sont
pas viables et ne peuvent donc pas infecter une autre cellule.
Les premiers inhibiteurs de protéase font leur
apparition, en France, vers le deuxième trimestre de l'année
1996. Leur introduction dans le traitement des patients correspond à une
véritable révolution dans la stratégie de traitement. A
présent, le but deviens d'inhiber au maximum la réplication
virale en s'attaquant aux différentes phases du cycle du VIH (voir
chapitre 1, 3.1.3. « Cycle de réplication du VIH et mode
d'action des traitements »).
On parle donc de «combinaisons
thérapeutiques », des associations thérapeutiques
incluant au moins une antiprotéase ou un inhibiteur non
nucléosidique de transcriptase inverse et deux antirétroviraux.
Ces « combinaisons thérapeutiques » vont soulever
d'immenses espoirs thérapeutiques.
Depuis l'introduction de ces associations de molécules
dans le traitement des patients, une amélioration importante de
l'état des malades a été observée. Ces
progrès se font aussi bien sur le plan de l'amélioration de
l'état général du patient que sur le plan des pathologies
opportunistes ou des symptômes comme les diarrhées
chroniques282. Le taux de survie des patients est ainsi fortement
améliorée.
Cependant, ces traitements montrent aussi des limites en ce
qui concerne le problème particulier de l'infection du système
nerveux central par le VIH-1. Dans un article postérieur à
l'apparition des antiprotéases, McArthur107 met notamment
l'accent sur le fait que les différentes molécules
antirétrovirales, et notament les inhibiteurs de la protéase,
passent peu la barrière hémato-encéphalique (de 10
à 40%) et que celles qui passent le mieux ont des effets secondaires
tels qu'elles sont probablement très peu utilisées aux doses
effectives sur le virus présent dans le cerveau (pour revue sur l'action
des différentes molécules dans le SNC, voir Enting et
coll.283). Cette question est essentielle car la force principale
des combinaisons thérapeutiques consiste en leur capacité
à inhiber de façon prolongée la réplication virale,
pour prévenir l'apparition de souches mutantes. Or, s'il se trouve que
la pénétration des médicaments est difficile dans le
système nerveux central, il pourrait alors subsister une activité
réplicative pourvoyeuse à terme de souches mutantes devenant
progressivement des virus résistants. Les avis à ce sujet restent
partagés car d'autres considèrent qu'en diminuant fortement la
charge virale plasmatique, les thérapies diminuent aussi le passage de
nouveaux virus dans le système nerveux central. Dans ce cas, beaucoup
d'autres paramètres vont intervenir comme l'ancienneté de la
maladie, la charge virale dans le système nerveux central avant
traitement, l'efficacité des traitements antérieurs, les souches
de virus existantes, et le délai avant traitement. Le risque de
pathologies neurologiques est aussi lié à l'intensité de
l'activation microgliale dans le système nerveux central, qui induit une
réactivation de l'infection latente dans ces cellules avec une possible
réinfection périphérique et une sécrétion
des cytokines inflammatoires. Ce risque n'est donc probablement pas totalement
écarté. De plus, le fait que seul un sous-groupe de patients
souffrent de pathologies neurologiques suggère une composante
génétique75. Ces considérations reposent le
problème du cerveau comme "réservoir" du VIH-1.
Nous avons vu, dans les chapitres précédents
(chapitre I, 3.4 « Neuroinvasion et aspects
neuropathologiques » ; chapitre II, 3.1.2
« Paramètres biologiques et troubles cognitifs » et
chapitre III) que les études d'imagerie cérébrale et de
neuropathologie post-mortem ne retrouvent pas de relation entre ces marqueurs
morphologiques et l'intensité des troubles cognitifs. Ceci rend
très complexe l'exploration du rôle joué par les
combinaisons thérapeutiques sur l'apparition ou sur l'évolution
des troubles cognitifs. Nous manquons notamment de recul pour évaluer
l'évolution future des troubles modérés vers la
démence associée au SIDA, puisque la valeur accordée aux
paramètres d'immunosuppression a été bousculée
depuis l'introduction des combinaisons thérapeutiques.
Il existe encore trop peu d'études pour avoir une
idée claire et objective de l'effet des combinaisons
thérapeutiques sur les troubles cognitifs. Cependant, les premiers
résultats sont très encourageants. La fréquence de ces
troubles, évaluée à 20,6 % avant trithérapie
(inclusions jusqu'en décembre 95)35 aurait
considérablement diminué, selon Ferrando et collaborateurs (22%
de patients ayant des troubles cognitifs sous combinaisons
thérapeutiques contre 54% antérieurement)284.
Cependant, ces auteurs n'ont pas exclu de cette étude les patients
présentant des pathologies opportunistes du SNC. Or, on sait que la
fréquence des pathologies opportunistes a elle-même diminué
avec les nouvelles thérapies. Cette étude, bien que
récente, nous renseigne donc assez peu sur l'effet du VIH lui-même
sur les troubles cognitifs lorsque les patients sont traités par
trithérapies.
Lors du dernier congrès de l'Américan Academy of
Neurology, Sacktor et coll.138 annonçaient cependant une
réduction de l'incidente de la démence du SIDA aux Etats Unis de
21.1/1000 personnes années en 90-92 à 17.4/1000 personnes
années en 93-95 et 14.7/1000 personnes années en 96-97. Soit des
résultats très favorables. Cependant il faut noter que la
diminution de l'incidence était antérieure à
l'arrivée des antiprotéases, ce qui laisse penser que d'autres
facteurs interviennent dans la prévention de cette pathologie.
Dans une autre étude très récente, Tozzi
et coll.285 décrit, sur 26 patients, une diminution de la
fréquence des troubles cognitifs de 80% (initialement) à 50% six
mois plus tard et à 21.7 quinze mois plus tard. Parmi les fonctions
explorées, les troubles de la concentration et de la rapidité des
processus centraux diminuaient de 65.4% à 21.7% et ceux de la
mémoire de 50 à 8.7%. Une amélioration significative des
niveaux de base était observée en ce qui concerne la
concentration, la rapidité des processus centraux, la flexibilité
mentale, la mémoire, le contrôle moteur fin, les habileté
visuospatiales et le habiletés visuo-constructives. Chez les patients
qui s'étaient améliorés, on constatait
parallèlement qu'ils avaient une baisse de la charge virale plus
importante.
Ces résultats sont concordants avec d'autres
études très récentes qui font état d'un
amélioration du ralentissement psychique et moteur sous
antirétroviraux286-288. Les résultats des
études sur la charge virale qui montrent que ces associations de
molécules peuvent permettre de rendre la charge virale plasmatique
indécelable (voir chapitre III.3) et ceux des techniques d'imagerie
(voir chapitre III.4) renforcent l'idée d'une action
bénéfique des combinaisons thérapeutiques sur le
système nerveux central. Enfin Price et coll. 137 soulignent
l'effet globalement bénéfique des combinaisons
thérapeutiques sur les troubles neurologiques et sur la survie des
patients, tout en précisant que des mauvaises performances neurologiques
restent très indicatrices de mauvais pronostic. Finalement Dore et coll.
289 remarquent que la diminution de l'incidence de la démence
du SIDA est inférieure à la diminution des autres pathologies
neurologiques, laissant penser que cette diminution est en grande partie
liée à l'amélioration de l'état
général des patients et l'augmentation du taux de lymphocytes
CD4. Les combinaisons thérapeutiques ayant en fin de compte moins
d'impact sur la démence que sur les pathologies neurologiques
opportunistes à cause de la mauvaise pénétration des
médicaments dans le système nerveux central.
Ces nouvelles données sur l'effet des combinaisons
thérapeutiques, et en particulier le fait qu'une amélioration des
troubles cognitifs est observée chez des patients ayant des associations
de médicaments incluant ou n'incluant pas d'antiprotéases,
laissent penser à un effet indirecte des traitement sur les troubles
cognitifs. C'est probablement l'efficacité globale des combinaisons
thérapeutiques sur l'inhibition de la réplication du virus et sur
l'amélioration de l'état général des patient qui
est la plus pourvoyeuse d'amélioration. Cependant, nous avons vu plus
haut les limites que ces molécules pourraient avoir dans le cas
particulier de l'affection du système nerveux central. Certains auteurs
proposent déjà des stratégies complémentaires dans
le traitement des troubles cognitifs. Swindells et coll., notamment, envisagent
que « l'avenir des traitements contre la démence, passera
peut-être par une surveillance des macrophages immuno-compétants,
plus encore que de la charge virale dans le cerveau, et le développement
de traitement anti-inflammatoires et
neuroprotecteurs »290.
2. Article V : Outcome of patients with HIV1-related
cognitive impairment on highly active antiretroviral therapy
2.1. Objectifs de
l'étude
Examiner l'effet des combinaisons thérapeutiques
incluant une molécule antiprotéase ou un inhibiteur non
nucléosidique de la transcriptase inverse sur l'évolution des
troubles cognitifs modérés et sévères,
associés à l'infection par le VIH-1. Apporter des
précisions sur le type d'évolution des différents domaines
cognitifs.
2.2. Patients et
méthodes
Nous avons réalisé une étude prospective
neuropsychologique et neurologique de 91 patients infectés par le VIH-1.
Quarante huit patients (53%) ont bénéficié d'au moins deux
examens neuropsychologiques. La moyenne de suivi était de 12.3 8.3
mois. L'examen neuropsychologique permettait de mettre en évidence les
troubles sous-corticaux-frontaux caractéristiques de l'infection par le
VIH-1 et d'apprécier l'évolution des différents domaines
cognitifs impliqués (mémoire antérograde, fonctions
exécutives et ralentissement psychomoteur). La batterie
neuropsychologique est détaillée dans l'article
n°1112. Un modèle d'équations
généralisées (GEE) permettant l'analyse de données
longitudinales répétitives sur des sujets identiques a
été utilisé pour prendre en compte les durées de
traitement par combinaisons thérapeutiques (HAART).
2.3.
Résultats
Les combinaisons thérapeutiques améliorent les
troubles cognitifs. Cependant les troubles cognitifs restent associés
à un mauvais pronostic (21% de décès). Les facteurs
impliqués dans un mauvais pronostic étaient un faible taux de CD4
à l'inclusion ainsi qu'un ralentissement psychomoteur
sévère. Le ralentissement psychomoteur récupérait
plus tardivement que les troubles du rappel et continuait à
s'améliorer plus longtemps après l'introduction des traitements
de type HAART.
2.4.
Conclusions
Nos
résultats montrent clairement un effet bénéfique des
combinaisons thérapeutiques sur les troubles cognitifs. Cependant ils
soulignent aussi que les troubles cognitifs restent malheureusement
associés à un mauvais pronostic. Enfin, les différences
dans la cinétique de récupération du ralentissement
psychomoteur et des autres domaines cognitifs suggèrent que ces troubles
puissent être associés à des mécanismes
neuropathologiques différents.
V. DEVENIR DES PATIENTS
AYANT PRESENTE DES TROUBLES COGNITIFS
1. Evolution de la mortalité et de la
morbidité
Depuis l'apparition des combinaisons thérapeutiques,
une baisse important de la mortalité et de la morbidité a
été constatée chez les patients séropositifs pour
le VIH. Cette diminution s'est tout d'abord traduite par une diminution des
taux d'hospitalisations de ces patients291.
Une étude américaine sur 1255 patients ayant
tous eu au moins une fois des lymphocytes CD4 inférieurs à
100/mm3292, a montré un déclin de la mortalité
de 29.4 pour cent personnes-année dans les trois premiers mois de 1995
à 8.8 pour cent personnes-années dans le deuxième
trimestre de l'année 1997. Cette diminution de la mortalité
concernait tous les patients, quelque soient leur âge, sexe, ou facteur
de risque pour la transmission du virus. L'incidence des trois pathologies
opportunistes les plus fréquentes dans ce pays (la pneumocystose, Les
mycobactérioses et la rétinite à cytomégalovirus) a
diminué de 21.9 pour cent personnes-années en 1994 à 3.7
pour cent personnes-années en 1997. Ces améliorations sont
imputables à l'augmentation de l'utilisation des traitements par
combinaisons thérapeutiques incluant une antiprotéase. La
diminution de la mortalité et de la morbidité, quelque soit le
facteur de risque initial pour l'infection par le VIH a été
retrouvée dans d'autres études293. Le risque de voir
apparaître de nouvelles pathologies opportunistes du SIDA diminue
progressivement après l'introduction des combinaisons
thérapeutiques294.
Des études récentes sur la
Leucoencéphalopathie Multifocale Progressive (LEMP), une pathologie
opportuniste due à l'infection par le virus JC dont le pronostic
était très sévère jusqu'ici montrent une
amélioration sensible du pronostic depuis la mise sous combinaisons
thérapeutiques de ces patients295-297.
La perte de poids (de 10% ou plus du poids prémorbide)
est aussi associée à une plus grande mortalité et est un
des facteur prédictif d'une mauvaise évolution
ultérieure298, 299. De nouveaux indices de l'état
nutritionnel sont proposés afin de mieux surveiller l'évolution
des patients300.
Enfin, les troubles cognitifs modérés et
sévères étaient associés, avant l'apparition des
antiprotéases à un taux de mortalité plus
élevé (44.9% de mortalité chez les sujets avec des
troubles cognitivo-moteurs modérés, sur un suivi de 2.4 ans),
soit un risque quatre fois plus élevé que celui des patients sans
troubles cognitifs301. Une étude ultérieure portant
sur 105 patients302, a confirmé ces premiers résultats
en montrant que les patients ayant des troubles modérés ont un
risque de mortalité plus élevé. Dans cette étude,
les auteurs soulignaient le fait que ce sont en particulier le ralentissement
psychomoteur et les troubles du rappel à long terme qui sont
associés à un risque de mortalité plus
élevé. Depuis l'apparition des antiprotéases, peu
d'études ont traité de l'évolution de l'impact sur les
troubles modérés. Dans l'étude que nous avons
réalisée sur le devenir des patients sous combinaisons
thérapeutiques139, nous observions un taux de 24% de
mortalité chez les patients ayant un trouble cognitif
modéré. La mortalité était en particulier
associée à un ralentissement psychomoteur plus
sévère. Ces résultats montrent que, bien que la
mortalité ait diminué, les troubles cognitifs restent un facteur
important de mortalité chez les patients séropositifs pour le
VIH.
2. Séquelles cognitives
Chez les personnes ayant eu une atteinte du système
nerveux central, on peut craindre que la récupération des
fonctions cognitives (grâce à la mise en place de nouveaux
traitements) laisse derrière elle des séquelles
consécutives aux lésions irréversibles qui ont eu lieu.
Typiquement, des séquelles ont été
décrites chez les personnes ayant eu un traumatisme crânien avec
de lourdes conséquences sur le devenir et la capacité de
réinsertion socioprofessionnelle de ces personnes. Ces séquelles
sont multiples, ce sont des troubles cognitifs, du type troubles de
mémoire, lenteur et troubles des fonctions exécutives
(capacités de concentration, planification, etc.) mais aussi des
troubles de l'humeur, avec des problèmes comportementaux du type
impatience, comportement infantile, refus d'admettre les difficultés,
dépression, repli sur soi, irritabilité et des incapacités
physiques303304.
Dans la pathologie VIH, de nombreuses atteintes du
systèmes nerveux ont été décrites. Elles sont soit
liées à l'infection par le VIH lui-même, comme
l'encéphalopathie VIH, soit la conséquence de pathologies
neurologiques opportunistes du type Leucoencéphalite Multifocale
Progressive, Lymphome, Toxoplasmose cérébrale,
Mycobactérie, etc.
Jusqu'à l'arrivée de nouveaux traitements plus
efficaces, le pronostic vital de ces patients était très mauvais,
rendant inopportun la question de leur devenir cognitif et de leur
qualité de vie.
Depuis l'arrivée des antiprotéases, la question
de l'évolution des troubles cognitifs s'est posée et si les
études réalisées ont pu montrer une amélioration
spectaculaire des troubles cognitifs, elles ont aussi montré les limites
de cette amélioration. Voir dans le Chapitre 3 « effet des
traitements », le paragraphe II « évolution des
troubles cognitifs sous antiprotéase ».
Les études prospectives en cours manquent encore de
recul pour savoir si les patients qui ont eu des pathologies neurologiques dues
au VIH ou opportunistes du VIH garderons des séquelles à long
terme (et il n'y a pas encore, a notre connaissance d'étude
détaillée de ces séquelles dans la littérature),
mais plusieurs auteurs s `accordent à penser qu'il est
malheureusement très probable qu'il y en ait107289. Dans
l'infection opportuniste du SNC par le JC virus, par exemple (la LEMP), une
équipe française a montré que si effectivement les
combinaisons thérapeutiques ont amélioré la survie des
patients, leur état neurologique, par contre est resté stable,
laissant les patients avec de graves handicaps neurologiques295.
3. Qualité de vie
La qualité de vie est devenue un enjeu important des
soins médicaux. Cela implique un élargissement important du champ
d'attention donné au patient, avec un questionnement spécifique
sur la manière dont il arrive à gérer sa vie quotidienne,
ses rapports sociaux (familiaux, amicaux et professionnels), sa capacité
à travailler et à s'insérer dans un environnement ainsi
que son état psychologique305. Dans le cas particulier de
l'infection par le VIH, ou des symptômes physiques de la maladie sont
associés à une détresse émotionnelle importante,
des échelles particulières d'évaluation de la
qualité de vie de ces patients se sont développées
récemment306-308.
Depuis l'arrivée des combinaisons
thérapeutiques, et leur effet sur l'augmentation de la durée de
la vie des patients, la question de la qualité de la vie de ces
personnes qui survivent se pose. Certains auteurs arguent déjà
d'une amélioration de la qualité de la vie des patients sous
combinaisons thérapeutiques309. Mais certains traitements
sont plus contraignants que d'autres avec plus ou moins d'effets
secondaires310.
Dans l'absolu, la qualité de vie des patients
infectés par le VIH reste significativement moins bonne que celle de la
population générale, en particulier si ces personnes ont des
symptômes physiques de la maladie311.
En effet ces patients ont souvent, parallèlement aux
autres manifestation de la maladie, des symptômes constitutionnels du
type myalgies, fatigue, anorexie, nausées, vomissements, insomnie,
fièvre et perte de poids. Tous ces facteurs influent négativement
sur la qualité de la vie des patients312 qui reste
significativement moins bonne que celle de la population générale
y compris dans la phase asymptomatique313. Pour Cunningham et coll.,
une fois atteint le stade symptomatique de la maladie, les symptômes
constitutionnels prédisent mieux une baisse de la qualité de la
vie que le taux de lymphocytes CD4312.
La fatigue est souvent un indicateur important du devenir du
patient en terme de morbidité et de perte de qualité de vie. Elle
se rencontre plus souvent dans la phase symptomatique, mais pas seulement. La
fatigue est notamment en relation avec les symptômes physiques,
l'anémie et la douleur, la perte de capacités physiques et la
dépression314. Cependant ce symptôme chronique ne
semble pas être une conséquence de la dépression des
patients mais un élément contribuant de façon
indépendante à la diminution physique des patients et devant
être traité à part entière315.
L'insomnie, la fatigue dans la journée et la diminution
des capacités cognitives sont existent dans tous les stades de la
pathologie VIH. Certains auteurs pensent qu'une dérégulation des
cycles du sommeil (aggravée par une dérégulation de
l'hormone de croissance) pourraient être en partie à l'origine de
ces symptômes316.
La prise en charge de la fatigue dans la démarche
thérapeutique nécessite une compréhension des multiples
facteurs qui interviennent dans ce symptôme et doit être plus
comprise dans la démarche thérapeutique317. Des prises
en charges parallèles de la fatigue et du stress ont été
proposées, avec notamment des traitements par des herbes, des vitamines,
de la gestion du stress, des massages ou de l'acuponcture. Finalement, les
méthodes « douces » les plus efficaces pour
améliorer la qualité de vie semblent être celles
attachées à la gestion du stress du patient318, le
soutient social, et un soutien psychologique du découragement ainsi que
les méthodes pour faciliter l'adaptation du patient à sa
situation319. Dans beaucoup de services recevant des patients
séropositifs pour le VIH, des soutiens psychologiques se sont ainsi
développés afin d'intervenir sur la détresse psychologique
et les symptômes physiques et psychosociaux qui en
résultent320.
Un autre facteur intervenant fortement dans la baisse de la
qualité de vie des patients est la baisse de la vision qui peut
intervenir321.
Enfin, plusieurs études ont montré l'importance
d'une perte de poids importante (10% du poids prémorbide) dans la baisse
de la qualité de la vie298, 299.
4. Article VI : Long term outcome of HIV1-infected
patients with neurological disability (en préparation)
4.1. Objectifs de
l'étude
Depuis l'introduction des combinaisons thérapeutiques,
le taux d'hospitalisation291, de mortalité et de
morbidité292, 293 a diminué chez les patients
présentant un SIDA et traités par combinaisons
thérapeutiques, y compris dans le cas particulier des complications
neurologiques liées au VIH, comme dans l'encéphalopathie
VIH272, 284, 285, 289, 292, 293 ou la LEMP295-297.
Cependant, la présence de troubles cognitifs reste un important
prédicteur d'évolution défavorable137, 139.
Chez les patients qui survivent, les séquelles et, par conséquent
la qualité de vie des patients, deviennent essentielles107,
289. Une étude effectuée chez des patients atteints de SIDA
et présentant des complications neurologiques sévères a
montré une plus longue survie alors que l'état neurologique ne
s'était pas amélioré295. Il nous est paru
essentiel d'enquêter sur les séquelles possibles et l'avenir des
patients séropositifs ayant des complications neurologiques pour
établir un pronostic et envisager les structures de soin ou
d'hébergement ultérieurement nécessaires. Dans cette
étude, nous avons examiné la survie et le handicap fonctionnel de
patients atteints de complications neurologiques et traités par des
combinaisons thérapeutiques modernes.
4.2. Patients et
méthodes
279 patients atteints de SIDA déclaré ont
été hospitalisés, une ou plusieurs fois, dans un service
de réadaptation des affections neurologiques liées au VIH entre
le premier janvier 1995 et le 30 juin 1999. Les données suivantes ont
été rétrospectivement recueillies , au moment de leur
première hospitalisation: l'âge, le régime
thérapeutique, la charge virale, le taux de lymphocytes CD4, l'indice de
Karnowsky et un indexe nutritionnel, le NRI (Nutrition Risk Index)298,
300. Les affections ont été classées comme suit,
présence : d'une encéphalopathie VIH (oui/non), toxoplasmose
cérébrale (oui/non), atteinte du système nerveux par le
cytomégalovirus (Neuro-CMV) (oui/non), autres atteintes opportunistes
à tropisme neurologique (lymphomes, encéphalite du virus
varicelle-zona, méningite à cryptoccoque, tuberculose
neuroméningée) (oui/non), autres atteintes neurologiques
(accident vasculaire cérébral, encéphalite
métabolique ou traumatisme crânien) (oui/non), infection
nosocomiale survenue pendant l'hospitalisation (oui/non),
mycobactérioses atypiques sans tropisme neurologique (oui/non).
Sur les 279 patients, nous avons retenu 137 patients
traités par combinaisons thérapeutiques modernes et pour lesquels
nous avons obtenu des renseignements sur l'évolution. Cette
enquête a été close au 31 décembre 1999. Les
patients qui étaient retournés à leur domicile et ne
nécessitaient pas d'aide étaient considérés comme
« autonomes ». Les patients qui, à leur domicile,
nécessitaient une aide médicale et paramédicale et les
patients institutionnalisés étaient considérés
« dépendants ». La durée moyenne du suivi a
été de 14.5 16.9 mois. Sur les 279 patients, 115 étaient
vivants. Nous avons envoyé à tous les patients vivants
l'échelle d'activité de la vie quotidienne (EAVQ) de Lawton et
Brody322 (adaptée en Français par Derouesné,
1990)(*). Les personnes pouvant
répondre au questionnaire étaient : le patient, une personne
de l'entourage médical, ou un proche. Nous avons reçu 52
réponses. Un patient ayant répondu, mais
décédé avant la fin de la période de surveillance,
a été exclu de l'analyse des résultats du questionnaire.
Une régression logistique a été
réalisée pour décrire le lien entre la survie à
long terme (décédé vs vivant), la dépendance
(dépendant vs autonome) et les variables étudiées. Des
analyses de variance et des régressions simples ont été
utilisées pour explorer le lien entre les variables numériques
(EAVQ, âge, taux de lymphocytes CD4, charge virale, NRI et score de
Karnowsky).
4.3.
Résultats
Les caractéristiques générales des 137
patients traités par combinaisons thérapeutiques sont
montrés dans les annexes $8 et $9. Sur les 81 patients qui survivaient,
47 % étaient autonomes dans la vie quotidienne, 28% étaient
à leur domicile mais nécessitaient une aide médicale ou
paramédicale et 25 % étaient institutionnalisés.
La survie et le handicap fonctionnel n'étaient
liés ni à l'âge, ni au taux de lymphocytes CD4, ni à
la charge virale. La survie était en revanche liée à
l'index nutritionnel (NRI), au score de Karnowsky et au score EAVQ. Les
variables qui contribuaient le plus à un risque de décès
étaient l'encéphalite du VIH, la toxoplasmose
cérébrale, la LEMP et la cachexie. Les variables qui
contribuaient le plus à une dépendance étaient la
toxoplasmose cérébrale, la LEMP et les « autres
atteintes neurologiques ». Les résultats étaient
similaires dans le sous-groupe de patients ayant répondu au
questionnaire de Lawton et Brody. Le score EAVQ chez les patients
traités par combinaisons thérapeutiques n'était lié
ni à l'âge, ni au taux de lymphocytes CD4, à la charge
virale ou au NRI. Il était, en revanche, lié au score de
Karnowsky, et inversement lié à la durée de suivi.
4.4.
Conclusions
Dans cette étude, nous avons confirmé les
résultats de la littérature sur l'augmentation du taux de survie
chez les patients ayant des affections neurologiques liées au SIDA
(24.7% des patients hospitalisés dans le service ont survécu
avant l'introduction des combinaisons thérapeutiques et 57.9%
après leur instauration). L'encéphalite du VIH, la LEMP, la
toxoplasmose cérébrale, l'infection neurologique par le CMV, la
cachexie et les infections opportunistes étaient associés
à un plus mauvais pronostique. Le facteur nutritionnel apparaissait plus
important dans la survie des patients traités par combinaisons
thérapeutiques que ne l'étaient le taux de lymphocytes CD4 ou la
charge virale.
Cependant, seuls 47% des patients n'avaient pas de handicap
dans leur vie quotidienne. 28% nécessitaient une aide à domicile
et étaient institutionnalisés. Les facteurs intervenant le plus
dans le handicap étaient la LEMP, la toxoplasmose
cérébrale, le score NRI et le score de Karnowsky. Le score EAVQ
étaient inversement liés à la durée de suivi. Deux
hypothèses pourraient l'expliquer: soit une amélioration
progressive ultérieure à la sortie des patients, soit un
meilleure pronostic chez les patients dont la complication neurologique
était récente, en raison de l'amélioration de la
qualité des soins. Seul un suivi longitudinal permettrait de
trancher.
En conclusion, dans cette étude de patients atteint des
principales affections neurologiques liées à l'infection par le
VIH, la moitié des patients survivent avec un handicap quotidien. Ces
résultats montrent clairement la nécessité d'anticiper sur
les besoins de ces patients en termes médicaux et paramédicaux (
de rééducation notamment) et de prévoir d'important
efforts pour améliorer leur vie quotidienne.
VI.
CONCLUSION
Nous avons étudié les troubles cognitifs des
patients infectés par le VIH, leur évolution depuis l'apparition
des combinaisons thérapeutiques modernes et les paramètres
neurobiologiques qui interviennent dans ces troubles. Nous nous sommes ensuite
interrogés sur l'avenir de ces patients, en terme de dépendance
quotidienne et sur les nouveaux enjeux thérapeutiques qu'ils doivent
faire prévoir.
Phénomène très rare dans l'histoire des
neurosciences, la démence du SIDA, qui est devenue dans certains cas, au
moins partiellement, réversible. La définition d'une
démence doit-elle prendre en compte l'irréversibilité des
troubles ? Ce point a été longtemps discuté Quoiqu'il
en soit la « démence du SIDA » doit être,
aujourd'hui, classée parmi les démences potentiellement
curables.
VII. REFERENCES
BIBLIOGRAPHIQUES
1. Zhu T, Korber B, Nahinias A, Hooper E, Sharp P. (ADS) An
african HIV-1 sequence from 1959 and implications for the origin of the
epidemic. Nature 1998. 391: 594-597.
2. CDC. Kaposi's sarcoma and pneumocystis pneumonia among
homosexual men. MMWR 1981. 30: 305-308.
3. CDC. Update on acquired immune immunodeficiency
syndrome. MMWR 1982. 36: 3-15.
4. Sellers T. CDC warns of possible pathogen as AIDS
cause. Emerg Dep News 1982. 4: 11.
5. Barré-Sinoussi F, Chermann J, Rey F, et al.
Isolation of a T-lymphotrophic retrovirus from a patient at risk for acquired
immune deficiency syndrome (AIDS). Science 1983. 220:
868-871.
6. Gallo R, Salahuddin S, Popovic M, et al. Frequent detection
and isolation of cytopathic retroviruses (HTLV-III) from patients with AIDS or
at risk of AIDS. Science 1984. 224: 500-502.
7. Clavel F, Guyader M, Guétard D, Sallé M,
Montagnier L, Alizon M. Molecular cloning and polymorphism of the human
immunodeficiency virus type 2. Nature 1986. 324:
691-695.
8. Clavel F, Guétard D, Brun-Vézinet F, et al.
Isolation of a new human retrovirus from West African patients with AIDS.
Science 1986. 233: 343-346.
9. Fultz P, McClure H, Anderson D, Swenson R, Anand R,
Srinivasan A. Isolation of a T-lymphotropic retrovirus from naturally infected
sooty mangabey monkeys (Cercocebus). Proc Natl Acad Sci 1986.
83: 5286-5290.
10. Marx P, Li Y, Lecherche N, et al. Isolation of a simian
immunodeficiency virus related to human immunodeficiency virus type 2 from a
west African pet sooty mangabey. J Virol 1991. 65:
4480-4485.
11. Gao F, Bailes E, Robertson D, et al. Origin of HIV-1 in
the chimpanzee Pan troglodytes. Nature 1999. 397:
436-441.
12. Corbet S, Muller-Trutwin M, Versmisse P, et al. env
sequences of simian immunodeficiency viruses from chimpanzees in cameroon are
strongly related to those of human immunodeficiency virus group N from the same
geographic area. J Virol 2000. 74: 529-534.
13. Britton C, Marquardt M, Koppel B. Neurological
complications of the gay immunosuppressed syndrome: clinical and pathological
features. Ann Neurol 1982. 12: 80.
14. Horowitz S, Benson D, Gottlieb M. Neurological
complications of gay-related immunodeficiency disorder. Ann Neurol
1982. 12: 80.
15. Snider W, Simpson D, Nielsen S, Gold J, Metroka C, Posner
J. Neurological complications of Acquired Immune Deficiency Syndrome: analysis
of 50 patients. Ann Neurol 1983. 14: 403-418.
16. Nielsen S, Petito C, Urmacher C, Posner J. Subacute
encephalitis in acquired immune deficiency syndrome: a post-mortem study.
Am J Clin Pathol 1984. 82: 678-682.
17. Epsein L, Sharer L, Joshi V, Fojas M. Progressive
encephalopathy in children with acquired immune deficiency syndrome. Ann
Neurol 1985. 17: 448-496.
18. Navia B, Cho E, Petito C, Price R. The AIDS dementia
complex. II. Neuropathology. Ann Neurol 1986. 19:
525-535.
19. Navia B, Jordan B, Price R. The AIDS Dementia Complex: I.
Clinical features. Ann Neurol 1986. 19: 517-524.
20. Miller D, Riccio M. Non Organic psychiatric and
psychosocial syndromes associated with HIV-infection and disease. AIDS
1990. 4: 381-388.
21. Maj M, Satz P, Janssen R, et al. Who Neuropsychiatric AIDS
Study, cross-sectional phase II. Arch Gen Psychiatry 1994.
51: 51-61.
22. Maj M, Janssen R, Starace F, et al. Who neuropsychiatric
AIDS study, cross-sectional phase I. Arch Gen Psychiatry 1994.
51: 39-49.
23. Lyketsos C, Treisman G. Psychiatric disorders in
HIV-infected patients: epidemiology and issues in drug treatment. CNS
Drugs 1995. 4: 195-206.
24. Lyketsos C, Federman E. Psychiatric disorders and HIV
infection: impact on one another. Epidemiol Rev 1995.
17: 152-164.
25. Epstein L, Sharer L, Cho E, Myenhofer M, Navia B, Price R.
HTLV-III/LAV-like retrovirus particles in the brains of patients with AIDS
encephalopathy. AIDS Res 1984. 1: 447-454.
26. Ho D, Rota T, Schooley R, et al. Isolation of HTLV-III
from cerebrospinal fluid and neural tissues of patients with neurologic
syndromes related to the acquired immunodeficiency syndrome. N Engl J
Med 1985. 313: 1493-1497.
27. Shaw G, Harper M, Hahn B, et al. HTLV-III infection in
brains of children and adults with AIDS encephalopathy. Science
1985. 227: 177-182.
28. Ho D, Hirsch M. Infection of monocyte/macrophages by human
T lymphotrophic virus type III. J Clin Invest 1986.
77: 1712-1715.
29. Gray F, Lescs M, Keohane C, et al. Early brain changes in
HIV infection: neuropathological study of 11 HIV seropositive, non-AIDS
cases. J Neuropathol Exp Neurol 1992. 51: 177-185.
30. Price R, Brew B. The AIDS dementia complex. J Infect
Dis 1988. 158: 1079-1083.
31. Janssen R, Cornblath D, Epstein L, Foa R, McArthur J,
Price R. Nomenclature and research case definitions for neurologic
manifestations of human immunodeficiency virus-type-1 (HIV-1) infection :
reports of a working group of the american academy of neurology AIDS task
force. Neurology 1991. 41: 778-785.
32. Murray C, Lopez A. Alternative projections of mortality
and disability by cause 1990-2020: global burden of disease study.
Lancet 1997. 349: 1498-1504.
33. Murray C, Lopez A. Mortality by cause for eight regions of
the world: global burden of disease study. Lancet 1997.
349: 1269-1276.
34. Mapou R, Rundell J, Kay G, Tramont E. Relating cognitive
function to military aviator performance in early HIV infection.
Vaccine 1993. 11: 555-559.
35. Marder K, Albert A, Dooneief G, et al. Clinical
confirmation of the American Academy of Neurology algorithm for
HIV-1-associated cognitive/motor disorder: The Dana Consortium on Therapy for
HIV Dementia and Related Cognitive Disorders. Neurology 1996.
47: 1247-1253.
36. Fauci A. Multifactorial nature of human immunodeficiency
virus disease:implications for therapy. Science 1993.
262: 1011-1018.
37. Kieny M. Structure and regulation of the human AIDS
virus. J Acquir Immune Defic Synd 1990. 3:
395-402.
38. Levy J. Pathogenesis of HIV infection. Microbiol
Rev 1993. 57: 183-289.
39. Greene W. AIDS and the immune system. Sci Am
1993. 269: 98-105.
40. McKeating J, Griffiths P, Weiss R. HIVsuceptibility
conferred to human fibroblasts by cytomegalovirus-induced Fc receptor.
Nature 1990. 343: 659-661.
41. Adamson DC, Kopnisky KL, Dawson TM, Dawson VL. Mechanisms
and structural determinants of HIV-1 coat protein, gp41-induced
neurotoxicity. J Neurosci 1999. 19: 64-71.
42. BrackWerner R. Astrocytes: HIV cellular reservoirs and
important participants in neuropathogenesis. AIDS 1999.
13: 1-22.
43. Feng Y, Broder C, Kennedy P, Berger E. HIV-1 entry
cofactor: functional cDNA cloning of a seven-transmembrane, G-protein-coupled
receptor. Science 1996. 272: 872-877.
44. He JL, Chen YZ, Farzan M, et al. CCR3 and CCR5 are
co-receptors for HIV-1 infection of microglia. Nature 1997.
385: 645-649.
45. Ghorpade A, Xia MQ, Hyman BT, et al. Role of the
beta-chemokine receptors CCR3 and CCR5 in human immunodeficiency virus type 1
infection of monocytes and microglia. J Virol 1998.
72: 3351-3361.
46. Albright AV, Shieh JTC, Itoh T, et al. Microglia express
CCR5, CXCR4, and CCR3, but of these, CCR5 is the principal coreceptor for human
immunodeficiency virus type 1 dementia isolates. J Virol 1999.
73: 205-213.
47. Banks WA, Kastin AJ. Characterization of lectin-mediated
brain uptake of HIV-1 GP120. J Neurosci Res 1998. 54:
522-529.
48. Cooj D, Fantini J, Spitalnik S, Gonzalez-Scarano F.
Binding of human immunodeficiency virus type 1 (HIV-1) gp120 to
galactosylceramide (GalCer): relationship to the V3 loop. Virology
1994. 201: 206-214.
49. Miller R, Meucci O. AIDS and the Brain: is there a
chemokine connection? Trends Neurosci. 1999. 22:
471-479.
50. Doms R, Peiper S. Unwelcomed guests with master keys: how
HIV uses chemokine receptors for cellular entry. Virology
1997. 235: 179-190.
51. Levy J. Infection by human immunodeficiency virus--CD4 is
not enough. N Engl J Med 1996. 335: 1528-1530.
52. Pantaleo G, Graziosi C, Fauci A. The immunopathogenesis of
human immunodeficiency virus infection. N Engl J Med 1993.
328: 327-335.
53. Seilhean D, Kobayashi K, He Y, et al. Tumor necrosis
factor-alpha, microglia and astrocytes in AIDS dementia complex. Acta
Neuropathol 1997. 93: 508-517.
54. Saag M, Hammer S, Lange J. Pathogenecity and diversity of
HIV and implications for clinical management: a review. J Acquir Immune
Defic Syndr 1994. 7: S2-S11.
55. Bryant M, Ratner L. Myristoylation-dependant replication
and assembly of human immunodeficiency virus 1. Proc Natl Acad Sci
1990. 87: 523-527.
56. Greene W. The molecular biology of human immunodeficiency
virus type 1 infection. N Engl J Med 1991. 324:
308-317.
57. Wei X, Ghosh S, Taylor M, et al. Viral dynamics in human
immunodeficiency virus type 1 infection. Nature 1995.
373: 117-122.
58. Furrer H, Wendland T, Minder C, et al. Association of
syncytium-inducing phenotype of HIV-1 with CD4 cell count, viral load and
sociodemographic characteristics. AIDS 1998. 12:
1341-1346.
59. Korber B, MacInnes K, Smith R, Myers G. Mutational trends
in V3 loop protein sequences observed in different genetic lineages of human
immunodeficiency virus type 1. J Virol 1994. 68:
6730-6744.
60. Cohen J. Differences in HIV strains may underlie disease
patterns. Science 1995. 270: 30-31.
61. Hu D, Dondero T, Rayfield M, et al. The emerging genetic
diversity of HIV. The importance of global surveillance for diagnostics,
research, and prevention. Jama 1996. 275: 210-216.
62. Robertson D, Sharp P, McCutchan F. Recombinaison in
HIV-1. Nature 1995. 374: 124-126.
63. Haynes B, Pantaleo G, Fauci A. Toward an understanding of
the correlates of protective immunity to HIV infection. Science
1996. 271: 324-328.
64. CDC. Update: trends in AIDS incidence, deaths, and
prevalence--United States. MMWR 1997. 46: 165-173.
65. Kahn J, Walker B. Acute human immunodeficiency virus type
I infection. N Engl J Med 1998. 339: 33-39.
66. Quinn T. Acute primary HIV infection. JAMA
1997. 278: 58-62.
67. Phillips A. Reduction of HIV concentration during acute
infection: independence from a specific immune response. Science
1996. 271: 497-499.
68. Pantaleo G, Graziosi C, Demarest J, et al. HIV infection
is active and progressive in lymphoid tissue during the clinically latent stage
of disease. Nature 1993. 362: 355-358.
69. Galai N, Park L, Wesch J, Visscher B, Riddler S, Margolick
J. Effect of smoking on the clinical progression of HIV-1 infection. J
Acquir Immune Defic Syndr Hum Retrovirol 1997. 14:
451-458.
70. CDC. From the Centers for Disease Control and prevention.
Update: trends in AIDS diagnosis and reporting under the expanded surveillance
definition for adolescents and adults--United States, 1993. JAMA
1994. 272: 1815-1816.
71. Glass J, Wesselingh S, Selnes O, McArthur J.
Clinical-neuropathologic correlation in HIV-associated dementia.
Neurology 1993. 43: 2230-2237.
72. Seilhean D. Correlations clinico-neuropathologiques dans
les troubles cognitifs associés à l'infection par le virus de
l'immunodéficience humaine (VIH). Thèse. Paris: Paris
VI, 1997.
73. Bouwman F, Skolasky R, Selnes O, et al. Variable
progression of HIV-associated dementia. Neurology 1998.
50: 1814-1820.
74. Seilhean D, Michaud J, Duyckaerts C, Hauw J.
Physiopathologie de l'infection du système nerveux par le VIH-1 et de la
démence du SIDA. Rev neurol 1998. 154:
830-842.
75. Tardieu M. HIV-1 related central nervous system
diseases. Curr Opin Neurol 1999. 12: 377-381.
76. Johnson. Questions and prospects related to HIV-1 and the
brain. Res Publ Assoc Res Nerv Ment Dis 1994. 72:
311-323.
77. Bell AB, JW Ironside, S Rebus, YK Donaldson, P Simmonds,
JF Peutherer. Human Immunodeficiency Virus and the brain - Investigation of
virus load and neuropathologic changes in pre-AIDS subjects. J Infect
Dis 1993. 168: 818-824.
78. McArthur J, Hoover D, Bacellar H, et al. Dementia in AIDS
patients: incidence and risk factors. Neurology 1993.
43: 2245-2252.
79. Banks W, Kastin A, Akerstrom V. HIV-1 protein GP120
crosses the blood-brain barrier: role of adsortive endocytosis. Life
Sci 1997. 61: 119-125.
80. Moses A, Bloom F, Pauza C, Nelson J. Human
Immunodeficiency Virus infection of human brain capillary endothelial cells
occurs via a CD4/galactosylceramide-independent mechanism. Proc Natl Acad
Sci USA 1993. 90: 10474-10478.
81. Hurwitz A, Berman J, Lyman W. The role of the blood-brain
barrier in HIV infection of the central nervous system. Adv
Neuroimmunol 1994. 4: 249-256.
82. Nottet H, Persidsky Y, Sasseville V, et al. Mechanisms for
the transendothelial migration of HIV-1-infected monocytes into brain. J
Immunol 1996. 156: 1284-1295.
83. Persidsky Y, Stins M, Way D, et al. Model for monocyte
migration through the blood-brain barrier during HIV-1 encephalitis. J
Immunol 1997. 158: 3499-3510.
84. Sebire G, Hery C, Peudenier S, Tardieu M. Adhesion
proteins on human microglial cells and modulation of their expression by IL-1
alpha and TNF alpha. Res Virol 1993. 144: 27-40.
85. Sasseville V, Lackner A. Neuropathogenesis of simian
immunodeficiency virus infection in macaque monkeys. J Neurovirol
1997. 3: 1-sep.
86. Seilhean D, Dzia-Lepfoundzou A, Sazdovitch V, et al.
Astrocytic adhesion molecules are increased in HIV-1-associated cognitive/motor
complex. Neuropathol Appl Neurobiol 1997. 23:
83-92.
87. Hesselgesser J, HalksMiller M, DelVecchio V, et al.
CD4-independent association between HIV-1 gp120 and CXCR4: Functional chemokine
receptors are expressed in human neurons. Current Biology
1997. 7: 112-121.
88. Lazarini F, Seilhean D, Rosenblum O, et al. Human
immunodeficiency virus type 1 DNA and RNA load in brains of demented and
nondemented patients with acquired immunodeficiency syndrome. J
Neurovirol 1997. 3: 299-303.
89. Corder E, Robertson K, Lannfelt L, Bogdanovic N, Eggertsen
G, Wilkin J. HIV-infected subjects with the E4 allele for APOE have excess
dementia and peripheral neuropathy. Nature Med 1998.
10: 1182-1184.
90. Harrison M, McArthur J, Johnson R. HIV-associated dementia
complex. AIDS and Neurology. NY: Churchill Livingston, 1995.
(vol 3).
91. Selnes O, Galai N, Bacellar H, et al. Cognitive
performance after progression to AIDS: A longitudinal study from the
multicenter AIDS cohort Study. Neurology 1995. 45:
267-275.
92. Association AP. Diagnostic and statistical manual of
mental disorders. In: Press A, ed. . Washington DC: American Psychiatric
Association, 1994.
93. Sidtis J. Evaluation of the AIDS dementia complex in
adults. In: Price R, Perry S, eds. AIDS and the brain. New York: Raven
Press, 1994.
94. Power C, Selnes O, Grim J, Arthur JM. HIV dementia scale:
a rapid screening test. J Acquir Immune Defic Syndr Hum Retrovirol
1995. 8: 273-278.
95. Baldeweg T, Gruzelier J, Stygall J, et al. Detection of
subclinical motor dysfunctions in early symptomatic HIV infection with
topographical EEG. Int J Psychophysiol 1993. 15:
227-238.
96. Gifford D, Jeffrey L, Cummings L. Evaluating dementia
screening tests: methodologic standards to rate their performance.
Neurology 1999. 52: 224-227.
97. Becker J, Sanchez J, Dew M, Lopez O, Dorst S, Banks G.
Neuropsychological abnormalities among HIV-infected individuals in a
community-based sample. Neuropsychology 1997. 11:
592-601.
98. Concha M, Selnes OA, Vlahov D, et al. Comparison of
neuropsychological performance between AIDS-free injecting drug users and
homosexual men. Neuroepidemiol 1997. 16: 78-85.
99. Dunlop O, Bjoklund R, Abdelnoor M, Myrvang B. Total
reaction time: a new approach in early HIV encephalopathy? Acta Neurol
Scand 1993. 88: 344-348.
100. Goodkin K, Wilkie F, Concha M, et al. Subtle
neuropsychological impairment and minor cognitive-motor disorder in HIV
infection : neuroradiological, neurophysiological, neuroimmunological, and
virological correlates. Neuroimaging Clin N Am 1997.
7: 561-579.
101. Heaton R, Velin R, McCutchan A, et al. Neuropsychological
impairment in Human Immunodeficiency Virus-infection: implications for
employment. Psychosom Med 1994. 56: 8-17.
102. Janssen R, Saykin A, Cannon L, et al. Neurological and
neuropsychological manifestations of HIV-1 infection: association with
AIDS-related complex but not asymptomatic HIV-1 infection. Ann Neurol
1989. 26: 592-600.
103. Jones B, Teng E, Folstein M, Harrison K. A new bedside
test of cognition for patients with HIV infection. Ann Intern Med
1993. 119: 1001-1004.
104. Karlsen N, Reinvang I, Froland S. A follow-up study of
neuropsychological function in asymptomatic HIV-infected patients. Acta
Neurol Scand 1993. 87: 83-87.
105. Marsh N, McCall D. Early neuropsychological changes in
HIV infection. Neuropsychology 1994. 8: 44-48.
106. Maruff P, Currie J, Malone V, McArthur-jackson C, Mulhall
B, Benson E. Neuropsychological characterization of the AIDS Dementia Complex
and rationalization of a test battery. Arch Neurol 1994.
51: 689-695.
107. McArthur J. NeuroAIDS: Diagnosis and management.
Hospital Practice 1997. August 15: 73-84.
108. Sahakian B, Elliott R, Low N, Mehta M, Clark R, Pozniak
A. Neuropsychological deficits in tests of executive function in asymptomatic
and symptomatic HIV-1 seropositive men. Psychol Med 1995.
25: 1233-1246.
109. Starace F, Baldassarre C, Biancolilli V, et al. Early
neuropsychological impairment in HIV-seropositive intravenous drug users:
evidence from the Italian Multicentre Neuropsychological HIV Study. Acta
Psychiatr Scand 1998. 97: 132-138.
110. Stern Y, MArder K, Todak G, Sano M, Ehrhardt A, Gorman J.
Neuropsychological changes in a prospectively followed cohort of homosexual and
bisexual men with and without HIV infection. Neurology 1995.
45: 467-472.
111. Stout J, Salmon D, Butters N, et al. Decline in working
memory associated with HIV infection. Psychol Med 1995.
25: 1221-1232.
112. Suarez S, Stankoff B, Conquy L, et al. Similar
subcortical pattern of cognitive impairment in AIDS patients with and without
dementia. Eur J Neurol 2000. : In press.
113. Villa DM, C Marra, A Bartoli, A Antinori, F Pallavicini,
E Tamburrini, I Izzi. Neuropsychological abnormalities in AIDS and asymptomatic
HIV seropositive patients. J Neurol Neurosurg Psychiatry 1993.
56: 878-884.
114. Gutierrez R, Atkinson J, Grant I. Mild neurocognitive
disorder: A needed addition to the nosology of cognitive impairment (organic
mental) disorders. J Neuropsychiatry Clin Neurosci 1993.
5: 161-177.
115. Selnes O, Miller E, McArthur J, al e. HIV-1 infection: no
evidence of cognitive decline during the asymptomatic stages.
Neurology 1990. 40: 204-208.
116. Damos D, John R, Parker E, Levine A. Cognitive functions
in asymptomatic HIV infection. Arch Neurol 1997. 54:
179-185.
117. Kokkevi A, Hatzakis A, Maillis A, et al.
Neuropsychological assessment of HIV-seropositive haemophiliacs. AIDS
1991. 5: 1223-1229.
118. Villa G, Solida A, Moro E, et al. Cognitive impairment in
asymptomatic stages of HIV infection. Eur Neurol 1996.
36: 125-133.
119. Lezak M. Neuropsychological assessment, third edition. .
New York: Oxford University Press, 1995.
120. Satz P, Morgenstern H, Miller E, et al. Low education as
a possible risk factor for cognitive abnormalities in HIV-1: finding from the
multicenter AIDS cohort study (MACS). J Acquir Immune Defic Syndr
1993. 6: 503-511.
121. Stern R, Silva S, Chaisson N, Evans D. Influence of
cognitive reserve on neuropsychological functionning in asymptomatic Human
Immunodeficiency Virus-1 infection. Arch Neurol 1996.
53: 148-153.
122. Selnes O, McArthur J, Nance-Sproson T, et al. HIV-1
infection and intravenous drug use: Longitudinal neuropsychological evaluation
of asymptomatic subjects. Neurology 1992. 42:
1924-1930.
123. Dal Pan G, McArthur J, Aylward E, et al. Patterns of
cerebral atrophy in HIV-1-infected individuals-results of a quantitative MRI
analysis. Neurology 1992. 42: 2125-2130.
124. Aylward E, Henderer J, McArthur J, al e. Reduced basal
ganglia volume in HIV-1-associated dementia: results from quantitative
neuroimaging. Neurology 1993. 43: 2099-2104.
125. Raininko R, Elovaara I, Virta A, valanne L, Haltiaa M,
Valle S. Radiological study of the brain at various stages of human
immunodeficiency virus infection: early development of brain atrophy.
Neuroradiology 1992. 34: 190-196.
126. Hestad K, McArthur J, Pan GD, et al. Regional brain
atrophy in HIV-1 infection: association with specific neuropsychological test
performance. Acta Neurol Scand 1993. 88: 112-118.
127. Di Sclafani V, Mackay S, Meyerhoff D, Norman D, Weiner M,
Fein G. Brain atrophy in HIV infection is more strongly associated with CDC
clinical stage than with cognitive impairment. J Int Neuropsychol Soc
1997. 3: 276-287.
128. Lubetzki C, Stankoff B, Suarez S, Calvez V, Lacomblez L,
Tourbah A. Troubles cognitifs du sida. La lettre du Neurologue
1998. : 65-67.
129. Bell J, Donaldson Y, Lowrie S, et al. Influence of risk
group and zidovudine therapy on the development of HIV encephalitis and
cognitive impairment in AIDS patients. AIDS 1996. 10:
493-499.
130. Seilhean D, Duyckaerts C, Vazeux R, et al.
HIV-1-associated cognitive/motor complex: Absence of neuronal loss in the
cerebral neocortex. Neurology 1993. 43: 1492-1499.
131. Everall I, Glass J, McArthur J, Spargo E, Lantos P.
Neuronal density in the superior frontal and temporal gyri does not correlate
with the degree of human immunodeficiency virus-associated dementia. Acta
Neuropathol 1994. 88: 538-544.
132. Dunbar N, Perdices M, Grunseit A, Cooper D. Changes in
neuropsychological performance of AIDS-related complex patients who progress to
AIDS. AIDS 1992. 6: 691-700.
133. Bornstein R, Nasrallah H, Para M, Whitacre C, Rosenberger
P, Fass R. Neuropsychological performance in symptomatic and asymptomatic HIV
infection. AIDS 1993. 7: 519-524.
134. Martin A, Heyes M, Salazar A, et al. Progressive slowing
of reaction time and increasing cerebrospinal fluid concentrations of
quinolinic acid in HIV-infected individuals. J Neuropsychiatry Clin
Neurosci 1992. 4: 270-279.
135. Beeker J, Salthouse T. Neuropsychological test
performance in the acquired immunodeficiency syndrome: independent effects of
diagnostic group on functionning. J Int Neuropsychol Soc 1999.
5: 41-47.
136. Sacktor N, Bacellar H, Hoover D, et al. Psychomotor
slowing in HIV infection: a predictor of dementia, AIDS and death. J
Neurovirol 1996. 2: 404-410.
137. Price R, Yiannoutsos C, Clifford D, et al. Neurological
outcomes in late HIV infection : adverse impact of neurological impairment on
survival and protective effect of antiviral therapy. AIDS
1999. 13: 1677-1685.
138. Sacktor N, Lyles R, McFarlane G, et al. HIV-1-Related
Neurological Disease Incidence Changes in the Era of highly Active
Antiretroviral Therapy. 51st Annual Meeting of American Academy of Neurology.
Toronto: Neurology (Suppl 2), 1999. (vol 52).
139. Suarez S, Baril L, Stankoff B, et al. Outcome of patients
with HIV1-related cognitive impairment on Highly Active Antiretroviral Therapy
(HAART). AIDS 2000. : Submitted.
140. Robbins T, Roberts A, Owen A, et al.
Monoaminergic-dependent cognitive functions of the prefrontal cortex in monkeys
and man. In: al AMTe, ed. Motor and cognitive functions of the prefrontal
cortex. Berlin: Springer Verlag, 1994.
141. Jasiukaitis P, Fein G. Differential association of
HIV-related neuropsychological impairment with semantic versus repetition
priming. J Int Neuropsychol 1999. 5: 434-441.
142. Nielsen-Bohlman L, Boyle D, Biggins C, Ezekiel F, Fein G.
Semantic priming impairment in HIV. J Int Neuropsychol Soc
1997. 3: 348-358.
143. Hinkin C, Castellon S, Hardy D, Granholm E, Siegle G.
Computerized and traditional stroop task dysfunction in HIV-1 infection.
Neuropsychology 1999. 13: 306-316.
144. Jaffard R. Neurobiologie de la mémoire. Rev
Prat 1991. 41: 2203-2213.
145. E Tulving tpBD. Les systèmes de mémoire
chez l'animal et chez l'homme. In: Shacter D-L, ed. . Marseille:
SOLAL, 1996.
146. Cohen N, Squire L. Preserved learning and retention of
pattern-analysing skill in amnesia: Dissociation of knowing how and knowing
that. Science 1980. 210: 207-210.
147. Tulving E. Elements of episodic memory. In: Press OU, ed.
. London, 1983.
148. Baddeley A. Working Memory. Science
1992. 255: 556-559.
149. Goldman-Rakic P. Cellular and circuit basis of working
memory in prefrontal cortex of non-human primates. In: H. B. M. Uylings CGVE,
J. P. C. DeBruin, M. A. Corner and M. G. P. Feenstra, ed. In Progress in Brain
Research. New York: Elsevier Science, 1990. (vol
85).
150. Owen A, Downes J, Sahakian B, Polkey C, Robbins T.
Planning and spatial working memory following frontal lobe lesions in man.
Neuropsychologia 1990. 28: 1021-1034.
151. Benton A. Differential behavioural effects in frontal
lobe disease. Neuropsychologia 1968. 6: 53-60.
152. Phelps E, Hyder F, Blamire A, Shulman R. FMRI of the
prefrontal cortex during overt verbal fluency. Neuroreport
1997. 8: 561-565.
153. Jonides J, Smith E, Koeppe R, Awh E, Minoshima S, Mintun
M. Spatial working memory in humans as revealed by PET. Nature
1993. 363: 623-625.
154. Becker J, Caldararo R, Lopez O, Dew M, Dorst S, Blank G.
Qualitative features of the memory deficit associated with HIV infection and
AIDS: cross-validation of a discriminant function classification scheme. J
Clin Exp Neuropsychol 1995. 17: 134-142.
155. Peavy G, Jacobs D, Salmon D, et al. Verbal memory
performance of patients with Human Immunodeficiency Virus infection: evidence
of subcortical dysfunction. J Clin Exp Neuropsychol 1994.
16: 508-523.
156. Pillon B, Michon A, Malapani C, Agid Y, Dubois B. Are
explicit memory disorders of progressive supranuclear palsy related to damage
to striatofrontal circuits? Comparison with Alzheimer's, Parkinson's, and
Huntington's diseases. Neurology 1994. 44:
1264-1270.
157. Hinkin C, Gorp WV, Satz P, et al. Actual versus
self-reported cognitive dysfunction in HIV-1 infection: memory-metamemory
dissociations. J Clin Exp Neuropsychol 1996. 18:
431-443.
158. Fox H, Gold L, Henriksen S, Bloom F. SIV : A model for
NeuroAIDS. Neurobiology and Disease 1997. 4:
265-274.
159. Phillips T, Propero-Garcia O, Wheeler D, et al.
Neurologic dysfunctions caused by a molecular clone of feline immunodeficiency
virus, FIV-PPR. J Neurovirol 1996. 2: 388-396.
160. Letvin N, Daniel M, Sehgal P, et al. Induction of
AIDS-like disease in macaque monkeys with T-cell tropic retrovirus
STLV-III. Science 1985. 230: 71-73.
161. Rausch D, Murray E, Eiden L. The SIV-infected rhesus
monkey model for HIV-associated dementia and implications for neurological
diseases. J Leukoc Biol 1999. 65: 466-474.
162. Westmoreland S, Halpern E, Lackner A. Simian
immunodeficiency virus encephalitis in rhesus macaques is associated with rapid
disease progression. J Neurovirol 1998. 4:
260-268.
163. Murray E, Rausch D, Lendway J, Sharer L, Eiden L.
Cognitive and motor impairments associated with SIV infection in Rhesus
monkeys. Science 1992. 255: 1246-1249.
164. Podell M, Hayes K, Oglesbee M, Mathes L. Progressive
encephalopathy associated with CD4/CD8 inversion in adults FIV-infected
cats. J Acquir Immune Defic Syndr Hum Retrovirol 1997.
15: 332-340.
165. Steigerwald E, Sarter M, March P, Podell M. Effects of
Feline Immunodeficiency Virus on cognition and behavioral function in cats.
J Acquir Immune Defic Syndr Hum Retrovirol 1999. 20:
411-419.
166. Podell M, Maruyama K, Smith M, et al. Frontal lobe
neuronal injury correlates to altered function in FIV-infected cats. J
Acquir Immune Defic Syndr 1999. 22: 10-18.
167. Meeker R, Thiede B, Hall C, English R, Tompkins M.
Cortical cell loss in asymptomatic cats experimentally infected with feline
immunodeficiency virus. AIDS Res Hum Retroviruses 1997.
13: 1131-1140.
168. Buck W, Podell M. Neuronal loss in FIV-MD infected
cats. J Neuro-AIDS 1998. 2: 69-77.
169. Kieburtz K, Zettelmaier A, Ketonen L, Tuite M, Caine E.
Manic syndrome in AIDS. Am J Psychiatry 1991. 148:
1068-1070.
170. Heaton R, Crowley T. The effects of psychiatric disorders
and their somatic treatments on neuropsychological test results. In: Boll SFT,
ed. Handbook of clinical neuropsychology. New York: Wiley and Sons,
1981.
171. Maj M. Depressive syndromes and symptoms in subjects with
Human Immunodeficiency Virus (HIV) Infection. Brit J PSychiatr
1996. 168 (suppl.30): 117-122.
172. Hinkin C, Gorp WV, Satz P, Weisman J, Thommes J,
Buckingham S. Depressed Mood and its Relationship to Neuropsychological Test
Performance in HIV-1 Seropositive Individuals. J Clin Exp Neuropsychol
1992. 14: 289-297.
173. Bornstein R, Pace P, Rosenberger P, et al. Depression and
neuropsychological performance in asymptomatic HIV infection. Am J
Psychiatry 1993. 150: 922-927.
174. Dufouil C, Fuhrer R, Dartigues J, Alperovitch A.
Longitudinal analysis of the association between depressive symptomatology and
cognitive deterioration. Am J Epidemiol 1996. 144:
634-641.
175. Silvestre D, Linard F, Desi M, et al. Statut
anxio-dépressif et déficit cognitif au cours de l'infection par
le VIH. L'Encéphale 1995. XXI: 285-288.
176. VanGorp W, Satz P, Hinkin C, et al. Metacognition in
HIV-1 seropositive asymptomatic individuals: self-ratings versus objective
neuropsychological performance. Multicenter AIDS Cohort Study (MACS). J
Clin Exp Neuropsychol 1991. 13: 812-819.
177. Wilkins J, Robertson K, Snyder C, Robertson W, Horst CVd,
Hall C. Implications of self-reported cognitive and motor dysfunction in
HIV-positive patients. Am J Psychiatry 1991. 148:
641-643.
178. Moore L, Gorp WV, Hinkin C, Stern M, Swales T, Satz P.
Subjective complaints versus actual cognitive deficits in predominantly
symptomatic HIV-1 seropositive individuals. J Neuropsychiatry Clin
Neurosci 1997. 9: 37-44.
179. Poutiainen E, Elovaara I. Subjective complaints of
cognitive symptoms are related to psychometric findings of memory deficits in
patients with HIV-1 infection. J Int Neuropsychol Soc 1996.
2: 219-225.
180. Stern Y, Marder K, Bell K, et al. Multidisciplinary
baseline assessment of homosexual men with and without human immunodeficiency
virus infection. III. Neurologic and neuropsychological findings. Arch Gen
Psychiatry 1991. 48: 131-8.
181. Mapou R, Law W, Martin A, Kampen D, Salazar A, Rundell J.
Neuropsychological performance, mood, and complaints of cognitive and motor
difficulties in individuals infected with the human immunodeficiency virus.
J Neuropsychiatry Clin Neurosci 1993. 5: 86-93.
182. Beason-Hazen S, Nasrallah H, Bornstein R. Self-report of
symptoms and neuropsychological performance in asymptomatic HIV-positive
individuals. J Neuropsychiatry Clin Neurosci 1994. 6:
43-49.
183. Grant I, Olshen R, Atkinson J, et al. Depressed mood does
not explain neuropsychological deficits in HIV-infected persons.
Neuropsychology 1993. 7: 53-61.
184. Castellon S, Hinkin C, Wood C, Yarema K. Apathy,
depression, and cognitive performance in HIV-1 infection. J Neuropsychiatry
Clin Neurosci 1998. 10: 320-329.
185. Boller F, Marcie P, Starkstein S, Traykov L. Memory and
depression in Parkinson's disease. Eur J Neurol 1998.
5: 291-295.
186. Perdices M, Dunbar N, Grunseit A, Hall W, Cooper D.
Anxiety, depression and HIV related symptomatology across the spectrum of HIV
disease. Aust NZJ Psychiatry 1992. 26: 560-566.
187. Poutiainen E. Cognitive deficits and emotional disorders
in HIV-1 infected individuals. Acta Psychiatr Scand 1995.
92: 429-435.
188. Leserman J, Jackson E, Petitto J, et al. Progression to
AIDS: the effects of stress, depressive symptoms, and social support.
Psychosom Med 1999. 61: 397-406.
189. Moore J, Schuman P, Schoenbaum E, Boland B, Solomon L,
Smith D. Severe adverse life events and depressive symptoms among women with,
or at risk for, HIV infection in four cities in the United States of
America. AIDS 1999. 13: 2459-2468.
190. Gordillo V, Amo JD, Soriano V, Gonzalez-Lahoz J.
Sociodemographic and psychological variables influencing adherence to
antiretroviral therapy. AIDS 1999. 13: 1763-1769.
191. Nath A, Booze R, Hauser K, et al. Interactions of drugs
of abuse and HIV dementia. NeuroAIDS 1999. 2: 1-9.
192. Heckman T, Kelly J, Bogart L, Kalichman S, Rompa D. HIV
risk difference between African-American and white men who have sex with
men. J Natl Med Assoc 1999. 91: 92-100.
193. Selnes O, Galai N, McArthur J, et al. HIV infection and
cognition in intravenous drug users: Long-term follow-up. Neurology
1997. 48: 223-230.
194. Leshner A, Koob G. Drugs of abuse and the brain. Proc
Assoc Am Physicians 1999. 111: 99-108.
195. Davies J, Everall I, Weich S, Laughlin JM, Scaravilli F,
Lantos L. HIV-associated brain pathology in the United Kingdom: an
epidemiological study. AIDS 1997. 11: 1145-1150.
196. Tyor W, Middaugh L. Do alcohol and cocaine abuse alter
the course of HIV-associated dementia complex? J Leukoc Biol
1999. 65: 475-481.
197. Grassi M, Perin C, Clerici F, et al. Do alcohol and
cocaine abuse alter the course of HIV-associated dementia complex? J Leukoc
Biol 1997. 65: 475-481.
198. Bell J, Brettle R, Chiswick A, Simmonds P. HIV
encephalitis, proviral load and dementia in drug users and homosexuals with
AIDS - Effect of neocortical involvement. Brain 1998.
121: 2043-2052.
199. Fiala M, Gan X, Zhang L, et al. Cocaine enhances monocyte
migration across the blood-brain barrier. Cocaine's connection to AIDS dementia
and vasculitis? Adv Exp Med Biol 1998. 437:
199-205.
200. Zhang L, Looney D, Taub D, et al. Cocaine opens the
blood-brain barrier to HIV-1 invasion. J Neurovirol 1998.
4: 619-626.
201. Berger J, Nath A. HIV dementia and the basal ganglia.
Intervirology 1997. 40: 122-131.
202. Scarponi M, Bernardi G, Mercuri N. Electrophysiological
evidence for a reciprocal interaction between amphetamine and
cocaïne-related drugs on rat midbrain dopaminergic neurons. Eur J
Neurosci 1999. 11: 593-598.
203. Mirsattari S, Power C, Nath A. Parkinsonism with HIV
infection. Mov Disord 1998. 13: 684-689.
204. Maher J, Choudhri S, Halliday W, Power C, Nath A. AIDS
dementia complex with generalized myoclonus. Mov Disord 1997.
12: 593-597.
205. Donahoe R, Vlahov D. Opiates as potential cofactors in
progression of HIV-1 infections to AIDS. J Neuroimmunol 1998.
83: 77-87.
206. Peterson P, Gekker G, Hu S, et al. Morphine Amplifies
HIV-1 expression in chronically infected promonocytes cocultured with human
brain cells. J Neuroimmunol 1994. 50: 167-175.
207. Peterson P, Gekker G, Hu S, Chao C. Opiate effects on HIV
infection in human brain cells. J Neuroimmunol 1996.
69: 44-45.
208. Peterson P, Gekker G, Hu S, Lokensgard J, Portoghese P,
Chao C. Endomorphin-1 potentiates HIV-1 expression in human brain cell
cultures: implication of an atypical mu-opioid receptor.
Neuropharmacology 1999. 38: 273-278.
209. Sclafani VD, Maackay R, Meyerhoff D, Norman D, Weiner M,
Fein G. Brain atrophy in HIV infection is more strongly associated with CDC
clinical stage than with cognitive impairment. J Int Neuropsychol Soc
1997. 3: 276-287.
210. Mattis S. Mental status examination for organic mental
syndrome in the elderly patient. In: Bellac L, Karasu T, eds. Geriatric
Psychiatry: A Handbook for Psychiatrists and Primary Care Physicians. New
York: Grune & Straton, 1976.
211. Folstein M, Folstein S, McHugh P. "Mini-Mental State". A
practical method for grading the cognitive state of patients for the
clinician. J Psychiat Res. 1975. 12: 189-198.
212. Reitan R. Validity of the Trail Making test as indicator
of organic brain damages. Percept Mot Skills 1958. 8:
271-276.
213. Costa L, Vaughan H, Levita J, Farber N. Purdue pegboard
as a predictor of the presence and laterality of cerebral lesions. J
consult psychol 1963. 27: 133-137.
214. Hamm N, Curtis D. Normative data for the Purdue Pegboard
on a sample of adult candidates for vocational rehabilitation. Percept Mot
skills 1980. 50: 309-310.
215. Grober E, Buschke H. Genuine memory deficits in
dementia. Developmental Neuropsychology 1987. 3:
13-36.
216. Montgomery S, Asberg M. A new depression scale designed
to be sensitive to change. Brit. J. Psychiatr 1979.
134: 382-389.
217. Hall M, Whaley R, Robertson K, Hamby S, Wilkins J, Hall
C. The correlation between neuropsychological and neuroanatomic changes over
time in asymptomatic and symptomatic HIV-1-infected individuals.
Neurology 1996. 46: 1697-1702.
218. Portegies P, Enting R, Gans Jd, et al. Presentation and
course of AIDS dementia complex: 10 years of follow-up in Amsterdam, The
Netherlands. AIDS 1993. 7: 669-675.
219. Albert M, Feldman R, Willis A. The "subcortical dementia"
of progressive supranuclear palsy. J Neurol Neurosurg Psychiatry
1974. 37: 121-130.
220. Pillon B, Deweer B, Agid Y, Dubois B. Explicit memory in
Alzheimer's, Huntinton's, and Parkinson's diseases. Arch Neurol
1993. 50: 374-379.
221. Stern R, Arruda J, Somerville J, et al. Neurobehavioral
functioning in asymptomatic HIV-1 infected women. J Int Neuropsychol
Soc 1997. 4: 172-178.
222. Dal Pan G, Farzadegan H, Selnes O, et al. Sustained
cognitive decline in HIV infection: relationship to CD4+ cell count, plasma
viremia and p24 antigenemia. J Neurovirol 1998. 4:
95-99.
223. McArthur J, McClernon D, Cronin M, et al. Relationship
between Human Immunodeficiency Virus-associated dementia and viral load in
cerebrospinal fluid and brain. Ann Neurol 1997. 42:
689-698.
224. Stankoff B, Calvez V, Suarez S, et al. Plasma and
cerebro-spinal fluid human immunodeficiency virus type-1 (HIV-1) RNA levels in
HIV-related cognitive impairment. Eur J Neurol 1999.
6: 669-675.
225. Stankoff B, Suarez S, Rosenblum O, et al. Troubles
cognitifs du SIDA : aspects cliniques, virologiques et neuroradiologiques.
Rev Neurol 1998. 154: 843-849.
226. Krivine A, Force G, Servan J, et al. Measuring HIV-1 RNA
and interferon-alpha in the cerebrospinal fluid of AIDS patients: insights into
the pathogenesis of AIDS Dementia Complex. J Neurovirol 1999.
5: 500-506.
227. Conrad PS, K Syndulko, EJ Singer, RM Nagra, JJ Russell,
WW Tourtellotte. Quantifying HIV-1 RNA using the polymerase chain reaction on
cerebrospinal fluid and serum of seropositive individuals with and without
neurologic abnormalities. J Acquir Immune Defic Syndr Hum Retrovirol
1995. 10: 425-435.
228. Garcia F, Niebla G, Romeu J, et al. Cerebrospinal fluid
HIV-1 RNA levels in asymptomatic patients with early stage chronic HIV-1
infection: support for the hypothesis of local virus replication. AIDS
1999. 13: 1491-1496.
229. Di Stephano M, Monno L, Fiore J, et al. Neurological
disorders during HIV-1 infection correlate with viral load in cerebrospinal
fluid but not with virus phenotype. AIDS 1998. 12:
737-743.
230. Foudraine N, Hoetelmans R, Lange J, et al. Cerebrospinal
fluid HIV-1 RNA and drug concentrations after treatment with lamivudine plus
zidovudine or stadivudine. Lancet 1998. 351:
1547-1551.
231. Eggers C, VanLunzen J, Buhk T, Stellbrink H. HIV
infection of the central nervous system is characterized by rapid turnover of
viral RNA in cerebrospinal fluid. J Acquir Immune Defic Syndr Hum
Retrovirol 1999. 20: 259-264.
232. Cinque P, Scarpellini P, Vago L, Linde A, Lazzarin A.
Diagnosis of central nervous system complications in HIV infected patients:
cerebro-spinal fluid analysis by the polymerase chain reaction. AIDS
1997. 11: 1-17.
233. Schmid P, Conrad A, Syndulko K, et al. Quantifying HIV-1
proviral DNA using the polymerase chain reaction on cerebrospinal fluid and
blood of seropositive individuals with and without neurological
abnormalities. J Acquir immune Defic Syndr 1994. 7:
777-788.
234. Chiodi F, Keys B, Albert J, et al. Human immunodeficiency
virus type 1 is present in the cerebrospinal fluid of a majority of infected
individuals. J Clin Microbiol 1992. 30: 1768-1771.
235. Ellis R, Hsia K, Spector S, et al. Cerebrospinal fluid
Human immunodeficiency virus type 1 RNA levels are elevated in neurocognitively
impaired individuals with acquired immunodeficiency syndrome. Ann
Neurol 1997. 42: 679-688.
236. Cinque P, Vago L, Ceresa D, et al. Cerebrospinal fluid
HIV-1 RNA levels: correlation with HIV encephalitis. AIDS
1998. 12: 389-394.
237. Lubetzki C, Dubard T, Suarez S, Katlama C, Turell E,
Calvez E. Cerebrospinal fluid HIV load and AIDS dementia. Neurology
1997. 48: A387.
238. Brew B, Pemberton L, Cunningham P, Law M. Levels of human
immunodeficiency virus type 1 RNA in cerebrospinal fluid correlate with AIDS
dementia stage. J Infect Dis 1997. 175: 963-966.
239. Meeker R, Robertson K, Barry T, Hall C. Neurotoxicity of
CSF from HIV-infected humans. J Neurovirol 1999. 5:
507-518.
240. Childs E, Lyles R, Selnes O, et al. Plasma viral load and
CD4 lymphocytes predict HIV-associated dementia and sensory neuropathy.
Neurology 1999. 52: 607-613.
241. Budka H, Willey CA, Kleihues P, et al. HIV-associated
disease of the nervous system: review of nomenclature and proposal for
neuropathology-based terminology. Brain Pathol 1991.
1: 142-150.
242. Spencer D, Price R. Human immunodeficiency virus and the
central nervous system. Ann Rev Microbiol 1992. 46:
655-693.
243. Manji H, Connolly S, Mcallister R, et al. Serial MRI of
the brain in asymptomatic patients infected with HIV - results from the
ucmsm/medical research council neurology cohort. J Neurol Neurosurg
Psychiatry 1994. 57: 144-149.
244. Miller RF, Lucas SB, HallCraggs MA, et al. Comparison of
magnetic resonance imaging with neuropathological findings in the diagnosis of
HIV and CMV associated CNS disease in AIDS. J Neurol Neurosurg
Psychiatry 1997. 62: 346-351.
245. Everall I, Chong W, Wilkinson I, et al. Correlation of
MRI and neuropathology in AIDS. J Neurol Neurosurg Psychiatry
1997. 62: 92-95.
246. Aylward E, Brettschneider P, Arthur JM, et al. Magnetic
resonance imaging measurement of gray matter volume reductions in HIV
dementia. Am J Psychiatry 1995. 152: 987-994.
247. Tracey I, Hamberg L, Guimaraes A, et al. Increased
cerebral blood volume in HIV-positive patients detected by functional MRI.
Neurology 1998. 50: 1821-1826.
248. Navia B, Gonzalez R. Functional imaging of the AIDS
dementia complex and the metabolic pathology of the HIV-1-infected brain.
Neuroimaging Clin N Am 1997. 7: 431-445.
249. Ollo C, Johnson R, Grafman J. Signs of cognitive change
in HIV disease: An event-related brain potential study. Neurology
1991. 41: 209-215.
250. Sakaie K, Gonzalez R. Imaging of NeuroAIDS.
NeuroAIDS 1999. 2: 1-11.
251. Lopez-Villegas D, Lenkinski R, Frank I. Biochemical
changes in the frontal lobe of HIV-infected individuals detected by magnetic
resonance spectroscopy. Proc Natl Acad Sci USA 1997.
94: 9854-9859.
252. Harrison M, Newman S, Hall-Craggs M, et al. Evidence of
CNS impairment in HIV infection: clinical, neuropsychological, EEG, and MRI/MRS
study. J Neurol Neurosurg
Psychiatry 1998. 63: 301-307.
253. Bencherif B, Rottenberg D. Neuroimaging of the AIDS
dementia complex. AIDS 1998. 12: 233-244.
254. Miller B, Chang L, Booth R, et al. In vivo 1H MRS
choline: correlation with in vitro chemistry/histology. Life Sci
1996. 58: 1929-1935.
255. Menon D, Ainsworth J, Cox I, et al. Proton MR
spectroscopy of the brain in AIDS dementia complex. J Comput Assist
Tomogr 1992. 16: 538-542.
256. Meyerhoff D, MacKay S, Bachman I, et al. Reduced brain
N-acetylaspartate suggests neuronal loss in cognitively impaired human
immunodeficiency virus-seropositive individuals: in vivo 1H magnetic resonance
spectroscopic imaging. Neurology 1993. 43:
509-515.
257. Mcconnell J, Swindells S, Ong C, et al. Prospective
utility of cerebral proton magnetic resonance spectroscopy in monitoring HIV
infection and its associated neurological impairment. AIDS Res and Hum
Retroviruses 1994. 10: 977-982.
258. Barker RL, JC Mc Arthur. AIDS dementia complex:
evaluation with proton MR spectroscopic imaging. Radiology
1995. 195: 58-64.
259. Tracey I, Carr C, Guimaraes A, Worth J, Navia B, Gonzalez
R. Brain choline-containing compounds are elevated in HIV-positive patients
before the onset of AIDS dementia complex: A proton magnetic resonance
spectroscopic study. Neurology 1996. 46: 783-788.
260. Ketzler S, Weis S, Haug H, al e. Loss of neurons in the
frontal cortex in AIDS brains. Acta Neuropathol 1990.
80: 92-94.
261. Masliah E, Ge N, Mucke L. Pathogenesis of HIV-1
associated neurodegeneration. Crit Rev Neurobiol 1996.
10: 57-67.
262. Masliah E, Heaton RK, Marcotte TD, et al. Dendritic
injury is a pathological substrate for human immunodeficiency virus-related
cognitive disorders. Ann Neurol 1997. 42: 963-972.
263. Vion-Dury J, Nicoli F, Salvan A, Confort-Gouny S, Dhiver
C, Cozzone P. Reversal of brain metabolic alterations with zidovudine detected
by proton localized magnetic resonance spectroscopy. Lancet
1995. 345: 60-61.
264. Alonso J, Rovira A, Capellades J, et al. Cerebral proton
spectroscopy of people infected with the human immunodeficiency virus [article
in Spanish]. Med Clin (Barc) 1996. 107: 316-365.
265. Wilkinson I, Miller R, Miszkiel K, et al. Cerebral proton
magnetic resonance spectroscopy in asymptomatic HIV infection. AIDS
1997. 11: 289-295.
266. Moller I, Vermathen P, Lentschig M, et al. Metabolic
characterization of AIDS dementia complex by spectroscopic imaging. J Magn
Reson Imaging 1999. 9: 10-18.
267. Meyerhoff DJ, Bloomer C, Cardena V, Norman D, Weiner MW,
Fein G. Elevated subcortical choline metabolites in cognitively and clinically
asymptomatic HIV+ patients. Neurology 1999. 52:
995-1003.
268. Stankoff B, tourbah A, Suarez S, et al. Clinical and
Spectroscopic improvement in HIV associated cognitive impairment: A
longitudinal study. Neurology 2000. : Submitted.
269. Chang L, Ernest T, Leonido-Yee M, Walot I, Singer E.
Cerebral metabolite abnormalities correlate with clinical severity of HIV-1
cognitive motor complex. Neurology 1999. 52:
100-108.
270. Wilkinson ID, Lunn S, Miszkiel KA, et al. Proton MRS and
quantitative MRI assessment of the short term neurological response to
antiretroviral therapy in AIDS. J Neurol Neurosurg Psychiatry
1997. 63: 477-482.
271. Vion-Dury J, Salvan AM, ConfortGouny S, Cozzone PJ.
Proton magnetic resonance spectroscopy of the brain - Indications for the
diagnosis and follow-up of HIV-related encephalopathy in adults. Presse
Medicale 1998. 27: 1398-1405.
272. Chang I, Ernst T, Leonido-Yee M, et al. Highly active
antiretroviral therapy reverses brain metabolite abnormalities in mild HIV
dementia. Neurology 1999. 53: 782-789.
273. Salvan A, VionDury J, ConfortGouny S, Nicoli F, Lamoureux
S, Cozzone P. Brain proton magnetic resonance spectroscopy in HIV-related
encephalopathy: Identification of evolving metabolic patterns in relation to
dementia and therapy. AIDS Res and Hum Retroviruses 1997.
13: 1055-1066.
274. Lubetzki C, B BS, Suarez S, et al. Proton magnetic
resonance spectroscopy in patients with AIDS-Associated Cognitive Disorders: a
longitudinal study. In: Neurology, ed. 51st Annual Meeting of American
Academy of Neurology. Toronto: Neurology, 1999. (vol
52).
275. Foster R, Faulds D. Abacavir. Drugs
1998. 55: 729-738.
276. Gray F, Bélec L, Keohane C, et al. Zidovudine
therapy and HIV encephalitis: a 10-year neuropathological survey. AIDS
1994. 8: 489-493.
277. Simpson D. Human immunodeficiency Virus-Associated
Dementia: Review of Pathogenesis, Prophylaxis, and Treatment Studies of
Zidovudine Therapy. CID 1999. 29: 19-34.
278. Cooper C, Pedersen C, Aiuti F, et al. The efficacity and
safety of zidovudine with or without acyclovir in the treatment of patients
with AIDS-related complex. The European-Australian Collaborative Group.
AIDS 1991. 5: 933-943.
279. Damos D, Richard M, John R, Parker E, Levine A.
Anti-retroviral therapy and cognitive function. Aviat Space Environ
Med 1997. 68: 900-906.
280. Brew B, Dumbar N, Druett J, Freund J, Ward P. Pilot study
of the efficacity of atevirdine in the treatment of AIDS dementia complex.
AIDS 1996. 10: 1357-1360.
281. Campiani G, Morelli E, Fabbrini M, et al.
Pyrrolobenzoxazepione derivatives as non-nucleoside HIV-1 RT inhibitors:
further structure-activity relationship studies and identification of more
potent broad-spectrum HIV-1 RT inhibitors with antiretroviral activity. J
Med Chem 1999. 42: 4462-4470.
282. Bini E, Cohen J. Impact of protease inhibitors on the
outcome of human immunodeficiency virus-infected patients with chronic
diarrhea. Am J Gastroenterol 1999. 94: 3553-3559.
283. Enting R, Hoetelmans R, Lange J, Burger D, Beijnen J,
Portegies P. Antiretroviral drugs and the central nervous system. AIDS
1998. 12: 1941-1955.
284. Ferrando S, Gorp WV, McElhiney M, Goggin K, Sewell M,
Rabkin J. Highly active antiretroviral treatment in HIV infection: benefits for
neuropsychological function. AIDS 1998. 12:
F65-F70.
285. Tozzi V, Balestra P, Galgani S, et al. Positive and
sustained effects of highly active antiretroviral therapy on HIV-1-associated
neurocognitive impairment. AIDS 1999. 13:
1889-1897.
286. Sacktor N, Lyles R, Skolasky R, et al. Combination
antiretroviral therapy improves psychomotor speed performance in
HIV-seropositive homosexual men. Neurology 1999. 52:
1640-1647.
287. Martin E, Pitrack D, Novak R, Pursell K, Mullane K.
Reaction times are faster in HIV-seropositive patients on antiretroviral
therapy: A preliminary report. J Clin Exp Neuropsychol 1999.
21: 730-735.
288. Martin E, Pitrak D, Pursell K, Andersen B, Mullane K,
Novak R. Information processing and antiretroviral therapy in HIV-1
infection. J Int Neuropsychol Soc 1998. 4:
329-335.
289. Dore G, Correll P, Li Y, Kaldor J, Cooper D, Brew B.
Changes to AIDS dementia complex in the era of highly active antiretroviral
therapy. AIDS 1999. 13: 1249-1253.
290. Swindells S, Zheng J, Gendelman H. HIV-associated
dementia: new insights into disease pathogenesis and therapeutic
interventions. AIDS Patient Care 1999. 13:
153-163.
291. Baum S, Morris J, Gibbons R, Cooper R. Reduction in human
immunodeficiency virus patient hospitalizations and nontraumatic mortality
after adoption of highly active antiretroviral therapy. Mil Med
1999. 164: 609-612.
292. Palella FJ, Delaney K, Moorman A, et al. Declining
morbidity and mortality among patients with advanced human immunodeficiency
virus infection. HIV outpatient Study Investigators. N Engl J Med
1998. 338: 853-860.
293. Mocroft A, Madge S, Johnson A, et al. A comparison of
exposure groups in the EuroSIDA study: starting highly active antiretroviral
therapy (HAART), response to HAART, and survival. J Acquir Immune Defic
Syndr 1999. 22: 369-378.
294. Ledergerber B, Egger M, Erard V, et al. AIDS-related
opportunistic illnesses after initiation of potent antiretroviral therapy: the
Swiss HIV Cohort Study. JAMA 1999. 282: 2220-2226.
295. Gasnault J, Taoufik Y, Goujard C, et al. Prolonged
survival without neurological improvement in patients with AIDS-related
progressive multifocal leukoencephalopathy on potent combined antiretroviral
therapy. J Neurovirol 1999. 5: 421-429.
296. Tassie J, Gasnault J, Bentata M, et al. Survival
improvement of AIDS-related progressive multifocal leukoencephalopathy in the
era of protease inhibitors. AIDS 1999. 13:
1881-1887.
297. Clifford D, Yiannoutsos C, Glicksman M, et al. HAART
improves prognosis in HIV-associated progressive multifocal
leukoencephalopathy. Neurology 1999. 52: 623-625.
298. Melchior J, Niyongabo T, Henzel D, Durack-Bown I, Henry
S, Boulier A. Malnutrition and wasting, immunodepression, and chronic
inflammation as independant predictors of survival in HIV-infected
patients. Nutrition 1999. 15: 865-869.
299. Testa M, Lenderking W. The impact of AIDS-associated
wasting on quality of life: qualitative issues of measurement and
evaluation. J Nutr 1999. 129: 282S-289S.
300. Brosseau J, Gasnault J, Sanchez I, et al. Nutritional
monitoring using body mass index and nutrition risk index in Neuro-AIDS
patients. 3rd International Conference on Nutrition and HIV infection. Cannes,
1999.
301. Ellis R, Deutsch R, Heaton R, et al. Neurocognitive
impairment is an independent risk factor for death in HIV infection. Arch
Neurol 1997. 54: 416-424.
302. Wilkie F, Goodkin K, Eisdorfer C, et al. Mild cognitive
impairment and risk of mortality in HIV-1 infection. J Neuropsychiatry
1998. 10: 125-132.
303. Masson F, Maurette P, Salmi R, al. e. Prevalence of
impairments 5 years after a head injury and their relationhip with disabilities
and outcome. Brain Injury 1996. 7: 487-497.
304. Mazaux J, Masson F, Levin H, al. e. Long term
neuropsychosocial outcome and loss of social autonomy after traumatic brain
injury. Arch Phys Med Rehabil 1997. 78: 1316-1320.
305. Billingham L, Abrams K, Jones D. Methods for the analysis
of quality-of-life and survival data in health technology assessement.
Health Technol Assess 1999. 3: 1-152.
306. Leplege A, Rude N, Ecosse E, Ceinos R, Dohin E, Pouchot
J. Measuring quality of life from the point of view of HIV-positive subjects:
the HIV-QL31. Qual Life Res 1997. 6: 585-594.
307. Holmes W, Shea J. Two approaches to measuring quality of
life in the HIV/AIDS population: HAT-Qol and MOS-HIV. Qual Life Res
1999. 8: 515-527.
308. Scott-Lennox J, Wu A, Boyer J, Ware J. Reliability and
validity of French, German, Italian, Dutch, and UK English translations of the
Medical Outcome Study HIV Health Survey. Med Care 1999.
37: 908-925.
309. Revicki D, Moyle G, Stellbrink H, Barker C. Quality of
life outcomes of combination zalcitabine-zidovudine, saquinavir-zidovudine, and
saquinavir-zalcitabine-zidovudine therapy for HIV-infected adults with CD4 cell
counts between 50 and 350 per cubic millimeters. PISCES (SV14604) Study
Group. AIDS 1999. 13: 851-858.
310. Chatterton M, Scott-Lemox J, Wu A, Scott J. Quality of
life and treatment satisfaction after the addition of lamivudine or lamivudine
plus loviride to zidovudine-containing regimens in treatment-experienced
patients with HIV infection. Pharmacoeconomics 1999.
15: 67-74.
311. Globe D, HAys R, Cunningham W. Associations of clinical
parameters with health-related quality of life in hospitalized persons with HIV
disease. AIDS care 1999. 11: 71-86.
312. Cunningham W, Shapiro M, Hays R, et al. Constitutional
symptoms and health-related quality of life in patients with symptomatic HIV
disease. Am J Med 1998. 104: 129-136.
313. O'Dell M, Hubert H, Lubeck D, O'Driscoll P. Pre-AIDS
physical disability: data from the AIDS Time-Oriented Health Outcome Study.
Arch Phys Med Rehabil 1998. 79: 1200-1205.
314. Breitbart W, McDonald M, Rosenfeld B, Monkman N, Passik
S. Fatigue in ambulatory AIDS patients. J Pain Symptom Manage
1998. 15: 159-167.
315. Ferrando S, Goggin K, Sewell M, Fishman B, Rabkin J.
Fatigue in HIV illness: relationship to depression, physical limitations, and
disability. Psychom Med 1998. 60: 759-764.
316. Darko D, Mitler M, Miller J. Growth hormone, fatigue,
poor sleep, and disability in HIV infection. Neuroendocrinology
1998. 67: 317-324.
317. Rose L, Lears K, Gordon D. The fatigue experience:
persons with HIV infection. J Adv Nurs 1998. 28:
295-304.
318. Ozoy M, Ernest E. How effective are complementaries for
HIV and AIDS?-A systematic review. Int J STD AIDS 1999.
10: 629-635.
319. Swindells S, Mohr J, Justis J, et al. Quality of life in
patients with human immunodeficiency virus infection: impact of social support,
coping style and hopelessness. Int J STD AIDS 1999.
10: 383-391.
320. Vogl D, Rosenfield B, Breitbart W, et al. Symptom
prevalence, characteristics, and distress in AIDS outpatients. J Pain
Symptom Manage 1999. 18: 253-262.
321. Scott I, Smiddy W, Schiffman J, Feuer W, Pappas C.
Quality of life of low-vision patients and the impact of low-vision
services. Am J Ophtalmol 1999. 128: 54-62.
322. Lawton M, Brody E. Assessment of older people:
self-maintening and instrumental activities of daily living.
Gerontologist 1969. 9: 179-186.
VIII. ANNEXES
* Sources : « Le point sur
l'épidémie du SIDA, programme commun des Nations Unies sur le
VIH/SIDA ». ONUSIDA et Organisation mondiale de la Santé,
déclaration du 1décembre 1999 sur Internet :
http://www.unaids.org.ONUSIDA
* Sources : Rapport 1999, n8 61 du
Centre européen pour la surveillance épidémiologique du
SIDA, Centre collaborateur OMS-ONUSIDA- UE sur le SIDA, Saint-Maurice, France.
Internet :
http://www.ceses.org
* Sur 72976 patients répartis dans
68 hôpitaux français.
* Les patients devaient indiquer si des
items présentés étaient des mots ou des non-mots. Tous les
mots avaient un antonyme évident et les deux tiers étaient
présentés comme des paires d'antonymes (successifs). Le temps de
réponse était relevé.
* L'IRM utilise, pour former l'image,
le signal électromagnétique émis par les noyaux
d'hydrogène des tissus soumis à un champ magnétique
intense. Les protons des tissus s'orientent alors dans une même direction
suivant une constante de temps caractéristique de chaque tissu. Pour
obtenir un signal, on introduit une onde de radiofréquence perturbant
l'orientation des protons. Lorsque l'onde s'interrompt, les protons reviennent
à leur position d'équilibre en libérant une onde
électromagnétique, transformée en une image ordinateur. La
décroissance de ce signal a une constante de temps
caractéristique de chaque tissu (T1, séquences rapides; T2,
séquences lentes).
* Après approbation par le
comité d `éthique médical.
| 

