|
UNIVERSITE DE TUNIS EL MANAR
FACULTE DES SCIENCES DE TUNIS

MEMOIRE
Pour l'obtention du
Mastère de Génétique et
Bio-ressourses
POLYMORPHISME DES ANTIGENES PLAQUETTAIRES HUMAINS DANS
LA POPULATION TUNISIENNE
Présenté et soutenu publiquement par
Melle : LASSOUED
Mabrouka
Le 31/12/2007
Soutenu devant le Jury :
- Mme. Saida BEN ARAB: Président de
Jury
- Mr. Slema HMIDA: Membre
- Mme. Najet MOJAAT: Encadreur
- Mr. Aly RAIES: Membre
Centre National de Transfusion Sanguine
![]()
Je dédie ce mémoire
A
La mémoire de mon père Mohamed
Tu m'étais le père et l'ami conseiller. Que vous
trouviez ici le témoignage de mon inaltérable amour et mon
profond dévouement pour vos sacrifices, vos encouragements et vos
prières qui ont été le meilleur gage de ma
réussite.
A
Ma chère mère : Hania
Que ce travail soit le témoignage de ma gratitude pour
l'amour et la sollicitude que vous avez su me prodiguer depuis toujours.
Que Dieu vous préserve et Vous prête longue
vie.
A
Toute ma famille et tous mes proches
Mes frères Abdallah, Lhédi, Hmed, Hmad et Ali.
Mes soeurs Fatma, Salha et Zina.
Mes chers Akrmi, Amor, Zied, Tarek, Mohamed, Ala, khmaies Dhia,
Abdel kader,
Rabii, Aziz, Beha et Anas.
Mes chères Leila, Maeyoufa, Saida, Houda, Hanen, Abir,
Chiraz et Safa.
Merci à mes parents et à ma famille pour leur
soutien et leur confiance indéfectibleçç
A
Mes chers amis Radhi, Sami, Mourad, Rabeh, Samir, Hichem,
Chouaib, Mohamed et Tarek.
Mes chères amies Amani, Fatma, Neziha, Wafa, Fatma,
Sana, Sameh, Nejwa, Rima, Yemina, Sessia, Abir, Nejet, Soumaya, Gleya et
Kawther, Semira et Moufida.
Je voudrais exprimer ma profonde reconnaissance
à mes directeurs de Mémoire
Monsieur Slema HMIDA, Professeur à la FPM & Chef
service au CNTS
Et
Madame Najet MOJAAT, Professeur à la FPM & Chef
service au CNTS de m'avoir accueilli au sein de leur équipe de
recherche, d'avoir mis à ma disposition les moyens matériels et
scientifiques pour réaliser ce travail de master.
Vous avez su me guider et conseiller tout au long de ce
travail.
Merci pour vos conseils, pour votre disponibilité
inconditionnelle malgré un emploi du temps toujours chargé et
merci pour la confiance que vous m'avez portée.
Bref, en un mot, merci pour votre
générosité.
Merci au personnel du laboratoire
pour l'ambiance agréable sans laquelle aucun travail serein n'est
envisageable.
Remerciements particuliers à Melle Houda
KAABI pour ses coups de mains et pour ses connaissances.
A tous ceux qui ont participé à l'avancement de
ce travail, et particulièrement Mr Hajjaj.
Mes remerciements s'adressent
à Mme Saida BEN ARAB, professeur
à la FMT en reconnaissance de l'honneur qu'elle m'accorde en acceptant de présider le jury du
mémoire.
Un grand merci également au professeur Aly RAIES pour l'honneur qu'il m'accorde en acceptant de juger ce
travail.
Je remercie le
Je remercie vivement le professeur et le coordinateur du
mastère de génétique et bio ressources à la
faculté des sciences de Tunis, Mohamed EL GAZZAH.
Liste des matières
INTRODCTION
1
I LES PLAQUETTES
SANGUINES
3
I.1 DÉFINITION
3
I.2 FORMATION
3
I.3 MORPHOLOGIES
4
I.4 ULTRASTRUCTURE
4
I.5 RÔLE
5
II LES
GLYCOPROTÉINES DE LA MEMBRANE PLAQUETTAIRE
7
II.1 GÉNÉRALITÉS SUR
LES GPS
7
II.2 LES GPS MAJEURES DE LA MEMBRANE
PLAQUETTAIRE
8
II.3 LES COMPLEXES
GLYCOPROTÉIQUES MAJEURS
9
II.3.1 Le complexe
GPIIb/IIIa
9
II.3.2 Le complexe GPIb-V-IX
(CD 42)
12
II.4 LES COMPLEXES
GLYCOPROTÉIQUES MINEURS
16
II.4.1 Complexe Ia-IIa ou
á2â1 (analogue à VLA-2)
16
II.4.2 Complexe Ic-IIa
17
II.4.3 Le complexe
GPIc*/IIa/VLA-6
17
III IMMUNOLOGIE
PLAQUETTAIRE
20
III.1 DÉFINITION DES
ALLO-ANTIGÈNES PLAQUETTAIRES
20
III.2 LES ALLO-ANTIGÈNES COMMUNS AVEC
D'AUTRES CELLULES
20
III.3 LES ALLO-ANTIGÈNES
SPÉCIFIQUEMENT PLAQUETTAIRES
20
III.3.1 Fréquences
alléliques des systèmes HPA-1-5 chez les différentes
populations
36
III.3.2 Les
allo-antigènes les plus impliqués dans les immunisations
inter-humaines
39
III.3.3 Les
allo-antigènes les moins fréquemment impliqués dans les
immunisations inter-humaines
40
III.4 ÉTUDE DE POLYMORPHISME
PLAQUETTAIRE
42
III.4.1 Méthodes de
phénotypage sérologique
42
III.4.2 Méthodes de
Typage moléculaire
43
OBJECTIFS
49
I. MATÉRIEL
49
II. MÉTHODES
49
II-1. PRÉLÈVEMENT DU SANG
50
II-2. EXTRACTION DE L'ADN GÉNOMIQUE
TOTAL
50
II-3. MISE AU POINT DE LA RÉACTION
PCR-SSP
52
III. CONTRÔLE DE L'AMPLIFICATION
57
III-1 Électrophorèse sur gel
d'agarose (Delpech. M et al. 1999).
57
III-2 Électrophorèse et
visualisation des produits d'amplification
58
IV. ANALYSES STATISTIQUES
59
RÉSULTATS
63
DISCUSSION :
77
CONCLUSION
89
PERSPECTIVES
90
RÉFÉRENCES
BIBLIOGRAPHIQUES
92
Liste des figures
FIGURE 1. A: ULTRA STRUCTURE PLAQUETTAIRE B :
GLYCOPROTÉINES MEMBRANAIRE ET LEUR LIGANDS :
6
FIGURE 2. ASSOCIATION OBSERVÉES POUR LES
DIFFÉRENTES SOUS-UNITÉS Á ET Â DES
INTÉGRINES.
7
FIGURE 3. LES GPS DE LA MEMBRANE PLAQUETTAIRE
(MILLER. 2005).
8
FIGURE 4. STRUCTURE DE L'INTÉGRINE
ÁIIBÂ3 (LAURENT. T. 2004).
10
FIGURE 5. LE COMPLEXE GPIB-IX-V
14
FIGURE 6. NOUVELLE VARIANTE DE L'ALLÈLE
HPA-1A.
24
FIGURE 7. LES COMPLEXES GLYCOPROTÉIQUES
SPÉCIFIQUEMENT PLAQUETTAIRE
41
FIGURE 8: PROFIL
ÉLÉCTROPHORÉTIQUE DES AMPLIFICATION DU SYSTÈME
HPA-1 EN GEL D'AGAROSE À 1.5%.
63
FIGURE 9: PROFIL
ÉLÉCTROPHORÉTIQUE DES AMPLIFICATIONS DES SYSTÈMES
HPA-2, HPA-3, HPA-4 ET HPA-5 EN GEL D'AGAROSE À 1.5%.
64
FIGURE 10: REPRÉSENTATION GRAPHIQUE DE LA
DISTRIBUTION ALLÉLIQUES DES YSTÈMES HPA-1, -2,- 3,-4 ET-5 DANS
L'ÉCHANTILLON ÉTUDIÉE DE LA POPULATION TUNISIENNE
66
FIGURE 11. REPRÉSENTATION GRAPHIQUE DE LA
RÉPARTITION DES FRÉQUENCES
67
FIGURE 12. REPRÉSENTATION GRAPHIQUE DE LA
RÉPARTITION DES FRÉQUENCES
71
FIGURE 13. REPRÉSENTATION GRAPHIQUE DE LA
RÉPARTITION DES FRÉQUENCES
72
FIGURE 14. REPRÉSENTATION GRAPHIQUE DE LA
RÉPARTITION DES FRÉQUENCES
73
FIGURE 15: FRÉQUENCE THÉORIQUE DES
DESCENDANTS À RISQUE ALLO-IMMUNS EN (%) POUR LES CINQ PREMIERS
SYSTÈMES CHEZ LA POPULATION D'ÉTUDE.
76
Liste des Tableaux
TABLEAU I : SPÉCIFICITÉ
DES GPS PLAQUETTAIRES.
19
TABLEAU II : IDENTIFICATION GÉNOMIQUES
ET PROTÉIQUES DES POLYMORPHISMES DES HPAS (KOUTSOGIANNI. P.
2004).
34
TABLEAU III :
LES DIFFÉRENTS ANTIGÈNES PLAQUETTAIRES
35
TABLEAU IV FRÉQUENCES
ALLÉLIQUES DES HPA-1,-2, -3, -4 ET -5 CHEZ DIFFÉRENTES
POPULATIONS (HTTP://WWW.EBI.AC.UK/IPD/):
38
TABLEAU V : LES DIFFÉRENTS
COUPLES D'AMORCES UTILISÉS POUR L'AMPLIFICATION SPÉCIFIQUE DES
SYSTÈMES HPA -1 À 5 (CAVANAGH.
G ET AL., 1997).
55
TABLEAU VI : COMPOSITION D'UN MILIEU
RÉACTIONNEL
56
TABLEAU VII :
PROGRAMME PCR UTILISÉ :
57
TABLEAU VIII : RÉSULTATS DU
GÉNOTYPAGE DES SYSTÈMES HPAS ÉTUDIÉS
65
TABLEAU IX : FRÉQUENCES
ALLÉLIQUES DES SYSTÈMES HPA-1, -2, -3, -4 ET 5.
65
TABLEAU X : FRÉQUENCES
GÉNOTYPIQUES DES SYSTÈMES HPA-1, -2, -3, -4 ET 5.
66
TABLEAU XI : FRÉQUENCES
GÉNOTYPIQUES EN % DES SYSTÈMES HPA-1, -2, -3, -4 ET 5
67
TABLEAU XII : SYSTÈME HPA-3:
ÉQUILIBRE DE H-W: ÷2 ET P
69
TABLEAU XIII : SYSTÈME
HPA-1 : ÉQUILIBRE DE H-W ; ÷2 CORRIGÉ
ET P
69
TABLEAU XIV : SYSTÈME HPA-2:
ÉQUILIBRE DE H-W; ÷2 CORRIGÉ ET P
70
TABLEAU XV : SYSTÈME
HPA-5 : ÉQUILIBRE DE H-W ; ÷2 CORRIGÉ ET
P
70
TABLEAU XVI : ÉQUILIBRE DE H-W:
÷2
74
TABLEAU XVII LES FRÉQUENCES
GÉNOTYPIQUES DES DÉSENDANTS ENTRE DIFFÉRENTES UNIONS
PARENTALES.
75
TABLEAU XVIII : FRÉQUENCES DES
ALLÈLES HPA-1 À 5 DANS QUELQUES POPULATIONS AFRICAINES
85
TABLEAU XIX FRÉQUENCES ALLÉLIQUES
HPA-1 À 5 DANS QUELQUES PAYS EUROPÉENS.
86
TABLEAU XX FRÉQUENCES ALLÉLIQUES DES
HPA-1 À 5 DANS QUELQUES POPULATIONS ASIATIQUES :
86
TABLEAU XXI FRÉQUENCES
ALLÉLIQUES DES HPA-1 À 5 DANS QUELQUES PAYS
AMÉRICAINS.
87
TABLEAU XXII: FRÉQUENCES
ALLÉLIQUES DES HPA-1 À 5 CHEZ QUELQUES PAYS DE
L'OCÉANIE.
87
LISTE DES ABREVIATIONS
ADN : Acide
Désoxyribonucléique
ADNc : Acide
Désoxyribonucléique Complémentaire
ARNm: Acide Ribonucléique
messager
aa: acide amine
ASO: Atherosclerosis Obliterans
ATP: Adénosine
Triphosphate
AMI: Acute Myocardial Infarction/ Infarctus
de myocarde
BET: Bromure d'Ethidium/ Ethiduim Bromide
SBS: Syndrome de Bernard et Soulier/
Bernard-Soulier Syndrome
CVD: Ischemic Cerebrovascular Disease
CNTS: Centre National de Transfusion Sanguine
CAD: Coronary Artery Disease
CD: Classe de Différenciation/Cluster
of Differenciation
d'NTPs: désoxyribonucleosde
tri-phosphate
DO: Densité Optique
EDTA: Ethylene Diamine Tetra-acetic Acid
ELISA: Enzyme Linked
Immunosorbent Assay.
FMAIT: Fetomaternal Alloimmune Thrombocytopenia/
Thrombopénie foeto-maternelle allo-immune
FvW: Facteur Von Willebrand/Von Willebrand
Factor
GPs: Glycoprotéines
GPI: Glycosylphosphatidylinositol
HPA : Human Platelet Antigen/Antigènes
Plaquetaires Humain
HEL: Human Erythroleukemia
HLA : Human Leucocyte Antigen
/Antigènes Leucocytaires Humain
HGNC: Human Genome Nomenclature Committee.
H.W: Hardy-Weinberg
ICAP-1: Integrin Cytoplasmic
domain-Associated Protein-1
ISBT: International Society of Blood
Transfusion
ISTH: International Society on Thrombosis and
Haemostasis/Société Internationale de Thrombose et
Hémostase
l'IS: Ischemic Stroke
kDa: Kilo Dalton
kb: Kilobase
LRR: Leucine-Rich Repeat
MAIPA: (Monoclonal Antibody-Specific
Immobilization of Platelet Antigens)
MI : Infractus Myocordique
Mr : Masse moléculaire
MO : Moelle Osseuse
ul : microlitres
ng : nanogrames
NA: Non Appreciable
NAIT : Neonatal Alloimmune
Thrombocytopenia/Thrombopénies Néonatale Alloimmune
NAION: Nonarteritic Anterior Ischemic Optic
Neuropathy
PCR-ASA: Polymerase Chain Reaction
Allel Specific Amplification
PCR: Polymerase Chain
Reaction
PCR-ASRA: Polymerase Chain Reaction
Allele-Specefic Restriction enzyme Analysis
PCR-SSCP: Polymerase Chain Reaction
Single-Strand Conformation Polymorphism
PHFA: Preferential Homoduplex Formation
PCR-OLA: Polymerase Chain Reaction
Oligonucleotide Ligation Assay
PCR-SSP: Polymerase Chain Reaction
Sequence-Specefic Primers
pb : Paire de base
PNC : Platelet Nomenclature Committee/
Comité de Nomenclature des Plaquettes
PSI : Plexins, Semaphorins, Integrins
PTR : Platelet Transfusion
Refractorines/États réfractaires à la transfusion des
PLT
PLTs : Les plaquettes
PCR: Polymerase Chain Reaction
PT : Polytransfused
PTP : Post-Transfusion
Purpura/ Purpura Post -Transfusionnel
SCO : Système Canaliculaire
Ouvert
SNP: Single-Nucleotide Polymorphism.
STD: Système Tubulaire Dense.
SAS: Statistical Analysis System
SAH: Subarachnoid Hemorrhage
SCD : Sudden Cardiac Death
SLR : Solution de Lyse des globules Rouges
SLB: Solution de Lyse des globules Blanc
trs/mn: tours/minute
VLA: Very Late Activation
receptor
VNTR : Variable Number of Tandem Repeat/
Variable Nombre de Répétition en Tandem
![]()
Introdction
Les plaquettes (PLTs) sont de petites cellules sanguines
anucléées. Elles sont formées et
individualisées dans les mégacaryocytes. Elles
jouent un rôle primordial dans l'hémostase. Comme toute autre
cellule sanguine humaine, les PLTs portent à leur surface leurs propres
systèmes alloantigéniques. Ces antigènes sont
portés par les GPs plaquettaires majeures de la membrane et sont
désignés, selon la nomenclature internationale, sous le terme de
«Human Platelet Antigens» (HPAs).
Actuellement, 24 allo-antigènes, spécifiquement
plaquettaires, sont définis, et sont reconnus par des sérums
alloimmuns. Douze de ces allo-antigènes sont groupés dans 6
systèmes bialléliques (HPA-1,-2,-3,-4,-5, et -15) avec des
anticorps sériques contre les deux allo-antigènes. Pour les 12
allo-antigènes restants, un seul allo-antigène par système
est identifié par méthodes sérologiques avec
l'allo-anticorps correspondant. Le polymorphisme
moléculaire de ces systèmes est simple. Dans la plupart des cas,
il résulte d'une substitution d'un seul nucléotide (SNP) au
niveau du gène codant se traduisant par la substitution d'un seul aa au
niveau de l'allo-antigène porté par la GP mature exprimée
à la surface plaquettaire.
Les allo-anticorps anti-HPAs sont impliqués dans
trois situations cliniques : les NAIT, le PTP et les PTR. La distribution
des HPAs sur les cellules et les organes est plus large qu'on ne pensait
initialement. Par conséquent, ils peuvent jouer d'autres rôles
à coté de leur rôle dans l'hémostase : dans la
transplantation des organes et des tissus, (antigènes mineures
d'histocompatibilité) telle que la transplantation de la MO. Ils peuvent
de même être impliqués dans la survenue d'accidents
cardiovasculaires mortelle.
Les antigènes plaquettaires sont codés par des
gènes dont l'expression est codominante et la distribution varie pour
chaque population ethnique. Ils pourraient ainsi constituer des marqueurs
génétiques des populations.
Notre présent travail vient compléter les
études antérieures touchant au polymorphisme plaquettaire. C'est
ainsi que nous nous sommes intéressés à l'étude du
polymorphisme moléculaire de cinq premiers systèmes
plaquettaires HPA-1, HPA-2, HPA-3, HPA-4 et HPA-5, dans un
échantillon de la population tunisienne, afin de :
- Définir les fréquences alléliques,
génotypiques, phénotypiques et haplotypiques dans la population
tunisienne des ces cinq systèmes de groupes plaquettaires cliniquement
les plus importants.
- De comparer ces fréquences á celles
rapportées dans la population tunisienne et dans d'autres
populations.
- D'évaluer le risque potentiel d'allo-umminisation
plaquettaire dans la population tunisienne.
![]()
I Les plaquettes sanguines
I.1 Définition
Les PLTs ou thrombocytes sont les plus petits
éléments figurés du sang. Elles ont été
découvertes tardivement en 1882 par Bizzozero à cause de leur
petite taille puis redécouvertes en 1960s après plusieurs
décennies d'oubli (Gaetano. G., 2001; Rozman. P. 2002).
Ce sont des fragments cellulaires anucléés. Les PLTs
naissent par le phénomène de
mégacaryocytopoïèse. Leur durée de vie est
courte; elles survivent dans la circulation chez l'homme pendant 7 jours
environ. Malgré leur absence de capacité de prolifération
et leur durée de vie limitée, ces cellules sont en nombre
constant dans le sang et perpétuellement renouvelées avec une
dynamique exceptionnelle.
I.2 Formation
La formation se déroule dans la MO, les PLTs sont
produites à partir de la prolifération et de la
différenciation des progénitures mégacaryocytaires
médullaires ussus des cellules souches hématopoetiques
(Hoffman. R. 1989; Vainchenker. W et al., 1995) par la mégacaryocytopoïèse. Leur nombre
usuel circulant dans le sang périphérique est de 150-400.109
PLTs/l. Environ 2x1011 PLTs doivent être produites
chaque jour chez l'homme adulte, chaque seconde, approximativement
2.106 PLTs sont produites dans le corps humain (Beutler. E. 2001).
Ce phénomène comprend plusieurs étapes :
· L'étape de
polyploïdisation
C'est l'étape où le précurseur
mégacaryocytaire passe d'un stade diploïde à une
ploïdie. IL subit des doublements successifs de son ADN en moyenne de 16 N
(peut arriver jusqu'à 180 N) par endomitose. Ce phénomène
s'accompagne de la production d'une vaste masse cytoplasmique, qui se
compartimente progressivement et se fragmente totalement en fin de maturation
pour former les PLT.
· L'étape de maturation
cytoplasmique
La maturation des mégacaryocytes n'a lieu que dans les
cellules 8N ou plus. La majorité des cellules donnent lieu à la
plaquettogenèse ayant une ploïdie de 8, 16 et 32 N, avec un pic
à 16 N. Un à huit milliers de PLTs naissent de chaque
mégacaryocyte. La maturation est marquée par l'augmentation de la
taille du cytoplasme, le développement d'un réseau de membranes
cytoplasmiques appelées membranes de démarcation et de la
production de granules sécrétoires, en particulier des granules
á.
· La production plaquettaire
C'est l'étape finale. Elle correspond à la
production plaquettaire par la fragmentation du cytoplasme du
mégacaryocyte en PLTs par un processus dynamique très original
différent d'une mitose appelée formation de proplaquettes.
Contrairement aux autres étapes de la
mégacaryocytopoèse qui se déroulent dans la MO, la
production des PLTs intervient dans la circulation sanguine. Ce processus prend
probablement place dans les capillaires du poumon (Beutler. E.
2001).
I.3 Morphologies
Les PLTs sanguines sont hétérogènes en
taille et en forme, souvent arrondies ou ovalaires. Leur diamètre est
approximativement de 1.5 à 3.3 micromètres. Les PLTs
représentent, à l'état de repos, une forme ovalaire d'un
volume plaquettaire moyen de 6 à 8 u maintenue par un réseau de
microtubules. Le cytoplasme est clair, légèrement basophile et
contient des granulations azurophiles.
I.4 Ultrastructure
En dépit de la simplicité de son apparence
externe, la PLT a une organisation interne très complexe.
· L'environnement
périplaquettaire : On trouve à la
périphérie le glycocalix, il est riche en protéines de
coagulation (fibrinogène, facteur V, facteur VIIIc, facteur XIII), en
amines vaso-actives, en FvW et en glucides qui appartiennent surtout à
la région extérieure des protéines membranaires.
· La membrane
plasmique : la membrane des PLTs obéit au
modèle général de mosaïque fluide. Elle constitue une
zone unique dans l'anatomie plaquettaire. Elle présente la structure
trilaminaire classique avec deux feuillets lipidiques de composition
différentes (Chap. H. J et al., 1977; Perret. B et al., 1979).
Elle est épaisse de 70 à 90 Å. À travers la bicouche
s'insère une série de récepteurs spécifiques
d'agents activateurs ou inhibiteurs plaquettaires. Les PLTs disposent d'un
réseau de profondes invaginations qui constituent des canalicules
connectés à la surface, ce sont des lieux d'échanges
importants, ces invaginations constituent le système canaliculaire
ouvert (SCO) qui a pour origine les restes des membranes de démarcation.
Outre la réserve de membranes qu'il constitue lors du changement de
forme des PLTs activées, le SCO est aussi le site de
sécrétion des différents granules plaquettaires. En plus
de la membrane plasmique et le SCO, le système membranaire plaquettaire
est constitué du système tubulaire dense (STD). Le STD est le
site majeur de stockage du Ca++ et contient des enzymes
impliquées dans la synthèse des prostaglandines. La membrane est
le support d'un grand nombre de GPs. Elles ont un rôle
dans la structure des PLT, un rôle fonctionnel de récepteurs
vis-à-vis d'un certain nombre de molécules et un rôle de
répulsion.
La couche externe du glycocalix contient des séries de
molécules complexes de GPs (Mohanty. D et al., 2004), dont
certaines d'ailleurs la traversent de part en part. Plus de 45 structures
membranaires différentes sont identifiées chez les PLTs
(Rozman. P. 2002) et plus de 50 GPs ont été
également identifiées (Newman. P. J et Goldberger. A.
1991).
· Le
cytoplasme : Le cytoplasme contient un cytosquelette formant
plusieurs systèmes fibrillaires tels que les microfilaments à
base d'actine et de myosine (White. J. G. 1969), les microtubules et
les filaments intermédiaires. Le cytoplasme contient des mitochondries,
des grains de glycogène, des microperoxysomes riches en catalase et
trois types d'organelles de stockage majeurs (Fig. 1).
· les granules plaquettaires :
formés par les granules denses, les granules á et les lysosomes.
Ces granules dont le contenu est secrété durant la
réaction de libération, et est nécéssaire au bon
fonctionnement des PLTs (Fig. 1).
· Les
microtubules : sont des structures rigides,
circonférentielles, périphérique, formées de
microfilaments. Ils sont responsables de la forme discoïde de la PLT au
repos. Après activation plaquettaire, ils se polymérisent ce qui
entraîne un changement de forme des PLTs, puis se reconstituent à
l'intérieur de pseudopodes.
· Le cytosquelette :
Il comprend les fibres et les filaments qui confèrent à
la PLT sa forme. Ils permettent aussi au cytoplasme et aux membranes plasmiques
de changer de forme.
I.5 Rôle
Les PLTs jouent un rôle primordial dans les
mécanismes de l'hémostase. Elles participent à de nombreux
autres processus physiopathologiques tels que, la thrombose et l'inflammation.
En fait, la fonction essentielle des PLTs réside dans le maintien de
l'intégrité du système circulatoire et la
prévention de la perte du sang en cas de blessure, que ce soit par leur
interaction avec les vaisseaux, leur participation à la coagulation et
à la fibrinolyse, ou par leur rôle dans la rétraction du
caillot. Ainsi, les PLTs manifestent plusieurs réponses
fonctionnelles : adhésion, activation, sécrétion et
agrégation qui dépendent des récepteurs
glycoprotéiques de surface (Blockmans. D et al., 1995).
A
B
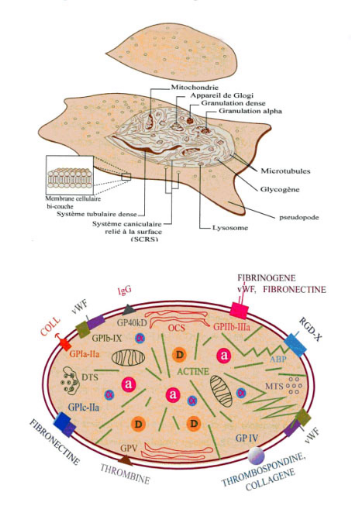
Figure 1. A: Ultra
structure plaquettaire B : Glycoprotéines membranaire et leur
ligands :
(PLT active avec le début de formation des filopodes).
Les complexes protéiques sont représentes avec leurs
éventuels ligands á : granules á, ABP :
Actin-binding protéine, a : grain de glycogènes, Coll :
collagène, D : granules dense, DTS : système tubulaire
dense, GP : glycoprotéine, MTS : système
microtubulaire, OCS : Système caniculaire ouvert, Vwf :
Facteur Von Willibrand (D'après White et al., 1979).
II
Les glycoprotéines de la membrane plaquettaire
II.1 Généralités sur les GPs
Les GPs sont importantes pour les fonctions plaquettaires.
Elles sont classées de la GPI à la GPX. Et agissent en tant que
récepteurs à des stimuli extérieurs pour beaucoup de
protéines adhésives telles que le collagène, le FvW,
l'ADP, etc (Mohanty. D et al., 2004). Parmi ces différents GPs,
seulement quelques unes sont polymorphes. Les GPs majeures possèdent des
chaînes polypeptidiques qui traversent la membrane de part en part. La
majeure partie, sinon toutes les chaînes oligosaccharidiques des GPs,
sont exposées vers la partie externe et interagissent avec
l'environnement plasmatique aqueux. Récemment, elles ont
été reconnues comme membres de la famille des intégrines.
Qui est une vaste famille de recepteurs d'adhesion membranaire
apparentés sous forme de complexes protéiques
hétérodimèriques avec deux sous-unités á et
â, liées de manière non-covalente (Gottschalk. K et
al., 2002).
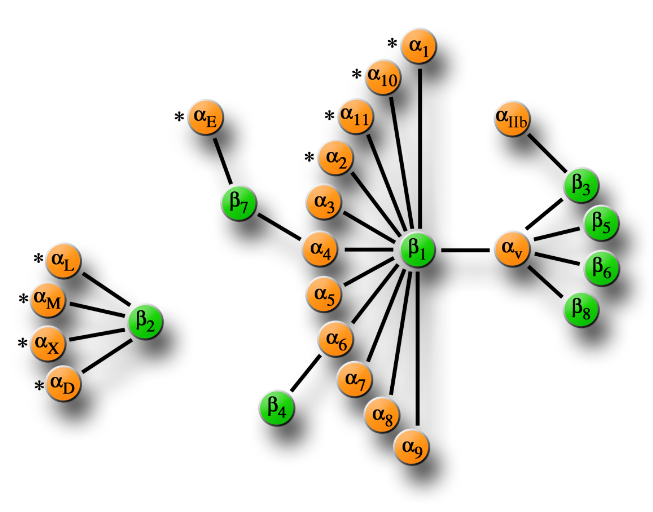
Figure 2. Association
observées pour les différentes sous-unités á et
â des intégrines.
De nos jours, 19 sous-unités á (orangé)
et 8 sous-unités â (vert) (Fig. 2) ont été
identifiées qui s'associent de manière non-covalente pour former
au moins 25 hétérodimèriques áâ
différents. Les sous- unités á marquées d'un
astérisque (?) contiennent un domaine I. Figure modifiée
d'après Shimaoka et al. (2002) (Laurent. T., 2004).
II.2 Les GPs majeures de la membrane plaquettaire
Six GPs majeures polymorphiques et
immunogènes (Newman. P. J et al., 1995; Kroll. H et al., 1998)
sont identifiées à la surface de la membrane plaquettaire
GPs : GPIIIa (CD61), GPIIb (CD41a), GPIb (CD42b); GPIbâ, GPIa et la
CD109 (Fig. 3) (Von dem Borne Aeg. K et Decary. H., 1990; Shih. M. C et
al., 2003). Elles jouent un rôle
vital dans la fonction des PLTs. Ces GPs portent les épitopes
allo-antigéniques génétiquement déterminés
qui peuvent provoquer une réponse alloimmune pendant la grossesse ou
après transfusion des PLTs. De même Plusieurs polymorphismes, sont
associés avec un risque élevé de thrombose
artérielle (Bray. P. F, 1999; Bussel. J. B et al., 2000; Reiner. A.
P et al., 2000). Un polymorphisme plaquettaire dans la région
régulatrice d'un gène, par exemple, peut changer l'expression du
récepteur à la surface de PLTs. De plus, les polymorphismes
résulte d'une substitution d'aa peuvent changez la structure tertiaire
du récepteur et par la suite une altération de la fonction
adhésive des PLTs (Charakida. M et al., 2003 ; Charakida. M et
al., 2003a). Les variations alléliques de ces GPs :
GPIbá, GPIbâ, GPIIb, GPIIIa (CD61), GPIa et la CD109 sont les
produits de six gènes, GP1BA (l7p12ter), GP1BB (22q 11.2), GP2B
(l7q21.3), GP3A (l7q21-22) (Jones. D. C et al., 2003), GP1A (5q23-31)
et (CD109) (Ch.6).
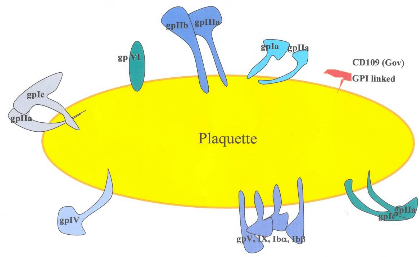
Figure 3. Les GPs de la membrane
plaquettaire (Miller. 2005).
II.3 Les complexes
glycoprotéiques majeurs
Les GPs majeures de la membrane plaquettaire sont
présentes sous forme de complexes multimoléculaires qui assurent
aux PLTs les fonctions adhésives indispensables au processus
d'hémostase. Ainsi l'adhésion des PLTs à la matrice
extracellulaire est médiée par des GPs spécifiques,
habituellement groupées dans les complexes glycoprotéiques
suivants
II.3.1 Le complexe
GPIIb/IIIa
a. Structure
Le complexe GPIIb-IIIa
(áIIbß3)/(CD41a) (Fig. 4) est un
récepteur hétérodimèriques Ca++
dépendant (Nurden. A.T. 1995). La GPIIb est
associée par des liaisons non covalentes avec une chaîne de la
GPIIIa pour former le complexe GPIIb/IIIa (Calvete. J. 1994). Ces deux
GPs sont reliées par un pont disulfure. C'est le représentant
majeur des complexes glycoprotéiques plaquettaires. Il y a
approximativement 80,000 copies par PLT (Nurden. A. T. 1995; Wager. C. L et
al., 1996; Gidwitz. S et al., 2004). Il est présent sur la membrane
plasmique à raison 50 000 copies par PLT non activée (Aiken.
M. L et al., 1986), avec un deuxième pool intra plaquettaire 30000
supplémentaires répartis essentiellement dans les membranes du
SCO et au niveau de la membrane des granules á (Von
dem born et al., 1991) et qui s'exprime à la surface plaquettaire
après l'activation plaquettaire.
b. Rôle physiologique de l'intégrine
áIIbß3
Ils constituent le récepteur des protéines
adhésives et cytoadhésives telles que le vitronectine
(Bray P.F et al., 1987) et le FvW
(Prandini. M. H. et al., 1988), il est le principal récepteur
du fibrinogène des PLTs.
La fixation du vWF et d'autres protéines
adhésives au GPIIb/IIIa se fait par l'intermédiaire de la
séquence RGD tripeptide (Arg-Gly-Asp) (Calvete. J. 1994). Il
joue un rôle majeur dans l'agrégation plaquettaire par interaction
avec l'extrémité C-terminale de la chaîne ã du
fibrinogène (Jallu. V et al., 2002; Unkelbach. K. 2005).
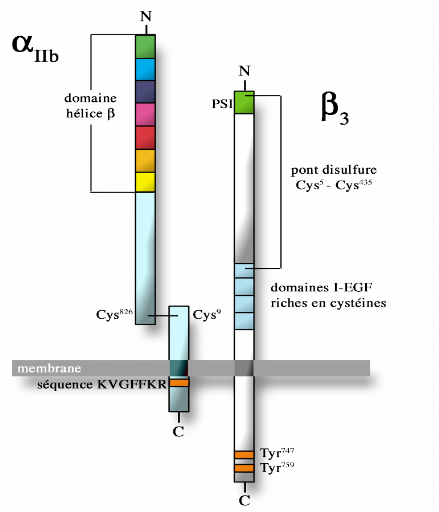
Figure 4.
Structure de l'intégrine
áIIbâ3 (Laurent. T.
2004).
c. La GPIIIa (CD61)
a.1 Gène
Le gène codant pour la sous-unité 3 a
été identifié au niveau de la région q 21-22 du
chromosome 17 et couvre une région de 63 kb. Le gène comporte 15
exons. L'ARNm transcrit a une taille de 3.17 kb (Fitzgerald. L. A et al.,
1987; Zimrin. A. B et al., 1988).
a.2 Structure protéique et fonction
La GPIIIa est formée d'une seule chaîne
polypeptidique de 762 aa (Fitzgerald. L. A et al., 1987; Zimrin. A. B et
al., 1988). Elle est composée de trois domaines : un large
domaine extracellulaire comportant plusieurs ponts disulfures extra
chaînes de l'extrémité N-terminale, fortement
associé par 28 liaisons disulfures ; un domaine transmembranaire et
une courte région cytoplasmique de l'extrémité C-terminale
correspondant au résidu d'aa 740 à 762 (Santoso. S. A et
al., 1998; Roúman. P. 2002).
Elle est associée à la GPIIb par des liaisons
non covalentes calcium dépendantes pour former un complexe
hétérodimérique au niveau membranaire (Jennings. L. K
et al., 1982; Bray. P. F et al., 1987). Le poids moléculaire est de
95 kDa (Calvete J. 1994) dans des conditions physiologiques. C'est un
récepteur pour le fibrinogène et le FvW, possède un
rôle important dans l'agrégation des PLTs (Bojesen. S. E et
al., 2003; Pamukcu. B. M. D et al., 2005). Elles sont impliquées
dans le maintien de la structure de l'intégrine
a.3 Distribution cellulaires de la GPIIIa (CD61)
C'est l'intégrine la plus polymorphe (Metcalfe. P
et al., 2003), elle porte les allo-épitopes HPA-1, -4, -6, -7, -8,
(Newman. P. J et al., 1989; Santoso. S. A et al., 1994) -10, -11,
Oea (Kroll. H et al., 1995) et Vaa. Les
alloantigènes et les antigènes privés sont
localisés sur les différents domaines de la GPIIIa, qui
apparaît donc très immunogène : HPA-1 et HPA-10 sur le
domaine terminal, HPA-4 sur le domaine de liaison des ligands, HPA-6 et
Oea dans le domaine riche en répétition
cystéine et HPA-8 et HPA-11 dans le domaine extra-cellulaire C-terminale
(Santoso. S. A et al., 2002) (Tableau II).
Les polymorphismes des gènes de la GPIa
C807T et de la GPIIIa (PlA1/A2) peuvent causer le
thromboembolisme artériel (Santoso. S. A et al., 1999a). La
région riche en Cys (50-kD) de la GPIIIa est fréquemment une
cible d'auto-anticorps dans la thrombocytopénie idiopathique
(Kekomaki. R et al., 1991).
La GPIIIa est distribuée sur les PLTs
(Phillips. D. R et al., 1988; Boldt. B et al., 1997),
les mégacaryocytes, les monocytes, les macrophages et sur des cellules
de l'endothélium.
d. La GPIIb (CD41,
áIIb)
a.1 Structure et expression du
gène
Le gène codant ITGA2B couvre une région de 17,2
kb et comportant une trentaine d'exons (Poncz. M et al., 1987; Lanza. F et
al., 1990; Poncz. M et Newman. P.J. 1990). Les gènes codant pour
les deux GPIIb (ITGA2B) (Cong. N. V et al., 1988) et GPIIIa
(ITGB3) qui sont localisés sur le bras long du chromosome 17
(q21=>23) sont approximativement physiquement proches (Sosnoski. D. M et
al 1988; Bray. P. F et al., 1988 ). Ils sont situés côte
à côte dans un seul segment de 260-kb (Bray. P. F et al.,
1988; Sosnoski. D. M et al., 1988; Rosa. J. P. et al., 1988; Thornton.
M. A et al., 1999). L'expression simultanée du GPIIb et du
GPIIIa pourrait dépendre de la proximité physique (Bray. P. F
et al., 1988).
a.2
Structure protéique
La GPIIb, a une structure de dimère avec deux
sous-unités á et â. La molécule non réduite a
une Mm de 142 kDa (Bray. P. F et al., 1987). C'est une protéine
transmembranaire avec une chaîne lourde extracellulaire de 125 kDa et une
chaîne légère de 23 kDa. Le domaine transmembranaire est
relié par un pont disulfure. Il y a 50 000-80 000 copies par PLT.
a.3 Distribution cellulaires de la GPIIb (CD41)
Elles sont spécifiques de la lignée
mégacaryocyto-plaquettaire et sont distinctes pour la sous-unité
á dans les autres tissus normaux (Bray. P.F et al.,
1987).
II.3.2 Le complexe
GPIb-V-IX (CD 42)
a.1 Structure
C'est un complexe transmembranaire
équimoléculaire qui comprend 2 molécules Ibá, 2
molécules Ibâ, 2 molécules IX, et une molécule
V : (Ibá Ibâ/IX) 2 V1 (un ratio de
2 :2 :2 :1) produites par des gènes distincts
(Bussel. J. B et al., 2000). Ces sous-unités possèdent
des traits structuraux semblables et appartenant à la famille des GPs
riches en Leu (Lopez. J. A et al., 1994; Shen. Y et al., 2000). Les
GPIbá et GPIbâ sont liées par une seule liaison de
covalence disulfure et sont liées par des liaisons non covalentes
à GPIX et à GPV.
La majeure partie du complexe GPIb-IX-V est extra-cellulaire
(fig. 5). Il y a approximativement 25 000 copies de GPIb/IX sur une PLT non
activée (Lopez. J A. et al., 1994; Ware. J. 1998; Cauwenberghs. K et
al., 2001) et 12 000 copies de GPV par PLT. Ce complexe est un
récepteur du FvW nécessaire aux premières étapes de
l'adhésion des PLTs aux parois du vaisseau lésé. Chacune
des GPs possède un segment hydrophobe pour l'insertion dans la membrane
(Berndt. M. C et al., 2001).
a.2 Rôle du complexe
GPIb-IX-V
C'est le principal récepteur responsable de
l'adhésivité plaquettaire (Afshar-Kharghan. V et al.,
1999). La partie intra cytoplasmique du complexe GPIb-IX-V joue un
rôle majeur dans l'activation de l'intégrine
áIIbß3 induite par la liaison GPIbX-FvW. La
fonction d'adhésion au sein de ce complexe est totalement assurée
par la GPIbâ.
Son rôle est prépondérant dans la
microcirculation, et elle est essentielle pour le maintien de
l'hémostase primaire normale. La GP Ibá est fonctionnellement
dominante de ce complexe. Le ligand se fixant sur la GPIb impliqué dans
l'adhésivité plaquettaire est le FvW (Lopez. J. A. 1994;
Chester. Q et al., 2000).
Les GPIbá, GPIbâ, GPIX et GPV sont toutes
déficientes ou affectées dans le syndrome thrombocytaire
hémorragique sévère : syndrome de Jean Bernard et
Soulier, avec désordres de saignement (Ruggeri. Z. M.
1991; Kelly. M. D et al., 1994).
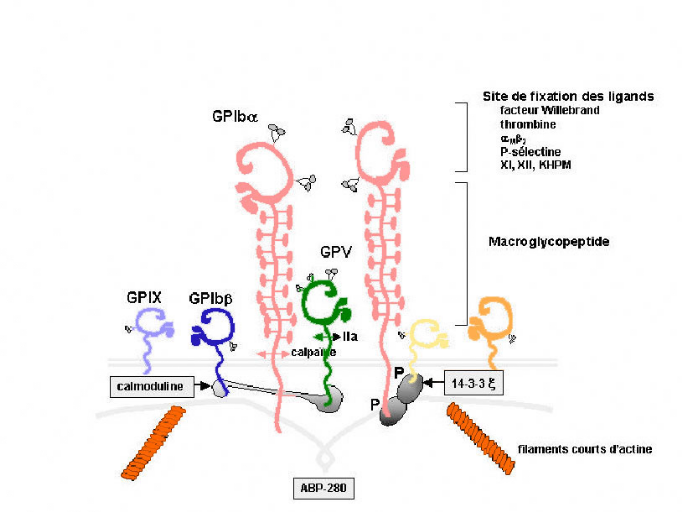
Figure 5. Le complexe
GPIb-IX-V
(D'après Lopez. 2002; Nathalie. H. 2004 avec
quelque modification).
L'organisation définitive du complexe n'est pas
élucidée, la figure 6 correspond donc à une
représentation schématique hypothétique, prenant en compte
la stoechiométrie du complexe et l'association
préférentielle de GPV à GPIb (Nathalie. H.
2004).
a. La glycoprotéine Ib; GPIb
(CD42b)
Cette GP hétérodimérique de haut poids
moléculaire 160 kDa, riche en carbohydrate (Lopez. J. A. et al.,
1988), la plus riche en acide sialique. Elle appartient à la
famille des protéines riches en répétitions Leu. Le
fragment extra cytoplasmique de la GPIb est de 140 kDa. Elle est
composée de deux chaînes polypeptidiques (Iba ou Ibá /Ibb
ou Ibâ) : une large sous unité á reliée par un
seul pont disulfure à une petite sous unité â
(Kuijpers. R. W. A. M et al., 1992; Murata. M et al., 1992; Aramaki. K. M.
et Reiner. A. P. 1999). La GPIbá a pour Mm de 143 kDa avec 610 aa)
et GPIbâ (CD42c, avec 181 aa) forment un complexe avec le GP IX (CD42a)
(160 aa), elles sont associées par des liaisons non covalentes (Wu.
G et al., 1996) en se reliant à la GPV (Ruggeri. Z. M.
1991; Lopez. J. A. 1994; Charakida. M et al., 2003).
- La GPIbá
C'est une protéine transmembranaire avec sept
répétitions en tandem de 24 aa (Lopez. J. A et al.,
1988). Elle possède dans sa partie externe les récepteurs
à la thrombine (Wicki. A. N et Clemetson. K. J. 1987; Katagiri. Y et
al., 1990), avec une très forte affinité et un site de
fixation par sa partie N-terminale du FvW (Handa. M et al., 1986) qui
joue un rôle essentiel dans l'adhésivité des PLTs. La
GPIbá a pour ligands aussi ámâ2 et plusieurs autres
facteurs de la coagulation (facteur XI, facteur XII, ... .). Les sites de
liaison pour ces ligands sont situés dans le domaine globulaire
N-terminal, constitué des 282 premiers aa. Ce domaine comprend 7
séquences riches en Leu et Tyr sulfates. La partie intra cellulaire de
GPIbá comprend 20 aa (Lopez. J. A. 1988; Berndt. M. C. et al.,
2001).
- GPIbâ
·
Gènes (GP1BA, GP1BB)
GPIbâ est la plus petite sous-unité (24-kDa)
(Ruggeri. Z. M. 1991) du complexe GPIb-V-IX
(Kelly. M. D et al., 1994; Roth. G. J. 1994). La région
terminale du GPIbâ contient une séquence riche en Leu de 24 aa.
Cette région est suivie par un segment transmembranaire de 25 aa et une
région intracellulaire de 34 aa à l'extrémité
carboxylique terminale (Lopez. J. A. 1988; Berndt. M. C et al., 2001).
·
Structure
GPIbâ est la plus petite sous-unité (24-kDa)
(Ruggeri. Z. M. 1991) du complexe GPIb-V-IX
(Kelly. M. D et al., 1994; Roth. G. J. 1994). La région
terminale du GPIbâ contient une séquence riche en Leu de 24 aa.
Cette région est suivie par un segment transmembranaire de 25 aa et une
région intracellulaire de 34 aa à l'extrémité
carboxylique terminale (Lopez. J. A. 1988; Berndt. M. C et al., 2001).
·
Distribution cellulaires de la GPIbâ
Les molécules ont été identifiées
sur les PLTs et les mégacaryocytes. Les GPIbâ se trouvent aussi au
niveau des cellules endothéliales (Roth. G. J. 1994). La GPIb
s'aperçoit d'ailleurs sur des cellules de l'épithélium
vasculaire, l'erythroleukemia (Rajagopalan. V et al., 1992) et
tonsilare.
b. La
glycoprotéine V (GPV) (CD 42 d)
a.1 Structure
La GPV est associée de manière non covalente
à la GPIbá, la GPIbâ et la GPIX pour former le complexe
GPIb-V-IX (Chester. Q et al., 2000). Elle est de poids
moléculaire de 83 kDa. La fonction exacte de la GPV n'est pas connue.
La GPV, est immunogène et des anticorps circulants ont
été détectés lors des thrombopénies. Des
polymorphismes de la GPV ont été observés dans une
population normale. Leur gènes codants (GP5*) sont localisés sur
le Chr 3q29 (Yagi. M et al., 1995).
a.2 Distribution cellulaires de la
glycoprotéine V
La GPV est fortement exprimée à la surface des
PLTs dont l'expression tissulaire est restreinte à la lignée
mégacaryocytaire et est exprimée dans les étapes tardives
de la différentiation de ces cellules.
Le complexe GPIb-IX-V est majoritairement exprimé par
la lignée mégacaryocytaire. Son expression par
l'endothélium, expression inductible en réponse aux cytokines,
reste controversée (Berndt. M. C et al., 2001).
c. La
glycoprotéine GPIX
La GPIX (CD42a, Mm 20 kDa) est formée par une seule
chaîne leur gène (GP9*) est localisé sur le chromosome 3q21
(Yagi. M et al., 1995).
II.4 Les complexes glycoprotéiques mineurs
II.4.1 Complexe
Ia-IIa ou á2â1 (analogue à VLA-2)
Le complexe Ia-IIa ou á2â1 (analogue à
VLA-2) est un hétérodimère constitué d'une
chaîne á2 et d'une chaîne â1. Membre de la famille des
intégrines qui sert comme un récepteur majeur pour le
collagène chez les PLTs (Chadderdon. R.C et al., 1999; Santoso. S. A
et al., 1993b) et chez d'autres types cellulaires. En plus de son
rôle dans l'adhésion plaquettaire au matrice extracellulaire, la
GPIa/IIa sert de médiateur d'adhésions plaquettaires au
collagène fibrillaire (type I et III) ou non fibrillaire (type IV et
VI). Leur fonction dépend des ions Mg2+ (Staatz. W. D. et
al., 1989). Il y a approximativement 800-2800 copies de GPIa/IIa par PLTs
et ne possèdent pas de stock interne secrété
(Clemetson. K. J. 2001 Roúman P. 2002).
a.1 La
glycoprotéine GPIa
La GPIa joue un rôle de récepteur pour le
collagène au sous endothélium elle exige les ions Mg++
(Staatz. W. D et al., 1989). Elle est
formée d'une chaîne polypeptidique unique. Et comporte une
distribution des résidus Cys et des domaines de liaison des cations. La
majeure caractéristique de GPIa est la présence de 191 aa
(domaine I) qui contient les sites potentiels pour l'interaction avec le
collagène (Santoso. S. A et al., 1993). Le nombre estimé
des molécules GPIa à la surface des PLTs est de 1842
(Pischel. K. D et al., 1988).
La GPIa est codée par le gène ITGA2,
localisé sur le bras long du chromosome 5 (5q23-q31) (Takada. Y et
Hemler. M. E. 1989; Jaspers. M et al., 1991). La densité de
GPIa-IIa à la surface des PLTs varie selon les polymorphismes
nucléotidiques dans le gène ITGA2 (Kunicki. T. J et al.,
1997).
a.2 La
glycoprotéine GPIIa (CD29)
Elle
est formée d'une chaîne polypeptidique unique. Elle participe
à l'adhésion comme récepteur de collagène et
à l'activation des PLTs. Le nombre
estimé des molécules GPIIa est de 4926. La GPIIa correspond
à la chaîne â VLA avec Mm (145 kDa). La GPIIa forme des
complexes hétérodimérique avec GPIc, GPIa (Pischel. K.
D et al., 1988).
II.4.2 Complexe
Ic-IIa
Le complexe Ic-IIa (analogue à VLA-5) impliqué
dans l'adhésion à la fibronéctine.
II.4.3 Le complexe
GPIc*/IIa/VLA-6
Le complexe GPIc*/IIa/VLA-6 récepteur de la
laminine.
Il existe d'autres
glycoprotéines polymorphes tels que la
- Protéine CD109 (GP 175) ou protéine
175-kD
·
Structure du gène
Le gène codant, formé par 32 exons, est
porté par le chromosome 6. L'ADNc de la CD109 est formé de 4335
pb (
http://www.gene.ucl.ac.uk/).
·
Structure et fonction protéique
La CD109 est un glycosylphosphatidylinositol-isoforme
monomérique (GPI) ancré à une GP de poids
moléculaire de 170 à 180 kDa (Brashem-Stein. C et al.,
1988) et de 1445 aa. C'est une protéine à 3 domaines. La
CD109 est ancrée à la membrane par un groupement phosphatidyl
inositol (Smith. J.W et al., 1995). Bien que sa fonction
précise n'est pas encore connue, elle a été
identifiée comme un nouveau membre de la famille des macroglobulines
(á2M)/C3, C4, C5 des protéines contenant des thio-esters
(Lin. M et al., 2002).
Elle est capable d'intervenir dans des interactions
covalentes cellule-substrat et cellule-cellule. Elle joue un rôle dans la
présentation d'antigène par les cellules T et dans l'interaction
des cellules B et T. De cette façon, la CD109 peut jouer un rôle
dans l'hèmatopoiese et l'hémostase primaire (Lin. M et al.,
2002). L'expression du CD109 sur les PLTs est faible :
approximativement 2000 #177; 400 molécules par PLT activée par la
thrombine (Schuh. A. C. 2002), mais il existe une variabilité
d'expression significative entre les individus (Berry. J. E et al.,
2000). La congélation ne permet pas de conserver cette
molécule CD109 (Cardone. J. D. B et al., 2004).
·
Distribution cellulaire de la CD109
Elle a été décrite d'abord sur les PLTs
et les cellules T activées (Suciu-Foca. N et al., 1985; Murray. L. J
et al., 1999). On l'a identifié par la suite sur les cellules
endothéliales (Murray. L. J et al., 1999), sur un sous-ensemble
de progéniteurs primitifs de la MO, sur les cellules souches
hématopoïétiques (Sutherland. D. R et
al., 1991; Schuh. A. C. 2002), sur les monocytes, sur les granulocytes,
sur les cellules CD34+ (Kelton. J.G et al., 1990) et sur
plusieurs lignées de cellules tumorales cultivées (Smith.
J.W. 1995).
Tableau I :
Spécificité des GPs plaquettaires.
|
GPs
|
CD
|
PM (kDa)
|
Chr
|
Gène
|
Nbr/d'exon
|
Nbr / PLT et distribution cellulaires
|
Rôle
|
|
GPIIb
|
CD 41,áIIb
|
142
|
17
|
GP2B
|
30
|
50000-80000
PLT, mégacaryocytes
|
Agrégation
plaquettaire
et
Adhésivité
|
|
GP IIIa
|
CD 61/á3
|
95
|
GP3A
|
15
|
PLT, mégacaryocytes, monocytes,
macrophages, cellules endothéliales, ostéoclastes, intima
synoviale, cellules de muscle lisse, anthérocytes, PLT des
foie cirrhotique
|
|
GPIa
|
CD49b/á2
VLA-2
|
150
|
5
|
GP1A
|
3
|
PLT, monocytes, lymphocytes B et T, cellules
NK,
cellules endothéliales vasculaires
|
Adhesion
et
Activation
|
|
GP IIa
|
CD 29
|
145
|
10
|
GP2A
|
23
|
1842-4926
PLT, tous les leucocytes, la plupart des
cellules
|
|
GPb
|
Ibá
|
CD42aá
|
143
|
17
|
GP1A
|
1
|
PLT, mégacaryocytes cellules endothéliale
vasculaires et tensilaires
|
Adhesion plaquettaire
|
|
Ibâ
|
CD42aß
|
24
|
22
|
GP1BB
|
30
|
PLT, mégacaryocytes
|
|
GP IX
|
CD 42c
|
20
|
3
|
GP9*
|
1
|
PLT, mégacaryocytes
|
|
GP V
|
CD 42d
|
83
|
3
|
GP5*
|
1
|
PLT, mégacaryocytes
|
Mal connu
|
|
CD 109
|
CD 109
|
170-180
|
6
|
CD 109
|
32
|
2000#177;400
|
- interactions
cellule-substrat/ cellule-cellule
- présentation d'antigène
- rôle dans l'hématopoïèse et
l'hémostaseprimaie
|
|
GPIIIb/GPIV
|
CD36
|
88
|
|
|
8
|
12000-19400
PLT, mégacaryocytes monocytes,
macrophages, précurseur erythroid,
adipocytes, cellule tumorale, kératinocytes
activé, quelques cellules endothéliales, et
épithéliales.
|
Récepteur pour le collagène et pour la
thrombo
spondine
|
III IMMUNOLOGIE
PLAQUETTAIRE
III.1 Définition des
allo-antigènes plaquettaires
Les
allo-antigènes sont des variétés antigéniques qui
différencient les individus d'une même espèce. Ils
constituent des allotypes, qui sont codés par différents
allèles que possèdent certains individus d'une même
espèce ce sont donc des marqueurs génétiques.
III.2 Les allo-antigènes communs
avec d'autres cellules
Plusieurs antigènes plaquettaires sont partagés
avec d'autres cellules sanguines : érythrocytes; les lymphocytes
(Bussel. J. B. 2000; Matei. D. E. 2002).
1- Les antigènes tissulaires HLA
Seuls les antigènes HLA de classe I classique
HLA-A,-B,-C (principalement A et B) sont détectables sur les PLTs des
sujets normaux. Cependant, chez certains patients de purpura auto-immun, des
antigènes HLA-DR ont été mis en évidence
(Boshkov. I. K et al., 1992). On a d'abord supposé que les
antigènes HLA étaient adsorbés sur les PLTs à
partir d'antigènes solubles plasmatiques (Kao. K. J. 1988).
Mais il a été montré que les antigènes HLA
pouvaient être synthétisés ; on a pu isoler de l'ARNm
spécifique des antigènes HLA-I dans les PLTs (Santoso. S. A
et al., 1993).
2- Les antigènes des groupes sanguins
Les antigènes du
système (A, B, O) (antigènes de Groupe A.B.H), (Von dem
Borne. A et Decary. F. 1990) Lewis (antigènes Lea
Leb), MN (antigènes M. N), l'antigène P et I peuvent
être présents à la surface plaquettaire. Ces
antigènes peuvent être soit adsorbés à partir du
plasma et sont alors présents sur les structures glycolipidiques, soit
être partie intégrante des GPs plasmatiques telles que la GPIb, la
GPIIa, la GPIIIa, la GPIIb (Santoso. S. A et al., 1991).
III.3 Les allo-antigènes spécifiquement
plaquettaires
1- Définition
Ces antigènes sont exprimés d'une manière
prédominante sur les PLTs. Ils ont été décrits pour
la première fois en 1959 (Van Loghem. J. J et al., 1959; Mohanty. D
et al., 2004). Ils ont été découverts suite à
des observations cliniques de NAIT ou des cas de PTP. Ils sont localisés
sur l'une ou l'autre des six GPs plaquettaires majeures Ia, Ibâ, Ib, IIb,
IIIa et la CD109, mais la GPIIIa porte la majorité de ces
alloantigènes (Von der Harst. D et al., 1994). Certains de ces
antigènes peuvent être exprimés sur d'autres cellules mais
à un degré moindre que sur les plaquettes. Ces épitopes
immunogènes sont responsables d'accidents d'alloimmunisation
foeto-maternelles (Shibfata. Y et al., 1986; Chantrain. C et al.,
2000) et post-transfusionnelles (Coffe. C et al., 1995; Crow. C,
1999), après grossesse ou transfusion incompatible à la
manière des groupes sanguins et des allo-antigènes du
systèmes alloantigèniques d'histocompatibilité HLA
(Von dem borne et al., 1995; Simpson. E et al., 1997). De tel
alloanticorps serait à l'origine de troubles hémorragiques
(Newman. P. J. 1994; Boldt. B et al., 1997).
2- Nomenclature des
antigènes spécifiquement plaquettaires
Initialement, les antigènes plaquettaires
étaient définis comme étant des groupes sanguins à
partir des sérums des patients allo-immunisés (Tseng L.H et
al., 1999; Kao. K.J et al., 1996) et portaient les noms des patients chez
qui ces allo-anticorps ont été découverts. Habituellement,
ils sont mis en évidence à l'aide de sérums provenant de
malades ayant présenté un purpura post-transfusionnel ou des
mères d'enfants atteintes de thrombopénie néo-natale
(Chabrenaud. J. L et al., 1995).
De ce fait, plusieurs antisérums découverts
indépendamment, portaient différents noms de patients
(exemple : Zw et P1A, Bak et Lek, Yuk et Peu...)
(Tableau III).
Pour pallier à ces différentes appellations, Von
dem Borne et Decary en 1990 ont proposé un système
simplifié dans lequel, chaque allo-anticorps spécifique d'un allo
antigène était désigné par le numéro de
l'allo-antigènes plaquettaire HPA correspondant ; ainsi
l'allo-antigènes P1A (Zw) (Van Loghem. J. J et al., 1959)
est désigné par HPA-1 (Newman P. J. 1994). Et les
systèmes plaquettaires sont classifiés dans le système
immunogénétique (Dem. Borne. A. E et Décary F.,
1990). Santoso et Kiefel ont révisé la nomenclature en 1998
dans le but d'éviter les confusions concernant ces systèmes
(Santoso. S. A et Kiefel. V. 1998). Cette nomenclature était
moins compliquée que la précédente, donnant les
données essentielles sur les nucléotides polymorphes aussi bien
que les polymorphismes correspondant aux aa au niveau protéique.
En 1990, the Working Party on Platelet Serology a
décidé de formuler un nouveau système de nomenclature
basé sur les points suivants :
· Les systèmes antigéniques
spécifiquement plaquettaires seront nommés HPA (human platelet
alloantigens).
· Les différents systèmes
antigéniques sont numérotés chronologiquement selon leur
date de découverte.
· On identifie actuellement six systèmes
dialléliques. Les formes alléliques seront indiquées
alphabétiquement par ordre de leur fréquence dans la
population : l'allèle a est le plus fréquent et
l'allèle b est le plus faible (Von dem Borne A .k et Decary. F.,
1990).
A côté de ces 6 systèmes pour qui on a
identifié des anticorps sériques contre les deux
allo-antigènes, d'autres systèmes n'ont été
identifiés par la sérologie qu'avec un seul allo-antigène.
Une désignation W est alors ajoutée après
le nom de l'antigène (après le chiffre arabe) si un
allo-anticorps contre l'antigène antithétique n'a pas
été rapporté (Metcafle. P et al., 2003).
· L'inclusion de nouveaux systèmes HPA devrait
être approuvée par The Working Party (Koutsogianni. P. 2004).
Plus tard, en 2003, le Comité de Nomenclature des
Plaquettes (PNC) a été créé comme une plate-forme
de collaboration entre les groupes qui travaillent sur les PLTs (Platelet
Working Party) de la Société Internationale de Transfusion
Sanguine (ISBT) et la sous-commission scientifique de l'immunologie
plaquettaire de la (ISTH). Ce comité sera le gardien pour la
nomenclature des systèmes plaquettaires HPA (Von dem Borne A .k et
Decary. F., 1990a).
· Un allo antigène spécifiquement
plaquettaire n'est dit HPA que quand sa base moléculaire est
définie.
· Les nouveaux antigènes ne seront inclus dans
la table de la nomenclature des HPA que lorsque l'approbation du PNC est
obtenue. Et avant que le PNC ne donne sa décision, il tient compte des
conditions suivantes :
(i) l'allo-antigènes devrait
être génétiquement déterminé. Une base de
données devrait être fournie : de préférence,
la séquence d'ADN ou au minimum la séquence d'ADNc.
(ii) L'association entre la mutation
génétique et la réactivité des allo-anticorps avec
les formes alléliques des protéines spécifiques par des
méthodes immunologiques.
(iii) Au moins deux laboratoires de
référence doivent confirmer les données
sérologiques et moléculaires.
(iiii) Le laboratoire de contribution devrait
fournir une base de données sur la population étudiée. Un
pedigree avec les individus de la famille index et/ou d'autres familles serait
d'une valeur additionnelle.
(iiiii) Le laboratoire de contribution
devrait faire tous ses efforts pour que les échantillons sanguins soient
disponibles aux laboratoires pour l'établissement des lignées
cellulaires lymphoblastoides (Metcalfe. P et al., 2003).
3- Polymorphisme et transmission
génétique des HPAs
A présent, 24 HPAs
allo-antigènes spécifiquement plaquettaires ont été
définis sérologiquement à l'aide d'allo-anticorps
allo-immuns correspondants, dont seulement 12 sont groupés dans six
systèmes bialléliques (HPA-1,-2,-3,-4,-5,-15) avec des anticorps
sériques contre les deux allo-antigènes. D'autres systèmes
antigéniques rares ou privés sont aussi détectés
avec un seul allo-anticorps correspondant. La base moléculaire de 22
antigènes parmi les 24 définis sérologiquement est
résolue. Pour les 21 HPAs, la différence entre les 2
allèles est due à la substitution d'un seul nucléotide
(SNP) au niveau du gène codant (Metcalfe. P et al., 2003 ;
Bugert. P et al., 2005). Sera à l'origine de la substitution d'un
seul aa aux niveaux de l'une des six GPs majeurs, matures de la membrane
plaquettaires (Koutsogianni. P. 2004). L'antigène
HPA-14w constitue la seule exception puisque son polymorphisme
est obtenu par une délétion de trois nucléotides, il en
résulte la perte d'un seul aa. Donc la plupart de ces polymorphismes est
un seul point de mutation de l'allèle de type sauvage (Newman. P. J
et Valentin N., 1995; Santoso. S. A et al., 1999). Tous les
allo-antigènes spécifiques aux PLTs sont hérités
par codominance autosomale (Kulkarni. B et al., 2002).
4- Les différents allo-antigènes
spécifiquement plaquettaires
1- Les systèmes les plus
impliqués en pathologies humaines
Le système HPA-1; (P1A,
Zw)
· Génétique du système
HPA-1
L'antigène Duzo, appelé également (PLA,
Zw), a été identifié en 1959 par Van Loghem (Van
Loghem. J. J et al., 1959), il est désigné par HPA-1
(Newman. P. J et al., 1985). C'est le système plaquettaire le plus
important cliniquement (Williamson. L. M et al., 1998).
L'expression bialléliques de HPA-1 donne les
antigènes allèles HPA-1a (PlAl) (GP3A*01) et HPA-1b
(PlA2) (GP3A*02) qui différent par un SNP 196T>C sur
l'exon 2 du gène codant ITGB3/GP3A. Ce SNP cause une substitution
Leu33/Pro33 de l'extrémité N-terminale de
la GPIIIa mature (Tableau IV). Ce qui donne des différences
considérables dans les structures secondaires des deux formes
alléliques (Newman. P. J et al., 1989).
Cette substitution mène à la formation de
l'épitope HPA-1 (Goldberger. A et al., 1991; Honda. S et al.,
1995), est localisée à l'apex Cys26-Cys38 du boucle AB du
domaine PSI (Xiong. J.P et al., 2004; Santoso. S. A et al.,
2006). Les déterminants antigéniques sont localisés
en outre sur un fragment non carbohydraté de 17-23-kDa (Newman. P. J
et al., 1985; Van Der Shoot. C. E et al., 1986). Les aa de 1 à 66
(Barron-Casella. E.A et al., 1999) et de 288 à 490 contribuent
à la formation de l'épitope HPA-1a. Il est possible que la
séquence 288-490 maintienne la boucle Cys26-Cys38
dans une orientation appropriée par rapport au reste de la
molécule â3 et de cette façon elle maintient la
présentation antigénique adéquate dans cette boucle
(Honda. S et al., 1995).
L'analyse de l'ADN génomique d'un individu a
révélé la présence d'un nouveau SNP
C175G dans le gène ITGB3 qui conduit à une
substitution Leu33Val pour donner un troisième allèle
HPA-1(ITGB3*001.1) (Fig.6). La mutation naturelle Leu33Val peut
interrompre quelques épitopes HPA-1a. Ces conclusions fournissent
l'évidence pour une réponse humorale
hétérogène contre l'antigène HPA-1a qui peut avoir
la possibilité d'être impliqué dans les désordres de
NAITP.
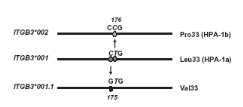
Figure 6. Nouvelle
variante de l'allèle HPA-1a.
La mutation C>G de l'ADNC de l'intégrine
â3 en position 175 (â3-Leu33; HPA-1a) et le résultat d'un
nouveau variant allélique différent ITGB3*001.1 codant pour
Val33. La mutation T>C à la position 176
résultat d'un allèle ITGB3*002 qui
code pour Pro33 (HPA-1b) (Santoso S. A et al.,
2006).
L'Arg 93 du GPIIIa contribue à la formation de
l'épitope HPA-1a au niveau des cellules B (Watkins. N. A et al.,
2002). Un nouveau SNP G376A du gène codant ITGB3 est le
résultat d'une substitution d'un seul aa Arg93Gln au niveau
de l'intégrine â3 remplaçant l'allèle
Leu33 (Watkins. N. A et al., 2002).
La configuration de l'épitope HPA-1 n'a pas
été complètement élucidée.
· Implications cliniques
La réponse humorale de l'antigène HPA-1a est
hétérogène (Valentin. N et al., 1995). Les
anticorps Anti-HPA-1a produits par les réponses immunes peuvent mener
à la destruction des PLTs (Mueller-Eckhard. C et al., 1989; Okamoto.
N et al., 1998) dans les désordres cliniques tels que NAIT
(Blanchette. V. S et al., 2000), le PTP et aux transfusions des les
PLTs. En plus de ces désordres immunologiques, le polymorphisme du
système HPA-1 est associé à plusieurs autres maladies.
D'ailleurs, l'altération de la structure moléculaire de la GPIIIa
au niveau des allèles HPA-1 peut affecter la fonction du GPIIb/IIIa.
De même, le polymorphisme du HPA-1b est associé
au risque de la thrombose coronaire (Weiss. E.J et al., 1996; Kadir et al.,
1999; Kroll. H et al., 2001).
Des différences dans la prédominance de ces
désordres génétiques ont été
observées chez les différents groupes ethniques. Carlsson et al.,
en 1997 ont montré qu'il n y'a pas association entre le polymorphisme
HPA-1 et l'IS (Carlsson. L. E et al., 1997). De même, van Goor
et al., en 2003 ont prouvé que la relation entre l'allèle HPA-1b
et ce désordre est incertaine (Van Goor. M.L.P.J et al., 2003).
Cependant, Jimmy Lim et al en 2003 ont affirmé que ces allèles
peuvent être associés positivement à l'IS (Jimmy Lim et
al., 2003). Le polymorphisme PlA est associé à
l'agrégation accrue des PLTs (Huang. T et Mervyn. A. S. 2003). Ainsi
l'agrégation plaquettaire joue un rôle central dans la
pathogénie de la thrombose aiguë, dans la maladie des coronaires,
l'IS et les maladies artérielles périphériques
(Ulrich. H. F et al., 2003). De ce fait, l'étude faite par
Weber et al en 2002 a démontré une hyperréactivité
considérable des PLTs chez les malades avec l'artère coronaire.
v Implications cliniques de l'allèle
HPA-1a
L'allo-antigène HPA-1a et le plus impliqué dans
les cas de NAIT, que les thrombopénies néonatales
résultantes sont profondes et que ça donne les formes les plus
sévères. En plus des trois principaux désordres
cliniques, le polymorphisme de HPA-1a été identifié comme
un facteur héréditaire du risque thrombotique (Weber. S et
al., 2002) tel que l'IM (Weiss. E. J et al., 1996; Walter et al.,
1997; Zotz. R. B et al., 1998; Nauck. M. S et al., 1999; Mathew. J. G et al.,
2001; Hohlagschwandtner. M. M. D et al., 2003). Le génotype
PlA1/A1 est associé avec la CAD sévère chez des
malades caucasiens de la région nord de la Pologne (Gruchala. M et
al., 2003). Normalement, les homozygotes PlA1/A1 ont un risque
hémorragique plus élevé (Morawski. W. MD. PhD. M et
al., 2005).
v Implications cliniques de l'allèle
HPA-1b
Les individus qui portent l'allèle HPA-1b
paraissent avoir un haut risque pour les événements
cardio-vasculaires (Weiss. E. J et al., 1996; Wagner.
K et al., 1998; Carter. A. M et al., 1996; Vijayan. K. V et al., 2000;
Santoso. S. A et al., 2002). Ainsi en 1996, Weiss et al été
les premiers à démontrer la grande prédominance de
l'allèle PlA2 dans l'IM chez les malades. Ce
même allèle constitue un risque pour les maladies de
l'artère coronaire (Weiss. E. J et al., 1996) (Jimmy Lim et al.,
2003). Il peut être associé positivement avec des
complications thrombotiques d'athérosclérose tels que l'IS.
Quelques études ont aussi montrés des résultats
contradictoires (Korral et al., 1997; Ghosh. K et al., 2002). Un rare
polymorphisme GPIIIa Leu 40/Arg 40 est lié à
l'antigène 1b (Walchshofer. S et al., 1994).
Le polymorphisme PlA1/A2 est fortement
associé avec les maladies ischémiques cardio-vasculaires et l'IM
aigu (Weiss. E. J et al., 1996; Feng. D. M. D et al., 2001; Huang. T et
Mervyn. A. S. 2003; Bojesen. S. E et al., 2003; Lopes. N. H. M
et al., 2004). Le variant allélique PIA2 est un facteur
héréditaire du risque des événements coronaires
aigus (Lim et al., 2003) et au restenosis
(Kastrati. A. M. D et al., 1999).
Il y a une interaction entre le polymorphisme des fumeurs
PlA2 et le CAD (Lopes. N. H. M et al., 2004). Les porteurs
de l'allèle PlA2 sont considérablement
protégés contre SAH. De même, ces allèles
réduisent largement le risque de SAH (Iniesta. J. A et al.,
2004). Les malades qui portent ces allèles présentent de
plus grands anévrismes, mais l'extension de leur hémorragie et le
niveau clinique sont considérablement inférieurs quand on les
compare avec les malades HPA-1a/a (Juan. A et al., 2004). Il est
remarquable que l'allèle HPA-1b paraît réduire la
sévérité de l'hémorragie chez les malades avec
Thrombasthénie de Glanzmann (Ghosh. K et al., 2002; Juan. A et al.,
2004) ou l'athérosclérose comme il peut réduire le
risque et la sévérité de l'hémorragie subarachnoid
(Iniesta. J. A et al., 2004; Corral. J et al., 2004).
Le système HPA-2; (Ko,
Sib)
· Génétique du système
HPA-2
Il a été décrit pour la première
fois par Van der Weerdt en 1961. C'est un système di-allélique
localisé sur un domaine globulaire de 45 kDa du coté N-terminal
du de la GPIbá (Kuijpers. R. W. A. M et al., 1989; Kuijpers. R. W.
A. M et al., 1992). Les deux formes alléliques (HPA-2a ou
Kob et HPA-2b ou Koa ou Siba) sont dus
à une substitution d'un seul nucléotide SNP (C/T Cyt/Thy
(ACG)/ (ATG)) (Kuijpers. R. W. A. M et al., 1992a) dans
l'exon 2 en position 434 du gène codant GP1BA. Ce polymorphisme se
traduisant par une substitution d'un seul aa 145 Thr/Met qui se trouve dans le
cinquième LRR (Murata. M et al., 1992).
· Liaison génétique du
polymorphisme HPA-2 avec d'autres polymorphismes
Le polymorphisme du système HPA-2 pourrait être
lié génétiquement à deux autres
polymorphismes : les polymorphismes VNTR (Simsek. S et al.,
1994; Afshar-Kharghan. V et al., 1999; Chester. Q et al., 2000) et les
polymorphismes de Kozaki (Kozak polymorphism) (Afshar-Kharghan. V et
al., 1999; Chester. Q et al., 2000).
Le polymorphisme HPA-2 est en déséquilibre de
liaison avec le polymorphisme de VNTR (Deckmyn. H et al,. 2004) auquel
il est fortement lié. Le HPA-2a (Thr145) est associé
au VNTR-C et au VNTR-D alors que l'allèle HPA-2b (Met145) est
lié au VNTR-A et au VNTR-B (Simsek. S et al., 1994; Aramaki. K. M.
et Reiner A. P. 1999). En effet, les allèles avec une ou deux
répétitions sont liés à l'isoforme Thr (HPA-a),
alors que les allèles avec trois ou quatre répétitions
sont liés à l'isoforme Met (HPA-b) (Ulrichts. H et al.,
2003). Tandis que d'autres études de génotypage des PLTs
étaient contradictoires en montrant un effet fonctionnel provoqué
par ce dimorphisme (Mazzucato et al., 1996; Boncler et al., 2002;
Jilma-Stohlawetz. P et al., 2003; Deckmyn. H et al.,
2004). Les distributions alléliques de la GPIbá VNTR et
HPA-2 diffèrent considérablement selon les différentes
populations. Par exemple, l'allèle VNTR-A est trouvé presque
exclusivement en Asie de l'est (Aramaki. A K. M. et Reiner. A. P.
1999).
Ces polymorphismes influent sur la structure et le niveau
d'expression de la GPIbá qui possède un effet potentiel sur les
maladies thrombotiques.
· Implications cliniques du polymorphisme du
système HPA-2
L'exposition au non soi des formes allo-antigèniques du
système HPA-2 au cours de la grossesse ou après transfusion peut
mener à la formation des allo-anticorps qui peuvent causer la NAITP
(Kuijpers. R. W. A. M et al., 1992), PTR (Kuijpers. R. W. A. M et al.,
1992a) et le PTP (Chester. Q et al., 2000).
De même, le rapport entre les allèles HPA-2b et
VNTR-B est considéré comme un facteur à risque
thrombotique pour les maladies cardio-vasculaires (Aramaki. K. M. et
Reiner. A. P. 1999), ce qui suggère que ces variantes ont pu
évoluer, au moins en partie, à cause de la pression
sélective pour activer l'hémostase (Aramaki. K. M. et Reiner.
A. P. 1999). Des études récentes ont montré qu'il
existe une association des génotypes HPA-2a/2b ou HPA-2b/2b avec L'IS ou
avec des maladies cardio-vasculaires. D'ailleurs, c'est parce que le
polymorphisme HPA-2 est localisé près des sites de fixation du
FvW et la thrombine qu'il pourrait causer une variation conformationnelle dans
la structure de GPIbá qui peut affecter le site de liaison des ligands.
Le système HPA-2 fait modifier la conformation au
niveau de la région aa-1-59 de GPIbá, ce qui résulte en
une interaction plus forte du FvW avec HPA-2a (Ulrichts. H et al.,
2003). L'association entre le polymorphisme VNTR et les
événements thrombotiques est vague parce que GPIbá est
aussi un récepteur pour P-sélectine, Mac-1, FXI, FXII et le
kininogène de haut poids moléculaire. Le polymorphisme HPA-2
pourrait affecter aussi ces interactions qui pourraient être à la
base de risques thrombotiques (Ulrichts H et al., 2003).
Un nombre limité d'études ont
démontré une association entre les allèles
Met145 (VNTR A ou B) et le risque des maladies cardio-vasculaires
(Murata. M et al., 1992; Murata. M et al., 1997; Gonzalez-Conejero. R et
al., 1998; Murata. M et al., 1998) tandis que d'autres études n'ont
pas trouvé cette association (Carlsson. L. E et al., 1997; Carter.
A. M et al., 1998). Dans une étude récente le HPA-2
Met/VNTR-B a été associé à des
événements importants de l'IM et la mort des vieillards malades
(Mikkelsson. J et al., 2001).
Les études ont des contradictions dans la
répartition du risque de polymorphisme du complexe GPIb/IX/V dans
différents groupes ethniques. Chez les populations européennes,
le génotype VNTR B/C est associé à une augmentation du
risque des maladies de l'artère coronaire dans la population espagnole,
mais aucune association n'a été détectée chez la
population française (Mercier. B et al., 2000). En outre, Chez
les Américains, le génotype VNTR C/C est associe avec un risque
faible des événements coronaires (Murata. M et al., 1998;
Afshar-Kharghan. V et al., 1999a). La présence de l'allèle
VNTRB possède un risque significatif de NAION
(Salomon. O et al., 2004).
Le système HPA-3
· Génétique du système
HPA-3
Le système antigénique
Baka/Bakb a été décrit pour la
première fois en 1980 par Von dem Borne et al suite à un cas de
NAITP. C'est un système bi-allélique localisé sur la
chaîne lourde de la GPIIb (Van der Schoot., 1986; Lyman et
al., 1990). Les deux -allèles HPA-3a et HPA-3b ce
diffèrent par un SNP près de l'extrémité 3' de
l'ARNm Au niveau de l'ADN, ce SNP est localisé dans l'exon 26
(Lyman. S et al., 1990) en position T2621G (Unkelbach.
K et al., 1995) du gène codant ITGA2B. Ce polymorphisme correspond
à la substitution de l'aa Ile/Ser843 du coté
C-terminal de la GP mature (Zain et al., 2002; Metcalfe. P et al.,
2003) dans les derniers 29 aa de la chaîne lourde de GPIIb
(Djaffar. I et al., 1993) (TableauIII). Le déterminant
antigénique est localisé sur un fragment de 76 et 60 Kd de cette
chaîne à l'extrémité riche en proline (Kieffer.
N et al., 1984).
· Implications cliniques
Le système HPA-3 est impliqué dans l'induction
de la NAIT, le PTP (Lyman. S et al., 1990 ; Kickler TS. 1988) et
PTR (Steffensen. R et al., 1996; Liu T.C et al., 2002; Wiwanitiki.,
2005). L'allèle Ser843 augmente
l'agrégation des PLTs in vitro et diminue la rétractation du
caillot en comparaison avec des PLTs qui ne possèdent pas cet
allèle (Honda. S et al., 1985).
Reiner et al ont rapporté un risque élevé
de l'MI chez les femmes qui possèdent au moins une copie de
l'allèle Ser843. Ce risque est présent seulement dans
un sous-groupe de femmes possédant des facteurs supplémentaires
du risque cardio-vasculaire (femmes fumeuses,
hypercholestérolémie ou avoir une histoire de famille avec IM)
(Reiner. A. P. et al., 2001).
Kroll et al ont démontré que le dimorphisme
T2622G de la GPIIb ne constitue pas un risque majeur ni pour le CAD
ni pour l'AMI (Kroll. H et al., 2001; Hato. T. M. D et al., 1997). Le
polymorphisme de délétion de 9 pb localisée au niveau de
l'intron 21 du gène codant pour la GPIIb et le polymorphisme de
l'antigène HPA-3b sont liées (Peyruchaud. O et al., 1995).
Le système HPA-4
C'est un système allo-antigénique
di-allélique, les deux allèles HPA-4a et HPA-4b correspondent
respectivement aux antigènes (Pena,
Penb/Yuka, Yukb) (Friedman. J.M et Aster.
R. H., 1985; Shibata. Y et al., 1986). Ces antigènes sont
localisés au niveau de la GPIIIa (Furihata. K et al., 1987). Le
polymorphisme de ce système est déterminé par un seul
SNPG526 A526 au niveau de l'exon 4 : du gène
codant ITGB3 (Tableau III). Les deux formes alléliques diffèrent
par un seul aa 143 (Arg/Glu) au niveau de la GP mature (Wang. R et al.,
1991; Wang. R et al., 1992).
Le système HPA-4 est le seul qui coïncide avec un
domaine fonctionnel connu (Wang Wang. R et al., 1992). Il est
situé dans la zone de fixation du ligand riche en Leu (Kuipers.,
1992). Ces polymorphies causent un changement structural
considérable dans la sous-unité Ibá, en affectant le site
de fixation du FvW (Ulrichts. H et al., 2003).
Il a été démontré que seulement
les anticorps anti-HPA-1a et anti-HPA-4a sont capables d'inhiber
l'agrégation des PLTs respectivement avec des antigènes HPA-la
(Van Leeuwen. E. F et al., 1982) et HPA-4a (Furihata. K et al.,
1987).
Le système HPA-5; (Br, Zav)
· Génétique du système
HPA-5
Deux antigènes HPA-5a et HPA-5b appartiennent à
ce système et correspondent aux antigènes Brb,
Zavb (HPA-5a) et Bra, Zava (HPA-5b) (Woods
V. L. Jr et al., 1989; Santoso. S . A et al., 1989; Santoso. S . A et al.,
1989a; Kiefel. V et al., 1989). L'SNP G/A1648 dans l'exon
13 du gène codant ITGA2 donne lieu à une substitution d'un
seul aa Glu505(GAG: Bra)/Lys505(AAG:
Brb) (Tableau III). Le Glu505Lys est localisé
entre le premier et deuxième site potentiel de fixation du calcium au
niveau de la GPIa (Santoso. S. A et al., 1993b; Kalb. R et al., 1994;
Simsek. S et al., 1994a; Juji. T et al., 1999).
· Implications cliniques du
système HPA-5
L'alloimmunisation contre le HPA-5 est associée aux
NAIT (Kiefel et al., 1988;
Mueller-Eckhardt.
C et al., 1989; Bettaieb. A et al., 1991; Kiefel et al.,
1991), le PTP (Christie. D. J et al., 1991) et les PTR
(Bierling et al., 1989). L'antigène HPA-5b est le
deuxième parmi tous les allo-antigénes impliqués dans la
pathogénie de la plupart des cas de NAIT (Mueller-Eckhardt. C et
al., 1989; Kaplan. C et al., 1991; Kretzschmar. E. 2001) et les cas de PTP
(Christie. D. J et al., 1991; Kretzschmar. E. 2001).
L'antigène HPA-5a est un déterminant dans les
cas de NAIT (Mueller-Eckhardt. C et al, 1989; Kaplan. C et al, 1991;
Kretzschmar. E. 2001). Le polymorphisme HPA-5 est aussi en rapport avec
des maladies immunes telles que le purpura thrombocytopénique
idiopathique aigu (Castro al., 2000; Kretzschmar. E. 2001). Dans une
grande étude, Kroll et al (2000) trouvez une association entre le
dimorphisme HPA-5 dans un sous-groupe de patient peu risqué avec les
maladies de l'artère coronaire. Le génotype HPA-5a/b
était considérablement inférieur chez les malades avec le
diabète de type 2 et chez les non diabétiques ASO-Positifs que
chez les contrôles. Ces conclusions suggèrent que l'études
génétiques de HLA, NA et HPA pourraient être utiles
à comprendre la pathogénie du diabète de type 2 et ASO
(Nomura. S et al., 2006).
· Liaison génétique du
polymorphisme HPA-5 avec d'autres polymorphismes
La résistance contre l'allo-immunisation anti-HPA 5b
pourraient être conférés par l'allèle DRB1*0301
(Koutsogianni. P. 2004). Une corrélation entre
l'allo-immunisation anti HPA-5 et HLA-DRW6 a été observée
(Mueller-Eckhardt. C et al., 1989).
Hallé et al., 2005 ont montré dans les
populations caucasiennes une association entre l'allo-immunisation anti HPA-5b
et un motif Glu-Asp en position 69-70 porté par la chaîne DRb1 des
molécules HLA-DR dont DR12 et DR13, antigènes fréquents
dans les populations d'Afrique sub-saharienne (Hallé. L et al.,
2005).
Le système HPA-15 (Gov)
· Génétique du système
Gov
Le système antigénique plaquettaire HPA-15 a
été décrit en 1990. C'est un système
biallélique (Gova, Govb) (Kelton. J.G et al.,
1990) exprimé par la GP CD109 (Smith. J.W et al., 1995). Les
deux allèles HPA-15a et HPA-15b se diffèrent par un SNP (A/C) en
position 2108 de la séquence codante. Le codant 703 change de TAC
(Gova) à TCC (Govb), il résulte alors une
substitution d'un seul aa Tyr/Ser703 au niveau de la protéine
mature. Ce polymorphisme (SNP) Tyr/Ser 703 se localise au niveau des
trois derniers nucléotides du coté de l'extrémité
5' de l'exon 19 et correspond à la fin de l'intron 18 (Schuh. A .C
et al., 2002).
· Immunogénécité du
système Gov et ses implications cliniques
L'immunisation anti-Gov après transfusions ou
grossesses a été rapportée en 1997
(Bordin. J.O et al., 1997). Les allo-anticorps réactifs aux
PLTs sont impliqués dans le développement de la NAIT (Bordin.
J. O et al., 1997; Kroll. H et al., 1998), en particulier chez les malades
qui reçoivent des transfusions multiples des PLTs (Ertel. K et al.,
2005), le PTP (Kelton. J. G et al., 1990) et les PTR (Kelton.
J.G et al., 1990) (Skouri H et al., 2003 ; Skouri H et
al., 2005), de même ils sont impliqués dans la
transplantation d'organes ou de la MO et sont associés à
l'hémorragie cérébrale et l'hémiplégie
(Berry. J.E et al., 2000). Les anticorps Gov ont été
associés plus fréquemment aux PTR qu'a la NAIT (Berry. J.E et
al., 2000). Malgré son importance, le rôle des
antigènes Gov dans la destruction des PLTs par les allo-anticorps n'a
pas encore été défini. Aucune association entre le
développement d'une allo immunisation anti-Gov et un haplotype HLA de
classe II particulier n'a été mise en évidence
(Mérieux et al., 2004).
2- Les systèmes les moins impliqués
en pathologies humaines
Le système HPA-6
Les deux allo-antigènes de ce système
correspondent aux antigènes (Caa, Tua). Il
dérive de la substitution A1564/G1564 (CAG)/(CGG)
conduisant à une substitution Arg489/Glu489 au
niveau de la GPIIIa mature. Ce système rare est impliqué dans la
NAIT (Wang. R et al., 1993 ; Kekomäki S et al., 1993).
Le système HPA-7 (Moa)
Ce système est composé de deux antigènes.
La substitution nucléotidique C1267/G1267 est le
résultat d'une substitution d'un seul aa Pro 407/Ala407
au niveau de la GPIIIa mature (Kuijpers. R. W. A. M et al.,
1993). Ce système rare est impliqué dans la NAIT (
Kroll. H et al.,
1990)
Le système HPA-8- (Sra)
C'est un système di-allélique correspondent
à l'antigène (Sra). Ils sont localisés sur la
GPIIIa au niveau de la région riche en répétition Cys
(Santoso. S. A et al., 2002) sur un fragment, de masse
moléculaire de 68Kd (Kroll. H. 1999). La substitution
T2004/C2004 conduit à une substitution d'un seul
aa Arg636/Cys636 au niveau de la GP mature.
Le système HPA-9 (Maxa)
Une substitution d'un SNP G2603A se produit au
niveau du gène de structure ITGA2B dans l'exon 26, entraînera une
substitution d'un aa Val/Mét 837 sur la chaîne lourde de la GPIIb
lors de la traduction. Le système Maxa est proche du HPA-3
(Lyma et al., 1990) (SNP G2603A/HPA-9 en amont du SNP
T2622G/HPA-3 de 19 bp), d'ailleurs il existe une association
génétique entre leurs polymorphismes et Maxa est une
variante de l'allèle HPA-3b. L'antigène Maxa doit
être pris en considération dans les PTR aussi bien que dans les
cas de NATP et de PTP (Noris. P. S et al., 1995).
Le système HPA-10, (Laa)
C'est un système di-allélique localisé
sur la région N-terminale du GPIIIa. La substitution d'un seul aa
Arg62/Glu 62 au niveau du GP mature, résulte d'une
substitution d'un seul SNP G281/A281 au niveau de l'exon
3 de l'ADNc du gène codant. Cette mutation est responsable de
l'expression de l'épitope Laa impliquée dans la
réponse immune à médiation humorale qui a pour
conséquence la NAIT (Peyruchaud. O et al., 1997).
Le système HPA-11 (Groa)
C'est un antigène privé localisé sur la
GPIIIa (Simsek. S et al., 1994a). Ce système est composé
de deux antigènes qui différent par un seul nucléotide
G1996 (CGT)/A1996 (CAT) au niveau du
gène codant se traduisant par une substitution d'un seul aa
Arg633/His633. Groa est identifié suite
à un cas de NAIT (Simsek. S et al., 1994b). La mutation
observée est en amont de 3 aa de la mutation Arg636Cys
(HPA-8/Sr) suggère que cette région de la GPIIIa est susceptible
pour les mutations (Simsek. S et al., 1997). Les polymorphismes des
deux SNPs T2004/C2004 et G1996/A1996
sont proches, ce qui montre que le système Sra (Arg 636
Cys) (Santoso. S. A et al., 1994) est associé à
l'antigène Groa (Simsek. S et al., 1997).
Le système HPA-12 (Lya)
C'est un système à deux allèles, qui
diffèrent par un seul SNP G/A (GGG/GAG) en position 141 de la
séquence codante. Il résulte alors une substitution d'un seul aa
Gly5/A Glu15 au niveau du domaine N-terminal de la
protéine mature GPIbâ (Sachs. UJH et al.,
2000). Le système Lya est responsable de cas
sévères de NAIT (Kiefel et al., 1995).
Le système HPA-13 (Sita )
L'allo-antigène Sita est de faible
fréquence (1/400 dans la population allemande) car il s'agit d'une
mutation récente. Il a été identifié chez un cas de
NAITP sévère. Le polymorphisme nucléotidique
C2531-T2531 au niveau de l'exon 20 du gène codant
donne lieu à un dimorphisme protéique qui se traduit par une
substitution de l'aa polaire, la Thr799, en un résidu
hydrophobe non polaire, la Met799 qui est responsable de la
formation du déterminant allo-antigénique
Sita, localisé sur la GPIa mature
(Muller-Eckhard. C et al., 1994). L'analyse de la structure
exon-intron a permis de définir le site polymorphique sur l'exon 20 avec
une longueur de 142 bp.
Le système HPA-14(Oea)
Ce système de faible fréquence est porté
par la GPIIIa. La délétion de 3 pb AAG en positions 1929-1931 au
niveau de l'exon 10 conduisant à une délétion
Lys611 est responsable de la formation de l'épitope
(Oea) (Kekomäki et al., 1992; Kekomäki et al.,
1993) et de l'altération de l'isoforme GPIIIa (PlA2). La
délétion Pro33 Lys611GPIIIa pourrait subir des
changements conformationnels et se lie au fibrinogène d'une
manière similaire à l'isoforme 33 Lys Pro 611. Par contraste avec
tous les autres antigènes de faibles fréquences, l'isoforme Lys
611 a été associé à l'antigène HPA-1b, et
non avec l'allèle HPA-1a.
Ces dernières années, plusieurs groupes ont
rapporté que le polymorphisme de GPIIIa n'intervient pas seulement dans
les réactions immunologiques, mais peut être aussi un facteur de
risque dans le développement des maladies coronaires (Santoso. S. A
et al., 2001).
Le système HPA-16
Le Duva est un antigène de faible
fréquence. Les deux allo-antigènes se diffèrent par un
seul SNP C/T en position 517 de la séquence codante. Il résulte
alors une substitution d'un seul aa Thr140/Ile140 au
niveau de la protéine mature GPIIIa. Le système Duva+
est impliqué dans la NAIT (Jallu. V et al., 2002).
3 - Les antigènes rare définis seulement
par la sérologie
D'autres allo-antigènes, ont été aussi
définis sérologiquement, mais leur base moléculaire n'est
pas encore bien élucidée (Johson-Hopson. C. N et al.,
2003). Sont des allo-antigènes privés. Les
allo-antigènes Vaa (GPIIb/IIIa) et Moua (GP
inconnue) (Kekomaki. R et al., 1992; Masters. R et al., 1998). Les
allo-antigènes Naka (Yamarnoto N. et al., 1994) et
Vis, sont portés par la GPIV (Tomiyama. Y et al., 1990; Wyler. B et
al., 1990; Yamamoto. N et al., 1990) alors que
l'allo-antigènes Pea est situe sur la GPIbá et
l'allo-antigènes Plt est localise sur la GPV.
Tableau II :
identification génomiques et protéiques des polymorphismes des
HPAs (Koutsogianni. P. 2004).
|
Antigènes
|
GPs
|
HGNC
|
Locus
|
Référence de la séquence
|
Nucléotide
polymorphe
|
protéine mature
|
Précurseur
|
|
HPA-1
|
GPIIIa
|
ITGB3
|
3690
|
NM_000212
|
176T > C
|
L33P
|
L59P
|
|
HPA-2
|
GPIbá
|
GP1BA
|
2811
|
NM_000173
|
482C > T
|
T145M
|
T161M
|
|
HPA-3
|
GPIIb
|
ITGA2B
|
3674
|
NM_000419
|
2621T > G
|
I843S
|
I874S
|
|
HPA-4
|
GPIIIa
|
ITGB3
|
3690
|
NM_000212
|
506G > A
|
R143Q
|
R169Q
|
|
HPA-5
|
GPIa
|
ITGA2
|
3673
|
NM_002203
|
1600G > A
|
E505K
|
E534K
|
|
HPA-6w
|
GPIIIa
|
ITGB3
|
3690
|
NM_000212
|
1544G > A
|
R489Q
|
R515Q
|
|
HPA-7w
|
GPIIIa
|
ITGB3
|
3690
|
NM_000212
|
1297C > G
|
P407A
|
P433A
|
|
HPA-8w
|
GPIIIa
|
ITGB3
|
3690
|
NM_000212
|
1984C > T
|
R636C
|
R662C
|
|
HPA-9w
|
GPIIb
|
ITGA2B
|
3674
|
NM_000419
|
2602G > A
|
V837M
|
V868M
|
|
HPA-10w
|
GPIIIa
|
ITGB3
|
3690
|
NM_000212
|
263G > A
|
R62Q
|
R88Q
|
|
HPA-11w
|
GPIIIa
|
ITGB3
|
3690
|
NM_000212
|
1976G > A
|
R633H
|
R659H
|
|
HPA-12w
|
GPIbâ
|
GP1BB
|
2812
|
NM_000407
|
119G > A
|
G15E
|
G40E
|
|
HPA-13w
|
GPIa
|
ITGA2
|
3673
|
NM_002203
|
2483C > T
|
T799M
|
T828M
|
|
HPA-14w
|
GPIIIa
|
ITGB3
|
3690
|
NM_000212
|
1909_1911 del AAG
|
K611del
|
K637del
|
|
HPA-15
|
CD109
|
CD109
|
135228
|
NM_133493
|
2108C > A
|
S682Y
|
S703Y
|
|
HPA-16w
|
GPIIIa
|
ITGB3
|
3690
|
NM_000212
|
497C > T
|
T140I
|
T166I
|
Tableau
III : Les différents antigènes
plaquettaires
(Norton. A et al., 2004 : avec quelque
modification):
|
Systèmes
|
Antigènes
|
Noms alternatifs
|
GPs
|
Polymorphisme nucléotidique
|
Polymorphisme d' aa
|
Date de découverte
|
|
HPA-1
|
HPA-1a
HPA-1b
|
Zwa, PIA1
|
GPAIIIa
|
T196 C196
|
Leucine33
Proline33
|
1959
|
|
HPA-2
|
HPA-2a
HPA-2b
|
Kob Koa,
Siba
|
GPIbá
|
C524 T524
|
Threonine145
Methinine145
|
1961
|
|
HPA-3
|
HPA-3a
HPA-3b
|
Baka, Leka
Bakb
|
GPIIb
|
T2621 G2621
|
Isoleucine843
Serine843
|
1980
|
|
HPA-4
|
HPA-4a
HPA-4b
|
Yukb, Pena Yuka,
Penb
|
GPIIIa
|
G526 A526
|
Arginine143
Glutamine143
|
1985
|
|
HPA-5
|
HPA-5a
HPA-5b
|
Brb, Zavb Bra,
Zava, Hca
|
GPIa
|
G1648 A1648
|
A.Glutamique505
Lysine505
|
1988
|
|
HPA-6
|
HPA-6bw
|
Caa, Tua
|
GPIIIa
|
A1564 G1564
|
Arginine489
Glutamine489
|
1992
|
|
HPA-7
|
HPA-7bw
|
Mo
|
GPIIIa
|
C1267 G1267
|
Proline407
Alanine407
|
1992
|
|
HPA-8
|
HPA-8bw
|
Sra
|
GPIIIa
|
T2004 C2004
|
Arginine636
Cysteine636
|
1993
|
|
HPA-9
|
HPA-9bw
|
Maxa
|
GPIIIa
|
G2603 A2603
|
Valine837
Methionine837
|
1995
|
|
HPA-10
|
HPA-10bw
|
Laa
|
GPIIIa
|
G281 A281
|
Arginine62
Glutamine62
|
1996
|
|
HPA-11
|
HPA-11bw
|
Groa
|
GPIIIa
|
G1996 A1996
|
Arginine633
Histidine633
|
1996
|
|
HPA-12
|
HPA-12bw
|
Iya
|
GPIb
|
G141 A141
|
Glycine15
A.Glutamique15
|
1999
|
|
HPA-13
|
HPA-13bw
|
Sita
|
GPIa
|
C2531 T2531
|
Threonine799
Methionine799
|
1999
|
|
HPA-14
|
HPA-14bw
|
Oea
|
GPIIIa
|
? AAG1929-1931
|
Lysine611
|
|
|
HPA-15
|
HPA-15a
HPA-15b
|
Govb Gova
|
CD109
|
C2108 A2108
|
Serine703
Thyrosine703
|
2000
|
|
HPA-16
|
HPA-16bw
|
Duva
|
GPIIIa
|
C517 T517
|
Threonine140
Isoleucine140
|
2002
|
|
|
Vaa
|
GPIIb/IIIa
|
|
|
|
|
|
Plt
|
GPV
|
|
|
|
|
|
Vis
|
GPIV
|
|
|
|
|
|
Pea
|
GPIbá
|
|
|
|
|
|
Dya
|
38 KD GP
|
|
|
|
|
|
Moua
|
inconnu
|
|
|
|
CD : terme définissent les antigènes de
surface des lymphocytes (reconnus grâce à des anticorps
monoclonaux) et qui caractérisent des étapes de la
différenciation dans les organes centraux et, éventuellement des
fonctions de ces cellules.
III.3.1 Fréquences
alléliques des systèmes HPA-1-5 chez les différentes
populations
Beaucoup d'études rapportent les fréquences
d'allo-antigènes plaquettaires dans différentes populations. La
plupart des études des polymorphismes alloantigèniques ont
analysé seulement les fréquences de quelques HPAs, surtout les
systèmes HPA-1 à HPA-5. Mais peu d'études ont
été exécutées sur les antigènes de faible
fréquence après qu'ils aient été identifiés
dans des familles individuelles. D'après ces études, on remarque
que les données précédentes chez les populations autre que
les blancs sont rares (Covas et al., 2000). Et que les
fréquences des systèmes HPAs varient dans différentes
populations (Santoso. S. A et al., 1993; Kim. H. O et al., 1995;
Klüter. H. 1996; Cardone. J. D. B et al., 2004). Des variations
simples de type mendélien peuvent être observées au sein
d'une même population aussi bien qu'entre distinctes populations. Chaque
population se caractérise par ces fréquences géniques pour
chaque système (Tableau IV). D'ailleurs on note, dans quelques pays,
généralement en Asie (Taiwan, les Philippines,
l'Indonésie, la Chine, le Japon, la Corée, le Vietnam), un grand
taux de stabilité raciale vu qu'ils n'ont pas eu d'échanges de
gènes avec d'autres populations. Par contre, d'autres populations
présentent des exemples types pour leur diversité ethnique et
leur mélange racial tel que les tunisiens (Moojat. N et al.,
1999 ; Saadi H et al., 2006). La fréquence des allèles
HPA-a est plus élevée que celle des allèles HPA-b presque
chez toutes les populations.
La fréquence de l'allèle HPA-1a est
élevée chez les Brésiliens, les Amérindiens et dans
la République Centrafricaine, par conséquent l'allèle b
est absent chez ces populations. La fréquence de l'allèle HPA-1b
varie de 0 (Amérindiens) à 0,330 (Grèce) (Cavas et
al., 1997; Castro et al., 1999; Cavas et al., 2000; Hallé. L et al.,
2005). Chez les asiatiques, la fréquence de l'allèle HPA-1a
est élevée (0.8-0.9983) (Arabie Saoudite-Taiwan). La
fréquence de l'allèle HPA-2a varie de 0,607 à la
République centrafricaine à 0,975 au Philippines. La
fréquence de l'allèle HPA-2b est élevée chez les
africains (0,393 République Centrafricaine).
Les fréquences des allèles HPA-3a et HPA-3b sont
élevées chez toutes les populations étudiées. Les
fréquences des allèles HPA-3b sont plus élevées que
celles des allèles HPA-3a ; en Grèce 0,595/0,405, au Vietnam
0,514/0,486 et en Indonésie 0,539/0,461. Dans la république
centrafricaine, les fréquences alléliques HPA-3a et HPA-3b sont
égales (0,500). L'allèle «b» du système HPA-4
est extrêmement rare (dem Borne, Décary et al., 1990;
Mueller-Eckhardt. C et al., 1990; Roúman. P. 2002). Cette
fréquence est estimée à 0-0.115 (0.115 pour les tunisiens)
(Saadi H et al., 2006) chez toutes les populations.
La fréquence de l'allèle HPA-5b est
élevée chez les africains (0,125 à 0,405) comparativement
à celle observée chez les asiatiques et les européens. La
fréquence de l'allèle HPA-5b dans l'Asie varie de 0,005
(Indonésie) à 0,155 (Arabie Saoudite). Chez les européens
HPA-5b varie de (0,050) (Finlande) à (0,150) Grèce. Chez les
américains la fréquence de l'allèle HPA-5a est
élevée chez les brésilien (Castro et al., 1999).
On remarque, d'après ces études, que les fréquences des
antigènes HPA-1b, -2b, -3b,-5b, pour la plupart des populations, sont
élevées. Les données sur la prévalence
d'antigènes plaquettaires dans une population donnée est
essentielle pour le diagnostic des cas des NAIT, pour l'organisation des
programmes de screening pour les femmes à risque de FMAIT, pour les
consultations génétiques et pour la provision de la
thérapie pour les malades avec des anticorps anti-HPA
(Mueller-Eckhardt. C et al., 1989; Bussel.B. J et al., 1997; Roúman.
P. 2002). Excepté leur impact pratique sur la provision des
transfusions des PLTs, elles pourraient contribuer aussi à la
compréhension des rapports évolutionnaires et offrent des
informations sur les expansions démographique (Cavalli, Piazza.,
1993; Roúman. P. 2002).
Tableau IV
Fréquences alléliques des HPA-1,-2, -3, -4 et -5 chez
différentes populations (
http://www.ebi.ac.uk/ipd/):
|
Pays
|
HPA-1a
|
HPA-1b
|
HPA-2a
|
HPA-2b
|
HPA-3a
|
HPA-3b
|
HPA-4a
|
HPA-4b
|
HPA-5a
|
HPA-5b
|
référence
|
|
Tunisiens
|
0,824
|
0,176
|
0,800
|
0,200
|
0,707
|
0,293
|
1,000
|
0,000
|
0,875
|
0,125
|
Présente étude
|
|
Tunisiens
|
0.73
|
0.27
|
0.680
|
0.320
|
0.765
|
0.235
|
0.885
|
0.115
|
0.665
|
0.335
|
Saadi H et al., 2006
|
|
Autrichiens
|
0,852
|
0,148
|
0,918
|
0,082
|
0,612
|
0,388
|
1,000
|
0,000
|
0,892
|
0,108
|
Holensteiner et al., 1995.
|
|
Autriche
|
0,830
|
0,170
|
0,870
|
0,130
|
0,620
|
0,380
|
|
|
0,920
|
0,080
|
Macher et al., 2004.
|
|
Bénin
|
0,896
|
0,104
|
0,708
|
0,292
|
0,679
|
0,321
|
1,000
|
0,000
|
0,818
|
0,182
|
Hallé et al., 2005.
|
|
Brésil
|
0,903
|
0,097
|
0,810
|
0,190
|
0,666
|
0,334
|
1,000
|
0,000
|
0,876
|
0,124
|
Castro et al., 1999.
|
|
Brésil
|
1,000
|
0,000
|
0,821
|
0,179
|
0,757
|
0,243
|
1,000
|
0,000
|
1,000
|
0,000
|
Castro et al., 1999.
|
|
Cameroun
|
0,907
|
0,093
|
0,763
|
0,237
|
0,614
|
0,386
|
1,000
|
0,000
|
0,746
|
0,254
|
Hallé et al., 2005.
|
|
Rép.centraf-ricaine
|
1,000
|
0,000
|
0,607
|
|
0,500
|
0,500
|
1,000
|
0,000
|
0,595
|
0,405
|
Hallé et al., 2005.
|
|
Congo
|
0,904
|
0,096
|
0,776
|
0,224
|
0,596
|
0,404
|
1,000
|
0,000
|
0,732
|
0,268
|
Hallé et al., 2005.
|
|
Croatie
|
0,830
|
0,170
|
0,880
|
0,120
|
0,640
|
0,360
|
|
|
0,890
|
0,110
|
|
|
République du czech
|
0,830
|
0,170
|
0,900
|
0,100
|
0,590
|
0,410
|
|
|
0,930
|
0,070
|
Korinkova et al., 1999
|
|
Danemark
|
0,830
|
0,170
|
0,920
|
0,080
|
0,630
|
0,370
|
1,000
|
0,000
|
0,920
|
0,080
|
Steffensen et al., 1998.
|
|
Polynésie française
|
0,975
|
0,025
|
0,913
|
0,087
|
0,599
|
0,401
|
1,000
|
0,000
|
0,975
|
0,025
|
Hallé et al., 2001.
|
|
France
|
0,880
|
0,120
|
|
|
0,650
|
0,350
|
|
|
0,830
|
0,170
|
Mérieux., 1997.
|
|
Français
|
0,848
|
0,152
|
0,920
|
0,080
|
0,620
|
0,380
|
0,992
|
0,008
|
0,874
|
0,126
|
Mérieux., 1997.
|
|
Grèce
|
0,670
|
0,330
|
0,750
|
0,250
|
0,405
|
0,595
|
|
|
0,850
|
0,150
|
|
|
Irlande
|
0,882
|
0,118
|
0,934
|
0,066
|
0,624
|
0,376
|
1,000
|
0,000
|
0,912
|
0,088
|
|
|
Italie
|
0,850
|
0,150
|
0,890
|
0,110
|
0,610
|
0,390
|
1,000
|
0,000
|
0,900
|
0,100
|
Mazzucco et al., 2000.
|
|
Luxembourg
|
0,869
|
0,131
|
0,933
|
0,067
|
0,616
|
0,384
|
|
|
0,902
|
0,098
|
|
|
Maroc
|
0,748
|
0,252
|
0,818
|
0,182
|
0,682
|
0,318
|
1,000
|
0,000
|
0,861
|
0,139
|
Ferrer et al., 2002.
|
|
Berbères (Maroc)
|
0,747
|
0,252
|
0,817
|
0,182
|
0,682
|
0,317
|
1
|
0,0
|
0,8616
|
0,1383
|
Ferrer et al., 2002
|
|
Algérie
|
0,827
|
0,173
|
|
|
|
|
|
|
0,837
|
0,163
|
Mercies et al., 1997.
|
|
Tunisiens
|
0,750
|
0,250
|
|
|
0,694
|
0,306
|
|
|
0,779
|
0,221
|
Mojaat. N et al., 1999.
|
|
Pologne
|
0,824
|
0,176
|
0,901
|
0,099
|
0,582
|
0,418
|
1,000
|
0,000
|
0,944
|
0,056
|
Rzewek et al., 1998.
|
|
Macédonie
|
0,865
|
0,135
|
0,852
|
0,148
|
0,578
|
0,422
|
|
|
0,909
|
0,091
|
Pavkovic et al., 2006.
|
|
Slovénie
|
0,832
|
0,168
|
0,899
|
0,101
|
0,667
|
0,333
|
|
|
0,892
|
0,108
|
Rozman. P et a.l, 1999.
|
|
Espagne
|
0,810
|
0,190
|
0,900
|
0,100
|
0,650
|
0,350
|
1,000
|
0,000
|
0,880
|
0,120
|
Muñiz-Diaz et al., 1998.
|
|
Suisse
|
0,809
|
0,191
|
0,918
|
0,109
|
0,591
|
0,407
|
0,997
|
0,000
|
0,934
|
0,066
|
Boehlen et al., 2003.
|
|
Taiwan
|
0,997
|
0,003
|
0,960
|
0,040
|
0,575
|
0,425
|
0,998
|
0,002
|
0,985
|
0,015
|
|
|
Vietnam
|
0,986
|
0,014
|
0,953
|
0,047
|
0,486
|
0,514
|
1,000
|
0,000
|
0,972
|
0,028
|
Hallé et al., 2001.
|
|
Le Pays de Gales
|
0,825
|
0,175
|
0,902
|
0,098
|
0,607
|
0,393
|
1,000
|
0,000
|
0,903
|
0,097
|
Sellers et al., 1999.
|
|
Caucasiens
|
0,810
|
0,190
|
0,915
|
0,085
|
0,520
|
0,480
|
1,000
|
0,000
|
0,905
|
0,950
|
Legler et al., 1996.
|
|
Arabie Saoudite
|
0,800
|
0,200
|
0,805
|
0,195
|
0,880
|
0,120
|
0,970
|
0,030
|
0,845
|
0,155
|
AL-SHEIKH et al., 2000.
|
|
Taiwan
|
0,998
|
0,002
|
0,961
|
0,039
|
0,595
|
0,405
|
0,998
|
0,002
|
0,981
|
0,019
|
SHIH. M. C et al., 2003.
|
|
Thaïlande
|
0,985
|
0,015
|
0,938
|
0,062
|
0,507
|
0,493
|
1,000
|
0,000
|
0,963
|
0,037
|
SHIH. M. C et al. , 2003.
|
|
Philippines
|
0,980
|
0,200
|
0,975
|
0,025
|
0,530
|
0,470
|
0,995
|
0,005
|
0,965
|
0,035
|
SHIH. M. C et al., 2003.
|
|
Danemark
|
0,831
|
0,169
|
0,917
|
0,083
|
0,626
|
0,374
|
1,000
|
0,000
|
0,922
|
0,079
|
Steffensen et al., 1996.
|
|
Pays Bas
|
0,846
|
0,154
|
0,934
|
0,066
|
0,555
|
0,445
|
1,000
|
0,000
|
0,902
|
0,098
|
SIMSEK, et al., 1993.
|
|
Finlande
|
0,860
|
0,140
|
0,910
|
0,090
|
0,590
|
0,410
|
|
|
0,950
|
0,050
|
Kekomaki et al., 1995.
|
|
Allemagne
|
0,820
|
0,180
|
0,920
|
0,080
|
0,635
|
0,365
|
|
|
0,900
|
0,100
|
Chen et al.1997.
|
|
Allemagne
|
0,840
|
0,160
|
0,930
|
0,070
|
0,6 0
|
0,400
|
|
|
0,920
|
0,080
|
Arlsson et al., 1997.
|
|
Allemands
|
0,846
|
0,154
|
0,934
|
0,066
|
0,555
|
0,445
|
1,000
|
0,000
|
0,902
|
0,098
|
1993
|
|
Américains (Noirs)
|
0,920
|
0,080
|
0,820
|
0,180
|
0,630
|
0,370
|
1,000
|
0,000
|
0,790
|
0,210
|
Kim et al., 1995.
|
|
Américains
|
0,890
|
0,110
|
0,920
|
0,080
|
0,670
|
0,330
|
1,000
|
0,000
|
0,890
|
0,110
|
Kim et al., 1995.
|
|
Amérindiens
|
1,000
|
0,000
|
0,957
|
0,042
|
0,27-0,75
|
0,25-0,73
|
|
|
1
|
0
|
Cavas et al., 1997; Cavas et al., 2000.
|
|
Corée
|
0,988
|
0,012
|
0,923
|
0,770
|
0,555
|
0,445
|
0,990
|
0,010
|
0,978
|
0,022
|
Seo et al., 1998.
|
|
Chine
|
0,995
|
0,005
|
0,975
|
0,025
|
0,525
|
0,475
|
1,000
|
0,000
|
0,965
|
0,035
|
Chang et al., 1998.
|
|
Indonésiens
|
0,991
|
0,009
|
|
|
0,461
|
0,539
|
0,997
|
0,003
|
0,954
|
0,046
|
Santosos. S et al. 1993.
|
|
Indonésiens
|
0,991
|
0,009
|
0,939
|
0,061
|
0,505
|
0,495
|
1,000
|
0,000
|
0,995
|
0,005
|
SHIH. M. C et al., 2003.
|
|
Japonais
|
0.998
|
0.002
|
0.898
|
0.102
|
0.594
|
0.406
|
0.990
|
0.010
|
|
|
Tanaka et al., 1995.
|
|
Japonais
|
0,990
|
0,010
|
0,910
|
0,090
|
0,715
|
0,285
|
1,000
|
0,000
|
0,980
|
0,020
|
Legler et al., 1996.
|
|
Aborigènes
|
0,992
|
0,008
|
|
|
0,937
|
0,063
|
|
|
0,847
|
0,153
|
Chen et al., 1997.
|
|
Australiens
|
0,838
|
0,162
|
|
|
0,648
|
0,352
|
|
|
0,930
|
0,070
|
Chen et al., 1997.
|
III.3.2 Les allo-antigènes les
plus impliqués dans les immunisations inter-humaines
La probabilité, pour développer une
réponse allo-immune chez les individus, dépend des
fréquences des antigènes qui peuvent varier d'une population
à une autre. Parmi tous les systèmes alloantigéniques
HPAs, le HPA-1a et le HPA-5b sont les plus immunogènes (Newman. P. J
et al., 1989; Watkins. N. A et al., 2002); ils sont le plus souvent
impliqués dans le processus d'allo-immunisation. En effet, les anticorps
anti HPA1-a et HPA5-b sont impliqués respectivement dans 80% et 10% des
cas d'
incompatibilité
foeto-maternels (Boldt. B et al., 1997). En Europe et en
Amérique, ces deux anticorps sont impliqués respectivement dans
78 % et 19% des cas de NAIT (Newman. P. J., 1994; Kaplan. C. 2001;
Kaplan. C. 2002; Davoren. A et al., 2002). Les anticorps dirigés
contre l'antigène Br(a)/(HPA-5b) est le deuxième
fréquemment impliqué dans les cas de NAIT chez les
européens (Mueller-Eckhardt. C et al., 1989).
De même, 75% des cas de NAIT chez les blancs ont pour
cause l'immunisation contre l'antigène HPA-1a (Van Loghem. J. J et
al., 1959; Noris. P. S et al., 1995; Krol. H, Kiefel et Santoso. S. A 1998;
Santoso. S. A 2002) et on la retrouve dans plus de 90% des cas de NAIT
(Kaplan. C. 2003). Moins fréquemment la réponse immune
est induite par une sensibilisation contre l'antigène HPA-5b, alors que
rarement il est causé par d'autres allo-antigènes
(Marcelli-Barge. A et al., 1973; Shibata. Y et al., 1986; Kekomki et al.,
1993; McFarland. J.Y et al., 1993). Au Japon, où la
quasi-totalité de la population est HPA-1a positif (PLA1 positif), le
système HPA-5 est le plus souvent impliqué dans les cas de NAITP
(Juji. T et al., 1999). De même pour l'antigène
HPA-4b.
D'après les résultats trouvés par Miadi
N et al., 2003 en Tunisie l'antigène HPA-1a est le plus
immunogène suivi par HPA-5b et HPA-3b (Miadi N et al.,
2003).
Les travaux de Berry et al. (2000) sur des échantillons
avec des cas de NAIT, de PTP ou de PTR montrent que les systèmes
antigéniques HPA-15 et HPA-5 possèdent une
immunogénécité similaire (Berey. J.E et al., 2000;
Cardone. J. D. B et al., 2004). L'immunisation contre le HPA-15b est
élevée (Panzer. S et al., 1995), et
l'allo-antigène HPA-15b pourrait être plus immunogène que
l'allo-antigène HPA-15a (Berry. J.E et al., 2000). André
C et al 2002 ont montré que la fréquence d'alloanticorps Gov,
surtout chez les malades PTR, est élevée avec une
fréquence dépassée seulement par ceux dirigés
contre l'antigène (HPA-l). Les anti-Gov sont apparus plus souvent en
cause dans les PTR, cependant la fréquence d'allo-immunisation de HPA-15
chez les malades PT était considérablement plus
élevée que celle chez les mères avec FNAIT (3,0% contre
0,22%). Le HPA-15 représente 6,2 ? des cas d'allo-immunisations
anti-plaquettaires. Le nombre des anticorps anti-Govb
observés est très proche de celui des allo-anticorps
dirigés contre les allo antigènes à faible
immunogénécité (anti HPA-2b et -3a). Ces observations
indiquent que l'allo-immunisation contre HPA-15 devrait être
considérée comme une cause de thrombocytopénie immune, en
particulier chez les malades qui reçoivent de multiples transfusions
plaquettaires (Ertel. K et al., 2005).
III.3.3 Les allo-antigènes les moins
fréquemment impliqués dans les immunisations
inter-humaines
Sont les antigènes rares ou privés tel
que : HPA-4b, HPA-6bw, HPA-7bw, HPA-8bw, HPA-9bw, HPA-10bw,
Duva, Vaa.
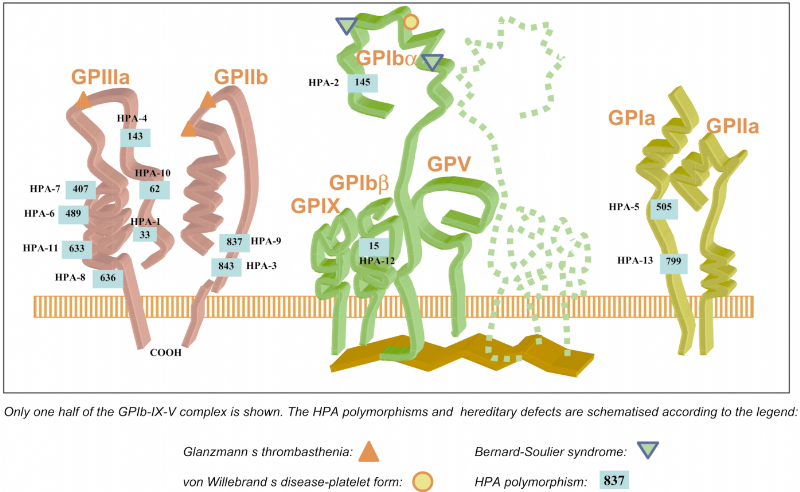
Figure 7. Les
complexes glycoprotéiques spécifiquement plaquettaire
avec leurs polymorphismes (Rozman. P.,
2002).
III.4 Étude
de polymorphisme plaquettaire
Le typage sérologique est
une méthode communément utilisée (Bessos. H et al.,
1996). Plusieurs méthodes sérologiques et
moléculaires ont été utilisées lors de
l'exploration des propriétés biologiques des systèmes
plaquettaires (Juji. T et al., 1999a).
III.4.1 Méthodes de phénotypage
sérologique
Habituellement les allo-antigènes plaquettaires sont
mis en évidence à l'aide d'antisérums provenant de malades
ayant présenté un PTP ou des mères d'enfants atteints de
NAIT (Chabrenaud. J. L et al., 1995). Plusieurs techniques peuvent
être utilisées, telles que l'agglutination (Teramura et al.,
1996), la fixation du complément, l'immunofluorescence ou encore
les essais radio immunologiques. Parmi les techniques les plus utilisées
pour la recherche des anticorps anti-plaquettaires, on cite la
cytométrie en flux, le MAIPA test (Weiss. H. J et al., 1995;
Cardone. J. D. B et al., 2004) et l'ELISA (Honda. S et al., 1995;
Barron-Casella. E.A et al., 1999; Garner et al., 2000).
- La cytométrie en
flux (C MF)
La technique d'immunofluorescence a
été adaptée à la cytométrie en flux qui
permet une interprétation objective de l'intensité de
fluorescence. Elle permet de combiner une appréciation quantitative de
l'antiglobuline fixée et une analyse qualitative de la fluorescence.
Pour cette technique, on utilise souvent une Ig de
chèvre anti-IgG humaine marquée à la fluorescéine.
Pour chaque série il faut inclure des sérums ne contenant pas
d'anticorps anti-plaquettaires et des sérums contenant des anticorps
antiplaquettaires et des sérums contenant des anticorps
antiplaquettaires. Les cellules sont analysées par un automate, avec un
seuil de négativité et de positivité
pré-établi.
La cytométrie en flux est particulièrement
adoptée à l'étude des GP, aussi bien dans le cadre des
syndromes hémorragiques que des états thrombotiques.
La cytométrie en flux et rapide pour rendre le
résultat et la possibilité de travailler sur des volumes sanguins
très faibles.
- La technique de
MAIPA
La technique de MAIPA est utilisée pour le
dépistage et l'identification des allo-anticorps spécifiquement
plaquettaires (Kiefel et al., 1987; Metcafle et al., 1997a; Berry. J.E et
al., 2000). Elle est sensible et spécifique et aide à
l'identification de nouveaux systèmes allo-antigèniques
plaquettaires (Simsek. S et al., 1994; Noris P. S et al., 1995; Berry. J.E
et al., 2000 ). Cette technique est extrêmement utilisée par
l'ensemble des laboratoires d'immunologie plaquettaire et est
considérè comme le test de référence pour le
dépistage et l'identification des anticorps antiplaquettaires.
Le principe de ce test repose sur la capture des
antigènes plaquettaires par des anticorps monoclonaux, chaque anticorps
reconnaîtra l'un des complexes glycoprotéiques polymorphes
GPIIb/IIIa, GPIb/IX/V et GPIa/IIa. C'est un test sensible est facile a
interpréter. Ainsi, il n'y pas d'interférence des anti-HLA.
Le MAIPA test a l'inconvénient de ne pas être un
test rapide.
- L'immunoblotting
Cette technique consiste à identifier les
allo-anticorps suivant leur réaction avec telle ou telle GP plaquettaire
pour ceci après lyse des PLTs on fait migrer les GPs sur un gel de
polyacrylamide, puis il y'aura transfert de ces GPs sur une membrane de
nitrocellulose. Ces GPs immobilisées peuvent réagir avec les
anticorps présents dans le sérum ou le plasma. L'immunoblotting a
l'avantage d'être un test très sensible (Emily A et al.,
1999) et de grande valeur si l'antigène est inconnu (Kaplan. C.
1988).
III.4.2 Méthodes de Typage
moléculaire
Plusieurs techniques de typage des systèmes
plaquettaires HPAs basées sur l'étude de l'ADN ont
été développées (Drzewek. K et al., 1998; Seo.
D. H et al., 1998; Liu T.C et al., 2002). Contrairement aux
méthodes décrites ci dessus ces méthodes de typage
décrivent le polymorphisme au niveau de l'ADN. L'introduction de la PCR
(Newman. P. J et al., 1989; Arlet., 2003) a rendu la
technique de typage moléculaire directe et rapide. Pour surmonter les
limites des testes de phénotypage HPA sérologiques, plusieurs
techniques basées sur l'ADN sont valables (Meyer. O et al., 1999;
Kretzschmar. E. 2001).
La plupart des techniques basées sur la PCR ont
été développées pour étudier les SNPs
responsable des HPAs (Hurd. C. M et al., 2002).
· La
PCR-RFLP
La PCR-RFLP (Newman. P. J et al., 1989; Chen. C. H et al.,
2004), est la première technique PCR utilisée (Von Dem
Borne et Decany., 1990; Carl. B et al., 2000; Shih. M. C et al., 2003).
C'est une méthode fiable, sensible et facile à
exécuter (Hatzidimitriou Gr. Zoulas D et al., 2001).
Elle permet d'identifier le polymorphisme au niveau des sites
de restriction des endonucléases au niveau de l'allèle. L'ADN,
est digéré par des enzymes de restriction. La coupure de l'ADN se
fait en un site particulier reconnu par l'enzyme, il existe alors un
polymorphisme de restriction allèle spécifique. Chaque enzyme
reconnaît une séquence d'ADN qui lui est spécifique. Les
enzymes de restrictions appartiennent à la classe des
endonucléases, elles sont capables de cliver les liaisons phosphodiester
entre deux nucléotides à l'intérieur d'un acide
nucléique. Pour les deux variants alléliques HPA-a et HPA-b, elle
consiste en une amplification moléculaire de la région polymorphe
du gène codant. Le produit d'amplification est soumis à une
digestion enzymatique. Le produit de digestion est visualisé sur un gel
de polyacrylamide afin de déduire le génotype correspondant.
Selon le nombre de bandes générées, il y
aura la déduction de l'allèle existant puis le génotype du
sujet en question.
· La
PCR-ASA
On l'appelle aussi PCR-SSP ou PCR-ASP; c'est la technique de
choix de typage plaquettaire. Cette technique est bien adaptée à
l'urgence et aux petites séries (Metcalfe. P et Waters. A. H. 1993;
Skogen. B et al., 1994; Kjaer et al., 2002). La PCR-SSP est
utilisées pour la première fois pour le typage des
antigènes HLA (Olerup et Zetterquist., 1992; Hato. T. M. D et al.,
1997), pour le groupage érythrocytaire (Lee et al., 1995;
Cavanagh. G et al., 1997) et récemment pour le typage des
systèmes HPAs (Cavanagh. G et al., 1997). Elle a l'avantage
d'être sensible, rapide, (Kroll. H et al., 2001a) de pouvoir
faire l'amplification de tous les systèmes en même temps
(Klüter. H et al., 1996; Lyon et al., 2002). La simplicité
et l'exactitude de cette méthode chez les malades et les donneurs
recommandent son application répandue.
Cette technique est basée sur le principe que
l'amplification par la Taq Polymérase, elle n'est effective que lorsque
l'amorce est parfaitement complémentaire de la séquence de l'ADN
cible. Les couples d'amorces sont définis pour être
spécifiques d'un seul allèle. Dans des conditions très
précises de PCR, le couple d'amorces spécifiques permet
l'amplification des séquences cibles (résultat positif), tandis
que les couples d'amorces non complémentaires ne donnent pas
d'amplification (résultat négatif). Après la PCR, les
fragments d'ADN amplifiés sont séparés par
électrophorèse sur un gel d'agarose et visualisés par
coloration avec le BET sous lumière UV. L'interprétation des
résultats PCR-SSP est basée sur la présence ou l'absence
de fragment spécifique amplifié. C'est pourquoi un couple
d'amorces de contrôle interne est intégré dans chaque
réaction PCR. Ce couple d'amorces témoin amplifie une
région conservée du gène de l'hormone HGH humaine par
exemple qui est présente dans tous les échantillons d'ADN et
permet de vérifier l'intégrité de la réaction PCR.
· La
PCR-SSCP
La PCR-SSCP, est une technique très performante de
génotypage moléculaire des systèmes plaquettaires. Cette
technique est caractérisée par sa rapidité et sa
sensibilité, simplicité comme elle est applicable pour une large
série (Fujiwara. K et al., 1995). Par comparaison à la
PCR-SSP. Elle repose sur le principe suivant : lorsqu'un segment d'ADN
double brin est dénaturé par la chaleur, puis refroidi
brutalement, les brins restent séparés. Si on les soumet ensuite
à une électrophorèse non dénaturante dans un gel de
polyacrylamide faiblement réticulé, chaque brin se
renaturé sur lui-même, et prend une conformation spécifique
conditionnant sa migration électrophorétique. Une variation de
séquence aussi minime qu'une mutation ponctuelle peut se manifester par
cette différence de conformation.
· La
QRT-PCR
Historique
C'est en 1991 que Holland et al. Montrent que
l'activité exo nucléasique 5' 3' de l'ADN polymérase
de Thermus aquaticus peut être utilisée lors de la PCR pour le
clivage d'une sonde marquée, spécifique d'une séquence
cible. L'hybridation de la sonde crée un signal fluorescent
proportionnel aux produits de l'amplification.
En 1992, Higuchi et al. Mettent au point un système en
temps réel qui détecte les produits de PCR dès leur
accumulation. Ce système est base sur le marquage de l'ADN par le BET
qui a la propriété de s'intercaler dans la molécule
nouvellement synthétisée. La détection est possible
grâce a une camera CCD reliée a un logiciel informatique.
En 1993, Lee et al. Créent la sonde Taq man dont
l'hydrolyse par l'activité 5' 3' exonucléasique de la Taq
polymérase produit un signal émis vers une camera CCD
collectrice. Ce système apporte un énorme progrès par
rapport aux autres méthodes de PCR classique qui ne donnaient que des
valeurs approximatives de la quantité d'amplifiat à la fin de la
réaction
La technologie PCR Taq man
La PCR quantitative en temps réel exige seulement
les produits PCR standard et le matériel (Nauck. M. S et al., 1999;
Jone. D. C et al., 2003). Comme la PCR classique la PCR quantitative a le
même principe, sauf que cette deuxième méthode exige une
sonde interne en plus des deux amorces. La sonde est un oligonucléotide
complémentaire de l'un des brins de ;la séquence cible,
à laquelle elle s'hybride pendant la phase d'hybridation, aussi bien que
les amorces. Cette sonde est doublement marquée elle possède
à son extrémité 5' une molécule fluorescente
(reporter), et à son extrémité 3' un autre fluorochrome
(quencher). A l'état initial, aucune fluorescence n'est émise,
puisque la proximité du Quencher provoque un transfert d'énergie
de résonance du Reporter vers le Quencher, ce qui inhibe
l'émission du fluorochrome Reporter. Ensuite, si la séquence
cible d'ADN est présente, la Taq polymérase catalyse la
polymérisation à partir deux amorces hybridées sur les
deux brins d'ADN dénaturé.
Quand la Taq polymérase rencontre la sonde
hybridée elle la clive nucléotide après nucléotide
grâce à son activité 5' 3' exonucléasique.
La molécule Reporter est alors libérée, et sa fluorescence
devient détectable. Ainsi, à chaque cycle d'amplification, lors
de l'étape d'extension, une molécule de Repoter est
libérée pour chaque molécule d'ADN amplifiée. La
fluorescence est enregistrée en temps réel, individuellement pour
chaque tube réactionnel grâce à un réseau de fibres
optiques (faisceau laser) qui acheminent le signal émis vers une
caméra collectrice. Elle est normalisée par rapport à un
fluorochrome présent en quantité constante dans la
réaction.  Rn représente l'intensité de fluorescence émise. Rn représente l'intensité de fluorescence émise.
 Rn = (Rn+) - (Rn-), Rn+ étant
l'intensité d'émission du Reporter/intensité
d'émission du Quencher, et Rn- le même rapport
mesuré avant la réaction PCR. Rn = (Rn+) - (Rn-), Rn+ étant
l'intensité d'émission du Reporter/intensité
d'émission du Quencher, et Rn- le même rapport
mesuré avant la réaction PCR.
Chaque échantillon est caractérisé par
son cycle seuil Ct, c'est-à-dire le délai nécessaire pour
que la fluorescence dépasse un seuil fixe correspondant au nombre des
cycles de PCR á partir desquels se différencie la valeur
d'intensité de la fluorescence du bruit de fond.
Il est mesuré pendant la phase exponentielle de la
réaction lorsque les réactifs sont présents en
quantité non limitante, d'où la précision et la
reproductibilité de ces mesures.
Il a été démontré que le Ct est
inversement proportionnel à la qualité initiale d'ADN
cible : plus le Ct est faible plus le nombre de copies d'ADN cible
présent avant l'amplification est important.
Les avantages de la technologie Taq man
La spécificité de la technique dépend
bien de la région étudiée et du choix des amorces et de la
sonde, mais elle est accrue puisque la fluorescence n'est émise que si
la sonde est hybridée à la cible puis clivée lors de
l'extension. Cette technique est également théoriquement
très sensible. On parvient en effet à mesurer un cycle seuil pour
un échantillon très faible dont l'amplification commence
seulement en fin de réaction, alors qu'il aurait été
indécelable par une technique de quantification en point final.
Outre la spécificité et la sensibilité de
la quantification, les avantages majeurs de la technologie Taq man sont la
précision et la reproductibilité de la mesure, ainsi qu'une
très large gamme de détection.
Cette précision est dûe au fait à la
quantification de la séquence cible qui est déterminée par
le cycle seuil Ct calculé pendant la phase exponentielle de la
réaction et non pendant la phase stationnaire, comme c'est le cas dans
la PCR semi-quantitative classique ou la quantification des produits de la PCR
se fait a la fin de la réaction. La PCR quantitative en temps
réel est basée sur la détermination du Ct à partir
duquel la fluorescence devient détectable. Il a été
démontré de façon formelle que c'est ce cycle seuil qui
est dépendant de la quantité ou qualité d'ADN initial. A
tous ces avantages s'ajoute la rapidité.
Les applications de la PCR quantitative en temps
réelle
Cette technique possède un très large champ
d'application puisque tout ARN (avec une étape préalable de
transcription inverse) ou ADN cible d'intérêt peut être
aisément quantifié.
La technologie Taq man est déjà utilisée
avec succès dans des domaines très variés de la biologie,
particulièrement en virologie pour la mesure de la charge virale d'un
grand nombre de virus tels que le virus des Hépatites B et C ; en
bactériologie, pour quantifier l'amplification génique dans les
cancers de sein ; ou pour l'étude du polymorphisme.
La PCR quantitative est par ailleurs utilisée dans
l'étude de l'expression des gènes en général. C'est
une méthode idéale pour le diagnostic de routine et la
prévention des conditions clinique associées (mismatching) aux
HPAs. Elle peut être aussi utilisée dans l'étude des
populations et pour la recherche d'autres effets potentiels de ces
polymorphismes ; par exemple, leurs rôles comme antigènes
d'histocompatibilité dans la transplantation (Kekomaki. S et al.,
2001; Jone. D. C et al., 2003), leurs effets sur le fonctionnement des
intègrines et leurs associations possibles avec les maladies
cardio-vasculaire (Weiss. E. J et al., 1996; Zotz. R.. B et al., 1998;
Ardissino et al., 1999; Kroll. H et al., 2000; Mikkelsson. J et al., 2001;
Jone. D. C et al., 2003). Récente, cette technique à grande
sensibilité et à forte exigence peut être optimisée
pour fournir la résolution maximale à faible coût
et peut
être mise à jour pour inclure d'autres allèles HPAs de
faibles fréquences, aussi bien aux allo-antigènes plaquettaires
génétiquement caractérisés tel que le
système Gov (Schuh. A. C et al., 2002).
· Les
techniques de séquençage automatique
Cette technique permet d'obtenir directement la
séquence des gènes et de mettre en évidence des mutations
ponctuelles. Elle est utilisée pour détecter des nouveaux
systèmes HPAs (Santoso. S. A et al., 2006) en cas d'obtention
des produits PCR anormales de point de vue tailles d'amplicons. Comme elle peut
être utilisé pour faire l'alignement des séquences obtenues
avec un BLASTN (NCBI) (http://www.ncbi.nlm.nih.gov/BLAST/), comparant
les séquences obtenues à toutes les séquences
nucléotidiques connues, déposées dans plusieurs banques de
données.
Il existe différentes méthodes de
séquençages, tel que la méthode de
séquençage enzymatique par les di-désoxynucléotides
développé à l'origine par Sanger et la méthode
chimique de Maxam et Gilbert.
Le séquençage repose sur le principe de
méthode de Sanger. Le marquage des fragments d'extension est
réalisé en 3' par l'incorporation de
di-déoxynucléotides triphosphates (ddNTPs) fluorescents. Son
principe est de réaliser, à partir d'un point fixe, des copies
incomplètes de la matrice d'ADN interrompues au hasard.
L'élongation de la chaîne est ainsi arrêtée à
cause de l'absence d'une extrémité 3' OH libre, ce qui aboutit a
l'obtention de fragments marqués de différentes tailles. On
utilise un mélange de 4 fluorophores correspondant chacun a une des
quatre bases.
Au cours de l'électrophorèse, la lecture se fait
en temps réel à travers une fenêtre de détection,
après extraction des fluorophores par un laser biochromatique. En effet,
la machine (séquenceur) intègre les données de migration
et les transforme en éléctrophorogrmmes sous forme de pic de
couleurs différentes et correspondant aux bases distinctes
![]()
OBJECTIFS
Dans la partie pratique de notre travail, nous nous
proposons :
1) D'étudier les bases moléculaires en
réalisant le génotype des systèmes plaquettaires HPA-1,
HPA-2, HPA-3, HPA-4 et HPA-5 sur un échantillon de la population
Tunisienne.
2) D'estimer les fréquences géniques,
génotypiques et phénotypiques des systèmes plaquettaires
HPA-1, HPA-2, HPA-3, HPA-4 et HPA-5 sur cet échantillon.
3) D'étudier l'équilibre de la population
tunisienne pour les loci des cinq premiers systèmes HPAs.
4) D'estimer les fréquences des associations
haplotypiques.
5) D'estimer le risque d'allo-immunisation chez notre
population.
6) De comparer les résultats trouvés avec ceux
rapportés dans la population tunisienne et d'autres populations.
I. MATÉRIEL
Notre étude a porté sur un échantillon de
111 donneurs de sang Tunisiens, pour le système HPA-1, 105 pour le
système HPA-2, 104 pour le système HPA-3, 102 pour le
système HPA-4 et de 100 donneurs de sang pour le système HPA-5.
Les échantillons sanguins choisis au hasard sont constitués de
donneurs de sang volontaires au (CNTS) de Tunis. Ces donneurs sont sans lien de
parenté, ne souffrant d'aucune pathologie apparente. Ils sont des deux
sexes, sont de différents âges et pourraient appartenir à
de différentes générations (l'age d'une
génération est de 25 ans).
II. MÉTHODES
Les polymorphismes des gènes des HPA-1, - 2, -3, -4 et
-5 résultant d'un SNP au niveau d'un brin codant de l'ADN, ont
été déterminés par la technique PCR-SSP.
II-1.
Prélèvement du sang
5 ml (1ml de sang renferme 30 à 50 ug d'ADN) dé
sang périphérique ont été prélevés
sur un anticoagulant EDTA, chez chaque sujet. Après
déplasmatisation, le culot globulaire a servi pour extraire l'ADN
génomique.
II-2. Extraction de L'ADN génomique
total
II-2.1. Principe :
L'ADN utilisé comme matrice d'amplification pour les
réactions PCR a été extrait par la technique du relargage
des protéines à force ionique élevée : la
technique de salting-Out (Technique au chlorure de calcium saturé)
(Miller. S. A et al., 1988). Le sang doit être initialement et
vigoureusement mélangé à une solution hypotonique pour
faire éclater les globules rouges par une solution hypotonique
(SLR : tris-HCl 10 Mm pH 7,5 ; MgCl2 5 Mm ; NaCl 10
Mm). Le lysat est centrifugé et, après élimination du
surnageant, le culot cellulaire contenant les leucocytes est traité par
une solution de lyse de GB (SLB) et une solution de protéinase
très active, la protéinase K qui digère les
protéines cellulaires. Ces dernières seront par la suite
relarguées par l'intermédiaire d'une force ionique du NaCl (6 M).
En fin, la précipitation de l'ADN génomique est effectuée
en utilisant une solution d'éthanol absolu à froid (-20 °C)
(Delpech. M et al., 1999; Raisonnier. A et al., 2002).
II-2. 2. Protocole expémental
1- Lyse des globules rouges
Après déplasmatisation de l'échantillon
sanguin, on complète les crachoirs à 50 ml avec la (SLR) en les
agitant avec le vortex afin de faciliter la lyse des globules rouges. On les
centrifuge 10 mn à 2600 trs/mn à température ambiante. On
élimine avec précaution, le surnageant. On répète
l'opération précédente 2 fois. S'il y a des
hématies qui persistent dans le culot, on effectue un lavage
supplémentaire jusqu à avoir un culot de blanc blanchâtre
dépourvu des globules rouges (sinon on met le culot au
congélateur pendant au moins 2 h). On élimine totalement le
surnageant de SLR.
2- Lyse des leucocytes
Au culot des blancs, on ajoute 600 ul de (SLB : tris-HCl
10mM pH 7,5 ; EDTA-Na2 10 mM ; NaCl 50 mM ; SDS 0,2%)
pour la micro méthode afin de solubiliser les membranes des leucocytes
et de la protéinase K à raison de 20 mg/ml. La suspension
obtenue, fortement agitée, et incubée une nuit à 42°C ou 3
heurs à 56°C sous agitation. Après refroidissement à
température ambiante, on ajoute à l'extrait 200 ul du NaCl (6
M).
3- Précipitation des protéines
La suspension obtenue est vortexée vigoureusement
jusqu'à avoir un aspect laiteux. Cette dernière est par la suite
centrifugée 30 mn à 15000 trs/mn afin de précipiter les
protéines. On obtient ainsi une phase inférieure organique
contenant les débris (protéines, lipides...), une phase
supérieure aqueuse contenant l'ADN et l'interface contient en
majorité des protéines et peu l'ADN. Le surnageant, contenant
l'ADN dispersé, est récupéré dans un tube de 5
ml.
4- Précipitation de
l'ADN
On ajoute un volume égal d'éthanol à
haute force ionique (95%) au surnageant récupéré
précédemment. On laisse l'ADN se précipiter par agitation
douce en retournant délicatement le tube jusqu'à ce que les
filaments d'ADN forment une méduse; on récupère l'ADN dans
un tube Eppendorf (0,5 ml) ; la méduse d'ADN ainsi condensée
est lavée 3 fois à l'éthanol 70% afin d'éliminer
les traces de sels : à chaque lavage, ajouter 1 ml l'éthanol
70% afin d'éliminer les traces de sel et elle est séchée
dans un lyophilisateur pendant 30 mn.
5- Dissolution de l'ADN
L'ADN séché est solubilise dans un volume d'eau
distillée qui sera choisi en fonction de la taille de la méduse.
Cette solubilisation nécessite une agitation continue pendant une nuit a
42 °C. Après dissolution complète, la DO de la solution d'ADN
obtenu est mesurée à 260 nm et à 280 nm afin de
déterminer la concentration et la pureté de l'ADN extrait. L'ADN
dissout peut être conservé à +4°C pendant quelques jours ou
à - 20°C pour une conservation à plus long terme. D'ailleurs,
l'ADN pure se conserve à - 20 °C, plus de 10 ans.
6- Estimation de la concentration de l'ADN
La méthode de spectrophotométrie est
utilisée pour une quantification précise de l'ADN. Ainsi, la
concentration en ADN est déterminée par mesure de la DO au
spectrophotomètre à la longueur d'onde de 260 nm pour des
solutions diluées au 1/50 ou 1/100, sachant que l'ADN a un spectre
d'absorption en UV maximum à 260 nm. Ce spectre d'absorption est
proportionnel à la concentration de l'ADN (une unité de
densité optique (DO) à 260 nm correspond à 50ug d'ADN
double brin ou à 25ug ADN ou d'ARN simple brin par ul) en tenant compte
de la dilution réalisée.
L'estimation de la concentration de l'ADN est
déterminée selon la formule suivante :
1 unité de DO
50 ng d'ADN
N unités de DO
50 × N ng d'ADN
|
[ADN] = DO 260 × facteur de dilution ×50 en
ng/ul
|
7- Estimation du degré de pureté
d'ADN
La mesure de la DO à 280 nm sert à
détecter les éventuels contaminants. La pureté de
l'échantillon est testée par comparaison des DO à 260 nm
et à 280 nm et cela pour détecter toute contamination
protéique. Ainsi les protéines absorbent à 280 tandis que
l'ADN absorbe à 260nm. Une préparation d'ADN est dite pure si
elle présente un rapport de DO 260/DO 280 entre 1.4 et 1.7.
8- Préparation des dilutions d'ADN
C'est une étape pré-PCR. On fait diluer un
volume de la solution mère d'ADN dans de l'eau bidistillée afin
d'obtenir une concentration finale de 100 ng/ul. Cette concentration est
optimale pour les réactions d'amplification moléculaire.
Dans le but de faire le
génotypage des cinq systèmes HPAs, six couples d'amorces (sens et
anti-sens) ont été utilisés : un couple d'amorces
pour amplifier une région du gène HGH, les autres pour amplifier
les régions spécifiques sont complémentaires à la
séquence à amplifier. Toutes ces amorces sont de type
séquences spécifiques (SSP), elles sont capables de
détecter les versions alléliques des différents
gènes codants. Dans notre étude, la Taq a été
diluée dans le tampon 1X a partir d'une solution mère dont la
concentration est de 5 U/ ul.
II-3. Mise au point de la
réaction PCR-SSP
Dans le but de faire le génotypage des cinq
systèmes HPAs, six couples d'amorces sens et anti-sens) ont
été utilisés : un couple d'amorces pour amplifier une
région du gène HGH. Les autres pour amplifier les régions
spécifiques sont complémentaires à la séquence
à amplifiés. Toutes ces amorces sont de types séquences
spécifiques (SSP), elles sont capables de détecter les versions
alléliques des différents gènes codants.
Dans notre étude, la Taq a été
diluée dans le tampon 1X a partir d'une solution mère dont la
concentration est de 5U/ul.
Nous avons utilisé une méthode basée sur
la réaction de (PCR-SSP) comme décrite par Cavanagh et al.
(Metcafle et al., 1993; Cavanagh. G et al., 1997),
avec quelques modifications. La mise au point de la PCR a
nécessité un changement chaque fois d'un paramètre, afin
d'optimiser et choisir le protocole standard pour chaque technique. Plusieurs
paramètres peuvent constituer des éléments critiques pour
le succès de la technique d'amplification. Une gamme de MgCl2
doit être testée afin de déterminer la concentration
optimale du magnésium permettant une bonne amplification. En effet, une
forte concentration en ions Mg++ peut engendrer des
polymérisations aspécifiques, alors q'une faible quantité
diminue le taux d'amplification. Les autres composants de la PCR peuvent aussi
influencer le déroulement de la polymérisation. Le pH doit rester
constant et correspondre au pH optimal de l'enzyme utilisée. De
même, la température de dénaturation de l'ADN matrice, de
l'hybridation et de l'élongation peuvent être des facteurs
limitants. Le chois des séquences oligonucléotidiques servant
comme amorces (% en GC) peut constituer à son tour un paramètre
essentiel intervenant surtout au moment de l'hybridation conduisant a la
formation de structures secondaires.
Cependant, nous avons rencontré des difficultés
pour l'amplification de tous les allèles, à cause de
l'amplification non spécifique car ces systèmes ont exigé
des conditions optimales pour leur amplification. Ces difficultés ont
été résolues par une légère modification de
la technique utilisée par Cavanagh et al. Nous avons donc essayé
d'améliorer la spécificité de la PCR-SSP en augmentant la
concentration du magnésium MgCl2 de 1,5 mM à 2,5 mM
dans le mélange de la réaction finale.
Chaque système bi-allélique est analysé
à l'aide de deux couples d'amorces spécifiques auxquels est
ajouté un couple d'amorces monomorphiques servant de témoin
d'amplification. Des ADN de référence portant les
différents allèles testés auparavant sont
nécessaires et ont été obtenus à partir d'individus
typés par PCR et avec d'autres méthodes. Au cours de la mise au
point, nous avons choisi des températures d'hybridation Th
spécifiques pour l'amplification des systèmes HPA-1 et HPA-3
(55°C), HPA-2 et HPA-5 (56°C) et (63°C) pour le système HPA-4. Les Tm
des amorces des allèles HPA-1a /HPA-1b (62 °C /64 °C) et
HPA-3a /HPA-3b (64 °C /66 °C) sont supérieures aux
températures d'hybridation. Quant au Th du système HPA-4 (63°C)
et au Th HPA-5 (56°C), ils sont supérieures au Tm (62°C pour HPA-4 et
42°C pour HPA-5. Concernant le système HPA-2, le Th (56 °C) est
inférieur au Tm (64°C). Nous savons que les réactions d'extension
d'amorce sont faites à des températures proches, mêmes
supérieures aux Tm des amorces qui en conséquence pourraient ne
pas avoir le temps de s'hybrider correctement et ainsi provoquer le
décrochage de l'enzyme après son arrivée.
Néanmoins, nous avons trouvé de bons résultats avec des Th
< Tm pour les trois systèmes HPA-1, HPA-2 et HPA-3. Nous avons choisi
des températures d'hybridation spécifiques identiques pour les
systèmes HPA-1 et HPA-3 (55°C) et pour les systèmes HPA-2 et
HPA-5 (56°C). Ainsi, nous pouvons amplifier les deux systèmes HPA-1 et
HPA-3 ensemble et les deux systèmes HPA-2 et HPA-5 ensemble avec le
même programme PCR.
Nous avons utilisé les mêmes concentrations du
mix pour tous les systèmes sauf pour les primers spécifiques. En
fait, nous avons utilisé la concentration 0,35 mM pour les trois
systèmes HPA-1, HPA-2, et HPA-4, tandis qu'on a augmenté cette
concentration à 0,5 mM pour les systèmes HPA-3 et HPA-5.
II-3-4- Préparation des solutions
stocks
Préparation des dNTPs : Pour
préparer le stock des dNTPs, on mélange volume à volume
les quatre solutions mères (100 mM) contenant chacune un type de base
(dATP, dCTP, dTTP, dGTP), ce qui donne une concentration de 25mM correspondant
à chaque
dNucléotides. La solution subira une dilution pour
obtenir la concentration adéquate de 2Mm (20ul + 920ul d'eau
bidistillée). Ce mélange préparé dans un tube
eppendorfs (1,5ml) est conservé à -20°C pour des utilisations
ultérieures.
Préparation de la solution de
MgCl2 : On mélange volume à volume MgCl2
à une concentration initiale de 25 mM et l'eau distillée pour
obtenir une solution de MgCl2 dont la concentration est de 12,5 mM.
Préparation des solutions d'amorces :
les amorces oligonucléotidiques nous ont
été fournies sous forme solubilisée. Les solutions
mères d'amorces doivent être diluées dans des tubes
eppendorfs (1,5ml) pour avoir une concentration de 2 ; 3,5 ou 5 uM suivant
le type d'amorce puis conservées à -20°C pour des utilisations
ultérieures 10X concentrées. IL faut faire attention à
toute contamination possible.
Pour chaque réaction PCR, on utilise une paire
d'amorces spécifiques à chaque système HPA (sens et
anti-sens). Ils doivent être apportés en excès afin de
favoriser leurs liaisons aux brins d'ADN dénaturés plutôt
que celles des brins d'ADN entre eux. Un deuxième couple d'amorces
désignées à l'amplification d'une région du
gène HGH, gène monomorphe codant l'hormone de croissance chez
l'homme, a été utilisé comme contrôle de
qualité (contrôle interne) pour les différentes
étapes d'extension d'ADN pour vérifier l'efficacité de la
réaction d'amplification témoignant le succès de la
réaction PCR.
Les amorces utilisées sont comparées aux
séquences connues dans la base de données NCBI selon les
étapes suivantes :
1- Aller sur le site NCBI.
http: www.ncbi.nlm.nih.gov/
2- Retrouver la séquence du gène.
3- Récupérer la séquence du gène
sous sa forma fasta.
4- ouvrir une page blast 2 séquences.
http://www.ncbi.nlm.nih.gov/BLAST/bl2seq/wblast2.cgi.
On a deux boites on colle la séquence de l'amorce
(forma fasta) dans l'une et celle du gène dans l'autre et on clique sur
le bouton Align.
Ces amorces utilisées pour chaque région
amplifiée sont les suivantes :
Tableau V :
Les différents couples d'amorces utilisés pour l'amplification
spécifique des systèmes HPA -1 à 5
(Cavanagh. G et al., 1997).
|
HPAs
|
spécificité
|
Amorces : sens 5' - 3'
|
Concentration finale (ìM/ìl)
|
Taille pb
|
Tm°C
|
|
HPA-1
|
HPA-1a
HPA-1b
HPA-As
|
5'TCACAGCGAGGTGAGGCCA 3'
5'TCACAGCGAGGTGAGGCCG 3'
5'GGAGGTAGAGAGTCGCCATAG 3'
|
0,35
|
90
|
62
64
50
|
|
HPA-2
|
HPA-2a
HPA-2b
HPA-As
|
5'GCCCCCAGGGCTCCTGAC 3'
5'GCCCCCAGGGCTCCTGAT 3'
5'TCAGCATTGTCCTGCAGCCA 3'
|
0,35
|
258
|
64
62
62
|
|
HPA-3
|
HPA-3a
HPA-3b
HPA-As
|
5'TGGACTGGGGGCTGCCCAT 3'
5'TGGACTGGGGGCTGCCCAG 3'
5'TCCATGTTCACTTGAAGTGCT 3'
|
0,5
|
267
|
64
66
50
|
|
H PA-4
|
HPA-4a
HPA-4b
HPA-As
|
5'GCTGGCCACCCAGATGCG 3'
5'GCTGGCCACCCAGATGCA 3'
5'CAGGGGTTTTCGAGGGCCT 3'
|
0,35
|
120
|
62
62
62
|
|
HPA-5
|
HPA-5a
HPA-5b
HPA-As
|
5'AGTCTACCTGTTTACTATCAAAG 3'
5'AGTCTACCTGTTTACTATCAAAA 3'
5'CTCTCATGGAAAATGGCAGTA 3'
|
0,5
|
246
|
42
42
50
|
|
Control positive HGH
|
HGH-s
HGH-As
|
5'GCCTTCCCAACCATTCCCTTA 3'
5'TCACGGATTTCTGTTGTGTTTC 3'
|
0,2
|
429
|
57
52
|
S: sens
As : antisens
v Préparation du master
Mix
Après la mise au point on prépare, dans un tube
eppendorf, un master mix (tampon, dNTP, les primers, MgCl2) pour le
typage de 100 échantillons, pour des raisons de bonne conservation du
produit, un aliquot est préparé selon les besoins. Ces tubes
doivent être conservés au congélateur dans le but
d'éviter la congélation et la décongélation des
produits.
v Préparation du Super-Mix
A chaque fois qu'on fait le typage, on laisse le master mix
à température ambiante jusqu'à la
décongélation puis on ajoute la Taq diluée dans le tampon
1X pour obtenir un super mix qui doit être réparti dans des tubes
PCR à raison de 8ul/tube.
De grandes précautions doivent être prises vis
à vis de la Taq polymérase qui demande à être
conservée à -20°C sans écart de température afin
d'éviter sa dégradation.
v Préparation d'un milieu
réactionnel PCR-SSP
En dernière étape, on ajoute à chaque
tube PCR 2ul d'ADN à 100ng/ul des échantillons à tester.
Le tout est mélangé doucement pour ne pas altérer
l'enzyme. Le volume final est de 10 ul contenant au moins 200 ng d'ADN
génomique. Les facteurs limitants l'amplification sont
l'épuisement des amorces et celui des nucléotides. La
spécificité d'amplification d'une séquence donnée
sera directement liée a celle des amorces choisies. Pour chaque PCR, un
tube sans ADN (à la place de l'ADN, on met de l'eau distillée),
qui sert comme témoin négatif, permet de s'assurer de l'absence
de contamination. Il faut bien choisir de «bons» témoins
positifs et négatifs. Puis, on amplifie l'ADN.
Le mélange réactionnel est composé comme
suit :
Tableau VI :
Composition d'un milieu réactionnel
|
Ingrédients
|
Concentration initiale
|
Concentration finale
|
Vo (ul) ×N
|
|
Tampon
|
5X
|
1X
|
2
|
|
MgCl2
|
12.5 mM
|
2.5 mM
|
0.8
|
|
dNTPs
|
2.5 mM
|
200 uM
|
0.2
|
|
Primers spécifiques sens
|
3.5* ou 5** uM
|
0.35* ou 0.5** uM
|
1
|
|
Primers spécifiques anti-sens
|
3.5* ou 5** uM
|
0.35* ou 0.5** uM
|
1
|
|
Primers
HGH-sens
|
2 uM
|
0.2 uM
|
1
|
|
Primers
HGH-antisens
|
2 uM
|
0.2 uM
|
1
|
|
Taq
|
3.5 U /ul
|
0.35 U
|
1
|
|
ADN
|
100 ng
|
200 ng
|
2
|
|
Volume total
|
|
|
10
|
*HPA-1, HPA-2 et HPA-4
ou
**HPA-3 et HPA-5.
II-3-5- Amplification des cinq premiers
systèmes plaquettaires
Nous avons adopté la technique PCR-SSP : c'est la
technique la plus utilisée pour l'étude du polymorphisme
allélique des gènes des différents systèmes HPAs.
Elle a l'avantage d'être une méthode en une seule étape
où la spécificité de primer simplifie la détection
du produit amplifié. C'est une technique simple, rapide et
précise (Skogen B et al. 1994). Cette méthode exige des
conditions particulières d'amplification pour les différents
systèmes.
II-3-6 - Programmes PCR
Chaque série d'amplification comporte
différentes étapes selon un programme bien
déterminé. Tous les éléments nécessaires
à la réaction sont regroupés dans un tube PCR qui sera
soumis aux différentes températures correspondant à chaque
étape. Ces cycles de température sont réalisés
automatiquement dans un thérmocycleur. L'amplification est
effectuée en 30 cycles qui sont précédés par une
étape de dénaturation de 5 mn à 95°C qui rend la
dénaturation initiale efficace. Elle consiste en une minute de
dénaturation à 95°C, une minute d'hybridation à une
température spécifique pour chaque système et une minute
d'extension à 72°C. Les Thyb (température d'hybridation) mises au
point pour chaque couple utilisé pendant la réaction
d'amplification sont mentionnées dans le (Tableau VIII). A la fin, ces
cycles sont suivis d'une étape d'élongation de 7 min à
72°C afin que la polymérase termine la synthèse des brins qu'elle
n'a pas eu le temps de finir pendant les cycles précédents. Cette
étape fixe donc la fin de l'élongation et achève ainsi la
réaction PCR. Ces cycles sont réalisés automatiquement
dans un thérmocycleur Perkin Elmer 2700 programmé pour effectuer
les cycles suivants. La température d'hybridation «Thyb »
Tableau
VII : Programme PCR utilisé :
|
Nbr de cycles
|
Température
|
Etape
|
Temps
|
|
1
|
95°C
|
Dénaturation
|
5
|
|
30
|
94°C
|
Dénaturation
|
1 min
|
|
HPA-1
|
55°C
|
Hybridation
|
1 min
|
|
HPA-2
|
56°C
|
|
HPA-3
|
55°C
|
|
HPA-4
|
63°C
|
|
HPA-5
|
56°C
|
|
72°C
|
Elongation
|
1 min
|
|
1
|
72°C
|
Elongation finale
|
7 min
|
|
+8
|
4 °C
|
Arrêt
|
+8
|
III. Contrôle
de l'amplification
III-1
Électrophorèse sur gel d'agarose (Delpech. M et al.
1999).
L'ADN, après extraction cellulaire et amplification par
PCR, peut être analysé sur gel d'électrophorèse.
a) Principe
Le contrôle des produits PCR se base sur le principe de
la séparation et l'identification des fragments d'ADN amplifié
des éventuels fragments non spécifiques en fonction de leur
taille et leur purification des restes des réactifs d'amplification dans
un champ électrique homogène. Les ADN sont des
macromolécules polyanioniques uniformément chargées
négativement, la charge relative de l'ADN étant constante. De ce
fait, l'ADN peut migrer dans un champ électrique de la cathode (-) vers
l'anode (+) dans un tampon de migration 1X, soit dans du TAE ou dans du TBE.
Les échantillons sont mélangés à
un colorant, le bleu de bromophénol de masse moléculaire faible
par rapport au fragment d'ADN pour marquer le fond de migration et de les
suivre au cours de l'électrophorèse. Pour nous, le tampon est
coloré par un colorant vert le green. Il s'avere inutile d'utiliser un
autre colorant lors de l'électrophorèse.
On additionne aussi au gel le BET, réactif intercalant
qui se fixe entre les bases nucléiques à l'intérieur de la
double hélice. Cette molécule naturellement non fluorescente
présente une fluorescence orange lorsqu'elle est intercalée entre
les bases nucléiques et après excitation aux longueurs d'ondes
ultraviolettes.
Après migration, on place le gel au dessus d'un
éclairage ultraviolet qui servira à exciter les molécules
de BET* fixées aux molécules d'ADN, lesquelles vont
émettre une lumière rougeâtre visible et photographiable,
permettant de visualiser la position des fragments d'ADN sur le gel.
b) Préparation du gel de
migration
Le contrôle a été
réalisé par électrophorèse horizontale sur un gel
d'agarose à 1,5%, colorés par le BET on ajoute 0,45 g d'agarose
à 30 ml de tampon TBE 1X (Tris-Boric-EDTA). Le mélange est
porté à ébullition sous agitation jusqu'à
dissolution totale de l'agarose et l'obtention d'un liquide transparent.
Après refroidissement, on additionne 10 ul de BET*. On fait couler 30 ml
de gel doucement à l'horizontale dans un bac à
électrophorèse muni d'un ou de plus de peignes de 8 dents selon
le nombre d'échantillons déposés, dont les empreintes
formeront des puits au sein du gel servant au dépôt des
échantillons à tester aussi bien pour le marqueur de taille et le
témoin négatif. On laisse le gel se refroidir jusqu'à ce
qu'il durcisse. Le gel polymérisé est recouvert de 10 ml de TBE
1X et le peigne est retiré.
III-2
Électrophorèse et visualisation des produits
d'amplification
On procède, enfin, à l'étape de
révélation des produits amplifiés. La
spécificité des bandes observées sur gel est
confirmée à l'aide des marqueurs de tailles M. Les produits PCR
doivent être visualisés afin de vérifier
l'efficacité de l'amplification et de déterminer le
génotype des échantillons étudiés. Pour cela, les
10 ul de chaque produit d'amplification des échantillons sont
déposés lentement dans les puits du côté de la
cathode selon l'ordre des échantillons dans les tubes PCR. Un marqueur
de taille de poids moléculaire connu est déposé aussi en
parallèle avec les échantillons préalablement
mélangés avec du bleu de bromophénol qui permet de suivre
leur migration au cours de l'électrophorèse pour estimer la
taille des amplicons.
Le gel de migration est recouvert de 20 ml de tampon basique
TBE 1X, de telle manière à assurer un contact entre l'anode et la
cathode. Après diffusion dans le gel, l'électrophorèse est
démarrée à 120V, 50 mA pendant 10 à 20 minutes.
Cette durée dépend toutefois de la taille des fragments
amplifiés.
A la fin de l'électrophorèse, le gel est
visualisé, puis photographié au dessus de l'éclairage UV
qui servira à exciter les molécules de BET fixées aux
molécules d'ADN, lesquelles vont mettre une lumière
rougeâtre visible, mais surtout photographiable pour vérifier la
visualisation puisque le BET permet de visualiser sous UV les fragments d'ADN
ainsi que la position des fragments du marqueur de taille qui doit
apparaître sous forme de bandes fluorescentes. Cette analyse permet de
vérifier la spécificité de la séquence
amplifiée. Pour cela, on doit s'assurer de la taille de la bande
correspondante à cette séquence, ce qui permet de
déterminer des génotypes et de vérifier que le produit PCR
du témoin négatif ne montre pas de bande amplifiée pouvant
correspondre à une contamination par un ADN étranger. Les
réactions ne sont considérées positives que lorsque le
contrôle positif (contrôle interne) est amplifié. Selon la
présence ou l'absence de la bande amplifiée, on peut
déduire le génotype de chaque individu. Par exemple, pour le
génotype a/a, on observe une bande spécifique qui correspond
à l'allèle a et l'absence de la bande correspondant aux produits
amplifies spécifique à l'allèle b. Pour tous les
échantillons, on observe la présence de bandes qui correspondent
au HGH (contrôle interne).
IV. ANALYSES STATISTIQUES
Cette étude statistique a été
menée dans le but de déterminer la distribution des
fréquences alléliques, génotypiques, phénotypiques,
les fréquences haplotypiques et le déséquilibre de
liaison des cinq premiers systèmes plaquettaires dans
l'échantillon étudié. Elle nous permet d'étudier
l'équilibre de la population Tunisienne pour les locus étudies en
utilisant l'approche de H-W.
Les moyens statistiques que nous avons utilisés sont
les suivants : Les tests statistiques et le logiciel SAS.
A- Les tests
statistiques
1. Calcul des
fréquences géniques et génotypiques en cas
d'équilibre panmictique :
ü Les fréquences
géniques
Il s'agit de la proportion des différents
allèles d'un gène dans une population.
Soient les effectifs suivants :
n1 = nombre des sujets de génotype a/a
n2 = nombre des sujets de génotype a/b
n3 = nombre des sujets de génotype b/b.
N = Effectif de l'échantillon.
Chaque individu, étant diploïde, à 2 loci
pour chaque système plaquettaire, soit en tout 2N loci pour tout
l'échantillon. Donc, les allèles présents dans les loci
sont l'allèle a et l'allèle b.
Les individus aa ont deux allèles a, les individus ab
ont un seul allèle a et un seul allèle b, ceux qui ont le
génotype bb n'ont évidemment aucun allèle a. La
fréquence de l'allèle a c'est donc la proportion de
l'allèle a parmi tous les gènes de la population qui peuvent
occuper le locus (a, b). De même, q est la proportion des allèles
b. puisque le locus (a, b) est nécessairement occupé soit par a
soit par b et que ces éventualités sont mutuellement exclusives,
on a donc en cas d'équilibre : p + q = 1
Soit :
La fréquence de l'allèle a : F (a) = p
La fréquence de l'allèle b : F (b) = q
Les fréquences des deux allèles a et b sont
obtenues par les formules suivantes :
F (a) = (2n1 + n2)/2N = n1/N
+ n2/2N
F (b) = (2n2 + n1)/2N = n2/N +
n1/2N
Donc il s'agit de calculer le nombre de sujets
présentant un allèle donné qu'on divise par le nombre
total des allèles dans chaque locus : chaque individu
possède deux allèles dans un locus donné.
ü Fréquences génotypiques
Soit l'échantillon étudié de N individus
et on a 3 génotypes (aa) n1, (ab) n2 et (bb)
n3
L'effectif total N = n1 + n2 +
n3
La fréquence des génotypes (a/a) =
n1/N
La fréquence des génotypes (a/b) =
n2/N
La fréquence des génotypes (b/b) =
n3/N
Par la suite, on a : n1/N+ n2/N +
n3/N= 1
Les
fréquences génotypiques observées sont obtenues par un
comptage direct. On calcule le nombre de sujets présentant un
phénotype donné qu'on divise par le nombre total de sujets
étudiés. Les fréquences
génotypiques calculées : sont estimées à
partir des fréquences alléliques en se basant sur la loi de
H-W :
2. Intervalle de confiance (IC) :
Il est appelé aussi intervalle au risque d'erreur
á. Cet intervalle a une probabilité de 1-á de contenir la
vraie valeur (inconnue) d'un paramètre donné. Pour un %, l'IC
(à 95%, au risque d'erreur 5%) est calculé de la manière
suivante :
IC: [pi, ps] = p #177; 1, 96 
pi : valeur inférieure de
l'IC
ps : valeur supérieure de
l'IC
Cet intervalle nous permet de dire que, pour un
échantillon de N individus, les résultats sont fiables à
#177; IC pour un résultat caractéristique à un % d'erreur
de 5%.
3.
Test de l'équilibre des locus et loi de H-W :
Une hypothèse génétique s'étend
d'ordinaire à plusieurs classes de génotypes. Pour l'estimer,
nous avons besoin d'un test qui nous permette d'associer une probabilité
aux écarts observés dans un échantillon par rapport aux
fréquences attendues et de décider si les écarts
relèvent ou non du seul hasard. Ce test doit également tenir
compte de la taille de l'échantillon et du nombre de variables
(degrés de liberté). Le test appelle du ÷2
satisfait à ces exigences.
ü Equilibre de H-W
(p +
q)2 = p2 + 2pq + q2 = 1
p + q = 1
ü Test chi-deux ( ÷2
) : La valeur x2 est calculée en
appliquant la formule
÷2 = ? i (Oi -
Ti)2
Ti
Avec :
Oi = valeur observée dans une classe.
Ti = valeur théorique dans une classe.
? i = Somme des valeurs ÷2 pour toutes les
classes d'indice i.
÷2 calculé < ÷2
théorique.
ü Le degré de
liberté
Nous avons : n-1
degrés de liberté (dl) pour fixer, au hasard, les valeurs de n
classes d'une expérience. Dans notre cas, on a 3 génotypes.
Ainsi, nous aurons : 3-1 =2 degrés de liberté.
Limitation de l'utilisation du
Test chi-deux : L'utilisation du Test chi-deux pour l'analyse des
résultats d'expérience génétique a deux limitations
importantes. (1) Les éléments du calcul
doivent être les effectifs de classe eux-mêmes et non les
pourcentages ou les proportions déduites de ces effectifs.
(2) On ne doit pas l'utiliser quand une des classes a un
effectif théorique inférieur a 5.
ü Correction pour les faibles
échantillons
Cette correction dite de Yates se
calcule ainsi :
÷2 (corrigé) = ? i (|Oi
- Ti| - 0,5)2
Ti
Cette correction n'entraîne habituellement qu'une
légère diminution de la valeur du ÷2, mais elle
peut jouer un rôle important au voisinage de la valeur critique.
B- Les
logiciels
1) Le SAS. On a utilisé ce logiciel
dans le but de comparer les fréquences alléliques des HPAs
étudiés à travers les différentes populations.
2) L'Arlequin. Pour calculer les
fréquences haplotypiques et le déséquilibre de
liaison :
![]()
Résultats
RESULTATS DE L'AMPLIFICATION PAR PCR DES ALLELES :
(GP3A*1/ GP3A*2), (GP1BA*1/GP1BA*2), (GP2B*1/GP2B*2), GP3A*1/GP3A*3 et GP1A*1/
GP1A*2
Les profils électrophorétiques des figures
représentent des exemples des génotypes HPA-1, -2, -3, -4, et -5
détectés sur gel d'agarose. Les produits amplifiés sont de
tailles distinctes : Les bandes les plus lourdes sont les bandes les plus
lentes et elles correspondent aux produits amplifiés du contrôle
positif HGH de taille (429 pb). Cette bande doit être présente
dans chaque échantillon étudié pour permettre de
vérifier l'intégrité de la réaction PCR. Concernant
les bandes spécifiques, la bande la plus légère correspond
au produit amplifié des amorces (90 pb) du système HPA-1 qui
migre le plus rapide. Les bandes spécifiques au HPA - 4 (120 pb),
ensuite par la bande spécifique HPA-5 (246 pb), puis HPA-2 (258 pb) et
enfin par la bande spécifique HPA-3 (267 pb).
Echantillon 1 Echantillon 2
Echantillon 3
100pb
Sens de migration
HGH 429 pb
Allèle spécifique
90 pb
a+
b+
a+
b-
a+
b-
M
T-
a+ b+
a+ b-
a+ b-
Génotypes
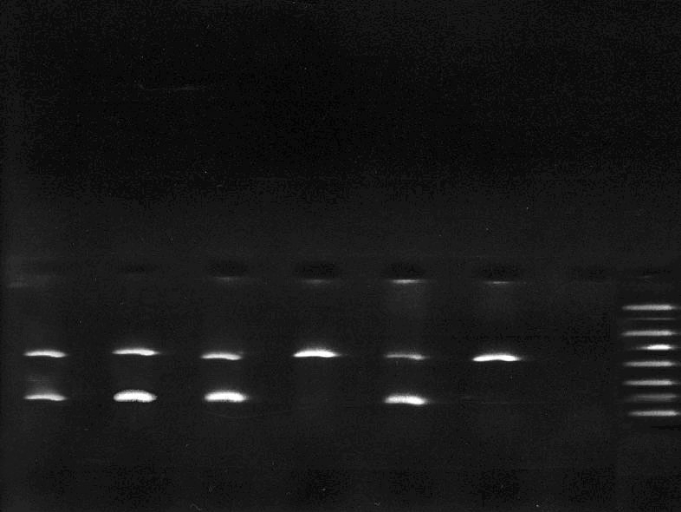
Figure 8: Profil
éléctrophorétique des amplifications du système
HPA-1 en gel d'agarose à 1.5%.
Les puits : M = marqueur de Taille, a = allèle
(a), allèle (b), T = témoin négatif (eau
distillée). La bonde supérieure présente dans tous les
puits correspondants aux produits amplifiés des amorces de
contrôle interne HGH. Les bondes inférieures correspondantes aux
produits amplifiés des amorces spécifiques des allèles
a/b
Sens de migration
a+
b-
a+
b-
M
HGH 429 pb
Allèles spécifiques
a+ b-
a+ b-
a+ b-
a+
b-
a+
b-
a+ b-
1 2 3
4 5 6 7 8 9
100pb
246pb
120pb
267pb
258pb
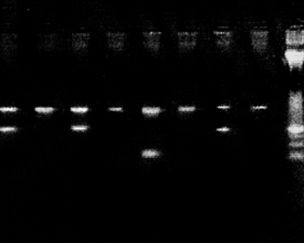
Figure 9: Profil
éléctrophorétique des amplifications des systèmes
HPA-2, HPA-3, HPA-4 et HPA-5 en gel d'agarose à 1.5%.
L'électrophorèse sur gel d'agarose montrant les
4 systèmes HPAs. Les puits 1 et 2; 3 et 4; 5 et 6; 7 et 8 sont pour les
systèmes HPA-2 (258 pb), -3 (267 pb), -4 (120 pb), et-5 (246 pb),
respectivement. Le puit 9 contient le marqueur de taille Chaque couple montre
des génotypes identiques HPA-2 (a+b-), HPA-3(a+b-), HPA-4 (a+b-) et HPA5
(a+b-). Noter que la bande migrant plus faiblement représente le produit
d'amplification de témoin d'amorces positif HGH (429pb).
Analyse statistique de la répartition des
fréquences géniques, et phénotypiques des cinq
systèmes plaquettaires étudiés
Chaque système est gouverné par un couple
d'allèles codominants (a, b). L'allèle a est l'allèle
fréquent, il est dit sauvage et il correspond à la
synthèse de l'antigène HPA-a. L'allèle b est
l'allèle rare, il est dit mutant et il correspond à la
synthèse de l'antigène HPA-b. Ces systèmes sont
déterminés par des gènes autosomaux.
Les résultats de typage obtenus chez 111 pour le
système HPA-1, 105 pour le système HPA-2, 104 pour le
système HPA-3, 102 pour le système HPA-4 et 100 donneurs pour le
système HPA-5, sont donnés dans le tableau VIII:
Tableau VIII :
Résultats du génotypage des systèmes HPAs
étudiés
|
Système
|
HPA-1
|
HPA-2
|
HPA-3
|
HPA-4
|
HPA-5
|
|
a/a
|
75
|
66
|
54
|
102
|
78
|
|
a/b
|
33
|
36
|
39
|
0
|
19
|
|
b/b
|
3
|
3
|
11
|
0
|
3
|
|
Total
|
111
|
105
|
104
|
102
|
100
|
1. Structure génétique de
l'échantillon étudiée de la population
Tunisienne
pour les systèmes HPA
étudiés
ü Fréquences
alléliques
Tableau IX :
Fréquences alléliques des systèmes HPA-1, -2, -3, -4 et
5.
|
Système
|
HPA-1
|
HPA-2
|
HPA-3
|
HPA-4
|
HPA-5
|
|
p
|
0,824
|
0,800
|
0,707
|
1
|
0,875
|
|
q
|
0,176
|
0,200
|
0,293
|
0
|
0,125
|
|
p + q
|
1
|
1
|
1
|
1
|
1
|
|
Système
|
Allèle
|
Nombre
|
Fréquence % (95% IC)
|
|
HPA-1
|
HPA-1a
HPA-1-b
|
183
39
|
82,432
17,568 (#177; 5)
|
|
HPA-2
|
HPA-2 a
HPA-2 b
|
168
42
|
80
20 (#177; 5,410)
|
|
HPA-3
|
HPA-3 a
HPA-3b
|
147
61
|
70,673
29.327 (#177; 5,181)
|
|
HPA-4
|
HPA-4a
HPA-4b
|
204
0
|
100 NA
0 NA
|
|
HPA-5
|
HPA-5 a
HPA-5b
|
175
25
|
87,5
12,5 (#177; 4,58)
|
On remarque d'après ces données une
distribution irrégulière des différentes formes
alléliques. Les fréquences des allèles HPA-a sont plus
élevées, pour tous les systèmes, que celles des
allèles HPA-b. Les allèles HPA-1a, HPA-2a, HPA-3a, HPA-4a et
HPA-5a ont les valeurs respectives de 0,824; 0,8; 0,707; 1 et 0,875. Les
allèles HPA-1b, HPA-2b, HPA-3 b, HPA-4 b et HPA-5 b ont les valeurs
respectives de 0,176; 0,2; 0,293; 0 et 0,125. D'après ces
données, on remarque l'importance des fréquences des
allèles HPA-b pour les quatre systèmes HPA-1, 2, 3 et 5.
D'ailleurs, la fréquence de l'allèle HPA-3b (0,293) est plus
élevée que celles de tous les autres allèles, elle est
suivie par les allèles HPA-2b (0,2), HPA-1b (0,176) et HPA-5b (0,125).
0,824
0,176
0, 8
0, 2
0,707
0,293
1
0
0,875
0,125
0
0,2
0,4
0,6
0,8
1
Fréquences alléliques
HPA-1
HPA-2
HPA-3
HPA-4
HPA-5
p
q
![]()
Figure 10:
Représentation graphique de la distribution alléliques des
systèmes HPA-1, -2,- 3,-4 et-5 dans
l'échantillon étudiée de la population
tunisienne
ü Fréquences
génotypiques
Les génotypes HPAs les plus communs en Tunisie sont les
homozygotes (a/a) : HPA-1 (a+b-), HPA-2
(a+b-), HPA-3 (a+b-), HPA-4
(a+b-) et HPA-5 (a+b-) (Voir
tableau ci-dessous). On remarque que le système HPA-4 est
homogène, tous les individus sont des homozygotes du génotype
HPA-4a (aa). Les homozygotes (b/b) sont faibles en comparaison avec les
homozygotes (a/a). On met en évidence une fréquence
élevée des génotypes homozygotes HPA-3b/b (10,576%) suivi
par le génotype b/b du système HPA-5 (de 3 %), le génotype
b/b du système HPA-2 (de 2,857 %), et enfin le génotype b/b du
système HPA-1 (de 2,702 %) contrairement aux génotypes b/b et a/b
du système HPA-4 qui sont totalement absents.
Tableau X :
Fréquences génotypiques des systèmes HPA-1, -2, -3, -4 et
5.
|
Système
|
HPA-1
|
HPA-2
|
HPA-3
|
HPA-4
|
HPA-5
|
|
a/a
|
0,67
|
0,63
|
0,520
|
1
|
0,78
|
|
a/b
|
0,30
|
0,34
|
0,375
|
0
|
0,19
|
|
b/b
|
0,03
|
0,03
|
0,105
|
0
|
0,03
|
|
Total
|
1
|
1
|
1
|
1
|
1
|
Tableau XI :
Fréquences génotypiques en % des systèmes HPA-1, -2, -3,
-4 et 5
|
Système
|
HPA-1
|
HPA-2
|
HPA-3
|
HPA-4
|
HPA-5
|
|
a/a
|
67
|
63
|
52
|
100
|
78
|
|
a/b
|
30
|
34
|
37,5
|
0
|
19
|
|
b/b
|
3
|
3
|
10,5
|
0
|
3
|
|
Total (%)
|
100
|
100
|
100
|
100
|
100
|
67
30
3
63
34
3
52
37,5
10,5
100
0
0
78
19
3
HPA-1
HPA-2
HPA-3
HPA-4
HPA-5
a/a
a/b
b/b
Fréquences génotypiques
![]()
Figure 11.
Représentation graphique de la répartition des
fréquences
génotypiques des cinq premiers systèmes
HPA dans l'échantillon étudié de la population
tunisienne
ü Fréquences
phénotypiques
Lorsqu'un système à deux allèles est en
situation de codominance, chacun des trois génotypes diploïdes
s'exprime par un phénotype distinct et il existe une correspondance
parfaite entre génotype et phénotype. Ainsi, les systèmes
plaquettaires se transmettent par codominance et les deux allèles vont
s'exprimer d'une manière équivalente chez les
hétérozygotes. Les phénotypes sont déduits des
génotypes correspondants, c'est pourquoi les fréquences
génotypiques sont égales aux fréquences
phénotypiques en %. Il existe donc trois phénotypes pour les
quatre systèmes HPA-1,-2 -3 et 5 : [aa], [ab] et [bb]. Tandis que
pour le système HPA-4 il existe un seul phénotype : [aa].
ü L'équilibre des locus des
systèmes plaquettaires étudiés
Montrer que la population tunisienne est en équilibre
de H-W?
Est-ce que la population tunisienne est en équilibre de
H-W?
Pour tirer des estimations valables et pour que ces
estimations aient un sens biologique, certaines conditions doivent être
remplies lors du prélèvement de l'échantillon afin
d'obtenir un échantillon représentatif.
Considérons que l'effectif de l'échantillon est
infiniment grand et que les unions se font totalement au hasard (panmixie) sans
mutation ni migration ni sélection, c.á.d les génotypes
HPAs n'influencent pas le choix du conjoint et les différents
génotypes sont également viables et féconds (ils sont
neutres du point de vue de la sélection naturelle, les individus sont
supposés avoir la même espérance de vie pour les
gènes HPAs et ne présentent aucune mortalité
différentielle). Dans notre cas, le test ÷2 n'est
applicable que pour le système HPA-3 dont les effectifs des trois
classes sont supérieurs à 5.
Le système HPA-3
Hypothèse
On suppose que la population tunisienne est à
l'état panmictique pour le locus du système HPA-3.
Les effectifs observés et les effectifs
théoriques attendus d'après H-W pour les génotypes du
système HPA-3 sont présentés dans le tableau XII.
Il est évident que l'adéquation entre effectifs
observés et effectifs théoriques est très bonne :
elle n'est pas parfaite à cause des fluctuations statistiques
aléatoires des effectifs observés des divers génotypes et
qui doivent normalement se traduire par des erreurs négligeables.
Pour vérifier cette hypothèse, on utilise un
test statistique qui permet de quantifier l'ajustement aux valeurs
attendues : c'est le test chi-deux : ÷2.
Degrés de liberté : (ddl)
ddl = (k-1)-(r-1) = k-r = Nombre de génotype -
Nombre d'allèles.
ddl = 1.
Il est évident que pour le système HPA -3,
l'adéquation entre les effectifs observes et les effectifs
théoriques est très bonne : elle n'est pas parfaite à
cause des fluctuations statistiques aléatoires des effectifs
observés des divers génotypes et qui doivent normalement se
traduire par des erreurs négligeables. Pour vérifier cette
hypothèse, on utilisé un test statistique qui permet de
quantifier l'ajustement aux valeurs attendues : c'est le test
chi-deux : ÷2
Tableau XII : Système HPA-3:
équilibre de H-W: ÷2 et P
|
Effectifs observés
Population
étudiée
|
Effectifs théoriques
Population en équilibre
|
H-W
|
|
Systèmes
|
Génotypes
|
Nombre
|
Fréquence
|
Nombre
|
Fréquence
|
÷2 (P)
|
|
HPA-3
|
a/a
|
54
|
51,920
|
52
|
50,0
|
0,893 (0,5 - 0,3)
p>á = 0,05
|
|
a/b
|
39
|
37,500
|
43
|
41,3
|
|
b/b
|
11
|
10, 580
|
9
|
8,7
|
On a : 0,3 <P< 0,5
P > 0,05 et le ÷2 calculé
= 0,893 < ÷2 théorique = 3,84, donc
l'hypothèse est vraisemblable. En conclusion, la population
tunisienne est en équilibre panmictique pour le locus du gène du
système HPA-3.
Système HPA-1
Tableau XIII : Système HPA-1 :
équilibre de H-W ; ÷2 corrigé et
P
|
Effectifs observés
Population
étudiée
|
Effectifs théoriques
Population en équilibre
|
H-W
|
|
Systèmes
|
Génotypes
|
Nombre
|
Fréquence
|
Nombre
|
Fréquence
|
÷2 (P)
|
|
HPA-1
|
a/a
|
75
|
67,57
|
75
|
67,567
|
0,736 (0,5 - 0,3)
p>á = 0,05
|
|
a/b
|
33
|
29,73
|
32
|
28,830
|
|
b/b
|
3
|
2,70
|
4
|
3,603
|
On a : 0,3 <P< 0,5
÷2 (corrigé) = 0,736<
÷2 théorique = 3,84, donc l'hypothèse est
vraisemblable. En conclusion, la population tunisienne est en
équilibre panmictique pour le locus du gène du système
HPA-1.
Système HPA-2
Tableau XIV : Système HPA-2:
équilibre de H-W; ÷2 corrigé et P
|
Effectifs observés
Population
étudiée
|
Effectifs théoriques
Population en équilibre
|
H-W
|
|
Système
|
Génotypes
|
Nombre
|
Fréquence
|
Nombre
|
Fréquence
|
÷2 (P)
|
|
HPA-2
|
a/a
|
66
|
62,857
|
67
|
63,81
|
0,0735 (0,8-0,7)
p>á = 0,05
|
|
a/b
|
36
|
34,285
|
34
|
32,38
|
|
b/b
|
3
|
2,857
|
4
|
3,81
|
On a : 0,7 <P< 0,8
÷2 (corrigé) = 0,0735<
÷2 théorique = 3,84, donc l'hypothèse est
vraisemblable. En conclusion, la population tunisienne est en
équilibre panmictique pour le locus du gène du système
HPA-2.
Système HPA-5
Tableau XV : Système HPA-5 :
équilibre de H-W ; ÷2 corrigé et P
|
Effectifs observes
Population étudiée
|
Effectifs théoriques
Population en équilibre
|
H- W
|
|
Système
|
Génotypes
|
Nombre
|
Fréquence
|
Nombre
|
Fréquence
|
÷2 (P)
|
|
HPA-5
|
a/a
|
78
|
78
|
77
|
78
|
2,53 (0,8-0,7)
p>á = 0,05
|
|
a/b
|
19
|
19
|
22
|
19
|
|
b/b
|
3
|
3
|
1
|
3
|
On a : 0,7 <P< 0,8
÷2 (corrigé) = 2,53 <
÷2 théorique = 3,84, donc l'hypothèse est
vraisemblable. En conclusion, la population tunisienne est en
équilibre panmictique pour le locus du gène du système
HPA-5.
Dans notre cas, il est inutile de faire un ÷2
pour le système HPA-4 pour voir que les effectifs réels ne sont
pas statistiquement différents de ceux prévus. Donc,
l'hypothèse est vraisemblable et la population tunisienne est en
équilibre panmictique pour le locus du gène du système
HPA-4.
2. Associations haplotypiques et
déséquilibre de liaison :
Haplotype : L'ensemble des allèles de deux
loci ou plus situés sur un même chromosome sont dits en cis sur ce
chromosome est appelé un haplotype. Le nombre d'haplotype possibles est
égal au produit du nombre d'allèles pour chaque gène. Les
différents allèles d'un haplotype ont d'autant plus tendance
à coségréger, à la méiose, que les locus des
différents gènes sont physiquement proches sur ce chromosome,
c'est-à-dire, génétiquement liés
Le déséquilibre de liaison :
Certaines associations de spécificités a et b sont plus
fréquemment associées que ne le voudrait le hasard. Elles sont
dites en déséquilibre de liaison. En absence de liaison
statistique entre deux allèles a, b (de fréquences respectives
Pa et Pb) à deux loci différents, la
fréquence du gamète portant ces deux allèles sera Pa
Pb.
On appelle déséquilibre de liaison ou (delta
Ä), l'écart entre la fréquence observée Pab
de l'haplotype ab et la fréquence attendue Pa
Pb
Äab = Pab- Pa
Pb
Pour 1 degré de liberté (ddl) au seuil á
= 5% la valeur de ÷2 est de 3.84 à partir de
cette valeur de ÷2, on dit que l'association est significative
et que les deux antigènes sont en déséquilibre de
liaison.
Haplotype HPA-2/HPA-3 Freq. s.d.
D' X² P
1 HPA-2a HPA-3a 0.53123 0.04201
-0.3228 1.9582 0.1617
2 HPA-2a HPA-3b 0.26997 0.03875
0.3228 1.9582 0.1617
3 HPA-2b HPA-3a 0.15551 0.03119
0.3228 1.9582 0.1617
4 HPA-2b HPA-3b 0.04328 0.02166
-0.3228 1.9582 0.1617
53%
27%
16%
4%
HPA2a/HPA3a
HPA2a HPA3b
HPA2b HPA3a
HPA2b HPA3b
Figure 12.
Représentation graphique de la répartition des
fréquences
Haplotypiques des HPA-2 et HPA-3 dans
l'échantillon étudié de la population
tunisienne.
Haplotype HPA-1/HPA-2 Freq. s.d.
D' X² P
1 HPA-1a HPA-2a 0.66683 0.03818
-0.2261 0.3911 0.5317
2 HPA-1a HPA-2b 0.17655 0.03194
0.2261 0.3911 0.5317
3 HPA-1b HPA-2a 0.13438 0.02810
0.2261 0.3911 0.5317
4 HPA-1b HPA-2b 0.02225 0.01621
-0.2261 0.3911 0.5317
P est sup. à 0.05 (÷2 est inf
à 3.84) ; pas de déséquilibre
s.d = déviation standard
D'= relative linkage disequilibrium ; paramètre
qui mesure la force de l'association haplotypique
67%
18%
13%
2%
HPA-1a HPA-2a
HPA-1a HPA-2b
HPA-1b HPA-2a
HPA-1b HPA-2b
![]()
Figure 13.
Représentation graphique de la répartition des
fréquences
Haplotypiques des HPA-1 et HPA-2 dans
l'échantillon étudié de la population
tunisienne.
Fréq. Haplotypiques : HPA-1, HPA-2,
HPA-3 :
Freq. s.d. Haplotype
1 0.45608 0.04252 HPA-1a HPA-2a HPA-3a
2 0.21179 0.03631 HPA-1a HPA-2a HPA-3b
3 0.14003 0.03304 HPA-1a HPA-2b HPA-3a
4 0.03546 0.02083 HPA-1a HPA-2b HPA-3b
5 0.07508 0.02828 HPA-1b HPA-2a HPA-3a
6 0.05825 0.02489 HPA-1b HPA-2a HPA-3b
7 0.01556 0.01545 HPA-1b HPA-2b HPA-3a
8 0.00774 0.01013 HPA-1b HPA-2b HPA-3b
45%
20%
14%
4%
8%
6%
2%
1%
HPA-1a HPA-2a HPA-3a
HPA-1a HPA-2a HPA-3b
HPA-1a HPA-2b HPA-3a
HPA-1a HPA-2b HPA-3b
HPA-1b HPA-2a HPA-3a
HPA-1b HPA-2a HPA-3b
HPA-1b HPA-2b HPA-3a
HPA-1b HPA-2b HPA-3b
![]()
Figure 14.
Représentation graphique de la répartition des
fréquences
Haplotypiques des HPA-1, HPA-2 et HPA-3 dans
l'échantillon étudié de la population
tunisienne.
Ø Estimation du risque d'alloimmunisation
antiplaquettaire chez la population tunisienne :
Est-ce que la population tunisienne est en équilibre de
H-W?
ü Calculer les fréquences alléliques chez
les mâles et les femelles.
Pour qu`une population soit en équilibre de
H-W, il faut que :
p? = p? = p pop.
q ? = q ? = q pop.
Tableau XVI :
Équilibre de H-W: ÷2
|
HPA-1
|
HPA-2
|
HPA-3
|
HPA-4
|
HPA-5
|
|
n?
|
38
|
35
|
34
|
31
|
31
|
|
n ?
|
72
|
70
|
70
|
71
|
69
|
|
Total
|
110
|
105
|
104
|
102
|
100
|
|
p?
|
0.847
|
0.785
|
0.72
|
1
|
0.791
|
|
q?
|
0.208
|
0.214
|
0.426
|
0
|
0.097
|
|
p?
|
0.847
|
0.807
|
0.671
|
1
|
0.855
|
|
q?
|
0.166
|
0.192
|
0.271
|
0
|
0.13
|
|
ppop
|
0 .824
|
0.800
|
0.707
|
1
|
0.875
|
|
qpop
|
0 .176
|
0.200
|
0.293
|
0
|
0.125
|
La population tunisienne est en équilibre de H-W. Elle
subit un cycle de reproduction panmictique.
Chez l'homme, chaque système plaquettaire HPA est
contrôlé par un gène autosomique codominant (a, b)
où les allèles vont s'exprimer d'une manière
équivalente.
Calcul des fréquences du risque
d'allo-immunisation d'un système :
Les fréquences alléliques et génotypiques
sont les mêmes dans les deux sexes, c.á.d. tous les adultes
reproducteurs possèdent une fertilité égale. La
fréquence des zygotes de la génération suivante est
calculée en faisant les produits des fréquences gamétiques
comme le montre le tableau suivant de rencontre des gamètes.
Les mères qui peuvent donner des descendants à
risque allo-immune ont des génotypes (aa) ou (bb) et les pères
sont de génotypes (aa), (ab) ou (bb) (transmission par codominance). Les
mères de génotypes (ab) possède les deux allèles a
et b donc leurs descendants ne sont pas à risques (Tableau XVII).
Tableau XVII Les
fréquences génotypiques des désendants entre
différentes unions parentales.
|
Génotypes ?
Génotypes ?
|
aa p2
|
ab 2pq
|
bb q2
|
|
aa p2
|
1aa p4
|
½aa 2p3q
½ab
|
1ab p2q2
|
|
bb q2
|
1ab p2q2
|
½ab 2pq3
½bb
|
1bb q4
|
Lorsque les pères et les mères sont de
même génotypes (aa) ou (bb) ? le risque est nul.
Lorsque les mères de génotypes (bb) et les
pères sont de génotypes (aa) ? la fréquence des
descendants à risque allo-immune est de p2q2.
Lorsque les mères de génotypes (aa) et
les pères sont de génotypes (ab) ? la fréquence des
descendants à risque allo-immune est de ½
(2p3q).
Lorsque les mères de génotypes (bb) et
les pères sont de génotypes (ab) ? la fréquence des
descendants à risque allo-immune est de ½
(2pq3).
Lorsque les mères de génotypes (aa) et
les pères sont de génotypes (bb) ? la fréquence des
descendants à risque allo-immune est de p2q2.
Donc d'après le tableau XVII, les
fréquences génotypiques attendues des descendants á risque
allo-immune sont :
F (ab) = Fréquence des descendants à risque
allo-immune.
F (ab) à risque allo-immune = p3q +
2p2q2 +pq3
= pq
(p2 + 2pq + q2)
Or, on a :
p2 + 2pq + q2 = (p + q) 2 = 1
F (ab)
à risque = pq
Les résultats obtenus sont une réalité
biologique car toutes les conditions envisagées ci-dessus sont
réalisées. Dans les conditions de H-W, les fréquences
d'allo-immunisation se maintiennent constantes de génération en
génération.
|
Système
|
HPA-1
|
HPA-2
|
HPA-3
|
HPA-4
|
HPA-5
|
|
Fréquences théoriques des génotypes dans
les descendants à risque allo-immuns (p.q)
|
0,145
|
0,16
|
0,207
|
0
|
0,109
|
|
Fréquences théoriques des descendants à
risquent allo-immuns en (%)
|
14,5
|
16
|
20,7
|
0
|
10,9
|
|
Effectifs théoriques des allo-immunisations attendues
par H.W
|
16,095
|
16,8
|
21,528
|
0
|
10,9
|
En se basant sur ces résultats, on remarque que le taux
d'immunisation est variable selon l'antigène considère et qu'un
nombre élevé de grossesses (jusqu'à 21,528 (20,7 %) pour
le HPA-3) serait incompatible pour les allo-antigènes HPAs
dans la population d'étude.
14,5
16
20,7
0
10,9
0
5
10
15
20
25
F(a/b)= pxq
HPA-1
HPA-2
HPA-3
HPA-4
HPA-5
Fréquences théoriques des descendants
à risquent allo-immuns en (%)
![]()
Figure 15:
Fréquence théorique des descendants à risque allo-immuns
en (%) pour les cinq premiers systèmes chez la population
d'étude.
Discussion :
Les HPAs sont des allo-antigènes spécifiquement
plaquettaires génétiquement déterminés. Chaque
système est codé par un gène bi-allélique d'un seul
locus. Les allèles sont indiqués alphabétiquement par
ordre de leur fréquence. L'allèle «a» est le plus
fréquent, l'allèle «b» est le plus rare. Le
polymorphisme de la plupart de ces systèmes est dû à une
substitution d'une seule paire de bases au niveau du gène codant.
Nous nous sommes intéressés dans notre
étude aux cinq premiers systèmes : HPA-1, HPA-2, HPA-3,
HPA-4 et HPA-5 qui se présentent dans l'échantillon
étudiée sous les formes alléliques suivantes :
HPA-1a, HPA-1b ; HPA-2a, HPA-2b; HPA-3a, HPA-3b; HPA-4a, HPA-5a,
HPA-5b. Ces allo-antigènes sont immunogènes et sont
impliqués dans la pathophysiologie de la thrombopénie allo-immune
qui peut être impliquée dans des syndromes cliniques bien
décrits tels que la NAIT et le PTP, mais aussi chez des malades
multi-transfusés qui sont réfractaires à la
thérapie de la transfusion plaquettaire. Ces systèmes jouent le
rôle d'antigènes d'histocompatibilité dans la
transplantation d'organes aussi bien que leurs associations avec les maladies
cardiaques, la thrombose coronaire et les événements
cèrebro-vasculaires. Les données sur la prédominance des
antigènes plaquettaires dans une population donnée sont
essentielles pour le diagnostic des cas des NAITP immunes, pour la
planification des programmes de criblage pour des femmes en danger de FMAIT,
pour la consultation génétique et pour la thérapie des
patients avec fourniture des anticorps anti-HPA (Mueller-Eckhardt. C et
al., 1989; Bussel. J. B et al., 1997; Roúman. P. 2002).
Excepté leur impact pratique sur la provision des transfusions des PLTs.
Elles contribuent également à la compréhension des
rapports évolutionnaires et offrent des informations sur les expansions
démographiques (Roúman. P. 2002). En effet les
allo-antigènes spécifiquement plaquettaires constituent des
marqueurs génétiques.
Le génotypage des HPAs peut être utile pour le
diagnostic, pour la thérapie et la prévention des syndromes
thrombocytopénques allo-immunes telles que la NAIT et le PTP (Nauck.
M. S et al., 1999), ainsi que dans les traitements de differentes maladies
(Liu T.C et al., 2002) et dans la prévision des composants du
sang en HPAs pour ces malades (Hatzidimitriou. Gr et al., 2001; Ficko. T
et al., 2004).
Le typage des allo-antigènes plaquettaires ont
été tout d'abord identifiés par des techniques
sérologiques telles que l'ELISA, la cytométrie en flux, la
technique du MAIPA (Kiefel V et al., 1987; Okomato N et al., 1998) et
l'agglutination plaquettaire.
Le typage sérologique des HPAs en pratique courante est
bien développé (Kekomäki., 1995; Chang. Y. W et al.,
1998). Mais au fil des années, on a remarqué qu'il a
plusieurs inconvénients.
D'abord, la présence à la fois d'anticorps
leucocytaires humains et d'anticorps de groupes sanguins. Le système
HPA-1, présente un risque de confusion (Okamoto. N et al., 1998).
De plus, on a remarqué qu'il y a un manque d'alloantiserums bien
caractérisés et des difficultés d'obtenir du sérum
de typage de bonne qualité (Simsek et Von Dem Borne., 1994;
Cavanagh. G et al., 1997), qui sont dus à la faible
immunogénécité des HPAs et la contamination de ces
sérums par des anticorps anti-HLA (Killick. S. B et al., 1997).
Les difficultés et l'insuffisance d'obtenir des PLTs qu'on observe chez
les malades avec thrombocytopénie (Boldt B et al., 1997) et
chez les foetus (NAIT) rend le diagnostic difficile et peut affecter le foetus
pendant le prélèvement du sang. Le phénotypage ne peut pas
être réalisé pour tous les antigènes plaquettaires
car on ne dispose pas d'allo-anticorps correspondants. De plus les techniques
de typage sérologique présente plusieurs
inconvénients ; le test de MAIPA où le risque d'un
résultat négatif est élevé, qui est dû
à la compétition des anticorps humains avec des anticorps
monoclonaux de capture, en plus ce test nécessite une quantité de
PLTs significative, et il est de réalisation longue et délicate.
Ainsi que la mise en évidence des allo-anticorps maternel n'étant
pas toujours facilement mis en évidence (Santoso. S . A et al.,
2003).
Les méthodes de typage basées sur l'ADN sont
devenues plus exactes (Kluter et al., 1996; Legler TJ et al., 1996; Bein G.
H et KLuter. H. 1997; Chang. Y. W et al., 1998), plus
performantes, plus rapides et sont plus préférables de ce fait,
plus utilisables que les méthodes sérologiques conventionnelles.
Ainsi, l'amplification de l'ADN génomique par PCR élimine le
besoin des PLTs et exige seulement un petit nombre de cellules
nucléées de différentes sources telles que le liquide
amniotique ou la villosité chorionique pour tester les cas de NATP
foetale (Boldt. B et al., 1997). Elle permet aussi d'obtenir des
résultats même dans les cas de thrombopénie extrême
(Cavanagh. G et al., 1997). En fait, plusieurs techniques de
génotypage moléculaire basées sur la PCR se sont
développées (Meyer. O et al., 1999). On cite parmi
elles : La PCR allele-specefic oligonucleotide (Bray. P. F et al.,
1994), PCR-ASRA : allele-specefic restriction enzyme analysis
(Kuijpers. R. W. A. M et al., 1992; Simsek S et al.,
2003 ; Kalb. R et al., 1994), PCR-SSCP : single-strand
conformation polymorphism (Jin. Y et al., 1993; Skogen. B et al., 1994;
Peyruchaud. O et al., 1995), PCR-PHFA : preferential homoduplex
formation (Fuijawara et al., 1996), PCR-OLA : Oligonucleotide
ligation assay (Zotz. R. B et al., 1997) (Nauck M. S et al., 1999). La
PCR-RFLP (Unkelbach. K et al., 1995), RT-PCR: (Simsek et al.,
1997), La PCR-SSP : sequence-specefic primers (Lyou. J.Yi et al.,
2002 ; Jones. D. C et al., 2003). Cependant, toutes ces
méthodes ont des inconvénients, d'ailleurs elles exigent un
processus étendu de post-amplification, y compris
l'électrophorèse (ASRA, SSP, SSCP, PHFA (Nauck M. S et al.,
1999).
La PCR en temps réel est une méthode
idéale pour le diagnostic de routine et la prévention des
conditions cliniques associées aux HPAs. Elle peut être
utilisée dans l'étude des populations et pour la recherche
d'autres effets potentiels de ces polymorphismes ; par exemple, leurs
rôles comme antigènes d'histocompatibilité dans la
transplantation (Kekomaki. S et al., 2001; Jones. D. C et al., 2003),
leurs effets sur le fonctionnement des intègrines et leurs associations
possibles avec les maladies cardio-vasculaires (Weiss. E. J et al., 1996;
Zotz. R. B et al., 1998; Ardissino et al., 1999; Jones. D. C et al., 2003).
Il est essentiel de combiner les analyses phénotypiques
et génotypiques pour éviter une erreur de typage plaquettaire et
rendre un diagnostic erroné dans le cadre d'un processus
d'allo-immunisation. Il reste important de signaler que les résultats de
typage, dans quelques cas, peuvent être confirmés par le
séquençage direct.
La méthode adoptée dans notre étude
est la PCR-SSP. Elle est simple, exacte et rapide (Chang. Y. W et al.,
1998) car elle se fait en une seule étape (á la
différence des autres techniques laborieuses comme la RFLP qui exige une
étape post-amplification par digestion enzymatique). De plus, la
spécificité des primers facilite la détection du produit
amplifié (Kulkarni. B et al., 2005). Elle est
également fiable, très sensible, complète et reproductible
à faible coût. Elle a aussi la possibilité d'être
appliquée sur les liquides amniotiques, ce qui mènera à un
typage plus étendu et plus complet des antigènes plaquettaires et
facilitera le diagnostic exact des thrombocytopénies allo-immunes et la
prévision de produits du sang pour ces malades. De même, elle
offre la valeur particulière dans l'identification rapide des donneurs
des PLTs convenables pour les patients qui exigent la transfusion des cellules
plaquettaires. Ce qui explique le fait qu'elle soit utilisée pour la
routine en cas de génotypage rapide dans les laboratoires et pour les
études de recherches de génétique des populations à
grande échelle. De même, elle peut être mise à jour
pour inclure d'autres allèles HPA de faible fréquence aussi bien
que d'autres allo-antigènes plaquettaires génétiquement
caractérisés (tel que le système Gov). Cette technique est
optimisée dans notre laboratoire pour fournir une résolution
maximale à coût minimum.
Pourtant, cette méthode présente quelques
inconvénients. Un inconvénient majeur de cette méthode est
que des conditions PCR différentes ont été exigées
pour définir chaque paire d'allèles (Skogen. B et al., 1994;
Tanaka. S et al., 1995 ; Cavanagh. G et al., 1997). De même,
l'amplification des deux allèles exige deux tubes séparés
ce qui nous coûte plus de dépenses et de temps (Boldt.
B et al., 1997). De plus, la PCR-SSP est un processus dynamique
qui exige des conditions optimum pour assurer une amplification
spécifique.
De nombreux facteurs peuvent affecter l'efficacité de
la PCR : erreurs de pipetage, mauvaise qualité de l'ADN,
présence d'inhibiteurs... . Ainsi, le protocole associé au
produit doit donc être rigoureusement respecté.
L'échantillon d'ADN extrait va fournir la matrice pour l'amplification
spécifique, d'où l'importance de sa qualité. La
concentration et la pureté doivent se situer dans la limite des valeurs
précisées dans le protocole. Il est important de noter que le BET
utilisé pour la coloration de l'ADN est cancérigène
potentiel et que tous les produits à base de sang doivent être
manipulés comme des produits potentiellement infectieux. Toutefois,
aucune méthode d'analyse connue ne peut garantir que des produits
dérivés du sang humain ne transmettent pas d'agents infectieux.
De même, il faut faire attention à l'UV susceptible d'affecter la
peau et les yeux. Cependant, pour installer un programme de criblage, la PCR en
temps réel serait préférée parce qu'elle prend
moins du temps et évite l'utilisation de BET et l'UV.
D'après les résultats obtenus, nous avons
remarqué que les fréquences des antigènes plaquettaires
varient entre les différents systèmes chez l'échantillon
étudié de la population tunisienne. Ainsi, bien que les
fréquences alléliques HPA-a soient élevées pour les
cinq systèmes HPA-1, HPA-2, HPA-3, HPA-4 et HPA-5, on note que les
fréquences des allèles HPA-b sont importantes pour les quatre
systèmes polymorphes HPA-1, HPA-2, HPA-3 et HPA-5 contrairement au
système mineur HPA-4 qui est monomorphe où l'allèle b est
totalement absent chez cet échantillon. Ce qui prouve que la population
tunisienne est manifestement hétérogène et que notre
population est à haut risque d'allo-immunisation pour ces 4
systèmes HPA-1, 2, 3 et 5.
En effet, la Tunisie a ses propres distributions
alléliques et génotypiques des systèmes HPAs qui lui sont
spécifiques. D'ailleurs, les résultas obtenus montrent que la
population tunisienne est hétérogène. Elle se
caractérise par un haut taux de mélange ethnique. Un fait
d'état qui peut être expliqué par beaucoup de raisons
géographiques, historiques et sociales.
D'autre part, La Tunisie occupe une zone géographique
stratégique. Elle s'ouvre sur la Méditerranée au nord et
à l'est. Elle tend par le sud vers le Grand Sahara et l'Afrique. Ainsi,
la Tunisie est vue comme un point de liaison entre l'Europe et l'Afrique et
entre l'est et l'ouest. C'est pour ces raisons qu'elle a été une
cible de conquêtes et de migrations de toutes les directions; d'abord les
Phéniciens, puis les Romains et les Byzantins, ensuite les Arabes, les
Turcs, la colonisation française jusqu'à l'apparition de
l'état national moderne.
Ainsi, le peuplement de la Tunisie est issu de la migration de
populations venues de tous les endroits (introduction de nouveaux
géniteurs). Les
tribus
berbères
(Amazigh) ont assisté à des échanges culturels et
génétiques avec les autres populations et civilisations qui se
sont soldés quelquefois par le refus et la révolte et par
l'acceptation, l'entraide et l'échange jusqu'à la dissolution
dans d'autres cas. Ces échanges entre la population autochtone et les
peuples venant de l'extérieur ont joué un rôle majeur dans
l'enrichissement génétique de la population.
Du coup, on parle de la diversité du peuple tunisien,
un peuple où vivent des personnes de plusieurs origines.
Parmi ces peuples qui ont vécu en Tunisie on
cite : Les berbères, les phéniciens, les romains, les
arabes, les turcs, les andalous (mourisquioun), les africains noirs et les
juifs (Hmida S et al 1995).
De plus, ce mélange peut être expliqué par
la transmission des gènes entre individus appartenant à ces
différents groupes ethniques par voie de reproduction sexuée (le
mariage).
Bref, la population tunisienne d'aujourd'hui est un ensemble
d'origines berbères, arabes, africaines et européennes. Ce qui
lui procure une propriété de mixage.
L'investigation génétique réalisée
nous a permis d'étudier l'équilibre de la population Tunisienne
pour les locis des systèmes HPAs étudiés. D'ailleurs, cet
équilibre a été vérifié en utilisant
l'approche décrite par H-W. L'application de la loi H-W nous a permis de
constater que l'échantillon étudié est en équilibre
panmictique pour les locis précités. Cet équilibre
s'explique par l'absence de grandes sélections orientées contre
les génotypes HPAs (a/b) malgré l'existence de quelques cas de
mort des nouveaux nés thrombopéniques à la naissance ou
dans les heures qui suivent. Cet équilibre a pour origine probable
l'absence de sélection orienté contres l'un des différents
génotypes et les flux géniques qui sont en grande partie
très anciens. De plus, la prévention, la prise en charge des
thrombopénies chez les nouveaux nés et les patients
polytransfusés et la thérapie des conditions cliniques dues
à l'immunisation plaquettaire sont bien développés en
Tunisie ; ce qui intervient directement dans la diminution des cas de
mort.
La probabilité pour développer une
réponse allo-immune chez les individus dépend des
fréquences des antigènes qui peuvent varier d'une population
à une autre.
La différence entre les fréquences
alléliques (a et b) des systèmes HPA-1, HPA-2, HPA-3 et HPA-5
montre que la population tunisienne est à haut risque
d'allo-immunisation.
D'après nos résultats, nous avons trouvé
que le système mineur HPA-4 n'est pas polymorphe et que l'allèle
4b est absent chez notre population, ces résultats suggèrent que
le HPA-4 n'a pas d'importance clinique.
De même, nous avons observé la forte
présence de HPA-3b suivi respectivement par HPA-2b, HPA-1b et HPA-5b.
Le système HPA-3 est donc le plus polymorphe, ce qui
signifie que cette population a un plus gros risque d'allo-immunisation pour ce
système. Ce système est en principe à implications
cliniques importantes. Théoriquement parlant, plus le degré de
polymorphisme dans un système antigénique est haut, plus il est
cliniquement important, bien que ce ne soit pas toujours vrai. Il existe
d'autres importants facteurs qui contribuent dans ce phénomène
dont l'immunogénicité et l'accessibilité de
l'antigène à l'anticorps formé.
Selon les résultats trouvés par Miadi N et
al., 2003 lors de son étude sur 860 femmes tunisiennes enceintes dans
son mémoire de DEA : "L'allo-immunisation anti-HPA chez les femmes
enceintes tunisiennes : étude prospective", l'allo-immunisation
réelle chez la population tunisienne est relativement
élevée (0,34 %). Par contre, nos résultats ont
donné les valeurs théoriques des fréquences des
descendants à risque allo-immuns en (%) suivantes : 14,5 pour
HPA-1, 16 pour HPA-2, 20,7 pour HPA-3, 10,9 pour HPA-5.
Nous remarquons que nos résultats théoriques
pour l'allo-immunisation chez l'échantillon étudiés de la
population tunisienne sont trop supérieurs à ceux trouvés
par Miadi. N et al, une telle différence serait due à ce que
l'allo-immunisation dépend de plusieurs autres facteurs.
D'ailleurs, le taux d'immunisation varie selon
l'antigène considéré et dépend, en partie, de la
fréquence relative des antigènes dans la population, de leur
immunogénicité (l'immunogénicité de la
protéine est liée à un polymorphisme naturel de la GP ou
à la structure particulière de la molécule), de leur
distribution cellulaire et tissulaire.
L'immunogénicité est déterminée,
en partie, par quatre propriétés de l'immunogène :
son caractère étranger (le degré de
l'immunogénicité d'un antigène dépend du
degré de son caractère étranger), sa taille
moléculaire, sa capacité à être apprêté
et associé à une molécule du CMH à la surface d'une
cellule présentatrice de l'antigène ou d'une cellule du Soi
altérée et enfin sa composition et sa complexité chimique.
D'ailleurs, chaque protéine est caractérisée par ses
propriétés physiques. Lorsqu'un des aa qui forme une
protéine est remplacé par un autre, ce remplacement influe sur
les propriétés physiques de cette dernière. Cinq aa
(l'acide glutamique, l'acide aspartique, l'arginine, la lysine et l'histidine)
présentent des chaînes latérales ionisables qui donnent
à la protéine une charge nette caractéristique,
dépendant du pH du milieu environnant. Des substitutions d'aa peuvent
remplacer directement l'un de ces aa chargés, ou bien une substitution
neutre près de l'un de ceux-ci peut affecter le degré
d'ionisation de l'aa chargé, ou encore, une substitution au niveau de la
jonction de deux hélices á peut entraîner une
légère modification de la configuration tridimensionnelle du
polypeptide replié. Dans tous ces cas, la charge nette du polypeptide
sera modifiée car la charge nette d'une protéine n'est pas
simplement la somme de toutes les charges individuelles de ses aa, mais elle
dépend également de leur exposition au milieu liquide qui les
entoure.
Quel que soit le système considéré,
l'absence d'un antigène chez la mère ne signifie pas
nécessairement qu'elle développera un anticorps lors d'une ou de
plusieurs grossesses incompatibles.
Ainsi le développement de NAIT dépend
également du type de HLA de la mère; néanmoins, le nombre
de grossesses dans lesquelles le foetus est en danger pour la NAITP.
Il sera donc important de suivre l'allo-immunisation anti-HPA
pour tous les systèmes, et si nécessaire, de rechercher une
éventuelle implication des antigènes HLA. La capacité
d'établir une immunisation foeto-maternelle est associée au
phénotype HLA-II de la mère.
L'allo-antigène plaquettaire le plus immunogène
dans la thrombocytopénie est la forme leucine 33 de la GPIIIa
(Giffin. H. N et Ouwehand. W. H., 1995). L'allèle HPA-1a
(PlA1) est responsable de la plupart des cas de purpura post
transfusionnel et de thrombocytopénie néo-natale allo immune dans
la population caucasienne (Okamoto. N et al., 1998).
Le polymorphisme
PlA1/PlA2 pourrait être en
déséquilibre de liaison avec beaucoup d'autres polymorphismes
(Marian et al.,
1996).
Une corrélation entre l'allo-immunisation anti HPA-1a et l'haplotype
HLA-B8, DR3 (HLA-DRB3*0101) (Williamson. L. M et al.,
1998) est fréquemment observée. L'allo-immunisation HPA-1a
est fortement associée au génotype de type maternel HLA de classe
II DRB3*0101 (phénotype DRW52a) (Décary. S. F et al.,
1991) et/ou DQB1*0201 (Décary et al., 1991; L'Abbé. D et
al., 1992; Cohen et al., 1995; Homans. A. 1996; Chantrain. C et al.,
2000 ; Murphy et al., 2002; Koutsogianni. P. 2004). Pour les
personnes de génotypes Leu33 négatifs (Pro33 homozygote), HLA
DRB3*0101-positifs, l'exposition du forme Leu33 est très
immunogène (Watkins. N. A et al., 2002). Les phénotypes
HLA DRw6 est associés à l'allo-immunisation des antigènes
HPA-5b (Bra) (Muller-Eckhard. C et al., 1989a). L'allo-immunisation
maternelle contre l'antigène HPA-6b (GPIIIa-GIn489) peut être
associé au différent haplotype CMH de classe II, c.-à-d.
HLA-DRB1*1501, DQA1*0102, DQAB1*0602 (Westman. P et al., 1997).
Ces antigènes sont considérés comme
marqueurs d'un haut risque d'allo-immunisation plaquettaire dans notre
population, et c'est pour cette raison qu'on est appelé alors à y
faire attention dans le cas de grossesse ou de transfusion du sang et des
transplantations d'organes et de MO. Ces données sont d'importance
majeure et elles ne doivent pas être négligées lors des cas
des la NAIT, le PTP, en cas de greffe d'organes et essentiellement dans les
traitements des différents cas des maladies associées aux
implications cliniques résultant de l'incompatibilité
plaquettaire telles que l'hémorragie cérébrale et
l'hémiplégie des génotypes HPAs (a/b) qui
présentent une allo-immunisation. C'est pourquoi Flug et al., (1994) ont
proposé que toutes les femmes en grossesse soient testées pour
leur système HPA-1 car les femmes HPA-1b homozygotes ont un risque
très significatif pour la NAIT et le PTP (Okamoto. N et al.,
1998).
De plus, ces résultats permettent la comparaison entre
les sous populations aussi bien qu'entre les populations.
Une variable fondamentale en génétique des
populations est la fréquence à laquelle les allèles sont
présents au niveau du locus donné. La fréquence d'un
allèle donné dans une population peut être modifiée
par mutation (le taux de la mutation est défini comme la
probabilité qu'une copie d'un allèle change de forme
allélique en une génération. Supposons que la
totalité d'une population soit homozygote pour a et que les mutations
transformant cet allèle en b se produisent à un taux de 1/100
000), la sélection et la migration des gènes. Il est ainsi pour
les systèmes HPAs. D'ailleurs, les fréquences des systèmes
HPAs varient chez les différentes populations. Et chaque population est
caractérisée par ses fréquences géniques. Ainsi,
chaque apport extérieur d'un allèle a ou b a la chance de
perturber la constitution génétique de la population.
Les êtres humains ne s'unissent pas au hasard puisque
les groupes ethniques et les populations diffèrent les uns des autres
par les fréquences génétiques et présentent des
taux élevés d'endogamie, non seulement au sein des principales
populations mais aussi dans les ethnies locales. Les Danois et les Italiens
différent par la fréquence de leurs groupes plaquettaires. Les
Italiens épousent des Italiennes et les Danois, des Danoises, de sorte
que les unions ne se réalisent plus au hasard, même si ce n'est
pas intentionnellement, en ce qui concerne les systèmes plaquettaires
ainsi que d'autre antigènes tels que les groupes sanguins ABO.
Tableau XVIII :
Fréquences des allèles HPA-1 à 5 dans quelques populations
africaines
|
Systèmes/Population
|
HPA-1a
|
HPA-2a
|
HPA-3a
|
HPA-4a
|
HPA-5a
|
|
Tunisie
(présente étude)
|
0,824
|
0,800
|
0,707
|
1
|
0,875
|
|
Tunisie
|
0,73
|
0,680
|
0,765
|
0,885
|
0,665
|
|
Algérie
|
0,827
|
|
|
|
0,837
|
|
Maroc
|
0,748
|
0,818
|
0,682
|
1
|
0,861
|
|
Berbères (Maroc)
|
0,747
|
0,817
|
0,682
|
1
|
0,8616
|
|
Bénin
|
0,896
|
0,708
|
0,679
|
1
|
0,818
|
|
Cameroun
|
0,907
|
0,763
|
0,614
|
1
|
0,746
|
|
Rép. Centrafricaine
|
1,000
|
0,607
|
0,500
|
1
|
0,595
|
|
Congo
|
0,904
|
0,776
|
0,596
|
1
|
0,732
|
On note, d'après le tableau XVIII, que les
fréquences alléliques des systèmes HPA-1 les plus proches
de celle de la tunisie sont respectivement : l'Algérie suivie du
Bénin. Pour le système HPA-2 on note que la fréquence
allélique est voisine à celles du Maroc, les berbères
Marocains, puis du Congo et du Cameroun. Pour le systéme HPA-4, la
Tunisie possède des fréquences alléliques HPA-4a qui
varient de (1 à 0,885) et par conséquent, l'allèle HPA-4b
varie de (0 à 0,115) (Saadi H et al., 2006; présente
étude). On remarque, d'après ces résultats, que la
Tunisie présente les fréquences des allèles HPA-4b les
plus élevées (0,115) comparée à celles obtenues
avec les autres études. Ceci peut être attribué aux
techniques utilisées dans cette étude (Saadi H et al.,
2006). Pour les systèmes HPA-3 et HPA-5 les fréquences
alléliques sont très proches de celles observées chez les
berbères Marocains et les Marocains. Cela s'explique par le fait qu'une
partie de notre population est d'origine berbère et aussi par
l'immigration d'un certain nombre d'algériens et de marocains à
notre pays. Pourtant, on a remarqué une certaine différence avec
les fréquences alléliques rapportées dans les pays
africains subsahariens. Les pays subsahariens présentent des valeurs
élevèes pour le HPA-1a, en comparaison avec ceux du nord du
continent. Il existe toujours un échange génique
plus ou moins important entre populations voisines.
Tableau XIX
Fréquences alléliques HPA-1 à 5 dans quelques pays
européens.
|
Population
|
HPA-1a
|
HPA-2a
|
HPA-3a
|
HPA-4a
|
HPA-5a
|
|
Tunisie (présente étude)
|
0,824
|
0,800
|
0,707
|
1,000
|
0,875
|
|
Croatie
|
0,830
|
0,880
|
0,640
|
|
0,890
|
|
Autriche
|
0,852
|
0,918
|
0,612
|
1,000
|
0,892
|
|
République du czech
|
0,830
|
0,900
|
0,590
|
|
0,930
|
|
Danemark
|
0,830
|
0,920
|
0,630
|
1,000
|
0,920
|
|
France
|
0,848
|
0,920
|
0,620
|
0,992
|
0,874
|
|
Grèce
|
0,670
|
0,750
|
0,405
|
|
0,850
|
|
Irlande
|
0,882
|
0,934
|
0,624
|
1,000
|
0,912
|
|
Italie
|
0,850
|
0,890
|
0,610
|
1,000
|
0,900
|
|
Luxembourg
|
0,869
|
0,933
|
0,616
|
|
0,902
|
|
Pologne
|
0,824
|
0,901
|
0,582
|
1,000
|
0,944
|
|
Macédonie
|
0,865
|
0,852
|
0,578
|
|
0,909
|
|
Slovénie
|
0,832
|
0,899
|
0,667
|
|
0,892
|
|
Espagne
|
0,810
|
0,900
|
0,650
|
1,000
|
0,880
|
|
Suisse
|
0,809
|
0,918
|
0,591
|
0,997
|
0,934
|
|
Le Pays de Gales
|
0,825
|
0,902
|
0,607
|
1,000
|
0,903
|
|
Caucasiens
|
0,810
|
0,915
|
0,520
|
1,000
|
0,905
|
|
Danemark
|
0,831
|
0,917
|
0,626
|
1,000
|
0,922
|
|
Pays Bas
|
0,846
|
0,934
|
0,555
|
1,000
|
0,902
|
|
Finlande
|
0,860
|
0,910
|
0,590
|
|
0,950
|
|
Allemagne
|
0,846
|
0,934
|
0,555
|
1,000
|
0,902
|
D'après la comparaison des frèquences des
differents systèmes etudiés faite à l'aide du logiciel
SAS, on remarque que les valeurs rapportées chez l'Espagne et la Suisse
sont les plus proches de celles observées chez notre population. Cela
peut etre demontré par les relations historiques trés anciennes
qui ont relié la Tunisie et l'Espagne.
Tableau XX
Fréquences alléliques des HPA-1 à 5 dans
quelques populations asiatiques :
|
Population
|
HPA-1a
|
HPA-2a
|
HPA-3a
|
HPA-4a
|
HPA-5a
|
|
Tunisie (présente étude)
|
0.824
|
0.800
|
0.707
|
1.000
|
0.875
|
|
Vietnam
|
0.986
|
0.953
|
0.486
|
1.000
|
0.972
|
|
Arabie Saoudite
|
0.8
|
0.805
|
0.88
|
0.97
|
0.845
|
|
Taiwan
|
0.9983
|
0.961
|
0.595
|
0.9983
|
0.9814
|
|
Thaïlande
|
0.985
|
0.938
|
0.507
|
1.000
|
0.963
|
|
Philippines
|
0.98
|
0.975
|
0.53
|
0.995
|
0.965
|
|
Corée
|
0.988
|
0.923
|
0.555
|
0.990
|
0.978
|
|
Chine
|
0.995
|
0.975
|
0.525
|
1.000
|
0.965
|
|
Indonésie
|
0.991
|
0.939
|
0.505
|
1.000
|
0.995
|
|
Japon
|
0.99
|
0.91
|
0.715
|
1.000
|
0.98
|
|
Taiwan
|
0.9967
|
0.96
|
0.575
|
0.9983
|
0.985
|
D'après la comparaison des frèquences des
differents systèmes etudiés faite à l'aide du logiciel
SAS, on note que, de tous les pays asiatiques, l'Arabie Saoudite
présente les fréquences les plus proches de celles
rapportés chez la population Tunisienne. Cela s'explique par
l'appartenance des deux populations frères au cadre de communauté
arabo-musulmane l'origine arabe de la plus part des tunisiens.
Tableau XXI
Fréquences alléliques des HPA-1 à 5 dans quelques pays
américains.
|
Population
|
HPA-1a
|
HPA-2a
|
HPA-3a
|
HPA-4a
|
HPA-5a
|
|
Tunisie (présente étude)
|
0,824
|
0,800
|
0,707
|
1,000
|
0,875
|
|
Polynésie française
|
0,975
|
0,913
|
0,599
|
1,000
|
0,975
|
|
Américains (Noirs)
|
0,920
|
0,820
|
0,630
|
1,000
|
0,790
|
|
Américains
|
0,890
|
0,920
|
0,670
|
1,000
|
0,890
|
|
Amérindiens
|
1,000
|
0,957
|
|
|
|
Après comparaison des
fréquences alléliques des systèmes étudiés,
on signale que les fréquences du système HPA-2 chez la population
tunisienne sont très proches de celles observées chez les
Américains Noirs.
Tableau XXII:
Fréquences alléliques des HPA-1 à 5 chez quelques pays de
l'Océanie.
|
Population
|
HPA-1a
|
HPA-3a
|
HPA-5a
|
|
Tunisie (présente étude
|
0,824
|
0,800
|
0,707
|
|
Aborigènes
|
0,992
|
0,937
|
0,847
|
|
Australie
|
0,838
|
0,648
|
0,930
|
On remarque que les
fréquences alléliques du système HPA-1 rapportées
chez la population Australienne sont proches de celles de la population
Tunisienne.
On a remarqué d'après les comparaisons des
fréquences alléliques des systèmes HPAs entre les
différentes populations que quelques populations vivant dans les
mêmes régions ont des fréquences très proches pour
quelques systèmes. De même, d'autres populations appartenant
à des régions tout à fait distinctes pourraient aussi
avoir des fréquences proches. Par exemple, on s'aperçoit que pour
le système HPA-2 les fréquences observées chez la
population tunisienne sont très proches de celles rapportées chez
les Américains Noirs malgré la distance qui sépare la
Tunisie de l'Amérique et l'absence de forte relations historiques
directes entre ces deux pays.
Il nous été montré que, pour les
populations africaines, les fréquences alléliques du Cameroun
sont les plus proches de celles trouvées chez la population tunisienne.
C'est le cas de l'Arabie Saoudite pour les populations asiatiques et de
l'Espagne pour les populations européennes.
Bien que les polymorphismes du gène de quelques
marqueurs génétiques tel que le polymorphisme du gène de
l'inhibiteur l'acétylcholinestérase, (Wiwanitkit. V. 2003;
Wiwanitkit. V. 2005), parmi les populations dans les mêmes
régions a été démontré, nous n'avons pas
trouvé cette observation dans le polymorphisme des gènes HPAs.
Décisivement, c'est tout à fait difficile d'utiliser les
gènes HPAs comme marqueurs génétiques pour
déterminer la ressemblance entre les différentes populations.
Bien qu'il y ait une légère variation entre les populations, ce
n'est pas une différence discrète.
Ces études du polymorphisme
plaquettaire parmi les différentes populations fourniraient de nouvelles
données pour l'hématologie géographique, ces études
qui si loin ont été basées sur les polymorphies
érythrocytaires, leucocytaires et du sérum.
![]()
Conclusion
Cette étude nous a permis de montrer que la population
tunisienne est à l'état d'équilibre panmictique pour les
locis des systèmes étudiés selon la loi de H-W.
Les données présentes indiquent que la
population tunisienne se caractérise par un haut taux de mélange
ethnique, ce qui montre que notre population fait face à un haut risque
d'allo-immunisation anti-plaquettaire pour les systèmes : HPA-1,
HPA-2, HPA-3 et HPA-5.
L'étude du polymorphisme allélique des HPAs
s'est avérée importante pour les études
génétiques, biologiques et anthropologiques. D'ailleurs, elle
permet la déduction du flux des gènes et la constitution
génétique de la population, mais surtout la prédiction du
risque d'allo-immunisation anti-plaquettaire. Elle aidera à anticiper la
dimension et les causes de la NAIT dans notre communauté par la
définition des allo-antigènes les plus probables qui soient
impliqués dans ce phénomène.
Ce travail nous a permis de montrer la nécessité
de vulgariser le génotypage des antigènes plaquettaires afin de
réduire la survenue de l'allo-immunisation transfusionnelle ou
foeto-maternelle d'origine plaquettaire. D'ailleurs, pour une population
donnée, la connaissance des fréquences alléliques nous
fournit des informations précises concernant les conditions cliniques
spécifiques telles que la NAIT et le PTP, sur les capacités du
diagnostic de la NAIT, la prévention de l'hémorragie
intracrânienne des foetus et nous procure plus d'informations pour des
traitements exacts des transfusions plaquettaires incompatibles. Ainsi la
caractérisation du système plaquettaire à l'origine de
l'incompatibilité foetomaternelle est susceptible d'améliorer la
connaissance de la distribution géographique de cette affection et sa
prise en charge.
Ainsi, les pédiatres, les biologistes et les
spécialistes en santé publique, les obstétriciens et les
personnels de la médecine transfusionnelle sont appelés à
exploiter ces résultats dans leurs laboratoires et services pour le
diagnostic définitif des cas de NAIT et PTP et leur traitement pendant
la période néo-natale. D'ailleurs, l'agrément reçu
pour la réalisation de tests de diagnostic prénatal dans le
domaine des thrombopénies et thrombopathies lui permet de participer
directement à la prise en charge de grossesses à haut risque.
Perspectives
Au cours de ce stage, j'ai pu faire un pas de plus en
direction du monde de la recherche en m'immergeant dans la vie d'un
laboratoire, en observant son fonctionnement, ses difficultés et ses
bons moments. Cette expérience me fut très enrichissante et a
confirmé mon goût pour la recherche scientifique.
Durant ce stage, je n'ai fait que commencer le travail que je
devrais continuer par la suite. Car je me propose de poursuivre et
d'approfondir cette étude en :
- Elargissant la taille de population et en introduisant
d'autres sous populations comme les berbères, les noirs et les juifs.
Ainsi, l'échantillon devient représentatif et les
résultats qui seront obtenus seront proches de la réalité.
Cela nous permettra d'évaluer plus précisément la
signification clinique des systèmes HPA en Tunisie.
- Introduisant des malades incluant les cas de (NAIT), de
PTP et de PTR pour dégager le ou les systèmes les plus
impliqués dans ces réactions immunologiques.
- L'étude de la distribution des fréquences des
HPAs et leurs associations avec NAIT, PTP et les autres désordres.
- La recherche des valeurs réelles du risque
d'alloimmunisation chez notre population on introduisant les autres facteurs
qui intervient dans les phénomènes d'alloimmunisation.
- Etudiant l'association des antigènes HPAs et les
antigènes HLA pour déduire l'implication de ces deux
systèmes dans les réactions alloimmunes tel que l'association
entre l'alloimmunisation anti HPAs et l'haplotype DRB13*.
- Etudiant les fréquences alléliques des
différents gènes des systèmes HPA qui restent et donc de
définir l'haplotype des systèmes plaquettaires en Tunisie par la
mise en oeuvre d'autres techniques telle que la PCR en temps réel.
- La recherche de nouveaux antigènes plaquettaires
à travers l'étude de l'ADN des nouveaux nés et des
patients qui montrent des réactions alloimmunes post transfusionnelles
par le séquençage et par la comparaison de l'ADN
étudié avec les séquences d'ADN fournies par les banques
de donnés disponibles.
- L'étude de la toute
nouvelle mutation découverte par Santoso en 2006 Leu33Val de
l'intégrine â3 chez la population tunisienne qui peut être
impliquée dans les cas de NAITP.
- Entrer en coopération avec d'autres groupes de
spécialistes tunisiens et étrangers/d'origines différentes
en la matière non seulement à l'échelle régionale
mais aussi nationale et internationale car cela est indispensable à la
progression des connaissances et des techniques dans les domaines d'immunologie
plaquettaire, de biologie moléculaire et d'immuno-pathologie
plaquettaire au cours de la grossesse et de son retentissement foetal, qui
évoluent sans cesse.
![]()
Références
bibliographiques
· Afshar-Kharghan V, Li CQ, Khoshnevis-Asl
M, Lopez JA. (1999). Kozak sequence polymorphism of the glycoprotein
(GP) Iba gene is a major determinant of the plasma membrane levels of the
platelet GPIb-IX-V complex. Blood, 94: 186-91.
· Afshar-Kharghan V, Aleksic N, Ahn, C.
(1999a). Prospective study of the variable number of tandem repeat
(VNTR) polymorphism in platelet glycoprotein (GP) Ib and incidence of coronary
heart disease. Thromb Haemost, 82: 1693a.
· Aiken Martha L., Mark H. Ginsberg, and Edward
F. Plow. (1986). Identification of a New Class of
Inducible Receptors on Platelets Thrombospondin Interacts with Platelets via a
GPIIb-IIIa-independent Mechanism. J. Clin. Invest, 78: 1713-1716.
· AL-Sheikh A, Rahi M. AL-Khalifa. (2000).
Genotyping human platelet alloantigens (HPA-1-5) in Saudis from
Eastern Province, Saudi Arabia. Eastern Mediterranean
Health Journal, 6: 168-175.
· Aramaki KM. et Reiner A P. (1999). A
novel isoform of platelet glycoprotein Ibá is prevalent in African
Americans. A. J.H, 60:
77-79.
· Barron-Casella Emily A., Nebbia Gaia, Rogers
Ophelia C., King Karen E., Kickier Thomas S et Casella James F. (1999).
Construction of a human platelet alloantigen-Ia epitope(s) within
murine glycoprotein IIIa: Identification of residues critical
to the conformation of the antibody binding site(s). Blood, 93:
2959-2967.
· Berndt MC, Shen Y, Dopheide S, Gardiner EE,
Andrews RK. (2001). The vascular biology of the
glycoprotein Ib-IX-V complex. Thromb Haemost, 86: 178 -188.
· Berry J.E, Murphy C M, Smith G.A, Ranasinghe E,
Finberg R, Walton J, Brown J, Navarette C, Metcalfe P et Ouwehand W.H. (2000).
Detection of Gov system antibodies by MAIPA reveals an immunogenecity
similar to the HPA-5 alloantigens. B. J. H, 110: 735-742.
· Bein G. Hackstein, H. KLuter. (1997).
DNA Typing of human platelet antigen system 1.2.3 and 5 in
B-lymphoblastoid cell lines of the international Histocompatibility Workshop.
Tissue Antigens, 49: 443-447.
· Bettaieb, A.; Fromont, P.; Rodet, M.; Godeau,
B.; Duedari, N.; Bierling, P. (1991). Br(b), a platelet alloantigen
involved in neonatal alloimmune thrombocytopenia.
Vox
Sang, 60: 230-234.
· Bessos H., Mirza S., McGill A., Williamson L.
m., Hadfield R., AND Murphy W. G. (1996). A Whole blood assay for
platelet HPA1 (PlA1) phenotyping applicable to large-scale screening. Br.J.
Haematel, 92: 221-225.
· Beutler E, Lichtman MA, Coller BS, Kipps TJ,
Seligsohn U. Williams.
( 2001).
Hematology. 6th ed. New York: McGraw-Hill Medical Publishing
Division.
· Blanchette VS, Johnson J, Rand M.
(2000). The management of alloimmune neonatal thrombocytopenia.
Bailliers Clin Hematol, 13: 365-390.
· Blockmans, D., Deckmyn, H. & Vermylen, J.
(1995). Platelet activation. Blood Rev 9: 143-156.
· Bojesen. Stig E, MD, PHD, Klaus Juul, MD, Peter
Schnohr, MD, Anne Tybjærg-Hansen, MD, DMSc, Børge G. Nordestgaard,
MD, DMSc. (2003). Platelet Glycoprotein IIb/IIIa
PlA2/PlA2 Homozygosity Associated With Risk of Ischemic
Cardiovascular Disease and Myocardial Infarction in Young Men.
JACC, 42: 661-7.
· Boldt B, Skogen B, Agostini T, Roscetti D et Mc
Farland. J. (1997). One-tube method for complete
HPA-1 genotyping by PCR using sequence-specific primers. B. J. H, 99:
968-973.
· Bordin JO, Kelton JG, Warner MN, Smith JW,
Denomme GA, Warkentin TE, McGrath K, Minchinton R et Hayward CPM.
(1997). Maternal immunization to Gov system alloantigens on
human platelets. Transfusion, 37: 823-828.
· Boshkov IK, Kelton JG et Halloram PF.
(1992). HLA-DR expression by platelets in acute idiopathic
thrombocytopenic purpura. Br J Haematol, 81: 552-7.
· Bray P.F, Rosa Jean-Philippe, Johnston Geoffrey
I., Shiu Donny T., Cook Richard G, Lau Chris, Yuet Wai Kan, McEvert Rodger P.
et Shuman Marc A. (1987). Platelet Glycoprotein IIb Chromosomal
Localization and Tissue Expression. J. Clin. Invest, 80:
1812-1817.
· Bray P F, Brash G, Rosa JP, Luo X Y, Magenis E
and Shuman M A. (1988). Physical linkage of the genes for platelet
menbrane glycoprotein IIIa. Proc Nalt Acad Sci USA, 85, (22):
8683-8687.
· Bray PF. Jin Y and Kickler T.(1994).
Rapid genotyping of the five major platelet alloantigens by reverse
dot-blot hybridisation. Blood, 84: 4361-4367.
· Bray. P.F. (1999). Integrin
polymorphisms as risk factors for thrombosis. Thrombosis and
Haemostasis, 82: 337.
· Brashem-Stein C, Nugent D, Bernstein
ID. (1988). Characterization of an antigen expressed
on activated human T cells and platelets. J. Immunol, 140:
2330-2333.
· Bugert P, McBride S, Smith G, Dugrillon A,
Kluter H, Ouwehand W H et Metcalfe P. (2005). Microarray-based
genotyping for blood groups: comparaison of gene array and 5'-nuclease assay
techniques with human platelet antigen as a model.
Transfusion, 45: 654-659.
· Bussel J. B., Thomas J. Kunicki, and Alan D.
Michelson. (2000). Platelets: New Understanding of Platelet
Glycoproteins and Their Role in Disease. Hematology, 221: 240.
· Calvete J.
(1994). Clues for understanding the structure and function of a
prototypic human integrin: The platelet glycoprotein IIb-IIIa complex.
Thromb Haemost, 72: 1-15.
· Cardone J. D. B., Chiba A. K.,
Boturão-Neto E., Vieira-Filho J. P. B. & Bordin J. O. (2004).
Gene frequencies of the HPA-15 (Gov) platelet alloantigen system in
Brazilians. Transfusion Medicine, 14: 433-437.
· Carl B, Kroll H, Bux J, Bein G, Santoso S.
(2000). B-lymphoblastoid cell lines as source of reference DNA for
genotyping of human platelet and neutrophil antigens. Transfusion, 40:
62-8.
· Carter AM, Ossei-Gerning N, Grant PJ.
(1996). Platelet glycoprotein IIIa P1A polymorphism in young men with
myocardial infarction. Lancet, 348: 485-6.
· Carter AM, Catto AJ, Bamford JM, et al.
(1998). Platelet GP IIIa PlA and GP Ib variable number tandem repeat
polymorphisms and markers of platelet activation in acute stroke.
Arterioscler Thromb Vasc Biol, 18, 1124-1131.
· Carlsson L. E MSc, Andreas Greinacher MD,
Carsten Spitzer MD, Reinhard Walther, PhD; Kessler Christof MD. (1997).
Polymorphisms of the human Platelet antigens HPA-1, HPA-2, HPA-3, and
HPA-5 on the platelet receptors for fibrinogen (GPIIb/IIIa), Von Willebrand
Factor (GPIb/IX), and collagen (GPIa/IIa) are not correlated with an increased
risk for stroke. Strok, 28: 1392-1395.
· Cavanagh G, Dunn A N, Chapman
C. E et Metcalfe P. (1997). HPA genotyping by PCR sequence-specific
priming (PCR-SSP): a streamlined method for rapid routine investigations.
Transfusion Medicine, 7: 41-45.
· Cauwenberghs N, Karen V, Stephan V, Douwe F. W,
Gabriel R, Eric G. Huizinga, José A. Lopez, Michael C. Berndt,
Jolàn Harsfalvi, and Hans Deckmyn. (2001). Epitope mapping of
inhibitory antibodies against platelet glycoprotein Iba reveals interaction
between the leucine-rich repeat N-terminal and C-terminal flanking domains of
glycoprotein Iba. Blood. 98: 652-660.
· Chen DF, Pastucha LT, Chen HY, Kaddar JG et
Stangel N. (1997). Simultaneous genotyping of human platelet antigens
by hot start sequence specific polymerase chain reaction with DNA polymerase
ampli Taq Gold. Vox sang, 72: 192- 196.
· Chang Y.W, Mytilineos J, Opelz G, et Hawkins
B.R. (1998). Distribution of human platelet antigens in Chinese
population. Tissue Antigens, 51: 391-393.
· Chabrenaud J.L.; Lacaze T., Zupan V., Boithias
C., Gross E., Dehan M. (1995). Transfusion
plaquetaires en neonatologie. T CB, 1: 17-25.
· Chadderdon Robert C. and Michael Cappello.
(1999). The Hookworm Platelet Inhibitor: Functional Blockade of
Integrins GPIIb/IIIa (áIIbâ3) and GPIa/IIa
(á2â1) Inhibits Platelet Aggregation and
Adhesion In Vitro. The Journal of Infectious Diseases, 179:
1235-41.
· Chantrain C., Debauche C., Langhendries J.P.,
Vermylen C. P. (2000). Perintal thrombocytopenia of maternal origin.
Arch Pediatr, 7: 756-62.
· Charakida Marietta, Tousoulis Dimitris,
Stefanadis Christodoulos et Toutouzas Pavlos. (2003). The role of
platelet glycoprotein Ib and IIb polymorphism in coronary artery
disease. Hellenic Journal of Cardiology, 44: 43-48.
· Charakida M, Tousoulis D, Christodoulos S,
Pavlos T. (2003a). The impact of platelet glycoprotein IIIa and Ia
polymorphisms in cardiovascular thrombotic disease. Ital Heart J, 4
(1): 17-22.
· Chap, H. J., Zwaal, R. F. and Van Deenen, L.L.
(1977). Action of highly purified phospholipases on blood platelet.
Evidence for an asymetric distribution of phospholipids in the surface
membrane. Biochem. Biophys. Acta. 467, 146-164.
· Chen Z, Lester S, Boetcher B and Mc Cheskeg J.
(1997). Platelet antigen allele frequencies in Australian, Aboriginal
and Caucasian populations. Pathology, 29 : 392-
· Chen Chih-Hung, Yuk-Keung Lo, Dershin Ke,
Chin-Kuan Liu, Chia-Wei Liou, Hua-Lin Wu, Ming-Liang Lai. (2004).
Platelet glycoprotein Ia C807T, Ib C3550T, and IIIa PlA1/A2
polymorphisms and ischemic stroke in young Taiwanese. Journal of the
Neurological Sciences. 227: 1-5.
· Chester Q. Li, Stephen F. Garner, Julian
Davies, Peter A. Smethurst, Mark R. Wardell, and Willem H. Ouwehand. (2000).
Threonine-145/Methinine-145 variants of baculovirus produced
recombinant ligand binding domain of GPIbá express HPA-2 epitopes and
show equal binding of von Willebrand factor. Blood, 95:
205-211.
· Christie. D. J., Pulkrabek S., Putnam J. L.,
Slatkoff M. L., and Pischel. K. D. (1991). Posttransfusion purpura
due to alloantibody reactive with glycoprotein Ia/IIa (anti-HPA-5b).
Blood, 77: 2785-2789.
· Clemetson K.J. (2001). Platelet
glycoproteins and their role in deseases. Transfusion Clinique et
bilogique ; 8 (3): 155-162.
· Covas DT, Biscaro TA, Nascuitti DC, Guerrerio
JF, Santos SE, Zagoma. (2000). Gene frequencies of
the HPA-3 and HPA-5 platelet antigen alleles among the Amerindians. Eur J
Haematol, 65: 128-131.
· Corral. J, Gonzalez-conejero. R, Vicente. R,
Vicente, V. (2004). Genetic determinants of platelet functionin
thromboembolic diseases. J Biol Regul Homeost Agents, 18: 166-71.
· Cong, N. V., Uzan, G., Gross, M. S.,
Jegou-Foubert, C., Frachet, P., Boucheix, C., Marguerie, G. & Frezal, J.
(1988). Assignment of human platelet GP2B (GPIIb) gene to chromosome
17, region q21.1-q21.3. Hum Genet, 80: 389-392.
· Coffe C., Bardiaux L., Couteret Y., Devillers
M., Leroy M., Morel P., Pouthier-stein F., Herve P. (1995). indication
et surveillance des transfusions plaquettaires en chirurigie. TCB, 2:
91-99.
· Crow A.R., Freedom J. Hannach B., Blanchette
V., Lazarus A.H. (1999). Antibody-medited inhbition of the human
alloimune response to platelet tranfusion in Hu-PBL-SCID mic. Br. J.
Hae, 104 (4): 919-924.
· Davoren A, McParland P, Barnes CA,
Murphy WG. (2002). Neonatal alloimmune thrombocytopenia in the Irish
population: a discrepancy between observed and expected cases. J Clin
Pathol, 55: 289-92.
· Deckmyn H, Ulrichts H, Van de Walle G. et
Vanhoorelbeke K. (2004). Platelet antigens and their
function, Vox Sanguinis, 87 (Suppl. 2), 105-111.
· Delpech M, Bienvenu T, Meunier C, Bousquet S,
Chiron S, Richard L. (1999). Les techniques d'extraction de L'ADN
à partir d'un échantillon sanguin. Annals de biologie
clinique, 57 (1): 77-84.
· Djaffar I, Vilette D,
Pidard D, Wautier JL, Rosa JP.
(1993). Human platelet antigen 3 (HPA-3): localization of the
determinant of the alloantibody Lek (a) (HPA-3a) to the C-terminus of platelet
glycoprotein IIb heavy chain and contribution of O-linked carbohydrates.
Thromb Haemost, 69: 485.
· Drzewek K, Brojer E and Zupanska B.
(1998). The frequency of human platelet antigen (HPA) genotypes in the
Polish population. Transfus Med. 8: 339-342.
· Emily A., Casella B. Nebbia G. (1999).
Construction of a human platelet allo-antige-la Epitope within murine
glycoproteine IIIa: identification of residues critical to the conformation of
the antibody binding site(s). Blood, 93(9): 2959-67.
· Ertel Katharina, Milad Al-Tawil, Sentot
Santoso, and Hartmut Kroll. (2005). Relevance of the HPA-15 (Gov)
polymorphism on CD109 in alloimmune thrombocytopenic syndromes.
Transfusion, 45: 366.
· Ferrer. G, Muñiz-Diaz .E, Aluja.
M. P, Arilla. M, Mar Tinez. C, Nogués. R, Servin. A and Balli .A.
(2002), Analysis of human platelet antigen systems in a Moroccan
Berber population. Transfusion Medicine, 12,
49-54.
· Feng Dali, M D ; klaus Lindpaintner, M D
martin G. Larson, SD;Christopher J. O' Donnell, M D; isabella Lipinsca PhD;
Patrice A . Suterland, BS ; Murray Mittleman, M D ; James E. Muller, M D ;
Ralph B. D' Agostino, PhD; Geoffrey H . Tofler, M D. (2001).
Platelet Glycoprotien IIIa PlA Polymorphism, Fibrinogen,
and platelet Aggregability The Framinogham Heart study.
Criculation, 104: 140-144.
· Ficko, T., Galvani, V., Rupreht, R., Dovc, T.
& Roúman, P. (2004).
Real-time PCR genotyping of human
platelet alloantigens HPA-1, HPA-2, HPA-3 and HPA-5 is superior to the standard
PCR-SSP method.
Transfusion
Medicine 14 (6): 425-432.
· Fitzgerald, L. A., Steiner, B., Rall, S. C.,
Jr., Lo, S. S. and Phillips, D. R. (1987). Protein
sequence of endothelial glycoprotein IIIa derived from a cDNA clone. Identity
with platelet glycoprotein IIIa and similarity to integrin.
J. Biol Chem, 262: 3936-3939.
· Friedman JM, Aster RH. (1985).
Neonatal alloimmune thrombocytopenic purpura and congenital porencephaly in two
siblings associated with a "new" maternal antiplatelet antibody.
Blood, 65: 1412.
· Fujiwara Koki, Katsushi Tokunaga, Kazumi Isa,
Masaki miyamoto, Li Wanga, Tatsuya Akaza, Kenji Tadokoro, Yoichi Shibata, Takeo
Juji. (1995). DNA- Based Typing of Human Platelet Antigen Systems by
Polymerase Chain Reaction Single-Strand Conformation Polymorphism Method.
Vox Sang. 69: 347-351.
· Fujiwara K, Isa K., Oka T, Maekawajiri S,
Yamane A, Akaza T, Tadokoro K, Juji T, Shibata Y, Tokunaga K. (1996).
Large-scale DNA typing for human platelet alloantigens by PCR-RHFA
(preferential homoduplex-formation assay). BJH. 95:
198-203.
· Furihata K, Nugent DJ, Bissonette A, Aster RH,
Kunicki TJ. (1987). On the association of the platelet-specific
alloantigen, Pena with glycoprotein IIIa. Evidence for heterogeneity
of the glycoprotein IIIa. J Clin Invest, 80: 1624.
· Gaetano G. (2001).
Historical overview of the role of platelets in haemostasis and
thrombosis. Haematological, 86(4): 349-356.
· Gidwitz S, Temple Brenda and White Gilbert C,
II. (2004). Mutations in and near the second calcium-binding domain of
integrin aIIb affect the structure and function of integrin aIIbb3.
Biochem. J. 379: 449-459.
· Goldberger A, Kolodziej M, Poncz M, Bennett JS,
Newman PJ. (1991). Effect of single amino acid substitutions on the
formation of the PlA and Bak alloantigenic epitopes. Blood, 78:
681-7.
· Gottschalk. Kaye, Pauld. Adams, Axelt. Brunger,
And Horst Kessler. (2002). Transmembrane signal transduction of the
áIIbâ3 integrin. Protein Science, 11: 1800-1812.
· Gruchala Marcin, Dariusz Ciecwierz, Karolina
Ochman, Radoslaw Targonski, Witold Dubaniewicz, Wojciech Sobiczewski, Bartosz
Wasag, Piotr Drewla, Pawel Skarzynski, Piotr Romanowski, Janusz Limon, Andrzej
Rynkiewicz. (2003). A ssociation between the Pl platelet glycoprotein
GPIIIa polymorphism and extent of coronary artery disease. International
Journal of Cardiology, 88: 229-237.
· Hatzidimitriou, Gr. Zoulas D.
(2001). Evaluation-standardization study of the PCR-RFLP (polymerase
chain reaction-restriction fragment length polymorphism) technique for human
platelet alloantigen (HPA) typing. Haema, 4(3), 181-184.
· Handa. M, Titani. K, Holland L. Z, and Ruggeri.
Z. M. (1986). The von Willebrand factor-binding domain of platelet
membrane glycoprotein Ib. Characterization by monoclonal antibodies and partial
amino-acid sequence analysis of proteolytic fragments. J. Biol. Chem.
261: 12579-12585.
· Hato Takaaki, MD, Yoko Minamoto, MD, Takaya
Fukuyama, MD, and Shigeru Fujita, MD. (1997). Polymorphisms of HPA-1
through 6 on platelet membrane glycoprotein receptors are not a genetic risk
factor for myocardial infraction in the Japanese population. Excerpta
Medica, Inc.. 28: 1222-1224.
· Hallé L, Bigot A, Mulen-Imandy G,
M'Badyo K, Jaeger G, Anani L, Martageix C, Bianchi F, Julien E and Kaplan
C. (2005). HPA Polymorphism in sub-Saharian African
populations: Beninense, Cameroonians, Congolese, and Pygmies. Tissue
Antigens, 65: 295-98.
· Hmida S., Gauther A., Dridi A. (1995).
HLA class II gene polymorphism in tunians. Tissue
antigens. 45: 63-68.
· Hoffman R. (1989). Regulation of
megakaryocytopoiesis. Blood; 74: 1196-212.
· Holensteiner A, Walchshofer S, Alder A, Kittle
M, Mayr WR, Panzer S. (1995). Human platelet antigen gene frequencies
in the Austrian population. Hoemostasis, 25: 133- 136.
· Hohlagschwandtner Maria, M.D., Gertrud Unfried,
M.D., Georg Heinze, Ph.D., Johannes C. Huber, M.D., Fritz Nagele, M.D., and
Clemens Tempfer, M.D. (2003). Combined thrombophilic polymorphisms in
women with idiopathic recurrent Miscarriage. Fertil Steril, 79:
1141-8.
· Honda Shigenori, Yumiko Honda, Barbara Bauer,
Changgeng Ruan, and Thomas J. Kunicki. (1995). The Impact of
Three-Dimensional Structure on the Expression of PIA Alloantigens on Human
Integrin â3, Blood, 86: 234-242.
· Homans A. (1996). Thrombocytopenia In
the neonate. Pedi Clin North Am, 43: 749-56.
· Huang Tao, Mervyn A. Sahud. (2003).
Association of C807T, PlA, and 5 C/T Kozak genotypes with density of
glycoprotein receptors on platelet surface. Thrombosis Research,112:
147-150
· Hurd CM, Cavanagh G, Schuh A, Ouwehand WH,
Metcalfe P. (2002). Genotyping for platelet-specific antigens:
techniques for the detection of single nucleotide polymorphisms. Vox
Sang, 83: 1-12.
· Iniesta JA., PhD; Rocio
González-Conejero, PhD; Claudio Piqueras, MD; Vicente Vicente, PhD
Javier Corral, PhD. (2004). Platelet GP IIIa Polymorphism HPA-1 (PlA)
Protects Against Subarachnoid Hemorrhage. Stroke, 35: 2282.
· Jallu Vincent, Marc Meunier, Maryline
Brément, and Cécile Kaplan. (2002). A
new platelet polymorphism Duva+, localized within the RGD binding
domain of glycoprotein IIIa, is associated with neonatal thrombocytopenia.
Blood, 99: 4449-4456.
· Jennings LK, Phillips DR. (1982).
Purification of glycoproteins IIb and III from human platelet plasma membranes
and characterization of a calcium-dependent glycoprotein IIb-III complex. J
Biol Chem, 257(17): 10458-10466.
· Jin Y, Dietz H, Nurden AT, Bray PF.
(1993). SSCP is a rapid and effective method for the identification of
mutations and polymorphisms in the gene for GPIIIa. Blood, 82:
2281-2288.
· Jimmy Lim, Suman Lal, Kenneth C. Ng,
Kheng-Siang Ng, Nilmani Saha, Chew-Kiat Heng. (2003). Variation of the
platelet glycoprotein IIIa PI A1 /A2 allele frequencies in the three
ethnic groups of Singapore. International Journal of Cardiology, 90:
269-273.
· Jilma-Stohlawetz P, Homoncik M, Jilma B,
Knechtelsdorfer M, Unger F, Mannhalter C, Santoso S, Panzer S.
(2003). Glycoprotein Ib polymorphisms influence platelet plug
formation under high shear rates. B. J. H, 120: 652.
· Johson-Hopson C.N., Arlet C.M. (2003).
Evidence against the long-term persistence of fetal DNA in maternel
plasma after pregnancy. Intern. J. Trans.
Med, 84(4).
· Jones D. C, Bunce. M, Fuggle. S. V, Young. N.
T. & Marshall. S. E. (2003). Human platelet
alloantigens (HPAs): PCR-SSP genotyping of a UK population for 15 HPA alleles.
European Journal of Immunogenetics, 30: 415-419.
· Juji T, Saji H, Satake M, Tokunaga K.
(1999). Typing for human platelet allo-antigens. Rev
Immunogenet, 1: 239-54.
· Juji T., Watanable Y., Ishikawa Y., Fuujiwara
K., Tonami H., Tanaka H., Satake M., Akaza T., Tadokoro K., Kodera Y.,
Sasazuki., Morishima Y., Takaku F. (1999a). Human platelet alloantigen
(HPA)-5a/b mismatch decreases disease-free survival in unrelated bone marrow
transplantation. Tissue Antigens. 54: 229-234.
· Kaplan C. (2001). Immune
thrombocytopenia in the foetus and the newborn: diagnosis and therapy.
Transfus Clin Biol, 8: 311- 4.
· Kaplan C. (2002). Alloimmune
thrombocytopenia of the foetus and the newborn. Blood Rev, 16:
69-72.
· Kaplan C. (2003). Fetal and neonatal
alloimmune thrombocytopenia. Orphanet Encyclopedia,
1-6.
· Kaplan C. (2003).
Thrombopénies foetales et néonatale d'origine immune,
orientations diagnostiques et thérapeutiques
Hématologie, 9(1).
· Kalb R, Santoso S, Unkelbach K, Kiefel V and
Mueller Eckhardt C. (1994). Localization of the Br polymorphism on a
144 exon of the GPIa gene and its application in platelet DNA typing.
Thrombosis and Haemostasis, 71: 651-654.
· Kastrati, Adnan MD; Albert Schömig, MD;
Melchior Seyfarth, MD; Werner Koch, MD; Shpend Elezi, MD; Corinna
Böttiger, MD; Julinda Mehilli, MD; Kathrin Schömig, MD; Nicolas von
Beckerath, MD. (1999). PlA Polymorphism of Platelet Glycoprotein IIIa
and Risk of Restenosis After Coronary Stent Placement. Circulation,
99: 1005-1010.
· Kao Kj. (1988). Selective elution of
HLA antigens and B2 microglobulin from human platelets by chlorochine
diphosphote. Transfusion, 28: 14-7.
· Kao K.J. (1996). Indiction of humoral
immune tolerance to major histocapatibility complex antigens by transfusions of
UVB-irradiated leukocytes. Blood, 88(11): 4375-4382.
· Katagiri. Y, Hayashi. Y, Yamamoto. K, Tanoue.
K, Kosaki. G and Yamazaki. H. (1990). Localization of von Willebrand
factor and thrombin-interactive domains on human platelet glycoprotein Ib.
Thromb. Haemostasis, 63: 122-126.
· Kelly. Michael. D, David W. Essex, Sandor S.
Shapiro, Frank J. Meloni, Teresa Druck, t Kay Huebner, and Barbara A. Konkle.
(1994). Complementary DNA Cloning of the Alternatively Expressed
Endothelial Cell Glycoprotein Ibâ (GPIbB) and Localization of the GPIbB
Gene to Chromosome 22. J. Clin. Invest, 93: 2417-2424.
· Kelton JG, Smith JW, Horsewood P, Humbert JR,
Hayward CP et Warkentin TE. (1990). Gova/b alloantigen
system on human platelets.
Blood, 75(11): 2172
-2176.
· Kekomaki, R.; Dawson, B.; McFarland, J.;
Kunicki, T. J. (1991). Localization of human platelet autoantigens to
the cysteine-rich region of glycoprotein IIIa. J. Clin. Invest. 88:
847-854.
· Kekomaki R, Raivio P, Kero P. (1992).
A new low-frequency platelet alloantigen, Vaa, on
Glycoprotein IIbIIIa associated with neonatal alloimmune thrombocytopenia.
Transfus Med, 2: 27-33.
· Kekomäki S., Jouhikainem T., Ollikainem
J.,Westman P., Laes M.A.(1993). New platelet alloantigen,
Tua, on glycoprotein IIIa families. Br.J. Hæmatol. 83(2):
306-310.
· Kekomaki S, Partanen J, Kekomaki R. (1995).
Platelet alloantigens HPA-1, -2, -3, -5 and 6 in Finns.
Transfus medecine, 5: 193-8.
· Killick S.B., Win N., Marsh J.C. (1997).
Pilot study of HLA allommunization after tranfusion with pre-storage
leucodepleted blood products in aplastic anæmia. Br.J.H. 97(3):
677-684.
· Kickler TS, Herman JH, Furihata K, Kunicki TJ,
Aster RH. (1988). Identification of Bakb, a new
platelet-specific antigen associated with post-transfusion purpura.
Blood. 71: 894.
· Kieffer N, Boizard B, Didry D, Wautier J-L,
Nurden AT. (1984). Immunochemical characterization of the
platelet-specific alloantigen Leka: A comparative
study with the PlA1 alloantigen. Blood, 64: 1212.
· Kiefel V, Santoso S, Weisheit M,
Mueller-Eckardt C. (1987). Mononisms clonal antibody-specific
immobilization of platelet antigens (MAIPA): A new tool for the identification
of platelet reactive antibodies. Blood, 70: 1722.
· Kiefel, V; Santoso, S; Katzmann, B;
Mueller-Eckhardt, C. (1988). A new platelet-specific alloantigen
Br(a): report of 4 cases with neonatal alloimmune thrombocytopenia.
Vox
Sang, 54,101-106.
· Kiefel, V., Santoso, S., Weisheit, M. and
Mueller-Eckhardt. C. (1989). The Bra/Brb
alloantigen system on human platelets. Blood, 733: 2219-2223.
· Kiefel, V.; Shechter, Y.; Atias, D.; Kroll, H.;
Santoso, S.; Mueller-Eckhardt, C. (1991). Neonatal alloimmune
thrombocytopenia due to anti-Br(b) (HPA-5a): report of three cases in two
families.
Vox
Sang. 60: 244-245.
· Kiefel V, Vicariot M, Giovangrandi Y, et al.
(1995). Alloimmunization against Iy, a low-frequency antigen on
platelet glycoprotein Ib/IX as cause of severe neonatal alloimmune
thrombocytopenic purpura. Vox Sang, 69, 250.
· Kim HO, Jin Y, Kickler TS, Blalemorek Kwoon OH,
Bray PF. (1995). Gene frequencies of the five major Human platelet
antigens in African American, white and Korean population.
Transfusion, 35, 863-867.
· Klüter, H., Fehlau, K., Panzer, S.,
Kirchner, H. & Bein, G. (1996). Rapid typing for human platelet
antigen systems-1-2-3 and 5 by PCR amplification with sequence- specific
primers. Vox Sanguinis, 71: 121-125.
· Koutsogianni Panagiota. (2004).
Nomenclature of human platelet antigens and clinical conditions.
Haema, 7: 82-88.
· Kretzschmar Evelyne, Von Ahsen Nicolas et
Trobisch Heiner. (2001). Rapid genotyping of human platelet antigen 5
with fluorophore-labelled hybridization probes on the LightCycler.
B.J.H, 114: 397-399.
· Kroll H. Kiefel V. Santoso S, Mueller-Eckherdt
C. (1990). Sr (a) I private platelet antigen on glycoprotein IIIa
associated with neonatal alloimmune thrombocytopenia. Blood, 76: 2296.
· Kroll H, Santoso S, Bohringer M, et al.
(1995). Neonatal alloimmune thrombocytopenia caused by immunization
against Oea: a new low-frequency alloantigen on platelet
glycoprotein IIIa. Blood, 86: 540a.
· Kroll H, Kiefel V, Santoso S. (1998).
Clinical aspects and typing of platelet alloantigens. Vox Sang.
74(Suppl 2): 345-54.
· Kroll H, Gardemann A, Fechter A, Haberbosch W,
Santoso S. (2000). The impact of the glycoprotein Ia collagen receptor
subunit A1648G gene polymorphism on coronary artery disease and
acute myocardial infarction. Thrombosis and Haemostasis, 83:
392.
· Kroll H, Andreas F, Andreas G. (2001).
The Role of the Glycoprotein IIb Fibrinogen Receptor Subunit T2622G
Gene Polymorphism (HPA-3) on Coronary Artery Diseaseand Acute Myocardial
Infarction. Thromb Haemost, 85: 182-3.
· Kroll H, Carl B, Santoso S, Bux J and Bein G.
(2001a). Workshops Report on the genotyping of Blood Cell
Alloantigens. Transfusion Medicine, 11: 211-219.
· Kuijpers, R. W. A. M., W. H. Ouwehand, P. M. M.
Blecker, D. Christie, and A. E. G. Kr. van den Borne. (1992).
Localization of the platelet-specific alloantigens
Koa/Kob on the N-terminal globular fragment of platelet
glycoprotein GPIbá. Blood. In press.
· Kuijpers R.W.A.M, Faber N.M, Cuypers H.Th.M,
Ouwehand W.H et von dem Borne A.E.G.Kr. (1992a). NH2 terminal globular
domain of human platelet glycoprotein Ibá has a methionine
145/threonine145amino acid polymorphism; which is associated with the HPA-2
(Ko) alloantigens. J. Clin. Invest, 89: 381-384.
· Kuijpers, R. W. A. M.; Simsek, S.; Faber, N.
M.; Goldschmeding, R.; van Wermerkerken, R. K. V.; von dem Borne, A. E. G. K.
(1993). Single point mutation in human glycoprotein IIIa is associated
with a new platelet-specific alloantigen (Mo) involved in neonatal alloimmune
thrombocytopenia. Blood, 81: 70-76.
· Kulkarni B, Mohanty D et Ghosh K.
(2005). Frequency distribution of human platelet antigens in the
Indian population. Transfusion Medicine, 15: 119-124.
· Kunicki, T. J.; Kritzik, M.; Annis, D. S.;
Nugent, D. J. (1997). Hereditary variation in platelet integrin
alpha-2-beta-1 density is associated with two silent polymorphisms in the
alpha-2 gene coding sequence.
Blood,
89: 1939-1943.
· L'Abbe D, Termblay L, Filion M et al.
(1992). Alloimmunization to platelet antigen HPA-1a (PlAl)
is strongly associated with both HLA-DRB3*0101 and HLA-DQB1*0201. Hum
Immunol, 34: 107-114.
· Lanza Francois, Nelly Kieffer, David R.
Phillips, and Laurence A. Fitzgerald. (1990). Characterization of the
Human Platelet Glycoprotein IIIa Gene Comparaison with therenchctine receptor
â subunit gene*. The Journal of biological chimestry, 265:
18098-18103.
· Lin, M., Sutherland, D.R., Rorsfall, W., Totty,
N., Yeo, E., Nayar, R., Wu, X.F. & Schuh, A.C. (2002). Cell
surface antigen CD109 is a novel member of á2
macroglobulin/C3, C4, C5 family of thioester-containing proteins.
Blood, 99: 1683-1691.
· Liu. T.C, Shih. M.C, Lin. C.L, Lin. S.F, Chen.
C.M and Chang. J.G. (2002). Gene frequencies of the
HPA-1 to HPA-8w platelet antigen alleles in Taiwanese, Indonesian, and Thai.
Ann Hematol, 81: 244-248.
· Lopez J. A., Dominic W. Chung, K F, Frederick
S. H, Earl W. Davie et Gerald J. Roth. (1988). The
á and â chains of human platelet glycoprotein Ib are both
transmembrane proteins containing a leucine-rich amino acid sequence.
Biochemistry, 85: 2135-2139.
· Lopez. J A (1994). The platelet
glycoprotein Ib-IX complex. Blood Coagulat Fibrinol, 5:
97-119.
· Lopes. N.H.M, Alexandre C. Pereira, Whady Hueb,
Paulo R. Soares, Jose R. Lanz, Bernard J. Gersh, Sergio de Oliveira, Luiz A.M.
Cesar, Jose FRamires, and Jose Eduardo Krieger. (2004). Effect of
Glycoprotein IIIa PlA2 Polymorphism on Outcome of Patients With Stable Coronary
Artery Disease and Effect of Smoking. The American Journal of
Cardiology, 93: 1469-1472.
· Lyman S, Richard H. Aster, Gian P. Visentin,
and Peter J. Newman. (1990). Polymorphism of Human Platelet Membrane
Glycoprotein IIb Associated With the Bak»/Bakb Alloantigen System.
Blood, 75: 2343-2348.
· Lyou, Jau-Yi, Chen, Ying-Ju, Hu, Hui-Yu, Lin,
Jeong-Shi & Tzeng, Cheng-Hwai. (2002). PCR with sequence-specific
primer-based simultaneous genotyping of human platelet antigen-1 to -13w.
Transfusion, 42 (8): 1089-1095
· Marcelli-Barge A, Poirier JC, Dausset J.
(1973). Alloantigens and all-antibodies of the Ko system, serological
and genetic studies. Vox Sang 24: 1.
· Masters R, Taaning E. (1998). Three
cases of platelet alloimmunisation associated with the presence of a novel
platelet-specific antibody. Vox Sang, 75: 242-6.
· Matei Daniela E, M.D. and Gary Schiller,
M.D. (2002). Immune-Mediated Thrombocytopenias.
Clinical Review. 6: 33-39.
· Mathew, Joseph P., MD Christine S. Rinder, MD,
J. Greg Howe, PhD, Manuel Fontes, MD, Jill Crouch, MHS, Mark F. Newman, MD,
Barbara Phillips-Bute, PhD, Brian R. Smith, MD and the Multicenter Study of
Perioperative Ischemia (McSPI) Research Group. (2001). Platelet PlA2
Polymorphism Enhances Risk of Neurocognitive Decline After Cardiopulmonary
Bypass. Ann Thorac Surg, 71: 663-6.
· McFarland JG. Blanchette V, Collins J, Newman
PJ, Wang R, Aster RH. (1993). Neonatal alloimmune thrombocytopenia due
to a new platelet-specific alloantibody. Blood, 81: 3318.
· Mercies P, Chike protiche C, Dabaniam-C Gamerre
M, Reviran D. (1994). Platelet antigen HPA-5 b Bra in an
Algerian population. Tissue Antigens, 43: 58-59.
· Mérieux Yves, Debost M, Bernard J,
Raffiss A ,Meyer F, Rigal D. (1997). Human platelet antigen
frequencies of platelet donors in the French population determined by
polymerase chain reaction with Sequence specific primers. Path Biol,
45: 697-700.
· Metcalfe P., Watkins N. A., Ouwehand, Kaplan
C., Newman P., Kekomaki R., M. de Haas, R. Aster, Y. Shibata, J. Smith, V.
Kiefel et S. Santoso. (2003). Nomenclature of human
platelet antigens. Vox Sanguinis, 85, 240-245.
· Metcalfe P, Waters
AH. (1993). HPA-1 typing by PCR amplification with sequence-specific
primers (PCR-SSP): a rapid and simple technique. Br J H. 85:
227-229.
· Meyer O., Hildebrandt M., Schulz B., Blasczyk
R., and Slama A. (1999). Simultaneous genotyping of human platelet
antigens (HPA) 1 through 6 using new sequence-specific primers for HPA-5.
Tranfusion, 39:1256-1258.
· Mikkelsson J, Perola M, Penttila A, et al.
(2001). Platelet glycoprotein Ibalpha HPA-2 Met/VNTR B haplotype as a
genetic predictor of myocardial infarction and sudden cardiac death.
Circulation, 104: 876-880.
· Miller SA,
Dykes DD, Polesky HF. (1988). A Simple salting-out
for extracting DNA from human nucleated cells. Nucleic Acids Res, 16:
1215.
· Mohanty D, Bipin K, Kanjaksha G, Sona N, Amit
K. (2004). Human Platelet Specific Antigens and their Importance.
Indian Pediatricts, 41: 797-805.
· Mojaat N, Halle I, Proule V, Ben Hamed L, Hmida
S, Boukef k, kaplan C. (1999). Gene frequencies of
Human platelet antigens in the Tunisian population. Tissue Antigens,
54: 201-204.
· Morawski. W, MD, PhD, M. Sanak, MD, PhD, M.
Cisowski, MD, PhD, M. Szczeklik, MD, W. Szczeklik, PhD, J. Dropinski, MD,
PhDWaclawczyk, MD, R. Ulczok, MD, and A. Bochenek, MD, PhD.
(2005). Predictions of the excessive perioperative
bleeding in patients undergoing coronary artery bypass grafting: Role of
aspirin and platelet glycoprotein IIIa polymorphism. J. of Thoracic and
Cardior Surgery, 130: 791-796.
· Mueller-Eckhardt C, Kiefel V, Grubert A, Kroll
H, Weisheit M, Weisheit M, Schmidt S, Mueller-Eckhardt G, Santoso S.
(1989). 348 cases of suspected neonatal alloimmune thrombocytopenia.
Lancet, 8634: 363-6.
· Mueller-Eckhardt C, Kiefel V, Kroll H and
Mueller-Eckhardt G. (1989a). HLA DRw6, a new immune response
marker for immunization against the platelet alloantigen Bra.
Vox Sang, 57: 90-91
· Mueller-Eckhardt C, Kiefel V, Santoso S.
(1990). Review and update of platelet alloantigen systems. Trans
Med Rev, 4: 98.
· Murata M, Furihata K, Ishida F, et al.
(1992). Genetic and structural characterization of an amino acid
dimorphism in glycoprotein Ib alpha involved in platelet transfusion
refractoriness. Blood, 79: 3086-3090.
· Murata M, Matsubara Y, Kawano K, Zamat T, Aoki
N, Yoshino H, Watanabe G, Ishikawa K, Ikeda Y. (1997). Coronary artery
disease and polymorphisms in receptor mediating shear stress-dependent platelet
activation. Circulation, 96: 3281-3286.
· Murata M, Kawano K, Matsubara Y, et al.
(1998). Genetic polymorphisms and risk of coronary artery disease.
Semin Thromb Hemost, 24: 245-250.
· Murray LJ, Bruno E, Uchida N, et al.
(1999). CD109 is expressed on a subpopulation of CD34+ cells enriched
in hematopoietic stem and progenitor cells. Exp Hematol. 27:
1282-1294.
· Nauck Markus S, Gierens H, Nauck Matthias A,
Marz W et Wieland H. (1999). Rapid genotyping of human platelet
antigen 1 (HPA-1), B.J.H, 105: 803-810.
· Nauck Markus S., Hedi Gierens, Matthias A.
Nauck, Winfried Marz and Heinrich Wieland Division of Clinical Chemistry, D
epartment of Medicine, University Hospital, Freiburg, Germany. (1999).
Rapid genotyping of human platelet antigen 1 (HPA-1) with fluorophore-labelled
hybridization probes on the LightCyclerTM. Br. J. H, 105:
803-810.
· Newman, P. J., Martin L. S., Knipp M. A, and
Kahn R. A. (1985). Studies on the nature of the human platelet
alloantigen, P1A1. Localization to a 17,000-dalton polypeptide.
Mol. Immunol. 22: 719-729.
· Newman. P J, Rebecca S. Derbes, and Richard H.
Aster. (1989). The Human Platelet Alloantigens, PlAl and
PLA2 Are Associated with a Leucine33/Proline33 Amino Acid
Polymorphism in Membrane Glycoprotein IIIa, and Are Distinguishable by DNA
Typing. J. Clin. Invest, 83: 1778-1781.
· Newman P.J and Goldberger A. (1991).
Molecular genetic aspects of human platelet antigen systems.
Baillières clinical haematology, 4: 869-88.
· Newman PJ, McFarland JG, Aster RH
(1994a). Alloimmune thrombocytopenias. In Thrombosis and hemorrage.
Cambridge, KA. Blackwell Scientific, 529-543.
· Newman P. J. (1994). Nomenclature of
human platelet alloantigens systems: a problem with the HPA system.
Blood, 83: 1447-1451.
· Newman P. J. et Valentin N.
(1995). Human platelet alloantigens: recent findings, new
perspectives. Thromb Hemost, 74: 234-239.
· Nomura. S, Shouzu. A, Omoto. S, Matsuzaki. T, M
Yamaoka, M Abe, M Hosokawa, M Nishikawa, T Iwasaka, and Fukuhara, S.
(2006). Genetic analysis of HLA, NA and HPA typing in type 2 diabetes
and ASO. Int J Immunogenet. 33(2) : 117-122.
· Norton A., Allen D.L and Murphy M.F.
(2004). Platelet alloantigens and antibodies and their clinical
significance. Review Immuno Heamatology, 20: 89-101.
· Noris P, S. Sirnsek, L.G. de Bruijne-Adrniraal,
L. Porcelijn, E. Huiskes, G.J. van der Vlist, E.F. van Leeuwen C.E. van der
Schoot, and A.E.G.Kr. Von dern Borne. (1995). Maxa, A New
Low-Frequency Platelet-Specific Antigen Localized on Glycoprotein IIb, Is
Associated With Neonatal Alloimmune Thrombocytopenia. Blood, 86:
1019-1026.
· Nurden. A.T. (1995). Polymorphisms of
human platelet membrane glycoproteins: structure and clinical significance.
Thromb Haemost, 74: 345-351.
· Okamoto N., Kennedy S.D, Barron-Casella
E.A, Casella J.F, Inoko H., Kickler T.S. (1998). Identification of a
human heavy chain antibody fragment directed against human platelet alloantigen
18 by phage display library. Tissue Antigens, 51: 156-163.
· Panzer S, Auerbach L, Cechova E, et al.
(1995). Maternal alloimmunization against fetal platelet antigens: a
prospective study. Br J Haematol, 90: 655-60.
· Pamukcu Burak, MD, Oflaz Huseyin, MD, and
Nisanci Yilmaz, MD. (2005). The role of platelet glycoprotein IIIa
polymorphism in the high prevalence of in vitro aspirin resistance in patients
with intracoronary stent restenosis. Am Heart J, 149: 675-80.
· Perret, B., Chap, H. and Douste-Blazy, L.
(1979). Asymmetric distribution of arachidonic acid in the plasma
membrane of human platelets. A determination using purified phospholipases and
a rapid method for membrane isolation. Biochem. Biophys. Acta. 556,
434-446.
· Peyruchaud, O. Alan N and François B.
(1995). Bilateral linkage between a new deletion ploymorphism in
intron 21 of the GP IIb gene and the HPA-3b (BaKb) Determinant.
B. J. H. 91: 747-751.
· Peyruchaud. O, Bourre. F, Morel-Kopp. M.-C,
Reviron. D, Mercier. P, A. Nurden, and Kaplan. C. (1997). HPA-10wb (Laa):
Genetic Determination of a New Platelet-Specific Alloantigen on
Glycoprotein IIIa and Its Expression in COS-7 Cells.
Blood, 89: 2422-2428.
· Phillips, D. R., I. F. Charo, L. P. Parise, and
L. A. Fitzgerald. (1988). The platelet membrane glycoprotein IIb-IIIa
complex. Blood, 71: 831-843.
· Poncz M, Eisman R, Heidenreich R, Silver SM,
Vilaire C;,Surrey S, Schwartz E, Bennet JS. (1987). Structure of the
platelet membran glycoprotein IIb. Homology to the subunits of the vitronectin
and fibronectin membrane receptors. J Biol Chem, 262: 8476.
· Poncz M. & Newman P. J. (1990).
Analysis of rodent platelet glycoprotein IIb: evidence for evolutionarily
conserved domains and alternative proteolytic processing. Blood, 75:
1282-1289.
· Prandini, M. H.; Denarier, E.; Frachet, P.;
Uzan, G.; Marguerie, G. (1988). Isolation of the human platelet
glycoprotein IIb gene and characterization of the 5- prime flanking region.
Biochem Biophys. Res. Commun, 156: 595-601.
· Qintanar Andre, Jallu Vincent, Legros
Yann and Kaplan Cecile. (1998). Human platelet antigen genotyping
using a fluorescent SSCP technique with an automatic
sequencer. British Journal of Haematology, 103:
437-444.
· Raisonnier A. (2002). Biologie
génique.
· Rajagopalan, V., Essex D. W., Shapiro S. S.,
and Konkle. B. A. (1992). Tumor necrosis factor-a modulation of
glycoprotein Iba expression in human endothelial and erythroleukemia cells.
Blood, 80: 153-16 1.
· Reiner AP, Kumar PN, Schwartz SM, Longstreh WT,
Pearce RM, Rosendaal F, Psaty BM, Siscovick DS. (2000). Genetic
variants of platelet glycoprotein receptors and risk of stroke in young women.
Stroke, 31: 1628
· Reiner AP, Schwartz SM, Kumar PN, et al
(2001). Platelet glycoprotein IIb polymorphism, traditional risk
factors and nonfatal myocardial infarction in young women. Br J
Haematol, 112: 632-636.
· Rosa, J-P., Bray, P.F. Gayet, 0. Johnston, G.
I. Cook, R.G. Jackson, K.W. Shuman, M.A. and McEver. R.P. (1988).
Cloning of glycoprotein IIIa cDNA from human erythroleukemia cells and
localization of the gene to chromosome 17. Blood. 72: 593-600.
· Roth. G. J. (1994). The Wanderings of
a Platelet Gene: What Is "Neo" Ibâ Telling Us?
Journal of Clinical Investigation, Inc. 93:
2301-2302.
· Rozman P, Drabbels J,
Schipper RF, Doxiadis I, Stein S, Claas FH. (1999). Genotyping for
human platelet-specific antigens HPA-1, -2, -3, -4 and -5 in the Slovenian
population reveals a slightly increased frequency of HPA-1b and HPA-2b as
compared to other European populations. Eur J Immunogenet, 26: 265.
· Rozman P. (2002).
Platelet antigens the role of human platelet alloantigens (HPA) in blood
transfusion and transplantation. Transpl Immunol, 10: 165-181.
· Ruggeri, Z. M. (1991). The platelet
glycoprotein Ib-IX complex. Prog. Hemostasis, Thromb, 10: 35-68.
· Sachs UJH, Kiefel V, Bohringer M,
Afshar-Kharghan V, Kroll H and Santoso S. (2000). Single amino acid
substitution in human platelet glycoprotein Ibß is responsible for the
formation of the platelet-specific alloantigen Iya. Blood,
95: 1849-1855.
· Salomon Ophira, MD, Nurit Rosenberg, PhD, David
M. Steinberg, PhD, Ruth Huna-Baron, MD, Joseph Moisseiev, MD, Rima Dardik, PhD,
Oren Goldan, MD, Shimon Kurtz, MD, Aviya Ifrah, MSc, Uri Seligsohn, MD.
(2004). Nonarteritic Anterior Ischemic Optic Neuropathy Is Associated
with a Specific Platelet Polymorphism Located on the Glycoprotein Ibá
Gene. Ophthalmology, 111: 184-188.
· Santoso, S, Kiefel V et Mueller-Eckhardt.
(1989). Immunochemical Characterization of the new platelet
alloantigen system Bra/Brb. Br. J. Haematol, 72:
191-198.
· Santoso, S., Kiefel, V., and Mueller-Eckhardt,
C. (1989a). Human platelet alloantigens Bra/Brb
are expressed on the very late activation antigen 2 (VLA-2) of T lymphocytes.
Hum. Immunol. 25: 237-246.
· Santos S, Kalb R., Kiefel V et Muller-Eckhardt
C. (1993). The presence of messenger RNA for HLA classe I in human
platelet and is compatibility for protein biosynthesis. Br J Haematol,
84: 451-6.
· Santoso S, Kiefel V, Masri R, Mueller
Eckhardt. (1993a). Frequency of platelet specific
antigens among Indonesians. Transfusion, 33: 739-741.
· Santoso S, Kalb R, Walka M, Kiefel V,
Mueller-Eckhardt C, and Newman P J. (1993b). The Human Platelet
Alloantigens Bra and Brb Are Associated with a Single
Amino Acid Polymorphism on Glycoprotein Ia (Integrin Subunit a2). J. Clin.
Invest. 92: 2427-2432.
· Santoso S, Kalb R, Kroll H, Walka M, Kiefel V,
Mueller-Eckhardt C, Newman PJ. (1994). A point mutation leads to an
unpaired cysteine residue and a molecular weight polymorphism of a functional
platelet b3 integrin subunit. J Biol Chem, 269: 8439.
· Santoso S, Kiefel V.
(1998). Human Platelet-Specific Alloantigens: Update. Vox
Sang, 74(suppl. 2): 249-253.
· Santoso S; Amrhein J, Hofmann H.A., Sachs U.
J.H., Walka M M., Kroll H et Kiefel V. (1999). A point mutation
Thr799Met on the á2 integrin leads to the formation of
new Human Platelet Alloantigen Sita and affects
collagen-induced aggregation. Blood, 94: 4103-4111.
· Santoso, S., Kunicki, T.J., Kroll, H.,
Haberbosch, W., Gardemann, A., (1999a). Association of the platelet
glycoprotein Ia C807T gene polymorphism with nonfatal myocardial infarction in
younger patients. Blood, 93: 2449-2453.
· Santoso S. (2001). Platelet
polymorphisms in thrombotic disorders. Transfus Clin.Biol. 8:
261-266.
· Santoso S, Kiefel Volker, Richter lna G, Ulrich
J. H. Sachs, Abdul Rahman, Bettina Carl, and Harmut Kroll.
(2002). A functional platelet fibrinogen receptor with a
deletion in the cysteine-rich repeat region of the â3 integrin: the
Oea alloantigen in neonatal alloimmune thrombocytopenia.
Blood, 99: 1205-1214.
· Santoso S, Zimmermann P, Sachs UJ, Gardemann A.
(2002a). The impact of the Kozak sequence polymorphism of the
glycoprotein Iba gene on the risk and extent of coronary heart disease.
Thromb Haemost, 87: 345-6.
· Santoso S, Hartmut Kroll, Cornelia L.
Andrei-Selmer, Ines Socher, Angela Rankin, Evelyne Kretzschmar, Nicholas A.
Watkins, and Willem H. Ouwehand. . (2006). A naturally occurring
Leu33Val mutation in b3-integrin impairs the HPA-1a epitope: the third allele
of HPA-1. Transfusion. 46:790-799.
· Schuh A.C, Watkins N.A, Nguyen Q, Harmer N.J,
Lin M., Prosper J.Y.A, Campbell K, Sutherland D.R, Metcalfe P, Horsfall W and
Ouwehand W.H. (2002). A tyrosine 703 serine
polymorphism of CD109 defines the Gov platelet alloantigens. Blood,
99: 1692-1698.
· Seo DH, Parks S, Kim DW, Furihata K, Ueno I and
Hank S. (1998). Gene frequencies of eigth Human platelet specific
antigens in Koreans. Transfus mede, 8: 129-132.
· Shibfata Y., Matsuda I., Miyajn. T. Ichikawa
Y., Yuka A. (1986). A new platelet antigen involved in two cases of
neonatal alloimmune thrombocytopenia. Vox Sang, 50: 177-180.
· Shen Y, Gabriel M., Dong Jing-fei, Schade A,
McLintire L V., Kenny D, Whisstock J. C., Berndt M. C., López
José A. and Andrews R. K. (2000). Requirement of leucine-rich
repeats of glycoprotein (GP) Ibá for shear-dependent and static binding
of von Willebrand factor to the platelet membrane GPIb-IX-V complex.
Blood, 95: 903-910.
· Shibata Y, Matsuda I, Miyaji T, Ichikawa Y.
(1986). Yuka, a new platelet antigen involved in two cases
of neonatal alloimmune thrombocytopenia. Vox Sang, 50: 177.
· Shih M.C, Liu T.C, Lin I-Ling, Lin S.F, Chen
C.M and Chang. J.G. (2003). Gene frequencies of the HPA-1 to HPA-13,
Oe and Gov platelet antigen alleles in Taiwanese, Indonesian, Filipino and Thai
populations. International Journal of Molecular Medecine, 12:
609-614.
· Simpson E., Roopenian D.
(1997). Minor histocomptability antigens. Curr. Opin.
Immunol. 9(5): 655-661.
· Simsek S., Bleeker P.M.M., Heeremans J.AndVon
DemBorne A.E.G.Kr. (1994). Human platelet antigen-5(Br) genotyping by
ASPA: allele-specific primer amplification (PCR-SSP). Br.J. Haemot.
88: 659-661.
· Simsek S, Vlekke ABJ, Kuijpers RWAM,
Goldenschmeding R, von dem Borne AEGK. (1994a). A new private platelet
alloantigen Gro (a), localized on glycoprotein IIIa, involved in neonatal
alloimmune thrombocytopenia. Vox Sang 67, 302.
· Simsek S., Gallardo D., RiberaA.AndVon DemBorne
A.E.G.Kr. (1994b). the human platelet alloantigens, HPA-5(a+, b-) and
HPA-5(a-, b+), are associated with a Glu505/Lys505
polymorphism of glycoprotein Ia (the á2 subunit of VLA-2).
Br.J of Haematol, 86: 671-674.
· Simsek S, Faber NM, Bleeker PM, Vlekke A.B.J,
Huiskes E, Goldschmeding R, and von dem Borne A.E.G.Kr. (1993).
Determination of human platelet antigen frequencies in the Dutch population by
immunophenotyping and DNA (allele-specific restriction enzyme) analysis.
Blood, 81(3): 835-840.
· Simsek S., Folman C., Van er Schoot C. E. and
Von Dem Borne A. (1997). The Arge633His substitution responsible for
the private platelet antigen Groa unravelled by SSCP analysis and
direct squencing. British Jornal of Haematology, 97, 330-335.
· Skogen B., Bellissimo D.B., Hesser M.J.,
Santoso S., Aster R.H., Newman P.J., Mc. Ferland J.C. (1994). Rapid
determination of platelet alloantigen genotypes by polymerase chain reaction
using allele-spacific primers. Transfusions; 34: 955-60.
· Skouri H., Soua H., Seket B., Elomri H.,
Mounastiri K., Ayadi A., Gueddiche M.N., Sfar M.T., Bierling P.
H. (2003). Thrombopénies néonatales dues
à un allo-anticorps anti-HPA-5b: à propos de deux cas
observés en Tunisie HPA-5b neonatal allo-immune thrombocytopenia : two
cases described in Tunisia. Archives de pédiatrie, 10,
887-890.
· Skouri H, Bettaieb A, Fromont P, Elomri H,
Ennabli S, Bierling P. (2005). Platelet and granulocyte
alloimmunisation in multitransfused Tunisian patients. Eur J Haematol,
75, 248-251.
· Smith JW, Hayward CP, Horsewood P, Warkentin
TE, Denomme GA and Kelton JG. (1995). Characterization and
localisation of the Gova/b alloantigens
to the glycosylphosphatidylinositol anchored protein CDw109 on
human platelets. Blood, 86 (7), 2807-2814.
· Sosnoski, D. M., Emanuel, B. S., Hawkins, A.
L., van Tuinen, P., Ledbetter, D. H., Nussbaum, R. L., Kaos, F. T., Schwartz,
E., Phillips, D. & Bennett, J. S. et al. (1988). Chromosomal
localization of the genes for the vitronectin and fibronectin receptors alpha
subunits and for platelet glycoproteins IIb and IIIa. J Clin Invest,
81: 1993-1998.
· Staatz, W. D., Rajpara, S. M., Wayner, E. A.,
Carter, W. G. and Santoro. S. A. (1989). The membrane glycoprotein
Ia-IIa (VLA-2) complex mediates the Mg2+-dependent
adhesion of platelets to collagen. J. Cell Biol. 108: 1917-1924.
· Steffensen R, Kaczan E, Varming K, Jersild C.
(1996). Frequency of platelet-specific alloantigen in a Danich
population. Tissue antigen, 48: 93-96.
· Suciu-Foca N, Reed E, Rubinstein P, MacKenzie
W, Ng AK, King DW. (1985). A late-differentiation
antigen associated with the helper inducer function of human T cells.
Nature, 318: 465-467.
· Sutherland DR, Yeo E, Ryan A, Mills GB, Bailey
D, Baker MA. (1991). Identification
of a cell-surtace antigen associated with activated T lymphoblasts and
activated platelets. Blood, 77: 84-93.
· Sykes T C F, Fegan C, Mosquera D.
(2000). Thrombophilia, polymorphisms, and vascular disease. J
Clin Pathol: Mol Pathol, 53: 300-306.
· Tanaka S, Ohnoki S, Shibata K, Okubo Y,
Yamaguchi H et Shibfata. (1995). Genes frequencies of human platelet
antigens on glycoprotein IIIa in Japanese. Transfusion, 36, 813817.
· Takada, Y.; Hemler, M. E. (1989). The
primary structure of the VLA-2/collagen receptor alpha-2 subunit (platelet
GPIa): homology to other integrins and the presence of a possible
collagen-binding domain.
J.
Cell Biol. 109: 397-407.
· Thude H., Gatzka E., Anders O., Barz D. (1999).
Allele frequencies of human platelet antigen 1, 2, 3 and systems in
patients with chronic refractory autoimmune thrombocytopenia and in normal
persons. Vox sang, 77: 149-153.
· Thornton MA, Poncz M, Korostishevsky M, et al.
(1999). The human platelet alphaIIb gene is not closely linked to its
integrin partner beta 3. Blood, 94, 2039-2047.
· Tomiyama Y, Take H, Ikeda H, et al.
(1990). Identification of the platelet-specific alloantigen,
Naka, on platelet membrane glycoprotein IV. Blood, 75(3),
684-687.
· Tseng L.H., Lin M.T., Hansen J.A. (1999).
Correlation between disparity for the minor histocomptability antigen
HPA-1 and the development of acute graft-versus-host disease after allo-geneic
marrow transplantation. Blood, 94(8): 2911-2914.
· Ulrichts H, Vanhoorelbeke K,
Cauwenberghs S, Vauterin S, Kroll H, Santoso S et Deckmyn H. (2003).
Von Willebrand factor but not á- thrombin binding to platelet
glycoprotein Ibá is influenced by the HPA-2 polymorphism.
Arterioscelerios thrombosis and vascular biology, 23: 1302-1307.
· Ulrich H. Frey, Nese Aral, Norbert Muller,
Winfried Siffert. (2003). Cooperative effect of GNB3 825C>T and
GPIIIa PI(A) polymorphisms in enhanced platelet aggregation. Thrombosis
Research, 109, 279- 286
· Unkelbach K., Kalb R., Kroll H.,
Muller-Eckhardt C. and Kiefel V. (1995). Genomic RFLP typing of human
platelet alloantigens Zw (PLA), Bak and Br (HPA-1, 2, 3, 5). B. J. H,
89, 169-176.
· Valentin N, Gian P. Visentin, and
Newman. P. J. (1995). Involvement of the Cysteine-Rich Domain of
Glycoprotein IIIa in the Expression of the Human Platelet Alloantigen,
PIA1: Evidence for Heterogeneity in the Humoral Response.
Blood, 85: 3028-3033.
· Vainchenker W, Debili N, Mouthon M-A, Wendling
F. (1995). Megakaryocytopoiesis: cellular aspects and regulation.
Oncol Hematol; 20; 165-92.
· Van Der Shoot, C. E., M. Wester, A. E. G. Kr.
von Dem Borne, and H. G. Huisman. (1986). Characterization of
platelet-specific alloantigens by immunoblotting: localization of Zw and Bak
antigens. Br. J. Haematol, 64: 715-723.
· Van Leeuwen EF, Leeksma OC, van Mourik JA,
Engelfriet CP. von dem Borne AEGKr. (1982). The effect of the binding
of anti-Zwa antibodies on platelet function. Blood, 59: 23.
· Van der Harst D., Goulmy E., Falkenburg J.H.
(1994). Recongnition of minor histocomptability antigens on
lymphocytic and myeloid leukemic cells by cytotoxic T-cell clones.
Blood, 83(4) : 1060-1066.
· Van Loghem JJ, Dorfmeijer H, van der Hart M,
Schreuder F. (1959). Serological and genetical studies on a platelet
antigen (Zw). Vox Sang, 4: 161.
· Van Goor Mary-Lou P.J., Encarna Gomez Garera,
Geert-Jan Brouwers, Frank W.G. Leebeek, Peter J. Koudstaal, Diederik W. J.
Dippel. (2003). PLA1/A2 polymorphism of the platelet
glycoprotein receptor IIb/IIIa in young patients with cryptogenic TIA or
ischemic stroke. Thrombosis Research, 108: 63-65.
· Von dem Borne Aeg. K et Decary H. (1990).
Nomenclature of platelet specific antigens.
Br. J. Hematol, 74: 2039-240.
· Von dem Borne AEG Kr, von Riesz E, Verheught
FWA, ten Cate JW, Koppe JG, Engelfriet CP, Nijenhuis LE. (1980).
Baka, a new platelet-specific antigen involved in neonatal
allo-immune thrombocytopenia. Vox Sang 39: 113.
· Von dem Borne A.E.G.K., Kaplan C. (1995).
Nomenclature of human platelet alloantigens. Blood.
85:1409-1410.
· Von dem. Borne A.E., Decary F. (1990a).
ISCH/ISBT Working Party on platlet serology. Nomenclature of
platelet-specific antigens. Vox sang, 58(2):176.
· Vijayan KV, Goldschmidt-Clermont PJ, Roos C,
Bray PF. (2000). The Pl(A2) polymorphism of integrin beta(3) enhances
outside-in signaling and adhesive functions.
J Clin Invest, 105(6) : 793-802.
· Walchshofer S., Ghali D., Fink M.,
Panzer-Grümayer E.R., Panzer S. (1994). A rare
leucine40/arginine40 polymorphism on platelet glycoprotein IIIa is linked to
the human platelet antigen Ib. Vox sang; 67(2): 231-234.
· Ware J. (1998). Molecular analyses of
the platelet glycoprotein Ib-IX-V receptor. Thromb Haemost, 79:
466-478.
· Wagner K., W. Giles, C. Johnson, P. Bray, P.
Goldschmidt-Clermont, J. Croft, V. Brown, B. Stern, B. Feeser, D. Buchholz, C.
Earley, R. Macko, R. McCarter, M. Sloan, P. Stolley, R. Wityk, M. Wozniak, T.
Price, and S. Kittner. (1998). Platelet Glycoprotein Receptor IIIa
Polymorphism PlA2 and Ischemic Stroke Risk: The Stroke Prevention in Young
Women Study. Stroke, 29: 581-5
· Wang R., Furihata K., Mc Farland J.G., Friedman
K., Aster R. H and Newman P.J. (1992). An amino acid polymorphism
within the RGD binding domain of platelet menbrane glycoprotein III ais
responsible fort he formation of the Pena / Penb alloantigen system.
Clinical invest. 90: 2038- 2043.
· Wang, R., McFarland, J.G., Kekomäki, R.
& Newman, P.J. (1993). Amino acid 489 is encoded by mutational
??hot spot? on the â3 integrin chain: the Ca/Tu human platelet
alloantigen system. Blood, 82, 3386-3391.
· Wager CL, Mascelli MA, Neblock DS, Weisman HF,
Coller BS, Jordan RE. (1996). Analysis of GPIIb-IIIa receptor number
by quantification of 7E3 binding to human platelets. Blood, 88: 907.
· Watkins N A., Elisabeth S-R, David L. A, Graham
J. H, Nicolaas H. C. B, Graham A. S, Paul Metcalfe, Michael F. Murphy, Nelly
Keiffer, and Willem H. O. (2002). HPA-1a phenotype-genotype
discrepancy reveals a naturally occuring Arg93Gln Substitution in the platelet
â3 integrin that disrupts the HPA-1a epitope. Blood, 99:
1833-1839.
· White. J. G. (1969). The submembrane
filaments of blood platelets. Am. J. Path. 56, 267-272.
· Weber S, Stefan A. Hummel, Artur-Aron Weber,
Rudolf F. Zirwes. Olaf H. Weiner et Bernadette E. Reuber.
(2002). Genotyping of human platelet antigen-1 by gene amplification
and labelling in one system and automated fluorescence correlation
spectroscopy. British Journal of Haematology, 116:
839-843.
· Weiss EJ, Bray PF, Tayback M, Schulman SP,
Kicker TS, Becker LC, Weiss JL, Gerstenblith G, Goldschmidt-Clermont PJ.
(1996). A polymorohism of a platelet glycoprotein receptor as an
inherited risk factor for coronary thrombosis. New England Journal of
Medicine, 334: 1090.
· Weiss, E. J. Goldschmidt-Clermont, P. J.
Grigoryev, D. Jin, Y. Kickier, T. S. Bray, P. E. (1995).
A monoclonal antibody (SZ21) specific for Platelet GPIIIa
distinguishes PlA1 from P1A2. Tissue
Antigens: 46: 374-381.
· Westman, P. Hashemi-Tavoularis, S. Blanchette,
V. Kekomäki, S. Laes, M. Porcelijn, L. and Kekomäki, R. (1997).
Maternal DRB1*1501, DQA1*0102, DQB1*0602 haplotype in fetomaternal
alioimmunization against human platelet alloantigen HPA-6b (GPIIIa-Gln489).
Tissue Antigens, 50: 113-118.
· Wicki, A. N., and Clemetson. K. J.
(1987). The glycoprotein Ib complex of human blood platelets. Eur.
J. Biochem, 163: 43-50.
· Williamson L M., Gerald H, Janet R, Christopher
R. Palmer, Caroline M, Ruth Hadifield, Darren H, Shirley J, and Willem H O.
(1998). The natural history of fetomatrnal alloimmunisation to
the platelet-specific antigen HPA-1a (Pl1A, Zwa) as
determined by anntenatal screening. Blood, 92:
2280-2287.
· Wiwanitkit Viroj. (2005). Gene
Frequencies of the Human Platelet Antigen-3 in Different Populations. Clin
Appl Thrombosis/Hemostasis, 11(1): 89-93.
· Woods, V. L., Jr.; Pischel, K. D.; Avery, E.
D.; Bluestein, H. G. (1989). Antigenic polymorphism of human very late
activation protein-2 (platelet glycoprotein Ia-IIa): platelet alloantigen
Hca.
J.
Clin. Invest, 83: 978-985.
· Wu G, Meloni FJ, Shapiro. S. S.
(1996). Platelet glycoprotein (GP) IX associates with GPIb in the
platelet membrane GPIb complex. Blood. 87: 2782.
· Wyler B., Daviet L., Bortkiewicz H., Bordet
J.C., Mc Gregor J.L. (1990). Cloning of the Cdna encoding human
platelet membrane glycoprotein IV. Blood, 75 (3): 684-687.
· Xiong JP, Stehle T, Goodman SL, Arnout MA.
(2004). A novel adaptation of the integrin PSI domain revealed from
its crystal structure. J Biol Chem., 279, 40252-4.
· Yamamoto N, Ikeda H, Tandon N. N.
(1990). A platelet membrane glycoprotein (GP) deficiency in healthy
blood donors: Naka-platelets lack detectable GPIV (CD36).
Blood, 76(9):1698-1703.
· Yagi M, Edelhoff S, Disteche CM, Roth GJ.
(1995). Human platelet glycoprotein V and IX: mapping of two
leucine-rich glycoprotein genes to chromosome 9 and analysis of structures.
Biochemistry. 34:16-132.
· Yamarnoto Naornasa, Noriko A, Hitoshi S, Hiroh
Y, and Kenjiro T. (1994). Platelet Glycoprotein IV (CD36) Deficiency
Is Associated With the Absence (Type I) or the Presence (Type II) of
Glycoprotein IV on Monocytes. Blood, 83. 392-3.
· Zimrin A. B., Eisman R., Vilaire G., Schwartz
E., Bennett J. S., and Poncz M. (1988). Structure of platelet
glycoprotein IIIa. A common subunit for two different membrane receptors.
J. Clin. Invest, 81: 1470-1475.
· Zotz R. B., Giers G., Maruhn-debowski B and
scharf. R. E. (1997). Genetic typing of human plalet antigen 1 (H P A
-1) by oligonucleotide ligation assy in a specific and reliable semi-automated
system. British journal of haematology, 96: 198-203.
Abstract :
· Jaspers, M.; Zhang, Z.; Marynen, P.; Vekemans,
S.; Aly, M. S.; Cuppens, H.; Hillicker, C.; Cassiman, J.-J. (1991).
Localization of the genes encoding the alpha-2 and alpha-4 subunits of the
human VLA-receptors to chromosome 5q23-31 and 2q31-32 respectively.
Cytogenet.
Cell Genet, 58: 1870.
· Décary S. F, L'Abbe' D, Tremblay L,
Chartrand P. (1991). The immune response to the HPA-1a antigen :
Association with HLA-DRw52a. Transfus Med, 1: 55.
· Kuijpers, R. W. A. M., P. W. Modderman, P. P.
M. Bleeker, W. H. Ouwehand, and A. E. G. Kr. von dem Borne. (1989).
Localization of the platelet-specific Ko-system antigens
Koa/Kob on GPIb/IX. Blood. 74(Suppl. 1)58a, 205.
· Thèses et
mémoires:
· Laurent Tremuth. (2004). Le rôle
de la taline dans la connexion des intégrines au cytosquelette-une
étude structurale et fonctionnelle.
· Nathalie Hezard. (2004). Apport de la
cytometrie en flux dans l'étude de la physiologie plaquettaire et de la
modulation pharmacologique des fonctions plaquettaires.
· Miadi Najla.
(2003-2004). " L'alloimmunisation anti plaquettaire chez
les femmes enceintes tunisiennes : étude prospective".
Mémoire de DEA.
· SAADI Hanene. (2005-2006).
Le polymorphisme des antigènes plaquettaires dans la population
tunisienne. Mémoire de mastere.
Adresses Internet
· http://www gene.ucl.ac.uk/nomenclature.
· http://www
ncbi.nlm.nih.gov/BLAST/.
· http://www ebi.ac.uk/ipd/hpa/.
· http: www.ncbi.nlm.nih.gov/.
·
http://www.ncbi.nlm.nih.gov/BLAST/bl2seq/wblast2.cgi.

|
|


