|
UNIVERSITE DE ROUEN
Ecole Doctorale Normande Chimie-Biologie
THESE
présentée par
Ludovic Carlier
pour obtenir le titre de
Docteur de l'Université de Rouen
Spécialité : Biologie Structurale et
Fonctionnelle
Production de domaines recombinants PRODH en vue de
l'analyse structurale
&
Caractérisation de la région 51-160 de la
protéine KIN17
humaine par RMN et Modélisation
Moléculaire
Soutenue le 10 Juillet 2006 devant le jury : Président
Patrice LEROUGE
Rapporteur Constantin T. CRAESCU Rapporteur Jean-Pierre SIMORRE
Examinateur Bernard GILQUIN Examinateur Laure GUILHAUDIS Directeur de
Thèse Daniel DAVOUST
préparée au Laboratoire de Résonance
Magnétique Nucléaire de
l'Equipe de Chimie Organique et Biologie Structurale (CNRS UMR
6014, IFRMP 23)
A la mémoire de ma belle-maman,
Belle tu l'as été, quand tu m'as
élevé comme ton propre fils, Belle tu l'es
restée, lorsque tu as fait face avec courage à la maladie, et
Belle, tu le seras toujours dans le coeur de tes enfants.
« Rêve de grandes choses, cela te permettra
au moins d'en faire de toutes petites » Jules Renard
(1864-1910)
Remerciements
Je tiens en premier lieu à remercier le
Docteur Constantin Craescu, Directeur de Recherche à l'Institut
Curie de l'Université d'Orsay, et le Docteur Jean-Pierre
Simorre, Directeur de Recherche à l'Institut de Biologie Structurale
Jean-Pierre Ebel de l'Université
de Grenoble, pour l'honneur qu'ils me font en
acceptant de juger ce travail. Je voudrais également remercier
le Professeur Patrice Lerouge, du Laboratoire de Structure/fonctions des
glucides de l'Université de Rouen, et le Docteur Bernard
Gilquin, du Département d'Ingénierie et d'Etude des
Protéines du CEA de Saclay, de participer à ce jury.
Voici donc le moment tant attendu des remerciements...
Après quatre années de thèse
où se sont alternés « des hauts
» et « des bas », il est temps pour moi de confirmer
une rumeur de plus en plus persistante : ces quatre années de
doctorat, et les deux sujets sur lesquels je me suis penché, n'ont
pas eu raison de ma passion pour la recherche qui en sort bien plus
ravivée. Alors, je le dis haut et fort : « la recherche, c'est
formidable ! », même si (ma famille et mes proches en
conviendront) cette passion demande parfois quelques sacrifices. J'ai eu
la chance d'avoir été accueilli dans pas moins de 4
laboratoires durant cette thèse, et d'avoir côtoyé
des chercheurs de différents horizons qui m'ont
énormément appris, tant sur le plan scientifique, que sur le plan
humain. A moi à présent de remercier toutes les personnes qui
m'ont aidées, de près ou de loin, à obtenir les
résultats présentés dans ce manuscrit. J'espère ne
pas en oublier !!!
J'exprime toute ma reconnaissance au Professeur
Daniel Davoust, Directeur de l'Equipe de Chimie organique et Biologie
Structurale de l'Université de Rouen, pour m'avoir accueilli dans son
laboratoire et m'avoir permis de réaliser cette thèse. Je vous
remercie de
la confiance que vous m'avez accordée depuis le
premier apprentissage que j'ai effectué dans votre laboratoire de RMN
dans le cadre d'un stage de maîtrise de chimie, il y a
déjà quelques années...
J'exprime mes profonds remerciements aux Docteurs
Laure Guilhaudis et Isabelle Milazzo pour avoir encadré ce
travail de thèse. Merci d'avoir partagé vos connaissances
scientifiques avec un « zouave » de mon espèce. La
qualité de vos rapports humains, le suivi constant de ce travail, et
l'émulation scientifique dont vous avez fait part, ont
largement contribué à la réussite de cette thèse :
« chapeaux bas !!! ».
Que les rencontres furent nombreuses et enrichissantes au
cours de ces quatre années! Des médecins aux modélistes,
en passant par les généticiens, les biochimistes, et bien
sûr les RMNistes, vous m'avez formé à
différentes techniques dans vos laboratoires respectifs, et
transmis un savoir extraordinaire à travers de nombreuses
discussions, qui se sont parfois prolongées au-delà des
heures réglementaires autour d'un petit verre sur une
terrasse...
? Au Laboratoire de Génétique
Moléculaire de l'EMI 9906 (INSERM U614) :
J'adresse mes remerciements au Professeur Thierry
Frébourg pour m'avoir accueilli dans son laboratoire et
intégré à son équipe pendant les huit premiers mois
de cette thèse. Je remercie également le Docteur Dominique
Campion pour l'intérêt qu'il a porté à mon
travail.
Un grand merci à Hélène Jacquet-Delmulle
pour m'avoir introduit dans l'univers de
la proline déshydrogénase et de la
schizophrénie. Ton aide et ton initiation à la biologie
moléculaire m'ont été très précieux ! Merci
également à Grégory Raux d'avoir construit le premier
vecteur de surexpression de PRODH même si, à priori, nous n'avons
pas la même vision de la recherche et des relations humaines.
Je tiens à remercier très
chaleureusement le Docteur Chahrazed El Hamel qui m'a prise sous son
aile pendant mon séjour dans ce laboratoire. Je souhaite te
témoigner toute mon admiration pour l'étendue de tes
connaissances, la qualité de ton encadrement, et l'immense
gentillesse qui te caractérise.
Un grand merci à Cécile, Magalie, Nathalie,
Jackie, Audrey, Olivier, Anne, et Isabelle pour votre amitié et votre
précieuse aide. Courage Olivier, tu vas y arriver !!!
? Au Laboratoire de Marquage des Protéines du
CEA de Saclay :
Difficile d'écrire ces quelques lignes sans une
certaine émotion car mon passage au LMP a été l'occasion
de rencontrer des personnes plus formidables les unes que les autres. Mes
remerciements s'adressent tout d'abord au Docteur Roger Genet, responsable de
cette équipe, qui m'a accueilli avec sympathie et enthousiasme avant de
partir vers des fonctions ministérielles.
J'exprime ma profonde reconnaissance aux Docteurs
Muriel Gondry et Sandrine
Braud, les « deux femmes de ma thèse ». Je
ne te remercierai jamais assez Muriel pour tout
ce que tu as fait pour moi : ton écoute et
tes encouragements m'ont redonné confiance lorsque le moral n'y
était plus. Tu m'as énormément appris dans le domaine de
la protéine recombinante que tu compares si joliment à de la
« cuisine » ! Un grand merci également à
ma p'tite mamie Braud qui m'a encadré avec tout le
sérieux et la rigueur qu'on lui connaît. J'ai eu la chance de
découvrir la femme formidable qui se cachait au fond de la «
working girl », et ça vaut le détour !!! (comme quoi il ne
faut jamais se fier aux apparences).
Mon passage au LMP n'aurait pas été si
plaisant sans la présence inattendue d'un fan club de groupies
présidé par Rachel Amouroux et Muriel Bahut. Merci
beaucoup Rachel, alias « super boomeuse », pour ton amitié et
ton soutien. Tu voulais du rêve, je t'ai offert un
Mc Do, que souhaiter de mieux ??? Grâce à toi,
je me souviendrais longtemps de ce chef- d'oeuvre du cinéma
américain intitulé « independence day »... Merci ma
p'tite Muriel, alias
« Mu-Mux », pour ta bonne humeur, ta joie
de vivre, tes fous rires communicatifs, et ton accent du sud qui te va
si bien. Je remercie Marie Courçon et Cédric Masson pour leur
aide précieuse et leur sympathie de tous les instants. Je salue
également Cathy, Marianne, Mireille, Carine, Jean, Christine, et
Jean-Baptiste, ainsi que les vigiles de la FLS avec qui j'ai eu l'occasion de
partager quelques courses-poursuites (qui a dit que la recherche n'était
pas sportive ?).
? Au Laboratoire de Structure des Protéines du
CEA de Saclay :
J'exprime ma profonde reconnaissance aux
Docteurs Bernard Gilquin, Sophie Zinn-Justin, et Joël Couprie pour
m'avoir permis de travailler sur la protéine KIN17. Avoir
été encadré par vous trois est pour moi une
véritable chance, tant sur le plan scientifique, que sur
le plan humain. Merci beaucoup Joël d'avoir
élargi mon horizon à la RMN 3D et d'avoir répondu
à mes nombreuses questions avec gentillesse et disponibilité. Un
immense merci au Monsieur Modélisation Moléculaire du LSP,
en la personne de Bernard Gilquin, pour ses longues discussions
passionnées sur le programme d'attribution automatique des nOe.
Quel
bel outil ! Je reste assurément votre premier fan !
Merci beaucoup Sophie de m'avoir guidé dans la rédaction de ce
manuscrit : tu as éclairé ma lanterne à de nombreuses
reprises !
Je salue également tous les étudiants
et post-docs du LSP que j'ai eu l'occasion de côtoyer en
commençant par Albane le Maire qui a finie par ne plus confondre mon
prénom après 12 mois de collaboration !!! Plus
sérieusement, merci Albane de m'avoir accepté sur KIN17 et
pour le temps que tu m'as consacré. Spéciale
dédicace à Sandrine Caputo : tiens tiens, un Winged Helix peut
en cacher un autre... Je te remercie chaleureusement pour
ton aide spontanée, ton extrême gentillesse,
et pour m'avoir offert deux nuits dans le New York parisien. Merci
également à Gaëlle, Cédric, Nathalie, et
Virginie avec qui j'ai partagé de belles discussions.
? Au Laboratoire de Résonance
Magnétique Nucléaire de l'Université de
Rouen
J'en reviens finalement à mon laboratoire d'accueil
qui a également été le théâtre de très
jolies rencontres. J'adresse mes profonds remerciements aux Docteurs Hassan
Oulyadi et Eric Condamine qui m'ont beaucoup appris dans le domaine de la RMN
haute résolution, et pas seulement des macromolécules biologiques
! Merci au Docteur Gaël Coadou, le « nouveau venu », pour ses
conseils frais et dynamiques.
Un grand merci à Nicole Roussel, véritable
pièce maîtresse de ce labo, qui s'occupe si bien de « ses
petits étudiants » avec une immense, que dis-je ? Une
péninsule de gentillesse.
Je remercie également toutes les « vieilles
canailles » avec qui j'ai passé de très bons moments pendant
ces quatre années. Aux anciens pour commencer : Karine courchay,
merci
de m'avoir fait découvrir des endroits chaleureux
avec de la musique ringarde remixée et des lumières de toutes
les couleurs. A Michel Auvray, alias « michou »,
l'homme le plus extraordinaire que j'ai rencontré dans un labo !
Merci d'avoir réinventé la recherche : si le CNAM en recrute
encore deux comme toi, alors il peut mettre la clef sous la porte, mais il en
sortira humainement grandi !!! A Didier Rivière, alias le «
créolais », merci pour les belles parties de tennis, et comme
l'a souligné « michou », pour ta vision optimiste de la
recherche...
A Pedro Lameiras, le footballeur portugais le plus
parisien, un immense merci pour avoir partagé tes connaissances de
la RMN avec autant de générosité, et pour ton
amitié qui m'a beaucoup touchée. A Franck Paté : qui
aurait cru, lorsque nous pratiquions le tarot intensif pendant les cours de
philo en terminale, que nous arriverions à ce niveau d'étude ?
(surtout ne
le dis à personne, ce sera notre secret). A Anne
Lautrette, j'ai une révélation à te faire : je
déteste ton café ! Quoi qu'il en soit, merci beaucoup pour ta
sympathie même si au début ce n'était pas gagné ! A
Romain Thuau, mon technicien supérieur préféré,
sans aucun doute le chercheur au plus grand coeur que je connaisse. Merci ma
poule pour ton amitié sans calcul.
Je te souhaite tous mes voeux de bonheur avec la belle
Nadège que je salue au passage.
Je tiens également à exprimer toute ma
sympathie aux membres du Laboratoire de Spectrométrie de Masse
Bio-Organique pour leur gentillesse et leurs conseils judicieux : le Professeur
Catherine Lange, les Docteurs Marie Hubert-Roux, Corinne Loutelier-Bourhis, et
Héléne Lavanant, ainsi qu'Albert Marcual. Je salue
également les joyeux étudiants de ce laboratoire : Julie
Hardouin, Delphine Oursel, Thomas Vincent (félicitations pour cet
heureux événement), et le non moins gigantesque Romain
Dolé, alias « p'tit bouchon », qui gagne à être
connu et reconnu !
Enfin, je ne pouvais terminer ces
remerciements sans témoigner ma profonde reconnaissance
à mes plus fidèles proches qui m'ont
énormément soutenus pendant ces quatre années de
thèse. Un grand merci à Sammy, Aurélie, Frédo,
Greg, et mon p'tit TD pour vos nombreux encouragements et votre
amitié.
MERCI A TOUS !!!
TABLE DES MATIERES
ABREVIATIONS
.....................................................................................................................
1
PREAMBULE
..........................................................................................................................
3
PREMIERE PARTIE
..............................................................................................................
9
PRODUCTION DE DOMAINES RECOMBINANTS PRODH EN
VUE DE L'ANALYSE STRUCTURALE
CHAPITRE I : INTRODUCTION
.......................................................................................
11
1 CONTEXTE BIOLOGIQUE
.................................................................................................
13
1.1 Le catabolisme de la proline
................................................................................................
13
1.2 Hypothèses sur les partenaires biologiques de
PRODH chez les organismes eucaryotes .. 16
1.3 Modèle de régulation du catabolisme de la
proline chez la bactérie E. coli ....................... 17
1.4 Les troubles de l'activité de PRODH chez les
eucaryotes supérieurs ................................. 21
1.5
Conclusions..........................................................................................................................
23
2 STRATEGIE D'ETUDE STRUCTURALE DE PRODH HUMAINE PAR
RMN............................... 24
2.1 Analyse bio-informatique préliminaire
................................................................................
24
2.2 Démarche
entreprise............................................................................................................
27
CHAPITRE II : EXPRESSION DES PROTEINES PRODH SAUVAGE ET
MATURE CHEZ E.
COLI.........................................................................................
29
1 PRODUCTION DE PRODH SAUVAGE
.................................................................................
31
1.1 Caractérisation de l'expression et de la
solubilité de la protéine hétérologue
................... 31
1.2 Optimisation de paramètres d'expression et
d'extraction ................................................... 33
2 PREDICTION DU PEPTIDE SIGNAL DE LA SEQUENCE PRODH HUMAINE
............................. 34
2.1 Les messages d'adressage mitochondrial
............................................................................
34
2.2 Résultats de la
prédiction.....................................................................................................
35
3 PRODUCTION DE LA PROTEINE MATURE PRO564
............................................................ 37
4 CONCLUSIONS
...............................................................................................................
38
5 MATERIELS ET METHODES
............................................................................................
40
5.1 Création des plasmides
d'expression...................................................................................
40
5.2 Production, extraction, et analyse de PRODH et PRO564
................................................. 42
CHAPITRE III : ETUDE BIO-INFORMATIQUE : SELECTION DE
3 DOMAINES
PRODH.....................................................................................................
45
1 DESCRIPTION DE LA STRUCTURE DU DOMAINE PRODH DE E. COLI
.................................. 47
2 CARACTERISATION DE L'ORGANISATION DE PRODH
HUMAINE....................................... 49
2.1 Report de la structure secondaire de PutA669 sur
l'alignement initial .............................. 49
2.2 Organisation de PRODH humaine
......................................................................................
49
3 SELECTION DES 3 DOMAINES A EXPRIMER
..................................................................... 55
4 MATERIELS ET METHODES
............................................................................................
57
4.1 Recherche d'homologie de séquence
...................................................................................
57
4.2 Alignements de séquences et prédictions
structurales .........................................................
57
CHAPITRE IV : EXPRESSION DE 4 PROTEINES PRODH DANS LE
CADRE D'UN PROGRAMME DE
PRODUCTION..................................................... 59
1 STRATEGIE DE PRODUCTION DES 4 PROTEINES PRODH
................................................... 61
1.1 Stratégie générale
................................................................................................................
61
1.2 Stratégie de construction des vecteurs
d'expression ...........................................................
63
1.3 Criblage des conditions d'expression en
microplaques....................................................... 64
1.4 Production à grande échelle et obtention de
la protéine d'intérêt
...................................... 65
2 PRODUCTION DES 4 DOMAINES PRODH
..........................................................................
68
2.1 Bilan du criblage des conditions d'expression en
microplaques ......................................... 68
2.2 Production, purification, et obtention des
protéines d'intérêt
............................................. 69
3 CONCLUSIONS
...............................................................................................................
82
4 MATERIELS ET METHODES
............................................................................................
83
4.1 Production à grande échelle et extraction
des protéines .....................................................
83
4.2 Purification des protéines de fusion et des
protéines d'intérêt
............................................ 84
4.3 Clivage du partenaire de fusion par la protéase
TEV ......................................................... 85
CHAPITRE V : CONCLUSIONS ET
PERSPECTIVES................................................... 87
SECONDE PARTIE
..............................................................................................................
93
CARACTERISATION DE LA REGION 51-160 DE LA
PROTEINE KIN17 HUMAINE PAR RMN ET MODELISATION MOLECULAIRE
CHAPITRE I : INTRODUCTION
.......................................................................................
95
1 GENERALITES SUR LE MAINTIEN DE L'INTEGRITE DU GENOME
...................................... 97
1.1 Les dommages de
l'ADN......................................................................................................
97
1.2 Les systèmes mis en jeu
........................................................................................................
99
1.3 Les voies de réparation des lésions de
l'ADN .....................................................................
99
2 LA PROTEINE KIN17
...................................................................................................
101
2.1 Les propriétes de la protéine KIN17
..................................................................................
101
2.2 Organisation des domaines structuraux de KIN17
............................................................ 103
3 CONCLUSIONS
.............................................................................................................
107
CHAPITRE II : PRODUCTION ET ANALYSES PRELIMINAIRES
DU DOMAINE K2 DE LA PROTEINE HUMAINE
KIN17........................................ 109
1 PREPARATION DES ECHANTILLONS POUR L'ANALYSE RMN
........................................ 111
1.1 Sélection et optimisation du système
d'expression ............................................................
111
1.2 Obtention de K2 simplement marquée 15N
et doublement marquée 15N / 13C
.................... 115
2 CARACTERISATIONS PRELIMINAIRES
...........................................................................
122
2.1 Caractérisation de la séquence primaire et
contrôle du marquage................................... 122
2.2 Caractérisation de l'état
oligomérique
..............................................................................
123
2.3 Caractérisation de la structure secondaire et
tertiaire...................................................... 124
2.4 Etude préliminaire par Résonance
Magnétique
Nucléaire................................................ 126
3 CONCLUSIONS
.............................................................................................................
131
CHAPITRE III : STRATEGIE D'ETUDE PAR RMN ET MODELISATION
MOLECULAIRE DU DOMAINE K2..........................................
133
1 METHODE D'ATTRIBUTION DES RAIES DE RESONANCE
................................................ 137
1.1 Attribution des carbones de la chaîne principale et
des 13Câ.............................................
138
1.2 Attribution des protons 1Há et
1Hâ
......................................................................................
143
1.3 Attribution des chaînes
latérales........................................................................................
145
2 DETERMINATION DE LA TOPOLOGIE ET RECUEIL DES CONTRAINTES
STRUCTURALES ... 146
2.1 L'effet Overhauser nucléaire
.............................................................................................
147
2.2 Détermination de la structure secondaire et de la
topologie............................................. 149
2.3 Recueil des contraintes structurales
..................................................................................
153
3 MODELISATION MOLECULAIRE SOUS CONTRAINTES RMN
......................................... 155
3.1 Principe de la mécanique moléculaire
adaptée aux systèmes biologiques........................
155
3.2 Le logiciel CNS
..................................................................................................................
160
3.3 Le programme d'attribution automatique des pics nOe du
LSP........................................ 166
CHAPITRE IV : CARACTERISATION STRUCTURALE DU
DOMAINE K2 PAR RMN ET MODELISATION MOLECULAIRE ..............................
181
1 DETERMINATION DE LA STRUCTURE DU DOMAINE K2 DE KIN17 HUMAINE
................ 183
1.1 Attribution des raies de
résonance.....................................................................................
183
1.2 Détermination de la topologie du domaine K2
.................................................................. 192
1.3 Calcul de la structure par Modélisation
Moléculaire sous contraintes RMN ................... 199
2 DESCRIPTION DE LA STRUCTURE DU DOMAINE
K2....................................................... 204
2.1 Structure secondaire
..........................................................................................................
204
2.2 Les éléments qui composent le coeur
hydrophobe ..............................................................
205
2.3 La boucle entre les hélices H2 et H3
.................................................................................
206
2.4 L'hélice C-terminale H4
....................................................................................................
208
CHAPITRE V : RELATIONS STRUCTURE-ACTIVITE : QUEL EST LE
ROLE DU DOMAINE K2 DE KIN17 HUMAINE ?
............................................ 211
1 LE DOMAINE K2 DE KIN17 ADOPTE UN REPLIEMENT DE TYPE WINGED
HELIX ............. 213
2 LE MOTIF WINGED HELIX DE KIN17 EST-IL CAPABLE DE LIER
L'ADN OU L'ARN ? ........ 216
2.1 Approche structurale
.........................................................................................................
217
2.2 Approche fonctionnelle
......................................................................................................
227
3 LE MOTIF WINGED HELIX DE K2 PRESENTE UNE SURFACE
ULTRA CONSERVEE.............. 229
4 CARACTERISATION DE LA POSITION DU MOTIF PREDIT EN « DOIGT
DE ZINC » AUTOUR DU DOMAINE WINGED HELIX
.............................................................................................
232
4.1 Stratégie employée
.............................................................................................................
233
4.2 Préparation de l'échantillon de
protéine K3 simplement marquée 15N
............................. 233
4.3 Résultats de la cartographie des variations de
déplacement chimique ............................. 234
CHAPITRE VI : CONCLUSIONS ET PERSPECTIVES
............................................... 239
ANNEXE : CRIBLAGE DES CONDITIONS D'EXPRESSION DES
PROTEINES PROENTIER, PROCATAL, PROTER, ET PROINSER ........................
247
1 MATERIELS ET METHODES
..........................................................................................
249
1.1 Construction des plasmides d'expression par recombinaison
homologue ........................ 249
1.2 Criblage des conditions d'expression en
microplaques..................................................... 253
2 RESULTATS
.................................................................................................................
254
2.1 Le domaine PROcatal
........................................................................................................
254
2.2 Le domaine
PROentier.......................................................................................................
256
2.3 Le domaine
PROter............................................................................................................
258
2.4 Le domaine PROinser
........................................................................................................
260
REFERENCES BIBLIOGRAPHIQUES
...........................................................................
263
ABREVIATIONS
1D, 2D, 3D, 4D Expérience RMN à
une, deux, trois, ou quatre dimension(s)
3PM Programme de Production et Marquage des
Protéines
ADN Acide DésoxyriboNucléique
ADNc Acide DésoxyriboNucléique
complémentaire
Ampi Ampicilline
ARN Acide RiboNucléique
ARNm Acide RiboNucléique messager
ATP Adenosine TriPhosphate
BCIP 5-Bromo-4-Chloro-3-Indolyl Phosphate
BER Base Excision Repair
CAM ChlorAMphénicol
CNS Cristallography and NMR System
COSY COrrelation SpectroscopY
CSD Chemical Shift Deviation (déplacement
chimique secondaire)
DC Dichroïsme Circulaire
DNase Désoxyribonucléase
DO Densité Optique
DTT 1,4-DiThioTréitol
EDTA Acide éthylène diamine
tétraacétique
ESI ElectroSpray Ionization
FAD Flavine Adénine
Dinucléotide
FADH2 Flavine Adénine Dinucléotide
réduit
FMN Flavine MonoNucléotide
GABA Acide gamma amino-butyrique
HCA Hydrophobic Cluster Analysis
HMQC Heteronuclear Multiple-Quantum Coherence
spectroscopy
HPLC High Pressure Liquid Chromatography
HR Homologous Recombinaison
HSQC Heteronuclear Single Quantum Coherence
spectroscopy
IMAC Immobilized Metal ion Affinity
Chromatography
INEPT Insensitive Nuclei Enhanced by
Polarisation Transfert
IPTG IsoPropyl -D ThioGalactopyranoside
IR Radiations ionisantes
1
ITD-MS Ion Trap Detector-Mass Spectroscopy
LB Luria-Bertani
MALDI-TOF Matrix Assisted Laser Desorption
Ionization-Time Of Flight
MMR MisMatch Repair
MMS Methyl Methane Sulfonate
NAD+ Nicotinamide Adénine
Dinucléotide
NADH Nicotinamide Adénine
Dinucléotide réduit
NADP+ Nicotinamide Adénine
Dinucléotide Phosphate
NADPH Nicotinamide Adénine
Dinucléotide Phosphate réduit
NBT Nitro Blue Tetrazolium
NER Nucleotide Excision Repair NHEJ
Non Homologous End Joining nOe nuclear Overhauser
effect
NOESY Nuclear Overhauser Effect SpectroscopY
PBS Phosphate Buffer Saline
PCR Polymerase Chain Reaction PMSF
PhenylMethylSulfonyl Fluoride PRODH PROline
DésHydrogénase
PVDF PolyVinylidine DiFluoride
P5C Pyrroline-5-Carboxylate
P5CDH Pyrroline-5-Carboxylate
DésHydrogénase
RMN Résonance Magnétique
Nucléaire RMSD Root Mean Square Deviation RNase
Ribonucléase
RT-PCR Reverse Transcription-Polymerase Chain
Reaction
SDS Sodium Dodecyl Sulfate
SDS-PAGE Sodium Dodecyl Sulfate-PolyAcrylamide
Gel Electrophoresis
SMART Simple Modular Architecture Research
Tool
TCEP Tris((2-CarboxyEthyl)Phosphine)
TEV Tobacco Etch Virus
TOCSY TOtal Correlation SpectroscopY
TRIS Tris(hydroxyméthyl) amino
méthane
TSP
3-(TriméthylSilyl)[2,2,3,3-2H4] Propionate
TST Tris Saline Tween
UV Ultraviolet
E. coli Escherichia coli
2
Préambule
Préambule
Les protéines sont des polymères d'acides
aminés qui, avec l'eau, représentent les
composants principaux des organismes vivants. A
l'image de l'ADN, support de l'information génétique et
de l'hérédité, elles peuvent être
considérées comme les molécules fondamentales de la vie.
Cependant, à la différence des acides nucléiques qui sont
à l'origine
de leur synthèse, les protéines se
caractérisent par une diversité fonctionnelle immense
associée à une diversité structurale considérable.
Bien que les êtres vivants n'utilisent qu'une vingtaine d'acides
aminés différents pour composer les protéines,
la grande diversité structurale de ces macromolécules
s'explique par la variabilité de l'enchaînement des acides
aminés dans la séquence primaire, qui guide
véritablement la nature du repliement. Avec l'essor de la biologie
structurale, il est aujourd'hui communément admis que la fonction d'une
protéine est intimement liée d'une part, à sa
structure tridimensionnelle, c'est-à-dire à l'organisation de
ses atomes dans l'espace, et d'autre part, à sa dynamique
intra-moléculaire, c'est-à-dire à l'amplitude des
mouvements de ses atomes. A l'heure où la caractérisation des
fonctions des protéines encodées par les génomes
constitue un défi majeur de l'ère post- génomique,
la détermination de la structure tridimensionnelle des
protéines à l'échelle de l'atome représente donc
un enjeu considérable dans l'optique d'identifier et de comprendre
leur(s) fonction(s). Cette approche, appelée « étude
structurale », a pour objet d'apporter des informations sur les
relations structure-activité, et structure-dynamique-activité,
et ainsi d'améliorer la compréhension des bases
moléculaires du rôle de ces molécules. Une telle
étude peut avoir deux objectifs différents :
- Dans le cas de protéines de fonction inconnue,
la connaissance de la structure tridimensionnelle va permettre, par
comparaison avec celles dont le repliement est proche, de proposer une ou
plusieurs activités pour la biomolécule étudiée.
- Lorsque la fonction est connue, l'objectif est d'identifier les
éléments de structure ou
les acides aminés essentiels à
l'activité dans le but de caractériser les
mécanismes réactionnels spécifiques de la
fonction. Ceci peut être initié, par exemple, en
comparant les structures native et mutée (induisant une
modification de l'activité) d'une même protéine, ou
en identifiant les sites d'interaction avec des partenaires biologiques
potentiels (ligand, substrat, cofacteur, partenaire protéique...).
C'est dans cette double optique que nous avons entrepris
l'étude structurale de deux protéines humaines : la
protéine mitochondriale proline déshydrogénase (PRODH)
qui fait
l'objet de la première partie de ce manuscrit, et la
protéine nucléaire KIN17 qui fait l'objet de
la seconde partie.
A ce jour, il existe plusieurs techniques qui offrent la
possibilité de caractériser la structure tridimensionnelle
d'une protéine. Cependant, seules la cristallographie des rayons X
et la Résonance Magnétique Nucléaire
(RMN) en solution permettent d'atteindre une résolution de
l'ordre de l'Angström qui est nécessaire pour comprendre le
fonctionnement d'objets moléculaires aussi petits que les
protéines. Quelle que soit la technique utilisée, la RMN ou la
cristallographie, le préliminaire à une étude
structurale est l'obtention d'une quantité importante
de la protéine à étudier (~ 1 umole dans 500 uL,
c'est-à-dire ~ 15 mg pour une protéine de 15 kDa) sous
forme soluble, pure, et
stable. Ces critères constituent
véritablement les exigences inhérentes à l'analyse
structurale. A l'heure actuelle, trois méthodes permettent
potentiellement de remplir ces conditions : la protéine
peut être directement extraite de son organisme naturel,
synthétisée chimiquement, ou surexprimée sous forme
recombinante dans un organisme hôte. Cependant, les
quantités obtenues par extraction sont souvent insuffisantes, et la
synthèse chimique devient difficilement réalisable pour des
polypeptides de plus de 50 acides aminés. Pour ces raisons, la
surexpression dans un système recombinant est de loin la technique la
plus utilisée, d'autant plus qu'elle permet de réaliser des
marquages isotopiques indispensables à l'étude d'une
protéine de taille élevée
(> 10 kDa) par RMN.
Les organismes de surexpression les plus courants sont les
bactéries, les levures, et les cellules d'insectes (baculovirus).
Pour des raisons essentiellement liées à sa
facilité d'utilisation, et à sa capacité à
produire des quantités importantes de protéine
marquée, la bactérie Escherichia coli est le
système le plus communément utilisé. C'est pourquoi, nous
avons entrepris de surexprimer des domaines protéiques de PRODH et KIN17
chez cet hôte bactérien. Cependant, bien que la protéine
d'intérêt soit produite in vivo, il n'est pas garanti
qu'elle adopte un repliement stable et biologiquement actif. En
d'autres termes, le comportement d'une protéine exogène
exprimée dans un organisme recombinant n'est pas
prévisible, et la préparation de l'échantillon,
véritable étape limitante de l'étude structurale, peut
demander de longues étapes d'optimisation, qui, dans certains cas,
peuvent se solder par
un échec. Ainsi, dans le cadre de ce travail de
thèse, de grandes difficultés ont été
rencontrées
pour préparer les échantillons de protéines
PRODH, ce qui n'a pas été le cas avec la protéine
KIN17 où la réussite de cette première
étape majeure a permis d'envisager une caractérisation
structurale par RMN. Par conséquent, ce manuscrit est
organisé en deux parties distinctes.
La première partie, consacrée à
la proline déshydrogénase PRODH, présente
conjointement les différentes stratégies que nous avons
employées pour surexprimer des protéines et domaines
structuraux PRODH chez E. coli en vue de l'analyse structurale, ainsi
que les résultats de la production et de l'optimisation de
l'expression de ces protéines. Le problème de la
délimitation des domaines structuraux sera notamment
évoqué. Ce travail a été réalisé
dans le cadre d'une collaboration avec le Laboratoire de
Génétique Moléculaire de l'EMI 9906 de la Faculté
de Médecine-Pharmacie de Rouen, et le Laboratoire de Marquage des
Protéines du CEA de Saclay. Les objectifs de cette étude seront
préalablement abordés
après avoir présenté l'intérêt
biologique suscité par la proline déshydrogénase PRODH.
Dans la seconde partie de ce manuscrit, je présenterai
l'étude structurale du domaine
K2 de la protéine KIN17 humaine par RMN et
Modélisation Moléculaire qui a été entreprise dans
le cadre d'une collaboration avec le Laboratoire de Structure des
Protéines du CEA de Saclay. Dans un premier chapitre, j'introduirai de
manière simplifiée et succincte le contexte biologique de la
protéine KIN17, puis les objectifs de ce travail seront exposés.
Les premiers résultats de l'étude expérimentale,
à savoir la préparation des échantillons de
protéine marquée 15N et 15N /
13C pour l'analyse par RMN, ainsi que les analyses
préliminaires, seront présentés dans le second
chapitre. Dans le troisième chapitre, sera détaillé
l'ensemble des méthodologies de RMN et modélisation
moléculaire utilisées dans cette étude. Les
résultats
de la caractérisation structurale proprement dite,
c'est-à-dire l'attribution des raies de résonance du
domaine K2, la détermination de la topologie de la
protéine, le recueil des contraintes expérimentales, et le
calcul de la structure par Modélisation Moléculaire, seront
décrits dans le chapitre 4. Les relations structure-activité du
domaine K2 de la protéine KIN17 humaine seront finalement
discutées dans le chapitre 5.
PREMIERE PARTIE
Production de domaines recombinants
PRODH en vue de l'analyse structurale
Chapitre 1 : Introduction
CHAPITRE 1
Introduction
1) Contexte biologique
1.1) Le catabolisme de la proline
Parmi les vingt acides aminés qui composent les
protéines, la proline représente une classe unique d'acide
aminé. L'incorporation du noyau azote du groupement amine au sein d'une
structure cyclique la distingue des autres acides aminés et lui
confère des propriétés particulières (Figure
1.1). Cette topologie unique contribue aux propriétés
physiques et structurales de plusieurs métabolites essentiels comme le
collagène, une glycoprotéine riche
en résidus glycine et proline.
H O
H
N+ C
O-
Figure 1.1 : Structure de la
proline sous forme zwitterionique.
Au-delà de sa fonction de module
élémentaire pour la biosynthèse des protéines,
la proline, comme tout autre acide aminé, est également
utilisée comme source d'énergie, d'azote et de carbone
pour la biosynthèse d'intermédiaires métaboliques
majeurs. C'est un acide aminé glucoformateur, c'est-à-dire
susceptible d'être converti en glucose par le biais des cycles
métaboliques de l'organisme.
Comme le montre la Figure 1.2, le catabolisme de la
proline empreinte la voie de
l'á-cétoglutarate du cycle de l'acide
citrique, communément appelé cycle de Krebs (pour revues :
Adams & Frank, 1980 ; Phang, 1985). Le point de départ de ce flux
métabolique fait intervenir la proline déshydrogénase
PRODH qui oxyde la proline en P5C (Pyrroline-5- Carboxylate). Cet
intermédiaire cyclique se linéarise de manière
spontanée en glutamate-ã- semialdéhyde, qui est ensuite
dégradé en glutamate par la P5C déshydrogénase
P5CDH via la consommation d'un dinucléotide NAD+. Le
glutamate subit une désamination oxydative par
une aminotransférase conduisant à l'ion
ammonium NH4+, qui entre alors dans le cycle de
l'urée. Cette réaction est catalysée par la
glutamate déshydrogénase qui forme l'á-
cétoglutarate. L'entrée de ce dernier dans le cycle de l'acide
citrique donne lieu à une série de
réactions, qui aboutit à un transfert de
plusieurs électrons de haute énergie vers des
dinucléotides de type FAD et NAD. Leur oxydation dans
la chaîne respiratoire conduit à la formation d'ATP. Toutes les
réactions enzymatiques du catabolisme de la proline ont lieu
dans la mitochondrie chez les eucaryotes, et dans le cytosol chez
les procaryotes. Il est toutefois à noter que le cycle de
l'urée se termine dans le cytoplasme chez les eucaryotes.
H H
N+ COO-
H
PRODH N+ COO- non
enzymatique H
COO-
+
NH3
O
proline P5C glutamate-ã- semialdéhyde
NAD+
O
-O
COO-
- aminotransférase
- glutamate déshydrogénase
P5CDH
-O
NADH
COO-
+
NH3
O
á-cétoglutarate
NH4+
O
glutamate
citrate
succinyl-CoA
Cycle de l'acide
citrique
Cycle de
l'urée
oxaloacétate
fumarate
malate
Figure 1.2 : Vue d'ensemble du
catabolisme de la proline.
De par sa forte biodisponibilité, la proline est un des
acides aminés les plus efficaces
en terme de glucogenèse. Chez la levure Saccharomyces
Cerevisiae, l'oxydation de la proline
en glutamate permet de maintenir la croissance cellulaire lorsque
cet aminoacide contient la seule source d'azote disponible (Wang &
Brandiss, 1986). Chez certaines plantes, la proline
est la première source d'énergie utilisée
après un choc osmotique (Blum & Ebercon, 1976),
ou pour la fabrication du pollen (Hong-qi et al., 1982). C'est
également le cas chez certains
insectes où la proline est très rapidement
oxydée dans les muscles impliqués dans les
mécanismes du vol (Holden, 1973).
Dans les années 1980, les travaux de Hagedorn
et Phang ont mis en évidence la capacité de la
proline à catalyser les cycles métaboliques mitochondriaux
via un cycle de transfert de potentiel redox (Hagedorn et al., 1982
;Hagedorn & Phang, 1983 ; Hagedorn & Phang, 1986). En effet, il existe
une enzyme appelée P5C réductase qui, dans le cytoplasme, conduit
à la réduction du P5C en proline via la
consommation d'un dinucléotide NADPH (Figure 1.3). Les
mécanismes de régulation de PRODH et de la P5C
réductase n'étant pas liés, il est proposé que la
proline et le P5C forment un couple redox utilisé pour transférer
des équivalents réducteurs du cytoplasme vers la
mitochondrie. Selon cette hypothèse, le dinucléotide
NADPH, nécessaire à la réduction du P5C en
proline, pourrait être fourni par l'oxydation du glucose dans la
voie cytoplasmique du pentose phosphate. Le cycle de transfert de
potentiel redox du couple proline/P5C serait donc une voie alternative
de l'utilisation de la proline, et servirait à relier le cycle
mitochondrial de l'acide citrique à celui
du pentose phosphate cytoplasmique.
Mitochondrie
Cytoplasme
H H
N+ COO-
H H
N+ COO-
voie du pentose phosphate
proline proline
glucose
H ? PRODH
P5C
réductase
NADP+
NADPH
H
N+ COO-
H
N+ COO-
ribulose
P5C P5C
Figure 1.3 : Mise en
évidence de la capacité du couple redox proline/P5C à
transférer des
potentiels réducteurs du cytoplasme vers la
mitochondrie.
1.2) Hypothèses sur les partenaires biologiques de
PRODH chez les
organismes eucaryotes.
Bien que toutes les enzymes intervenant dans le
catabolisme de la proline aient été identifiées, la
littérature fait état de très peu de données
fonctionnelles concernant les modes d'action et de régulation des
proline déshydrogénases eucaryotes. En effet, cette enzyme
responsable de la première étape de la dégradation de la
proline en glutamate a fait l'objet de peu d'études à
l'échelle de la protéine chez les organismes eucaryotes. Ceci
pourrait en partie être expliqué par les difficultés qui
ont été rencontrées pour extraire une quantité
suffisante d'enzyme PRODH soluble et biologiquement active à
partir de mitochondries (Johnson & Strecker, 1962 ; Brosemer &
Veerabhadrappa, 1965 ; Brunner & Neupert, 1969)
La présence de la proline oxydase (ou
déshydrogénase) a été détectée pour
la première fois dans des mitochondries de foie de rat (Johnson
& Strecker, 1962) à partir d'un test d'activité qui
repose sur la réactivité spécifique du produit P5C avec
l'O-aminobenzaldéhyde (Strecker, 1960). Les méthodes d'extraction
de protéines mitochondriales, réalisées avec des
successions de gradient de sucrose et d'ultracentrifugation, ont
révélé que cette protéine appartient à
la matrice mitochondriale et qu'elle est fortement liée à
la membrane interne (Brunner & Neupert, 1969). D'autre part, il a
été montré que l'activité enzymatique de
PRODH nécessite la présence de dioxygène et d'un accepteur
d'électrons de type cytochrome
c ou ubiquinone (Johnson & Strecker, 1962 ; Erecinska, 1965),
qu'elle est inhibée par le KCN
et l'antimycine A (Brosemer & Veerabhadrappa, 1965),
et qu'elle est indépendante en dinucléotide de type NAD+
ou NADP+ (Kramar, 1967). Toutes ces caractéristiques
suggèrent
que la proline déshydrogénase eucaryote est
une flavoenzyme (c'est-à-dire une enzyme de cofacteur flavinique
FAD ou FMN) qui intervient dans la chaîne respiratoire
de la mitochondrie via un accepteur d'électrons de la
membrane interne. Cependant, à ce jour le cofacteur de la proline
oxydase n'a jamais été clairement identifié chez un
organisme eucaryote. D'autres études, menées sur des
mitochondries extraites d'intestin de porc et de foie de rat, ont mis
en évidence la capacité du lactate à
réduire l'activité de PRODH de manière drastique (de
50 % à 95 %), et à diminuer l'affinité de l'enzyme pour
son substrat proline (Kowaloff et al., 1977 ; Dillon et al., 1999). Ces
observations supportent l'hypothèse que le lactate est un inhibiteur
compétitif des proline déshydrogénase eucaryotes qui
pourrait
être impliqué dans la régulation de
l'enzyme.
Contrairement aux organismes eucaryotes, les
partenaires biologiques et les
mécanismes de régulation de la proline
déshydrogénase sont en partie connus chez la bactérie
E. coli. La mise au point de méthodes
efficaces de purification de la protéine endogène sous forme
soluble et biologiquement active (Menzel & Roth, 1981 ; Brown
& Wood, 1992) a permis de réaliser plusieurs études
in vitro, et ainsi de caractériser d'un point de vue
biochimique plusieurs aspects de ses mécanismes d'action. Sur la
base de ces études fonctionnelles, il est possible de proposer
un modèle de régulation du catabolisme de la proline chez
les organismes procaryotes.
1.3) Modèle de régulation du catabolisme de la
proline chez la bactérie E. coli
Chez les organismes procaryotes (qui ne possèdent pas
d'organelles), la dégradation
de la proline en glutamate a lieu à la
périphérie de la membrane interne et met en jeu des
intermédiaires réactionnels identiques à ceux
décrits dans le paragraphe 1.1. Alors que chez
les eucaryotes la proline déshydrogénase
PRODH et la P5C déshydrogénase P5CDH sont encodées
par deux gènes différents, chez la bactérie, les deux
fonctions sont assurées par une seule protéine encodée
par le gène PutA, dont l'évolution
phylogénétique a conduit à la
séparation de ce gène en deux distincts (Figure
1.4).
H H
H COO-
N+ COO-
N+ COO-
NAD+ NADH
-O
3
NH +
O
proline P5C glutamate
PRODH
PutA
P5CDH
Eucaryotes
Procaryotes
Figure 1.4 : Evolution
phylogénétique du gène encodant les enzymes proline
déshydrogénase
PRODH et P5C déshydrogénase P5CDH. La
conversion intermédiaire spontanée et non enzymatique du P5C
en glutamate-ã-semialdéhyde n'est pas
représentée.
PutA (proline utilization) est une flavoenzyme
multifonctionnelle bactérienne qui
présente quatre types d'activités pour un seul
polypeptide : la fonction PRODH, la fonction P5CDH, la capacité à
lier l'ADN, et la capacité à fixer la membrane interne. Chez
E. coli, PutA est une protéine de 1320 acides aminés,
qui adopte une structure quaternaire dimérique
en solution, et qui contient un cofacteur FAD lié
de manière non covalente à chaque monomère (Brown
& Wood, 1992). Le domaine PRODH oxyde la proline en P5C et catalyse
le transfert de 2 électrons du substrat proline
vers son cofacteur flavine (Surber & Maloy,
1999). L'association de PutA à la membrane permet alors
de transférer ces 2 électrons à un accepteur de la
chaîne respiratoire, ce qui régénère la forme
oxydée du FAD. Après linéarisation spontanée
du P5C, le domaine P5CDH catalyse l'oxydation de glutamate-ã-
semialdéhyde en glutamate par un mécanisme NAD-dépendant
(Figure 1.5).
H H
N+ COO-
FAD FADH2
H
N+ COO-
NAD+ NADH
-O
COO-
+
NH3
O
proline P5C glutamate
Figure 1.5 : Mécanismes de
dégradation de la proline en glutamate par la protéine PutA
de
la bactérie E. coli. L'étape
intermédiaire non enzymatique n'est pas
représentée.
PutA est également un répresseur de
transcription de la région inter-génique put. Cette
région comporte le gène PutA, ainsi que le
gène PutP qui encode une perméase dont la
fonction est d'assurer l'entrée et le transport de la proline dans la
cellule (Chen et al., 1985). L'expression des gènes put
dépend de la biodisponibilité de la proline dans la cellule
et de la localisation intracellulaire de PutA. En absence de proline, la
protéine PutA est localisée dans
le cytoplasme et inhibe la transcription des gènes
PutA et PutP en fixant la région promotrice
du régulon Put (Ostrovsky De Spicer et
al., 1991). En présence de proline, PutA rejoint la membrane, ce
qui lève l'inhibition de la transcription des gènes put
et déclenche le processus d'oxydation de la proline par le domaine
PRODH (Wood, 1987 ; Surber & Maloy, 1999).
La proline réduisant le cofacteur FAD, il est
proposé que l'état d'oxydation du FAD soit
l'élément clé qui gouverne la localisation
cellulaire de PutA, et par conséquent sa fonction
(répresseur de transcription ou déshydrogénase). En
effet, il a été montré que la
forme réduite FADH2 est primordiale pour
la liaison de PutA à la membrane (Wood, 1987).
Surber et Maloy ont confirmé cette
hypothèse en démontrant que la réoxydation du cofacteur
FADH2 conduisait à la rupture de cette liaison (Surber & Maloy,
1999). En revanche, d'autres études ont mis en évidence que
la liaison à l'ADN dépend peu de l'état
d'oxydation du cofacteur (Ostrovsky & Maloy, 1995 ; Becker & Thomas,
2001). Sur la base de ces données,
il apparaît donc que la localisation intracellulaire et la
fonction de PutA sont intimement liées
à des modifications d'affinité de liaison
de PutA à la membrane. Des études de digestion
chymotripsique suivie sur gel SDS-PAGE ont été
réalisées dans l'optique de caractériser la nature de ces
modifications. Les travaux de Brown et Wood montrent que la
protéine PutA présente des susceptibilités aux
protéases différentes selon la présence ou l'absence de
proline (Brown & Wood, 1992). Ceci indique que la réduction
du FAD par la proline induit une modification structurale
significative de la protéine. Ces résultats ont
été récemment confirmés par une étude
similaire, réalisée sur un échantillon de protéine
recombinante PutA purifiée, qui suggère que les changements
d'état d'oxydation du FAD sont capables de provoquer des
changements de conformation au-delà du domaine catalytique PRODH (Zhu
& Becker, 2003). L'ensemble de ces données permet de proposer un
modèle fonctionnel de la régulation du catabolisme de la proline
chez les procaryotes où le cofacteur flavinique joue un rôle
central (Figure 1.6).
2 e
FADH2
PutA
P5C
FADH2
PutA
PutP
proline
PutP
PutP








proline
FAD
PutA
FAD
PutA
PutP
put
PutA
Figure 1.6 : Modèle de
régulation du catabolisme de la proline chez E. coli par la
protéine
PutA. La protéine de transport PutP permet
l'entrée de la proline dans le cytosol de la bactérie. En
absence de proline, la protéine PutA fixe le régulon put et
inhibe la transcription des gènes PutP et PutA. La dégradation de
la proline en P5C par PutA s'accompagne d'une réduction du cofacteur
FAD en FADH2. Ce changement d'état d'oxydation du
cofacteur
induit une modification structurale de PutA qui
conduit d'une part, à un repliement plus
favorable à la liaison de PutA à la
membrane interne, et d'autre part, à la rupture de la liaison
au régulon put. Cette rupture lève l'inhibition de la
transcription des gènes PutP et PutA. Les 2 électrons provenant
de l'oxydation de la proline sont transférés à un
accepteur de
la chaîne respiratoire après fixation de PutA
à la membrane.
La caractérisation du cofacteur flavinique chez
E. coli supporte l'hypothèse de l'existence d'un
cofacteur FAD ou FMN chez les organismes eucaryotes. Cependant, le
modèle de régulation du catabolisme de la proline par PutA ne
peut être transposé chez les eucaryotes où la
présence de PRODH dans la matrice mitochondriale ne permet pas
une
régulation de son propre gène situé dans le
noyau.
1.4) Les troubles de l'activité de PRODH chez les
eucaryotes supérieurs
Au cours de ces 15 dernières années, la
séquence primaire de la protéine PRODH a été
identifiée chez tous les organismes eucaryotes les plus
communément étudiés. En parallèle,
un certain nombre d'études médicales,
réalisées chez des eucaryotes supérieurs, ont
récemment permis de caractériser les bases moléculaires
cliniques des troubles de l'activité de
la proline déshydrogénase.
1.4.1) L'hyperprolinémie de type I
La première séquence primaire de proline oxydase
(baptisée PUT1) a été découverte
en 1986 chez la levure Sacchamoryces Cerevisiae (Wang
& Brandriss, 1986). La séquence de cette protéine a ensuite
été identifiée chez la mouche Drosophila Melanogaster
(Hayward et
al., 1993), puis chez la plante Arabidopsis
Thaliana (Verbruggen et al., 1996) sur la base d'une homologie de
séquence avec la protéine PUT1. Ce n'est qu'en 1997 que
le gène PRODH humain a été localisé au
niveau de la région q11 du chromosome 22 (Campbell et al.,
1997). Ce gène est principalement exprimé au
niveau du foie, des reins, et du cerveau
(Maynard et al., 2003).
Une perte d'activité de la proline oxydase se
caractérise sur le plan biochimique par une hyperprolinémie de
type I, c'est-à-dire un taux anormalement élevé de
proline dans l'organisme (Efron, 1965). Chez les patients
hétérozygotes, cette maladie rare autosomale récessive
est dite « silencieuse » et conduit généralement
à une hyperprolinémie bénigne associée à
des désordres mineurs. Cependant, des manifestations
neurologiques sévères (retard mental et épilepsie)
ont récemment été rapportées chez plusieurs
sujets atteints d'hyperprolinémie de type I (Humbertclaude et al.,
2001), et notamment chez deux enfants porteurs d'une délétion
homozygote du gène PRODH, ou de la mutation rare L441P à
l'état homozygote (Jacquet et al., 2003 ; Jacquet et al., 2002). Ces
deux enfants souffraient d'une forme sévère
d'hyperprolinémie de type I associée à des retards
psychomoteurs (Jacquet et al.,
2003). Les souris Pro/Re et les mouches slgA
représentent des modèles animaliers intéressants
pour étudier les bases moléculaires de l'hyperprolinémie
de type I. La lignée de souris Pro/Re comporte une mutation faux
sens homozygote du gène PRODH qui entraîne une
terminaison précoce de la traduction de la région C-terminale de
la protéine (Gogos et al.,
1999). Ces souris sont spontanément
hyperprolinémiques avec un niveau de proline 7 fois
supérieur à la normale. L'activité de PRODH
s'avère déficiente au niveau du foie, des reins,
et notamment du cerveau. Les souris Pro/Re présentent
des anomalies d'apprentissage et de la réaction de sursaut
associées à une diminution des taux de glutamate, de
GABA, et d'aspartate dans le cortex frontal. De manière
intéressante, ces types de trouble sont également
observés chez les souris hyperprolinémiques slgA, qui
comportent plusieurs mutations du gène PRODH, et qui
présentent un comportement psychomoteur léthargique (Hayward
et al., 1993). Le glutamate étant un neurotransmetteur de
jonctions musculaires dans le cerveau, il est proposé que les
troubles neurologiques constatés chez l'homme, la souris, et la
mouche, soient dus à une diminution du taux de glutamate dans
le cerveau, induite par une réduction d'activité de la proline
oxydase (Gogos et al., 1999 ; Hayward et al.,
1993). Selon cette hypothèse, le catabolisme de la
proline serait une des principales voies métaboliques conduisant
à la formation de glutamate dans le cerveau.
1.4.2) PRODH et schizophrénie
L'hypothèse de l'implication du gène PRODH
dans le déterminisme génétique de la
schizophrénie a relancé l'intérêt suscité
par cette enzyme mitochondriale. La schizophrénie
est une maladie qui affecte les fonctions supérieures du
cerveau et qui est caractérisée par la présence
d'hallucinations, de délires, et d'une dissociation mentale,
symptômes se traduisant
par un comportement atypique ou inadapté du sujet
atteint (pour revue : Murphy, 2002). Cette pathologie constitue une
préoccupation majeure de santé publique en raison de sa
prévalence (environ 1 % de la population), de son âge de
début précoce (dans 50 % des cas avant 23 ans),
et des troubles du comportement qu'elle implique
(prévalence élevée des suicides, de la toxicomanie,
et de comportements agressifs). Des études de jumeaux, qui
consistent à comparer le taux de concordance de la maladie au sein de
paires de jumeaux monozygotes par rapport à celui retrouvé au
sein de paires de jumeaux dizygotes, montrent que les facteurs
génétiques sont pour la plus grande part à
l'origine de la schizophrénie (McGuffin et al.,
1994). Toutefois, aucun gène de susceptibilité
n'est actuellement identifié avec certitude.
L'implication de la région chromosomique 22q11
(qui comporte le gène PRODH) dans le déterminisme
génétique de la schizophrénie a été
suggéré par la fréquence élevée de traits
schizophrènes retrouvée chez les patients atteints du syndrome de
Digeorge (incidence environ 20 fois supérieure à celle
observée dans la population générale) (Murphy et al.,
1999).
Ce syndrome se caractérise chez 95 % des sujets atteints
par une microdélétion hétérozygote
de la région q11 du chromosome 22 qui affecte le
gène PRODH (Hoffmann & Vadstrup,
2000). De manière intéressante, certains
troubles associés à l'hyperprolinémie de type I,
comme la diminution de l'inhibition de la réaction de sursaut
chez la souris Pro/Re, sont également présents dans la
schizophrénie (Chakravarti, 2002). Sur la base de ces
observations, le gène PRODH a été
défini comme candidat dans l'étiologie de la
schizophrénie. Dans l'optique d'établir le lien entre
l'hyperprolinémie de type I et la schizophrénie, des
études de recherche de variations nucléotidiques du gène
PRODH ont été réalisées chez des patients
schizophrènes et chez des sujets témoins. Certaines d'entre
elles
ont mis en évidence une augmentation de
la prévalence de mutations conduisant à
l'hyperprolinémie de type I dans des échantillons de patients
schizophrènes (Liu et al., 2002 ; Jacquet et al., 2002). En revanche,
d'autres études uniquement basées sur des statistiques de
variations nucléotidiques n'ont révélé aucun
lien entre le gène PRODH et la schizophrénie
(Williams et al., 2003 ; Fan et al., 2003). L'association entre la proline
déshydrogénase et la schizophrénie est donc une
hypothèse qui, à ce jour, reste très
controversée.
1.5) Conclusions
Jusqu'en 1995, les proline déshydrogénases
eucaryotes étaient peu étudiées que ce soit
à l'échelle du gène ou de la
protéine. Les quelques données biochimiques disponibles dans la
littérature permettent d'émettre des hypothèses sur
la nature du cofacteur et de l'inhibiteur compétitif naturel de
cette enzyme mitochondriale. Cependant, les mécanismes d'action et de
régulation de PRODH restent à ce jour inconnus chez les
organismes eucaryotes, bien qu'ils soient en partie connus chez la
bactérie E. coli. Sur le plan médical, plusieurs
études récemment menées montrent que certaines
mutations du gène PRODH peuvent induire des troubles de
l'activité enzymatique qui, dans certains cas, sont associés
à des manifestations neurologiques sévères. Il est
également proposé que ces mutations puissent être des
facteurs
de risque de la schizophrénie. Aucune donnée
structurale n'étant publiée au moment où nous
entreprenions cette étude, il apparaissait que la résolution de
la structure tridimensionnelle de
la proline déshydrogénase humaine serait d'un
grand intérêt. La caractérisation structurale de cette
enzyme constituerait une amorce essentielle vers la compréhension
de son mode de fonctionnement. A terme, elle permettrait d'étayer
les hypothèses émises sur la nature de ces partenaires, et de
caractériser d'un point de vue structural les bases
moléculaires de
l'hyperprolinémie de type I en déterminant les
conséquences des mutations sur la structure de
PRODH. C'est pourquoi, nous avons entrepris l'étude de la
proline déshydrogénase humaine
par Résonance Magnétique Nucléaire (RMN),
qui est une des deux techniques permettant de résoudre la structure
d'une protéine à l'échelle de l'atome.
2) Stratégie d'étude structurale de PRODH
humaine par RMN
La RMN est une méthode de choix pour étudier une
protéine en solution et caractériser son interaction avec ses
partenaires biologiques. Cependant, c'est une technique qui possède une
limitation majeure : la masse moléculaire de la biomolécule
étudiée. Bien que le record de détermination de
structure tridimensionnelle ait été enregistré sur
un monomère de 48 kDa (Williams et al., 2005), la taille limite
actuelle pour une étude de routine se situe aux alentours
de 20 kDa, soit environ 180 résidus. Dans le
cas de la protéine PRODH humaine, dont le poids
moléculaire est de 70 kDa pour 600 résidus, il n'est donc pas
concevable d'envisager une étude structurale par RMN. Toutefois,
les protéines de masse moléculaire élevée
sont généralement composées de plusieurs domaines de
repliement autonome qui sont plus ou moins indépendants les uns des
autres. L'organisation de la structure tertiaire des protéines de grande
taille en domaines structuraux offre ainsi la possibilité de
caractériser la structure de
ces macromolécules domaine par domaine. Toute la
difficulté de cette approche repose sur l'identification de ces domaines
de repliement autonome. La stratégie communément utilisée
consiste à recourir à des outils bio-informatiques afin de
prédire leur délimitation.
2.1) Analyse bio-informatique préliminaire
Nous avons réalisé dans un premier temps un
alignement de 9 séquences de PRODH
connues d'origine eucaryote et procaryote avec le logiciel
clustalw (Figure 1.7).
Human
1-MALRRALPALRPCIPRFVQLSTAPASR----------EQPAAGPAAVPGGGSA-----------------------TAVR
Drosophila
MALLRSLSAQRTAISLVYGRNSSKSSNSVAVAACRSFHQRGNGSTSIAGEGAASESTRGVNGARFLHSGDRPLQASTLVQ
CAEEL
------------------------------------------------------------------------------MK
Arabidopsis
-------------------------------------------------------------------------------M
Oryza
-------------------------------------------------------------------------------M
POMBE
--------------------------------------------------------------------------------
Emericella
--------------------------------------------------------------------------------
Cerevisiae
--------------------------------------------------------------------------------
PutA
------MGTTTMGVKLDDATRERIKSAATRIDRTPHWLIKQAIFSYLEQLENS------------------------DTL
Human
48-PPVP------------------------------AVDFGNAQEAYRSRRTWELARSLLVLRLCAWPALLARHEQLLY-VS
Drosophila
PEVVSSETVKRSMKQESSQEKNPSPAGSPQRDPLDVSFNDPIAAFKSKTTGELIRAYLVYMICSSENLVEHNMTLMK-WS
CAEEL
IPVA-------------------------------LVLTIIIEFFQSKSNTELVRALVVLRLCGIQTLVNQNQIILN-TM
Arabidopsis
ATRLL------------------------------R-TNFIRRSYRLPAFSPVGPPTVTASTAVVPEILSFGQQAPEPPL
Oryza
AIASR------------------------------I-QKRVLASFAAAAAAKLPEAAVAAAGGAAEAVEEVASSVQE---
POMBE
-----------------------------------------MRAFRLAS-GVLRNRKVILGIGAGSLITAGNIKIRN---
Emericella
--------------------------------------------MKAATPRPSVRALSSGRSYRTARFVSRTSNARSSLA
Cerevisiae
-----------------------------------------MIASKSSLLVTKSRIPSLCFPLIKRSYVSKTPTHSN---
PutA
PELPALLSG-----------------------AANESDEAPTPAEEPHQPFLDFAEQILPQSVSRAAITAAYRRPETEAV
Human
97-RKLLGQRLFNKLMKMTFYGHFVAGEDQESIQPLLRHYRAFGVSAILDYGVEEDLSPEEAEHKEMES----------CTSA
Drosophila
KNVLGQRLFTLLMKATFYGHFVAGEDQIKIIPTLERLRSFGVKPILDYSVEEDITQEEAEKREVESS---------VSSA
CAEEL
RRVLGKNLFKKTLKNTFFGHFVAGETEEEVRHVVEKLRNYGVKSILDYSVEADITSQEATDKTVKGTSVATVKPAAMTPV
Arabidopsis
HHPKPTEQSHDGLDLSDQARLFSSIPTSDLLRSTAVLHAAPIGPMVDLGTWVMSSKLMDASVTRGMV-----LGLVKSTF
Oryza
---QVQAQGAQVLEFGDTERLFAGERSTSLVRTLAVLQALSVGPLVDVATAALRSPAVAGSAA-G-------RAAARATA
POMBE
---DSK--FDAFFAKGFPDELQHR-SLFSVLRSAFVYEICSRAWLVKLSLGAMSLCDVFHLSFLYN-------PFCRYTF
Emericella
ADTNSLLQQAPPSPKKQLASPLAKLPLSSVLRSLLILSVSSSSILLKPCIYTLSALAHPKTALLDVAKNPLLNLLVKHTI
Cerevisiae
--TAANLMVETPAANANGNSVMAPPNSINFLQTLPKKELFQLGFIGIATLNSFFLNTIIKLFPYIP------IPVIKFFV
PutA
SMLLEQARLPQPVAEQAHKLAYQLADKLRNQKNASGRAGMVQGLLQEFSLSSQEGVALMCLAEALLR--IPDKATRDALI
:
Human
167-AERDGSGTNKRDKQYQAHRAFGD-RRNGVISARTYFYANEAKCDSHMETFLRCIEAS-GRVSDD-GFIAIKLTALGRPQF
Drosophila
GDKKEEGSMP---QYHVDKSFAD-RRYKVSSARTYFYLNEATCERNMEIFIKCLEAVSGATFGT-GITAIKLTALGRPQL
CAEEL
VDAKTLETTR--ERYTVHEEFGD-RRQGVSSARTYFYEGEEQCDKNRDIFKDSINAVASATKNE-GFVAVKITALGRPQL
Arabidopsis
YDHFCAGEDADAAAERVRSVYEATGLKGMLVYGVEHADDAVSCDDNMQQFIRTIEAAKSLPTSHFSSVVVKITAICPISL
Oryza
YQHFCAGETAEEAAAAVRRLWRG-GMGGILDYGIEDAEDGPACDRNAAGFLAAIDVAAALPPGS-ASVCIKITALCPVAL
POMBE
YKHFCGGETPQAVMATMDTLQAAGITSCLNYSREVDLDGDMDVNKIASQGVVPPQVPVPSEKNQKVLRQIADKAFESNMH
Emericella
YKQFNAGENKLEVQRSINAIKELGYRGVLLGYAREVLVGESKTD----------------PRDEQASRQEIQTWLDGTLQ
Cerevisiae
SSLYCGGENFKEVIECGKRLQKRGISNMMLSLTIENSEGTKSLSS------TPVDQIVKETISS--VHNILLPNIIGQLE
PutA
RDKISNGNWQSHIGRSPSLFVNAATWGLLFTGKLVSTHNEASLSRSLNRIIGKSGEPLIRKGVDMAMRLMGEQFVTGETI
. : . . .
Human
244-LLQFSEVLAKWRCFFHQMAVEQGQAGLAAMDTKLEVAVLQESVAKLGIASR-AEIEDWFTAETLGVSGTMDLLDWSSLID
Drosophila
LLQLSEVIMRTRKYMEDMVGGQG----NVLTHHKTIKDLEKYYATLGDNK---DVKEFLNNVTSDKEGILHLFPWSGIVD
CAEEL
LLKLSEAIVQTQNFFKALTGGMS-----LQEGRLTSQEFYKRLGELGVKTDTESVKKFFDEVDFDSDGIVDLHGWNHILD
Arabidopsis
LKRVSDLLR--------------------WEYKSPNFKLSWKLKSFPVFS------------------------------
Oryza
LEKASDLLR--------------------WQQKHPATKLPWKVHGFPVLC------------------------------
POMBE
IIDMATYKP--------------------GTVCAVKLTPFINPLVLQRYN--------------------------SILN
Emericella
TVDMAQEGD---------------------------FVALK----FTGMG------------------------------
Cerevisiae
SKPINDIAP--------------------G-YIALKPSALVDNPHEVLYN------------------------------
PutA
AEALANARKLEEKGFRYSYDMLGEAALTAADAQAYMVSYQQAIHAIGKAS--------------------------NGRG
Human
323-SRTKLSKHLVVPNAQTGQLEPLLSRFTEEEELQMTRMLQRMDVLAKKATEMGVRLMV---DAEQTYFQP-AISRLTLEMQ
Drosophila
EDSQLSDTFRVPDPQTGQMRRLISQIPPKEEEMFRNMIRRLNTIVKAAADLDVRIMV---DAEQTYFQP-AISRITLEMM
CAEEL
DHVKLGQLFQVLNIKTGSLEPLIQNLSNEEEQEFRNMVRRTLDVAEYAIEKGVRIMV---DAEQTYLQP-AISKITIEMM
Arabidopsis
----ESSPLYHTNSEP-------EPLTAEEERELEAAHGRIQEICRKCQESNVPLLI---DAEDTILQP-AIDYMAYSSA
Oryza
----VSSPLYLTAAEP-------PALEAEEERELEMAHGRLLAIGERCAEYDIPLLV---DAEYATVQP-AIDYFTFAGA
POMBE
QYPVESACNYLEHLKS-------PELSTYEVSELKKFWEYADKLCQFAKEKQIPLFI---DAEQTYFQD-CMHAVTVDLM
Emericella
----IQALEYLQNQAP-------P---------SPFMDEAIKQVCDLAISRNVRLLV---DAEEQAVQP-GIEEWATMYQ
Cerevisiae
----FSNPAYKAQRDQ---------LIENCSKITKEIFELNQSLLKKYPERKAPFMVSTIDAEKYDLQENGVYELQRILF
PutA
IYEGPGISIKLSALHP-------RYSRAQYDRVMEELYPRLKSLTLLARQYDIGINI---DAEEADRLEISLDLLEKLCF
: . : : *** :
Human
399-RKFNVE---KPLIFNTYQCYLKDAYDNVTLDVELARREGWCFGAKLVRGAYLAQE-------RARAAEIGYEDPINPTYE
Drosophila
RKYNKD---KAIVFNTYQCYLRETFREVNTDLEQAKRQNFYFGAKLVRGAYMDQE-------RDRAKSLGYPDPVNPTFE
CAEEL
KKYNKG---RGNIFNTYQAYLKGTLQNMEADMQVARREGWHFGAKLVRGAYMEQE-------RARAKAIGYEDPINDNFE
Arabidopsis
IMFNADKD-RPIVYNTIQAYLRDAGERLHLAVQNAEKENVPMGFKLVRGAYMSSE-------ASLADSVGCKSPVHDTIQ
Oryza
LAFNGG-G-RPIVHGTVQAYLRDARDRLEAMARAAQGERVCLALKLVRGAYLARE-------ARLAASLGVPSPVHRSIQ
POMBE
RKYNKE---VAIVHNTYQLYLKKSRKIMDDHIKKCVAEGWLMGAKLVRGAYLNSEPRFLIHDTKAETDKDFDSAVEAIIA
Emericella
KYCNSRTPGRAIFYNTYQAYLCSTPATLARHLEISRKEGYTLGVKLVRGAYLKTEPRHLIWAKKEQTDECYDGIVEALLT
Cerevisiae
QKFNPTSSKLISCVGTWQLYLRDSGDHILHELKLAQENGYKLGLKLVRGAYIHSE------KNRNQIIFGDKTGTDENYD
PutA
EPELAG---WNGIGFVIQAYQKRCPLVIDYLIDLATRSRRRLMIRLVKGAYWDSEIKR----AQMDGLEGYPVYTRKVYT
. * * : . . : :**:*** *
Human
469-ATNAMYHRCLDYVLEELKHN-------AKAKVMVASHNEDTVRFALRRMEELG-LHPADHR-VYFGQLLGMCDQISFPLG
Drosophila
ATTDMYHRTLSECLRRIKLMKDCDDDARKIGIMVASHNEDTVRFAIQQMKEIG-ISPEDKV-ICFGQLLGMCDYITFPLG
CAEEL
ATSKMYESCLTRIADEVHRR-----GKTNVSVMVASHNEDTVRFALNLMKEKC-ISPSERV-MCMAQLYGMCDQVSFSLG
Arabidopsis
DTHSCYNDCMTFLMEKASNGS-------GFGVVLATHNADSGRLASRKASDLG-IDKQNGK-IEFAQLYGMSDALSFGLK
Oryza
DTHDCYNGCAAFLLDRVRRG--------AAAVTLATHNVESGQLAAARALELG-IGGGGDRGLQFAQLMGMADGLSLGLR
POMBE
AAAKFAPGDPASASDPIASRK------GKWGIMVASHNKKTMFESVNLAETKK-VDFTKTS-FYLAQLLGMADDITYALA
Emericella
RRYNHMLKPASAEHTTELPP---------VSVIVATHNRDSVRKAHALRLEQASRGEKSDVELTYAQLQGMADEISCELL
Cerevisiae
RIITQVVNDLIINGEDSYFG----------HLVVASHNYQSQMLVTNLLKSTQDNSYAKSN-IVLGQLLGMADNVTYDLI
PutA
DVSYLACAKKLLAVPNLIYP------------QFATHNAHTLAAIYQLAGQNY-----YPGQYEFQCLHGMGEPLYEQVT
.*:** .: * **.: : :
Human
540--------------QAGYPVYKYVPYGPVMEVLPYLSRRALENSSLMKGT--HRERQLLWLELLRRLRTGNLFHRPA----
Drosophila
-------------QAGYSAYKYIPYGPVEEVLPYLSRRAQENKGVLKKI--KKEKRLLLSEIRRRLMRGQLFYKPKGNYV
CAEEL
-------------QAGFSVYKYLPYGPVEEVLPYLSRRALENGSVLKKA--NKERDLLWKELKRRISSGEFKARSSSSS-
Arabidopsis
-------------RAGFNVSKYMPFGPVATAIPYLLRRAYENRGMMATG--AHDRQLMRMELKRRLIAGIA---------
Oryza
-------------NAGFQVSKYLPYGPVEQIIPYLIRRAEENRGLLSSS--SFDRQLLR---------------------
POMBE
Y-------SQRNQQPNFCIVKYVSCGPISEVLPYLVRRARENIDALDRC--KEERAYYRQALRRRIF-------------
Emericella
QGFQTAGPENTKVAESPNVYKLLTWGSVKECMGFLLRRAVENTEAVGRT--KQSQEAMFSELRRRARRAFGLRY------
Cerevisiae
TN-----------HGAKNIIKYVPWGPPLETKDYLLRRLQENGDAVR----SDNGWPLIKAIAKSIPKRVGL--------
PutA
G-------KVADGKLNRPCRIYAPVGTHETLLAYLVRRLLENGANTSFVNRIADTSLPLDELVADPVTAVEKLAQQEGQT
. *. :* ** ** .
Figure 1.7 : Alignement de 9
séquences de proline déshydrogénase
réalisé avec l'aide du logiciel clustalw. Les
séquences alignées sont relatives aux espèces
eucaryotes, humaine (Human, 600 résidus), Drosophila Melanogaster
(Drosophila, 669 résidus), Caenorhabditis elegans (CAEEL, 564
résidus), Arabidopsis thaliana (Arabidopsis, 499 résidus), Oryza
sativa (Oryza, 475 résidus), Schizosaccharomyces pombe (POMBE,
492 résidus), Emericella nidulans (Emericella, 478
résidus), Saccharomyces cerevisiae (Cerevisiae, 476 résidus), et
à l'espèce procaryote PutA (607 résidus N-terminaux).
Les résidus sont colorés en rouge lorsqu'ils sont
conservés (sigle *), en vert lorsqu'il sont fortement similaires (sigle
:), et en bleu lorsqu'ils sont faiblement similaires (sigle .). La
numérotation est relative à la séquence humaine.
D'une manière générale, la proline
déshydrogénase est une protéine très peu
conservée
de la bactérie jusqu'à l'homme (3 %
d'identité, et 4 % de similarité). Comme le montre la
Figure 1.7, les 9 séquences ne s'alignent que dans la
moitié C-terminale de PRODH humaine
qui s'étend des résidus 340 à 600.
Dans cette région, les pourcentages d'identité et de
similarité atteignent respectivement 8 % et 10 %. Sur la base de cet
alignement, il apparaît donc que la fonction proline oxydase,
commune à toutes ces protéines, est assurée par un
domaine catalytique situé entre les résidus 340 et 600 dans la
séquence humaine.
Nous avons soumis la séquence de PRODH humaine
aux logiciels SMART (Simple Modular Architecture Research Tool)
(Schultz et al., 1998) et Pfam (Finn et al., 2006) qui
permettent de détecter des domaines de repliement connu. Aucune
prédiction n'a été proposée avec un degré de
confiance suffisant par ces 2 programmes, ce qui était attendu dans la
région
C-terminale dans la mesure où il n'existait aucune
structure connue de proline oxydase lorsque nous avons abordé
cette étude. Une étude de prédiction de structure
secondaire a
également été réalisée et
suggère une structuration de la protéine PRODH humaine en
hélice á
et feuillet â. Devant le peu d'informations
apportées par ces logiciels, il nous est apparu
déraisonnable d'envisager de sélectionner des
domaines à partir d'un alignement de séquence
et d'une prédiction de structure secondaire.
C'est pourquoi, nous avons opté pour une stratégie
différente qui consiste à isoler des domaines
structurés, et dont la taille soit compatible avec une analyse
par RMN, par protéolyse ménagée à partir de
la protéine PRODH humaine sauvage.
2.2) Démarche entreprise
La première étape de cette stratégie
a pour objectif de surexprimer la proline déshydrogénase
humaine sauvage sous forme soluble et repliée dans un système
recombinant. Dans l'optique d'une étude par RMN, nous avons
choisi d'utiliser la bactérie E. coli qui présente
l'avantage d'être bien caractérisée, d'avoir une
croissance rapide, et de pouvoir produire des quantités importantes
de protéines marquées en isotopes 15N,
13C, ou 2H. Une fois purifiée par chromatographie
d'affinité, la protéine repliée sera soumise à une
digestion enzymatique ménagée en conditions non
dénaturantes par des endoprotéases clivant
spécifiquement les chaînes polypeptidiques après
certains acides aminés. Les fragments protéiques seront
séparés sur colonne HPLC, puis leur séquence sera
déduite de leur analyse
par spectrométrie de masse MALDI-TOF. En optimisant les
conditions de digestion, il sera possible de limiter la protéolyse aux
sites exposés et non enfouis reconnus par les différentes
endoprotéases, et ainsi d'identifier potentiellement les domaines
structuraux globulaires de la protéine PRODH. Les domaines dont le poids
moléculaire sera compatible avec une analyse
par RMN seront ensuite clonés, puis surexprimés
chez E. coli, dans l'optique de résoudre leur
structure par Résonance Magnétique Nucléaire
et Modélisation Moléculaire.
Chapitre 2 : Expression des protéines PRODH
sauvage et mature chez E. coli
CHAPITRE 2
Expression des protéines PRODH
sauvage et mature chez E. coli
L'expression des protéines PRODH sauvage et mature a
été réalisée au Laboratoire de
Génétique Moléculaire de l'EMI 9906
dirigé par le Professeur Thierry Frebourg, et en étroite
collaboration avec les chercheurs du Groupe d'Etude des Bases
Moléculaires de la Schizophrénie. Le détail des
protocoles expérimentaux relatifs au clonage, à la production,
et
à l'analyse de ces protéines figure à la fin
de ce chapitre dans la partie Matériels et Méthodes.
1) Production de PRODH sauvage
Comme nous l'avons définie dans le chapitre
précédent, la première étape de la stratégie
d'étude structurale de PRODH consiste à surexprimer la
totalité de la protéine sauvage
(1-600) chez la bactérie E. coli.
Pour cela, nous avons choisi d'utiliser le plasmide d'expression
pQE-31 de la société Qiagen qui permet de
produire des protéines recombinantes fusionnées à
une étiquette 6xHis en N-terminal (succession de 6
résidus histidine). Ce vecteur comporte dans sa région
promotrice un promoteur fort de type T5 à induction directe
à l'IPTG, suivi de 2 sites opéron lactose qui permettent sa
régulation par la protéine LacI. Après clonage du
plasmide par l'ADNc de PRODH sauvage, des souches
bactériennes BL21 contenant le vecteur pREP-4 (Qiagen) ont
été transformées par le plasmide pQE-31 cloné. Le
vecteur pREP-4 comporte le gène qui encode la protéine
de régulation LacI.
1.1) Caractérisation de l'expression et de la
solubilité de la protéine hétérologue
Les premiers tests de surexpression ont
été entrepris dans le but de caractériser
l'expression et la solubilité de la protéine recombinante.
La production de PRODH a été induite en erlenmeyer de 300
mL contenant 30 mL de milieu de culture LB par ajout de 1 mM d'IPTG à
une densité optique (DO) de 0.5. Des prélèvements
de 1 mL ont été réalisés à intervalles
de temps réguliers afin de suivre l'évolution de
l'expression. Pour chaque prélèvement, les cellules
bactériennes ont été lysées dans un tampon
phosphate à pH neutre et
les fractions contenant les protéines solubles et
insolubles ont été séparées par centrifugation.
Le culot contenant le matériel insoluble a
été repris avec un tampon phosphate contenant de l'urée 8
M à pH neutre, puis incubé à 4°C sous agitation
pendant 6 heures. Après une seconde étape de centrifugation, les
agrégats du culot final ont été finalement
solubilisés avec du SDS,
un détergent ionique dénaturant. Toutes les
fractions prélevées ont été analysées
par SDS-
PAGE et Western-blot (Figure 2.1). L'analyse Western-blot a
été réalisée avec un anticorps
anti-6xHis qui permet de révéler les
protéines qui présentent un amas de 6 résidus histidine en
surface.
t0 2H 4H 5H
t0 2H 4H 5H
(kDa)
91
50
fraction soluble
fraction urée 8M
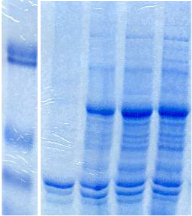




t0 2H 4H 5H
2H 4H 5H
(kDa)
91
PRODH
PRODH
50
culot final
Western-blot
Figure 2.1 : Expression de PRODH
sauvage chez E. coli. Analyses SDS-PAGE des fractions
contenant les protéines solubles, les
protéines insolubles solubilisées dans l'urée et le culot
final repris avec du SDS, après 2 heures (2H), 4 heures (4H), et 5
heures d'induction (5H), et avant induction (t0). Les résultats de
l'analyse Western-blot du culot final sont également
présentés.
L'analyse SDS-PAGE du culot final fait apparaître une
bande de surexpression à une masse apparente de 70 kDa à partir
de 2 heures d'induction. L'intensité de cette bande, qui correspond
à la masse attendue de PRODH sauvage, est maximale lorsque le
temps d'induction atteint 4 heures. La révélation de cette
bande protéique par un anticorps anti-
6xHis confirme la présence de PRODH sauvage dans le culot
final (Figure 2.1). En ce qui concerne les fractions contenant les
protéines solubles et les protéines insolubles reprises dans
l'urée, aucune bande de surexpression correspondant
à cette masse apparente n'est clairement
discernable sur le gel SDS-PAGE. De plus, l'analyse Western-blot
par l'anticorps anti-6xHis,
qui est plus sensible qu'une coloration au bleu de
Coomassie, ne révèle aucune bande d'expression de
manière significative dans ces fractions (non
présenté). L'ensemble de ces résultats indique
clairement que la protéine PRODH sauvage est exprimée
sous forme insoluble chez E. coli dans ces conditions
d'expression.
1.2) Optimisation de paramètres d'expression et
d'extraction
Comme nous l'avons évoqué
précédemment, les données de la littérature
indiquent que PRODH humaine est une enzyme mitochondriale localisée dans
la matrice et fixée à la membrane interne. Par conséquent,
la structure tertiaire de la forme native de cette protéine pourrait
présenter des surfaces hydrophobes exposées qui lui
permettrait de fixer les phospholipides de la membrane interne.
Lorsqu'elle est exprimée chez E. coli, ces surfaces hydrophobes
pourraient accrocher les membranes bactériennes, et ainsi
retenir la protéine hétérologue dans les fractions
contenant les protéines insolubles. Nous avons essayé de
recueillir la protéine recombinante PRODH dans la fraction soluble en
introduisant différents détergents non dénaturants dans le
tampon de lyse. Ces types de détergents amphiphiles, qui
dégradent les membranes bactériennes, interagissent
également avec les surfaces hydrophobes exposées des
protéines, et favorisent ainsi leur solubilisation en
présentant une extrémité polaire au solvant aqueux.
Trois détergents différents ont été
testés dans des gammes de concentration s'échelonnant de 1 %
à 5 % (v/v). Il s'agit du Triton X-100, du NP 40, et du Tween 20. Dans
tous les cas, nous ne sommes jamais parvenus à détecter PRODH
sauvage dans la fraction soluble. Les profils d'expression obtenus étant
tout à fait identiques à celui obtenu en absence de
détergent, nous en avons conclu que la protéine recombinante
PRODH humaine est produite sous forme de corps d'inclusion chez E.
coli.
Les corps d'inclusion sont des agrégats
insolubles composés principalement de protéine recombinante
mal repliée et biologiquement inactive. Leur formation est en
partie liée au taux d'expression du gène
hétérologue dans la bactérie. Ainsi, un taux d'expression
trop élevé favorise les interactions hydrophobes
intermoléculaires par rapport aux interactions internes, et
empêche le repliement correct de la protéine au profit de la
formation d'agrégats (Georgiou & Valax, 1999). L'urée
est un chaotrope puissant qui permet de dénaturer et solubiliser
ces corps d'inclusion. Dans certains cas, des concentrations
modérées d'urée de l'ordre de 2 à 3 M sont
suffisantes pour resolubiliser des protéines agrégées dont
le repliement
n'est pas trop éloigné du repliement natif (Clark,
2001). Dans le cas de PRODH la reprise de
la fraction insoluble par de l'urée 8 M pendant 6 heures
n'a pas été suffisante pour dénaturer
les corps d'inclusion contenant la protéine
recombinante ne serait-ce que partiellement. Ceci laisse supposer que plusieurs
segments hydrophobes de PRODH sont exposés en surface et
établissent de fortes interactions intermoléculaires au
sein des agrégats. Dans l'optique de limiter ce type d'interaction,
nous avons essayé de réduire les taux d'expression de PRODH,
d'une part en ralentissant le métabolisme bactérien par
une diminution de température de
37°C à 29°C, et d'autre part en diminuant la
quantité d'inducteur IPTG de 1 mM à 0.1 mM.
En parallèle, nous avons également
réalisé une optimisation de la DO d'induction en testant
les valeurs suivantes : 0.3, 0.4, 0.5, 0.6, 0.7, et 0.8. Dans
tous les cas de figure testés, aucune amélioration de
solubilité de la protéine recombinante n'a été
constatée.
2) Prédiction du peptide signal de la
séquence PRODH humaine
Nous avons montré que la production de la
protéine PRODH humaine chez E. coli
conduit à l'unique formation de corps d'inclusion. La
stratégie d'étude structurale de PRODH
qui a été initialement définie repose sur
une recherche de domaines structurés à partir de la
protéine entière repliée. Cette protéine de 70
kDa n'étant pas exprimée sous forme soluble chez E.
coli, nous avons été contraint de modifier notre
stratégie de départ. Nous avons ainsi entrepris d'identifier et
de produire la forme mature de cette protéine humaine mitochondriale.
2.1) Les messages d'adressage mitochondrial
La proline déshydrogénase humaine est une
protéine mitochondriale encodée par un gène porté
par de l'ADN nucléaire. Comme toutes les protéines
mitochondriales synthétisées
au niveau du ribosome du cytoplasme, elle possède dans sa
séquence primaire l'information
qui lui permet de rejoindre et d'intégrer la
mitochondrie (pour revue : Pfanner & Neupert,
1990). Cette information, appelée message
d'adressage ou peptide signal, est toujours localisée dans
l'extrémité N-terminale de la séquence. Ces motifs sont au
nombre de 1 ou 2 dans la séquence primaire en fonction de la
localisation précise de la protéine dans la mitochondrie.
Ils n'intègrent pas la forme mature de la protéine et sont
clivés après leur entrée dans la mitochondrie. D'un
point de vue structural, les peptides signaux sont des hélices
amphipatiques de longueur variable qui comportent, une
région N-terminale chargée positivement, une région
centrale très hydrophobe, et une région C-terminale
plutôt neutre
(Nielsen et al., 1997). Ils se caractérisent
également par la présence dans leur extrémité C-
terminale d'un résidu neutre de type alanine en
position -3 et -1 du site de clivage. La présence de tels
motifs à caractère hydrophobe dans une région qui
n'appartient pas à la protéine mature peut favoriser
la formation de corps d'inclusion dans le cas d'une
surexpression chez E. coli. L'identification de ces peptides signaux
s'avère donc utile dans l'optique d'améliorer les profils
d'expression de PRODH recombinante.
2.2) Résultats de la prédiction
Plusieurs programmes bio-informatiques disponibles sur le
web ont été récemment développés avec
pour objectif la prédiction de peptides signaux à partir
d'une séquence primaire. A ce jour, il n'existe pas de
séquence consensus de peptide signal d'adressage mitochondrial.
Aussi, ces programmes utilisent les caractéristiques topologiques de ces
motifs (citées ci-dessus) pour prédire leur présence
dans une séquence. Nous avons soumis la séquence de PRODH
humaine à plusieurs de ces programmes. Les résultats
obtenus sont reportés dans le tableau suivant :
|
Programme
|
Longueur du peptide signal N-terminal
|
|
Predotar (Small et al., 2004)
|
non détecté
|
|
iPSORT (Bannai et al., 2002)
|
30
|
|
MitoProt II 1.0 (Claros & Vincens, 1996)
|
49
|
|
PSORT II (Nakai & Horton, 1999)
|
57
|
|
TargetP V1.0 (Emanuelsson et al., 2000)
|
114
|
|
SignalP V1.0 (Nielsen et al., 1997)
|
35 et 24
|
Tableau 2.1 : Bilan de l'analyse de
la séquence primaire de PRODH humaine par 6 logiciels
de prédiction de peptide signal d'adressage
mitochondrial.
De manière inattendue, les prédictions obtenues
sont très différentes d'un programme
à l'autre. Ainsi, la présence d'un message
d'adressage mitochondrial est bien prédite chez PRODH par la
majorité des logiciels, mais la longueur du peptide signal
varie de manière significative selon le programme de 30 à 114
résidus. La figure 2.2 présente le diagramme de prédiction
obtenu avec le logiciel SignalP qui est présenté comme
l'algorithme le plus fiable d'après une récente étude
comparative (Emanuelsson et al., 2001). Ce diagramme suggère la
présence de 2 peptides signaux dans la région N-terminale de
PRODH, qui pourraient peut- être expliquer la divergence des
résultats obtenus avec les autres programmes. Le premier est
prédit au niveau des 35 premiers résidus et le
second serait situé entre les résidus 65 et 88.
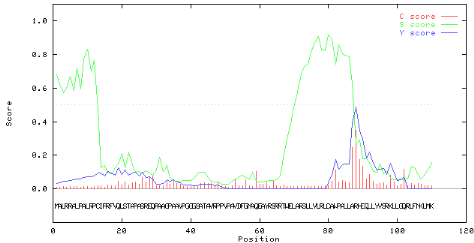
Figure 2.2 : Diagramme de
prédiction de peptide signal de PRODH par le logiciel SignalP.
La prédiction de l'existence de 2 peptides
signaux dans l'extrémité N-terminale de PRODH humaine est
surprenante dans la mesure où les protéines mitochondriales
destinées à intégrer la matrice ne contiennent qu'un seul
de ces motifs (Pfanner & Neupert, 1990). Nous avons soumis la
séquence primaire de PRODH humaine au logiciel HMMTOP (Tusnady &
Simon, 1998) qui permet de prédire les hélices transmembranaires.
De manière intéressante, l'algorithme HMMTOP suggère la
présence d'une hélice transmembranaire unique entre les
résidus 70 et 87, c'est-à-dire au niveau du second
peptide signal potentiel. Dans leur étude comparative, Emanuelsson
et al., ont mis en évidence la grande difficulté des
programmes de prédiction de peptide signal à distinguer les
motifs transmembranaires des hélices d'adressage mitochondrial
(Emanuelsson et al., 2001). La prédiction d'un second peptide signal
potentiel n'est donc pas surprenante et correspond plus vraisemblablement
à un segment hydrophobe.
Au final, nous avons utilisé une partie des
résultats de SignalP pour estimer la longueur du peptide signal
N-terminal de PRODH à 35 résidus. La longueur moyenne
d'un signal d'adressage mitochondrial étant de 23 résidus, cette
prédiction semble tout à fait réaliste.
L'analyse de la séquence de PRODH humaine par des outils
bio-informatiques a donc permis de prédire la présence d'un
peptide d'adressage mitochondrial dans l'extrémité
N-terminale 1-35 de la protéine. Ce motif
à caractère hydrophobe n'incluant pas la forme mature de
la protéine, nous avons par conséquent entrepris
de réaliser une nouvelle construction PRODH délestée
de ce peptide signal. Cette forme mature est nommée PRO564
et correspond à la région 37-600 de PRODH humaine
(66 kDa).
3) Production de la protéine mature PRO564
Le vecteur d'expression pQE-31 encodant la protéine PRO564
a été obtenu à partir du plasmide pQE-31 contenant le
gène PRODH par délétion. Après
vérification de sa séquence,
ce nouveau vecteur a été intégré dans
des souches BL21 thermocompétentes préalablement
transformées par le plasmide pREP-4.
Les protocoles d'expression et d'extraction que nous
avons appliqués pour tester la solubilité de la nouvelle
construction sont tout à fait identiques à ceux utilisés
pour produire PRODH sauvage. Les cultures d'expression de 30 mL de PRO564 ont
été induites à une DO mesurée à 600 nm de
0.5 par ajout de 1 mM d'IPTG, puis incubées à 37°C pendant 3
heures. Après lyse des bactéries, les protéines
cytosolubles ont été extraites par centrifugation, et le
matériel insoluble a été repris avec de l'urée 8 M.
Après une seconde étape de centrifugation,
les agrégats du culot final ont été
solubilisés avec du SDS.
A l'instar de PRODH sauvage, nous avons
également réalisé une optimisation de plusieurs
paramètres comme la quantité d'inducteur IPTG (0.1 mM, 0.5
mM, et 1 mM), la température d'expression (29°C et 37°C), et
la nature du détergent utilisé dans le tampon de lyse (Triton
X-100, NP 40, et Tween 20). Les meilleurs taux d'expression pour l'ensemble des
fractions ont été obtenus avec une concentration d'IPTG de 0.1
mM, une température de
29°C, et en utilisant du Triton X-100 à 2 %.
L'analyse SDS-PAGE de l'expression de
PRO564 dans ces conditions est présentée dans la
Figure 2.3.
Comme le montre la Figure 2.3, une bande de surexpression dont
la masse apparente correspond à la masse attendue (66 kDa) est
observable dans les fractions contenant le culot final et les
protéines insolubles reprises dans l'urée. Toutefois, cette
bande d'expression n'apparaît pas dans le surnageant de lyse contenant
les protéines solubles. D'autre part, nous avons constaté que
l'optimisation des conditions expérimentales citées
ci-dessus n'a eu aucune influence sur la solubilité de la
protéine recombinante. Par conséquent, à l'image de la
protéine hétérologue PRODH sauvage, la surexpression
de PRO564 chez E. coli conduit à l'unique formation
de corps d'inclusion. Nous avons toutefois noté une
différence significative entre le profil d'expression de la forme
mature et celui de la forme sauvage. Lorsque nous avons produit PRODH
humaine, la reprise du matériel insoluble par de l'urée
8 M pendant 6 heures n'a pas été suffisante
pour dénaturer ne serait-ce qu'une partie de
protéine insoluble PRODH. Dans le cas de PRO564, plus d'un
tiers de protéine insoluble a été dénaturée
par l'urée dans les mêmes conditions. Par conséquent, ceci
laisse supposer la part
de responsabilité du peptide signal dans
l'agrégation de PRODH recombinante.

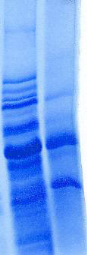

SN U C
(kDa)
91
PRO564
50
Figure 2.3 : Expression de la
protéine PRO564 chez E. coli. Analyse SDS-PAGE des
fractions contenant, les protéines solubles (SN), les
protéines insolubles resolubilisées dans l'urée 8 M (U),
et le culot final repris dans du SDS (C).
4) Conclusions
Dans le travail qui a été
présenté, nous avons abordé la première
étape de l'étude structurale de PRODH humaine en la
surexprimant chez la bactérie E. coli. Nous avons
montré que la production des protéines PRODH sauvage et
mature dans cet organisme conduit à l'unique formation de corps
d'inclusion, et ceci malgré une optimisation de quelques
paramètres d'expression et d'extraction.
Lorsque l'expression d'une protéine sous forme soluble
n'est pas possible chez E. coli, une des stratégies
alternatives d'obtention de la forme repliée communément
utilisée consiste
à renaturer la protéine in vitro
à partir de corps d'inclusion purifiés. Cette
technique, qui repose sur la renaturation de la protéine par dialyse de
l'agent dénaturant, présente cependant
un caractère tout autant incertain. Elle
nécessite ainsi de contrôler que le repliement final de la
protéine soit natif et biologiquement actif à l'issu de
la renaturation. Lorsque nous avons entrepris de produire PRODH chez E.
coli, il n'existait aucune donnée structurale connue de
proline déshydrogénase d'une espèce
eucaryote suffisamment proche qui permette de vérifier
l'efficacité d'une éventuelle renaturation in
vitro. Les données de la littérature font état de
2
tests d'activité de PRODH in vitro, dont l'un
repose sur le dosage du produit de dégradation
de la proline par l'O-aminobenzaldehyde (Johnson &
Strecker, 1962). Ces tests d'activité, qui n'étaient pas mis en
place au Laboratoire de Génétique de Rouen, nécessitent
généralement une mise au point qui d'une part, peut être
coûteuse en temps et d'autre part, demande une certaine expérience
en la matière. Par conséquent, ne disposant d'aucun moyen de
vérification
de l'efficacité d'une éventuelle renaturation,
nous n'avons pas envisagé d'obtenir la forme repliée de
PRODH par renaturation in vitro des corps d'inclusion.
A ce stade du projet, il apparaissait donc que seule
l'expression de PRODH sous forme soluble nous permette d'accéder
à l'étape supérieure de l'étude structurale. Deux
choix stratégiques différents étaient alors possibles :
changer d'organisme de production, ou intégrer une structure qui
permette un criblage à haut ou moyen débit de plusieurs
conditions d'expression chez E. coli. Nous avons opté
pour la deuxième stratégie en initiant une collaboration
avec le Laboratoire de Marquage des Protéines (LMP) du
Département d'Ingénierie et d'Etude des Protéines
(DIEP, Direction Docteur André Menez) du CEA de Saclay. Dans le
cadre de cette collaboration, j'ai été accueilli au sein
du LMP afin de bénéficier des infrastructures d'une
plate-forme de production à moyen débit de
protéines recombinantes chez E. coli.
Outre la possibilité de tester un grand nombre
de conditions expérimentales, le programme de production du LMP
permet également de surexprimer en parallèle plusieurs
protéines ou domaines dans un délai raisonnable. C'est
pourquoi, nous avons entrepris de produire la forme mature de PRODH
humaine, ainsi que 3 autres domaines de cette protéine, dans le cadre
de cette plate-forme. En effet, de nouvelles données sont
apparues dans la littérature lorsque nous produisions PRO564. La
publication récente de la première structure
du domaine catalytique de proline déshydrogénase de
E. coli (Lee et al., 2003) nous a permis
de modifier notre stratégie d'étude structurale de
PRODH. Sur la base de cette structure, nous avons mené une analyse
bio-informatique afin de sélectionner 3 domaines potentiels de
PRODH. La description de cette étude fait l'objet du
prochain chapitre.
5) Matériels et Méthodes
5.1) Création des plasmides d'expression
5.1.1) Clonage de PRODH sauvage dans le vecteur
d'expression pQE-31
L'ADNc de PRODH sauvage a été
obtenu au laboratoire par RT-PCR à partir d'ARNm extraits de
cellules humaines, puis cloné dans un plasmide eucaryote pcDNA3
(Invitrogen). Le clonage des 1800 nucléotides de la région
codante de PRODH dans le vecteur d'expression procaryote pQE-31
(Qiagen) se déroule en plusieurs étapes. Des souches E. coli
DH5á transformées par le plasmide pcDNA3 sont mises
en culture et sont utilisées pour réaliser une
minipréparation d'ADN plasmidique. L'ADNc de PRODH sauvage,
encadré par
les sites de restriction reconnus par les enzymes BamHI
et KpnI, est linéarisé par digestion avec ces 2 enzymes
à 37°C pendant 2 heures. En parallèle, le vecteur pQE-31,
qui contient
ces 2 sites de restriction au niveau de son site de clonage
multiple, est également digéré par
BamHI et KpnI dans les mêmes conditions.
Les produits de digestion sont séparés et purifiés
sur gel d'agarose à 0.9 % en découpant les
bandes correspondantes avec le kit QIAquick Gel Extraction (Qiagen).
Après estimation de la concentration finale des produits par mesure des
absorbances à 260 et 280 nm, la ligation entre l'insert PRODH
sauvage et le plasmide pQE-31 linéarisé est
réalisée avec la T4 DNA ligase (New England Biolabs)
à 16°C pendant 24 heures.
Le produit de ligation est transformé dans des souches
chimiocompétentes E. coli BL21 par choc thermique à
42°C pendant 45 secondes. Le vecteur pQE-31 possédant un
gène de résistance à l'ampicilline, le produit de
transformation est étalé sur boîte LB/agar contenant de
l'ampicilline à 50 ug/mL, puis incubé à 37°C pendant
toute une nuit. Le lendemain, plusieurs colonies isolées sont
mises en culture en milieu LB et une minipréparation
d'ADN plasmidique est effectuée pour chacune de ces cultures. Les
produits de ces minipréparations sont séquencés sur
séquenceur automatique Applied Biosystem 373A afin de
sélectionner les colonies qui possèdent le vecteur pQE-31
correctement cloné par PRODH sauvage. La dernière
étape consiste à transformer des souches BL21 contenant
le vecteur pREP-4, qui encode la protéine de régulation
LacI, par le vecteur pQE-31 cloné. Le plasmide pREP-4 contenant
un gène de résistance à la kanamycine, les
bactéries qui possèdent les vecteurs pQE-31 et pREP-4 sont
sélectionnées par ajout de 50 ug/mL d'ampicilline et de 30 ug/mL
de kanamycine dans les milieux de culture. Ces cultures sont finalement
mélangées à 25 % de
glycérol puis stockées à -80°C.
5.1.2) Construction du vecteur pQE-31 contenant
PRO564
La protéine PRO564 correspond à la
protéine PRODH sauvage délestée de 36 acides aminés
à son extrémité N-terminale. De ce fait, nous avons choisi
de réaliser le vecteur de surexpression de PRO564 à partir
du vecteur pQE-31 cloné par PRODH sauvage par une
technique apparentée à la mutagenèse dirigée.
La stratégie consiste à amplifier par PCR la
totalité du vecteur pQE-31 cloné à l'exception de la
région qui correspond à l'extrémité 1-36
de PRODH. Pour cela, nous avons utilisé le kit de
mutagenèse QuikChange Site-Directed
Mutagenesis de la société Stratagene.
Une minipréparation d'ADN plasmidique est
préalablement réalisée à partir de cultures de
BL21 uniquement transformées par le plasmide pQE-31 cloné
par PRODH. Ce vecteur est amplifié par PCR à
l'aide d'amorces sens et anti-sens complémentaires des
extrémités des régions d'ADN à dupliquer. Les
amorces utilisées sont del-PRODH-40-For
(5'-GTGCCAGGAGGTGGGTCGGCCAC-3') et del-PRODH-Rev
(5'-ATGGTGATGGTGA TGGTGAGATCCTCTCATAG-3'). Le mélange
réactionnel de PCR est composé de 2.5 unités
d'enzyme Pfu Turbo (Stratagene), de 5 uL de tampon Pfu 10x,
de 125 ng de chaque amorce, de 0.2 mM de mélange
nucléotidique dNTP, et de 30 ng de vecteur pQE-31. Le volume
final est complété à 50 uL avec de l'eau MilliQ
stérile. Les conditions d'amplification
par la polymérase haute fidélité Pfu
Turbo sont résumées dans la figure suivante :
? 95°C ? 4 min
1) ? 95°C ? 30 sec
? 55°C ? 1 min
? 68°C ? 10 min
20x
? 95°C ? 30 sec
2) ? de 64°C à 55°C ?
1 min
(-1°C à chaque cycle)
? 68°C ? 10 min
10x
Figure 2.4 : Description du
protocole d'amplification PCR utilisé pour réaliser
la
construction PRO564.
Une fois les 30 cycles de PCR terminés, les amplicons sont
vérifiés sur gel d'agarose
1%, puis digérés avec l'enzyme de restriction
DpnI à 37°C pendant 1 heure 30. L'action de DpnI
permet de linéariser de manière spécifique le
plasmide circulaire parental pQE-31, et ainsi de le neutraliser avant
transformation dans E. coli. Les produits de PCR sont purifiés
sur
gel d'agarose à 0.9% avec le kit QIAquick Gel
Extraction en découpant les bandes qui migrent aux tailles
attendues. Après estimation de la quantité de
matériel, la ligation des
extrémités de l'insert par « bout franc »
est entreprise en mélangeant 50 ng d'amplicon avec 3
unités de T4 DNA ligase pendant 30 heures à
16°C. Plusieurs colonies BL21
chimiocompétentes contenant le plasmide pREP-4
sont transformées avec le produit de ligation par choc thermique
à 42°C pendant 45 secondes, puis étalées sur
boîte LB/agar contenant 50 ug/mL d'ampicilline et 30 ug/mL de kanamycine
à 37°C pendant toute une nuit.
Le lendemain, plusieurs colonies isolées sont mises en
culture avec les mêmes concentrations d'antibiotiques et une
minipréparation d'ADN plasmidique est effectuée pour chacune
d'entre elles. La construction du nouveau vecteur pQE-31 encodant la
protéine PRO564 est contrôlée
par digestion avec les enzymes BglI et
EcoRV. L'amplification d'un fragment par PCR pouvant engendrer
des erreurs inhérentes à l'utilisation des
polymérases, la séquence des clones
sélectionnés a été minutieusement
vérifiée avec le séquenceur automatique Applied
Biosystem 373A. Finalement, les cultures bactériennes
qui contiennent la construction attendue sont mélangées
à 25 % de glycérol puis stockées à -80°C.
5.2) Production, extraction, et analyse de PRODH et
PRO564
5.2.1) Surexpression des protéines
recombinantes
Les différents tests de surexpression des
protéines PRODH sauvage et PRO564 ont été
réalisés en utilisant le protocole décrit
ci-après. Certains paramètres ont fait l'objet d'une
optimisation et sont discutés dans les paragraphes 1.2 et 3.
Pour chaque production, l'ensemencement initial des
boîtes de pétri est effectué à partir des
stocks glycérol de bactéries conservés à
-80°C. Plusieurs colonies BL21 transformées par le vecteur
pREP-4 et un plasmide pQE-31 contenant un des gènes
d'intérêt sont étalées sur boite LB/agar contenant
50 ug/mL d'ampicilline et 30 ug/mL de kanamycine. Après une nuit
d'incubation à 37°C, une colonie isolée est mise en
préculture dans un falcon
de 12 mL contenant 5 mL de milieu LB, 100 ug/mL
d'ampicilline, et 30 ug/mL de kanamycine. L'ensemble est incubé
à 37°C pendant 3 heures sous une agitation de 250 rpm.
La croissance bactérienne est suivie par mesure de la DO
à 600 nm. Une culture de 30 mL de milieu LB contenant la même
concentration d'antibiotiques est introduite dans un erlenmeyer
de 300 mL, puis ensemencée avec un volume de
préculture de manière à obtenir une DO initiale
égale à 0.05. Cette culture est ensuite placée à
37°C sous une agitation de 250 rpm. L'expression des protéines
recombinantes est induite par ajout de 1mM d'IPTG dans les
milieux de culture lorsque la DO mesurée à 600 nm
atteint 0.5. La culture est alors poursuivie
pendant 3 heures à 37°C sous une agitation de 250
rpm. A l'issu de ce délai, la croissance
bactérienne est stoppée en introduisant les
erlenmeyers dans la glace.
5.2.2) Extraction des protéines
hétérogènes
La lyse des cellules bactériennes et l'extraction
des protéines sont réalisées de la manière
suivante. Les bactéries sont récoltées par centrifugation
à 6000xg pendant 20 minutes
à 4°C. Après élimination des
surnageants, les culots bactériens sont resuspendus dans 3 mL
d'un tampon contenant, 50 mM de phosphate (pH=7.1), 500 mM de
NaCl, et 2 mM d'EDTA.
La rupture des membranes bactériennes est
initiée en introduisant du lysozyme à une concentration
finale de 100 ug/mL. Le mélange est laissé sur glace pendant 15
minutes puis porté à 37°C pendant 10 minutes. 5 mM de MgCl2,
10 ug/mL de DNase I, et 50 ug/mL de RNase sont ensuite introduits dans le but
de dégrader l'ADN et l'ARN libérés dans le lysat.
L'ensemble est incubé à température ambiante pendant 10
minutes avant d'être soumis à 2 cycles de sonication afin de
compléter la lyse des membranes. Les fractions soluble et
insoluble sont séparées par centrifugation à 14000xg
pendant 45 minutes à 4°C. Il est à noter que l'utilisation
de DNase I, qui digère l'ADN endogène visqueux de E.
coli, permet d'éviter d'entraîner des protéines
solubles qui accrocheraient cet ADN dans la fraction des protéines
insolubles. Le culot contenant le matériel insoluble est
resuspendu avec 3 mL de tampon dénaturant composé de 50
mM de phosphate (pH=7.1), 500 mM de NaCl, et 8 M d'urée.
L'urée est un puissant chaotrope qui permet de dénaturer et
solubiliser les corps d'inclusion. L'ensemble est mis sous agitation
à 4°C pendant 6 heures, puis de nouveau centrifugé
à
14000xg pendant 45 minutes à 4°C. La
fraction soluble de cette seconde centrifugation contenant les corps
d'inclusion dénaturés est appelée « fraction
urée 8M ». Le culot final est finalement repris avec 3 mL de SDS
2%, un détergent anionique dénaturant très puissant.
5.2.3) Analyse de l'expression des protéines
recombinantes
Une fois la totalité des fractions de
centrifugation collectées, les surnageants des fractions soluble et
« urée 8M » sont aliquotés, congelés dans
l'azote liquide, puis stockés à -
80°C. La préparation des échantillons
pour l'analyse par électrophorèse SDS-PAGE se déroule
de la manière suivante. 30 uL de chaque aliquot sont
prélevés, puis mélangés avec 30
uL de « bleu de charge » (0.2 mM de Tris-HCl, 40 % de
glycérol, 2.6 % de SDS, 2.6 % de â- mercaptoéthanol, et
0.05 % de bleu de Coomassie G-250). Les échantillons ainsi
préparés
sont chauffés à 95°C pendant 5 minutes, puis
centrifugés 1 minute à 6000xg. Environ 15 uL
de ce mélange sont finalement prélevés et
déposés sur gels de polyacrylamide pour l'analyse
SDS-PAGE.
Les échantillons analysés par Western-blot
sont préparés de la même manière et sont dans
un premier temps soumis à une analyse SDS-PAGE. Une fois
séparées sur gel de polyacrylamide, les bandes
protéiques sont transférées sur membrane de PVDF
avec un appareil de transfert semi sec. La membrane est ensuite
incubée à 37°C pendant une heure avec une solution saline
contenant l'anticorps anti-6xHis greffé à la phosphatase
alcaline (Sigma). La révélation des protéines qui
possèdent une étiquette 6xHis est finalement menée
en déposant la membrane dans une solution contenant
le substrat BCIP/NBT. Ce substrat interagit de manière directe
avec la phosphatase alcaline, ce qui induit une coloration des
bandes protéiques fixées par l'anticorps
anti-6xHis.
Chapitre 3 : Etude bio-informatique : sélection de
3 domaines PRODH
CHAPITRE 3
Etude bio-informatique :
sélection de 3 domaines PRODH
Lorsque nous avons débuté l'étude
structurale de PRODH, il n'existait aucune
structure connue de proline déshydrogénase
d'un quelconque organisme. La publication en février 2003 de la
première structure d'un domaine PRODH chez E. coli (PutA669)
nous a permis d'aborder une stratégie structurale alternative qui
consiste à sélectionner des domaines PRODH humains à
partir de cette structure et de prédictions bio-informatiques.
La démarche consiste dans un premier temps
à prédire la délimitation du domaine catalytique
humain en reportant les éléments de structure secondaire
de PutA669 sur l'alignement de séquences qui a été
présenté dans le chapitre 1. Ceci permet de caractériser
l'organisation des domaines de la protéine humaine de
manière prédictive, et ainsi de restreindre notre champ
d'investigation à un ou plusieurs domaines supposés
structuralement indépendants. Un certain nombre d'outils
bio-informatiques de prédiction de structure secondaire, et de
visualisation des résidus hydrophobes, sont alors utilisés afin
de délimiter de manière précise les domaines
structuraux sélectionnés. Avant de présenter les
résultats de cette étude bio-informatique, qui a
été menée en étroite collaboration avec le
Dr. Sophie Zinn-Justin du DIEP, la structure du domaine proline
déshydrogénase de E. coli sera brièvement
décrite.
1) Description de la structure du domaine PRODH de E.
coli
Comme je l'ai évoqué dans le premier chapitre, la
protéine de liaison à l'ADN PutA
est l'orthologue procaryote de PRODH humaine chez E.
coli. La structure des 669 résidus
N-terminaux de cette protéine (PutA669), qui
correspondent au domaine proline déshydrogénase de E.
coli, a été résolue par cristallographie des rayons X
(Lee et al., 2003). Dans cette structure, PutA669 est un homodimère
composé de 3 domaines : un domaine N- terminal de dimérisation
(87-139), un second domaine N-terminal de fonction inconnue (140-
260), et un domaine catalytique C-terminal de près
de 350 résidus qui assure la fonction proline oxydase
(261-612).
Le domaine de dimérisation est constitué d'un bras
de 3 hélices á qui embrasse les 3
hélices homologues de l'autre sous-unité. Le second
domaine N-terminal comporte 6 hélices
á qui n'établissent aucun contact significatif avec
le domaine catalytique ou la seconde sous- unité. Lee et al.,
ont dans un premier temps suggéré que ce domaine, dont le
repliement est
proche des motifs caractéristiques de liaison
à l'ADN « hélice-coude-hélice », puisse
être
responsable de la fixation aux acides nucléiques. Plus
récemment, Gu et al., ont montré que le domaine de
liaison à l'ADN est en réalité localisé dans
l'extrémité N-terminale 1-90 de PutA
qui est déstructurée dans la structure
cristalline de PutA669 (Gu et al., 2004). Au vu de l'alignement de
séquences initial qui a été réalisé
dans le chapitre 1, il apparaît que les 2 domaines N-terminaux de
PutA669 ne sont présents ni chez l'Homme, ni chez aucune espèce
eucaryote. En effet, la longueur des séquences PRODH est
très différente d'une espèce à l'autre (de
475 résidus chez la plante Oryza à 669
résidus chez la mouche) et elles ne s'alignent que dans la
moitié C-terminale des séquences eucaryotes qui correspond
au domaine catalytique. De plus, contrairement à PutA qui est capable de
réguler la transcription
de son gène dans le cytoplasme, il n'a jamais
été montré qu'une proline oxydase eucaryote,
localisée dans la mitochondrie, soit capable de lier l'ADN.
La superstructure du domaine catalytique de PutA669 est
un tonnelet de type á8â8 dans lequel 8 feuillets â
forment un coeur hydrophobe autour d'un cofacteur FAD et d'un
inhibiteur lactate (Figure 3.1). Cette poche hydrophobe est
protégée du solvant par 10 hélices
á situées en périphérie du domaine.
Les molécules de FAD et de lactate sont fortement liées
à
ce domaine catalytique par de nombreuses interactions non
covalentes avec l'hélice C- terminale á8 et les
extrémités C-terminales des feuillets â.
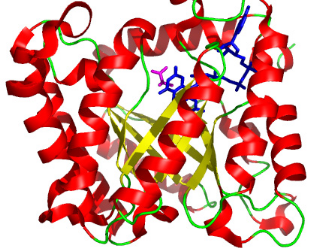
Figure 3.1 : Structure du
tonnelet á8â8 du domaine catalytique de PutA669. Les
hélices apparaissent en rouge, les feuillets en jaune, et les
boucles en vert. Le cofacteur FAD est coloré en bleu, et
l'inhibiteur lactate en violet.
La comparaison de séquence de la région C-terminale
conservée de PRODH humaine
(340-600) avec PutA669 met en évidence une faible
identité de séquence d'à peine 15 %. De manière
intéressante, ce pourcentage atteint 47 % lorsqu'on restreint la
comparaison aux résidus situés à moins de 5 Å du
cofacteur FAD ou de l'inhibiteur lactate. Ainsi, sur les 40 résidus
à proximité de ces 2 molécules, 19 sont rigoureusement
identiques et 6 sont similaires. Sur la base de cette forte conservation
au niveau de ces résidus critiques, il apparaît que
PutA669 et PRODH humaine comportent le même site actif et par
conséquent, un domaine catalytique qui pourrait être de repliement
similaire. La présence d'une molécule de lactate dans le coeur
hydrophobe de PutA669 conforte cette hypothèse. En effet, le lactate est
connu pour sa capacité à inhiber l'activité de la
proline déshydrogénase chez les organismes eucaryotes
(Kowaloff et al., 1977), ce qui n'avait jamais été
constaté chez les PRODH procaryotes.
2) Caractérisation de l'organisation de PRODH
humaine
2.1) Report de la structure secondaire de PutA669 sur
l'alignement initial
La délimitation du domaine catalytique humain
peut être prédite en reportant les éléments de
structure secondaire de PutA669 sur l'alignement initial (cf. chapitre
1) mené avec 9 séquences eucaryotes et la séquence
de PutA. Sur la base de ce report de structure secondaire, il
apparaît que le domaine catalytique humain est situé entre les
résidus 270 et 600 (non montré). Cette délimitation
est en accord avec les prédictions réalisées
par les programmes de threading de type 3D-PSSM, qui
confirment l'alignement initial en construisant un modèle par
pseudo homologie du domaine catalytique humain qui s'étend des
résidus 267 à 570.
2.2) Organisation de PRODH humaine
Dans l'optique de caractériser l'organisation des
domaines de PRODH humaine, nous avons voulu tester la
véracité de l'alignement initial en vérifiant
l'état de conservation des résidus impliqués dans
l'interaction avec le cofacteur ou l'inhibiteur qui sont supposés
être fortement conservés. De fortes identités
sont effectivement observables au niveau de l'ensemble des
feuillets â du tonnelet à l'exception des 2 brins
N-terminaux â1 et â2 qui apparaissent non conservés. Ce
résultat semble surprenant dans la mesure où 2 résidus du
brin
â2 établissent des contacts avec le cofacteur FAD
dans la structure de PutA669. Nous avons
soumis les séquences correspondant aux brins
â1 et â2 de PutA669 au programme PSI- BLAST afin de
déterminer les homologues séquentiels de ces
régions. De manière intéressante, les résultats
de cette recherche montrent des homologies significatives avec des segments de
la région 110-240 de PRODH humaine qui ne sont pas alignés avec
la séquence
de PutA sur l'alignement initial (cf. chapitre 1). Le
programme PSI-BLAST met également en évidence des homologies avec
des fragments N-terminaux de plusieurs séquences de proline
déshydrogénase eucaryote qui sont également non
alignés avec PutA sur l'alignement initial.
Sur la base de cette analyse, nous avons
corrigé l'alignement de séquences qui s'avérait
inexact dans la région N-terminale du domaine catalytique. La
totalité de ce nouvel alignement de 10 séquences eucaryotes avec
PutA669 qui s'étend sur près de 850 résidus est
présenté dans la figure 3.2. Cet alignement corrigé
fait apparaître 2 insertions au sein du domaine catalytique chez
les eucaryotes supérieurs comme le ver Caenorhabditis elegans,
la mouche Drosophila Melanogaster, et l'Homme. La
première de ces insertions, composée d'une cinquantaine de
résidus, est située entre le feuillet â1 et l'hélice
á1 du tonnelet á8â8. La seconde, d'une taille deux fois
plus importante, est localisée entre le feuillet â2 et
l'hélice á2.
Figure 3.2 :
Alignement corrigé de 10 séquences de
proline déshydrogénase réalisé avec l'aide du
logiciel clustalw. Les éléments de structure secondaire
reportés sont relatifs à la structure
tridimensionnelle du domaine catalytique de PutA669, résolue par
diffraction des rayons X (Lee et al., 2003). Les feuillets â sont en
vert, et les hélices á en rouge. Les 2 insertions dans
la région N-terminale du domaine catalytique
sont mises en évidence. Les résidus sont colorés en
rouge lorsqu'ils sont conservés (sigle *), en vert lorsqu'il sont
fortement similaires (sigle :), et en bleu lorsqu'ils sont faiblement
similaires (sigle .).
Les 10 séquences alignées correspondent aux
espèces suivantes :
Human : proline oxydase humaine (600
résidus)
CAEEL : proline oxydase de Caenorhabditis
elegans (564 résidus) Drosophila : proline oxydase
de Drosophila Melanogaster (669 résidus) POMBE
: proline oxydase de Schizosaccharomyces pombe (492
résidus) Arabidopsis : proline oxydase de
Arabidopsis thaliana (499 résidus)
|
Tabacum
|
:
|
proline oxydase de Nicotiana tabacum (493
résidus)
|
|
Oryza
|
:
|
proline oxydase de Oryza sativa (475 résidus)
|
|
Cerevisiae
|
:
|
proline oxydase de Saccharomyces cerevisiae (476
résidus)
|
|
Emericella
|
:
|
proline oxydase de Emericella nidulans (478
résidus)
|
|
PutA
|
:
|
proline dehydrogenase de E.coli (648 premiers
résidus)
|
Chapitre 3 : Etude bio-informatique : sélection de
3 domaines PRODH
Human
1-----------------MALRRALPALRPCIPRFVQLSTAPASR----------EQPAAG
CAEEL
1--------------------------MKIPVALVL-------------------------
Drosophila
1-----------------MALLRSLSAQRTAISLVYGRNSSKSSNSVAVAACRSFHQRGNG
POMBE
1----------------MRAFRLASGVLRNRKVILGIGAGSLITAG---------NIKIRN
Arabidopsis
1----------------MATRLLRTNFIRRSYRLPAFSPVGPPTVTAS-------TAVVPE
Tabacum
1----------------MANKVVCPKAFRDLRSFVRCLNTAPTVPPMN-------FTGAYD
Oryza
1-------------------MAIASRIQKRVLASFAAAAAAKLPEAA--------VAAAGG
Cerevisiae
1----------------MIASKSSLLVTKSRIPSLCFPLIKRSYVSKT-------PTHSNT
Emericella
1--------------MKAATPRPSVRALSSGRSYRTARFVSRTSNAR--------SSLAAD
PutA
1-MGTTTMGVKLDDATRERIKSAATRIDRTPHWLIKQAIFSYLEQLENSDTLPELPALLSG
51
Human
34-PAAVPGGGSAT------------A---------------------VRP-----------PVPA---------VD-FGNAQ--------------------
CAEEL
-T-------------------------------------------III-------------------------E-F------------------------
Drosophila
STSIAGEGAASESTRGVNGARFLHSGDRPLQASTLVQPEVVSSETVKRSMKQESSQEKNPSPAGSPQRDPLDVS-FNDPI--------------------
POMBE
DSKFDAFFAKG----------------------------------------------------------------FPD----------------------
Arabidopsis
ILSFGQQAPEPP---------------------------------LHHP---------KPTEQSHDG-----LD-LSDQA--------------------
Tabacum
ATTVTTPALIP-----------------------------------TD----------QVITADKKV-----IN-FEDVK--------------------
Oryza
AAEAVEEVASS----------------------------------VQEQ-----------VQAQG----AQVLE-FGDTE--------------------
Cerevisiae
AANLMVETPAAN----------------------------------------------------------------ANGN--------------------
Emericella
TNSLLQQAPPS--------------------------------------------------------------P-KKQLA--------------------
PutA
60-AANESDEAPTPAEEPHQPFLDFAEQILPQSVSRAAITAAYRRPETEAVSMLLEQARLPQPVAEQAHKLAYQLADKLRNQKNASGRAGMVQGLLQEFSLSS
Human
60---------------EAYRSRRTWELAR---------------SLLVLRLCAWPALLARHEQLLY--V-----SRKLLGQRLFNKLMK------MTFYGH
CAEEL
----------------FQSKSNTELVR---------------ALVVLRLCGIQTLVNQNQIILN--T-----MRRVLGKNLFKKTLK------NTFFGH
Drosophila
--------------AAFKSKTTGELIR---------------AYLVYMICSSENLVEHNMTLMK--W-----SKNVLGQRLFTLLMK------ATFYGH
POMBE
---------------ELQHRSLFSVLR---------------SAFVYEICSRAWLVKLSLGAMS--L-----CDVFHLSFLYNPFCR------YTFYKH
Arabidopsis
--------------RLFSSIPTSDLLR---------------STAVLHAAAIGPMVDLGTWVMSSKL-----MDASVTRGMVLGLVK------STFYDH
Tabacum
--------------ELFTGVSTLKLIR---------------STLTLQMAATEPMVDVGIWVMNSKL-----MHMPIVKEVILGFVK------GTFYEH
Oryza
--------------RLFAGERSTSLVR---------------TLAVLQALSVGPLVDVATAALR--------SPAVAGSAAGRAAAR------ATAYQH
Cerevisiae
--------------SVMAPPNSINFLQT-------------LPKKELFQLGFIGIATLNSFFLN--------TIIKLFPYIPIPVIK------FFVSSL
Emericella
--------------SPLAKLPLSSVLR---------------SLLILSVSSSSILLKPCIYTLSALAHPKTALLDVAKNPLLNLLVK------HTIYKQ
PutA
160-QEGVALMCLAEALLRIPDKATRDALIRDKISNGNWQSHIGRSPSLFVNAATWGLLFTGKLVSTHNEASLSRSLNRIIGKSGEPLIRKGVDMAMRLMGEQ
51
. : . : :
á0 â1
Chapitre 3 : Etude bio-informatique : sélection de
3 domaines PRODH
Human
117-FVAGEDQ-ESIQPLLRHYRAFGVSA-ILDYGVEEDLSPEEAEHKEMES----------CTSAAERDGSGTNKRDKQYQAHRAFGDRRNGVISARTYFYAN
CAEEL
FVAGETE-EEVRHVVEKLRNYGVKS-ILDYSVEADITSQEATDKTVKGTSVATVKPAAMTPVVDAKTLETTR--ERYTVHEEFGDRRQGVSSARTYFYEG
Drosophila
FVAGEDQ-IKIIPTLERLRSFGVKP-ILDYSVEEDITQEEAEKREVESS---------VSSAGDKKEEGSMP---QYHVDKSFADRRYKVSSARTYFYLN
POMBE
FCGGETP-QAVMATMDTLQAAGITS-CLNYSREVDLDGDMDVNKIAS--------------------QG-------------------VVPPQVPVPSEK
Arabidopsis
FCAGEDADAAAERVRSVYEATGLKG-MLVYGVEHAD---------------------------------------------------------------D
Tabacum
FCAGKDL-IEVRRTVTKLSDVGLKG-MLDYGVEHAT---------------------------------------------------------------E
Oryza
FCAGETAEEAAAAVRRLWRG-GMGG-ILDYGIEDAE---------------------------------------------------------------D
Cerevisiae
YCGGENF-KEVIECGKRLQKRGISNMMLSLTIENSEGTKSLSSTP------------------------------------------------VDQIVKE
Emericella
FNAGENK-LEVQRSINAIKELGYRGVLLGYAREVLVGESKTDPR-------------------------------------------------------D
PutA
259-FVTGETI-AEALANARKLEEKGFRY-SYDMLGEAAL---------------------------------------------------------------T
: *: * *
1ère insertion
á1 â2
52
Human
205-EAKCDSHME--TFLRCIEA--SGRVSDDGF----IAIKLTALGRPQFLLQFSEVLAKWRCFFHQMAVEQGQAGLAAMDTKLEVAVLQESVAKLG-IASRA
CAEEL
EEQCDKNRD--IFKDSINAV-ASATKNEGF----VAVKITALGRPQLLLKLSEAIVQTQNFFKALTGGMS-----LQEGRLTSQEFYKRLGELGVKTDTE
Drosophila
EATCERNME--IFIKCLEAV-SGATFGTGI----TAIKLTALGRPQLLLQLSEVIMRTRKYMEDMVGGQGN----VLTHHKTIKDLEKYYATLG---DNK
POMBE
NQKVLRQIADKAFESNMHIIDMATYKPGTV----CAVKLTPFINPLVLQRYNSILN--------------------------------------------
Arabidopsis
AVSCDDNMQ--QFIRTIEAAKSLPTSHFSS----VVVKITAICPISLLKRVSDLL---------------------------------------------
Tabacum
NESCDQSMK--VFLQTAESTKSLPSSSVSF----VVVKITAICTPKLLKRMSDLL---------------------------------------------
Oryza
GPACDRNAA--GFLAAIDVAAALPPGSAS-----VCIKITALCPVALLEKASDLL---------------------------------------------
Cerevisiae
TISSVHNILLPNIIGQLESK-PINDIAPGY----IALKPSALVDNP-----HEVLY--------------------------------------------
Emericella
EQASRQEIQ--TWLDGTLQT-VDMAQEGDF----VALKFTGMGI--------QAL---------------------------------------------
PutA
294-AADAQAYMV--SYQQAIHAIGKASNGRGIYEGPGISIKLSALHP--------------------------------------------------------
52
:* : :
2ème insertion
á2 â3 á3
Human
296-EIEDWFTAETLGVSGTMDLLDWSSLIDSRTKLSKHLVVPNAQTGQLEPLLSRFTEEEELQMTRMLQRMDVLAKKATEMGVRLMV---DAEQTYFQP-AIS
CAEEL
SVKKFFDEVDFDSDGIVDLHGWNHILDDHVKLGQLFQVLNIKTGSLEPLIQNLSNEEEQEFRNMVRRTLDVAEYAIEKGVRIMV---DAEQTYLQP-AIS
Drosophila
DVKEFLNNVTSDKEGILHLFPWSGIVDEDSQLSDTFRVPDPQTGQMRRLISQIPPKEEEMFRNMIRRLNTIVKAAADLDVRIMV---DAEQTYFQP-AIS
POMBE
---QYP-------------------VESACNYLEHLK---------SP--E-LSTYEVSELKKFWEYADKLCQFAKEKQIPLFI---DAEQTYFQD-CMH
Arabidopsis
---RWEYKSPNFK------LSWK--LKSFPVFSESSPLYHTNS---EP--EPLTAEEERELEAAHGRIQEICRKCQESNVPLLI---DAEDTILQP-AID
Tabacum
---RWEHKNPSFN------LPWK--QKSLPLFSDSSPFYHTPQ---KP--EPLTVEEEHDLQLAHERLMTICKKCLELDVDLLI---DAEDTAIQP-AID
Oryza
---RWQQKHPATK------LPWK--VHGFPVLCVSSPLYLTAA---EP--PALEAEEERELEMAHGRLLAIGERCAEYDIPLLV---DAEYATVQP-AID
Cerevisiae
---NFSN----------------------PAYKAQ------------R--DQLIENCSKITKEIFELNQSLLKKYPERKAPFMVSTIDAEKYDLQENGVY
Emericella
---EYL------------------------------------------------QNQAPPSPFMDEAIKQVCDLAISRNVRLLV---DAEEQAVQP-GIE
PutA
336----RYS-------------R----------------------------------AQYDRVMEELYPRLKSLTLLARQYDIGINI---DAEEADRLEISLD
: : . : : *** :
á3 â4 á4 â5
á5A á5B
Chapitre 3 : Etude bio-informatique : sélection de
3 domaines PRODH
Human
392-RLTLEMQRKFNVEKP---LIFNTYQCYLKDAYDNVTLDVELARREGWCFGAKLVRGAYLAQERAR-AAEIGYEDPINPTYEATNAMYHRCLDYVLEELKH
CAEEL
KITIEMMKKYNKGRG---NIFNTYQAYLKGTLQNMEADMQVARREGWHFGAKLVRGAYMEQERAR-AKAIGYEDPINDNFEATSKMYESCLTRIADEVHR
Drosophila
RITLEMMRKYNKDKA---IVFNTYQCYLRETFREVNTDLEQAKRQNFYFGAKLVRGAYMDQERDR-AKSLGYPDPVNPTFEATTDMYHRTLSECLRRIKL
POMBE
AVTVDLMRKYNKEVA---IVHNTYQLYLKKSRKIMDDHIKKCVAEGWLMGAKLVRGAYLNSEPRF-LIHDTKAETDKDFDSAVEAIIAAAAKFAPGDPAS
Arabidopsis
YMAYSSAIMFNADKDR-PIVYNTIQAYLRDAGERLHLAVQNAEKENVPMGFKLVRGAYMSSEASL-ADSLGCKSPVHDTIQDTHSCYNDCMTFLMEKASN
Tabacum
YFAYSAAIKYHKDDD--PMIFGTIQAYLKDSKERMVIAKKAAEKMGVPMGFKLVRGAYMSSEREL-ASRLGVQSPIHDSIEQTHDCFNSCAEFMLDEISN
Oryza
YFTFAGALAFNGGGR--PIVHGTVQAYLRDARDRLEAMARAAQGERVCLALKLVRGAYLAREARL-AASLGVPSPVHRSIQDTHDCYNGCAAFLLDRVRR
Cerevisiae
ELQRILFQKFNPTSSKLISCVGTWQLYLRDSGDHILHELKLAQENGYKLGLKLVRGAYIHSEKNRNQIIFGDKTGTDENYDRIITQVVNDLIINGEDSYF
Emericella
EWATMYQKYCNSRTPGRAIFYNTYQAYLCSTPATLARHLEISRKEGYTLGVKLVRGAYLKTEPRH-LIWAKKEQTDECYDGIVEALLTRRYNHMLKPASA
PutA
383-LLEKLCFEPELAGWN---GIGFVIQAYQKRCPLVIDYLIDLATRSRRRLMIRLVKGAYWDSEIKRAQMDGLEGYPVYTRKVYTDVSYLACAKKLLAVPNL
. * * : . : :**:*** *
â6 á6 â7 á7 â8
á8
53
Human
488-N--------AKAKVMVASHNEDTVRFALRRMEELG-LHPADHR-VYFGQLLGMCDQISFPLGQ-------------AGYPVYKYVPYGPVMEVLPYLSRR
CAEEL
R------GKTNVSVMVASHNEDTVRFALNLMKEKC-ISPSERV-MCMAQLYGMCDQVSFSLGQ-------------AGFSVYKYLPYGPVEEVLPYLSRR
Drosophila
MKDCDD-DARKIGIMVASHNEDTVRFAIQQMKEIG-ISPEDKV-ICFGQLLGMCDYITFPLGQ-------------AGYSAYKYIPYGPVEEVLPYLSRR
POMBE
ASDPIASRKGKWGIMVASHNKKTMFESVNLAETKK--VDFTKTSFYLAQLLGMADDITYALAYS-------QRNQQPNFCIVKYVSCGPISEVLPYLVRR
Arabidopsis
GSGF--------GVVLATHNADSGRLASRKASDLG-IDKQNGK-IEFAQLYGMSDALSFGLKR-------------AGFNVSKYMPFGPVATAIPYLLRR
Tabacum
GSG---------AVVLATHNIDSGKLAASKAIDLG-IRKDSQK-LQFAQLYGMAEGLSFGLRN-------------AGFQVSKYLPFGPVEQVMPYLIRR
Oryza
GAA---------AVTLATHNVESGQLAAARALELG-IGGGGDRGLQFAQLMGMADGLSLGLRN-------------AGFQVSKYLPYGPVEQIIPYLIRR
Cerevisiae
G-----------HLVVASHNYQSQMLVTNLLKSTQ-DNSYAKSNIVLGQLLGMADNVTYDLITN-----------HGAKNIIKYVPWGPPLETKDYLLRR
Emericella
EHTT---ELPPVSVIVATHNRDSVRKAHALRLEQASRGEKSDVELTYAQLQGMADEISCELLQGFQTAGPENTKVAESPNVYKLLTWGSVKECMGFLLRR
PutA
480-IYP-----------QFATHNAHTLAAIYQLAGQNY----YP-GQYEFQCLHGMGEPLYEQVTGKVADG-------KLNRPCRIYAPVGTHETLLAYLVRR
.*:** .: * ** : : : . *. :* **
á8
Human
565-ALENSSLMKGT--HRERQLLWLELLRRLRTGNLFHRPA-600
// CAEEL ALENGSVLKKA--NKERDLLWKELKRRISSGEFKARSSSSS-564
//
Drosophila
AQENKGVLKKI--KKEKRLLLSEIRRRLMRGQLFYKPKGNYVPI-669 // POMBE
ARENIDALDRC--KEERAYYRQALRRRIF-492 //
Arabidopsis AYENRGMMATG--AHDRQLMRMELKRRLIAGIA-499
// Tabacum AEENRGLLSTS--AFDRQLMRKELTRRFKVATS-493 //
Oryza AEENRGLLSSS--SFDRQLLR-475 //
53
Cerevisiae LQENGDAVR----SDNGWPLIKAIAKSIPKRVGL-476
// Emericella AVENTEAVGRT--KQSQEAMFSELRRRARRAFGLRY-478
//
PutA
557-LLENGANTSFVNRIADTSLPLDELVADPVTAVEKLAQQEGQTGLPHPKIPLPRDLYGHGRDNSAGLDLANEHRLASLSSALLNSALQKWQAL-648
...............
** .
Notre alignement corrigé met ainsi en évidence la
présence de domaines entremêlés
chez PRODH humaine, et permet de prédire que les
segments qui constituent le domaine catalytique humain sont situés
dans les régions 113-150, 204-240, et 346-571. Les prédictions
de structure secondaire réalisées sur la
séquence humaine sont globalement en accord avec le report des
éléments de structure secondaire de PutA669 sur le nouvel
alignement, et notamment au niveau des brins supposés â1
(139-144) et â2 (231-233) de PRODH humaine
qui sont effectivement prédits en feuillet. Cette analyse
confirme ainsi l'exactitude de notre alignement comparé à
l'alignement initial.
Les prédictions de structure secondaire
suggèrent une structuration des 2 insertions,
qui apparaissent dans le domaine catalytique, en hélice
á. Sur notre alignement corrigé, ces 2 insertions sont
respectivement localisées sur la séquence de PutA autour
des résidus L292 pour la première, et R336 pour la seconde
(Figure 3.2). De manière intéressante, ces 2 résidus
appartiennent à des boucles situées sur une même surface
exposée au solvant de la structure tridimensionnelle de PutA669
(Figure 3.3). Cette caractéristique structurale laisse supposer
que ces 2 insertions pourraient former un domaine unique
entremêlé à la périphérie du tonnelet
á8â8 chez les eucaryotes supérieurs.
1ère
insertion
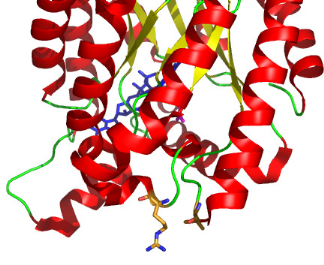
R336
L292
2éme
insertion
Figure 3.3 : Mise en
évidence des 2 sites d'insertion sur la structure du tonnelet
á8â8 de PutA669. Les éléments de structure
secondaire et les molécules de FAD et lactate sont
colorés comme dans la figure précédente. Les
résidus L292 et R336 sont représentés par des sticks. Les
atomes d'azote sont colorés en bleu, et d'oxygène en
rose.
Outre les 2 insertions, l'alignement de la Figure 3.2
fait également apparaître une région N-terminale non
conservée de la bactérie à l'homme dans
l'extrémité 1-112 de PRODH humaine. Cette région, de
fonction inconnue et uniquement conservée chez les
eucaryotes supérieurs, semble structurée au vu des
prédictions de structure secondaire, et du
nombre de résidus hydrophobes qu'elle comporte. Par
conséquent, elle pourrait contenir un domaine de repliement autonome
indépendant du domaine catalytique et des 2 insertions.
3) Sélection des 3 domaines à
exprimer
Lorsque nous avons entrepris l'étude structurale
de PRODH humaine, nous avions envisagé une stratégie qui
consiste à isoler des domaines structurés de PRODH par digestion
enzymatique à partir de la protéine sauvage repliée, puis
à résoudre par RMN la structure de ceux dont la taille est
compatible avec une telle étude. A présent, nous avons
montré à partir
de la connaissance de la structure de
l'hétérologue bactérien PutA669 que le domaine
catalytique humain est très probablement constitué d'un tonnelet
á8â8 de près de 350 résidus. Nous avons
également mis en évidence la présence d'un ou deux
domaines entremêlés dans
ce domaine catalytique (Figure 3.4). Sur la base de
ces prédictions, il est clair que la caractérisation
structurale du domaine catalytique humain n'est envisageable que par
la résolution de structure de la large région 113-571 de
PRODH humaine qui comporte le domaine catalytique et les 2 insertions.
Aussi, l'étude structurale d'une telle région de 59 kDa
n'est pas réalisable par RMN.
domaine
PS N-terminal
1ère
insertion
2ème





insertion
domaine catalytique
1 36 113 151 203 241 345 600
Figure 3.4 : Prédiction
des domaines potentiels de la proline déshydrogénase
humaine à
partir du bilan consensus de l'ensemble des
analyses bio-informatiques. Le sigle PS
correspond à « peptide signal ».
La Figure 3.4 résume la prédiction des
domaines potentiels de PRODH humaine réalisée à partir
de l'ensemble des analyses bio-informatiques. Ces prédictions
suggèrent que
la proline déshydrogénase humaine comporte :
un peptide signal d'adressage mitochondrial
(1-36), une région N-terminale hydrophobe (37-112), et un
large domaine catalytique (113-
600) entremêlé avec 2 insertions de
respectivement 52 et 104 résidus (151-203 et 241-345). Sur la base de
cette analyse bio-informatique, nous avons décidé de modifier
notre stratégie d'étude structurale initialement définie,
et de sélectionner 3 domaines de PRODH humaine à
exprimer. En effet, au-delà du domaine catalytique, il
serait intéressant de connaître les rôles
structural et fonctionnel de la région N-terminale
hydrophobe et de chacune des 2 insertions. Cependant, la résolution de
structure du domaine catalytique n'en demeure pas moins un de nos objectifs
dans l'optique de caractériser son mode d'action chez les organismes
eucaryotes. C'est pourquoi, nous avons sélectionné 3
régions de PRODH humaine afin de les soumettre
au programme de production de protéines du LMP.
Ces 3 domaines protéiques sont les suivants :
- PROcatal (59 kDa) : région
115-600 de PRODH qui comporte le domaine catalytique
prédit et les 2 insertions potentielles.
- PROinser (16 kDa) : région
239-353 de PRODH qui correspond à la seconde
insertion.
- PROter (13 kDa) : région
39-125 de PRODH qui comporte le domaine N-terminal
potentiel délestée du peptide signal.
La délimitation de ces 3 régions a
été réalisée en utilisant les diagrammes de
prédiction de structure secondaire, et de visualisation des amas
de résidus hydrophobes (analyse HCA). Pour chaque domaine, la
démarche a consisté à choisir le premier et le dernier
résidu situés entre 2 éléments de structure
secondaire prédits, et d'éviter que ces résidus soient
hydrophobes, afin de limiter le risque d'agrégation lors de la
surexpression chez E. coli. D'autre part, ayant conscience que
la sélection de domaines à partir d'une analyse bio-
informatique présente un caractère spéculatif, nous avons
également entrepris de produire la forme mature de PRODH dans le cadre
du programme de la plate-forme du LMP. Le choix du premier résidu de ce
domaine a également été guidé en se basant sur le
diagramme HCA et les prédictions de structure secondaire. Cette
construction est nommée :
- PROentier (68 kDa) : région
39-600 de PRODH qui correspond à la
protéine entière délestée du peptide signal
potentiel.
Dans le cas où ces 4 protéines seraient
exprimées sous forme soluble, les domaines de faible masse
moléculaire PROinser et PROter seraient étudiés en
solution par RMN, alors que
les protéines PROcatal et PROentier, de poids
moléculaire supérieur ou égal à 59 kDa,
seraient plutôt destinées à une étude de
croissance cristalline dans l'optique de résoudre la structure par
radiocristallographie. Il serait également possible d'entreprendre une
digestion enzymatique des protéines PROcatal et PROentier
repliées afin d'isoler des domaines
structuraux de faible taille qui pourraient alors être
étudiés par RMN.
4) Matériels et Méthodes
4.1) Recherche d'homologie de séquence
Les recherches d'homologie de séquence protéique de
PRODH ont été menées avec
les logiciels BLAST et PSI-BLAST sur le serveur du NCBI
(http://www.ncbi.nlm.nih.gov/BLAST/)
à partir des bases de données suivantes :
Genbank (NCBI) :
http://www.psc.edu/general/software/packages/genbank/genbank.html
RefSeq (NCBI) :
http://www.ncbi.nlm.nih.gov/RefSeq/://www.ncbi.nlm.nih.gov/RefSeq/
SwissProt :
http://www.ebi.ac.uk/swissprot/p://www.ebi.ac.uk/swissprot/
PDB :
http://www.rcsb.org/pdb/Welcome.do://www.rcsb.org/pdb/Welcome.do
PIR :
http://pir.georgetown.edu/://pir.georgetown.edu/
PRF :
http://www.genome.jp/dbget-bin/www_bfind://www.genome.jp/dbget-bin/www_bfind?prf
4.2) Alignements de séquence et prédictions
structurales
L'alignement des séquences protéiques, le calcul
de quelques paramètres intrinsèques aux protéines, et les
prédictions de structure secondaire et tertiaire nécessitent
l'utilisation de différents logiciels d'analyse de
séquence primaire. De nombreux programmes sont disponibles
gratuitement sur Internet. Parmi ceux-ci, nous avons utilisé :
?
Clustalw
(http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_clustalw.html)
:
Logiciel d'alignement de séquences
protéiques. La version du Pôle Bio-Informatique
Lyonnais permet de visualiser facilement les résidus
conservés grâce à un code couleur.
? Consensus Secondary Structure Prediction du Pôle
Bio-Informatique Lyonnais
(http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_sspred.html)
:
Logiciel de prédiction de structure secondaire qui
établit un consensus à partir de l'analyse de
la structure primaire par plusieurs algorithmes (Predator, HNN,
GorIV...).
?
HCA
(http://bioserv.rpbs.jussieu.fr/RPBS/cgi-bin/Ressource.cgi?chzn_lg=fr&chzn_rsrc=HCA)
:
Logiciel de visualisation des amas de résidus
hydrophobes sur une séquence primaire. Ce programme permet
notamment de distinguer les régions structurées des
régions déstructurées.
?
3D-PSSM
(http://www.sbg.bio.ic.ac.uk/3dpssm/) :
Logiciel de prédiction du repliement du squelette d'un
polypeptide à partir de la structure de protéines dont la
séquence présente plus de 25 % d'homologie avec la
protéine d'intérêt.
Chapitre 4 : Expression de 4 protéines PRODH dans
le cadre d'un programme de production
CHAPITRE 4
Expression de 4 protéines PRODH dans le
cadre d'un programme de production
L'expression des protéines PRODH sauvage et mature
chez E. coli conduisant à
l'unique formation de corps d'inclusion, nous
avons modifié notre stratégie d'étude structurale
initialement définie. Sur la base de nouvelles données apparues
dans la littérature, nous avons sélectionné 3 domaines
PRODH humains (PROcatal, PROter, et PROinser) à partir d'une
étude bio-informatique. L'expression sous forme soluble
constituant l'étape limitante de la stratégie, nous avons
choisi de produire ces 3 domaines, ainsi que la forme mature de PRODH
(rebaptisée PROentier), dans le cadre du programme de production 3PM
du LMP du CEA de Saclay, dédié à la
surexpression de protéines solubles chez E. coli. Avant
de présenter les résultats de ce travail qui a
été mené sous la direction des Dr. Sandrine Braud
et Muriel Gondry, la stratégie de production des 4
protéines PRODH dans le cadre d'une telle structure sera
exposée.
1) Stratégie de production des 4
protéines PRODH
1.1) Stratégie générale
Parmi les différents hôtes d'expression
disponibles, la bactérie Escherichia coli est
généralement choisie pour sa facilité d'utilisation, sa
croissance rapide, sa génétique simple, son faible coût de
production, et ses taux d'expression élevés. De plus, dans
l'optique d'une caractérisation par RMN, il existe des milieux enrichis
en isotopes 15N et 13C qui permettent
de produire des protéines marquées avec des taux
d'expression tout aussi élevés. Cependant,
l'expression chez E. coli présente
des inconvénients majeurs, comme l'absence de modifications
post-traductionnelles, l'instabilité des ARNm, et la
différence d'usage de codons avec les espèces eucaryotes.
Ces inconvénients peuvent compromettre la qualité de
l'expression en favorisant l'apparition de corps d'inclusion, comme
nous l'avons constaté avec les protéines PRODH sauvage et
mature.
Au cours de ces dernières années, il a
été montré que la formation de corps d'inclusion,
inhérente à l'utilisation de l'hôte bactérien E.
coli, peut être limitée en exprimant
les gènes d'intérêt en fusion avec
un partenaire protéique fortement soluble (Kapust & Waugh,
1999). Les mécanismes avec lesquels ces partenaires favorisent
le repliement des protéines d'intérêt sont à ce
jour encore peu connus. Aussi, une récente étude comparative
des
7 partenaires décrits comme les plus efficaces a mis en
évidence qu'il n'existe aucune « règle
universelle » permettant de prévoir l'effet d'un
partenaire de fusion sur la solubilité d'une
protéine (Hammarström et al., 2002). En d'autres
termes, certains partenaires qui se montrent très efficaces pour
certaines protéines, se révèlent médiocres pour
d'autres. Par conséquent, le criblage du plus grand nombre d'entre eux
semble être la méthode qui offre le plus de chance d'obtenir une
protéine de fusion exprimée sous forme soluble (Vincentelli et
al., 2003).
Outre le partenaire de fusion, d'autres paramètres
comme la souche bactérienne d'E. coli peuvent influencer
l'expression et la solubilité des protéines recombinantes.
Les grandes avancées dans le domaine du génie
génétique, observées au cours de ces dernières
années, ont abouti à l'émergence de nouvelles
souches qui présentent des caractéristiques
intéressantes dans l'optique de produire des protéines
hétérogènes eucaryotes (stabilité accrue des ARNm,
présence d'ARNt correspondant à des codons rares chez E.
coli, surproduction de
membranes, etc.) (Lopez et al., 1999 ; Jonasson et al., 2002).
Sur la base de ces études récentes, il
nous est apparu intéressant d'intégrer une structure qui
permette de tester l'expression des 4 protéines PRODH en fusion avec
différents partenaires et dans plusieurs souches
bactériennes. C'est pourquoi, nous avons entrepris de produire
ces 4 protéines dans le cadre du Programme de Production et
Marquage des Protéines 3PM du LMP (Braud et al., 2005). Pour pallier la
formation des corps d'inclusion,
la plate-forme 3PM propose un criblage systématique de
plusieurs conditions expérimentales
(7 partenaires de fusion, 3 souches d'expression, et 2
températures d'expression) afin de déterminer les
meilleures conditions d'expression relatives à chaque
protéine testée. Cependant, le criblage d'un grand nombre de
conditions ne peut être réalisé dans un temps raisonnable
sans recourir à une miniaturisation et un traitement en parallèle
des échantillons.
Par conséquent, le processus d'expression des
protéines se déroule en 2 étapes distinctes :
- Une première étape de criblage des conditions
d'expression réalisée à petite échelle sur
microplaques en volume de 250 uL.
- Puis, une fois les meilleures conditions d'expression de
protéine soluble déterminées, une production à
grande échelle pour obtenir les protéines en quantité
appréciable.
Outre la possibilité de tester un grand nombre de
conditions, ce processus d'expression offre également l'avantage de
pouvoir produire plusieurs domaines simultanément, ce qui est
difficilement réalisable avec des techniques d'expression
classiques. La stratégie de
production de protéines solubles dans le cadre du
programme 3PM peut se décomposer en
trois principales phases que je vais brièvement
présenter dans les prochains paragraphes.
1.2) Stratégie de construction des vecteurs
d'expression
La production de protéines en fusion avec
de multiples partenaires nécessite la réalisation de
nombreux plasmides. Aussi, il est indispensable de recourir à une
méthodologie permettant de minimiser l'effort de clonage. Dans cette
optique, l'utilisation d'un système de clonage par recombinaison
homologue a été choisie : il s'agit de la technologie
Gateway. La stratégie consiste dans un premier temps
à cloner le gène d'intérêt à partir
d'amplicons de PCR dans un plasmide appelé vecteur
d'entrée (Figure 4.1). Le système Gateway
permet alors, à partir de ce seul plasmide, d'obtenir in vitro
et en parallèle, une multitude de vecteurs d'expression
portants chacun le gène d'intérêt et un
partenaire de fusion différent. Tout l'intérêt de cette
technique de recombinaison homologue repose sur la rapidité et
l'efficacité
avec lesquelles sont construits les vecteurs
d'expression.
produit de PCR
Vecteur
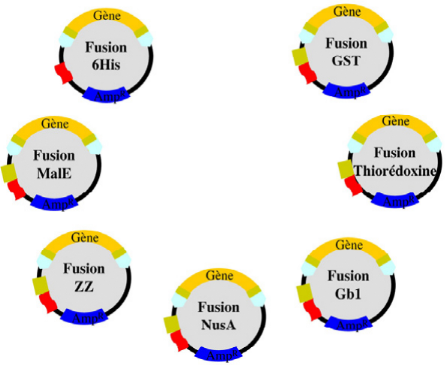
d'entrée
Fusion
MBP
Figure 4.1 : Stratégie de
clonage par recombinaison homologue du programme 3PM.
1.3) Criblage des conditions d'expression en
microplaques
Le programme 3PM doit permettre l'analyse de l'influence de
conditions expérimentales sur l'expression et la solubilité de
protéines de fusion recombinantes. Au vu
du nombre de conditions choisies, les techniques
d'expression habituelles ne sont pas adaptées, et constituent un
facteur limitant. Ainsi, à l'exception de l'analyse sur gel
SDS- PAGE, toutes les étapes relatives au criblage des
conditions d'expression, à savoir la transformation de E.
coli, la culture d'expression, la lyse des bactéries, et la
séparation des fractions soluble et insoluble, sont traitées
sur microplaques 96 puits via l'utilisation de pipettes
multicanaux. Outre le traitement plus facile d'échantillons en
parallèle, l'utilisation
de microplaques 96 puits permet de diminuer les volumes
de culture (250uL), de réactifs utilisés, et de rendre
possible une automatisation partielle du processus.
1.3.1) Les partenaires de fusion
Sur la base de l'étude comparative récente des 7
partenaires décrits comme les plus efficaces (Hammarström et
al., 2002), il a été décidé que les
séquences protéiques de l'étiquette 6xHis et des six
partenaires suivants seront systématiquement utilisées pour
être fusionnées en N-terminal avec les protéines
d'intérêt. Il s'agit de :
- GST (26 kDa) :
glutathion-S-transférase de Schistosoma japonicum
- Gb1 (7,5 kDa) : domaine de liaison aux
anticorps de la protéine G de Streptococcus
- ZZ (17 kDa) : double domaine de liaison aux
anticorps de la protéine A de Staphylococcus
- MBP (43 kDa) : protéine de fixation
au maltose de E. coli
- Trx (13 kDa) : thiorédoxine de E.
coli
- NusA (55 kDa) : protéine de E.
coli
1.3.2) Les souches bactériennes
Les souches d'expression utilisées dans le cadre du
programme 3PM dérivent de la souche BL21 déficiente en
protéases lon et ompT. Elles possèdent une
copie chromosomique
du gène codant pour l'ARN polymérase T7 sous le
contrôle d'un promoteur lac inductible par l'IPTG
(désignation DE3). Elles sont destinées à la surexpression
des plasmides d'expression dont la transcription est
contrôlée par un promoteur de type T7 inductible par
cette
polymérase. La production des protéines de fusion
est testée dans les 3 souches suivantes :
· BL21 STAR (DE3) : cette souche
possède un gène rne mutant qui code pour une
RNase E tronquée incapable de dégrader les
ARNm, d'où une augmentation de stabilité de ces derniers.
· ROSETTA pLysS (DE3) : la
souche Rosetta est désignée pour augmenter l'expression
de protéines recombinantes d'origine eucaryote dont les
gènes correspondant contiennent des codons rarement utilisés chez
E. coli. Cette souche contient ainsi un plasmide fournissant les
ARNt correspondant à 6 codons rares (AUA, AGG, AGA, CUA, CCC, GGA).
La souche pLysS exprime le lysozyme T7, qui inhibe l'activité basale
de l'ARN polymérase T7, optimisant de ce fait la
régulation du niveau d'expression. La sélection
spécifique de cette souche est possible en ajoutant du
chloramphénicol dans les milieux de culture.
· C41 (DE3) : recommandée
pour l'étude de protéines toxiques, cette souche
comporte une ou plusieurs mutation(s) de gène(s) inconnue(s)
conduisant à la surproduction de membranes.
1.3.3) Les températures d'expression
D'autres paramètres, comme la température
de la culture d'expression, peuvent influencer et améliorer la
solubilité des protéines recombinantes. Ainsi, une diminution de
la température conduit à une réduction des taux
d'expression. La diminution de ces niveaux d'expression permet d'une part,
de limiter les interactions hydrophobes intermoléculaires qui provoquent
l'agrégation, et d'autre part, d'éviter de saturer les
systèmes d'assistance au repliement formés par les
protéines chaperonnes de E. coli. Pour des raisons pratiques,
et afin
de limiter le nombre de conditions, il a
été décidé de tester l'expression des
protéines de
fusion aux 2 températures suivantes :
37°C et 20°C
1.4) Production à grande échelle et obtention
de la protéine d'intérêt
Une fois les meilleures conditions
déterminées pour chaque protéine
d'intérêt, la troisième phase du programme 3PM consiste
dans un premier temps à valider ces conditions dans le cadre d'une
production à grande échelle, et ainsi produire
les quantités de protéine désirées (Figure
4.2).
culture
grande échelle
lyse
E. coli
6xHis
Fusion
TEV
Protéine
E. coli
Ni2+ X
purification
6xHis Trx NusA Gb1
GST MBP ZZ
lavage




élution
E. coli
E. coli
6xHis
Fusion
TEV
Protéine
Ni2+
coupure par
la TEV
partenaire et
TEV retenus
protéine d'intérêt
Figure 4.2 : Stratégie de
production à grande échelle et d'obtention de la protéine
d'intérêt
Toutes les protéines de fusion sont marquées en
N-terminal par une étiquette 6xHis, ce
qui permet potentiellement de les purifier par
chromatographie de pseudo affinité IMAC (Immobilized Metal
Ion Affinity) sur colonne à nickel. Le principe de
ce type de chromatographie repose sur les interactions ioniques qui se
forment entre les ions nickel et plusieurs noyaux imidazoles de
résidus histidines. L'élution de la protéine
hétérologue est
réalisée par un mécanisme
compétitif en ajoutant des quantités croissantes
d'imidazole.
Cependant, il existe des résines qui
présentent une affinité plus spécifique pour
certains partenaires de fusion. C'est le cas de la résine de glutathion
pour GST, de la résine d'amylose pour MBP, et de la
résine d'anticorps IgG pour ZZ. Ces résines sont
utilisées préférentiellement pour purifier les
protéines hétérologues fusionnées avec l'un de
ces partenaires. Dans le cas de la résine d'amylose, qui a
été choisie pour purifier plusieurs domaines PRODH,
l'élution est également menée par compétition en
introduisant du maltose,
qui est le ligand naturel de la MBP (Maltose Binding
Protein).
Si l'incorporation d'un partenaire de fusion peut
améliorer la solubilité de la protéine associée,
celui-ci représente cependant une gêne, notamment dans le cas
d'études structurales. C'est pourquoi, le système
génétique utilisé permet la production de protéines
recombinantes,
qui possèdent une séquence de
reconnaissance de la protéase TEV (Tobacco Etch Virus),
insérée entre la protéine d'intérêt et
le partenaire de fusion. L'action de la TEV conduit à
l'élimination de tous les acides aminés relatifs au
partenaire de fusion et au site de reconnaissance de la protéase,
à l'exception d'une glycine qui devient alors le premier
résidu
N-terminal de la protéine cible. Cette étape peut
intervenir en solution, après purification de la protéine
hétérologue, ou bien « sur colonne »,
c'est-à-dire lorsque la protéine est accrochée à
la résine de la colonne d'affinité.
Après clivage du partenaire de fusion, la protéine
d'intérêt est finalement isolée après
un nouveau passage sur résine de nickel. Cette
deuxième étape de purification permet de retenir toutes les
protéines qui possèdent une étiquette 6xHis comme
notamment la protéine
de fusion résiduelle, le partenaire de fusion
clivé, et également la protéase TEV qui comporte une
étiquette 6xHis à son extrémité N-terminale. La
protéine d'intérêt ne possédant plus de
séquence 6xHis après coupure enzymatique, elle ne doit
donc pas accrocher la résine de nickel, et peut ainsi être
séparée de son partenaire.
Le succès du criblage des conditions en
microplaques, certes prédispose, mais ne garantit en aucun cas
la réussite de la production en grand volume. En effet, il va
falloir vérifier si le changement d'échelle induit une
modification des profils d'expression et de solubilité des
protéines hétérologues. D'autre part, une fois le
partenaire de fusion clivé par
coupure enzymatique, le comportement en terme de
solubilité de la protéine d'intérêt n'est
pas prévisible car c'est un phénomène qui
dépend de sa nature intrinsèque. Par conséquent,
cette opération constitue véritablement
l'étape critique de la stratégie.
2) Production des 4 domaines PRODH
2.1) Bilan du criblage des conditions d'expression en
microplaques
Chacun des gènes encodant les protéines PROentier,
PROcatal, PROter, et PROinser a
été cloné dans 7 plasmides
d'expression différents par recombinaison homologue. Les domaines
PROcatal et PROentier ont été exprimés dans 28
conditions expérimentales différentes (7 partenaires de
fusion, 2 souches d'expression, et 2 températures), contre 42
conditions pour les domaines PROter et PROinser (7 partenaires,
3 souches, et 2 températures). Au total, 140 cultures ont
été réalisées. Pour chacune d'elles, les
fractions contenant les protéines solubles et insolubles ont
été déposées sur gel SDS-PAGE. L'analyse
de l'expression des protéines de fusion à partir
de l'interprétation des profils électrophorétiques des
gels SDS-PAGE a été reportée, domaine par domaine, dans un
tableau récapitulatif afin de déterminer les meilleures
conditions d'expression et de solubilité pour chacune des 4
protéines.
Par souci de rendre fluide la lecture de ce manuscrit, je me
contenterai de présenter dans ce paragraphe un bilan du criblage des
conditions d'expression des 4 protéines PRODH
en microplaques. Le détail des résultats,
à savoir l'ensemble des gels SDS-PAGE et leur analyse, est
présenté en annexe. Les matériels et
méthodes, spécifiques au clonage et au criblage des
conditions d'expression des 4 protéines PRODH dans le cadre du
programme
3PM, sont également reportés en annexe.
De manière générale, les
résultats du criblage des conditions d'expression montrent que la
nature du partenaire de fusion est essentielle sur l'expression et
la solubilité des protéines hétérologues PRODH.
Ainsi, seuls les partenaires MBP et NusA permettent d'obtenir des
taux suffisants de protéine soluble, contrairement aux autres
partenaires, et notamment à l'étiquette 6xHis, qui conduisent
à la formation quasiment exclusive de corps d'inclusion. Si la
souche bactérienne et la température d'expression ne
semblent pas influencer de manière directe la solubilité des
protéines hétérologues PRODH, les criblages
de ces deux paramètres n'ont pas été pour
autant inutiles. En effet, nous avons constaté que
l'expression des domaines PRODH, en fusion avec les
meilleurs partenaires, et en souche
Rosetta cultivée à 20°C, permet dans la
plupart des cas d'augmenter de manière significative
les niveaux d'expression de protéine soluble.
Le criblage des conditions d'expression en microplaques du
programme 3PM a donc rempli son objectif. En effet, après avoir
testé en moyenne près d'une trentaine de conditions
par domaine PRODH, nous avons pu déterminer,
pour chaque domaine, au moins une condition qui permette une
expression suffisante de protéines de fusion solubles
(Tableau 4.1). Cependant, cette première étape a
également mis en évidence le caractère toxique du
domaine PROter qui laisse présager la rencontre de
difficultés lors de sa
production à grande échelle.
|
domaine
|
partenaire de fusion
|
Souche d'expression
|
Température d'expression
|
|
PROcatal
PROentier
PROter
PROinser
|
MBP
MBP
NusA
MBP
|
Rosetta
|
20°C
|
Tableau 4.1 :
Récapitulatif de la meilleure condition d'expression obtenue pour
chaque
domaine PRODH à l'issu du criblage en
microplaques.
2.2) Production, purification, et obtention des
protéines d'intérêt
Le détail des matériels et méthodes
relatifs à la production à grande échelle, à
la purification, et à l'obtention finale des 4 domaines PRODH figure en
fin de ce chapitre dans
le paragraphe 3.
2.2.1) Obtention du domaine PROcatal
La transposition des meilleures conditions d'expression de
PROcatal à grande échelle
a été entreprise en fernbach contenant 1
litre de culture. Les souches Rosetta ont été
transformées par le vecteur d'expression encodant
la protéine PROcatal fusionnée au partenaire MBP, puis
mises en préculture afin d'ensemencer la culture de 1 litre.
L'expression
de la protéine hétérologue a
été induite à une DO600 de 1.2 par ajout de 1 mM d'IPTG,
puis
maintenue pendant 14 heures à 20°C sous agitation.
Les culots bactériens ont été lysés dans
un tampon Tris-HCl à pH 8.0, et les fractions contenant
les protéines solubles et insolubles
ont été séparées par
centrifugation.
Comme le montre le Tableau 4.2, les résultats de la
transposition à grande échelle en fernbach sont tout à
fait satisfaisants : le taux de protéine hétérologue
PROcatal obtenu dans
la fraction soluble est similaire, voire légèrement
supérieur, à celui obtenu en microplaques.
|
PROcatal
|
Microplaque (250 uL)
|
Fernbach (1 L)
|
|
MBP/Rosetta/20°C
|
S I
++(+) ++
|
S I
+++ +++
|
Tableau 4.2 : Résultats
de la transposition à grande échelle des meilleures
conditions
d'expression PROcatal. Après dépôt
des fractions soluble (S) et insoluble (I) sur gel SDS- PAGE, chaque
bande de surexpression est analysée, et une valeur
semi-quantitative est associée à son intensité, de faible
(+) à très forte (++++). Les fractions se confondant aux
protéines endogènes de E. coli et constituant un doute, sont
associées au sigle +/-. La légende
de ce tableau s'applique également pour les
résultats de la transposition des autres protéines
PRODH (cf. Tableaux 4.3, 4.4, et 4.5).
Sur la base de cette transposition très favorable, la
purification de la protéine de fusion PROcatal a pu être
entreprise. Celle-ci a été menée par chromatographie
d'affinité sur résine d'amylose Amylose Resin (Biolabs)
spécifique au partenaire MBP. La moitié du volume de surnageant
de cassage, provenant de 500 mL de culture, a été
chargée dans la résine d'amylose. Après rinçage,
l'élution a été menée en introduisant un tampon
contenant 10 mM
de maltose. L'ensemble des fractions recueillies ont
été déposées sur gel
d'électrophorèse
SDS-PAGE, et les fractions d'élution ont été
quantifiées par mesure de l'absorbance à 280 nm sur un
spectromètre UV.
L'analyse SDS-PAGE de la fraction principale
d'élution fait apparaître une bande majoritaire dont la
masse apparente correspond à celle de la protéine de
fusion MBP- PROcatal (Figure 4.3A). La quantité de protéine
recombinante présente dans cette fraction est estimée à
environ 29 mg, ce qui correspond à une production de près de 60
mg par litre de culture. Cependant, nous avons remarqué la
présence de précipités troubles dans les
différentes fractions lors de l'élution de la
protéine. L'analyse SDS-PAGE de la fraction principale,
après centrifugation et reprise du culot insoluble par du SDS,
révèle en effet qu'une partie de la protéine
hétérologue s'est agrégée (Figure 4.3B).

(kDa)
150
100
75
S I
B)
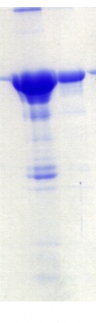
MBP
- PROcatal
50
37
25

A)
Figure 4.3 : Purification de la
protéine de fusion MBP-PROcatal (99 kDa) sur
résine
d'amylose. A) Analyse SDS-PAGE de la fraction principale
d'élution. B) Mise en évidence de l'agrégation partielle
de MBP-PROcatal : analyse SDS-PAGE des fractions soluble (S) et
insoluble (I) après centrifugation de la fraction principale
d'élution.
Nous avons mené une étude de stabilité
à 4°C sur plusieurs jours qui montre que la protéine de
fusion MBP-PROcatal présente une propension lente à
l'agrégation. En effet, alors que la fraction principale de
protéine purifiée contient 29 mg de protéine
soluble en sortie de purification, elle n'en contient plus que 23 mg au bout
de 24 heures, et 18 mg au bout de 48 heures. D'autre part, la conservation de
la protéine à -80°C, après congélation dans
l'azote liquide, n'est pas recommandée car elle provoque une
agrégation drastique à la décongélation. La
stabilité de la protéine de fusion a également
été testée sur plusieurs gammes de concentration. La
dilution des échantillons au 1/2, au 1/5, ou au 1/10 avec du
tampon d'équilibration (NaCl 200 mM, Tris-HCl 100 mM, pH
8.0) n'induit aucune modification du profil de stabilité, ce qui
suggère que la propension à l'agrégation n'est pas
dépendante de la concentration. Dans certains cas, ce type de
comportement peut être observé lorsqu'une protéine est
mise en solution avec un tampon non adapté qui provoque son
agrégation lente. Nous avons donc testé la
solubilité de la protéine MBP-PROcatal avec 9 solutions
tampons différentes : phosphate de sodium, hepès, MES,
acetate de sodium, phosphate de potassium, acétate d'ammonium,
citrate, imidazole, et Tris-HCl (étude non
présentée). Aucun de ces tampons ne permet de ralentir
le phénomène d'agrégation de la protéine de
fusion. D'autre part, l'oxydation de cystéines libres favorise la
formation de ponts disulfures intra ou intermoléculaires non natifs
qui peuvent conduire à l'agrégation. Le domaine PROcatal
possédant 8 cystéines, nous avons ajouté
différents réducteurs dans les surnageants de lyse et dans
les fractions d'élution : DTT, â-mercapto-éthanol, et
TCEP.
L'ajout de ces réducteurs n'a aucune influence sur
le profil de stabilité de la protéine de
fusion. Au vu de l'ensemble des résultats de ces tests, il
semble donc que cette propension à
l'agrégation soit intrinsèque à la
protéine hétérologue MBP-PROcatal.
En parallèle de ces études de stabilité,
nous avons entrepris de cliver le partenaire de fusion MBP par coupure
enzymatique avec la protéase TEV. Cette opération a
été réalisée en solution en ajoutant 640 ug de
protéase à un échantillon contenant 9 mg de
protéine de fusion. Après une incubation à 4°C sous
agitation pendant 14 heures, l'échantillon a été
centrifugé et
la fraction insoluble a été reprise avec du SDS.
Comme le montre le gel SDS-PAGE de la Figure 4.4, le clivage de la
protéine de fusion par la TEV n'a pas accentué le
phénomène d'agrégation. Trois bandes de forte
intensité dont les masses apparentes correspondent à celles
de MBP-PROcatal, MBP, et PROcatal, apparaissent dans la fraction soluble sur le
gel d'acrylamide. La coupure en solution est donc partielle, et le
rendement peut être estimé à
plus de 60% en comparant les intensités de ces 3 bandes
protéiques.
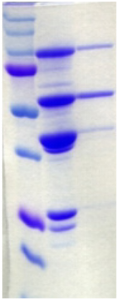
S I
(kDa)
100
75
50
37
MBP-PROcatal
PROcatal
MBP
25
20
Figure 4.4 : Clivage du
partenaire de fusion de MBP-PROcatal (99 kDa). Analyse de la
solution de clivage par la TEV après centrifugation.
Mise en évidence des bandes protéiques
de MBP-PROcatal, PROcatal (59 kDa), et MBP (43 kDa).
Sur la base de ce bon résultat, nous avons envisagé
de séparer le partenaire de fusion
de la protéine d'intérêt par chromatographie
de pseudo affinité sur résine de nickel HisTrap
HP (Amersham). La solution de clivage, issue de la
réaction de coupure enzymatique par la TEV de 9 mg de protéine de
fusion, a été intégralement chargée sur une colonne
HisTrap HP. Après lavage de la résine avec un
tampon contenant 20 mM d'imidazole, l'élution a été
réalisée avec un gradient linéaire en imidazole de 20 mM
à 300 mM. Les fractions récoltées
ont été analysées sur gel SDS-PAGE
(Figure 4.5), et la concentration d'imidazole dans les fractions
d'élution a été vérifiée par mesure de
l'absorbance à 300 nm sur un spectromètre
UV.
concentration en imidazole (mM)
20
100
200
chargement lavage élution
SC
MBP- PROcatal
1 2 3 4
5 6 7 8 9
10 11 12
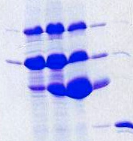
13 14 15

(kDa)
150
100
75

PROcatal
50
MBP
MBP coli 37
TEV
25
Figure 4.5 : Suivi par SDS-PAGE
de la purification de PROcatal sur résine de nickel.
Analyse des fractions de chargement, de lavage,
d'élution, et de la solution de clivage (SC). Les protéines
MBP-PROcatal (99 kDa), MBP clivée (43 kDa), et PROcatal (59
kDa) sont mises en évidence par des flèches rouges, et la
protéase TEV (27 kDa) et la MBP endogène à
E. coli (40 kDa) par des flèches vertes.
L'analyse SDS-PAGE met en évidence une bande
protéique, dont la masse apparente
est très proche de celle du partenaire MBP
clivé, dans les fractions contenant les protéines non retenues
(fractions 1 à 9). Cette bande correspond à la MBP
endogène de E. coli, qui ne possède pas
d'étiquette 6xHis, et qui s'est concentrée sur la
résine d'amylose lors de la première étape de
purification. La faible différence de poids moléculaire
entre les deux formes de MBP est suffisante pour les différencier sur
les gels d'acrylamide (respectivement
43 kDa pour le partenaire clivé, et 40 kDa pour la
protéine endogène).
De manière inattendue, le profil d'élution de
PROcatal, qui ne possède pas d'étiquette
6xHis, est similaire à celui de MBP-PROcatal et MBP
clivée qui en comportent une. Ainsi,
ces trois protéines fixent la résine de nickel
jusqu'à des concentrations d'imidazole de l'ordre
de 100 mM. La chromatographie de pseudo affinité IMAC
est une technique de purification peu spécifique. Ainsi, il est
communément observé que certaines protéines, qui
présentent à leur surface un amas de plusieurs résidus
histidine, sont capables de fixer la résine de nickel
de manière non spécifique à des
concentrations d'imidazole relativement faibles. PROcatal est
un domaine de 486 résidus qui comporte 14
résidus histidine dispersés dans la séquence primaire
; il est donc tout à fait possible qu'il interagisse de manière
non spécifique avec les ions nickel. Cependant, ce type d'interaction ne
suffit pas pour accrocher la résine jusqu'à des
concentrations d'imidazole de l'ordre de 100 mM. Par
conséquent, nous proposons une autre
hypothèse expliquant le profil d'élution de
PROcatal.
En effet, les gels d'acrylamide de la Figure 4.5 montrent que
la présence de la protéine d'intérêt PROcatal dans
les différentes fractions est toujours accompagnée du partenaire
de fusion clivé MBP, ou de la protéine de fusion
résiduelle MBP-PROcatal (fractions 10 à 15). Ceci suggère
que l'élution de PROcatal, à des concentrations d'imidazole de
l'ordre de 100 mM, puisse être due à un effet
d'entraînement de grande envergure, qui s'expliquerait par
l'existence de fortes interactions entre PROcatal et MBP. Ce type
d'interactions de fortes intensités, entre la protéine
d'intérêt et son partenaire de fusion après clivage
par la TEV, suggère un repliement incorrect ou instable du domaine
PROcatal, qui pourrait expliquer la propension à l'agrégation
de la protéine de fusion. De manière intéressante,
ce type de comportement a déjà été
rencontré au LMP avec d'autres protéines dont la
séparation de leur partenaire de fusion, pourtant clivé, n'a
jamais été obtenue.
Dans l'optique de vérifier notre hypothèse,
nous avons envisagé de séparer le partenaire de fusion
MBP de la protéine PROcatal sur résine d'amylose dont
l'affinité est beaucoup plus spécifique. Cependant, lorsque nous
avons préparé cette opération 48 heures après la
purification sur résine de nickel, nous avons constaté que plus
de 80% de la protéine PROcatal s'était agrégée dans
les fractions d'élution.
2.2.2) Obtention du domaine PROinser
Les meilleures conditions d'expression de PROinser obtenues
à l'issu du criblage en microplaques sont identiques à celles
obtenues pour PROcatal (MBP, Rosetta, 20°C). Par conséquent,
nous avons eu recours aux mêmes stratégies et
méthodologies pour produire, purifier, et isoler la protéine
d'intérêt PROinser. Comme le montre le tableau 4.3,
l'expression
de la protéine de fusion MBP-PROinser, en fernbach
contenant 1 L de culture, conduit à un taux de protéine soluble
tout à fait comparable à celui obtenu en microplaques. Sa
purification
est donc envisageable.
|
PROinser
|
Microplaque (250 uL)
|
Fernbach (1 L)
|
|
MBP/Rosetta/20°C
|
S I
++(+) +
|
S I
++ +(+)
|
Tableau 4.3 : Résultats
de la transposition à grande échelle des meilleures
conditions
d'expression PROinser.
La moitié du volume de surnageant de lyse,
provenant de 500 mL de culture, a été
déposé sur résine d'amylose. L'analyse
SDS-PAGE des différentes fractions, après élution
par un tampon maltose, met en évidence une bande
majoritaire dans la fraction principale dont
la masse apparente correspond bien au poids moléculaire de
MBP-PROinser (56 kDa) (Figure
4.6A). Le rendement de l'expression de la protéine de
fusion, obtenu à l'issu de cette première étape de
purification, est d'environ 30 mg de protéine par litre de culture, ce
qui est tout à fait satisfaisant. Cependant, nous avons
également constaté que la protéine MBP-PROinser
présente une tendance à l'agrégation comparable
à celle de MBP-PROcatal. En effet, l'analyse SDS-PAGE de la
fraction principale, après 24 heures d'incubation à
4°C, montre qu'une fraction importante de la protéine s'est
agrégée (Figure 4.6B). Une étude de stabilité,
identique à celle décrite précédemment, a
donc été entreprise dans l'optique d'améliorer la
solubilité de la protéine hétérologue. Les
résultats de cette étude indiquent que MBP- PROinser
présente une propension lente à l'agrégation quels
que soient les paramètres expérimentaux testés
(concentration, solution tampon, réducteur). Ainsi, aucune
condition permettant de ralentir ce phénomène d'agrégation
n'a été déterminée.

(kDa)
150
100
75
50
37
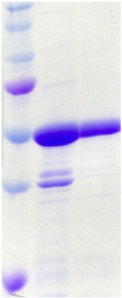
MBP
- PROinser
S I S I


(kDa)
75
50
37
25
20
15
10
MBP-PROinser
MBP
PROinser
25
A) B) C)
Figure 4.6 : Purification et
clivage du partenaire de fusion de MBP-PROinser (56 kDa)
suivis par SDS-PAGE. A) Purification sur résine
d'amylose : analyse de la fraction principale d'élution. B) Mise en
évidence de l'agrégation partielle de MBP-PROinser :
analyse des fractions soluble (S) et insoluble (I) après centrifugation
de la fraction principale 24 heures après l'élution. C) Analyse
de la solution de clivage par la TEV après centrifugation. Mise en
évidence des bandes protéiques de MBP-PROinser, PROinser (13
kDa), et MBP (43 kDa).
Nous avons tout de même entrepris de cliver le
partenaire de fusion par coupure enzymatique avec la TEV. 680 ug de
protéase ont été introduits dans la fraction principale
d'élution de 10 mL contenant environ 8.5 mg de protéine de
fusion. L'analyse SDS-PAGE de
la solution de clivage fait apparaître dans le surnageant
une bande protéique de faible poids
moléculaire à une masse apparente qui
correspond à celle du domaine PROinser (Figure
4.6C). En se basant sur les intensités des bandes
correspondant à MBP-PROinser, MBP, et PROinser, le rendement de coupure
peut être estimé à une valeur proche de 70%. De plus,
l'analyse de la fraction insoluble montre que l'introduction de la
TEV n'a pas accentué le phénomène
d'agrégation.
La quantité de protéine hétérologue
clivée étant suffisante, la séparation du partenaire
de fusion peut être envisagée sur résine
de nickel. La solution de clivage a été intégralement
chargée sur une colonne HisTrap HP. Après
rinçage avec un tampon contenant 20 mM d'imidazole,
l'élution a été menée avec un gradient
linéaire en imidazole de 20 mM à 300 mM. L'analyse des
éluats par SDS-PAGE fait apparaître 2 bandes
protéiques dans les fractions contenant les protéines non
retenues par la résine de nickel (fractions 1 à 6) (Figure
4.7). La première traduit la présence d'une
faible quantité de protéine de fusion MBP-
PROinser, et la seconde correspond à la MBP
endogène de E. coli.
concentration en imidazole (mM)
20
100
200
chargement lavage élution
MBP- PROinser
MBP MBP coli
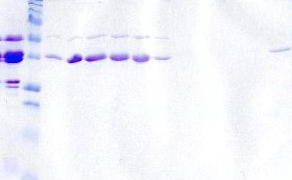
TEV
SC 1 2 3 4
5 6 7
8 9 10 11
12 13 14
15 16 17
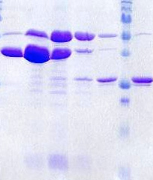

(kDa)
75
50
37
25
20
15
PROinser
10
Figure 4.7 : Suivi par SDS-PAGE
de la purification de PROinser sur résine de nickel.
Analyse des fractions de chargement, de lavage,
d'élution, et de la solution de clivage (SC). Les protéines
MBP-PROinser (56 kDa), MBP clivée (43 kDa), et PROinser (13
kDa) sont mises en évidence par des flèches rouges, et la
protéase TEV (27 kDa) et la MBP endogène à
E. coli (40 kDa) par des flèches vertes.
De manière intéressante, le profil d'élution
du domaine PROinser sur résine de nickel
est tout à fait comparable à celui de
PROcatal. En effet, PROinser, qui ne comporte pas d'étiquette
6xHis, est retenu dans la colonne jusqu'à des concentrations
d'imidazole de
l'ordre de 130 mM ; ce qui est également le cas de la
majeure partie de la protéine de fusion
MBP-PROinser, et de MBP clivée, qui en
possèdent une. Le domaine PROinser ne comportant que 2
résidus histidine dans sa séquence primaire, il est donc
exclu que cette liaison à la résine de nickel soit due à
des interactions non spécifiques. De plus, la présence de
PROinser dans les différentes fractions est toujours accompagnée
du partenaire MBP ou de la protéine de fusion résiduelle
(fractions 13 à 15). A l'instar de PROcatal, il semble donc que le
domaine PROinser interagisse avec son partenaire clivé ou la
protéine de fusion via des liaisons non covalentes de fortes
intensités. L'analyse des profils d'élution sur résine de
nickel suggère donc un repliement incorrect ou instable du
domaine PROinser, qui pourrait expliquer la propension à
l'agrégation de la protéine de fusion, avant et après la
coupure par la protéase TEV.
2.2.3) Obtention de la protéine
PROentier
La production du domaine PROentier a également
été entreprise avec le partenaire de fusion MBP et en souche
Rosetta cultivée à 20°C. Les résultats de la
transposition à grand volume, rassemblés dans le Tableau 4.4,
montrent que le taux de protéine soluble, obtenu en fernbach ou en
erlenmeyer, est très inférieur à celui obtenu en
microplaques. Ainsi, l'expression dans la fraction soluble est à
peine détectable en fernbach contenant 1 L de culture, et elle
est un peu plus prononcée en erlenmeyer de 3L contenant 300 mL de
culture.
|
PROentier
|
Microplaque
(250 uL)
|
Fernbach
(1 L)
|
Erlenmeyer
(300 mL)
|
|
MBP/Rosetta/20°C
|
S
++
|
I
++
|
S
+/-
|
I
++++
|
S
+
|
I
+(+)
|
Tableau 4.4 : Résultats
de la transposition à grande échelle des meilleures
conditions
d'expression PROentier.
Ces quantités de protéine soluble n'étant
pas satisfaisantes, nous avons voulu tester la production à grande
échelle du domaine PROentier en fusion avec le partenaire NusA,
qui conduit au deuxième meilleur taux de protéine soluble
en microplaques. Cependant, un certain nombre de difficultés
inhérentes au vecteur d'expression ont été
rencontrées lors des étapes de transformation et de
précultures, et n'ont pas permis d'exprimer la protéine
hétérologue NusA-PROentier.
La purification de la protéine de fusion MBP-PROentier a
donc été entreprise à partir
de la culture de 300 mL réalisée en
erlenmeyer. La totalité du surnageant de lyse a
été déposée sur résine d'amylose. L'analyse
SDS-PAGE de la fraction principale d'élution fait apparaître une
bande protéique majoritaire, dont la masse apparente correspond bien
à celle de MBP-PROentier (Figure 4.8A), ce qui confirme la surexpression
de la protéine de fusion. La quantité de protéine
recombinante présente dans cette fraction de 11 mL est estimée
à environ
2.3 mg, ce qui correspond à une production relativement
faible d'environ 8 mg par litre de culture. Cependant, contrairement aux
domaines PROcatal et PROinser, aucune tendance à
l'agrégation de la protéine de fusion n'a été
constatée.
Le clivage du partenaire de fusion a donc été
réalisé en ajoutant 180 ug de protéase
TEV dans la fraction d'élution contenant 2.3 mg de
protéine de fusion. Comme le montre le
gel d'acrylamide de la Figure 4.8B, le rendement de
coupure du partenaire MBP est très satisfaisant et atteint
quasiment 90 %. En effet, 2 bandes de moyenne intensité, dont
les masses apparentes correspondent à celles de PROentier et
MBP, apparaissent sur le gel d'acrylamide, alors que la bande
protéique correspondant à MBP-PROentier a quasiment
disparu.

(kDa)
150
100
75
50
37
25

A)
MBP-PROentier
(kDa)
150
100
75
50
37
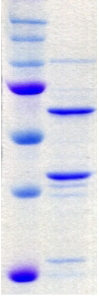
25
B)
MBP-PROentier
PROentier
MBP
Figure 4.8 : Purification et
clivage du partenaire de fusion de MBP-PROentier (107 kDa) suivis par
SDS-PAGE. A) Purification sur résine d'amylose : analyse de la fraction
principale d'élution. B) Analyse de la solution de clivage par
la TEV. Mise en évidence des bandes protéiques de
MBP-PROentier, PROentier (67 kDa), et MBP (43 kDa).
Au vu de ce bon résultat, nous avons
abordé avec confiance la dernière étape de
séparation du partenaire de fusion. La solution de clivage de
11 mL a été déposée sur une colonne
HisTrap HP. La résine a été dans un premier temps
lavée avec un tampon contenant
20 mM d'imidazole, puis l'élution a
été menée avec un gradient linéaire en
imidazole de
20 mM à 200 mM. L'analyse des fractions par SDS-PAGE
montre clairement que le domaine
PROentier n'est pas retenu sur la résine de nickel
(Figure 4.9). En effet, la bande protéique correspondante
n'apparaît que dans les fractions qui contiennent moins
de 40 mM d'imidazole (fractions 1 à 8). On retrouve également
dans ces fractions la MBP endogène de
E. coli issue de la première purification sur
résine d'amylose. Comme attendu, la protéine de fusion
résiduelle MBP-PROentier, et le partenaire clivé MBP,
fixent la résine de nickel jusqu'à des concentrations
d'imidazole de l'ordre de 120 mM. Par conséquent, la
chromatographie de pseudo affinité IMAC sur résine de nickel
permet de séparer le domaine PROentier de son partenaire de fusion, ce
qui n'était pas le cas avec PROcatal, et PROinser.
Au vu de l'analyse SDS-PAGE des fractions contenant
PROentier, un second passage sur résine d'amylose apparaît
cependant nécessaire pour éliminer la MBP endogène de
E. coli, et ainsi atteindre un degré de pureté
satisfaisant. En se basant sur les fractions 6, 7 et 8 contenant
la protéine relativement pure, le rendement obtenu,
à l'issu de cette seconde étape de purification, atteint
à peine 0.9 mg de domaine PROentier purifié par litre
de culture. Il
faudrait donc plus de 10 litres de culture pour espérer
obtenir 10 mg de protéine purifiée.
concentration en imidazole (mM)
20
100
200
chargement lavage élution
MBP- PROentier
PROentier
MBP MBP coli
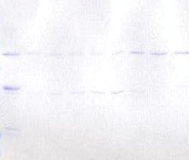
SC 1 2 3 4
5 6 7 8

9 10
11 12








13 14

(kDa)
150
100
75
50
37
TEV
25
20
Figure 4.9 : Suivi par SDS-PAGE
de la purification de PROentier sur résine de nickel.
Analyse des fractions de chargement, de lavage,
d'élution, et de la solution de clivage (SC). Les protéines
MBP-PROentier (107 kDa), MBP clivée (43 kDa), et PROentier (67 kDa) sont
mises en évidence par des flèches rouges, et la protéase
TEV (27 kDa), et la MBP endogène à
E. coli (40 kDa) par des flèches vertes.
Nous avons enregistré un spectre d'absorbance entre 400 et
600 nm sur l'échantillon
N° 7 contenant 83 ug de protéine PROentier pure
à plus de 90 % (estimation à partir du gel
d'acrylamide). De manière intéressante, ce spectre
fait apparaître un léger pic à 450 nm qui
correspond à la longueur d'onde d'absorbance du
FAD dans le visible (non montré). Ce résultat
suggère donc l'incorporation du cofacteur FAD au sein du domaine
PROentier dont le repliement serait proche de la forme native. Cette
analyse ne fournit cependant qu'une première indication et doit
être confirmée avec un échantillon de protéine plus
concentrée.
2.2.4) Obtention du domaine PROter
A l'issu des tests d'expression en microplaques, nous avons
retenu comme meilleures conditions d'expression de PROter : le partenaire NusA,
la souche Rosetta, et une température
de 20°C. Comme le met en évidence le tableau 4.5,
les résultats de la transposition à grande échelle,
menée en erlenmeyer et en fernbach, ne permettent pas de valider ces
conditions. En effet, l'expression dans la fraction soluble est quasiment nulle
en erlenmeyer de 3L contenant
300 mL de culture, et elle est très faible en fernbach
contenant 1 litre de culture. De plus, la diminution de la vitesse de
croissance bactérienne observée en microplaques, a
également été constatée à grande
échelle ; ce qui confirme l'hypothèse du caractère
toxique du domaine PROter. En parallèle, la production en grand volume
avec le partenaire NusA a été testée en souche C41, qui
permet d'améliorer l'expression des protéines
hétérologues PROter en microplaques. Que ce soit en erlenmeyer
ou en fernbach, aucune expression n'a été
détectée
dans la fraction soluble ou insoluble avec cette souche.
|
PROter
|
Microplaque
(250 uL)
|
Fernbach
(1 L)
|
Erlenmeyer
(300 mL)
|
|
NusA/Rosetta/20°C
|
S
++
|
I
++
|
S
+
|
I
+
|
S
+/-
|
I
+
|
Tableau 4.5 : Résultats
de la transposition à grande échelle des meilleures
conditions
d'expression PROter.
Nous avons tout de même entrepris la purification
de la protéine de fusion NusA- PROter à partir de la
culture menée en fernbach. Aucune résine d'affinité
spécifique à NusA n'étant disponible au laboratoire,
celle-ci a été réalisée sur une colonne de nickel
HisTrap HP.
La moitié du surnageant, provenant de 500 mL de
culture, a été déposée sur la résine.
L'élution a été menée avec un gradient
linéaire d'imidazole de 20 mM à 400 mM. L'analyse SDS-PAGE fait
apparaître une très légère bande à peine
discernable, dont la masse apparente correspond à celle de
NusA-PROter, dans les fractions contenant entre 100 et 200 mM
d'imidazole (fractions 9 à 12) (Figure 4.10A).
concentration en imidazole (mM)
20
100
200
chargement lavage élution
(kDa)
150
100
75
50
37
1 2 3 4
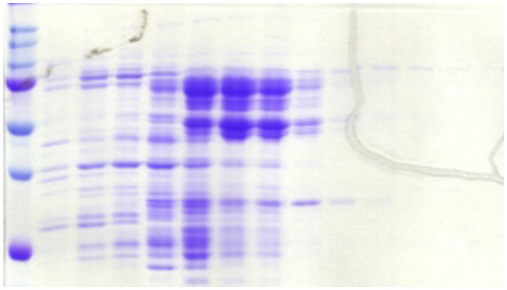
5 6 7 8
9 10 11 12 13

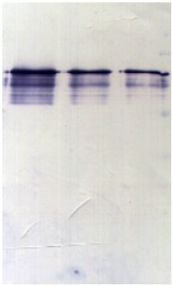
NusA- PROter
10 11 12
(kDa)
150
100
75
50
37
25
25
A) B)
Figure 4.10 : Purification de
la protéine de fusion NusA-PROter (68 kDa) sur résine
de
nickel. A) Analyse des fractions de chargement, de lavage,
et d'élution par SDS-PAGE. B) Analyse des fractions d'élution 10
à 12 par Western-blot avec un anticorps anti-NusA.
Afin de confirmer la présence de la protéine de
fusion, une hybridation Western-blot, beaucoup plus sensible que la
coloration au bleu de Coomassie, a été menée avec
des anticorps anti-NusA (Figure 4.10B). Cette analyse montre que la
bande protéique de faible intensité correspond bien à
la protéine NusA-PROter, mais elle révèle également
un profil de dégradation de la protéine
hétérologue. En effet, la présence de «
traînées » en dessous d'une bande majoritaire sur une
membrane de Western-blot est caractéristique d'une
protéolyse partielle. D'autre part, la quantité de
protéine étant trop faible dans les fractions d'élution,
il
n'a pas été possible de quantifier le rendement de
production par mesure de l'absorbance à
280 nm ; d'autant plus que l'imidazole possède
une absorbance résiduelle à cette longueur d'onde. Au vu de
ces résultats, il n'a pas été envisagé d'aborder
l'étape suivante de clivage du partenaire de fusion.
3) Conclusions
De manière générale, la deuxième
phase de production et purification du processus d'expression du
programme 3PM n'a pas permis de valider les meilleures conditions issues
du criblage en microplaques.
En ce qui concerne les domaines PROcatal et PROinser, bien que
la transposition à grande échelle soit favorable, la
tendance à l'agrégation constatée de ces 2 domaines
a considérablement compliqué leur purification. De plus,
malgré la réussite de la coupure enzymatique en solution par
la protéase TEV, nous n'avons pas été en mesure de les
séparer
de leur partenaire de fusion. Ces résultats
mettent ainsi en évidence le rôle complexe et ambigu du
partenaire de fusion qui semble, dans certains cas, capable
d'augmenter la solubilité générale d'une protéine
en fusion, sans pour autant favoriser son repliement natif et stable, et par
conséquent, indépendant. De manière intéressante,
les profils de stabilité et de purification de ces 2 domaines, de taille
très différente, sont tout à fait identiques. Ceci laisse
supposer que l'instabilité du domaine catalytique (PROcatal) pourrait
être due à la présence
de l'insertion (PROinser) dans sa région N-terminale.
Le caractère toxique du domaine PROter,
révélé à l'issu du criblage en microplaques,
a été confirmé lors de la
production à grande échelle : le vecteur d'expression
PROter en fusion avec NusA induit d'une part, une expression très
faible de protéine soluble et d'autre part, une diminution de la
vitesse de croissance bactérienne. De plus, l'analyse de cette
expression fait apparaître une protéolyse partielle de la
protéine de fusion. Au vu de ces résultats, la production
de ce domaine n'est donc pas adaptée chez l'organisme E.
coli.
Dans le cas de la protéine entière, la
stratégie d'expression du domaine PROentier en fusion avec le
partenaire MBP est à première vue une réussite.
Les différentes étapes de purification de la protéine
de fusion, de clivage par la TEV, et de séparation du partenaire, ont
globalement rempli leur objectif. Le seul désagrément se situe au
niveau de la transposition à grande échelle qui, conduisant
à une expression très faible de protéine, constitue un
véritable obstacle dans le cas d'une étude structurale.
4) Matériels et Méthodes
4.1) Production à grande échelle et
extraction des protéines
4.1.1) Production à grande
échelle
La production à grand volume est réalisée en
culture de 300 mL dans un erlenmeyer
de 3L, ou en fernbach de 1 L, à partir des meilleures
conditions définies à l'issue du criblage. Pour chaque
construction, 50 uL de bactéries thermocompétentes Rosetta
pLysS sont préalablement transformées par 20 ng de
vecteur d'expression par choc thermique à 42°C
pendant 45 secondes. Le produit de transformation est ensuite
mélangé avec 200 uL de SOC, incubé 1 heure à
37°C, puis étalé sur boîte LB/agar contenant
100 ug/mL d'ampicilline et
35 ug/mL de chloramphénicol. Après une nuit
d'incubation à 37°C, une préculture de 30 mL contenant les
mêmes concentrations d'antibiotiques est inoculée avec plusieurs
colonies, puis placée sous une agitation de 250 rpm à 37°C.
Les cultures de bactéries Rosetta contenant les 2 antibiotiques sont
ensemencées avec un volume de préculture de manière
à obtenir une DO600 initiale de 0.05, puis incubées à
37°C sous une agitation de 250 rpm. Après induction avec
1 mM d'IPTG lorsque la DO600 atteint 1.2, les cultures sont
placées à 20°C pendant 14 heures sous une agitation de 250
rpm. A l'issu de ce délai, la croissance bactérienne est
stoppée par centrifugation à 2830xg pendant 20 minutes et
à 4°C.
4.1.2) Lyse des bactéries et séparation des
fractions soluble et insoluble
Les culots bactériens sont congelés dans
l'azote liquide, repris au 1/20e dans un tampon de lyse (100
mM de Tris-HCl pH 8.0, 150 mM de NaCl, 5 % de glycérol, 1 uM de
phosphoramidon, et 1 mM de PMSF), puis cassés à la
presse d'Eaton (technique de lyse mécanique par application d'une
pression de 6 tonnes sur des cellules congelées à
-80°C, dirigées vers un orifice de petit diamètre). 0.5 U/mL
de benzonase et 10 mM de MgCl2 sont ajoutés au broyat. Après une
incubation de 15 minutes à 30°C, une centrifugation à
40000xg pendant 30 minutes à 4°C permet de séparer les
fractions soluble et insoluble. Le culot est repris dans le même
volume que le surnageant avec du SDS 2%. Les protocoles d'analyse
SDS-PAGE et Western-blot sont identiques à ceux
présentés dans la partie Matériels et Méthodes
du chapitre 2 (cf. § 4.2.3).
4.2) Purification des protéines de fusion et des
protéines d'intérêt
4.2.1) Purification sur résine
d'amylose
La purification des protéines en fusion avec le partenaire
protéique MBP est réalisée
sur résine d'amylose Amylose Resin
(Biolabs) de capacité théorique 3 milligramme par
millilitre de résine. Le volume de résine introduit dans la
colonne est choisi en fonction de la quantité de protéine de
fusion estimée dans les surnageants de lyse. La colonne est
préalablement équilibrée avec 10 volumes de tampon
d'équilibration contenant, 30 mM de Tris-HCl (pH 7.8), 200 mM de
NaCl, 1 mM de PMSF, et 2 mM de â-mercapto-éthanol. Le débit
est de 1 mL/min pour toute la purification. Après chargement des
surnageants de lyse, la résine est lavée avec le tampon
d'équilibration jusqu'à retour de l'absorbance mesurée
à 280
nm à la ligne de base (LKP Control Unit
UV-1, Pharmacia). L'élution est réalisée avec
ce même tampon contenant 10 mM de maltose. Le volume des fractions
recueillies est de 5 mL
ou 10 mL. Elles sont analysées par SDS-PAGE et
quantifiées par mesure de la DO280.
4.2.2) Purification sur résine de
nickel
Des colonnes HisTrap HP (Amersham) de 1 mL ou 5 mL,
pré-chargées en résine de nickel, et de capacité
théorique 12 mg/mL, sont utilisées pour séparer le
partenaire de fusion après clivage ou purifier la protéine
hétérologue NusA-PROter. Après équilibration de
la colonne, à un débit de 1 mL/min, avec 10 volumes de tampon de
fixation contenant, 100 mM
de Tris-HCl (pH 7.5), 300 mM de NaCl, 1 mM de PMSF, 2 mM de
â-mercapto-éthanol, et 20 mM d'imidazole (pour limiter les
interactions non spécifiques), les échantillons sont
introduits à un débit de 0.5 mL/min. La valeur du débit
est ensuite portée à 1 mL/min jusqu'à
la fin de la purification. La résine est lavée
avec du tampon de fixation jusqu'à retour de la DO600 à la ligne
de base initiale. La composition du tampon d'élution est identique
à celle du tampon de fixation et contient en plus de l'imidazole
à une concentration de 500 mM. L'élution est menée
avec un gradient linéaire d'imidazole de 20 à 200, 300,
ou 400 mM (selon les protéines purifiées) sur 10 volumes
de colonne. Un lavage est finalement réalisé avec un
tampon contenant 1 M d'imidazole sur 5 volumes de colonne. Le volume
des fractions recueillies est de 1 mL ou 2 mL (collecteur
GradiFrac, Pharmacia). Elles sont analysées par SDS-PAGE et
quantifiées par mesure de la DO280. La quantité d'imidazole
dans
les fractions d'élution est vérifiée en
comparant leur absorbance mesurée à 300 nm à celle de
solutions contenant des concentrations d'imidazole connues.
4.3) Clivage du partenaire de fusion par la
protéase TEV
La présente du motif ENLYFQG, reconnu par la
protéase TEV, en aval de la séquence
de la protéine d'intérêt permet le clivage
du partenaire de fusion. Par souci de réduction des coûts, cette
protéase est produire au LMP sous forme recombinante chez E.
coli. Les fractions issues de la première étape de
purification sont soumises à une coupure en solution en
introduisant 8% de TEV, soit 8 mg pour 100 mg de protéine de
fusion. Elles sont ensuite
incubées à 4°C pendant environ 14 heures sous
légère agitation.
Chapitre 5 : Conclusions et Perspectives
CHAPITRE 5
Conclusions & Perspectives
Lorsque j'ai abordé ce projet de thèse, le premier
objectif fixé était de déterminer et
produire un domaine structuré soluble de la proline
déshydrogénase humaine, en espérant que
sa taille soit compatible avec une étude par
RMN. Aucune donnée structurale de proline
déshydrogénase n'étant disponible, nous avons
abordé une première approche, qui consistait à exprimer
l'intégralité de la protéine sous forme native dans un
organisme recombinant, puis à caractériser ses domaines
structuraux par protéolyse ménagée. Pour des raisons
essentiellement basées sur la simplicité de mise en oeuvre,
l'organisme producteur qui a été choisi est la bactérie
E. coli. Cependant, et à l'image de plus de 50 % des
protéines eucaryotes exprimées dans cet organisme, la
production de PRODH chez E. coli conduit à l'unique
formation d'agrégats de protéine insoluble et inactive : les
corps d'inclusion.
Nous avons par conséquent entrepris de produire la forme
mature de PRODH, c'est-à- dire délestée de son peptide
d'adressage mitochondrial N-terminal. Sur la base de prédictions
bio-informatiques, le peptide signal de la séquence de
PRODH humaine a été ciblé au niveau
de l'extrémité N-terminale 1-36. L'expression de la
construction PRO564, correspondant aux
564 résidus C-terminaux de PRODH, a ainsi
été initiée chez E. coli. Cette production s'est
également soldée par l'unique formation de corps d'inclusion.
L'expression sous forme soluble constituant
l'étape limitante de notre étude structurale, il nous
fallait par conséquent modifier notre stratégie. Ces
dernières années ont vu l'apparition d'un certain nombre
d'innovations dans le domaine de l'expression de protéines
recombinantes, qui ont notamment abouti à l'émergence de
plates-formes de production dédiées à l'expression de
protéines solubles chez E. coli. Nous avons ainsi
entrepris de produire PRODH dans le cadre d'un de ces programmes, qui
offre la possibilité de tester plusieurs partenaires de fusion et
plusieurs souches d'expression différents. En parallèle, de
nouvelles données sont apparues dans la littérature et
nous ont permis d'envisager une stratégie alternative
d'étude structurale. La publication récente de la
première structure de proline déshydrogénase a mis
en évidence l'existence d'un domaine catalytique PRODH
replié en tonnelet á8â8 chez E. coli, et dont la
taille n'est pas adaptée à une étude par RMN. Nous avons
par conséquent abordé une seconde approche, qui
consiste à prédire l'organisation des domaines de la
protéine humaine sur la base de cette structure, et en
utilisant des outils bio-informatiques. A partir d'alignements de
séquence, de recherche
d'homologie de séquence, et de prédiction de
structure secondaire, nous avons prédit les
segments qui constituent le domaine catalytique humain.
Cette analyse suggère la présence
d'un large domaine catalytique C-terminal de 59 kDa,
entremêlé avec deux insertions, de fonction inconnue, de
respectivement, 50 et 100 résidus, et d'un domaine N-terminal
également de fonction inconnue. Dans l'optique de
caractériser les rôles structural et fonctionnel de ces
domaines potentiels, 3 régions de PRODH humaine ont été
sélectionnées afin de les soumettre au programme de
production 3PM du Laboratoire de Structure des Protéines du CEA
de Saclay. Il s'agit du domaine catalytique (PROcatal), du domaine N-
terminal (PROter), et de la deuxième insertion (PROinser). La
forme mature de PRODH humaine rebaptisée PROentier a également
été soumise à la plate-forme de production 3PM.
Malgré la possibilité de tester un grand
nombre de conditions d'expression, la production des 3 protéines
PROcatal, PROter, et PROinser dans le cadre du programme 3PM s'est
soldée par un échec. En effet, le criblage de ces conditions
d'expression à petite échelle
a, certes dans un premier temps, permis d'identifier un
partenaire protéique qui conduit à une expression suffisante de
protéine soluble. Cependant, nous avons été
confrontés à plusieurs difficultés lors de la
deuxième phase du processus d'expression à grand volume
(absence d'expression, agrégation, protéolyse,
incapacité à éliminer le partenaire), qui ne
permettent pas de satisfaire les exigences inhérentes à une
étude structurale par RMN ou cristallographie des rayons X
(protéine soluble, native, stable, pure, et en grande quantité).
En ce qui concerne
la protéine mature PROentier, nous avons réussi
à produire cette protéine sous forme soluble
et pure. Toutefois, les meilleures conditions
d'expression ne conduisent qu'à un rendement très faible
de 0.9 mg par litre de culture, qui nécessiterait plus de 10
litres de culture pour espérer obtenir ne serait-ce que 10 mg d'une
protéine de 67 kDa.
Au vu de l'ensemble des résultats, il ne
semblait pas raisonnable de poursuivre ce projet dans le cadre de mon
travail de thèse. En effet, la production de protéines et
domaines recombinants PRODH a été très coûteuse en
temps. Près de 20 mois ont été nécessaires pour
cloner, produire, et optimiser les conditions d'expression des
protéines PRODH, PRO564, PROentier, PROcatal, PROter, et PROinser. Au
final, seul le domaine PROentier fait l'objet
de résultats encourageants qui sont cependant
insuffisants pour envisager l'analyse structurale. De plus, cette large
région de 67 kDa n'est pas adaptée à une étude par
RMN, qui représente un des objectifs de mon projet scientifique.
La mise au point du protocole
d'expression de la protéine PROentier n'a pas
été pour autant inutile. En effet, la production
de plusieurs litres de culture serait suffisante pour
entreprendre des études biochimiques sur la
protéine PROentier, et ainsi caractériser pour
la première fois la nature du cofacteur et/ou de l'inhibiteur
compétitif naturel d'une proline déshydrogénase
eucaryote. Pour ma part, j'ai entrepris de mettre à profit
l'expérience acquise dans le domaine de l'expression de protéines
recombinantes pour produire, puis caractériser la structure d'une
autre biomolécule : la protéine KIN17, impliquée dans
la réparation des dommages de l'ADN.
SECONDE PARTIE
Caractérisation de la région 51-160 de la
protéine KIN17 humaine par RMN et Modélisation
Moléculaire
Chapitre 1 : Introduction
CHAPITRE 1
Introduction
1) Généralités sur le maintien de
l'intégrité du génome
L'Acide Désoxyribose Nucléique, connu sous
le nom d'ADN, est un polymère de nucléotides qui contient
sous forme codée une grande partie des informations relatives à
la vie d'un organisme vivant. La protection de l'intégrité du
génome est un défi capital et constant pour les cellules. En
effet, la survie des organismes dépend de la transmission
totale et correcte de l'information génétique lors des processus
de réplication de l'ADN. Les cellules doivent donc être capable
de détecter et réparer les dommages de l'ADN, de toute
nature qu'ils soient, afin de préserver leur patrimoine
génétique.
1.1) Les dommages de l'ADN
1.1.1) Sources
Une cellule doit lutter en permanence pour protéger
l'intégrité de son génome. Toute altération de ce
précieux matériel nucléaire constitue un dommage de l'ADN.
Les origines de
ces altérations moléculaires sont aussi
différentes que nombreuses. Ainsi, elles peuvent provenir
d'agents exogènes, on parle alors d'altération
environnementale, ou d'agents endogènes qui induisent des dommages
dits spontanés.
Parmi les sources exogènes, figurent les rayonnements,
les radiations ionisantes et les substances chimiques mutagènes. Les
ultraviolets (UV), les radiations ionisantes (IR), ou le rayonnement ã,
produisent des lésions sur l'ADN notamment dues à la
radiolyse des molécules d'eau qui forme des espèces
réactives de l'oxygène (Ames et al., 1993). Ces
espèces réactives peuvent modifier aussi bien les bases de l'ADN,
que les sucres, et créent des cassures au niveau du squelette phosphate
(Hutchinson, 1985). Les agents chimiques comme des drogues, les analogues de
base, ou les inhibiteurs de processus biologiques peuvent, une fois
passée la membrane nucléaire, causer d'importants
dégâts à la structure moléculaire de l'information
génétique. C'est notamment le cas du Méthyl
Methane Sulfonate (MMS) qui
bloque la séparation des brins d'ADN.
Les dommages spontanés peuvent provenir d'erreurs
générées durant les processus de réplication,
recombinaison, ou réparation de l'ADN. Des modifications
chimiques peuvent également avoir lieu dans certaines conditions
de pH et de température. D'autre part, le
métabolisme cellulaire induit la formation endogène
d'espèces réactives de l'oxygène. Elles
proviennent notamment de la chaîne respiratoire
mitochondriale (Ames et al., 1993), des peroxysomes (compartiments
cellulaires où sont dégradés les acides gras)
(Yeldandi et al.,
2000), et de la détoxification de certains
composants de la cellule comme la vitamine D (Bondy & Naderi,
1994).
1.1.2) Nature et conséquences
Les différents types de dommage causés par
des agents endogènes ou exogènes peuvent être
classés en quatre catégories distinctes :
- Les cassures d'ADN simple ou double brin
-
- Les mésappariements
- Les dégradations des bases, comme la déamination,
l'oxydation, ou la création de sites abasiques
- Les modifications encombrantes telles que pontages
intra- et inter- brins et la formation de dimères de pyrimidine.
Ainsi, les rayons UV sont connus pour introduire des dimères de
pyrimidines et des pontages ADN-protéine ou ADN-ADN qui causent des
distorsions importantes dans la double hélice pouvant amener
à la rupture de la chaîne polynucléotidique.
Le renouvellement de l'ADN peut provoquer la
dépurination spontanée de certaines bases à raison de 2000
à 10000 sites par cellule humaine quotidiennement (Lindahl, 1993). La
cassure double brin est la plus dangereuse éventualité qu'une
cellule peut rencontrer car elle peut susciter la perte d'un morceau de
chromosome ou une translocation chromosomique (Thompson & Limoli, 2003;
Tong et al., 2001). Si une cellule ne peut réparer les quelques
10000 altérations de l'ADN qu'elle s'impose
quotidiennement (Lindahl, 1993), elle s'expose alors à de graves
problèmes car l'instabilité génomique est l'un des
évènements pouvant mener à la perturbation de la fonction
cellulaire. En effet, l'accumulation de dommages peut avoir de lourdes
conséquences s'ils surviennent à des endroits critiques dans la
structure de l'ADN. Elle peut mener à l'apoptose (mort cellulaire
programmée) ou à la formation de tumeurs, et ainsi
augmenter le risque de développer des maladies graves telles que le
cancer
(Khanna & Jackson, 2001). Lorsque les mécanismes de
réparation échouent ou commettent
des impairs, il y a apparition de mutations. Si celles-ci
induisent une modification structurale
significative, elles peuvent modifier l'activité d'une
protéine ou la guider vers la dégradation.
1.2) Les systèmes mis en jeu
La cellule dispose de systèmes moléculaires
complexes qui lui permettent de réagir rapidement à
l'endommagement de son ADN. L'existence de pathologies humaines liées au
dysfonctionnement de ces systèmes a permis de les
caractériser et de souligner leur importance dans le maintien de
l'intégrité du patrimoine génétique. Ils ont pour
objectif :
- La réparation de l'ADN
- La signalisation des dommages
- La réponse transcriptionnelle (induction de gènes
de la réparation par exemple)
- L'apoptose
Les quatre types de voies citées ci-dessus sont
interconnectées : certaines protéines participent à
plusieurs types de réponses aux dommages de l'ADN. Un défaut dans
l'une de
ces voies provoque l'instabilité du génome (Sancar
et al., 2004).
1.3) Les voies de réparation des lésions de
l'ADN
Suite à la détection d'un dommage de l'ADN,
différentes voies de réparation peuvent être
activées : la réparation directe, la réparation par
excision de base (BER), la réparation par excision de nucléotide
(NER), la réparation des mésappariements (MMR), et la
réparation des cassures double brins qui peuvent être pris
en charge soit par la voie de recombinaison homologue (HR), soit par le
mécanisme de recombinaison non homologue (NHEJ pour Non Homologous
End Joining). Ces différentes voies sont récapitulées
dans la Figure 1.1.
La recombinaison homologue est sans doute le système le
plus efficace de réparation d'une cassure double brin (Szostak et al.,
1983). Ce mécanisme, principalement utilisé chez la levure et les
bactéries, assure la conservation intégrale de l'information
génétique. Il est basé
sur l'échange de brins provenant d'un chromosome
endommagé vers un chromosome homologue intact. La recombinaison non
homologue ou illégitime (NHEJ) se fait par ligature
des extrémités de la cassure de l'ADN. La
réparation des cassures double brins par ce système
s'accompagne fréquemment de délétions
(Jeggo, 1998). Ainsi, le gène n'est, en général, plus
fonctionnel lorsqu'il est réparé par ce
mécanisme.
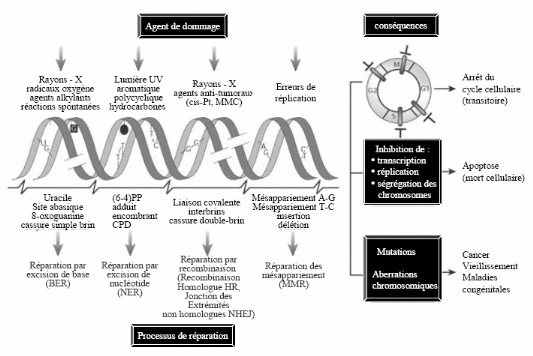
Figure 1.1 : Schéma
récapitulant la nature et les conséquences des dommages de l'ADN,
et
les différentes voies de réparation
(d'après Hoeijmakers, 2001).
Les mésappariements peuvent survenir au cours de la
réplication ou lors de la recombinaison homologue. Leur
réparation par le mécanisme MMR (Marti et al., 2002) peut
se décomposer en trois principales étapes: la
reconnaissance des mésappariements, le recrutement des
protéines du MMR, et l'excision-resynthèse de l'un des deux
brins.
La réparation par excision de bases (BER)
découverte en 1974 (Lindahl, 1974) reconnaît le plus
fréquemment des bases modifiées : déamination des
cytosines en uracile, alkylation de base induite par des
métabolites cellulaires, oxydation des bases de l'ADN et certains
mésappariements induits par les erreurs de réplication de
l'ADN. Plusieurs de ces lésions sont dues à des agents
environnementaux comme les radiations ionisantes mais certaines
résultent d'une hydrolyse spontanée. Dans ce
mécanisme, seule la base qui a été
modifiée est clivée, puis remplacée.
Enfin, la réparation de l'ADN par excision de
nucléotide (NER) (Friedberg, 2001) est
le mécanisme majeur de l'élimination des
lésions simple brin. Cette voie de réparation corrige
préférentiellement les modifications des nucléotides
telles que les dimères de pyrimidine et autres lésions produits
par les rayons UV, mais également les mésappariements de type
C?C. Contrairement au BER, cette voie de réparation clive
plusieurs nucléotides du brin d'ADN endommagé et
libère un fragment simple brin de 24 à 32 bases. Un nouveau brin
est alors synthétisé par des polymérases en utilisant
comme matrice le brin non endommagé. C'est ce mécanisme de
réparation qui intervient dans la régulation de la
protéine KIN17.
2) La protéine KIN17
Le Laboratoire de Génétique de la
Radiosensibilité (LGR) du CEA de Fontenay-aux- Roses s'intéresse
aux effets biologiques produits par les stress génotoxiques sur les
cellules, et notamment à la réponse des cellules de
mammifères aux radiations ionisantes (RI). En 1991,
les chercheurs du LGR ont découvert une nouvelle
protéine nucléaire de 45 kDa appelée KIN17 (Angulo
et al., 1991). Les fonctions précises et les partenaires
protéiques de cette protéine, uniquement présente
chez les organismes eucaryotes et exprimée de manière
ubiquitaire, sont à ce jour inconnues. Cependant, un certain
nombre d'études génétiques in vitro et
in vivo ont permis de caractériser l'implication de
cette protéine dans différents mécanismes
nucléaires majeurs, et notamment la réplication et la
réponse cellulaire aux dommages de l'ADN.
2.1) Les propriétés de la protéine
KIN17
La protéine KIN17 a été initialement
identifiée sur la base de sa capacité à réagir
avec
les mêmes anticorps que ceux dirigés contre RecA,
une protéine bactérienne impliquée à la fois dans
la réparation et la réplication de l'ADN (Angulo et al.,
1991). KIN17 possède effectivement dans sa séquence une
région d'environ 40 résidus qui présente 47 %
d'identité avec un segment situé en C-terminal de RecA.
De manière intéressante, dans RecA, cette région est
impliquée dans la régulation de la liaison de la
protéine à l'ADN et dans le mécanisme SOS, une des
réponses aux lésions de l'ADN chez les organismes
procaryotes (Kurumizaka et al., 1996).
2.1.1) Implication de KIN17 dans la
réplication et la réponse aux dommages
de l'ADN
Le gène humain encodant KIN17, situé sur
le chromosome 10, fait partie de l'ensemble des gènes de
l'organisme très faiblement exprimés. Cependant, les
cellules en division rapide contiennent 3 fois plus d'ARNm KIN17 que les
cellules au repos (Kannouche
et al., 1998). De plus, la protéine KIN17 est
distribuée dans les noyaux des cellules de mammifères
sous forme de structures discrètes appelées «
foyers intra-nucléaires » (Kannouche et al., 1997). Ce type de
foyers reflète la compartimentation de l'ADN lors des processus
complexes comme la réplication, la réparation, la
transcription, ou l'épissage. Toutes ces observations suggèrent
fortement que KIN17 appartient à un réseau de protéines
intranucléaires requises lors de la prolifération cellulaire.
L'irradiation par des rayons UV ou ã et des
radiations ionisantes provoquent une augmentation significative de la
concentration d'ARNm KIN17 dans les 13 heures qui suivent l'irradiation de
cellules de souris en culture (Kannouche et al., 2000 ; Biard et
al., 2002). Cette augmentation s'accompagne d'une accumulation de la
protéine dans le noyau des cellules. KIN17 est donc
régulée de manière positive suite à une
exposition à des sources rayonnantes qui induisent des dommages de
l'ADN. De manière intéressante, cette régulation positive
après irradiation par des UV-C dépend de la
présence des protéines XPA et XPC (Masson et al., 2003). Ces
deux protéines sont impliquées dans les mécanismes de
réparation
de l'ADN par excision de nucléotide (NER) (Wakasugi
& Sancar, 1999). Sur la base de ces observations, il apparaît donc
que la protéine KIN17 est impliquée dans la
réponse aux dommages cellulaires de l'ADN.
Par ailleurs, des études de gel filtration,
réalisées sur un extrait cellulaire humain de protéine
totale, ont révélé la présence de KIN17 dans 3
complexes multi-protéiques de très haute masse
moléculaire (respectivement : 400 kDa, 600 kDa, et 1800 kDa)
contenant la protéine de réplication RPA (Miccoli et al.,
2005). De plus, une interaction physique entre KIN17 et l'antigène
T du virus SV40 a été démontrée (Miccoli et al.,
2002). Ces observations confortent l'hypothèse de l'implication de KIN17
dans la réplication de l'ADN.
2.1.2) Liaison de KIN17 aux acides
nucléiques
En 1994, Mazin et al., ont mis en
évidence la capacité de KIN17 à lier l'ADN et
l'ADN courbe in vivo et in vitro chez l'homme et la souris
(Mazin et al., 1994). Ainsi, une fraction de KIN17 est fortement et directement
associée avec l'ADN chromosomique dans les cellules humaines (Biard et
al., 2002). L'ADN courbe est une forme de l'ADN, riche en bases adénine,
retrouvée dans les sites de recombinaison illégitime des cellules
de mammifère. Chez
les organismes procaryotes, l'ADN courbe occupe
une fonction importante dans la transcription des gènes
(Nishikawa et al., 2003). L'affinité de KIN17 pour cette forme
de l'ADN a été confirmée in vivo en surexprimant
la protéine KIN17 de souris chez la bactérie
E. coli (Timchenko et al., 1996). Dans cette
étude, il est montré que KIN17 est capable de complémenter
les fonctions du facteur de transcription H-NS, qui lie l'ADN courbe
et contrôle l'expression d'au moins 36 gènes.
Par ailleurs, une étude protéomique à grande
échelle a mis en évidence la présence de
KIN17 dans le spliceosome humain, un large complexe
protéine-ARN (Rappsilber et al.,
2002). L'interaction de KIN17 avec l'ARN a été
récemment caractérisée par Pinon-Lataillade
et al., qui ont montré d'une part, que la
protéine KIN17 de souris était capable de fixer l'ARN
in vivo, et d'autre part, que les
protéines humaines et de souris reconnaissent de manière
directe différents types d'homopolymères d'ARN in vitro
(Pinon-Lataillade et al., 2004).
2.2) Organisation des domaines structuraux de KIN17
Depuis sa découverte en 1991, parallèlement aux
études génétiques, la protéine KIN17
a fait l'objet d'analyses bio-informatiques qui ont
permis de caractériser une organisation segmentée en
domaines structuraux (Tissier et al., 1995 ; Pinon-Lataillade et al.,
2004 ; Ponting, 2002). Sur la base de ces analyses, les chercheurs du LGR ont
caractérisé quelques propriétés de ces domaines qui
sont présentées dans ce paragraphe.
La protéine KIN17 humaine est un polypeptide de
393 acides aminés. Comme le montre l'alignement de la Figure
1.2, KIN17 est remarquablement conservée de la levure
jusqu'à l'homme dans la moitié N-terminale correspondant aux
résidus 1-165 de la séquence humaine. Dans cette
région, les pourcentages d'identité et de
similarité atteignent respectivement 14 % et 22 %. Cependant, les
séquences de KIN17 ne s'alignent pas dans la
moitié C-terminale. En effet, la séquence de KIN17
ne compte qu'entre 250 et 300 résidus
chez les eucaryotes inférieurs telle que la levure,
alors qu'elle est proche de 400 résidus chez tous les eucaryotes
supérieurs. Ceci laisse supposer l'existence d'un domaine
structural supplémentaire chez les eucaryotes supérieurs.
Human
1-MGK-SDFLTPKAIANRIKSKGLQKLRWYCQMCQKQCRDENGFKCHCMSESHQRQLLLASENPQQFMDYFSEEFRNDFLEL
Mouse
MGK-SDFLSPKAIANRIKSKGLQKLRWYCQMCQKQCRDENGFKCHCMSESHQRQLLLASENPQQFMDYFSEEFRNDFLEL
CAEEL
MGK-HEKGSSKDLANRTKSKGLQKLKFFCQMCQKQCRDANGFKCHLTSEAHQRQLLLFAENSNSYLRQFSNDFEKNFMQL
Drosophila
MGR-AEVGTPKYLANKMKSKGLQKLRWYCQMCEKQCRDENGFKCHTMSESHQRQLLLFADNPGKFLHSFSKEFSDGYMEL
Arabidopsis
MGK-NDFLTPKAIANRIKAKGLQKLRWYCQMCQKQCRDENGFKCHCMSESHQRQMQVFGQNPTRVVDGYSEEFEQTFLDL
POMBE
MGR-AEAGTPKAISNALKSKGLQRLRWYCSACQKQMRDENGFKCHTQSEGHIRQMNVIAMNPGKRIQDFSNQFLRDFISL
Cerevisiae
---MADYDSAKYWSKQGARRGLQKTRYYCQICQRQCKDANGFQSHNKSPSHLRKISQVTAEDAR---RYNIQFEKGFLQL
: : :** : :::*. *::. :* **:: * : .* :. * ::
Human
80-LRRRFGTKRVHNNIVYNEYISHREHIHMNATQWETLTDFTKWLGREGLCKVDETP-KGWYIQYIDRDPETIRRQLELEKK
Mouse
LRRRFGTKRVHNNIVYNEYISHREHIHMNATQWETLTDFTKWLGREGLCKVDETP-KGWYIQYIDRDPETIRRQLELEKK
CAEEL
LRTSYGTKRVRANEVYNAFIKDKGHVHMNSTVWHSLTGFVQYLGSSGKCKIDEGD-KGWYIAYIDQ--EALIRKEEDQRK
Drosophila
LRRRFGTKRTSANKIYQEYIAHKEHIHMNATRWLTLSDYVKWLGRTGQVIADETE-KGWFVTYIDRSPEAMERQAKADRK
Arabidopsis
MRRSHRFSRIAATVVYNEYINDRHHVHMNSTEWATLTEFIKHLGKTGKCKVEETP-KGWFITYIDRDSETLFKERLKNKR
POMBE
LRTAHGEKKIHFNQFYQEYIRDKNHVHMNATRWHTLSEFCKFLGRQGMCRVEENE-KGFFISYIDKNPANILRNEANKKR
Cerevisiae
LKQRHGEKWIDANKVYNEYVQDRDHVHMNATMHRSLTQFVRYLGRAGKVDVDMDI---------DDTSENVEGPLLIRIH
: . . .* : : * *:*:* :*: : * :
Human
159-KKQDLDDEEKTAKFIEEQVRRGLEG-KE------------------QEVPTFTELSREN------------DEEKVTFNL
Mouse
KKQDLDDEEKTAKFIEEQVRRGLEG-KE------------------QETPVFTELSREN------------EEEKVTFNL
CAEEL
QQQEKDDEERHMQIMDGMVQRGKEL-AGD----------------DEHEYEATELIRDT------------PDQKIQLDL
Drosophila
EKMEKDDEERMADFIEQQIKNAKAKDGEE----------------DEGQEKFTELKRE-------------ENEPLKLDI
Arabidopsis
VKSDLAEEEKQEREIQRQIERAAEKLNGG-----------GGEGETSGNDEVVDDGDDERKKDEDLR--LKSGVKVGFAL
POMBE
ERQEKSDEEQRLRLLDEQIKRA----------------------------------------------------------
Cerevisiae
PSSLSSPSE------DGMLRSQ----------------------------------------------------------
: :
Human
208-SKGACSSSGA----TSSKSSTLGPSALKTIGSS--------------------------ASVKRKESSQSSTQSKEKKKK
Mouse
NKGAGGSAGA----TTSKSSSLGPSALKLLGSA--------------------------ASGKRKESSQSSAQP--AKKK
CAEEL
NLGILDRKLD----VKSGVASAKISIFDMPKVKKEDPDEPG------------PSQPSRKSGKKRSRSRSPAAK--KFSK
Drosophila
RLEKKFQPDT----VLGKSALAKRPAPEAEE---------------------------KVFKKPKSVAGDSQTR------
Arabidopsis
GGGVKQVATGK---ERGESSKLLFGDEENDKVERGEK-------------------------RKRSGDSGRSEK----ER
POMBE
---------------YESAQNNEDNKDGSSREQ--------------------------P--VLHEIDLSKKGN------
Cerevisiae
-------------------QEEQE--------------------------------------------------------
Human
258-KSALDEIMEIE-EEKKRTA------------------------RTDYWLQPEIIVKIITKKLGEK-YHKKKAIVKEVIDK
Mouse
KSALDEIMELE-EEKKRTA------------------------RTDAWLQPGIVVKIITKKLGEK-YHKKKGVVKEVIDR
CAEEL
KSALDEIKEMEERKKERKN------------------------RKDYWMREGIVVKVITKSLGSE-YYKAKGVVRKVVDD
Drosophila
-SVLDEIIKQEESKKERAN------------------------RKDYWLHKGIVVKFISKSMGEK-FFKQKAVVLDVIDR
Arabidopsis
RSALDELMKEEEKKKERMN------------------------RKDYWLFEGIIVKVMSKALAEKGYYKQKGVVKKVIDN
POMBE
PIQLNLSSSSDSHSAQNEFF----------------------------QTRNTPTFSFSSSSSQTSLKHKPKNVFAELNK
Cerevisiae
-VIAAELLKRQLNRAKRQT--------------------------------------------EKVYQPEMKSEISGD--
.
Human
312--YTAVVKMIDSG----DKLKLDQTHLETVIPAPGKRILVLNGGYRGNEGTLESINEKTFSATIVIETGPLKGRRVEGIQY
Mouse
-YTAVVKMTDSG----DRLKLDQTHLETVIPAPGKRVLVLNGGYRGNEGTLESINEKAFSATIVIETGPLKGRRVEGIQY
CAEEL
-YTAQVKLDD-G----TVVKLDQEHVETVIPSLGRQMMIVNGAYRGQEATLESIDEKRFSLRLKIASGPTRGRQID-VPY
Drosophila
-YQGKIKFLETG----EKLKVDQAHLETVIPALDKPVMVVNGAYRGSEALLRKLDERRYSVSVEILHGPLKGRIVDNVQY
Arabidopsis
-YVGEIKMLDSK----HVLRVDQKELETVLPQIGGMVKIVNGAYRGSNARLLGVDTEKFCAKVQIEKGVYDGRVIKSIEY
POMBE
---------------SRKKNNKDSLDQGQNVKRPRSAVEDIIAQETMRE-------KRRNIKL-----------------
Cerevisiae
----------------STLKRVQVTFHG-NGRVNKKKKKVPPRKDGIKFR------------------------------
:
Human
387-EDISKLA-----------------------------
Mouse EDISKLA----------------------------- CAEEL
EDASKLA----------------------------- Drosophila
EDISKLHGA--------------------------- Arabidopsis
EDICKLA----------------------------- POMBE ------------------------------------
Cerevisiae ------------------------------------
Figure 1.2 : Alignement de 7
séquences de KIN17 réalisé avec l'aide du logiciel
clustalw. Les séquences alignées sont relatives aux
espèces eucaryotes humaine (Human, 393 résidus), Mus
Musculus (Mouse, 391 réidus), Caenorhabditis elegans (CAEEL,
404 résidus), Drosophila Melanogaster (Drosophila, 390
résidus), Arabidopsis thaliana (Arabidopsis, 411 résidus),
Schizosaccharomyces pombe (POMBE, 304 résidus), et Saccharomyces
cerevisiae (Cerevisiae, 232 résidus). Les résidus sont
colorés en rouge lorsqu'ils sont conservés (sigle *), en
vert lorsqu'il sont fortement similaires (sigle :), et en bleu
lorsqu'ils sont faiblement similaires (sigle .). La numérotation est
relative à la séquence humaine.
L'utilisation des programmes SMART (Schultz et al.,
1998) et Pfam (Finn et al., 2006)
de reconnaissances de domaines suggère l'existence de 4
motifs dans la séquence primaire de
KIN17 humaine :
- Un motif « doigt de zinc » également
appelé C2H2 (résidus 28-50)
- Un domaine de repliement FF (résidus 50-150)
- Une séquence signale de localisation nucléaire
(région 239-256)
- Un motif KOW (335-373)
? Les motifs « doigts de zinc » sont des domaines de
liaison aux acides nucléiques (ADN et ARN). Ils se caractérisent
par la présence d'un ion zinc complexé entre 2 résidus
cystéine et 2 résidus histidine (Figure 1.3). Lorsqu'ils ne sont
pas retrouvés en plusieurs exemplaires dans une séquence, la
liaison à l'ADN n'est en général pas
spécifique d'une séquence nucléotidique donnée
(Böhm et al., 1997). La présence des 4 résidus
ultra conservés C28, C31, H44, et H50 (relatifs à la
séquence humaine) suggère fortement que la région
N-terminale de KIN17 comporte un motif C2H2 de la
levure jusqu'à l'homme. Cette hypothèse a été
confirmée par Mazin et al., qui ont montré que le «
doigt de zinc » potentiel
de la protéine KIN17 de souris était capable de
lier l'ADN de manière dépendante aux ions
Zn2+ (Mazin et al., 1994).






























Figure 1.3 : Représentation
schématique d'un motif « doigt de zinc »
? Entre les résidus 50 et 150, toutes les
protéines KIN17 possèdent une région, qui, chez la levure
Schizosaccharomyces pombe et le ver Caenorhabditis
elegans, est prédite comme adoptant un repliement de type FF par
le programme SMART. Les domaines de repliement FF sont des modules
de liaison à des peptides phosphorylés trouvés dans
de nombreuses protéines eucaryotes comme le facteur de
transcription CA150 (Allen et al., 2002). Ceci suggère que les
protéines KIN17 pourraient posséder un domaine de liaison
à un motif phosphorylé. Cependant, ce type de repliement n'est
pas prédit chez toutes les espèces qui ne contiennent pas tous
les résidus décrits comme importants pour la structure en 3
hélices des domaines FF. D'autre part, la surexpression de
plusieurs formes tronquées de KIN17 de souris chez E. coli
a montré que la région 71-281 de KIN17 (relative à la
séquence humaine) comportait un second domaine de liaison à l'ADN
(Mazin et al., 1994). Or, les modules FF ne sont pas des domaines de
liaison à l'ADN. Sur la base de ces résultats,
l'existence d'un domaine FF chez KIN17 apparaît donc
hypothétique.
? Enfin, les logiciels de détections de domaines ont mis
en évidence l'existence d'un domaine additionnel d'environ 100 acides
aminés contenant un motif appelé KOW (Kyrpides et
al.,
1996) dans la région C-terminale de KIN17 humaine absente
chez les eucaryotes inférieurs.
Ce module, également prédit dans les
séquences KIN17 de ver, de plante, et de mammifère,
est retrouvé dans une sous-famille des domaines
TUDOR (Selenko et al., 2001). Les domaines TUDOR sont présents dans
des protéines s'associant à l'ARN. Cependant, le rôle
biologique des domaines TUDOR à motif KOW demeure obscur. De
manière intéressante, les
chercheurs du LGR ont montré que la région
C-terminale de KIN17 contenant le module
KOW est impliquée dans la liaison à l'ARN
(Pinon-Lataillade et al., 2004). Ceci conforte la prédiction des
logiciels SMART et Pfam. Il est également à
noter que la région C-terminale de KIN17 n'apparaît pas
impliquée dans la liaison à l'ADN (Mazin et al., 1994).
3) Conclusions
Bien que plusieurs études aient montré
l'importance de KIN17 dans la réplication et la réponse
cellulaire aux dommages de l'ADN, les fonctions précises
et les partenaires biologiques de cette protéine nucléaire
demeurent à ce jour inconnus. Dans le cas de KIN17, l'utilisation de
logiciels bio-informatiques a permis de caractériser son
organisation en plusieurs domaines structuraux qui semblent
associés à différentes fonctions. Dans l'état
actuel des connaissances, la caractérisation structurale de KIN17
apparaît indispensable dans l'optique de progresser vers la
détermination de son rôle dans la régulation et la
maintenance
de l'ADN, de ses partenaires, et de ses modes d'action.
C'est pourquoi, le Laboratoire de Structure des Protéines du
CEA de Saclay (LSP) a entrepris une approche structurale par RMN et
cristallographie des rayons X qui a pour objectif de
résoudre la structure tridimensionnelle de la protéine
KIN17 humaine. En raison de difficultés rencontrées pour
surexprimer la protéine entière, cette caractérisation a
été abordée par domaine. Sur la base des analyses
bio-informatiques présentées précédemment, et
à partir des diagrammes de prédiction de structure
secondaire et de visualisation des amas de résidus
hydrophobes
(analyse HCA), trois domaines de la protéine KIN17 humaine
ont été sélectionnés :
- Domaine K1 : région 270-390 contenant le module
prédit KOW
- Domaine K2 : région 51-160 contenant le motif
prédit FF
- Domaine K3 : région 1-160 contenant le motif
prédit C2H2 et le domaine K2
En parallèle, une approche biochimique fonctionnelle a
également été initiée afin de rechercher les
partenaires biologiques de KIN17 et de chacun de ses 3 domaines structuraux.
Dans le cadre de ce projet, nous avons entrepris une
collaboration avec les chercheurs
du LSP afin de contribuer à améliorer la
connaissance du mode de fonctionnement de la protéine KIN17
humaine, en nous intéressant plus particulièrement au
domaine K2 correspondant à la région 51-160. La
résolution de la structure tridimensionnelle de ce
domaine a ainsi été envisagée par
Résonance Magnétique Nucléaire en solution. Cette
étude a
pour principal objectif d'émettre des hypothèses
sur les fonctions potentielles adoptées par le domaine K2 de la
protéine KIN17 humaine à partir de la connaissance de sa
structure. Des études fonctionnelles d'interaction,
réalisées en parallèle par les chercheurs du LSP
sur les domaines K2 et K3, permettront d'étayer ou d'infirmer nos
hypothèses structurales. A terme, l'interaction du domaine K2 avec un
partenaire biologique pourra être facilement
caractérisée
par RMN, qui est une méthode de choix pour
étudier les dynamiques moléculaires et les
interactions entre molécules.
Chapitre 2 : Production et analyses préliminaires
du domaine K2 de la protéine humaine KIN17
CHAPITRE 2
Production et analyses préliminaires
du domaine K2 de la protéine humaine KIN17
1) Préparation des échantillons pour
l'analyse RMN
1.1) Sélection et optimisation du système
d'expression
Le Laboratoire de Marquage des Protéines du CEA
de Saclay (LMP) a récemment élaboré un Programme de
Production et Marquage des Protéines (3PM) qui a pour objectif de
produire en grande quantité, chez la bactérie E.
coli, des protéines solubles, pures, et sous forme native en
vue de leur caractérisation structurale (Braud et al., 2005). La
stratégie mise
en oeuvre pour obtenir de tels résultats a
été présentée en détail dans le
chapitre 4 de la première partie de ce manuscrit,
consacrée à l'étude structurale de la
protéine PRODH humaine. Elle repose notamment sur un criblage
systématique de plusieurs conditions expérimentales. Toutes
les étapes relatives au clonage, au criblage des
conditions d'expression, à la production à grande échelle
en milieu non marqué, et à la purification du domaine K2 ont
été menées avec succès par les chercheurs du LMP
dans le cadre de cette plate-forme de production. Ainsi, les
quantités de protéine K2 soluble et pure obtenues
à l'issue du processus 3PM étaient suffisantes pour
envisager la préparation des échantillons pour l'analyse
structurale.
La stratégie d'attribution de l'ensemble des raies de
résonance RMN d'une protéine de
111 acides aminés comme K2 repose sur
l'enregistrement d'expériences hétéronucléaires
triple résonance (1H, 15N, 13C),
et nécessite la préparation d'échantillons
uniformément marqués 15N et 15N /
13C. En contrôlant de manière stricte les sources
d'azote et de carbone d'un milieu de culture, il est possible de produire des
protéines simplement ou doublement marquées de façon
uniforme avec des rendements de marquage(s) isotopique(s) proches de
100%. Le contrôle total du milieu impose
généralement des milieux pauvres (milieu minimum), dont le
plus fréquent est le M9 (Maniatis et al., 1982). Par comparaison avec le
LB, ce type de milieu est appauvri en métabolite, nécessite
l'adaptation de l'hôte bactérien, et entraîne par
conséquent une diminution de la croissance bactérienne, ainsi
qu'une baisse du rendement de production. Pour pallier ces
inconvénients, l'une des solutions consiste à enrichir le
milieu M9 avec des vitamines et des oligo-éléments qui
contiennent peu d'atomes
de carbone et d'azote (Jansson et al., 1996). D'autre
part, il existe des milieux riches contenant une réserve de
métabolites bio-disponibles et adaptés au(x)
marquage(s) isotopique(s) uniforme(s) (Reilly & Fairbrother, 1994). Ces
milieux sont pour le plus souvent
issus d'un hydrolysat de microorganismes
photosynthétiques, ce qui est le cas du milieu
Algone préparé au LMP à partir de cultures
de cyanobactéries Spirulina maxima doublement
marquées 15N / 13C de manière
uniforme.
La modification d'une condition expérimentale telle que le
milieu de culture peut nuire
à la qualité du profil d'expression
d'une protéine. Par conséquent, il est
nécessaire d'entreprendre une seconde étape d'optimisation de
l'expression de K2 en milieux marqués avant de préparer les
échantillons RMN. Celle-ci a été réalisée en
milieu minimum sur la base des meilleurs résultats obtenus
à l'issue du criblage des conditions d'expression en
microplaques, puis en milieu Algone.
1.1.1) Résultats du criblage des conditions
d'expression en microplaques
Le domaine K2 de la protéine KIN17 a été
exprimé dans 30 conditions expérimentales différentes : 5
partenaires de fusion (His, Gb1, ZZ, GST, et Trx), 3 souches
d'expression (BL21 Star, BL21 AI, et Rosetta DE3), et 2
températures (37°C et 20°C). Bien que les 3 souches
testées conduisent globalement à de bons taux d'expression
et de solubilité, les meilleurs résultats ont
été obtenus en souche Rosetta (DE3). Le tableau 2.1
présente les taux
de protéines soluble et insoluble obtenus dans cette
souche après analyse sur gel SDS-PAGE.
|
K2
|
His
|
Gb1
|
ZZ
|
GST
|
Trx
|
|
S
|
I
|
S
|
I
|
S
|
I
|
S
|
I
|
S
|
I
|
|
37°C
|
+
|
+
|
++(+)
|
++
|
++
|
+++
|
+
|
+++
|
+
|
+(+)
|
|
20°C
|
++
|
+++
|
+
|
+
|
+++
|
+
|
+++
|
++
|
++++
|
+(+)
|
Tableau 2.1 : Analyse de
l'expression et de la solubilité des protéines de fusion K2 en
souche
Rosetta. Après dépôt des fractions
soluble (S) et insoluble (I) sur gel SDS-PAGE, chaque bande de
surexpression a été analysée, et une valeur
semi-quantitative a été associée à son
intensité, de faible (+) à très forte (++++). Les
meilleures conditions sont mises en évidence par une coloration
orange.
Au vu de ces résultats, on constate que la nature du
partenaire de fusion combinée à la température
d'expression a un effet notable sur la solubilité des protéines
hétérologues K2. Ainsi, les meilleurs taux d'expression de
protéine soluble ont été obtenus à 20°C et
lorsque
K2 est en fusion avec les partenaires ZZ, GST, ou Trx. Au final,
l'association partenaire Trx,
souche Rosetta, et température de 20°C constitue la
meilleure condition d'expression à l'issue
du criblage à moyen débit proposé par le
programme 3PM.
1.1.2) Optimisation de l'expression en milieu minimum et
milieu riche
a) Optimisation en milieu minimum non
marqué
L'expression de K2 en milieu minimum M9 non-marqué a
été menée en volume de
100 mL dans des erlenmeyers de 1 L. Les 3 meilleures conditions
déterminées précédemment
ont été testées ; la protéine K2 a
été exprimée en fusion avec respectivement, GST, Trx, et
ZZ,
en souche Rosetta (DE3) cultivée à 20°C. Pour
chaque construction, les étapes principales du protocole d'expression
sont les suivantes :
La souche Rosetta (DE3) est transformée par le plasmide
d'expression d'intérêt, puis mise en culture en milieu LB
à 37°C. Après quelques heures, une préculture
en milieu minimum est ensemencée puis incubée à 37°C
sous agitation. La culture d'expression de 100 mL est inoculée avec
un volume de préculture, et l'expression de K2 est induite
à 20°C pendant 14 heures par ajout de 1mM d'IPTG lorsque
la DO600 atteint 1.2. Les culots bactériens sont repris et
lysés dans un tampon phosphate, et après centrifugation, les
fractions soluble et insoluble sont analysées sur gel SDS-PAGE (Figure
2.1).
GST
S I
Trx ZZ
S I S I
(kDa)
75
50

37
·
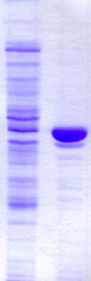
25
·
·
20
Figure 2.1 : Expression des
protéines de fusion GST-K2 (43.2 kDa), Trx-K2 (28.7 kDa), et
ZZ-K2 (33.3 kDa) en souche Rosetta cultivée
à 20°C en milieu minimum non marqué. Analyse SDS-PAGE des
fractions soluble (S) et insoluble (I) pour les 3 partenaires
testés. Les protéines de fusion sont mises en évidence
par des points rouges.
De manière intéressante, les profils
d'expression obtenus en milieu minimum
divergent significativement de ceux obtenus en milieu LB. La
différence la plus spectaculaire concerne la fusion GST qui conduit
à une expression de protéine très majoritairement
insoluble en milieu minimum. L'expression de K2 en fusion avec Trx
est tout à fait remarquable en milieu minimum, aussi bien dans
la fraction soluble, que dans la fraction insoluble. On retrouve ce
résultat pour le partenaire ZZ, bien que l'expression sous
forme soluble soit plus importante que sous forme insoluble.
Les fusions ZZ et Trx conduisant à des taux
d'expression de protéine soluble comparables, le choix du partenaire
a été guidé par d'autres considérations. Ainsi,
l'apparition d'un trouble persistant dans la fraction soluble de K2
fusionnée à Trx a révélé une propension
lente à l'agrégation de cette construction. De plus, en
anticipant sur la purification de la protéine
hétérologue, les résines d'affinité
spécifique à ZZ s'avèrent moins coûteuses que
celles spécifique au partenaire Trx. Par conséquent, nous
avons choisi de préparer les échantillons marqués
pour l'analyse RMN en produisant le domaine K2 en fusion avec le
partenaire ZZ en souche Rosetta (DE3) cultivée à 20°C.
b) Optimisation en milieu Algone doublement
marqué 15N / 13C
Bien que préparé au LMP, l'Algone est un milieu
riche très onéreux. Afin de réduire
les coûts de production de l'échantillon
doublement marqué 15N / 13C, ce milieu peut
être dilué avec de l'eau MilliQ. Toutefois et à l'image
des milieux pauvres, une diminution de la réserve métabolique
du milieu de culture, entraînée par une dilution, peut
avoir pour conséquence une modification du profil
d'expression de la protéine recombinante. L'optimisation de
l'expression de K2 en milieu Algone a ainsi pour objectif de déterminer
le milieu le plus dilué permettant d'obtenir le meilleur taux de
protéine soluble.
La production de K2 fusionnée à ZZ en souche
Rosetta (DE3) cultivée à 20°C a été
menée en volume de 30 mL dans des erlenmeyers de 300 mL. Trois dilutions
d'Algone 1/3,
1/2, et 2/3 ont été testées, ainsi
que le milieu Algone 100 %. Les résultats de cette optimisation
mettent en évidence que le profil d'expression de la protéine de
fusion ZZ-K2 en milieu Algone est quasiment identique à celui obtenu en
milieu minimum non marqué, quelle
que soit la dilution (Figure 2.2).
1/3
1/2
2/3
100%
S I S I
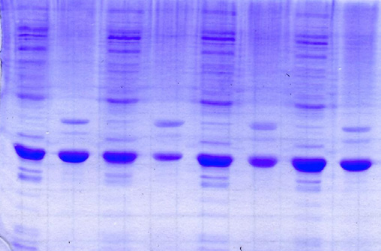
S I S I
ZZ-K2
Figure 2.2 : Optimisation de
l'expression de K2 en milieu Algone (fusion ZZ, souche Rosetta
cultivée à 20°C). Analyse SDS-PAGE des
fractions soluble (S) et insoluble (I) correspondant
à différentes dilutions du milieu Algone avec
de l'eau milliQ (1/3, 1/2, 2/3, et 100 % Algone).
Au vu des résultats de l'optimisation de
l'expression de K2 en milieu minimum et milieu riche, il a
été décidé de produire la protéine K2
doublement marquée 15N / 13C en milieu Algone
dilué au 1/3, et K2 simplement marqué 15N, en milieu
minimum.
1.2) Obtention de K2 simplement marquée
15N et doublement marquée 15N /
13C
1.2.1) Stratégie
générale
Cette stratégie a été
présentée en détail dans le chapitre 4 de la
première partie de ce manuscrit. Par conséquent, je me
contenterai de rappeler ses points essentiels.
Le plasmide d'expression de ZZ-K2 encode une protéine qui
contient, une étiquette de
6 résidus histidine (6xHis), le partenaire de fusion ZZ,
le site de reconnaissance TEV (sTEV),
et le domaine K2. La construction obtenue est de la forme
6xHis-ZZ-sTEV-K2 pour un poids moléculaire de 33 kDa. La
préparation de l'échantillon débute par la production de
plusieurs litres de protéine de fusion. Après lyse des
membranes bactériennes et extraction des protéines
cytosolubles, les surnageants sont déposés sur colonne
d'affinité spécifique au fragment protéique ZZ. Cette
étape permet, dans un premier temps, d'éliminer les
protéines endogènes de E. coli, puis de cliver le
partenaire ZZ par une coupure sur colonne avec la protéase
recombinante 6xHis-TEV (produite au laboratoire). Lors de l'élution de
la protéine
K2 clivée, le partenaire ZZ est retenu par la
résine d'affinité. Une seconde étape de
purification est alors réalisée sur
résine de nickel. Contrairement aux protéines
résiduelles
6xHis-ZZ-sTEV-K2 et 6xHis-ZZ, et à la
protéase 6xHis-TEV, la protéine K2 ne comporte
plus d'étiquette 6xHis après clivage de son
partenaire. Elle ne doit donc pas être retenue par la résine de
nickel et peut être séparée de la protéase
6XHis-TEV, ainsi que des 2 protéines résiduelles. Au
final, les échantillons RMN sont obtenus après
concentration sur cellule Amicon.
1.2.2) Production de la protéine de fusion en
milieu minimum et milieu Algone
La première étape de préparation des
échantillons de protéine K2, enrichie en isotopes
15N et 15N / 13C, a
consisté à produire la protéine de fusion dans 3
L de chaque milieu de culture marqué (milieu minimum marqué
15N, et milieu Algone doublement marqué 15N /
13C dilué au 1/3). Le protocole d'expression
résumé en Figure 2.3 a été
préalablement mis au
point en petit volume.
transformants
ensemencement avec plusieurs colonies
Préculture LB (37°C)
50 mL en erlenmeyer
+ Ampi 100 ug/mL
+ CAM 20 ug/mL
+ glucose 0.5 %
ensemencement pour
obtenir DO595 = 0.1
Culture milieu
marqué (20°C)
3 L en fernbach
+ Ampi 50 ug/mL
+ CAM 20 ug/mL
+ IPTG 1mM à DO595 = 1.2
ensemencement pour obtenir DO595 = 0.05
Préculture milieu
marqué (37°C)
100 mL en erlenmeyer
+ Ampi 50 ug/mL
+ CAM 20 ug/mL
Figure 2.3 : Protocole de
surexpression de K2 en milieu marqué (milieu minimum
marqué
15N, ou milieu Algone doublement
marqué 15N / 13C dilué au 1/3).
La souche Rosetta transformée par le vecteur
d'expression encodant la protéine de fusion est étalée
sur boîte LB-agar contenant 100 ug/mL d'ampicilline (Ampi) et 20 ug/mL de
chloramphénicol (CAM), puis incubée à 37°C
pendant 14 heures. Plusieurs colonies sont mises en préculture
dans un erlenmeyer de 500 mL contenant 50 mL de LB, 100 ug/mL
d'Ampi, 20 ug/mL de CAM, et 0.5 % de glucose. L'ensemble est incubé
à 37°C pendant 3 heures sous une agitation de 250 rpm. 100 mL de
milieu marqué contenant 50 ug/mL d'Ampi
et 20 ug/mL de CAM sont introduits dans un erlenmeyer de 1 L,
puis ensemencés avec un
volume de préculture LB de manière à obtenir
une DO initiale à 600 nm égale à 0.1. Cette
deuxième préculture est ensuite incubée à 37°C
sous agitation (250 rpm) pendant 6 heures.
3 L de milieu marqué contenant 50 ug/mL d'Ampi et 20 ug/mL
de CAM sont alors répartis dans 3 fernbachs de 1 L,
préchauffés à 37°C, puis ensemencés pour
obtenir une DO initiale à
595 nm égale à 0.05. Les cultures d'expression sont
ensuite placées à 37°C sous une agitation
de 250 rpm. L'expression de la protéine de fusion K2
est induite par ajout de 1mM d'IPTG dans les milieux de culture lorsque la DO
mesurée à 595 nm atteint 1.2. Les fernbachs sont ensuite
incubés à 20°C sous une agitation de 250 rpm pendant 14
heures. A l'issu de ce délai,
la croissance bactérienne est stoppée en
introduisant les fernbachs de culture dans la glace.
La lyse des cellules procaryotes et l'extraction des
protéines ont été menées de la manière
suivante. Les bactéries sont récoltées par centrifugation
à 2830xg pendant 20 minutes
à 4°C. Après élimination des
surnageants, les culots bactériens sont soumis à un cycle
de congélation-décongélation dans l'azote liquide. Ils
sont ensuite resuspendus dans un tampon Tris contenant, 100 mM de Tris-HCl
(pH=8.0), 150 mM de NaCl, 5 % de glycérol, 1 mM d'EDTA, et 1 mM de
PMSF. La rupture des membranes bactériennes est
réalisée par lyse mécanique en utilisant une presse
d'Eaton. 10 mM de MgCl2, 10 mM de MgSO4, et
0.5 uL/mL de benzonase sont ensuite introduits dans les
lysats. Les fractions soluble et
insoluble sont finalement séparées par
centrifugation à 40000xg pendant 30 minutes à 4°C.
L'expression soluble de K2 en fernbach a
été contrôlée sur gel SDS-PAGE pour chacune
des 3 cultures contenant 1 L de milieu marqué, et pour chaque
milieu de culture. Comme le montre la Figure 2.4, les niveaux d'expression
obtenus sont très satisfaisants en milieu minimum 15N et en
milieu Algone 15N / 13C. Ces profils d'expression sont
tout à fait comparables à ceux obtenus lors des tests
d'optimisation.
(kDa)
75
50
37
A B C
(kDa)
75
50
37
A B C
ZZ-K2 25
25
20 20
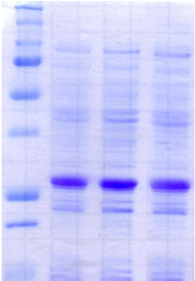
K2 15N Milieu Minimum
ZZ-K2
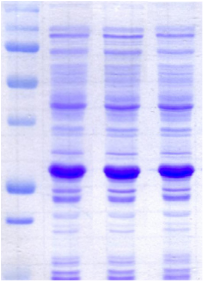
K2 15N / 13C Milieu
Algone
Figure 2.4 : Contrôle de
la production de la protéine de fusion K2 sous forme soluble
en
milieu minimum marqué 15N et en milieu
Algone 15N / 13C pour chaque litre de culture en
fernbach A, B, et C (analyse SDS-PAGE).
1.2.3) Purification de la protéine de fusion et
clivage de son partenaire
La purification de la construction 6xHis-ZZ-sTEV-K2
(ZZ-K2) a été entreprise par chromatographie
d'affinité sur résine IgG Sepharose 6 Fast Flow
(Amersham). Cette résine d'anticorps humains IgG greffés lie
de manière spécifique les fragments protéiques de type ZZ.
La colonne est préalablement équilibrée avec la solution
tampon Tris Saline Tween (TST) contenant, 50 mM de Tris (pH=7.5), 150 mM de
NaCl, 0.05 % de Tween 20, 1 mM de PMSF,
et 0.2 mM d'EDTA. Après injection des surnageants de lyse
bactérienne, la colonne est rincée plusieurs fois avec du tampon
d'équilibration TST jusqu'à retour de l'absorbance mesurée
à
280 nm à la ligne de base. La résine est alors
resuspendue dans du tampon TST et 2.6 mL de protéase recombinante TEV
à 1.8 mg/mL sont introduits dans la colonne. Après une nuit sous
agitation à 4°C, la solution de clivage contenue dans la
colonne est éluée avec du TST en fractions de 30 mL.
L'élution des protéines immobilisées à la
résine est menée par introduction de plusieurs volumes
d'acide acétique 0.5 M (pH=3.4). Toutes les fractions
d'élution sont déposées sur gel
d'électrophorèse SDS-PAGE et quantifiées par mesure
de l'absorbance à 280 nm sur un spectromètre UV. Le pH
des fractions d'acide acétique est préalablement
neutralisé avant analyses.
L'analyse SDS-PAGE des fractions de clivage fait
apparaître une bande de forte intensité à une masse
apparente de 13 kDa (Figure 2.5). Cette masse correspond au poids
moléculaire du domaine K2 (13.6 kDa), ce qui indique que
la protéine de fusion a été clivée
par la protéase TEV. Le tampon acide acétique
n'étant pas très adapté à la migration sur gel
d'électrophorèse, les bandes protéiques des fractions
d'élution apparaissent floues et peu intenses. Il est donc
difficile de réaliser une observation fine des protéines
présentes dans ces fractions et de calculer un rendement de coupure. On
peut toutefois observer des nuances de coloration à des masses
apparentes qui correspondent au partenaire ZZ clivé (20 kDa), et
à la protéine de fusion (33 kDa), ce qui démontre que la
coupure sur colonne n'est pas totale.
(kDa)
SN A B EL
(kDa)
SN A B EL
50
50
37
37
ZZ-K2



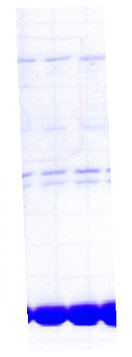
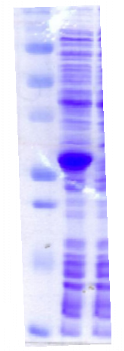

25 25
20 20
ZZ
15 15
K2
10
10
ZZ-K2
ZZ
K2
K2 15N Milieu Minimum
K2 15N / 13C Milieu
Algone
Figure 2.5 : Purification de la
protéine de fusion K2 sur IgG Sepharose et coupure sur
colonne du partenaire ZZ. Analyse SDS-PAGE du surnageant de
lyse bactérienne (SN), des fractions de clivage (A et B), et des
fractions d'élution à l'acide acétique (EL).
Au final, les rendements obtenus sont très
satisfaisants et atteignent, 30 mg/L en milieu minimum, et 25 mg/L en
milieu Algone. A l'issu de cette première étape, le degré
de pureté de K2 est estimé à 90% bien que
quelques bandes contaminantes de faible intensité soient observables
dans les fractions de clivage.
1.2.4) Purification du domaine K2
La seconde étape de purification de la protéine K2
a été réalisée par chromatographie
de pseudo-affinité sur colonne Ni-NTA Agarose
chargée en ions Ni2+ (Qiagen). La colonne est
équilibrée avec un tampon phosphate (50 mM, pH=7.2) contenant,
300 mM de NaCl, 4mM de
â-mercapto-éthanol, et 1mM de PMSF. Les
échantillons de K2 en solution dans du TST sont
directement chargés dans la colonne, et plusieurs
volumes de tampon phosphate sont
introduits jusqu'à retour de l'absorbance mesurée
à 280 nm à la ligne de base. L'élution est
menée par gradient linéaire d'imidazole avec du
tampon phosphate contenant de 10 à 500 mM d'imidazole. Les fractions
d'élution sont analysées sur gel SDS-PAGE (Figure 2.6) et
quantifiées par mesure de l'absorbance à 280 nm.
concentration en
imidazole (mM)
30
0
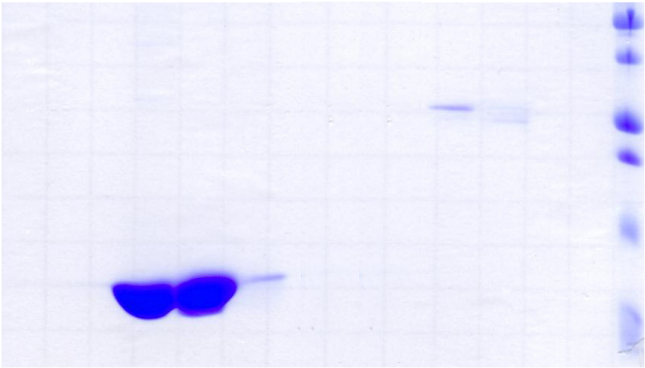
100
200
50
37



100
40
20
0
200


50
37
25 25
20 20
15
15
10 10
K2 15N Milieu Minimum
K2 15N / 13C Milieu
Algone
Figure 2.6 : Purification de K2
sur Ni-NTA Agarose. Analyse SDS-PAGE des fractions
d'élution par gradient linéaire d'imidazole (de
0 à 200 mM). La présence de « traînées »
en dessous de la bande correspondant à la protéine K2 sur le gel
de droite est due à un problème
de migration.
L'analyse SDS-PAGE des fractions d'élution fait
apparaître que la protéine K2 est retenue par la
résine de nickel jusqu'à des concentrations d'imidazole de
l'ordre de 30 mM. Sur les 4 histidines que compte K2, les 3
résidus H52, H55, et H57 sont proches dans la séquence
primaire. Par conséquent, ce type de fixation non spécifique,
à des concentrations d'imidazole relativement faibles, s'explique
probablement par la présence d'un amas, contenant ces 3
histidines, situé en surface de K2. Deux bandes
protéiques, de masse apparente autour de 30 kDa, sont observables dans
les fractions contenant une concentration d'imidazole d'environ 150 mM, et
correspondent à la protéase 6xHis-TEV (27kDa), et à la
protéine de fusion 6xHis-ZZ-sTEV-K2 (33 kDa).
Les rendements de l'expression du domaine K2 obtenus à
l'issu de cette seconde étape
de purification sont très satisfaisants car peu de
protéine a été perdue (Tableau 2.2). De plus,
aucune bande contaminante n'est clairement discernable sur
l'analyse SDS-PAGE des
fractions de K2. Par conséquent, nous pouvons
estimer avoir préparé des échantillons simplement et
doublement marqués avec un degré de pureté
supérieur à 95%.
|
Milieu
|
Milieu Minimum 15N
|
Milieu Algone 15N / 13C
|
|
A) Rendement IgG
|
30 mg/L
|
25 mg/L
|
|
B) Rendement final
|
26 mg/L
|
22 mg/L
|
Tableau 2.2 : Rendements de
l'expression de K2, après purification sur IgG Sepharose
et
coupure sur colonne du partenaire (A), et à l'issu de
la 2nde purification sur résine Ni2+ (B).
1.2.5) Préparation du tube RMN
La dernière étape de préparation de
l'échantillon pour l'analyse RMN a consisté à
concentrer la protéine à une valeur proche de 1 mM.
L'ultrafiltration de K2 a été menée sur cellule Amicon de
10 mL équipée d'une membrane YM3 avec un seuil de coupure de 3
kDa. Afin de réduire la concentration en NaCl et d'éliminer
l'imidazole, plusieurs étapes de lavage- concentration ont
précédé la concentration finale de K2. A l'issue
de l'ultrafiltration, la protéine est en solution dans un tampon
phosphate (50 mM, pH=7.0) contenant, 150mM de NaCl, 1 mM d'EDTA, 1 mM de
TCEP, et 1 mM de PMSF. L'échantillon RMN est alors obtenu en
ajoutant 10% de D2O, 2 mM de TSP, 2 mM de cocktail d'inhibiteurs (SIGMA), et
0.01% de NaN3. 600 uL de cette préparation
ont été introduits dans un tube RMN de 5 mm de
diamètre placé ensuite sous atmosphère
d'argon.
2) Caractérisations préliminaires
2.1) Caractérisation de la séquence
primaire et contrôle du marquage
Avant de débuter l'analyse structurale de K2 par RMN, nous
avons voulu nous assurer
de la fiabilité et de l'efficacité de la
méthode employée pour obtenir les échantillons de
protéine. La séquence primaire des protéines K2
simplement et doublement marquée a été
contrôlée par spectrométrie de masse ITD-MS (Ion Trap
Detector-Mass Spectroscopy) sous ionisation par ESI (ElectroSpray
Ionization).
Après concentration sur cellule Amicon, 2 uL d'un
échantillon de K2 ont été prélevés
et introduits dans une solution contenant, 100 uL d'H2O, 100
uL d'acétonitrile, et de l'acide formique (0.25% pour K2 15N,
et 0.75% pour K2 15N / 13C). 10 uL de ce mélange
contenant environ 15 ng/uL de K2 ont ensuite été injectés
dans la source ESI, puis analysés avec un enregistrement de 20
accumulations. Les spectres obtenus sont présentés en Figure 2.7
et les
résultats sont regroupés dans le Tableau 2.3.
15N
9+
1538.42
15N /
13C
13+
1107.09
14+
1028.12
12+
1199.32
10+
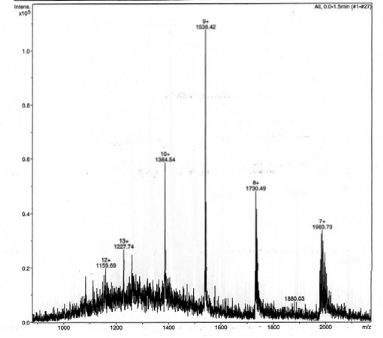
1384.54
8+
1730.42
11+
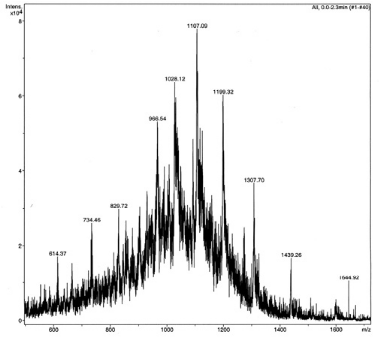
1307.70
7+
1978.73
10+
1439.26
Figure 2.7 : Analyse ESI-ITD-MS des
échantillons de K2 15N et K2 15N /
13C.
Le spectre de masse de K2 simplement marqué
15N fait apparaître une enveloppe d'ions
multichargés centrée autour d'un ion nonachargé. La
déconvolution de cette enveloppe permet de conclure à la
présence d'une forme unique de masse expérimentale 13836 #177; 3
Da. Cette masse correspond tout à fait à celle attendue
et indique que la protéine simplement marquée 15N
a été produite avec un enrichissement isotopique total ou
quasi-total.
Sur le spectre de l'échantillon doublement marqué,
on observe la présence d'un massif
isotopique pour chaque multichargé. L'apparition de ces
amas traduit l'existence de plusieurs formes isotopiques de K2, et
suggère que le double marquage n'est pas total. La
déconvolution automatique d'un tel spectre n'étant pas
possible, nous avons réalisé une déconvolution
manuelle de chaque pic majoritaire correspondant aux 4 ions multichargés
les plus intenses. Les résultats obtenus indiquent que la forme
isotopique prédominante possède une masse expérimentale de
14379 #177; 3 Da pour une masse attendue de 14445 Da. Le taux d'enrichissement
isotopique de l'échantillon 15N / 13C peut donc
être estimé à 92%.
|
Echantillon
|
masse théorique [Da]
|
masse expérimentale
(déconvolution) [Da]
|
taux d'enrichissement
|
|
K2 15N
|
13838,9
|
13836 #177; 3
|
~ 100 %
|
|
K2 15N / 13C
|
14445,3
|
14379 #177; 3
|
~ 92 %
|
Tableau 2.3 :
Caractérisation de la production des échantillons de K2
simplement et
doublement marqués par analyse ESI-ITD-MS. Les
masses théoriques sont indiquées en considérant un
enrichissement isotopique de 100%.
2.2) Caractérisation de l'état
oligomérique
Une fois le poids moléculaire du domaine
protéique vérifié par spectroscopie de masse, nous
avons entrepris une analyse par chromatographie de gel filtration afin
de caractériser l'état oligomérique de K2 en
solution. Cette technique chromatographique d'exclusion permet de
séparer des molécules en fonction de leur poids
moléculaire, et cela en conditions non dénaturantes en
choisissant un tampon d'équilibration adapté.
La colonne de filtration sur gel que nous avons utilisée
est une colonne Superdex75
HR analytique (Pharmacia) de 24 mL dont la gamme de
résolution se situe entre 70 et 3 kDa.
200 uL d'un échantillon concentré de
protéine doublement marquée 15N / 13C (soit
environ
300 ug) ont été introduits dans cette
colonne préalablement équilibrée avec une solution
tampon de PBS (Phosphate Buffer Saline : 150 mM NaCl, 10 mM phosphate, 2.5 mM
KCl,
pH 7.4). L'élution des protéines a
été détectée par suivi de l'absorbance à 280
nm. En amont
de l'analyse de K2, la Superdex75 a été
étalonnée dans les mêmes conditions avec un kit de
calibration contenant un mélange de protéines standard.
Le chromatogramme de l'analyse de K2 doublement
marquée fait apparaître un pic
ultra majoritaire à un volume d'élution de
12.22 mL (Figure 2.8A). En se basant sur les volumes de
rétention des protéines standard, ce volume correspond
à un poids moléculaire d'environ 18 kDa pour une masse
théorique monomérique attendue de 14.5 kDa. Il semble donc que
la structure quaternaire du domaine K2 soit monodisperse et
sous forme monomérique dans ces conditions d'analyse très
proches des conditions RMN. Ce résultat est cependant nuancé
par l'apparition d'un petit pic à 10.39 mL (~38 kDa) qui
pourrait correspondre à une forme dimérique très
minoritaire, ou à une impureté non observable sur le
gel SDS-PAGE (Figure 2.8B). Par ailleurs, la chromatographie
de gel filtration est certes une technique simple et rapide, mais
elle ne fournit qu'une indication de l'organisation quaternaire
d'une protéine dans un tampon donné. Ce résultat doit donc
être considéré avec précaution ou
confirmé par des études plus précises de
diffusion de la lumière ou
d'ultracentrifugation analytique.
A) 12.22 B)
(kDa)
50
37
25
20
15
10
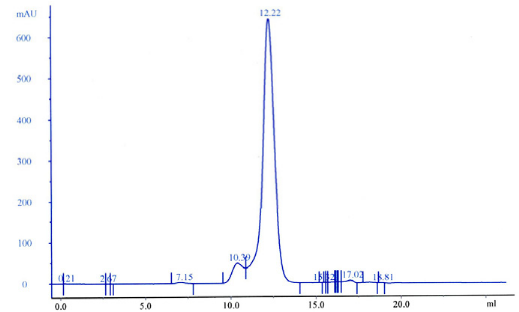
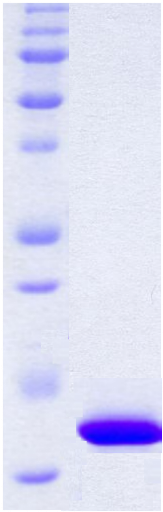

10.39
Figure 2.8 : Caractérisation
de la structure quaternaire de K2 15N / 13C. A)
chromatogramme
de l'analyse par gel filtration en conditions non
dénaturantes. B) analyse SDS-PAGE du même échantillon
(conditions dénaturantes).
2.3) Caractérisation de la structure secondaire et
tertiaire
Le dichroïsme circulaire (DC) est une méthode simple
et sensible qui permet d'évaluer
le niveau de structuration d'un polypeptide. La
technique repose sur les propriétés spectroscopiques des
molécules chirales qui absorbent les composantes polarisées
circulaires droite et gauche de la lumière de manière
inégale. Dans le cas des protéines, le phénomène de
dichroïsme circulaire est essentiellement dû à leur
structure secondaire dans l'UV lointain
(région dominée par l'absorption des liaisons
peptidiques), et à leur structure tertiaire dans
l'UV proche (région dominée par l'absorption des
cycles aromatiques). Aussi, chaque type de conformation présente un
profil d'absorption qui lui est propre (Yang et al., 1986).
Selon ce principe, l'analyse des spectres de DC permet
de distinguer sans aucune ambiguïté une protéine
repliée d'une protéine non structurée. C'est dans
cette optique que nous avons entrepris l'analyse du domaine K2 par
dichroïsme circulaire. Les spectres de DC
ont été enregistrés sur un
échantillon de K2 non marquée afin de vérifier la
structuration de la protéine avant de la produire en grande
quantité en milieu minimum et Algone.
L'analyse a été menée à 25°C en
solvant H2O contenant 50 mM de tampon phosphate
à pH 7.0, et 200 mM de NaCl. Le spectre enregistré
dans l'UV lointain présente un minimum
à 208 nm spécifique de la conformation
hélicoïdale (Figure 2.9). Cependant, aucun autre minimum
n'est clairement discernable dans la région autour de
222 nm. Ce profil d'absorption est caractéristique d'une
protéine structurée en partie en hélice á et en
feuillet â (Venyaminov & Yang, 1996). Par ailleurs, le signal
d'un spectre DC enregistré dans l'UV proche est
généralement très faible en absence de conformation
ordonnée. Dans le cas de la protéine K2, deux minima intenses
apparaissent à 279 et 288 nm et correspondent à
l'absorption de cycles aromatiques de tyrosine et de tryptophane d'une
protéine adoptant une structure tertiaire stable.
30
20
10
0
-10
èx10-3
[degré.cm2.dmole-1]
-20
4


UV lointain UV proche
2
0
-2
-4
-6
-8
-30
200 210 220 230 240
Longueur d'onde ë [nm]
-10
260 280 300 320 340
Figure 2.9 : Spectres de
dichroïsme circulaire enregistrés sur un échantillon de
protéine K2
non marquée solubilisée dans un tampon
phosphate à pH 7.0 et à 25°C.
2.4) Etude préliminaire par Résonance
Magnétique Nucléaire
2.4.1) Enregistrement des premières
expériences
Dans l'optique de confirmer les résultats obtenus par
dichroïsme circulaire, nous avons débuté l'étude
RMN par l'enregistrement des spectres 1D 1H et 2D HSQC
15N-1H sur un échantillon de protéine
simplement marquée 15N. Ces expériences ont
été réalisées sur un spectromètre 600
MHz équipé d'une cryosonde TXI triple
résonance dans les conditions initiales suivantes : une
température de 20°C, un tampon phosphate de pH 7.0, et
une concentration de protéine d'environ 0.7 mM.
Une protéine structurée présente une
signature très différente d'une protéine peu ou
pas structurée sur un spectre HSQC
15N-1H du fait de la variation de l'environnement
chimique induite par les structures secondaire et tertiaire. La dispersion
globale des pics de corrélation sur le spectre HSQC de K2
confirme que la protéine est repliée (Figure 2.10). Ainsi,
les pics situés à gauche de la région centrale, à
un déplacement chimique 1H supérieur à
9 ppm, sont généralement
caractéristiques d'une structuration en feuillet â. D'autre
part, le spectre 1D 1H fait apparaître des raies de
résonance à un déplacement chimique négatif dans
la région des protons aliphatiques. Ces observations
sont typiques d'une protéine repliée
adoptant des éléments de structure secondaire et
tertiaire.













ppm
104
106
108
110
112
114
116
118
120
122
124
126
128
130
10.5
10.0
9.5
9.0
8.5
8.0
7.5
7.0
6.5
ppm
Figure 2.10 : Spectre 2D HSQC
15N-1H de K2 simplement marqué 15N
(tampon phosphate, pH=7.0) enregistré sur un spectromètre 600 MHZ
et à 20°C.
Le nombre de pics de corrélation présents sur ce
spectre est difficile à comptabiliser en
raison d'une superposition importante dans la région
centrale. Il est d'autant plus difficile à déterminer que la
largeur des raies de résonance est importante à cette
température. On peut toutefois estimer ce nombre proche de 100
sachant que le nombre de groupements amides attendu s'élève
à 107 (111 résidus dont 3 prolines, et en considérant que
le groupement amide NH2 du premier résidu est
généralement non observable).
Afin de déterminer si les conditions initiales
choisies étaient adaptées à une étude
complète par RMN, nous avons enregistré une
première série de 4 expériences 3D
hétéronucléaires sur un échantillon
15N / 13C de K2. Il s'agit des expériences
3D HNCO, HNCA, CBCA(CO)NH, et CBCANH, qui permettent l'attribution de la
chaîne principale et
des Câ. Pour chaque expérience, le nombre de pics
qui apparaissent dans les spectres a été comptabilisé et
figure dans le tableau 2.4. Bien que la HNCO soit l'expérience
3D la plus sensible, dans ces conditions d'analyse, 16
déplacements chimiques de carbonyle sont manquants. Les
expériences HNCA et CBCA(CO)NH ne contiennent qu'environ 80% de
données attendues. CBCANH, la moins sensible des 4 expériences,
mais néanmoins majeure dans la stratégie d'attribution, ne
contient qu'un peu plus du tiers de données attendues. Au final, la
quantité d'information recueillie sur les spectres semble insuffisante
pour envisager une attribution quasi-complète du squelette de K2. Par
conséquent, nous avons conclu que les conditions initiales d'analyse
(pH=7.0 et T=20°C) ne sont pas les plus adaptées pour attribuer
l'ensemble des résonances 1H, 15N,
et 13C de la protéine K2.
Expériences
nombre de pics
attendus
nombre de pics
%
observés
HNCO 108 92 85 %
HNCA 216 174 80 %
CBCA(CO)NH 211 162 77 %
CBCANH 420 157 37 %
Tableau 2.4 : Analyse des
spectres HNCO, HNCA, CBCA(CO)NH, et CBCANH de K2
doublement marquée 15N / 13C
(pH=7,0) enregistrés sur un spectromètre 600 MHZ et à
20°C.
2.4.2) Optimisation des conditions de l'analyse par
RMN
A ce stade de l'étude, une optimisation des
conditions de l'analyse par RMN est apparue indispensable avant de
débuter l'analyse d'une longue série d'expériences
pouvant s'échelonner sur plusieurs semaines. Les deux principaux
paramètres qui influent sur la qualité et la quantité
de signal des spectres RMN sont le pH et la température.
L'absence d'une dizaine de corrélations de groupement
amide sur le spectre HSQC de
K2 peut être expliqué par un échange rapide
de protons amides accessibles avec les protons de l'eau. La plupart des
expériences 3D hétéronucléaires utilisées
pour l'attribution reposent sur
la corrélation d'un ou plusieurs atomes avec les
noyaux 1H et 15N amides. Par conséquent,
l'échange rapide des protons amides avec le solvant
affecte également la quantité de signal
sur ces spectres. Une diminution du pH de l'échantillon
permet de réduire la vitesse de cet échange (Figure 2.11).
Cette vitesse est minimum entre pH 3 et 4. Cependant, un pH trop
acide peut déstabiliser le repliement et conduire à une
déstructuration partielle ou totale de la protéine.
L'optimisation de ce paramètre a donc pour objectif de déterminer
la valeur de pH à laquelle l'information enregistrée est maximum
et le risque de dénaturation minimum.
600 ns
60 us
Tk 6 ms
H
0.6 s
1 min
10 min
N
0 2 4 6 8 10
pH
Figure 2.11 : Influence du pH
sur l'échange des protons amides accessibles au solvant. Tk
représente le temps moyen d'échange de proton amide labile de
polypeptide dans un solvant H2O et à 25°C (d'après
Wüthrich, 1986).
Le spectre HSQC de K2 enregistré à 20°C
présente une région centrale encombrée et peu
résolue. La cause de ce manque de résolution est notamment
imputable à la largeur de raie des pics de corrélation. Une
augmentation de la température permet de réduire la
viscosité
de l'eau, et de raccourcir le temps de réorientation
globale ôc de la macromolécule. Ceci se
traduit par un affinement des raies, et donc par une
amélioration de la résolution. De plus,
l'augmentation de l'agitation brownienne peut influer sur la
fréquence de résonance propre de certains noyaux et permettre
l'éclatement d'un massif. Cependant, les échantillons de
protéine sont généralement moins stables à
haute température car la tendance à la
dénaturation, et l'activité des protéases
résiduelles issues de E. coli, sont alors plus
prononcées. Comme pour
le pH, le choix de la température doit donc
résulter d'un compromis entre l'amélioration de la
résolution spectrale et le risque associé de
dénaturation.
L'influence du pH et de la température a
été évaluée sur un échantillon de K2
simplement marqué 15N concentré à 0.8 mM.
Une série d'expériences HSQC 15N-1H a
été entreprise sur un spectromètre 500 MHz dans 6
conditions expérimentales différentes. Chaque spectre a
été enregistré et traité de la même
façon et le nombre de pics (hors groupements NH
ou NH2 de chaîne latérale) a été
comptabilisé pour chaque condition testée (Tableau 2.5).
|
Condition
|
pH=7.0
|
pH=6.5
|
pH=6.0
|
|
T=25°C
|
T=30°C
|
T=25°C
|
T=30°C
|
T=25°C
|
T=30°C
|
|
nb de pics
|
89
|
98
|
101
|
103
|
110
|
110
|
Tableau 2.5 : Analyse des spectres
2D HSQC 15N-1H de K2 simplement marquée
enregistrés
sur un spectromètre 500 MHz dans différentes
conditions de température et de pH.
Tous les spectres HSQC enregistrés présentent le
même profil, similaire à celui obtenu dans les conditions
initiales. Par conséquent, la protéine K2 conserve un repliement
stable aux différents pH et températures testés.
L'effet du pH sur la vitesse d'échange des protons amides
de K2 est clairement visible
sur les spectres HSQC. On observe bien une augmentation du
nombre de résonances avec la diminution du pH. Jusqu'à 110
pics peuvent être comptabilisés à pH 6.0 pour un
nombre attendu de 107. La figure 2.12 illustre l'influence de ce
paramètre sur la région gauche du spectre qui fait
apparaître de nouvelles taches de corrélation à pH plus
acide. D'autre part, nous avons constaté que la diminution du pH permet
d'accroître l'intensité de la majorité des raies de
résonance de K2. Ce gain de signal est particulièrement
intéressant dans l'optique de
recueillir davantage d'information sur les spectres
3D hétéronucléaires. L'effet de la



































température est également visible sur l'allure
générale des spectres. La résolution de la région
centrale croît avec l'augmentation de la température, ce qui
permet de mieux distinguer les pics de corrélation.
pH=6.0 pH=6.5 pH=7.0
T=25°C
114
116
118
120
122
T=30°C
124
126
9.3
9.0
8.7
Figure 2.12 : Extrait de spectres
2D HSQC 15N-1H de K2 simplement marquée
15N enregistrés
sur un spectromètre 500 MHz dans différentes
conditions de température et de pH.
Finalement, à la vue de l'ensemble de ces
résultats, nous avons retenu comme conditions
expérimentales, une température de
30°C, et un pH de 6.0 pour mener
l'étude structurale de K2 par RMN.
3) Conclusions
La stratégie de surexpression du domaine K2
dans le cadre d'une facilité de production à moyen
débit s'est avérée très fructueuse. En effet, le
criblage systématique de plusieurs conditions d'expression en amont
de la production à grande échelle a permis d'augmenter les
chances de déterminer une condition qui réponde aux exigences
inhérentes à l'étude par RMN. Ainsi, nous avons
préparé une quantité suffisante de protéine
soluble simplement marquée 15N, et doublement
marquée 15N / 13C, pour envisager une
attribution complète des noyaux 1H, 15N, et
13C du domaine K2 (3 tubes simplement marqués 15N
à ~0.8 mM, et 3 tubes doublement marqués 15N /
13C à ~ 0.7 mM). De plus, les différentes analyses
qui ont été menées montrent que la
protéine est pure, stable, et structurée. D'un point de vue
matériel, l'optimisation de l'expression en milieux marqués,
associée aux choix portés sur les méthodes de
purification, ont permis de réduire le coût de production
des échantillons, qui reste cependant très onéreux.
Au cours de cette étude préliminaire,
nous avons également caractérisé une organisation
monomérique du domaine K2 dans des conditions chimiques proches
de l'échantillon RMN. Et enfin, nous avons optimisé les
conditions de l'analyse RMN avant d'aborder une caractérisation
structurale complète par RMN et modélisation moléculaire
qui
fait l'objet du prochain chapitre.
Chapitre 3 : Stratégie d'étude par RMN et
Modélisation Moléculaire du domaine K2
CHAPITRE 3
Stratégie d'étude par RMN et
Modélisation
Moléculaire du domaine K2
La radiocristallographie et la Résonance Magnétique
Nucléaire sont à ce jour les deux
techniques qui permettent d'obtenir la structure 3D
d'une protéine à l'échelle atomique. Comparée
à la RMN, les avantages principaux de la radiocristallographie sont
d'une part, la précision de l'information obtenue et d'autre part,
l'accès à des masses moléculaires élevées
(> 200 kDa). Il en découle que la grande
majorité des structures publiées sont résolues par cette
technique moins restreinte, et souvent moins coûteuse en terme de temps
d'interprétation des données. La spectroscopie de RMN n'est
donc, à priori, pas la méthode de choix pour
déterminer la structure tridimensionnelle d'une protéine.
Cependant, elle est le seul recours lorsque l'obtention d'un
monocristal ou la détermination des phases est impossible. Sa
principale qualité se situe dans l'étude des dynamiques
moléculaires et d'interactions entre les molécules, étape
clef de l'exploration des relations structure activité. Ces
quinze dernières années ont fait l'objet d'importants
développements techniques et méthodologiques qui rendent
la spectroscopie de RMN plus efficace dans la résolution
de structures macromoléculaires. Ces progrès ont notamment
visé à améliorer la résolution et la
sensibilité de la technique.
La fréquence de résonance du proton, qui traduit
l'intensité des aimants supraconducteurs, peut atteindre à
présent jusqu'à 950 MHz. L'augmentation de résolution
qui
en découle a été accompagnée par
l'apparition de sondes de mesure, à une température proche
de celle de l'hélium liquide (cryosondes), qui offrent un
gain de sensibilité d'un facteur 2 à 4
comparée aux sondes classiques. Ces grandes
avancées technologiques n'auraient cependant
pas été suffisantes sans le développement,
de séquences RMN 3D et 4D (Marion et al., 1989 ; Clore & Gronenborn,
1991), et de méthodes de marquage(s) isotopique(s) des noyaux
15N, 13C,
et 2D (Redfield et al., 1989 ; Xu et
al., 1999), qui permettent l'étude structurale de
biomolécules de taille de plus en plus importante.
Parmi ces progrès méthodologiques, figure notamment la
spectroscopie TROSY (Transverse Relaxation Optimized spectroscopY)
(Pervushin et al., 1997) utilisée à hauts champs pour
des protéines de masse supérieure à
20 kDa, et dont la combinaison à un marquage au
deutérium 2D, apporte un gain considérable
en résolution et en sensibilité. En amont du calcul
de la structure, l'émergence de programmes d'attribution automatique ou
semi-automatique, des résonances (Zimmerman et al., 1997), ou
des pics nOe (Nilges et al., 1997), rend la collecte
des contraintes structurales RMN moins lourde, et donc moins coûteuse
en temps. L'application de l'ensemble de ces développements permet
aujourd'hui de trouver dans la littérature des reports d'attribution de
protéines de plus
de 80 kDa (Tugarinov et al., 2004), ainsi que des structures
tridimensionnelles de protéine de
48 kDa résolues par RMN (Williams et al., 2005). Cela
étant, pour des protéines de cette taille,
les moyens employés en termes de production
d'échantillons (nécessité de différents
marquages spécifiques et onéreux), temps
d'enregistrement des expériences, et temps
d'interprétation des données, deviennent alors
considérables.
Stratégie générale de l'étude
structurale de K2 par RMN et Modélisation Moléculaire
La stratégie adoptée pour caractériser la
structure d'une protéine par spectroscopie de
RMN et Modélisation Moléculaire
dépend de sa masse moléculaire, ainsi que des outils
d'analyse et de traitement à disposition du structuraliste. Le
domaine K2 de KIN17 est une protéine de 111 acides aminés,
que l'on peut qualifier de taille moyenne pour une étude par RMN. Les
différentes étapes de la méthodologie employée pour
résoudre la structure de K2 sont schématisées en Figure
3.1, et sont détaillées dans ce chapitre.
Préparation des échantillons
de protéine marquée 15N,
et
15N /
13C
Attribution des raies de résonance de la
protéine
Détermination de la topologie
- chaîne principale
- chaînes latérales
- déplacements chimiques
- nOe 1H-1H caractéristiques des
structures secondaires
- constantes de couplage 3JHN-Há
- nOe hétéronucléaire
1H-15N
- vitesses d'échange 1H-2D
Calcul de la structure par
Modélisation
Moléculaire sous contraintes RMN

- attribution automatique des nOe
- dynamique moléculaire sous CNS
Recueil des contraintes structurales
- nOe 1H-1H
- angles dièdres et
- liaisons hydrogène
Structure 3D
Figure 3.1 : Résumé
des principales étapes de la stratégie d'étude structurale
du domaine K2
1) Méthode d'attribution des raies de
résonance
Après avoir préparé un échantillon
de protéine pur, concentré, et stable, l'enregistrement des
expériences RMN peut débuter. Les informations et contraintes
structurales ne peuvent être directement extraites des spectres RMN. Il
est tout d'abord nécessaire de déterminer les déplacements
chimiques de chacun des atomes de la biomolécule. Le choix de la
stratégie à employer pour attribuer les raies de
résonance est dicté par la taille de la protéine
étudiée. Pour une protéine de 111 résidus
comme le domaine K2, l'attribution peut être menée par
deux méthodes distinctes qui dépendent du type de
marquage isotopique de l'échantillon.
La première de celles-ci est basée sur
l'enregistrement d'expériences 3D hétéronucléaires
de type 15N-TOCSY-HSQC et 15N-NOESY-HSQC.
L'attribution des résonances 1H et 15N est alors
réalisée selon la stratégie décrite dans l'ouvrage
de Wüthrich (Wüthrich, 1986) qui repose sur les couplages
homonucléaires scalaire et dipolaire 1H-1H.
Cette méthode est généralement applicable à
des protéines contenant jusqu'à 130 acides aminés, et
présente l'avantage de ne nécessiter qu'un simple marquage
15N. Toutefois, pour
des polypeptides de cette taille, qui adoptent une structure
tertiaire, ce type de stratégie basée
sur l'identification du nOe présente un
caractère ambigu. De plus, dès 100 résidus,
l'augmentation de la taille peut conduire à une réduction de
l'efficacité du transfert TOCSY
1H-1H, qui engendre une diminution du
nombre de résonances, et notamment dans les régions
structurées en hélice où les
constantes de couplage 3JHN-Há sont faibles. C'est le
cas de la protéine K2 où la quantité d'information
recueillie sur le spectre 15N-TOCSY-HSQC est insuffisante pour
envisager l'attribution complète des fréquences 1H et
15N par cette stratégie.
Il existe pour les protéines doublement marquées
15N / 13C une attribution séquentielle alternative
basée uniquement sur des transferts de cohérence via
les couplages scalaires homonucléaires et
hétéronucléaires. La méthode repose sur
l'enregistrement d'une combinaison d'expériences 3D
hétéronucléaires de double ou triple résonance
qui corrèlent chacune 2 ou 3 types de noyaux différents. Cette
stratégie, utilisée pour attribuer l'ensemble des
résonances de la chaîne principale de la protéine
K2, est présentée dans le paragraphe
suivant.
1.1) Attribution des carbones de la chaîne
principale et des 13Câ
1.1.1) Stratégie
générale
Les expériences 3D de triple résonance
utilisées classiquement pour l'attribution du squelette d'une
protéine doublement marquée sont, pour la plupart d'entre elles,
basées sur le même principe : la corrélation du proton
amide d'un résidu i avec respectivement, l'azote qui
le porte, et un carbone Ci (CO, Cá, ou Câ)
du résidu précédent et/ou du même
résidu. Ces corrélations, dépendant du chemin de
cohérence sélectionné, se font par le transfert
successif
de l'aimantation à travers plusieurs liaisons
covalentes via des couplages scalaires 1,2J
relativement élevés (1JC-C,
1JC-N, 1JH-N, 1JH-C, 2JC-N,
et 2JC-C). Les six expériences 3D couramment
utilisées pour l'attribution complète des noyaux
1HN, 15N, 13CO, 13Cá, et
13Câ sont : HNCA, HN(CO)CA, HNCO, HN(CA)CO, CBCANH, et
CBCA(CO)NH (Pour revues : Cavanagh et al., 1996 ; Sattler et al., 1999 ;
Kanelis et al., 2001). Le nom de ces expériences indique les noyaux qui
sont impliqués dans les chemins de cohérence suivis. Les noyaux
mis entre parenthèses ne sont pas édités en
fréquence et servent uniquement à relayer
l'aimantation. On peut classer ces six expériences en deux
catégories distinctes :
- La première corrèle le groupement amide du
résidu i avec les carbones 13C du résidu i-1,
et permet ainsi d'obtenir les déplacements chimiques du
triplet (Ni ; HN ; C ).
i i-1
- Dans la seconde catégorie, le groupement amide du
résidu i est relié aux carbones 13C
du résidu i et du résidu i-1. L'information obtenue
est alors double, et les déplacements
chimiques recueillis constituent le quadruplet (Ni ; HN ; C
; C ).
i i-1 i
La combinaison de ces deux types d'expériences peut donc
permettre de déterminer
sans ambiguïté les déplacements chimiques des
carbones 13CO, 13Cá, et 13Câ des
résidus i et
i-1 relatifs à chaque groupement amide. Il est alors
possible de connecter les résidus deux à deux en comparant les
valeurs de déplacements chimiques Ci des uns aux valeurs de Ci-1 des
autres (Figure 3.2). Finalement, le rapprochement de cette attribution
séquentielle à la séquence primaire est initié en
recherchant les acides aminés particuliers qui présentent des
valeurs de déplacement chimique caractéristiques. C'est le cas de
la glycine qui ne possède
pas de Câ, et des résidus thréonine,
sérine et alanine, dont les valeurs de déplacement
chimique de Cá et Câ sont spécifiques.
Liste de déplacements chimiques (Ni ;
HNi ; Ci-1 ; Ci)
C C
N C C N C
C
C C
N C C N C C
C C
N C C N C C
H H O H H O
H H O H H O
H H O H H O
|
C
|
|
|
C
|
|
|
C
|
|
|
C
|
|
|
C
|
|
|
C
|
|
|
N
|
C
|
C
|
N
|
C
|
C
|
N
|
C
|
C
|
N
|
C
|
C
|
N
|
C
|
C
|
N
|
C
|
C
|
|
H
|
H
|
O
|
H
|
H
|
O
|
H
|
H
|
O
|
H
|
H
|
O
|
H
|
H
|
O
|
H
|
H
|
O
|
C C C
C C C
N C C N
C C N
C C N
C C N
C C N C C

H H O H
H O H
|
i
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
i+6
|
|
C
|
|
|
C
|
|
|
C
|
|
|
C
|
|
|
C
|
|
|
C
|
|
|
|
N
|
C
|
C
|
N
|
C
|
C
|
N
|
C
|
C
|
N
|
C
|
C
|
N
|
C
|
C
|
N
|
C
|
C
|
|
|
H
|
H
|
O
|
H
|
H
|
O
|
H
|
H
|
O
|
H
|
H
|
O
|
H
|
H
|
O
|
H
|
H
|
O
|
|
H O H
H O H
H O H H O
Figure 3.2 : Principe de
l'attribution séquentielle basée sur l'enregistrement
d'expériences 3D
i
de triple résonance 1H, 15N,
13C. Chaque groupement amide (HNi ; NH ) est
associé aux triplets
de déplacements chimiques (13COi ; 13Cái
; 13Câ ) et (13CO
; 13Cá
; 13Câ
) relatifs aux
i i-1
i-1
i-1
résidus i, et i-1. Les connexions de la
séquence peptidique sont retracées résidu par
résidu en comparant les valeurs de déplacements chimiques
13Ci aux valeurs de 13Ci-1 triplet par triplet.
Les valeurs de déplacement chimique des
noyaux 13CO, 13Cá, et
13Câ sont respectivement déterminées, par les
couples d'expériences HNCO et HN(CA)CO, HNCA et HN(CO)CA, CBCANH et
CBCA(CO)NH. Ces expériences s'interprètent par paire et chaque
couple fournit un chemin d'attribution potentiel.
1.1.2) Description des expériences 3D triple
résonance
Les séquences d'impulsion des expériences
3D de triple résonance utilisées pour l'attribution
séquentielle sont toutes du même type : la séquence
débute par un transfert de polarisation de type INEPT (Insensitive
Nuclei Enhanced by Polarisation Transfert) du proton
1H vers l'hétéroatome (15N ou
13C) qui le porte (Figure 3.3). L'aimantation évolue ensuite
en
fonction de différents couplages scalaires et du
déplacement chimique 13Ci avant d'être
transférée vers le noyau amide 15NH via le
couplage 15N-13Cá ou
15N-13CO. Finalement, un
transfert de polarisation de type INEPT-inverse permet de
détecter l'aimantation sur le noyau
amide 1HN.
J
1
C-H
(130 Hz)
J
1
X-H,
13CO
13Cá
1
1JN-CO
2
1JN-H (90 Hz)
2JX-H, ...
13Câ
JN-Cá ,
JN-Cá
1JN-H (90 Hz)
1HN/á/â
INEPT
15NH 13
C
13 Ci
á/â
15NH
INEPT-inverse
1HN
(t1) (t2) (t3)
Figure 3.3 : Représentation
schématique du transfert de l'aimantation dans une
expérience
3D triple résonance de type HNCA,
HN(CO)CA, HNCO, HN(CA)CO, CBCANH, ou
CBCA(CO)NH.
a) Attribution des Cá
L'expérience HNCA corrèle le groupement amide
au carbone Cá via le couplage scalaire
15N-13Cá. Sur les spectres correspondants, deux
pics peuvent apparaître par résidu :
une première corrélation du groupement amide
avec le Cái, et une seconde avec le
Cái-1
(Figure 3.4). Ceci s'explique par les valeurs de constante de
couplage 1JN-Cá (11 Hz) et 2JN-Cá
(-7 Hz) qui sont du même ordre de grandeur. La
différence d'intensité des deux pics, due au
fait que |1JN-Cá| >
|2JN-Cá|, pourrait permettre de distinguer le
déplacement chimique provenant
du Cái, de celui du Cái-1.
Il est cependant préférable de vérifier une telle
attribution à l'aide de l'expérience HN(CO)CA
complémentaire. Celle-ci corrèle le groupement amide au
seul carbone Cá-1 en deux étapes via le carbonyle
13COi-1 non édité. La différence de constante
de couplage 1JN-CO (15 Hz) et 2JN-CO (1 Hz) permet
d'orienter sélectivement l'aimantation vers le
13COi-1, puis vers le Cái-1 via la
liaison 1JCO-Cá (55 Hz).
Ces deux expériences fournissent un premier
chemin d'attribution séquentielle et pourraient suffire à
connecter tous les résidus deux à deux. Cependant, pour la
majorité des acides aminés, les déplacements
chimiques de Cá ne sont pas caractéristiques, et ne
permettent une attribution sans ambiguïtés. De plus, le
recouvrement des pics de corrélation dans les régions
encombrées des spectres peut induire une incertitude sur certaines
valeurs de
déplacement chimique de Cá.
HN(CO)CA
HNCA
|
|
H
|
O
|
H
|
H
|
O
|
|
H
|
O
|
H
|
H
|
O
|
|
A)
|
|
C
|
C
|
N
|
C
|
C
|
|
C
|
C
|
N
|
C
|
C
|
|
H
|
C
|
H
|
H
|
C
|
H
|
H
|
C
|
H
|
H
|
C
|
H
|
13Cá
B)
CáX-1
CáY-1
15N
13Cá
CáY
CáX
CáY-1
CáX-1
15N
HX HY
NY
NX
1HN
HX HY
NY
NX
1HN
Figure 3.4 : Expériences 3D
de triple résonance utilisées pour l'attribution des
13Cá d'une protéine. A) Schéma du transfert de
l'aimantation dans les expériences HN(CO)CA et HNCA.
Les noyaux édités en fréquence
sont indiqués par des cercles noirs, alors que ceux uniquement
relayés, sont indiqués par des cercles blancs. B) Exemple
illustré de spectres HN(CO)CA et HNCA correspondants à un
segment de 2 résidus consécutifs X-Y ; mise en
évidence de la connectivité séquentielle.
b) Attribution des CO
Selon le même principe, les expériences
HNCO et HN(CA)CO procurent les corrélations entre les
résonances 1HN et 15NH et les 13CO
intra- et inter-résidus (Figure 3.5). L'expérience HNCO fournit
uniquement une corrélation avec le carbonyle 13COi-1
via la constante de couplage 1JN-CO. La HN(CA)CO utilise
les couplages scalaires 1JN-Cá, 2JN-Cá,
et
1JN-CO pour relier le 13COi et le
13COi-1 au groupement amide via le noyau (CA) non
édité. A
l'instar de l'expérience HNCA, l'intensité du pic
inter-résidu est généralement plus faible que celle du pic
intra-résidu du fait que |1JN-Cá| >
|2JN-Cá|.
Alors que la HNCO est l'expérience de
triple résonance la plus sensible, la HN(CA)CO est la moins
sensible. Toutefois, il existe d'autres expériences, comme la
(HCA)CO(CAN)NH, qui permettent d'obtenir les corrélations des
13CO intra- et inter-résidus,
et qui sont plus sensibles que la HN(CA)CO. Cependant, la
quantité d'information présente
sur les spectres n'est pas toujours suffisante pour
retracer les connexions de la chaîne
peptidique résidu par résidu. D'autre part,
la dispersion des déplacements chimiques des
carbonyles étant faible (de l'ordre de 10 ppm),
le risque d'ambiguïtés et de recouvrement spectral en est plus
important. Ces deux types d'expériences ne constituent donc pas le
couple privilégié permettant l'attribution
séquentielle, mais apportent cependant une information
supplémentaire pour lever d'éventuelles
ambiguïtés.
HNCO
HN(CA)CO
|
|
H
|
O
|
H
|
H
|
O
|
|
H
|
O
|
H
|
H
|
O
|
|
A)
|
|
C
|
C
|
N
|
C
|
C
|
|
C
|
C
|
N
|
C
|
C
|
|
H
|
C
|
H
|
H
|
C
|
H
|
H
|
C
|
H
|
H
|
C
|
H
|
13CO
COX-1
13CO
COX-1
COY-1
B)
15N
COX
COY-1
COY
15N
HX HY
NY
NX
1HN
HX HY
NY
NX
1HN
Figure 3.5 : Expériences
3D de triple résonance utilisées pour l'attribution des
13CO d'une protéine. A) Schéma du transfert de
l'aimantation dans les expériences HNCO et HN(CA)CO. Les noyaux
édités en fréquence sont indiqués par des
cercles noirs, alors que ceux uniquement relayés, sont
indiqués par des cercles blancs. B) Exemple illustré de
spectres HNCO et HN(CA)CO correspondants à un segment de 2
résidus consécutifs X-Y ; mise en évidence de la
connectivité séquentielle.
c) Attribution des Câ
Les expériences CBCA(CO)NH et CBCANH sont sans aucun
doute les expériences majeures dans la stratégie
d'attribution. Elles fournissent les mêmes informations que le
couple HN(CO)CA et HNCA et de plus, mettent en évidence les
corrélations du groupement amide avec les 13Câ intra-
et inter-résidus (Figure 3.6). Outre l'apport d'une information de
fréquence supplémentaire, la connaissance du
déplacement chimique du 13Câ permet une
identification partielle du type de résidu, et de relier les
chaînes latérales à l'attribution séquentielle du
squelette.
Les deux expériences débutent de la même
façon : après transfert de l'aimantation des
protons aliphatiques vers leur carbone, l'aimantation provenant
du 13Câ est transférée au
13Cá
via la constante de couplage
1JCá-Câ (35 Hz). Elle est ensuite orientée
sélectivement vers le
15NH pour la CBCANH, ou vers le 13CO
(puis vers le 15NH) pour la CBCA(CO)NH. Au final,
l'expérience CBCANH fait apparaître les corrélations du
groupement amide avec les noyaux
13Cá et 13Câ des
résidus i et i-1, et la CBCA(CO)NH procure uniquement les
corrélations avec
les noyaux 13Cá et 13Câ du
résidu i.
CBCA(CO)NH
CBCANH
|
|
H
|
O
|
H
|
H
|
O
|
|
H
|
O
|
H
|
H
|
O
|
|
A)
|
|
C
|
C
|
N
|
C
|
C
|
|
C
|
C
|
N
|
C
|
C
|
|
H
|
C
|
H
|
H
|
C
|
H
|
H
|
C
|
H
|
H
|
C
|
H
|
13Cá/â
B)
CâX-1
CáX-1
HX HY
CâY-1
CáY-1
1HN
15N
NY NX
13Cá/â
CâX
CâX-1
CáX
CáX-1
HX HY
CâY
CâY-1
CáY
CáY-1
1HN
15N
NY NX
Figure 3.6 : Expériences
3D de triple résonance utilisées pour l'attribution des
13Câ d'une protéine. A) Schéma du transfert
de l'aimantation dans les expériences CBCA(CO)NH et CBCANH. Les
noyaux édités en fréquence sont indiqués par des
cercles noirs, alors que ceux uniquement relayés, sont
indiqués par des cercles blancs. B) Exemple illustré de
spectres CBCA(CO)NH et CBCANH correspondants à un segment de 2
résidus consécutifs X-Y ; mise
en évidence des connectivités
séquentielles.
1.2) Attribution des protons Há et
Hâ
Les 3 couples d'expériences décrits
précédemment ne sont pas toujours suffisants pour reconstituer
l'ensemble de la chaîne peptidique. En effet, certains
inconvénients inhérents à l'analyse d'une
protéine par RMN comme le recouvrement spectral, et la
dégénérescence partielle du signal, nuisent à la
qualité et à la quantité d'information présente sur
les spectres,
et ne permettent généralement pas de
relever la totalité des déplacements chimiques
13Cá,
13Câ, et 13CO des
résidus i et i-1 relatifs à chaque groupement
amide. L'attribution
séquentielle peut cependant être
complétée et vérifiée à partir du
déplacement chimique 1Há
fournit par les expériences 3D HNHA et HBHA(CO)NH.
L'expérience HNHA (Vuister & Bax, 1993) est
classiquement utilisée pour corréler le
groupement amide (1HN ; 15NH ) au seul proton 1Há
intra-résiduel. Cette expérience de double
i i i
résonance 1H, 15N ne
nécessite qu'un échantillon de protéine simplement
marquée 15N. Cependant, l'intensité des pics
observés sur les spectres est proportionnelle à la constante de
couplage 3JHN-Há, et pour des valeurs faibles, il est parfois
difficile d'observer une tache de corrélation. Cet
inconvénient ne se retrouve pas avec l'expérience
triple résonance HBHA(CO)NH (Grzesiek & Bax, 1993), qui permet de
relier chaque groupement amide aux noyaux 1Há et
1Hâ du résidu précédent (Figure 3.7). En
effet, cette expérience, plus sensible
que la HNHA, est tout à fait comparable à la
CBCA(CO)NH et met en jeu un transfert de
l'aimantation, via des couplages scalaires forts, de la
chaîne latérale i-1 au groupement amide
i via le noyau 13COi-1 non édité.
La combinaison des expériences HNHA et HBHA(CO)NH
permet donc d'identifier le déplacement chimique des protons
1Há des résidus i et i-1 relatifs à
chaque groupement amide, et ainsi de consolider l'attribution
séquentielle. Le déplacement chimique des protons
Hâi-1, dont l'ordre de grandeur fournit une
indication du type d'acide aminé, peut également
être utilisé pour contrôler l'attribution.
HBHA(CO)NH
1Há/â
Hâ2i-1
Hâ1i-1
H O
A) C C
H C H
H H O
N C C B)
H C H
Hái-1
15N
NHi
HNi
1HN
Figure 3.7 : Attribution des
protons 1Há et 1Hâ avec
l'expérience 3D de triple résonance HBHA(CO)NH. A)
Schéma illustrant le transfert de l'aimantation : les noyaux
édités en fréquence sont indiqués par des cercles
noirs, et ceux uniquement relayés, sont indiqués par
des cercles blancs. B) Illustration des corrélations
observables sur un spectre HBHA(CO)NH
pour un groupement amide i.
1.3) Attribution des chaînes latérales
1.3.1) Description des expériences 3D HCCH-COSY et
HCCH-TOCSY
Les expériences utilisées pour l'attribution des
chaînes latérales sont généralement des
expériences 3D de double résonance 1H et
13C. C'est le cas de la HCCH-COSY, et de la HCCH-TOCSY, qui
permettent d'identifier les protons et carbones des chaînes aliphatiques
et aromatiques (Kay et al., 1993 ; Majumdar et al., 1993) . Ces deux
expériences peuvent être interprétées comme des
2D 1H-1H COSY et TOCSY, et la troisième
dimension disperse l'information selon la fréquence du carbone
13C. Les chemins de cohérence suivis sont
cependant très différents de ceux
observés dans des expériences 2D homonucléaires
1H.
Ainsi, la séquence d'impulsions HCCH-TOCSY contient un
motif TOCSY 13C-13C pendant lequel l'aimantation est
transférée le long de la chaîne carbonée
(Figure 3.8). Comparé au TOCSY classique 1H-1H
basé sur les constantes de couplage 2JH-H et
3JH-H (4-10 Hz), ce type
de transfert est bien plus efficace car il repose sur le
couplage fort 1JC-C (35-55 Hz). Il en est
de même pour le transfert COSY
13C-13C de l'expérience HCCH-COSY qui est
basé sur cette même constante de couplage 1JC-C.
La bonne sensibilité de ces expériences s'explique
également par des transferts de cohérence rapides entre les
noyaux 1H et 13C via la constante
de couplage 1JC-H (125-250 Hz).
HCCH-TOCSY
1H
Häi
Hãi
H
A) H
N H H H
C C C C B)
Hâi
Hái
13C
á â ã ä
Cãi
O C H H H
Hái
Hãi 1H
Cái
Figure 3.8 : Attribution
des noyaux 1H et 13C des chaînes
latérales aliphatiques et aromatiques. A) Description du transfert
de l'aimantation dans une expérience 3D de double résonance
HCCH-TOCSY. Les noyaux édités en fréquence sont
indiqués par des cercles noirs
et le transfert TOCSY 13C-13C est
indiqué par un trait ondulé. B) Exemple illustré de
spectre HCCH-TOCSY. Mise en évidence du système de spin de 2
protons Há et Hã appartenant au même résidu et
respectivement portés par les carbones Cá et Cã.
1.3.2) Attribution des chaînes latérales
aliphatiques
Les fréquences de résonance des noyaux
1H et 13C de chaîne latérale
aliphatique se déterminent à partir des spectres HCCH-TOCSY
et HCCH-COSY édités sur la région
aliphatique. Dans l'optique d'obtenir des spectres de
bonne qualité, il est préférable
d'enregistrer ces 2 expériences sur un échantillon
de protéine doublement marquée 15N / 13C,
préalablement lyophilisé, et repris dans 100 % de D2O. Les
valeurs de déplacement chimique
de 13Cá, 13Câ,
1Há et 1Hâ servent de point de
départ pour identifier les autres résonances
aliphatiques de chaîne latérale.
1.3.3) Attribution des chaînes latérales
aromatiques
Les raies de résonance des protons et
carbones de chaîne latérale aromatique s'attribuent en
deux étapes. Dans un premier temps, les systèmes de spin
aromatiques se déterminent à partir de l'expérience
HCCH-TOCSY, éditée sur la région aromatique, et
enregistrée sur un échantillon doublement marqué
en solvant D2O. Les valeurs de déplacement chimique
13C des cycles aromatiques sont spécifiques du type d'acide
aminé, ils permettent ainsi la distinction entre les
différents cycles. L'attribution de ces systèmes se
réalise ensuite en recherchant les nOe intra-résiduels qui
corrèlent les noyaux 1Há et 1Hâ aux
noyaux 1H aromatiques sur le spectre 13C-NOESY-HSQC,
édité sur la région aliphatique et enregistré sur
le même type d'échantillon.
Dans le cas du domaine K2, les expériences 3D
HCCH-COSY et HCCH-TOCSY suffisent pour identifier la
quasi-totalité des résonances aliphatiques et
aromatiques. Cependant, la forte similarité de
déplacement chimique de certains types de noyaux
aliphatiques 1H et 13C induit généralement
la présence de nombreuses zones de recouvrement
sur les spectres, qui ne permet pas toujours une attribution sans
ambiguïté. Pour une protéine
de cette taille, la stratégie peut alors consister
à enregistrer d'autres expériences de type 3D ou
4D, qui corrèlent les noyaux 1H ou
13C aliphatiques au groupement amide du résidu suivant
(Grzesiek et al., 1993 ; Logan et al., 1993).
2) Détermination de la topologie et recueil des
contraintes structurales
Une fois les déplacements chimiques de la
protéine attribués, la seconde étape de
l'étude consiste à extraire un certain nombre de
paramètres des spectres RMN, puis à les convertir en
contraintes structurales. Pour une protéine de taille moyenne comme le
domaine K2, deux types de contraintes géométriques sont
classiquement utilisés pour le calcul de la
structure. Il s'agit de la distance inter-atomique
1H-1H, et des angles dièdres et qui
définissent le squelette peptidique. Les contraintes de
distance sont principalement issues de
l'effet Overhauser homonucléaire 1H. Aussi, la
détermination de la structure de biomolécules
par RMN repose essentiellement sur ce paramètre.
2.1) L'effet Overhauser nucléaire
2.1.1) Aspect expérimental
Deux noyaux proches dans l'espace présentent entre
eux des interactions dipolaires
qui peuvent être mises en évidence par
saturation ou inversion d'un des deux spins. Le couplage dipolaire
résultant correspond à un transfert d'aimantation à
travers l'espace, appelé relaxation croisée ou effet Overhauser
nucléaire (nOe). L'intensité ç d'un pic de
corrélation dipolaire entre deux atomes est fonction de plusieurs
paramètres caractéristiques de la molécule
étudiée. Elle dépend notamment de la longueur du vecteur
(ou distance) entre les deux atomes (en r -6), et du
temps de corrélation ôc de ce vecteur en
fonction du champ magnétique ù du spectromètre
:
ç = f ( r -6 ; ùôc )
(2.1)
Dans l'approximation d'un mouvement isotrope d'une
molécule sans mobilité interne, le
temps de corrélation ôc représente
le temps nécessaire à la molécule pour se
réorienter de un radian. Ce temps de corrélation est directement
lié à la masse moléculaire de l'objet analysé.
Par conséquent, l'effet Overhauser nucléaire est
fonction de la taille de la protéine étudiée.
Les expériences RMN qui contiennent des séquences
NOESY mettent en évidence des corrélations dipolaires
homonucléaires 1H entre protons distants de moins de
6 Å. Dans ce type d'expérience, le transfert de
l'aimantation entre deux protons proches dans l'espace a lieu pendant
le temps de mélange expérimental ôm. Dans
le cas d'une protéine de 14 kDa comme le domaine K2, le
produit ùôc implique un nOe négatif pouvant
atteindre un accroissement de 100%. Aussi, comparé aux petites
molécules, l'effet Overhauser est plus efficace et s'établit
à des faibles temps de mélange ôm. Toutefois, pour
des protéines de cette taille, le nOe est accompagné d'effets
indirects, telle que la diffusion de spin, qui apparaît à
des temps de mélange intermédiaires, et dont le
poids augmente avec ôm. Dans ces conditions, l'effet Overhauser
est biaisé et n'est plus représentatif de la proximité
entre protons. Le temps
de mélange ôm doit donc être
choisi de manière à obtenir le maximum d'informations
dipolaires tout en évitant le phénomène de
diffusion de spin. En pratique, il serait possible
d'estimer le ôm optimal
en réalisant pour chaque expérience des études classiques
de « build-
up » sur les échantillons de protéine
K2. Dans notre cas, le choix du ôm a été
guidé en se basant sur des études similaires menées sur
des protéines de taille comparable à fréquence de champ
comparable.
Une fois ces considérations prises en compte, on peut
alors estimer que le volume d'un
pic de corrélation dipolaire entre deux protons est
proportionnel à la distance r qui sépare les deux noyaux
en 1/r6. Cette dépendance du nOe en distance est
d'une importance considérable pour le calcul de la structure à
haute résolution car elle va permettre la conversion du volume
Vij de chaque pic en contrainte de distance rij
via une référence (Vref, et rref), et
ainsi d'accéder
potentiellement à l'ensemble des proximités
spatiales entre protons au sein de la protéine :
rij = rref
Vref
Vij
1/6
(2.2)
2.1.2) Le problème de l'attribution des pics
nOe
L'interprétation du nOe 1H en
contrainte de distance nécessite au préalable
l'attribution des pics de corrélation dipolaire,
c'est-à-dire de déterminer pour chaque pic associé
à un proton de la protéine, le (ou les) noyau(x) en interaction
dipolaire avec celui-ci. Dans le cas de l'étude d'un peptide n'adoptant
pas de structure tertiaire, la majorité des nOe inter-résidus
observés résulte de proximités à courte et moyenne
distances, qui traduisent la structure secondaire dans laquelle chaque acide
aminé est engagé. En pratique, ce type de nOe correspond
à la corrélation d'un proton d'un résidu avec un
ou plusieurs protons de résidu proche dans la séquence,
ce qui limite le nombre d'attributions possibles. De plus, la
connaissance de la structure secondaire, fournie par l'analyse des
paramètres structuraux RMN (couplage scalaire, déplacement
chimique...), facilite considérablement l'attribution de
ces pics. Dans le cas de l'étude d'une protéine
repliée, la structure tertiaire induit l'apparition
de nombreux effets à longue distance entre protons
appartenant à des résidus éloignés dans la
séquence. Ces effets sont d'une importance capitale car
ils traduisent l'organisation tridimensionnelle de la protéine, et
notamment celle du coeur hydrophobe. Aussi, l'attribution des pics nOe
devient alors beaucoup plus délicate car elle nécessite
une connaissance préalable d'éléments de structure
tertiaire, difficilement appréciables pour des protéines
structurées majoritairement en hélice. D'autre part, plus la
taille de la protéine est importante,
et plus les possibilités d'attribution sont nombreuses. La
collecte des contraintes de distance
peut alors devenir une étape lourde et coûteuse en
temps.
Pour contourner ces difficultés, il existe des
programmes d'attribution automatique (ou semi-automatique) des pics nOe
couplés aux logiciels classiques de modélisation
moléculaire. Dans le cadre de l'étude structurale du domaine K2,
nous avons pu bénéficier des ressources informatiques du DIEP
du CEA de Saclay, et notamment du programme d'attribution automatique
des nOe développé au Laboratoire de Structure des
Protéines par le Dr. Bernard Gilquin. Ce programme fonctionne en
interface avec le logiciel de modélisation CNS et sera
présenté dans le paragraphe 3.3. Son utilisation nécessite
la connaissance de la topologie de la protéine.
2.2) Détermination de la structure secondaire et de
la topologie
L'extraction des paramètres structuraux RMN est
la méthode classique qui permet d'identifier rapidement la
structure secondaire d'une protéine. L'analyse consensus de ces
paramètres conduit à l'obtention des premiers
éléments de structure tertiaire, utilisés par le
programme d'attribution automatique en amont du calcul de la structure.
2.2.1) Les déplacements chimiques
secondaires
Le déplacement chimique des noyaux de la chaîne
principale d'un résidu est influencé
par plusieurs facteurs dont la structure secondaire dans
laquelle celui-ci est engagé. Wishart et Sykes ont mis au point
une méthode qui permet d'apprécier les types, et les positions
dans la séquence, des éléments de structure secondaire
à partir du déplacement chimique des noyaux
13CO, 13Cá, 13Câ, et
1Há (Wishart et al., 1992 ; Wishart & Sykes, 1994). La
procédure consiste
à calculer la différence entre le
déplacement chimique expérimental (äobs) et une valeur
de référence (äref) correspondant au même
noyau pour le même acide aminé dans une conformation
aléatoire. La valeur obtenue est appelée déplacement
chimique secondaire (CSD
ou Chemical Shift Deviation). Les indices CSD n'ont
pas la même signification selon la nature du noyau. Ainsi, pour le
proton 1H, une succession d'indices supérieurs à 0.1
ppm en valeur absolue reflète une structure secondaire stable et
le signe en précise le type : positif pour un feuillet â, et
négatif pour une hélice á ou 310. Dans le cas
des 13Cá, il s'agit
exactement du contraire, et la valeur significative de CSD est de
1 ppm.
2.2.2) La constante de couplage
3JHN-Há
Les angles et sont deux des trois angles de
torsion qui décrivent le squelette peptidique d'une protéine.
La combinaison d'effets stériques, au sein d'un même
résidu, ou entre les chaînes latérales, ne permet
à ces angles de prendre que certaines valeurs qui
définissent les différents types de structure secondaire. La
constante de couplage 3JHN-Há est reliée
indirectement à l'angle dièdre via l'équation de
Karplus (2.3) avec les coefficients semi-empiriques A, B, et C (Karplus,
1959). Plusieurs jeux de coefficients ont été proposés, et
ceux utilisés par Pardi et al., sont les plus
communément admis (2.4) (Pardi et al., 1984). La résolution de
l'équation fait apparaître que les valeurs idéales de
constante 3JHN-Há sont de 4Hz pour l'hélice
á (-57°), et de 9 Hz pour le feuillet â
antiparallèle (-139°). Plusieurs études statistiques,
comme celle menée par Smith et al., sur 85
structures résolues par radiocristallographie, ont cependant
conduit à considérer des valeurs corrigées de
3JHN-Há (Smith et al., 1996). Sur la base de ces
études, il est généralement admis que plusieurs valeurs
de 3JHN-Há consécutives
inférieures à 5.5 Hz indiquent une conformation
hélicoïdale, alors que
des valeurs supérieures à 8 Hz reflètent une
structure en feuillet ou étendue.
O C) 3JHN-Há = 6,4.cos2
è - 1,4.cos è + 1,9
A) N
C'
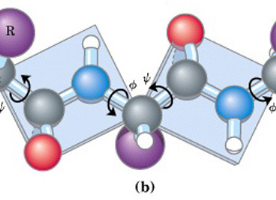
C'
Cá N
10
8
J
3
HN-Há 6
avec è = |-60°| (2.4)
O [Hz]
4
3JHN-Há = A.cos2
è - B.cos è + C
B) avec è = | - 60°|
(2.3)
2
-180 -120 -60
0 60
[°]
120
180
Figure 3.9 : Estimation de
l'angle dièdre à partir de la constante de
couplage 3JHN-Há.
A) Définition des angles de torsion et
du squelette peptidique. B) Relation de Karplus
liant la constante 3JHN-Há aux angles
è et . C) Résolution de l'équation de Karplus
selon les coefficients de Pardi.
Dans le cas des protéines, les constantes de
couplage 3JHN-Há sont généralement
estimées à partir de l'expérience 3D HNHA, qui met
en évidence la corrélation intra- résiduelle entre un
groupement amide (1HN, 15NH) et son proton alpha
(1Há). Sur les spectres correspondants, le rapport
d'intensité entre le pic croisé négatif (Ic) et
le pic diagonal positif
(Id) est relié à la constante
3JHN-Há via une constante å (Vuister
& Bax, 1993) :
Ic / Id = -tan2 (2ðå .
3JHN-Há )
(2.5)
2.2.3) Recherche des nOe
caractéristiques
Comme suggéré dans le paragraphe 2.1.2, chaque type
de structure secondaire donne naissance à des pics nOe
spécifiques en nature et en intensité sur les spectres NOESY.
Aussi,
la méthode classique proposée par
Wüthrich pour identifier les éléments de structure
secondaire consiste à reporter sur un diagramme les nOe
caractéristiques entre protons appartenant à des résidus
proches dans la séquence (Wüthrich, 1986) (Figure 3.10A). Ces nOe
sont facilement identifiables sur les spectres NOESY-HSQC éditée
15N, et un simple examen
de ce diagramme suffit en théorie à
discerner de manière univoque les différents types de
conformation au sein de la protéine.
A) B)
H
H
Figure 3.10 : Identification des
éléments de structure secondaire et de la topologie
d'une
protéine à partir de l'attribution de
pics nOe spécifiques. A) Diagramme des nOe
caractéristiques des différents types de structure
secondaire selon Wüthrich (Wüthrich,
1986). B) Description schématique d'un feuillet
â antiparallèle. Les liaisons hydrogène sont
représentées par des pointillés jaunes.
Les traits rouges représentent le type de connectivité longue
distance recherchée.
Une fois les éléments de structure secondaire
identifiés à partir du résultat consensus
de l'analyse des paramètres structuraux, la
topologie de la protéine est déterminée en
recherchant des effets spécifiques à longue distance de
type HN-HN, HN-Há, et Há-Há au niveau des
résidus n'adoptant pas de structure hélicoïdale. La
caractérisation de ces effets permet d'une part, de distinguer les
régions étendues de celles impliquées dans un feuillet
â et d'autre part, de caractériser l'organisation des brins
au sein des feuillets (parallèle ou antiparallèle) (Figure
3.10B). Ces éléments de structure tertiaire facilement
identifiables sont extrêmement précieux car ils fournissent
les premières informations du repliement de la protéine, et
vont guider le programme d'attribution automatique des nOe.
2.2.4) Le nOe hétéronucléaire
1H-15N
Comme nous l'avons évoqué
précédemment, le phénomène de relaxation
croisée entre deux spins dépend essentiellement de la
distance r entre les deux noyaux, et du temps de
corrélation effectif ôc entre les deux spins,
c'est-à-dire de l'amplitude de leur mouvement. Dans
l'approximation d'une distance fixe entre les atomes 15NH et
1HN (rHN ~ 1.01 Å), le paramètre nOe
hétéronucléaire 1H-15N ne
dépend plus que des mouvements rapides de la liaison NH-HN (de
la picoseconde à la nanoseconde). D'autre part, ce type
d'effet est indépendant des protons environnants (Kay et al., 1989).
Par conséquent, la mesure du nOe
1H-15N peut se révéler
très utile pour distinguer les parties flexibles de la protéine,
telles que
les boucles exposées ou les parties
déstructurées (mouvements rapides), de la partie
repliée
(mouvements plus lents).
En pratique, ce paramètre dynamique est
quantifié à partir de deux expériences 2D similaires
qui permettent d'observer la corrélation scalaire
1H-15N de chaque résidu. Le premier spectre
est enregistré avec une saturation préliminaire des
1H qui engendre un transfert d'aimantation vers le 15N
via le couplage dipolaire. Le second spectre est obtenu par
la même séquence d'impulsion, mais en absence de
saturation des 1H (Farrow et al., 1994).
Pour chaque pic de corrélation, le rapport entre
l'intensité du pic saturé (Isat) et l'intensité
du
pic non saturé (Iref) détermine
alors le nOe hétéronucléaire (2.6). L'incertitude
sur chaque valeur est calculée à partir des erreurs de mesure
des intensités qui sont estimées par le bruit spectral sur chacun
des deux spectres.
nOe (1H-15N) =
Isat
Iref
(2.6)
Les régions dans lesquelles les résidus ont une
valeur de nOe 1H-15N supérieure à 0.5
sont
considérées comme rigide, entre 0.2 et 0.5
comme relativement flexibles (boucles), et les acides aminés pour
lesquels ce nOe est inférieur à 0.2 ou négatif
appartiennent à des régions déstructurées de la
protéine. Dans l'optique du calcul de la structure, la mesure
du nOe hétéronucléaire fournit une information
supplémentaire car elle permet d'identifier avec précision
les résidus non structurés des extrémités N- et
C-terminales. Aussi, sur les spectres
de type NOESY, les nOe homonucléaires 1H
observables correspondent à une moyenne
pondérée des nOe correspondants à chaque
population de l'espace conformationnel. Dans le
cas de résidus en échange conformationnel
très rapide, le poids de chaque population est
faible et les contacts de type longue distance sont incompatibles
avec une telle flexibilité. Par
conséquent, la connaissance de ces résidus peut
être utilisée par le programme d'attribution automatique des pics
nOe afin de restreindre le nombre de possibilités d'attribution et donc
de faciliter celle-ci.
2.3) Recueil des contraintes structurales
Les contraintes expérimentales de distance et d'angle
dièdre utilisées pour le calcul de
la structure de K2 sont issues de quatre observables RMN :
l'effet Overhauser nucléaire 1H, le couplage scalaire, le
déplacement chimique, et les vitesses d'échange des protons
amides.
2.3.1) Collecte des pics nOe
La grande majorité des contraintes de distance est obtenue
à partir des expériences 3D NOESY-HSQC éditée
15N, et 13C. Les pics de corrélation dipolaire
1H-1H sont sélectionnés
sur les spectres, puis leur volume est mesuré
à l'aide du logiciel Felix (Accelrys). La démarche
consiste dans un premier temps à définir des «
boites » d'intégration autour de chaque pic nOe. Avant de
mesurer le volume de ces boites, leur taille doit être
optimisée, l'une après l'autre, afin d'éviter
d'intégrer du bruit spectral ou un pic voisin, qui conduirait à
des distances inter-atomiques erronées (souvent sous-estimées).
Cette étape est donc cruciale dans l'optique de préparer un jeu
de contraintes expérimentales de qualité. Selon la
stratégie définie, l'attribution des pics nOe n'est pas
nécessaire à ce stade de l'étude. Seuls les nOe
inter-résidus qui traduisent la topologie des feuillets
â sont attribués et convertis en
contraintes de distance.
2.3.2) Les contraintes d'angle dièdre
Le déplacement chimique des noyaux
13Cá, 13Câ, 13CO,
1Há, et 15N peut être utilisé
pour déterminer les angles et sur la base d'une
prédiction empirique réalisée par le logiciel TALOS
(Cornilescu et al., 1999). Ce programme recherche dans une base de
données, actuellement constituée de 78 protéines, des
triplets de résidus consécutifs ayant une homologie de
déplacements chimiques et de type de résidu avec les
triplets de la protéine étudiée. Pour chaque triplet de
la protéine, les 10 tripeptides les plus proches en séquence
et
en déplacements chimiques sont sélectionnés.
Aussi, si les angles et des résidus centraux
de 9 de ces 10 triplets sont similaires, la prédiction est
considérée comme fiable. Leur valeur
moyenne et l'écart type correspondant sont alors
utilisés pour constituer les contraintes
d'angle dièdre sous forme d'un intervalle de valeurs
permises.
Pour les résidus dont la prédiction TALOS
n'est pas jugée satisfaisante, le jeu de données angulaires
peut être complété à partir de la constante de
couplage 3JHN-Há pour des valeurs inférieures
à 5.5 Hz ou supérieures à 8 Hz (cf. §
2.2.2). L'intervalle des valeurs permises d'angle est alors déduit
de la courbe de Karplus intégrée par les coefficients de
Pardi (Tableau 3.1) :
|
3JHN-Há
(Hz)
|
contrainte sur l'angle
(°)
|
|
3J > 9.0
8.0 < 3J < 9.0
5.5 < 3J < 8.0
5.0 < 3J < 5.5
3J < 5.0
|
-120 #177; 30
-120 #177; 40
Ø
-70 #177; 30
-60 #177; 30
|
Tableau 3.1 : Intervalles de
contraintes sur l'angle en fonction de la constante
3JHN-Há.
2.3.3) Les contraintes de distance déduites des
liaisons hydrogène
Dans toute solution aqueuse, l'autodissociation de l'eau en
ions H3O+ et OH- catalyse l'échange des
protons amides avec les protons de l'eau (Englander et al., 1972).
Selon ce principe, l'incubation d'une protéine dans une solution
à 99% d'eau lourde va mener à la substitution progressive
de la majorité des protons 1HN par le deutérium de
l'eau lourde. La cinétique de cet échange dépend d'une
part, de l'accessibilité au solvant et d'autre part, de la liaison
hydrogène dans laquelle un proton amide peut être impliqué
en se liant à un oxygène
de carbonyle. Un proton 1HN exposé au
solvant et non engagé dans une liaison hydrogène
s'échangera très rapidement, alors que dans
le cas d'un proton amide impliqué dans une liaison
hydrogène, la vitesse d'échange sera d'autant plus lente que
cette liaison est enfouie dans le coeur hydrophobe de la protéine.
Par conséquent, une expérience de spectroscopie
d'échange 1H-2D suivie sur spectres HSQC
1H-15N peut permettre de localiser une partie des protons
amides impliqués dans une liaison hydrogène. Ces liaisons sont
spécifiques de chaque structure secondaire. Aussi, la connaissance
de la topologie de la protéine est utilisée
conjointement pour identifier les deux atomes
impliqués dans chaque liaison. Cette donnée
structurale est finalement convertie en contraintes de distance
de la manière suivante :
|
2.1 Å < dHN-O (i, i-4) < 2.5 Å
|
et
|
3.1 Å < dN-O (i, i-4) < 3.5 Å
|
dans une hélice á
|
|
2.1 Å < dHN-O (i, i-3) < 2.5 Å
|
et
|
3.1 Å < dN-O (i, i-3) < 3.5 Å
|
dans une hélice 310
|
2.1 Å < dHN-O (i, j) < 2.5 Å et 3.1 Å
< dN-O (i, j) < 3.5 Å dans un feuillet â
3) Modélisation Moléculaire sous
contraintes RMN
Les contraintes structurales issues de
l'interprétation des paramètres RMN ne permettent pas
d'accéder directement à la structure tridimensionnelle de
la protéine. Ces données expérimentales sont introduites
dans des procédures de modélisation moléculaire afin
de construire des modèles à l'échelle
atomique. Il s'agit de la modélisation moléculaire sous
contraintes RMN, étape indispensable pour obtenir une
représentation graphique de la structure 3D d'une protéine.
La méthode s'appuie sur des calculs
théoriques de mécanique moléculaire et de dynamique
moléculaire pour déterminer un ensemble de conformations, qui
correspondent à des minima énergétiques, et qui sont en
accord avec les données expérimentales RMN. Dans
le cadre de l'étude de la protéine K2,
nous avons utilisé le logiciel de modélisation CNS
adapté au traitement de données expérimentales
obtenues par radiocristallographie ou par RMN. Les principes de
mécanique et dynamique moléculaire sur lesquels repose ce
logiciel seront évoqués en première partie de ce
paragraphe. La seconde partie sera consacrée à la
présentation du programme d'attribution automatique des pics nOe,
qui fonctionne en interface avec le logiciel CNS.
3.1) Principe de la mécanique moléculaire
adaptée aux systèmes biologiques
3.1.1) Notion de champ de force
Une grande part des systèmes auxquels la
modélisation moléculaire s'intéresse ont une taille bien
trop importante pour pouvoir être étudiés par des
méthodes classiques de mécanique quantique de type ab
initio ou semi-empiriques. En effet, malgré le
perpétuel développement du domaine de l'informatique et
notamment de la capacité de calcul, les
équations de mécanique quantique, utilisées
pour représenter la fonction d'énergie de petites
molécules, ne peuvent être résolues
dans le cas de macromolécules. C'est pourquoi, les simulations
de mécanique et dynamique moléculaire s'appuient sur une
représentation simplifiée de la fonction d'énergie
appelée « champ de force ». Selon ce principe, à chaque
état conformationnel correspond une énergie potentielle
empirique définie par le champ de force, et qui est fonction
des interactions attractives et répulsives entre les atomes. La
conformation la plus stable est alors associée à l'état
pour lequel la fonction d'énergie est la plus basse. Cependant, et avec
ce seul terme d'énergie potentielle, une protéine qui fait
l'objet d'une simulation de mécanique moléculaire peut adopter
une multitude de conformations qui correspondent à des minima
énergétiques locaux. L'intégration de contraintes
expérimentales dans les protocoles de modélisation
moléculaire permet de restreindre l'espace conformationnel à
explorer, et de favoriser des configurations qui sont en accord avec
les données expérimentales. Pour prendre en compte ces
données, le champ de force est alors constitué, d'un
terme d'énergie potentielle empirique Epot
représentant les contraintes physiques internes à la
molécule entre atomes liés et non liés, et d'un
terme énergétique supplémentaire Econt qui
tient compte des contraintes expérimentales (2.7). C'est le cas
du champ de force du programme CNS qui sera présenté
succinctement dans le paragraphe 3.2.
E = Epot + Econt
3.1.2) Les algorithmes de minimisation
(2.7)
Une fois le champ de force choisi, l'objectif de la
modélisation moléculaire sous
contraintes est de déterminer les conformations
associées aux surfaces d'énergie les plus basses, et
satisfaisant un maximum de contraintes RMN. Pour cela, il existe de
nombreux algorithmes qui se différencient par leur efficacité et
leur rapidité d'exécution.
Une minimisation d'énergie, par des algorithmes dits
de mécanique moléculaire, est
un processus itératif qui a pour but d'optimiser la
géométrie d'une molécule à partir d'une
conformation initiale. Tous les paramètres définissant la
géométrie du système (degrés de
libertés) sont systématiquement modifiés par petits
incréments jusqu'à ce que l'énergie atteigne un
minimum, ou pendant un nombre de pas fixé préalablement. Les
algorithmes de minimisation les plus couramment utilisés recherchent un
minimum énergétique en se basant
sur le gradient de la surface d'énergie
potentielle, c'est à dire sur sa dérivée
première ou
seconde par rapport aux coordonnées atomiques.
Ce gradient indique la direction du
minimum, tandis que sa valeur renseigne sur la « pente
» locale de la fonction d'énergie. C'est
Energie potentielle
le cas de la méthode de la plus grande pente
(steepest descent ; Arkfen, 1985), efficace lorsque la
conformation est proche de la structure initiale, et de la
méthode de gradient conjugué Powell (Powell, 1977),
utilisée dans le protocole CNS, et plus adaptée aux
systèmes dont le minimum énergétique est proche.
Figure 3.11 : Illustration
de l'inconvénient des algorithmes de minimisation par la
représentation schématique d'une surface d'énergie
potentielle. La boule grise représente l'énergie potentielle
d'une conformation initiale. La minimisation de l'énergie en se
basant
sur des gradients de surface ne permet de
déterminer qu'un minimum local proche de la structure de
départ et souvent différent du minimum global.
Même si les algorithmes de minimisation basés sur
le gradient de l'énergie potentielle permettent d'optimiser rapidement
la géométrie d'un édifice moléculaire, ils
conduisent le plus souvent au minimum énergétique local, le plus
proche de la structure de départ, et très souvent
éloigné du minimum global (Figure 3.11). Par conséquent,
ces méthodes de calcul ne constituent certainement pas l'outil principal
pour déterminer la structure d'une protéine par
modélisation sous contraintes. La solution idéale serait
une méthode capable d'explorer efficacement l'espace conformationnel
d'un composé, c'est-à-dire l'ensemble des conformations qui lui
sont accessibles. La manière la plus simple et la plus
sûre serait de générer et d'optimiser des
modèles en combinant toutes les possibilités
conformationnelles imaginables. Cette approche, appelée recherche
systématique, est cependant difficilement envisageable dans le cas
de l'étude de macromolécules, où les temps de
calcul exigés constitueraient alors un facteur limitant.
3.1.3) La dynamique moléculaire
La dynamique moléculaire est une des méthodes
qui permet d'explorer l'espace conformationnel d'une structure complexe.
Elle a pour objectif de simuler les mouvements
internes d'une molécule en fonction du temps par
le calcul du déplacement de chacun des
atomes. Elle repose sur les principes de la mécanique
classique Newtonienne et la trajectoire
de chaque atome est définie par la relation
fondamentale de la dynamique (2.8), qui relie la force Fi
s'exerçant sur chaque atome à sa masse mi, son
accélération ai, et sa position ri en fonction
du temps t.
? ?
Fi = miai =
mi
i
d2r?
dt2
(2.8)
?
Fi = -
?
dEpot
i
dr?
(2.9)
?r(t+ät) = 2?r(t)
-
?r(t - ät) + ät2
a?(t) (2.10)
?v(t+ät) = ?v(t) +
0.5ät[a?(t) +
a?(t+ät)]
(2.11)
La force Fi étant
reliée à la fonction d'énergie potentielle (2.9), il est
donc possible de
calculer à tout instant t
l'accélération ai s'exerçant sur chaque
atome. Plusieurs algorithmes mathématiques sont proposés pour
résoudre ces équations. Parmi ceux-ci, figure celui de
Verlet (Verlet, 1967) dans lequel est utilisé un développement en
série de Taylor du second ordre du vecteur position. Ainsi,
connaissant la position à l'instant t, il est alors
possible d'obtenir les positions r(t+ät) (2.10) et les vitesses
v(t+ät) (2.11) des différents atomes. Cet algorithme
implique un pas d'intégration ät très court de
l'ordre de la femtoseconde (10-15 s) afin de maintenir une
trajectoire stable.
Dans une simulation de dynamique moléculaire,
l'énergie cinétique totale d'un système de N
atomes peut être reliée à la température T
via la constante de Boltzman kB :
Ecinétique
= 1
Ó
i 2
2
mivi =
3
2 NkBT
(2.12)
Cette équation introduit la notion de
température au sein des méthodes de dynamique
moléculaire. Plus cette température est
élevée, plus la vitesse communiquée aux atomes sera
importante. Par l'intermédiaire d'une hausse « fictive » de
température, il est alors possible de fournir au système une
certaine quantité d'énergie cinétique qui lui permet
de franchir les barrières d'énergie potentielle, et donc
d'explorer un espace conformationnel beaucoup plus large. Si la
température est suffisamment importante, une molécule est alors
potentiellement capable d'adopter toutes les conformations possibles.
Une simulation de dynamique
moléculaire se réalise généralement
en trois temps : après avoir attribué une vitesse initiale
aléatoire aux atomes par une distribution de type
Maxwell-Bolzman, une augmentation de
température est simulée afin d'atteindre rapidement
une valeur finale choisie (thermalisation).
La température est alors maintenue à cette valeur
constante afin de répartir l'énergie cinétique
sur toute la molécule (équilibration). Finalement,
la configuration du système est enregistrée à intervalles
de temps réguliers durant une phase de recueil de données
(collecte).
3.1.4) Le recuit simulé
Le recuit simulé est une méthode de dynamique
moléculaire couramment utilisée pour déterminer la
structure d'une protéine par modélisation sous contraintes
(Figure 3.12). Le nom de ce protocole s'inspire de la technique industrielle
de « recuit » qui consiste à refroidir très lentement
un matériau fondu, afin de lui permettre d'organiser au mieux
sa structure moléculaire.
1) thermalisation et équilibration
Energie potentielle
2) refroidissement
3) minimisation
1000 K
100 K
Figure 3.12 : Illustration d'une
procédure de recuit simulé par la représentation
schématique
d'une surface d'énergie potentielle. La
dynamique à haute température permet d'explorer l'espace
conformationnel et de s'éloigner de la structure initiale (non
représentée). Les structures générées
sont lentement refroidies de manière à occuper les zones
stables de la surface d'énergie. Une dernière étape de
minimisation permet d'optimiser les conformations
et d'obtenir finalement des minima énergétiques
plus profonds.
La méthode du recuit simulé consiste dans un
premier temps à mener une dynamique à haute température
(de 100 à 1000 K) à partir d'une structure initiale
aléatoire étendue préalablement minimisée. Cet
apport d'énergie cinétique permet d'explorer l'espace
conformationnel et de se dégager des minima locaux proches des
structures initiales. Après une période d'équilibration,
la température est lentement diminuée jusqu'à 100
K afin de réduire l'énergie cinétique et de
contraindre le système à occuper les états de plus
basse
énergie. On peut comparer schématiquement cette
étape à la phase de refroidissement de la
technique industrielle de « recuit » où une
diminution très lente de la température conduit à
l'obtention de l'espèce moléculaire la plus stable et qui
possède une énergie libre minimale. Finalement, les
dernières étapes du recuit simulé consistent à
minimiser puis à collecter les structures ainsi calculées. Il est
cependant important de noter que cette méthode ne garantit pas
l'identification du minimum énergétique global, mais s'en
approchera probablement dès lors que le jeu de contraintes
expérimentales est optimisé (diminution cohérente des
degrés de
liberté), et qu'un ensemble suffisant de configurations
est collecté.
3.2) Le logiciel CNS
Le logiciel CNS (Cristallography and NMR system ; Brünger et
al., 1998) est dédié à
la détermination et au raffinement de structures
tridimensionnelles à partir de données expérimentales
obtenues par RMN ou radiocristallographie. Il repose sur les principes
de mécanique et dynamique moléculaire et notamment celui du
recuit simulé.
3.2.1) Description du champ de force
Comme nous l'avons évoqué
précédemment, pour tenir compte des contraintes
expérimentales, les champs de force utilisés par le
programme CNS sont composés d'un terme d'énergie potentielle
empirique Epot, et d'un terme supplémentaire Econt
qui permet de contrôler le respect de ces contraintes :
E = Epot + Econt
a) La fonction d'énergie potentielle
Epot
(2.13)
L'énergie potentielle empirique d'une
molécule est définie par l'ensemble des
composantes qui décrivent sa géométrie. Elle
est la somme de deux termes, l'un représentant
les interactions entre atomes liés Eliés
et l'autre, les interactions entre atomes non liés
Enon_liés :
Epot = Eliés +
Enon_liés
(2.14)
La description succincte des différents termes de la
fonction Epot est réalisée à
partir
des composantes du champ de force CHARMM (Brooks, 1983) dont
dérive le champ de force
CHARMM22 utilisé pour le raffinement des structures
finales sélectionnées.
Terme des atomes liés :
Eliés =
Eliaisons +
Eangles
+ Eimpropres
+ Edièdres
(2.15)
2 Ó 2 Ó 2 Ó
Eliés =
Ó kr (r - r0) +
kè (è - è0) +
kù (ù - ù0) +
k (1 +
cosn)













r è ù
Les termes énergétiques associés aux
interactions entre atomes liés permettent en
premier lieu de maintenir la géométrie covalente
de la molécule. Ils rendent compte du coût
énergétique de l'élongation des liaisons covalentes
(r), des fluctuations, de l'angle è formé par
deux liaisons covalentes, de l'angle dièdre impropre ù
pour maintenir la chiralité d'un groupe d'atomes, ou bien encore
de l'angle de torsion formé par un groupe de quatre atomes (n
correspondant à la période de la fonction cosinus). Les
trois premiers termes de la fonction Eliés sont des potentiels
harmoniques centrés sur une position d'équilibre de
géométrie idéale
(r0,
è0, et
ù0). Ces valeurs de
référence dérivent de modèles
moléculaires obtenus, soit
expérimentalement par diffraction des rayons X ou
de neutrons, soit par des calculs théoriques de type ab
initio. Les différentes valeurs des constantes de force
kr, kè, et kù sont quant à
elles obtenues à partir d'analyses vibrationnelles de
molécules en phase gazeuse. Ainsi, par cette représentation de
la fonction d'énergie Eliés, la géométrie
moléculaire peut être considérée comme une
série d'oscillateurs composés de billes et de ressorts. La somme
des contributions de ce terme traduit alors les écarts des
structures calculées par rapport à des
géométries de référence.
Terme des atomes non liés :
Enon_liés =
Evan_der_Waals
+ Eélectrostatique
(2.16)
r
Enon_liés =
Ó
i,j
Aij
-
12
ij
Bij
6
r
ij
+ Ó
i,j
1
4ð år
. qiqj
å0 rij




qi




rij qj
Le terme d'énergie des interactions entre
atomes non liés Enon_liés est
la somme des
contributions correspondant aux interactions de type
électrostatiques et de van der Waals entre couples d'atomes. En
mécanique moléculaire, chaque atome est considéré
comme une masse possédant une charge ponctuelle. Selon ce principe,
l'interaction électrostatique entre deux charges partielles qi
et qj distantes de rij est définie par un
potentiel de type coulombien. L'intensité de l'interaction dépend
notamment de la distance rij, et de la constante
diélectrique
du milieu år. Le
paramètre år sert à
reproduire les effets d'écran du solvant entre les charges.
Aussi, un choix judicieux de la valeur år
permet de mimer de manière implicite l'effet du solvant et
de prendre en compte sa présence dans le calcul des interactions
électrostatiques.
Les interactions de van der Waals traduisent l'attraction
et la répulsion entre deux atomes séparés par une
distance rij et constituant un dipôle. Le terme
d'énergie correspondant
6
est un potentiel de Lennard-Jones qui contient une
composante attractive en 1/rij
12
due aux
forces de dispersion de London, et une composante
répulsive en 1/rij
due aux couches
électroniques des atomes les plus proches. Les
coefficients Aij et Bij dépendent de la nature des
atomes. Lorsque la distance rij est inférieure à la
somme des rayons de van der Waals, c'est le terme répulsif qui
prédomine et inversement pour une distance supérieure,
c'est le terme attractif qui agit principalement.
Selon la description des termes de la fonction
Enon_liée, des interactions de type électrostatiques
et de van der Waals peuvent en théorie être observées entre
deux atomes très éloignés en distance. La prise en
compte de l'ensemble des combinaisons possibles entre paires d'atomes
d'une macromolécule impliquerait un temps de calcul
considérable. Pour réduire la durée de ces calculs,
il est possible de limiter la portée des interactions en
définissant un « seuil de coupure ». Selon ce principe,
seules les interactions entre atomes séparés par une
distance inférieure à ce seuil seront prises en compte par le
champ de force. Aussi, pour éviter des discontinuités
d'énergie induites par l'introduction de ces seuils, la fonction
d'énergie Enon_liée est multipliée par une
fonction d'amortissement ou de « switch » (Brünger, 1992). Ce
type de fonction permet de faire tendre progressivement les
énergies électrostatiques et de van der Waals vers zéro,
et donc d'éviter des coupures brusques pouvant
générer des erreurs de calcul.
b) La prise en compte des contraintes RMN
Les contraintes expérimentales, introduites dans les
protocoles de recuit simulé sous forme d'intervalles de valeurs
permises, sont associées à des termes
énergétiques supplémentaires dans le champ de force de
CNS. Ces termes traduisent le degré de prise en compte de ces
données. Le non respect d'une contrainte conduit à une
pénalité énergétique, appelée violation, qui
informe de l'écart entre une valeur de distance ou d'angle dièdre
dans une structure calculée, et l'intervalle de valeurs
autorisées correspondant.
Terme des contraintes de distance :
Le logiciel CNS propose plusieurs types de fonctions
potentielles pour exprimer le
terme énergétique des contraintes de distance
EnOe. Dans le cadre du calcul de la structure du domaine K2, nous
avons utilisé les deux fonctions suivantes :
· Le potentiel carré (square) :
EnOe = KnOe . ä
n
avec :
ä = rmin - d pour d <
rmin
ä = d - rmax pour d >
rmax
(2.17)
où rmin et
rmax représentent les limites de
l'intervalle de distances permises pour une contrainte
donnée, d est la distance correspondante dans le
modèle, KnOe est la constante de force, et n
est généralement égal à 2.
Notons par ailleurs que le terme EnOe est
systématiquement nul lorsque la valeur de distance d est
encadrée par les valeurs rmin et rmax.
· Le potentiel biharmonique adoucie (softsquare)
Lorsque la distance d mesurée dans un
modèle se situe nettement en dehors de l'intervalle de valeurs
permises, le potentiel carré conduit à une
pénalité énergétique importante qui
pénalise le système et ralentit la convergence des structures.
Pour éviter que
les énergies de contraintes ne soient trop
élevées, le potentiel biharmonique adoucie est utilisé
lorsque l'écart entre la distance d et
l'intervalle de valeurs atteint un certain seuil. L'introduction
d'une asymptote à la courbe (kasym), et des constantes
a et b qui assurent la continuité de potentiel,
permet ainsi d'adoucir la fonction EnOe dans les cas extrêmes
(2.18).
EnOe = a + b/ä +
kasym.ä (2.18)
Terme des contraintes angulaires :
Le terme énergétique Edih
lié aux contraintes angulaires (ou ) est
représenté par une
fonction potentielle carrée comparable à celle
définie ci-dessus et de la forme suivante :
Edih = Kdih
( - x )2
(2.19)
avec x la limite minimum ou
maximum de l'intervalle de valeurs autorisées pour une
contrainte donnée, la valeur d'angle dans le
modèle, et Kdih la constante de force associée.
3.2.2) Description du protocole de recuit
simulé
Le paragraphe suivant décrit les différentes
étapes de la simulation de dynamique moléculaire
utilisée pour calculer la structure du domaine K2 de la protéine
humaine KIN17.
a) Génération des structures initiales
aléatoires
L'étape préalable aux calculs de dynamique
moléculaire consiste à générer des structures
initiales par tirage aléatoire des angles et . La topologie
de la protéine est obtenue à partir d'un champ de force
simplifié défini par les paramètres standard des fichiers
parallhdg et topallhdg. Le fichier topallhdg
décrit tous les acides aminés avec le type d'atome,
de liaison, et d'angle qui les constituent, ainsi que la charge partielle et la
masse des atomes. Ce fichier est utilisé par CNS pour créer le
fichier de topologie PSF spécifique à la séquence
du domaine K2. Les paramètres du champ de force sont
définis dans le fichier parallhdg qui contient toutes
les valeurs d'équilibre des liaisons et des angles de valence
dièdres et impropres relatives aux différents termes
énergétiques. La fonction d'énergie
potentielle Epot de ce champ de force simplifié
est de la forme suivante :
Epot = Eliaisons +
Eangles + Eimpropres + Erepel
(2.20)
Cette fonction exclue donc les termes
Edièdres et
Eélectrostatique uniquement pris en compte
dans
le champ de force CHARMM22 utilisé pour le
raffinement des structures. Par ailleurs, le terme des interactions de
van der Waals est remplacé par un potentiel continu purement
répulsif (Erepel) similaire à la composante
répulsive du potentiel de Lennard-Jones. Ce terme
est associé à une constante de force
kvd_Waals, et à un facteur multiplicateur
du rayon minimum
de van der Waals krepel.
b) Recuit simulé
Lors du recuit simulé, la fonction d'énergie du
système est recalculée après chaque pas
de simulation à partir de la position des atomes via les
équations de Newton. Les positions des protons géminés
sont régulièrement échangées pendant la
procédure (coordonnées et vitesses)
de manière à choisir la configuration dans laquelle
l'énergie est minimum. Il en est de même pour les
méthyles chiraux. Les valeurs des principaux
paramètres utilisés pendant la
simulation de dynamique moléculaire sont reportées
dans le Tableau 3.2.
|
Recuit simulé
|
T° [K]
|
Nombre
de pas
|
kvd_Walls
|
krepel
|
knOe
|
kdih
|
kr
|
kè
kù
|
|
i) Minimisation
ii) Chauffage
iii) Echantillonnage
iv) Refroidissement
v) Minimisation
|
-
2000
entre 1000
et 2000
1950?100
-
|
50
1300
2500
1000
2500
|
0.003
0.003?4
4
|
0.9
0.9?0.75
0.75
|
10?50
50
50
|
5
5
200
|
1000
1000
1000
|
500
500
500
|
Tableau 3.2 : Evolution des
principaux paramètres au cours du recuit simulé. Les
constantes
de force sont exprimées en kcal.mol-1.rad-2
à l'exception de kr et knOe
exprimé en kcal.mol-1.Å-2.
Le protocole de recuit simulé peut être
décomposé en 5 étapes distinctes :
i) La première étape consiste en une
minimisation de la structure initiale étendue par
un algorithme de type Powell.
ii) Le recuit simulé débute par une dynamique de
type Verlet où le système est porté à une
température de 2000 K. Les constantes de force relatives aux termes
d'énergie des élongations de distance, des angles de
valence, et des angles impropres sont maintenues à une forte
valeur pendant toute la durée de la simulation afin de
conserver la géométrie covalente de la protéine. A
contrario, la valeur de constante de force kvd_Waals est dans un
premier temps choisie faible dans le but d'autoriser les atomes
à s'interpénétrer, ce qui permet d'explorer l'espace
conformationnel. Aussi, le facteur krepel est fixé
à 0.9 afin d'éviter le collapse de la protéine.
Lors de cette première phase de chauffage, les
contraintes expérimentales sont prises en compte par introduction
des termes EnOe et Edih. Les contraintes de distance sont
par ailleurs
privilégiées via une valeur de
knOe progressivement augmentée de 10
à 50.
iii) La troisième étape du protocole est une phase
d'échantillonnage où la température
oscille entre 1000 et 2000 K. La constante kvd_Waals est
progressivement augmentée de
0.003 à 4 pour restreindre la
pénétration des atomes alors que le facteur
krepel est
diminué de 0.9 à 0.75.
iv) La dernière étape de dynamique consiste en un
refroidissement du système de 1950
à 100 K où les contraintes d'angles dièdres
sont pleinement prises en compte via une valeur de constante de force
fixée à 200.
v) La simulation s'achève par une
minimisation des structures calculées par un
algorithme de type Powell.
3.3) Le programme d'attribution automatique des pics nOe
du LSP
Développé récemment au Laboratoire de
Structure des Protéines du CEA de Saclay (LSP), le programme
d'attribution automatique des nOe est un module de gestion de scripts et
logiciels dédiés à la résolution de
structures tridimensionnelles de biomolécules par RMN (Savarin et
al., 2001). Outre l'attribution des pics nOe, les quatre autres principales
fonctions
du programme sont : la conversion des données
expérimentales en contraintes, l'utilisation et
le contrôle des calculs de CNS, l'exploitation des fichiers
de sortie de CNS, et l'analyse des structures. La succession de ces
différentes actions constitue un cycle de calcul.
L'attribution automatique des nOe est un processus qui n'est
efficace que s'il est mené
de manière itérative. De ce fait, le
programme procède au début de chaque cycle à une
nouvelle attribution qui dépend des modèles de plus basse
énergie de l'itération précédente.
La répétition des cycles de calcul permet
d'optimiser l'attribution des pics au fil des itérations, et
conduit à une convergence des structures vers un repliement
unique. Les paragraphes suivants sont consacrés à la
description des méthodologies utilisées par le programme
pour attribuer et convertir les volumes des pics nOe en contraintes de
distance.
3.3.1) Gestion des contraintes de distance
a) Notion de contrainte de distance
ambiguë
Comme nous l'avons évoqué dans le chapitre 2.1.1,
le volume d'un pic de corrélation dipolaire Vij entre deux
protons i et j peut être relié à la
distance rij séparant les deux noyaux
via un facteur de calibration (ou de
référence) Rcal :
rij = Rcal
1
Vij
1/6
(2.21)
La distance rij constitue
alors une contrainte de distance non ambiguë. En revanche,
lorsque
plusieurs attributions sont possibles pour un
même pic, le programme d'attribution automatique considère
le volume de ce pic Vx comme la somme des contributions dipolaires
entre plusieurs paires de protons :
Vx =
n
6
1
r
Ó
x=1 x
6
Rcal
(2.22)
avec rx l'ensemble des distances entre paires d'atomes
i et j relatif au n nombre d'attributions possibles.
Selon ce principe, la contrainte de distance ambiguë r introduite
par Nilges en 1995
(Nilges, 1995) peut s'écrire de la manière suivante
:
n 1 -1/6
6
r
r = Ó
x=1 x
Rcal
(2.23)
Cette notion de contrainte ambiguë est fondamentale car elle
va permettre d'intégrer au calcul
de la structure des informations nOe dont l'attribution
est incertaine. La gestion des attributions ambiguës et non
ambiguës est un des principes sur lequel repose la stratégie du
programme d'attribution automatique.
b) Calibration des distances inter-atomiques
La conversion des volumes nOe en distance nécessite de
déterminer préalablement la valeur du facteur de calibration
Rcal relative à chaque expérience NOESY-HSQC. La
méthode classique consiste à relever le volume de plusieurs pics
référents Vref qui correspondent à la
corrélation de 2 protons géminés aliphatiques, ou
de 2 protons aromatiques appartenant au même cycle, et dont la
distance rref les séparant est connue et fixe. La valeur de
Rcal peut alors être obtenue par la relation suivante :
Rcal =
rref
1/6
Vmoy
(2.24)
avec Vmoy la moyenne des
volumes Vref. Cette méthode manuelle
et simple permet une
première estimation approximative de la valeur de
facteur de calibration utilisée lors des premières
itérations. Le programme d'attribution automatique offre la
possibilité d'obtenir une calibration plus exacte à partir
des premières structures repliées de la protéine.
La procédure consiste à comparer les distances calculées
à partir du volume nOe (rnOe) avec ces mêmes
distances mesurées dans les modèles
(robs) (2.25). Selon ce principe, une valeur de
rapport C proche de 1 indique que la valeur de facteur
de calibration Rcal utilisée est adaptée à
la quantification des volumes nOe. Dans le cas contraire, le
paramètre Rcal doit être
réajusté
selon que la valeur de C est inférieure ou
supérieure à 1. Cette méthode est avantageuse car elle
donne la possibilité d'affiner la calibration au fil des
itérations.
Óij
C =
Óij
nOe
rij
obs
rij
(2.25)
c) Les intervalles de distances permises
Comme nous l'avons évoqué dans le paragraphe
2.1.1, le rapport de proportionnalité entre le volume nOe et la
distance inter-atomique 1H repose sur l'approximation d'un
mouvement isotrope de la molécule étudiée (ôc
moyenné). Aussi, la différence de ôc
résultant
de mouvements internes de la protéine, et le
phénomène de diffusion de spin, rendent la quantification
du nOe approximative. Pour tenir compte de cette imprécision,
les distances inter-protons sont classiquement introduites dans les
protocoles de calcul CNS sous forme
d'un intervalle de distances permises :
rij - ? < rij < rij + ?
(2.26)
avec ? représentant l'erreur sur la contrainte de
distance rij. Le programme d'attribution automatique propose
à l'utilisateur 3 expressions distinctes pour calculer le
paramètre ? :
mode 1 : ? = 0.25.rij
mode 2 : ? = 0.125.(rij)2
mode 3 : ? = 0.15.rij
(2.27)
Ces expressions peuvent être utilisées pour
définir l'erreur sur les contraintes de distance entre protons de
la chaîne principale (variable ER_MODE_SQE), et sur les
contraintes d'autre nature (variable ER_MODE). Dans le cas de
l'étude du domaine protéique K2, l'incertitude ?
sur la distance rij séparant deux
protons de la chaîne principale a été estimée
à 25% de rij (ER_MODE_SQE = 1). L'amplitude
des mouvements de chaîne latérale pouvant influer de
manière significative sur l'intensité du nOe, l'erreur sur
la distance séparant un proton de chaîne latérale
avec un autre noyau est généralement plus importante et a
été estimée à 12.5%
du carré de la distance rij (ER_MODE =
2).
Par ailleurs, les intervalles de valeurs permises doivent
tenir compte de la réalité physique du nOe. Ainsi, le
programme tolère une limite maximale définie par la
variable
DRMNMAX et contrôlée par le mode
DRMNMAX_MODE. Le mode suppression conduit au
rejet d'une contrainte dont la limite maximale est
supérieure à la valeur de DRMNMAX. Pour notre part, la
valeur de DRMNMAX a été fixée à 5.3
Å, et nous avons préféré utiliser le mode
truncature qui réduit systématiquement la limite
maximale d'une contrainte à la valeur de DRMNMAX lorsque
celle-ci est supérieure à DRMNMAX.
3.3.2) Préparation des données
initiales
L'utilisation du programme d'attribution automatique du LSP
nécessite dans un premier temps de préparer un certain nombre
de données initiales contenant :
- Une table des déplacements chimiques des noyaux
1H, 15N, et 13C de la protéine pour
chaque expérience NOESY-HSQC éditée 15N, et
13C.
- Une liste des pics nOe de chaque expérience où
sont reportés le volume de chaque pic
et ses coordonnées en déplacement chimique.
- Un fichier de contraintes de distances non ambiguës
attribué manuellement et qui renseigne sur la topologie de la
protéine (fichier ss.cons).
- Un fichier de contraintes dihédrales sur les angles et
Le fichier de topologie, appelé ss.cons,
contient des informations de distance qui renseignent sur la structure
secondaire et tertiaire de la protéine. Ces éléments
obtenus après analyse des paramètres structuraux RMN indiquent
principalement, la topologie des feuillets
â déduite des nOe caractéristiques
inter-brins, et la formation de liaisons hydrogène déduites des
expériences de spectroscopie d'échange. Il est à
noter que les fichiers de contraintes dièdres et de topologie ne
sont pas utilisés de manière directe pour attribuer les pics nOe
dans
le programme d'attribution automatique. Ces
données sont converties au format CNS, et systématiquement
introduites dans les protocoles de calcul CNS à chaque itération,
et quelles que soient les attributions choisies. Par conséquent,
un certain nombre d'informations de structure tertiaire sont
dupliquées dans les fichiers de contraintes de CNS.
3.3.3) Stratégie d'attribution des
pics
a) Collecte des possibilités
d'attribution
La première phase du processus d'attribution
automatique consiste à comparer les valeurs des tables de
déplacements chimiques de la protéine aux déplacements
chimiques des
pics nOe afin d'établir la liste de toutes les
attributions possibles de chaque pic. Pour cela, un
intervalle de tolérance est défini par
l'utilisateur pour chacune des trois coordonnées des nOe.
Les gammes de tolérance classiquement testées
sont : #177; 0.02 ppm dans la dimension 1H
d'acquisition, #177; 0.25 ppm sur la fréquence de
résonance de l'hétéroatome 15N ou
13C, et
#177; 0.04 ppm dans la dimension 1H
indirecte. Les nOe pour lesquels aucune possibilité d'attribution
n'est déterminée constituent la liste des non
attribués, et sont soumis à un nouveau processus
d'attribution lors de l'itération suivante.
Par ailleurs, il est parfois constaté pour certains
protons une légère différence entre la valeur de
déplacement chimique définie dans les tables et la valeur
réelle sur les spectres NOESY-HSQC. Aussi, le programme
d'attribution automatique repère ces « décalages »
systématiques afin que l'utilisateur puisse modifier en
conséquence les tables de déplacements chimiques de la
protéine en fin d'itération.
b) Le paramètre de seuil
Une fois établie la liste de toutes les
possibilités d'attribution relatives à chaque pic, chacune des
corrélations potentielles entre 2 protons i et j est
associée à une valeur de distance
dx. Le paramètre dx correspond
à la distance moyenne qui sépare les 2 protons i et
j dans les modèles de plus basse énergie de
l'itération précédente (ou dans les structures
initiales aléatoires pour la première itération). Il
constitue la donnée principale utilisée par le programme
pour choisir le nombre d'attributions de chaque pic. La méthode
consiste à convertir chaque moyenne de distance dx,
relative à une possibilité d'attribution, en une
intensité nOe théorique moyenne
Ix selon la relation suivante :
1
Ix = 6
dx
(2.28)
Par cette définition, la contribution
Ix traduit véritablement le poids
d'une attribution par
rapport à une autre : plus la distance moyenne
dx entre 2 protons est courte, et plus la contribution
moyenne associée Ix sera importante. Ainsi, à
l'issue de cette seconde étape, chaque possibilité
d'attribution d'un pic donné est associée à un poids
différent.
A ce stade du traitement des données, le programme utilise
un paramètre de seuil p, compris entre 10-4 et 0.3,
pour déterminer le nombre d'attributions retenues pour chaque pic :
les contributions Ix sont
classées par ordre décroissant, puis chacune d'entre elles
est
comparée à la précédente en
commençant par la contribution la plus forte (systématiquement
sélectionnée). Si pour une possibilité donnée,
la valeur de son poids Ix-1 est supérieure au produit
pIx, alors l'attribution est retenue et le poids Ix-2 est
ensuite comparé au produit pIx-1
de la même manière. Dans le cas contraire, si
Ix-1 < pIx, alors
l'attribution est rejetée ainsi que
toutes les autres dont la valeur de contribution
Ix-n est inférieure à Ix-1. Le processus
de sélection du nombre d'attributions est alors terminé. Au
final, ce nombre ne peut excéder 20 attributions par pic.
Prenons l'exemple d'un pic pour lequel 5 attributions
sont possibles. Dans le cas le plus simple, considérons les
corrélations potentielles d'un proton A avec respectivement, un proton
B, C, D, E, ou F. A chacune de ces possibilités d'attribution
correspond, une distance moyenne séparant chacun des 2 protons
respectifs dans les modèles précédents (dAB,
dAC, dAD, dAE, et dAF), et une contribution
moyenne associée (IAB, IAC, IAD,
IAE, et IAF). La figure 3.13 représente l'histogramme
des contributions classées par ordre décroissant.

Ix
pIAB
pIAC
pIAD
AB AC AD AE AF
Figure 3.13 : Principe de la
sélection du nombre d'attributions d'un pic nOe en fonction du
paramètre de seuil p. Les contributions sont dans un premier
temps triées par ordre décroissant puis chacune d'elles est
comparée à la précédente. Une possibilité
d'attribution
est sélectionnée si et seulement si Ix-1 >
pIx.
Dans notre exemple, le poids IAB constitue la
contribution la plus importante et sert donc de point de départ. La
valeur de contribution IAC étant supérieure au produit
pIAB, la possibilité d'attribution AC est par
conséquent sélectionnée. La valeur de IAC
est ensuite utilisée pour définir le seuil
d'attribution suivant et conduit à la sélection de la
possibilité AD car
IAD > pIAC. Chaque contribution est ainsi
comparée à la précédente tant que la valeur de
Ix-1 est
supérieure au produit pIx. Dans notre exemple, ce
n'est plus le cas pour la possibilité AE car
IAE <
pIAD, ce qui amène à rejeter les
attributions AE et AF. Finalement, seules les attributions
AB, AC, et AD sont maintenues après application du
paramètre de seuil.
Lors des premières itérations, l'utilisation d'une
valeur faible de paramètre de seuil p
favorise les attributions multiples et par conséquent, la
constitution de contraintes ambiguës.
Au fil des calculs, l'augmentation de la valeur de
p permet de restreindre le nombre d'attributions possibles pour
chaque pic, et donc de diminuer la quantité de contraintes
ambiguës au profit des contraintes non ambiguës.
La sélection du nombre d'attributions par un
paramètre de seuil présente plusieurs atouts. Le premier
réside dans la capacité du programme à
intégrer un maximum de possibilités d'attribution lors
des premières itérations, et donc à explorer
« l'espace conformationnel des nOe ». En effet, l'ajout
d'attributions ambiguës à une contrainte de distance ne
conduit pas à une violation énergétique tant que la
véritable attribution figure dans
les différentes possibilités. La multiplication
des contraintes ambiguës ne pénalise donc pas le système
lors des premières itérations, mais elle contribue à une
dilution de l'information, et limite la convergence des modèles.
C'est pourquoi, la valeur de paramètre de seuil est
incrémentée au fil des calculs afin de réduire
progressivement le nombre d'attributions de chaque pic. Par ailleurs,
notons ici le rôle important du fichier de topologie ss.cons qui
permet d'orienter le repliement de la protéine dès les premiers
calculs et par conséquent, de guider indirectement le programme vers les
attributions les plus probables d'un point de vue de la structure 3D.
Finalement, l'augmentation progressive du paramètre de seuil
conduit à la convergence des structures
générées vers un repliement unique dont les
distances intra-
moléculaires 1H correspondent le mieux à
un jeu de données expérimentales nOe.
c) Nombre de structures contribuant à une
attribution
Comme nous venons de l'évoquer, la sélection
du nombre d'attributions par pic est obtenue à partir de l'analyse
des 10 structures de plus basse énergie de l'itération
précédente
via le paramètre dx. Aussi, la distance rij
relative à une possibilité d'attribution peut être
très variable d'un modèle à l'autre, et notamment lors des
premières itérations où la convergence des structures est
faible. C'est pourquoi, le programme utilise une valeur seuil cut-off
de 20 Å pour considérer une possibilité d'attribution
dans un modèle. Dès lors, seules les possibilités
qui répondent à la condition rij
< 20 Å dans un nombre suffisant de structures sont
sélectionnées. On parle ainsi de nombre de
structures contribuant à une attribution. La valeur
de ce paramètre est progressivement augmentée au
fil des calculs (de 4 à 6), ce qui contribue à
la convergence des attributions, et donc des structures au fil
des itérations.
d) Gestion des pics intra-résiduels
Deux modes sont proposés par le programme d'attribution
automatique pour traiter les nOe relatifs aux corrélations
intra-résiduelles. En mode intra, chaque possibilité de
type intra- résiduel est associée à un
paramètre dx de 1.0 Å, ce qui favorise
considérablement ce type d'attribution par rapport aux autres
possibilités via une valeur forte de Ix. En mode
non-intra,
le paramètre dx est
utilisé comme défini dans le paragraphe précédent,
et quel que soit le type
de corrélation. Le mode intra,
utilisé pour la plupart des itérations, permet de limiter
le nombre de contraintes ambiguës au profit d'attributions plus
évidentes, ce qui conduit à une convergence plus rapide des
structures. Ce mode favorise toutefois à outrance les attributions
intra-résiduelles. Aussi, le mode non-intra est utilisé
lors des 3 dernières itérations, lorsque la protéine est
repliée, afin de tenir compte du repliement de la protéine de
manière plus réaliste pour réaliser l'attribution
automatique des pics.
e) Gestion des pics symétriques
Sur les spectres NOESY-HSQC éditée
13C, la présence d'un pic de corrélation entre 2
protons aliphatiques i et j sur un plan
13Ci peut être accompagnée d'un pic
réciproque ou symétrique sur le plan 13Cj
lorsque la résolution spectrale le permet. Dans le cas d'une
corrélation de type inter-résiduelle, l'identification de 2
pics réciproques est un argument supplémentaire qui
conforte leur attribution. Aussi, lorsque 2 pics symétriques sont
repérés
par le programme, l'attribution de type «
réciproque » est favorisée par rapport aux autres
possibilités en recevant une valeur de paramètre dx de
1.5 Å. Après application du paramètre
de seuil, si ces 2 pics sont dotés de la même
attribution, le programme supprime alors une des
2 informations et convertit la distance moyenne en contrainte.
3.3.4) Les filtres d'attribution
Une fois établie la liste des attributions
sélectionnées, le programme met à disposition
de l'utilisateur un certain nombre de filtres
d'attribution dont l'objectif est d'améliorer la qualité
des attributions, et donc des structures générées,
en favorisant les conformations
vraisemblables. L'application de ces différents
filtres conduit à une diminution du nombre
d'attributions et constitue la dernière étape avant
la conversion en contraintes de distance au format CNS.
a) Le filtre dRMN trop grande
Malgré la notion d'intervalle de distances
permises introduite pour tenir compte de l'incertitude de la
quantification du nOe, il est possible que les distances
associées aux volumes de certains pics soient démesurées
par rapports aux distances correspondantes dans
les structures calculées. Ceci est notamment
observé, pour des effets à longue distance, lors des
premières itérations où la structure n'est encore
que partiellement repliée. Aussi, l'intégration de ce type de
contraintes peut conduire à des violations et pénalités
énergétiques importantes. Le filtre dRMN trop grande
repère et supprime ces pics trop intenses avant leur conversion en
contraintes. Lors des itérations finales, l'application de ce
filtre ne doit conduire qu'à rejeter un très faible nombre de
pics pour lesquels la distance déduite du nOe apparaît trop courte
en raison de problèmes d'homogénéité de la
calibration ou de dynamique intra-moléculaire inhérents à
la technique RMN.
b) Le filtre DIAG
Le filtre DIAG (pour diagonal plot) est
utilisé pour éviter le calcul de conformations
qui correspondent à un repliement inexact ou
incohérent d'une partie de la protéine. Il recherche et
supprime les attributions longue distance dites « isolées ».
En pratique, lorsque l'attribution d'un nOe est associée à
la corrélation entre 2 protons appartenant à 2
résidus éloignés dans la séquence primaire, si
aucun autre pic ne correspond à un contact longue distance
entre protons appartenant aux mêmes résidus ou à
des résidus voisins, alors l'attribution est considérée
comme suspecte. Le pic est alors filtré mais l'attribution n'est pas
définitivement supprimée. Au fil des itérations, le
pic peut être à nouveau considéré si
l'évolution du repliement fait apparaître d'autres nOe
longue distance qui impliquent les mêmes régions de la
protéine.
c) Le filtre SUPRES
L'option SUPRES permet d'utiliser les informations
de flexibilité des résidus non structurés des
extrémités N- et C-terminales pour diminuer le nombre
d'attributions. En effet,
ces résidus en échange conformationnel très
rapide ne peuvent donner naissance à des nOe
longue distance sur les spectres de type NOESY
où l'information est moyennée sur l'ensemble de l'espace
conformationnel. Par conséquent, à partir d'une liste de
ces résidus fournie par l'analyse du nOe
hétéronucléaire 1H-15N (cf.
§ 2.2.4), l'option SUPRES rejette systématiquement
chaque attribution de type longue distance qui corrèle un proton de
résidu quelconque avec un autre appartenant à un résidu de
cette liste.
d) Le filtre SUPRES_LAT
Selon le même principe, cette option supprime
l'ensemble des attributions inter- résiduelles qui associent un
proton de résidu quelconque avec un proton de chaîne
latérale d'un résidu de la liste SUPRES_LAT. Ainsi, les
filtres SUPRES et SUPRES_LAT permettent
de tenir compte de données dynamiques obtenues par
RMN, et par conséquent, d'éviter le calcul de structures
incohérentes avec ces paramètres.
e) Le filtre SUP2_ini
Le fichier SUP2_ini est une liste arbitraire
qui définit l'ensemble des attributions interdites pour chaque pic
donné. Cette option offre à l'utilisateur la possibilité
de contrôler partiellement l'attribution de certains pics nOe. Aussi, les
filtres d'attribution ne sont utilisés qu'après application du
paramètre de seuil p. Par conséquent, le filtre
SUP2_ini ne permet pas d'influencer de manière directe
l'attribution des nOe, mais conduit au rejet des pics dont une certaine
attribution n'est pas souhaitée.
3.3.5) Gestion des violations
Les violations de distance systématiques
retrouvées à chaque itération sont souvent dues
à des erreurs d'attribution de résonance de la
protéine, ou à l'intégration de pics qui
correspondent en réalité à du bruit spectral. Aussi, les
contraintes qui engendrent ce type de violations supportent une information
erronée, qui peut ralentir la convergence du repliement
au fil des itérations, ou conduire à des
conformations inexactes. Pour pallier ce problème, le programme
d'attribution exploite les fichiers de sortie du logiciel CNS à
la fin de chaque cycle, et établit la liste SUP2 des
violations de distance supérieures à 0.5 Å communes
à au moins 4 des 10 meilleures structures. Chaque attribution
qui figure sur cette liste est alors
supprimée lors de l'itération suivante
après application du paramètre de seuil. Notons par
ailleurs que la composition du fichier SUP2 est
différente à chaque itération : aucune
attribution n'est supprimée définitivement et c'est
à l'utilisateur que revient cette tâche.
3.3.6) Analyse et validation des structures
Les ressources informatiques du Laboratoire de Structure des
Protéines permettent le calcul d'un grand nombre de structures. Une
sélection s'impose donc pour ne conserver que
les meilleures, du point de vue de la
géométrie covalente et du respect des contraintes
expérimentales. Plusieurs critères sont classiquement
utilisés pour évaluer leur qualité :
a) Le logiciel PROCHECK
Les valeurs des angles dièdres et des modèles
générés doivent occuper les zones permises du diagramme
de Ramachandran qui définit les régions favorables de
l'espace conformationnel (Ramachandran et al., 1971). Le logiciel
PROCHECK (Laskowski et al.,
1996) permet d'observer la répartition de ces couples
d'angles, et facilite l'identification des structures secondaires. On
considère généralement que les structures finales
sont de bonne qualité lorsque moins de 1% des valeurs d'angles et
occupent les régions interdites.
b) Les termes énergétiques
La fonction d'énergie est un paramètre
fondamental pour valider les structures calculées. Les valeurs des
différents termes énergétiques reflètent la
qualité de la géométrie covalente, et renseignent sur
le degré de prise en compte des contraintes
expérimentales. Aussi, l'objectif de la répétition
des processus itératifs n'est autre que de générer
des structures dont l'énergie associée est la plus basse
possible. Selon ce principe, les structures finales doivent
présenter une bonne géométrie covalente, des
violations de distance inférieures à 0.5 Å, et des
violations d'angles dièdres inférieures à 10°, pour
être considérées comme étant de bonne
qualité.
3.3.7) Description des cycles
itératifs
La phase préliminaire du processus itératif
consiste à générer 10 structures aléatoires
à partir des fichiers de paramètres CNS qui définissent le
champ de force et la géométrie de la protéine.
L'attribution automatique de la première itération est
réalisée à partir de ces structures aléatoires,
et des différentes listes de déplacements chimiques et de pics
nOe. Le nombre de structures contribuant à une attribution doit
être supérieur à 4, et la valeur de p est
fixée à 0.0001. Les facteurs de calibration sont dans un
premier temps estimés à partir du volume de pics
référents sur les différents spectres NOESY-HSQC.
Après application des filtres d'attribution, le premier jeu de
distances inter-atomiques 1H est constitué. Les
contraintes dièdres et de distance (dont les données de
topologie) sont converties au format CNS, puis intégrées aux
protocoles de dynamique moléculaire. Lors de la première
itération,
un premier jeu de 100 structures est
généré par le logiciel CNS. Le programme d'attribution
prend alors en charge le traitement de ces 100 modèles. Les
20 structures de plus basse énergie sont
sélectionnées, triées par ordre croissant
d'énergie, puis les 10 premières font l'objet d'analyses
systématiques : bilan des énergies moyennes pour chaque terme du
champ
de force, analyse des angles et par le logiciel
PROCHECK, et calcul des écarts quadratiques moyens sur le
squelette. Le programme permet également d'obtenir une
synthèse des violations de distance systématiques sur ces 10
meilleurs modèles à partir des fichiers de sortie du logiciel
CNS.
Après traitement des résultats de la
première itération, le programme entre dans un processus
itératif de 22 cycles où se succèdent l'attribution des
nOe, le calcul des modèles, et l'analyse des 10 meilleures structures
(Figure 3.14). A l'issue de chaque cycle, les 10 modèles
de plus basse énergie, ainsi que la liste de
leurs violations, sont utilisés pour réaliser l'attribution
de l'itération suivante. Ces 10 meilleurs modèles
constituent également les structures de départ des
protocoles de dynamique moléculaire. Au fil des
itérations, les valeurs de paramètre de seuil et de
nombre de structures contribuant à une attribution sont
progressivement augmentées (Figure 3.15). Il en est de même pour
le nombre de structures
calculées qui est fixé à 200 à partir
du treizième cycle, puis à 300 à la vingtième
itération.
Structures 3D aléatoires

Données initiales
· Listes des pics nOe (déplacements chimiques +
volume)
· Tables de déplacements chimiques 1H,
15N, et 13C
· Fichier de topologie ss.cons
· Fichier Talos de prédiction des angles et

début du cycle itératif
Attribution des pics
· Collecte des possibilités d'attribution :
- intervalles de tolérance
- nombre de structures contribuant à une attribution
· Mesure des distances moyennes dx
sur les modèles de l'itération
n-1
· Calcul des contributions associées Ix
· Application du paramètre de seuil p
fin du cycle itératif :
utilisation des structures 3D des
10 meilleurs modèles
Analyse des 10
meilleures structures

Filtres d'attribution
· dRMN trop grande
· DIAG
· SUPRES, SUPRES_LAT
· SUP2_ini
· SUP2 (violations n-1)

création des contraintes
· PROCHECK ( et )
· Bilan des énergies
· RMSD sur le squelette
· Violations systématiques
sélection des 20 structures de plus basse
énergie

Calcul des structures sous CNS
· Dynamique à haute température sous
contraintes RMN (recuit simulé)
· Création des fichiers de sortie (énergies,
violations...)
Figure 3.14 : Description d'un
cycle itératif du programme d'attribution automatique des
nOe développé au Laboratoire de Structure des
Protéines du CEA de Saclay (Savarin et al.,
2001).
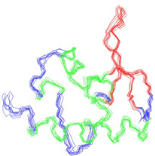
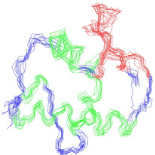
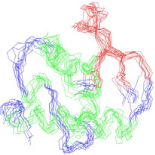
Chapitre 3 : Stratégie d'étude par RMN et
Modélisation Moléculaire du domaine K2
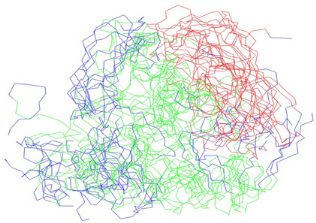
179
p 10-4
Na 4
Ns
10-3
5
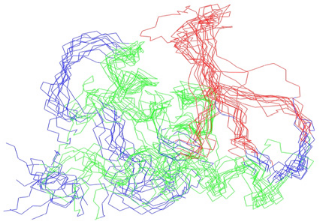
3.10-3
100
5.10-3
10-2
0.1
6
0.2
200
0.3
300
It
1 4 7
179
10 13 16 19 22
Figure 3.15 : Evolution de
paramètres du programme d'attribution au fil des 22 cycles (It) du
processus itératif. Les différentes annotations
correspondent au nombre de structures calculées (Ns),
au nombre de structures contribuant à une attribution (Na), et au
paramètre de seuil (p)
Chapitre 3 : Stratégie d'étude par RMN et
Modélisation Moléculaire du domaine K2
Finalement, les 20 meilleures structures du dernier cycle sont
soumises à une étape de
raffinement dans le champ de force CHARMM22 en solvant
implicite. Les structures convergentes obtenues lors des dernières
itérations sont utilisées pour réajuster les valeurs de
facteur de calibration via le calcul du rapport C. Ces valeurs pourront
être appliquées lors des processus itératifs suivants, puis
à nouveau optimisées en recalculant le rapport C.
Comme nous l'avons évoqué
précédemment, les violations sont dans la plupart des cas dues
à des erreurs d'attribution de résonance de la protéine,
ou à des inexactitudes inhérentes
à la spectroscopie de RMN (recouvrement
spectral, dégénérescence du signal, bruit,
dynamique interne...). L'analyse manuelle des violations en fin de chaque
processus itératif
de 22 cycles est par conséquent nécessaire
afin d'identifier les informations erronées en contradiction avec
le repliement de la protéine. La correction progressive du jeu de
données expérimentales est alors concomitante avec la
baisse des énergies, la convergence des
structures, le respect du diagramme de Ramachandran, et
l'élimination des violations.
Chapitre 4 : Caractérisation structurale du
domaine K2 par RMN et Modélisation Moléculaire
CHAPITRE 4
Caractérisation structurale du domaine K2
par RMN et Modélisation Moléculaire
1) Détermination de la structure du domaine K2
de KIN17 humaine
1.1) Attribution des raies de
résonance
L'attribution des raies de résonance des 111
acides aminés du domaine K2 a été
réalisée selon la stratégie définie dans le
chapitre précédent. Les différentes expériences 3D
hétéronucléaires utilisées pour
déterminer le déplacement chimique de chaque noyau, et
recueillir les contraintes expérimentales, ont été
enregistrées à 30°C et à pH 6.0 sur un
spectromètre 600 MHz équipé d'une cryosonde triple
résonance 1H, 15N, 13C.
Un échantillon unique de protéine doublement
marquée 15N / 13C concentrée à 0.7
mM
a suffi pour mener à bien l'attribution
séquentielle du squelette peptidique. La combinaison des
expériences HNCO, (HCA)CO(CAN)NH, HNCA, CBCANH, et
CBCA(CO)NH a conduit à l'identification des valeurs de
déplacement chimique des carbones 13CO,
13Cá, et
13Câ des résidus i et i-1 relatifs
à chaque groupement amide. Il est à noter que
l'expérience
(HCA)CO(CAN)NH, plus sensible que HN(CA)CO, a
été préférée pour obtenir les
corrélations des 13CO intra- et inter-résidus.
De plus, l'expérience HN(CO)CA, qui a également
été enregistrée, n'a pu être
exploitée en raison d'un problème d'artéfacts.
L'interprétation de ces 5 expériences par paire a permis dans un
premier temps de connecter
les résidus deux à deux, puis de reconstituer
l'ensemble de la séquence peptidique à partir des acides
aminés facilement identifiables tels que les glycines,
thréonines, sérines, alanines, et prolines. La Figure 4.1
présente un exemple d'attribution séquentielle de la
région F23-L28. L'attribution de la chaîne principale a
finalement été complétée par l'analyse des
spectres HNHA et HBHA(CO)NH.
L'optimisation préalable des conditions d'analyse a permis
d'obtenir des spectres de qualité qui contiennent une quantité
d'information satisfaisante. Dès lors, la bonne dispersion
des pics sur le spectre HSQC 15N-1H a facilité
l'attribution de la totalité des résonances
1HN,
1Há, 15NH, 13CO, et
13Cá du domaine K2 à l'exception des résidus
G1, Q2, et R53 dont les déplacements chimiques du groupement amide
sont manquants. L'absence de corrélation
15N-1H de résidu appartenant
à une extrémité flexible N- ou C-terminale est un
phénomène classique qui s'explique par l'échange
rapide du proton amide exposé au solvant avec les
protons de l'eau. Aussi, en anticipant sur la
résolution de la structure tridimensionnelle,
F23 F23 R24 R24 N25 N25 D26 D26 F27 F27 L28 L28
176.5
177.2
|
13CO E22
|
|
13CO F23
|
|
13CO R24
|
|
13CO N25
|
|
13
CO
D26
|
|
13CO F27
|
|
A) 178.0
178.8
179.5
ppm
13CO
F23 F23 R24 R24 N25 N25 D26 D26 F27 F27 L28 L28
B) 13Cá
E22
13Cá
F23
13Cá
R24
13Cá
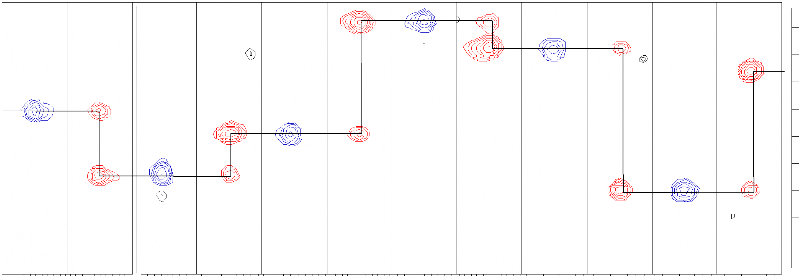
N25
13Cá
D26
13Cá
F27
56.0
58.0
60.0
62.0
64.0
ppm
F23 F23 R24 R24 N25 N25 D26 D26 F27 F27 L28 L28
26.0
13Câ
E22
C)
13Câ
R24
30.0
34.0
13Câ
13Câ
F23
13Câ
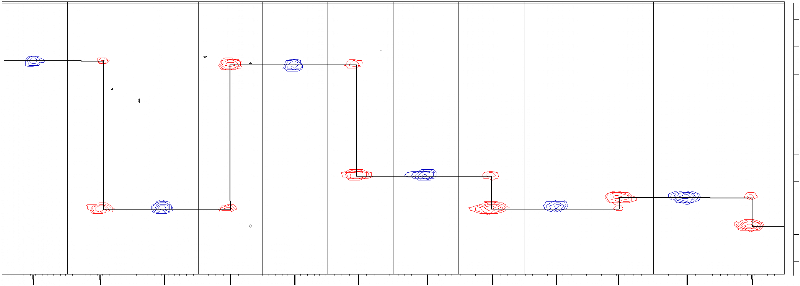
N25
13Câ
D26
13Câ
F27
42.0
8.61
8.61
8.48 8.48 8.72 8.72 8.97 8.97 8.81 8.81 8.43 8.43
1HN
ppm
Figure 4.1 : Attribution des
fréquences 13CO, 13Cá, et
13Câ de la région F23-L28 du domaine
K2. Les bandes sont extraites des plans
15N des spectres, A) HNCO (en bleu) et (HCA)CO(CAN)NH
(en rouge), B) CBCA(CO)NH (en bleu) et HNCA (en rouge), et C)
CBCA(CO)NH (en bleu) et CBCANH (en rouge) au déplacement chimique
1HN des résidus correspondants. Le chemin d'attribution est
indiqué par des lignes horizontales fléchées.
l'échange rapide du proton amide R53 peut être
expliqué par son appartenance à un résidu
situé dans une boucle, et à sa forte exposition au
solvant.
Les fréquences de résonance des noyaux
1H et 13C des chaînes latérales ont
été déterminées à partir des spectres
HCCH-TOCSY, HCCH-COSY, et 13C-NOESY-HSQC. Un échantillon
unique de protéine doublement marquée 15N /
13C concentrée à 0.7 mM, préalablement
lyophilisée, puis solubilisée dans du D2O (99.9 %) a
suffi pour enregistrer l'ensemble de ces expériences sur les
régions aliphatiques et aromatiques. La totalité des noyaux
des chaînes latérales aliphatiques des 111 acides aminés de
K2 a été attribuée. En ce
qui concerne les chaînes aromatiques, toutes les
fréquences de résonance des résidus histidine, tyrosine,
et tryptophane ont été identifiées à l'exception
des proton et carbone Z2 du résidu W63. Le bilan de l'attribution
est plus mitigé pour les phénylalanines, et seules 3 des
6 chaînes latérales ont été totalement
attribuées (F19, F23, et F15). Par ailleurs,
l'expérience
15N-TOCSY-HSQC a été
enregistrée sur un échantillon uniformément
marqué 15N afin de
vérifier l'attribution. Cependant, compte tenu de la
faible efficacité du transfert TOCSY 1H notamment due
à la taille de la protéine K2, moins de la moitié des
valeurs de déplacement chimique aliphatique 1H a pu
être contrôlée. La dernière étape de
l'attribution du domaine K2
a consisté à enregistrer le spectre
15N-NOESY-HSQC sur l'échantillon 15N, ce qui a
permis
d'attribuer la totalité des 13 groupements NH2
de résidus asparagine et glutamine à l'exception de
N42.
L'identification des fréquences de résonance
du domaine K2 de la protéine KIN17 humaine a fait l'objet d'une
note d'attribution soumise et acceptée dans la revue Journal of
Biomolecular NMR (Carlier et al., 2006). Cet article,
présenté dans les pages suivantes, est accompagné d'un
supplément technique (Supplemental Material) disponible
sur le site de J.Biomol.NMR. à l'URL suivante :
http://dx.dio.org/10.1007/10858-006-0013-y.
Journal of Biomolecular NMR (2006) (c) Springer 2006
Letter to the Editor
NMR assignment of region 51-160 of human KIN17, a DNA and
RNA-binding protein
DOI 10.1007/s10858-006-0013-y
KIN17 is a 45 kDa nuclear protein which is remarkably
conserved in eukaryotes, ubiquitously expressed in mammals, and forms
intra-nuclear foci in proliferating cells. Major features of KIN17 are its
ability to bind DNA and RNA and to be up-regulated in response to UV
and irradiation, suggesting a role in DNA replication, the DNA damage
response or RNA processing (Pinon-Lataillade et al., 2004; Miccoli et
al.,
2005). Human KIN17 comprises an N-terminal zinc finger (27-50)
and a C-terminal KOW motif (335-373). Here we report the 1H,
15N, 13C assignments of the region 51-160 of human KIN17
as a preliminary step toward obtaining the atomic structure of this region and
elucidating its biological function.
Residues corresponding to the KIN17 domain were all identified:
99% of the backbone chemical shifts, and
for the side chains, all the aliphatic and 88% of the aromatic
1H-13C nuclei were assigned. BMRB deposit with
accession number 6938.
References: Miccoli et al. (2005) Mol. Cell. Biol.,
25, 3814-3830; Pinon-Lataillade et al. (2004) J. Cell.
Sci.,
117, 3691-3702.
Ludovic Carliera, Albane le Maireb,
Sandrine Braudb, Cedric Massonb, Muriel
Gondryb, Sophie Zinn-Justina, Laure
Guilhaudisa, Isabelle Milazzoa, Daniel
Davousta, Bernard Gilquinb & Joël
Couprieb,*
aEquipe de Chimie Organique et Biologie
Structurale, CNRS UMR 6014, IFRMP 23, Université de Rouen, France;
bDépartement d'Ingénierie et d'Etude des
Protéines, CEA Saclay, 91191, Gif-sur-Yvette, France
*To whom correspondence should be addressed. E-mail: jo
el.couprie@cea.fr
Supplementary material is available in
electronic format at
http://dx.dio.org/10.1007/10858-006-0013-y.
Chapitre 4 : Caractérisation structurale du
domaine K2 par RMN et Modélisation Moléculaire
Letter to the Editor (Supplemental Material)
NMR assignment of region 51-160 of human
KIN17,
a DNA and RNA-binding protein
Ludovic Carliera, Albane le Maireb,
Sandrine Braudb, Cédric Massonb, Muriel
Gondryb, Sophie Zinn-Justinb, Laure
Guilhaudisa, Isabelle Milazzoa, Daniel
Davousta,
Bernard Gilquinb and Joël
Couprieb,*
a Equipe de Chimie Organique et
Biologie Structurale, IFRMP 23, CNRS UMR 6014, Université de
Rouen
b Département d'Ingénierie
et d'Etude des Protéines, CEA Saclay, 91191 Gif-sur-Yvette,
France.
187
* To whom correspondence should be addressed
: joel.couprie@cea.fr
Keywords: NMR assignments, KIN17, DNA-binding, RNA-binding,
nuclear metabolism
Chapitre 4 : Caractérisation structurale du
domaine K2 par RMN et Modélisation Moléculaire
Biological context
KIN17 is a 45 kDa nuclear protein conserved in eukaryotes and
initially identified on
the basis of its cross-reactivity with antibodies
raised against bacterial RecA, a recombination-repair enzyme
(Kannouche et al., 2000). In mammals, the KIN17 protein is
ubiquitously expressed. It forms intranuclear foci in proliferating
cells and is up-regulated after UV or ã irradiation (Biard et al.,
2002; Kannouche et al., 1998). The other features of KIN17 are its ability to
bind DNA and RNA in vitro and in vivo (Biard et al., 2002;
Pinon- Lataillade et al., 2004), to associate with multiprotein DNA replication
complexes (Miccoli et
al., 2005), and to complement the functions of the
bacterial transcriptional factor H-NS (Timchenko et al., 1996), suggesting a
role in nuclear metabolism.
Human KIN17 is a modular protein comprising four motifs: a zinc
finger (28-50) at
the N-terminus, a core domain homologous to RecA protein
(163-201), a nuclear localization signal (240-257), and a KOW motif (335-373)
found in several bacterial transcription factors (Kannouche et al., 2000;
Ponting et al., 2002). On the basis of sequence alignment and
secondary structure prediction, we identified a globular region located between
the zinc finger
and the core domain. We here report the 1H,
15N, 13C assignments of the region 51-160 as a
preliminary step toward obtaining the atomic structure of this
KIN17 domain and elucidating
its biological function.
Methods and experiments
188
The gene encoding the region 51-160 of human KIN17 was
amplified by polymerase chain reaction (PCR) and cloned into several expression
vectors using the Gateway system (Invitrogen). A protein expression screening
using different strains and fusion partners was performed (Braud et al.,
in press) and highest levels of expression and solubility
were obtained when transforming E. coli Rosetta(DE3)
strain with plasmid pEXP-TH5 (Invitrogen). This vector encodes a 6xHis
tag, the Z-domain from Staphylococcal Protein A (ZZ), a TEV protease
(Tobacco Etch Virus) cleavage site, and the KIN17 51-160 domain.
Protein expression was induced by the addition of 1 mM of
isopropyl-1-thio-â-D- galactopyranoside (IPTG) to the bacterial
culture at a cell density of 1.2 absorbance units measured at 595 nm.
The bacterial culture was further grown for 14 hours at 20°C.
The fusion protein was purified using IgG Sepharose Fast Flow
(Amersham) in order
Chapitre 4 : Caractérisation structurale du
domaine K2 par RMN et Modélisation Moléculaire
to separate the fusion protein from other bacterial proteins
and to remove the (His)6-ZZ tag after an on-column cleavage using
recombinant (His)6-TEV protease. A second step of purification was
performed using Ni-NTA column (Qiagen) to remove (His)6-TEV protease and
residual (His)6-ZZ tag.
Uniformly labelled 15N protein was produced in
minimal medium M9 containing
1 g.L-1 of (15NH4)2SO4 (Boehringer) as
the sole nitrogen source. Uniformly labelled 13C/ 15N
protein was produced in a rich medium prepared from uniformly labelled
13C/15N Spirulina maxima cyanobacteria. NMR
samples containing about 0.7 mM protein were prepared in 50 mM phosphate
buffer pH 6.0 containing 150 mM NaCl, 1 mM EDTA, a protease inhibitor cocktail
(SIGMA), 1 mM TCEP, and 1 mM NaN3 in either 90% H2O/10% D2O or in 100% D2O.
3-(trimethylsilyl)[2,2,3,3-2H4] propionate (TSP) was added as an
internal 1H chemical shift reference. 13C and
15N chemical shifts were referenced indirectly to TSP, using
the absolute frequency ratios (Wishart et al., 1995).
NMR experiments were performed at 303 K on Bruker
AVANCE DMX-600 or Varian INOVA-800 spectrometers equipped with
triple-resonance (1H, 15N, 13C) cryoprobes.
Spectra used for sequential backbone assignment were as follows: 2D
15N-1H HSQC, 3D HNCO, 3D HNCA, 3D CBCA(CO)NH, 3D
CBCANH, 3D HNHA, 3D (HCA)CO(CAN)NH experiments. Aliphatic and aromatic
1H and 13C side chain assignments were obtained using
3D HBHA(CO)NH, 3D HCCH-TOCSY, 3D HCCH-COSY, and 3D
13C-HSQC NOESY
experiments. All NMR data were processed with NMRPipe software
(Delaglio et al., 1995)
and analysed with Felix software (Molecular Simulations).
189
Extent of assignments and data deposition
For cloning strategy reasons, this KIN17 domain contains the
region 51-160 of human KIN17 plus an additional glycine residue at the N
terminus. Figure 1 shows the 15N-1H HSQC spectrum of
the 111 residues protein numbered from 1 to 111. All the amide
1H and 15N backbone resonances were assigned except for
N-terminal amino acids G1 and Q2, and R53 whose NH is unobservable presumably
due to loop mobility or chemical exchange since this residue is located between
secondary structure elements.
Chapitre 4 : Caractérisation structurale du
domaine K2 par RMN et Modélisation Moléculaire
Compared to the expected number of resonances, nine
additional weakened cross- peaks were observed in the
15N-1H HSQC spectrum. However, backbone and side
chain assignments, as well as 3D 15N HSQC-NOESY patterns revealed
that 6 spin systems split in
two (R3, Q4, L5, L6, A8, S9). Those residues are
located before the proline P12 and we suppose that the minor form is
due to a partial isomerisation of P12. The observation of a
characteristic strong NOE between Hä of P12 and Há of N11
indicated that the predominant form of this proline is in a trans
conformation.
Finally, all residues corresponding to the region 51-160 of
KIN17 were identified: the backbone assignment is complete for
1Há, 13Cá and 13CO, and it
reaches 98% for 1HN and 15N nuclei. All the aliphatic
side chain resonances were assigned and 88% of the aromatic 1H
and
13C resonances were identified, including all Tyr and
His aromatic rings, 3 Phe out of 6, and 2
Trp out of 3. Assignments of NH2 side chain resonances of
asparagines and glutamines were also completed except for N42. The
1H, 13C and 15N chemical shifts have been
deposited in
the BioMagResBank
(http://www.bmrb.wisc.edu) under accession number 6938.
Acknowledgement
The Bruker DMX-600 spectrometer was supported by grants of the
Conseil Regional
de Haute-Normandie (France). We are grateful to Dr Adrien
Favier who kindly recorded 3D HSQC NOESY experiments on Varian INOVA-800
spectrometer at I.B.S Jean-Pierre Ebel (Grenoble, France), and to Marie
Courçon and Mireille Moutiez for the protein expression
screening. We also thank the Centre de Ressources Informatiques de
Haute-Normandie (France) for NMR software facilities.
References
Braud, S., Moutiez, M., Belin, P., Abello, N., Drevet,
P., Zinn-Justin, S., Courçon, M., Masson, C., Dassa, J.,
Charbonnier, J.B., Boulain, J.C., Ménez, A., Genet, R., Gondry,
M. (2005). J. Proteome Res., In press.
Biard, DS., Miccoli, L., Despras, E., Frobert, Y.,
Creminon, C. and Angulo, JF. (2002)
190
J. Biol.Chem., 277(21), 19156-19165.
Delaglio, F., Grzesiek, S., Vuister, G. W., Zhu, G., Pfeifer J.
and Bax, A. (1995) J. Biomol. NMR., 6, 277-293.
Chapitre 4 : Caractérisation structurale du
domaine K2 par RMN et Modélisation Moléculaire
Kannouche, P., Mauffrey, P., Pinon-Lataillade, G., Mattei, MG.,
Sarasin, A., Daya-Grosjean,
L. and Angulo, JF. (2000) Carcinogenesis,
21(9), 1701-1710.
Kannouche, P., Pinon-Lataillade, G., Tissier, A.,
Chevalier-Lagente, O., Sarasin, A., Mezzina,
M. and Angulo, JF. (1998) Carcinogenesis,
19(5), 781-789.
Miccoli, , L., Frouin, I., Novac, O., Di Paola, D., Harper, F.,
Zannis-Hadjopoulos, M., Maga, G., Biard, D.S.F., Angulo, J.F. (2005). Mol.
Cell. Biol., 25(9), 3814-3830.
Pinon-Lataillade, G., Masson, C., Bernardino-Sgherri, J.,
Henriot, V., Mauffrey, P., Frobert, Y., Araneda, S. and Angulo, JF. (2004)
J. Cell. Sci., 117(16), 3691-3702
Ponting, CP. (2002) Nucleic Acids Res.,
30(17), 3643-3652.
Timchenko, T., Bailone, A. and Devoret, R. (1996) EMBO
J., 15, 3986-3992.
Wishart, DS., Bigam, CG., Yao, J., Abildgaard, F., Dyson, HJ.,
Oldfield, E., Markley, JL. and
Sykes, BD. (1995) J. Biol. NMR, 6,
135-140
R33
N11 E22
L73 K71
G74
T65
Q13
Y46
L31
E29
R102 E105
E107
T84
Q4
D17
T67
E10
Q91
Y18
L6
Q103
L104
M16
F15 Q4
R101
K109
K38
K108
L106
T61
G87
G77
D96
E83
R39
Y49
L7
R95
K110
L5 W63
A8
I90
E21
N59
N47
T37
R34
F35
I50
G36
S9 K86
E76
T70
Q62
C79
S51
E54 N25
S20
R24
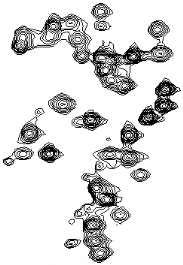

L28 F19
R32
T99
L78
H55
V45
E64
D26
E98
F23
R3
F69
L30
I100
A60
E48
I44
H52
Y89
N43
F27
W88 L66
D82 V81 I56
K80
M58
V40
R75
D94
D68
W72
I93
N42
H57
191
H41
K111
W63 å
W88 å
Y92
W72 å
Figure 1: 2D 15N-1H HSQC
spectrum of the region 51-160 of human KIN17. The
assignments are indicated with the one-letter amino acid
code. NH2 side chain resonances of asparagines and glutamine are connected
by purple horizontal lines.
Chapitre 4 : Caractérisation structurale du
domaine K2 par RMN et Modélisation Moléculaire
1.2) Détermination de la topologie du domaine
K2
La première étape de la
caractérisation structurale du domaine K2 a consisté
à déterminer les éléments de structure
secondaire à partir de paramètres structuraux et
dynamiques RMN afin d'établir la topologie de la protéine.
1.2.1) Analyse du nOe
hétéronucléaire 1H-15N
Nous avons dans un premier temps réalisé une
expérience de mesure des nOe hétéronucléaires
1H-15N dans le but d'identifier les résidus non
structurés des extrémités N- et
C-terminales. Les spectres ont été
enregistrés à 600 MHz sur un échantillon
uniformément
marqué 15N concentré à 0.8 mM.
1
0.8
0.6
0.4
0.2
0 ** *
* * * * *
-0.2
10 20 30 40 50 60 70 80 90 100
-0.4
-0.6
-0.8
-1
Figure 4.2 : nOe
hétéronucléaires 1H-15N du domaine
K2 de la protéine humaine KIN17. Les astérisques correspondent
aux résidus dont la valeur n'a pas été
déterminée ou aux résidus proline.
Les faibles valeurs de nOe hétéronucléaire
(<0.25) observées au niveau des extrémités
N- et C-terminales mettent en évidence que les
régions R3-N11 et K108-K111 sont flexibles
et déstructurées (Figure 4.2). L'échange
rapide du proton amide de Q2 avec l'eau n'ayant pas permis de mesurer sa
valeur de nOe 1H-15N, et le résidu 12
étant une proline, il semble raisonnable de conclure que les
segments G1-P12 et K108-K111 sont déstructurés.
192
Chapitre 4 : Caractérisation structurale du
domaine K2 par RMN et Modélisation Moléculaire
1.2.2) Analyse des paramètres structuraux
classiques
La caractérisation des éléments de
structure secondaire a été initiée en calculant les
indices de déplacement chimique CSD à partir des valeurs de
déplacements chimiques Há et
Cá, et des valeurs de
référence correspondantes dans la BioMagResBank. La
figure 4.3 montre
le diagramme des indices ?äCá et ?äHá
du domaine K2. La majorité des résidus présente une
valeur positive de ?äCá conjuguée à une
valeur négative de ?äHá, ce qui suggère une
structuration hélicoïdale importante. Pour être
significatives, les valeurs de ?äCá et ?äHá
doivent atteindre respectivement, 1, et 0.1 ppm. Sur la base de ces valeurs
seuils, 4 hélices sont clairement identifiables au niveau des
régions D17-R33, H41-S51, L66-R75, et P97- E107. A contrario, la
région C79-Q91 se caractérise plutôt par des valeurs
négatives de ?äCá conjuguées à des valeurs
positives de ?äHá, ce qui indique une conformation en feuillet
â ou étendue. Cependant, le signe des indices CSD dans cette
région est irrégulier, et l'amplitude
des index ?äCá négatifs est relativement
faible.
Afin d'approfondir la caractérisation des structures
secondaires, nous avons entrepris
une analyse partielle du spectre 15N-NOESY-HSQC
enregistré à 600 MHz sur un échantillon
uniformément marqué 15N concentré à 0.8
mM. Selon la stratégie définie dans le chapitre 2, nous nous
sommes contentés de collecter 3 types de nOe non
ambiguës facilement identifiables. Il s'agit des corrélations
caractéristiques de type dáN(i, i+1), dáN(i, i+2),
et dáN(i, i+3). Nous avons également estimé les
constantes de couplage 3JHN-Há à partir des
expériences 3D HNHA et 2D HMQC_J enregistrées sur
l'échantillon 15N. L'analyse combinée des nOe
caractéristiques et des constantes de couplage 3JHN-Há
(Figure 4.4) permet
de préciser la nature des éléments de
structure secondaire mis en évidence précédemment :
L'observation de contacts dáN(i, i+3)
consécutifs associés à de faibles intensités
de nOe dáN(i, i+1), et à une absence d'effets
dáN(i, i+2), caractérise une structuration des 4
régions D17-L31, N42-S51, L66-G77, et P97-Q103 en hélice
á. En ce qui concerne l'hélice
C-terminale, la forte similarité des valeurs de
déplacement chimique Há dans la région L104- K110 ne
permet pas de relever d'effets supplémentaires non ambiguës au
delà du résidu 103.
Les valeurs consécutives de 3JHN-Há
inférieures à 5.5 Hz au niveau des régions F15-R33,
H41-
Y49, D68-R75, et D96-E105 confirment l'existence de ces 4
hélices. Notons toutefois que 3
des valeurs de constante 3JHN-Há du segment
H41-Y49 (I44, V45, et E48) se situent entre 5.5
et 6 Hz.
193
5 10 15 20 25 30 35
Chapitre 4 : Caractérisation structurale du
domaine K2 par RMN et Modélisation Moléculaire
7
|
?äCá
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
5
|
10
|
15
|
20
|
25
|
30
|
35
|
40
|
45
|
50
|
55
|
60
|
65
|
70
|
75
|
80
|
85
|
90
|
95
|
100
|
105
|
110
|
|
?äHá
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
5
|
10
|
15
|
20
|
25
|
30
|
35
|
40
|
45
|
50
|
55
|
60
|
65
|
70
|
75
|
80
|
85
|
90
|
95
|
100
|
105
|
110
|
6
5
40 45 50 55 60 65
70 75
80 85 90 95 100 105 110
4
3
2
1
0
-1
-2
0 5 10 15 20 25 30 35 40 45 50 55 60 65 70 75 80 85 90 95
100 105 110
194
-3
1.2
0.9
0.6
0.3
0
-0.3
-0.6
-0.9
-1.2
-1.5
0 5 10 15 20 25 30 35 40 45 50 55 60 65 70 75 80 85 90 95
100 105 110
-1.8
Figure 4.3 : Déplacements
chimiques secondaires du domaine K2 de la protéine KIN17 humaine
calculés à partir de valeurs de référence de
la
194
BioMagResBank
(http://www.bmrb.wisc.edu/)
Chapitre 4 : Caractérisation structurale du
domaine K2 par RMN et Modélisation Moléculaire
5 10 15 20 25 30 35 40 45 50 55 60 65 70 75 80 85 90
95 100 105 110
| | | | | | | | | | | | | | | | | | | | | |
GQRQLLLASENPQQFMDYFSEEFRNDFLELLRRRFGTKRVHNNIVYNEYISHREHIHMNATQWETLTDFTKWLGREGLCKVDETPKGWYIQYIDRDPETIRRQLELEKKKK
dáN(i,i+1)
dáN(i,i+3)
195
dáN(i,i+2)
J
3 9
HN-Há 7
[Hz]
7
6
5
4
3
2
1
0
5
3
0 5 10 15 20 25 30 35 40 45 50 55 60 65 70 75 80 85 90 95 100 105
110
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
5
|
10
|
15
|
20
|
25
|
30
|
35
|
40
|
45
|
50
|
55
|
60
|
65
|
70
|
75
|
80
|
85
|
90
|
95
|
100
|
105
|
110
|
GQRQLLLASENPQQFMDYFSEEFRNDFLELLRRRFGTKRVHNNIVYNEYISHREHIHMNATQWETLTDFTKWLGREGLCKVDETPKGWYIQYIDRDPETIRRQLELEKKKK
Figure 4.4 : Diagramme
récapitulatif de 3 effets Overhauser caractéristiques et des
constantes de couplage 3JHN-Há du domaine K2 de KIN17
humaine. L'épaisseur des traits rouges, bleus, et verts indique
respectivement, l'intensité des effets à courte et moyenne
distance de type dáN(i, i+1), dáN(i, i+3), et dáN(i,
i+2) relevés sur le spectre NOESY-HSQC édité
15N. Les lignes horizontales violettes représentent les
valeurs
seuil de constante de couplage 3JHN-Há
dans les structures secondaires (<5.5 Hz dans l'hélice
á ou 310, >8 Hz dans le feuillet â). Les
traits
195
inclinés correspondent aux résidus dont la
valeur de 3JHN-Há n'a pas été
déterminée.
De manière générale, peu de connexions
non ambiguës de type dáN(i, i+2) ont été
relevées sur le spectre 15N-NOESY-HSQC. Ces
effets sont caractéristiques d'une structuration en hélice 310
ou en coude. L'observation de 2 contacts consécutifs de moyenne
intensité de type dáN(i, i+2) et d'un effet dáN(i, i+3) au
niveau du segment M58-T61 suggère l'existence potentielle d'un tour
d'hélice 310. Cette hypothèse est consolidée par
une faible valeur de 3JHN-Há (<4.5 Hz), et une valeur
significative d'indice ?äCá des résidus M58 et N59.
La succession d'effets dáN(i, i+1) de forte
intensité associés à une absence quasi-totale d'effets
dáN(i, i+3) et dáN(i, i+2) au niveau des régions C79-T84
et W88-D94 confirme la présence d'une zone étendue ou
structurée en feuillet â dans la région C79-D94. Les fortes
valeurs de constante 3JHN-Há (>8 Hz) au niveau des
résidus K80 à D82, T84, K86, Y89 à Q91,
et I93 sont en accord avec ce résultat. D'autre
part, le diagramme de la Figure 4.4 fait
apparaître 4 fortes intensités dáN(i,
i+1), un contact dáN(i, i+2), et 3 valeurs de
3JHN-Há supérieures à 8 Hz dans la
région T35-N42. Ce profil laisse présager la
présence d'un ou plusieurs types conformationnels : coude(s), brin
â, ou zone étendue. Aussi, il est difficile de caractériser
la structure secondaire de cette zone à partir de ces seules
données. Les valeurs significatives d'indice ?äHá des
résidus T37, R39 et V40 (> +0.2 ppm), et la forte valeur de
?äCá du résidu V40 (-1.9 ppm)
supportent l'hypothèse de l'existence d'un brin â.
1.2.3) Prédiction des angles et
Le déplacement chimique des noyaux
13Cá, 13Câ, 13CO,
1Há, et 15NH a été utilisé
pour estimer les angles dièdres et caractéristiques des
structures secondaires à l'aide du logiciel TALOS. Dans le cas du
domaine K2, 68 des 111 valeurs de couple d'angles et prédites sont
considérées comme fiables. Une grande partie de ces
valeurs se situe dans la région spécifique aux
hélices á du diagramme de Ramachandran, ce qui confirme une
structuration hélicoïdale importante. Sur la base de cette
prédiction, 4 hélices á sont identifiables au niveau
des régions Y18-R34, N42-I50, T67-R75, et
E98-L106. Le programme TALOS prédit également la
présence de 3 zones étendues ou structurées en brin
â au niveau des segments R39-H41, C79-T84, et W88-Y92. De plus, les
valeurs d'angles dièdres prédites pour les résidus
M58 et N59 sont caractéristiques d'une hélice 310, ce qui
conforte l'hypothèse de la présence d'un tel motif dans cette
région du domaine K2. Par ailleurs, les valeurs particulières
d'angles et créditées aux résidus isolés R53 et
E54, ainsi qu'aux résidus E76, P85, et K86,
suggèrent une implication de ces acides aminés dans
un coude régulier.
1.2.4) Identification et organisation des brins
â
Afin de préciser la structure secondaire des
régions qui présentent une préférence
conformationnelle étendue, nous avons entrepris une recherche d'effets
spécifiques à longue distance de type HN-HN, HN-Há,
et Há-Há sur les spectres 15N- et
13C-NOESY-HSQC. L'analyse de ces effets met en évidence
l'existence d'un feuillet â anti-parallèle triple brin dans
lequel le brin central G87-I93 est encadré par un second de même
taille qui le précède dans la séquence (L78-T84), et par
un troisième beaucoup plus court (V40-N42) (Figure 4.5).
Les résultats de l'expérience de spectroscopie
d'échange 1H ? 2D menée sur un
échantillon
15N / 13C solubilisé dans
100% D2O sont en accord avec une telle organisation : le
ralentissement constaté de la vitesse d'échange des protons
amides T84, Y89, I90, Q91, et I93 suggère une implication de ces
protons dans une liaison hydrogène, et confirme leur appartenance
à une structure secondaire régulière (Figure 4.5).
L78
C79 V81
K80 D82
E83
T84
P85
I93
Y92
Q91
I90
Y89
W88
G87
K86
V40
N42
H41
Figure 4.5 : Reconstitution de la
topologie du feuillet â anti-parallèle du domaine K2 à
partir
de l'analyse des nOe longue distance Há-Há
(traits oranges), HN-HN (traits bleus), et HN-Há (traits verts). Les
lignes hachurées correspondent aux liaisons hydrogène
déduites de l'analyse des vitesses d'échange des protons amides
et des nOe inter-brins.
1.2.5) Conclusions : structure secondaire du domaine
K2
La prise en compte de l'ensemble des informations structurales
obtenues à partir des indices CSD, des constantes
3JHN-Há, des nOe 1H caractéristiques, des
nOe hétéronucléaires, de l'échange des protons
amides, et de la prédiction des angles et , permet
finalement de caractériser la topologie des structures secondaires
(Figure 4.6). Le consensus réalisé à partir
de toutes ces informations met clairement en évidence
l'existence de 4 hélices á (H1 à H4) et
de 3 brins â (S1 à S3). De plus, comme nous l'avons
suggéré dans les paragraphes précédents,
plusieurs éléments sont en faveur de
l'existence d'un tour d'hélice 310 dans la région
M58-
T61. A ce stade de l'étude structurale, la présence
de ce motif demeure une simple hypothèse
qui doit être confirmée par le calcul de la
structure par modélisation moléculaire. Il en est de même
pour l'orientation et la délimitation précise des
structures secondaires, uniquement accessibles par la détermination de
la structure tridimensionnelle à l'échelle atomique.
|
5
|
10
|
15
|
20
|
25
|
30
|
35
|
40
|
45
|
50
|
55
|
60
|
65
|
70
|
75
|
80
|
85
|
90
|
95
|
100
|
105
|
110
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
GQRQLLLASENPQQFMDYFSEEFRNDFLELLRRRFGTKRVHNNIVYNEYISHREHIHMNATQWETLTDFTKWLGREGLCKVDETPKGWYIQYIDRDPETIRRQLELEKKKK
nOe 15N
CSD
3JHN-Há
TALOS
nOe 1H
Consensus
|
H1
|
S1
|
H2
|
?
|
H3
|
S2
|
S3
|
H4
|
|
* *******
|
|
*
|
|
*****
|
*
|
*** *
|
|
GQRQLLLASENPQQFMDYFSEEFRNDFLELLRRRFGTKRVHNNIVYNEYISHREHIHMNATQWETLTDFTKWLGREGLCKVDETPKGWYIQYIDRDPETIRRQLELEKKKK
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
5
|
10
|
15
|
20
|
25
|
30
|
35
|
40
|
45
|
50
|
55
|
60
|
65
|
70
|
75
|
80
|
85
|
90
|
95
|
100
|
105
|
110
|
Figure 4.6 :
Caractérisation de la structure secondaire du domaine K2 par
RMN. Un
cylindre bleu correspond à une hélice á,
un cylindre vert à une hélice 310, une flèche rouge
à
un brin â, et un rectangle rouge à une
région étendue ou à un brin â. Les traits
noirs
prononcés indiquent l'étendue des
extrémités flexibles et déstructurées et
une étoile correspond à un résidu dont la vitesse
d'échange de proton amide est ralentie.
1.3) Calcul de la structure par Modélisation
Moléculaire sous contraintes RMN
La structure du domaine K2 de la protéine KIN17
humaine a été calculée avec le logiciel CNS
couplé au programme d'attribution automatique des nOe du
Laboratoire de Structure des Protéines. Les méthodes et
principes sur lesquels reposent ces logiciels sont exposés dans le
chapitre 3 de cette seconde partie.
1.3.1) Préparation des contraintes
expérimentales RMN
Les spectres NOESY-HSQC utilisés pour déduire
les contraintes de distance ont été enregistrés à
l'IBS de Grenoble sur un spectromètre 800 MHz équipé d'une
cryosonde triple résonance, avec un temps de mélange de 80 ms
pour l'expérience éditée 15N, et de 100 ms pour
l'expérience éditée 13C. Les pics de
corrélation dipolaire ont été sélectionnés,
intégrés, puis collectés à l'aide du logiciel
Felix (Accelrys). Aucun de ces pics n'a été
attribué manuellement à l'exception des 9 effets à longue
distance de type HN-HN, HN-Há, et Há-Há
qui définissent la topologie du feuillet â de K2.
Les 136 valeurs d'angles et prédites par le logiciel
TALOS ont été utilisées pour former les contraintes
d'angle dièdre. Les intervalles de valeurs permises associés
à ces angles
ont été établis en multipliant
l'écart type estimé par TALOS pour chaque prédiction par
un coefficient d'une valeur de 1.5. Trois contraintes supplémentaires
d'angle ont été ajoutées dans les protocoles de
calcul sur la base d'une valeur de constante de couplage
3JHN-Há caractéristique des résidus F35,
H55, et D96. Les intervalles de valeurs permises correspondants
ont été déduits de la courbe de Karplus lissée en
utilisant les coefficients de Pardi (Pardi et al., 1984).
Enfin, l'expérience de spectroscopie
d'échange 1H ? 2D et la
caractérisation des éléments de structure secondaire ont
permis de mettre en évidence l'existence de 18 liaisons hydrogène
principalement localisées dans les hélices á (Figure 4.6).
Ces informations ont été interprétées en contrainte
de distance (dHN-O ~ 2.3 Å et dN-O ~ 3.3 Å) et introduites dans
les protocoles de calcul.
1.3.2) Bilan de l'attribution automatique des pics
nOe
Le calcul de la structure du domaine K2 a
nécessité la répétition de 21 processus
itératifs de 22 cycles où chaque itération a fait l'objet
d'une attribution des pics nOe et d'un calcul de 200 structures. L'analyse
conjointe des violations de distance et d'angles dièdres, ainsi que de
la liste des pics nOe non attribués en fin de chaque
processus a permis d'identifier, puis de corriger, les informations
erronées. Ainsi, les pics qui correspondent à du bruit spectral
ont été progressivement supprimés, et les erreurs
d'attribution des fréquences de résonance de la
protéine ont été rectifiées. Le jeu final de
données expérimentales RMN retenu a conduit à
l'attribution de 3347 pics nOe par le programme d'attribution automatique.
Moins de 2% des pics de ce jeu final n'ont pas été
attribués. Après application des différents filtres et
suppression des nOe redondants, une liste de 2716 contraintes de distance
contenant,
2192 contraintes non ambiguës, et 524 contraintes
ambiguës, a finalement été établie, ce qui
représente une vingtaine de contraintes de distance par résidu
(Tableau 4.1).
Attribution automatique des pics nOe :
Nombre de pics du jeu final 3413
Nombre de pics attribués 3347
Nombre de pics non attribués 66
Nombre de contraintes expérimentales :
Contraintes de distance 2743
Non ambiguës 2219
nOe attribués par le programme 2192
nOe de topologie attribués manuellement 9
Distances déduites de l'échange 1H ?
2D 18
Ambiguës (générés par le programme)
524
Contraintes dihédrales et 139
Prédictions TALOS 136
Angles déduits de la constante de couplage
3JHN-Há 3
Tableau 4.1 : Contraintes
expérimentales utilisées pour le calcul final de la
structure.
1.3.3) Evaluation de la qualité des structures
calculées
Les 20 structures de plus basse énergie de
l'itération finale ont été sélectionnées
et soumises à un protocole de raffinement dans le champ de force
CHARMM22. A l'issue de cette dernière étape, les 12 meilleurs
modèles ont été retenus et constituent l'ensemble final
de
la structure du domaine K2 résolue par modélisation
moléculaire sous contraintes RMN. Le
tableau 4.2 présente un résumé des
statistiques structurales de ces 12 meilleures structures.
Dans l'ensemble, les faibles valeurs des différents termes
de la fonction d'énergie suggèrent
que les modèles générés sont de
bonne qualité. Aucune violation de distance supérieure
à
0.5 Å ou d'angle dièdre et supérieure
à 10° n'est observée, ce qui indique une bonne prise
en compte des contraintes expérimentales RMN. Ces
structures présentent également une bonne
géométrie covalente avec de faibles valeurs de
déviation par rapport à la géométrie
idéale des longueurs de liaisons, des angles de valence, et des angles
dièdres impropres.
Contraintes expérimentales :
Contraintes de distance 2743
Contraintes dihédrales et 139
Nombre de violations de distance > 0.5 Å 0
Nombre de violations d'angle et > 10° 0
Energies (kcal/mol) :
Liaisons 141 #177; 6
Angles 599 #177; 15
Impropres 20 #177; 2
Van der Waals 146 #177; 15
Electrostatique -671 #177; 25
nOe 143 #177; 10
Dièdres et 5.6 #177; 0.7
Totale 258 #177; 5
Déviations à la géométrie
idéale :
Liaisons (Å) 0.0159
Angles (°) 3.35
Impropres (°) 2.74
nOe (Å) 0.032
Analyse du diagramme de
Ramachandran*
Résidus dans les régions favorables 88.6 %
Résidus dans les régions additionnelles permises 10.1 %
Résidus dans les régions
généreusement permises 0.8 %
Résidus dans les régions interdites 0.5 %
*réalisée avec le logiciel
Procheck dans la région 13-107
Tableau 4.2 : Statistiques
structurales de l'ensemble des 12 structures finales de K2.
La répartition des angles et sur le
diagramme de Ramachandran confirme la qualité des structures.
Plus de 99 % des valeurs de ces angles occupent les régions
énergétiquement favorables ou permises, et seules 0.5 %
sont situées dans les régions interdites. La distribution des
angles dièdres et pour chaque résidu est
présentée en Figure
4.8A. Les éléments de structure secondaire
également reportés ont été définis à
partir de ces
valeurs d'angle.
La convergence des modèles
générés constitue également un critère
de qualité. La
superposition des 12 structures finales sur les atomes de la
chaîne principale (N, CO, et Cá) montre la bonne définition
du squelette peptidique entre les résidus F15 et L106 (Figure 4.7). Pour
les acides aminés situés entre ces 2 résidus, la moyenne
des écarts quadratiques « rmsd » calculée sur le
squelette de l'ensemble final par rapport à une structure
moyenne est de
0.43 Å. Cette faible valeur indique une bonne
convergence des modèles vers une structure
tridimensionnelle unique entre les résidus F15 et L106.
N-ter
C-ter F15
L106
Figure 4.7 : Superposition des
12 structures finales du domaine K2 sur les atomes du
squelette (N, CO, et Cá) entre les résidus Q13
et E107. Les hélices á sont représentées en bleu,
les feuillets â en rouge, l'hélice 310 en vert pastel, et les
régions déstructurées en violet.
La Figure 4.8B montre les valeurs des écarts rmsd
calculés sur le squelette et les atomes lourds de chaîne
latérale pour chaque résidu. On constate une dispersion des
angles
et associée à de fortes valeurs de rmsd au niveau
des extrémités N- et C-terminales G1-P12
et K109-K111. Ceci est en accord avec l'analyse du nOe
hétéronucléaire 1H-15N (cf. §
1.2.1)
qui indique que ces 2 extrémités sont
flexibles et déstructurées. A contrario, la bonne
convergence des angles dièdres et les faibles valeurs de rmsd
sur les atomes du squelette
(< 1 Å) des résidus F15 à L106
suggèrent que ces acides aminés constituent le corps
globulaire de la protéine. Ainsi, on observe une bonne définition
du squelette peptidique dans
les régions structurées en hélice et
feuillet, mais également dans les boucles. D'autre part, la
majorité des valeurs de rmsd calculées sur les atomes lourds de
chaîne latérale sont inférieures
à 1.5 Å, ce qui démontre une orientation
privilégiée de la plupart des chaînes latérales dans
la région F15-L106. La moyenne des écarts rmsd sur les atomes
lourds entre les résidus F15 et
L106 est de 1.03 Å.
Chapitre 4 : Caractérisation structurale du
domaine K2 par RMN et Modélisation Moléculaire
A)
[°]
200
150
100
50
0
-50
-100
-150
-200
H1 S1 H2
310
H3 S2 S3 H4
203
[°]
200
150
100



50
0
-50
-100
-150
-200
10 20 30 40
50 60
70 80
90 100 110
B)
5
4.5
4
3.5
rmsd [Å]
3
2.5
2
1.5
1
0.5
0
10 20 30 40
50 60
70 80
90 100
110
203
0 5 10 15 20 25 30 35 40 45 50 55 60 65 70 75 80 85 90 95 100 105
110
Figure 4.8 : Analyse de la
convergence des 12 structures finales sélectionnées. A)
Distribution des angles dièdres et pour chaque acide
aminé de ces 12 structures. Les traits oranges correspondent aux
intervalles de contrainte d'angle définis dans CNS. B) Déviation
quadratique moyenne rmsd pour chaque résidu calculée par rapport
à la structure moyenne des 12 modèles sélectionnés
(en bleu : superposition sur les atomes N, Cá, et CO du squelette ; en
violet : sur tous les atomes lourds de chaîne latérale). Les
résidus 36, 74, 77, et 87 sont des glycines.
2) Description de la structure tridimensionnelle du
domaine K2
Le bilan de l'analyse des écarts rmsd et des
angles dièdres et montre une convergence des structures finales
vers une structure unique dans la région F15 à L106. Par
conséquent, nous avons choisi le modèle raffiné de plus
basse énergie (ET = 196.1 kcal.mol-1) comme le plus
représentatif de la structure du domaine K2 de la protéine KIN17
humaine.
2.1) Structure secondaire
Le calcul de la structure du domaine K2 par
modélisation moléculaire confirme l'existence de l'ensemble des
éléments de structure secondaire mis en évidence
précédemment par l'analyse des paramètres RMN. K2
adopte un repliement de type á/â constitué de 4
hélices á (H1: F15-R34, H2: N42-I50, H3: L66-R75, H4:
E98-E107), de 3 brins â (S1: R39-H41, S2: C79-T84, S3: G87-Y92),
et d'un tour d'hélice 310 (H2.5: M58- A60) situé dans une
large boucle entre H2 et H3 (Figure 4.9). La topologie du domaine est de
la forme H1-S1-H2-H2.5-H3-S2-S3-H4. La partie N-terminale de K2
est donc principalement hélicoïdale alors que la région
C-terminale est dominée par 2 brins â anti-parallèles (S2
et S3)
reliés par un coude â de type I (T84-G87).
H2.5
N
H3
S3
S2 H2
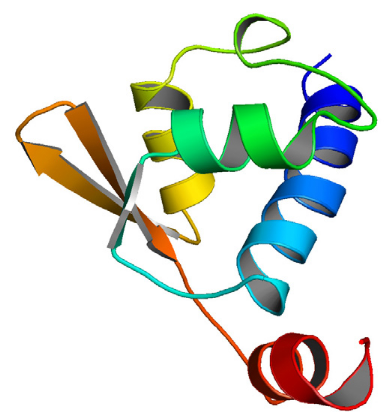
H1
S1
C
H4
Figure 4.9 : Structure
tridimensionnelle du domaine K2 de la protéine humaine
KIN17
résolue par RMN et Modélisation
Moléculaire. Les hélices sont représentées par des
rubans
et les brins â par des flèches.
2.2) Les éléments qui composent le coeur
hydrophobe
Les hélices H1, H2, et H3 constituent la zone centrale de
la protéine. Elles forment un
« tonnelet orthogonal » compact où la valeur
d'angle entre chacune d'entre elles est proche de
90°. Avec une longueur de 20 résidus,
l'hélice H1 est l'élément de structure secondaire le plus
imposant du domaine K2. Elle contient 5 tours réguliers d'hélice
á, soit deux fois plus que les hélices H2 et H3
(respectivement 9 et 10 résidus, soit environ 2.5 tours).
Chacune de ces hélices est stabilisée par un réseau
régulier de liaisons hydrogène qui se forment entre
l'oxygène du CO d'un résidu i et le proton amide d'un
résidu i+4. Ces 3 hélices canoniques présentent
un caractère amphipatique : elles projettent la
quasi-totalité de leurs chaînes latérales polaires
vers l'extérieur du domaine et la majorité de leurs
chaînes latérales hydrophobes sont orientées vers
l'intérieur et définissent ainsi une poche hydrophobe qui
stabilise le tonnelet. Cette poche dense et riche en acides aminés
aromatiques est composé des résidus M16, F23, F27, L28, L30, et
L31 de H1, des résidus V45, Y46, et Y49 de H2, et des
résidus L66, F69, W72, et L73 de H3 (Figure 4.10).
L66 H3
F69
W72 M16
Y46
L73
H2
Y49
V45
L30
F27
L31
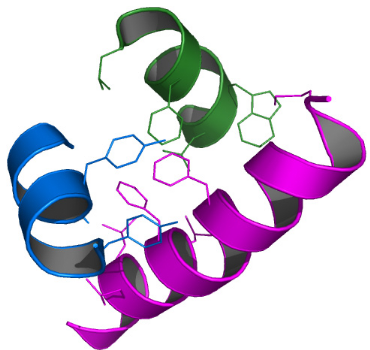
F23
L28
H1
Figure 4.10 : Description de la
poche hydrophobe du tonnelet orthogonal du domaine K2.
Les chaînes latérales des résidus
qui forment cette poche sont représentées par des sticks
violets pour l'hélice H1, bleus pour l'hélice H2, et verts pour
l'hélice H3.
Les 3 brins S1, S2, et S3 forment un feuillet â
anti-parallèle à la périphérie du tonnelet
orthogonal dans lequel le brin central S3 est stabilisé par 8 liaisons
hydrogène régulières de type COi - HNj. Le brin S1
qui ne compte que 3 résidus est principalement maintenu au
feuillet â via le résidu V40 qui forme 2 liaisons hydrogène
et plusieurs contacts hydrophobes
avec l'isoleucine 90 de S3. Le brin S2 est relié à
l'hélice H3 par un coude â de type I entre les
résidus G74 et G77. L'analyse de cette
région du domaine fait également apparaître une
seconde interface d'interactions hydrophobes entre les brins S2 et S3,
et les hélices du tonnelet. Cette interface est principalement
constituée des résidus F27, L28 et L31 de H1, et des
résidus L66, T70, L73, et G74 de H3 qui établissent des contacts
avec la face hydrophobe
du feuillet â (C79, et V81 de S2, et W88, I90, et Y92 de
S3) (Figure 4.11).
S2
W88
S3
T70
V81
L66
H3
G74
I90
Y92
C79

F27
L73
L28
L31
H1
Figure 4.11 : Description de la
seconde interface hydrophobe du domaine K2. Les chaînes
latérales des résidus qui forment cette
interface sont représentées par des sticks violets pour
l'hélice H1, verts pour l'hélice H3, et oranges pour les brins
S2 et S3.
Les valeurs de nOe hétéronucléaire
1H-15N obtenues au niveau des brins â et
des hélices du tonnelet sont pour la plupart des résidus proches
de 0.8 (Figure 4.2 du paragraphe
1.2.1). Cet ordre de grandeur montre que le feuillet â et
le tonnelet orthogonal constituent un ensemble rigide et stable au sein du
domaine K2.
2.3) La boucle entre les hélices H2 et
H3
A l'opposé du feuillet â anti-parallèle,
le domaine K2 présente une large boucle de 15 acides aminés
(S51-T65) située entre les hélices H2 et H3. Cette région
peu encombrée est très exposée au solvant, ce qui
explique probablement la faible intensité des pics de
corrélation 1H-15N des résidus I56, H57,
M58, N59, et E64 due à un échange rapide de leur proton amide
avec l'eau. Aussi, le proton amide du résidu R53, dont le pic de
corrélation est absent sur l'HSQC 15N-1H, est
orienté vers l'extérieur de la protéine et se situe dans
une zone
très fortement exposée. La boucle S51-T65
n'est pas dénuée de structure secondaire. Elle
comporte en effet un tour d'hélice 310 canonique de 3
résidus (H2.5) stabilisée par une liaison
hydrogène entre le proton amide de A60 et l'oxygène
du carbonyle de H57. La thréonine T61
joue un rôle majeur dans le maintien de cette
hélice via le groupement OãH de sa chaîne
latérale qui forme 2 liaisons hydrogène avec le proton
HN de W63 et l'oxygène du CO de M58 (Figure 4.12). L'existence
de ce réseau de liaisons hydrogène est en accord avec
les résultats de l'étude par RMN où le proton du
groupement OãH de T61 est observable sur le spectre
15N-NOESY-HSQC à un déplacement chimique
caractéristique de 5.6 ppm. Les chaînes latérales
hydrophobes des résidus I56, M58, et W63 sont orientées vers
l'intérieur de
la protéine et établissent des contacts avec
plusieurs acides aminés du coeur hydrophobe (F23, L66, et F69). Ces
3 résidus contribuent ainsi au positionnement de l'hélice
310 H2.5 à proximité du tonnelet orthogonal.
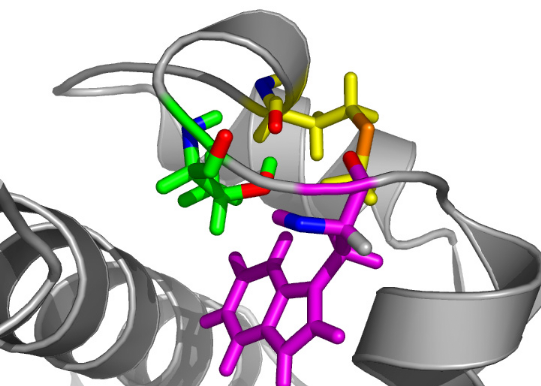
H2.5
M58
H2
T61
W63
H3
H1
Figure 4.12 : Rôle de la
thréonine T61 dans la stabilisation de l'hélice
310 H2.5. Les atomes
et liaisons des résidus M58, T61, et W63 sont
respectivement représentés par des sticks
jaunes, verts, et roses. Les hétéroatomes O, N,
et S sont différenciés et sont respectivement
représentés en rouge, bleu, et orange. Les liaisons
hydrogène apparaissent en pointillés.
L'analyse de la structure tridimensionnelle de K2 par
l'algorithme Stride (Frishman & Argos, 1995) met
également en évidence la présence de 2 coudes
â de type IV et VIII (respectivement I50-R53 et W63-L66) au niveau
des extrémités de la boucle S51-T65. Ces 2 coudes ne
présentent pas de liaison hydrogène de type CO-HN entre le
résidu i et le résidu i+3, ce qui est
fréquemment observé dans ces types de coude non classiques
(Hutchinson & Thornton, 1994). Il apparaît cependant 2 liaisons
hydrogène dans la région Y49-H55 qui semblent stabiliser
le motif I50-R53. La première implique l'oxygène du CO
de E54 et le proton du groupement OH de chaîne latérale de Y49,
et la seconde relie l'oxygène du CO de
R53 au proton amide de H55.
Le bilan de l'analyse des structures secondaires de la
région S51-T65 fait apparaître un
bon niveau de structuration de cette boucle avec la
présence d'un coude de type IV entre I50
et R53, d'une hélice 310 entre M58 et T60, et d'un
coude de type VIII entre W63 et L66. De plus, le nOe homonucléaire
1H correspondant à une moyenne de l'espace conformationnel en
solution, les nombreux effets observés entre les résidus I56,
M58, T61, et W63 et plusieurs résidus participant au coeur hydrophobe de
la protéine suggèrent une faible flexibilité de la
boucle S51-T65. Ainsi, dans le cas de l'isoleucine I56 située
entre le coude I50-R53 et l'hélice 310, près d'une quinzaine
de nOe de moyenne intensité témoignent de la proximité des
protons de sa chaîne latérale avec les protons du
résidu F23 impliqué dans la poche hydrophobe du domaine
K2. De manière intéressante, les valeurs de nOe
hétéronucléaire
1H-15N relevées dans la
région S51-T65 sont globalement comprises entre 0.7 et 0.85.
Ces valeurs caractéristiques de régions rigides sont tout
à fait comparables à celles obtenues dans
les hélices du tonnelet orthogonal et dans les
brins â. Ces résultats indiquent donc que la région
S51-T65 incorpore le corps rigide de la protéine.
2.4) L'hélice C-terminale H4
L'hélice H4 C-terminale s'étend du
résidu E98 à E107. Elle est constituée de 9
résidus, soit environ 2.5 tours d'hélice á
régulière, et présente un réseau régulier de
liaisons hydrogène qui contribuent à sa stabilité. Dans la
structure de K2, l'hélice H4 se positionne à
la périphérie du domaine au niveau de
l'extrémité C-terminale de l'hélice H1 et de
manière quasi perpendiculaire à H1. L'interface d'interactions
entre H4 et le tonnelet orthogonal est constituée des résidus
I100, Q103, et L104 de H4, des résidus R32 et R33 de H1, et du
résidu G36 de la boucle entre H1 et S1 (Figure 4.13). Cette
interface exclusivement hydrophobe apparaît comme relativement fragile
en raison du faible nombre de contacts observés entre ces acides
aminés. De plus, sur les spectres NOESY-HSQC, seule une dizaine d'effets
à longue distance de faible ou moyenne intensité ont
été collectés dans la région E98-E107. Avec ce
faible nombre de nOe, le programme d'attribution automatique a
rencontré quelques difficultés pour attribuer de
manière reproductible les nOe longue distance de l'hélice H4
au
fil des itérations. Ainsi, ce n'est que lors des derniers
processus itératifs que l'hélice H4 dans
les modèles générés a
adopté une position préférentielle unique autour de
la partie C- terminale de H1.
S2 S3 S1 H2
G36 H1
R33
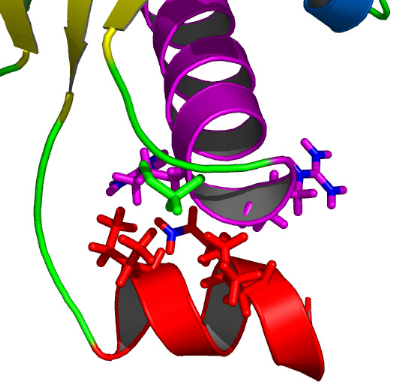
R32
I100
L104
H4
Q103
Figure 4.13 : Description de
l'interface d'interactions entre l'hélice H4 et l'hélice H1.
Les
résidus impliqués sont
représentés par des sticks rouges pour l'hélice H4, roses
pour l'hélice H1, et verts pour la boucle entre H1 et S1. Les
noyaux azote des chaînes latérales sont
différenciés et apparaissent en bleu.
De manière intéressante, les valeurs de nOe
hétéronucléaire 1H-15N
relevées dans la région E98-E107 sont globalement comprises
entre 0.4 et 0.5 (Figure 4.2 du paragraphe
1.2.1). Ces valeurs mettent en évidence des mouvements
rapides (sur une échelle de temps de
la picoseconde à la nanoseconde) au niveau de
l'hélice H4 et suggèrent un équilibre conformationnel
entre une forme structurée et une forme
déstructurée autour d'une conformation moyenne. Les valeurs
de déplacements chimiques secondaires CSD dans cette région sont
globalement inférieures à celles rencontrées au
niveau des hélices du tonnelet orthogonal. Ceci confirme
l'hypothèse de l'existence d'un échange conformationnel rapide et
explique probablement le peu de nOe 1H longue distance
observés dans cette région. Par conséquent,
l'hélice H4 du domaine K2 présente un caractère flexible
et n'intègre pas le corps rigide de la protéine.
Chapitre 5 : Relations structure-activité : quel
est le rôle du domaine K2 de KIN17 humaine ?
CHAPITRE 5
Relations structure-activité : quel est le
rôle
du domaine K2 de KIN17 humaine ?
Comme je l'ai exposé dans le premier chapitre, la
protéine KIN17 présente plusieurs
caractéristiques fonctionnelles qui
suggèrent son implication dans la régulation et la
maintenance du génome. Ses fonctions précises, ses
partenaires biologiques, et ses modes d'action sont toujours inconnus en ce
début d'année 2006. A présent, la connaissance de la
structure tridimensionnelle de la région 51-160 de KIN17 humaine
(domaine K2) apporte des informations supplémentaires qui pourraient
nous aider à identifier une (ou plusieurs) de ses fonctions biologiques.
Ceci nécessite dans un premier temps de répondre à un
certain nombre
de questions : quelle est la nature du repliement du domaine
K2 ? Quels sont les fonctions, les partenaires, et les mécanismes
d'action des protéines qui présentent ce type de repliement ?
Dans le cas où les bases moléculaires de ces modes d'action
seraient connues, le domaine K2 possède-t-il les
propriétés structurales nécessaires à la fonction
de ces protéines structuralement homologues ? Ce paragraphe a pour objet
d'apporter des éléments de réponse
à ces questions. La recherche de fonction d'une
protéine par une approche purement structurale présente
cependant un caractère spéculatif. Aussi, parallèlement
à cette étude, une recherche des partenaires biologiques du
domaine K2 a été entreprise au Laboratoire de Structure
des Protéines par une approche biochimique. Ces résultats
seront également présentés dans ce paragraphe dans
le but d'approfondir la connaissance des relations
structure-activité de la région 51-160 de KIN17 humaine.
1) Le domaine K2 de KIN17 adopte un repliement de type
Winged Helix
Nous avons recherché les homologues structuraux du
domaine K2 à l'aide du programme DALI disponible sur le web (Holm
& Sander, 1993) afin de caractériser la nature
de son repliement. Le serveur DALI compare la
structure tridimensionnelle d'une protéine cible avec chacune des
structures enregistrées dans la Protein Data Bank
(www.rcsb.org), puis établit un
score en fonction du degré d'homologie. Dans le cas du domaine
K2, des scores significatifs ont été obtenus avec des
protéines qui appartiennent à une famille structurale unique
: la famille des Winged Helix.
Le motif Winged Helix est une sous-classe de la
superfamille des domaines « hélice- coude-hélice » de
liaison à l'ADN (Pour revue : Gajiwala & Burley, 2000). Ce
motif, commun aux organismes procaryotes et eucaryotes, a été
découvert pour la première fois en
1993 chez le facteur de transcription eucaryote HNF-3
(Clark et al., 1993). La topologie
canonique d'un domaine Winged Helix est de la
forme H1-S1-H2-T-H3-S2-W1-S3-W2 où
S1, S2, et S3 forment un feuillet â
anti-parallèle situé en périphérie du motif «
hélice-coude- hélice » H2-T-H3 (Figure 5.1). W1 et W2 sont
deux boucles situées en région C-terminale et dont la
disposition autour de l'hélice H3 évoque les ailes d'un
papillon, ce qui explique le nom Winged Helix.
W1
H2
S2 S3 S1 N
H1
H3
C
W2
Figure 5.1 : Description de la
topologie d'un motif Winged Helix canonique.
Au-delà de la présence du feuillet â, la
longueur de la région contenant le coude qui relie H2 et H3
différencie les motifs Winged Helix des motifs «
hélice-coude-hélice » classiques. Ainsi, cette longueur est
très variable dans les Winged Helix, alors qu'elle se limite
généralement au nombre de résidus qui forment le coude T
dans les motifs « hélice-coude- hélice » (Brennan
& Matthews, 1989). D'autre part, la topologie d'un repliement de
type Winged Helix se révèle très
différente d'une protéine à l'autre. Les
éléments N-terminaux H1
et S1 sont fréquemment absents du motif. C'est
le cas par exemple des facteurs de transcription TorI (Elantak et al.,
2005) et MuR (Wojciak et al., 2001) dont la structure a été
résolue par RMN en solution et où l'hélice H1 est
manquante. La longueur du brin S1 dépasse rarement 4 acides
aminés et dans de nombreux cas, comme dans la structure du
domaine
C-terminal du facteur de transcription TFIIEâ
résolue par RMN (Okuda et al., 2000), seul un résidu hydrophobe
de la région reliant H1 et H2 est en contact avec les brins
S2 et S3 du feuillet â. La topologie de la région
contenant le coude qui relie les hélices H2 et H3 est
également variable et peut comporter des éléments de
structure secondaire additionnels. C'est
notamment le cas des facteurs de transcription AphA (De
Silva et al., 2005) et Genesis
(Marsden et al., 1997) qui contiennent une hélice á
supplémentaire dans cette région. Sur la
base de ces observations, il apparaît que le motif
minimum qui caractérise un repliement de type Winged Helix est
constitué du motif « hélice-coude-hélice »
H2-T-H3 et de la région C- terminale contenant les 2 brins â.
Au vu de l'homologie de topologie existante entre le domaine
K2 et le motif Winged Helix canonique, il apparaît que la
région 51-160 de la protéine humaine KIN17 adopte un repliement
de type Winged Helix. Le domaine K2 contient un tour d'hélice
310 atypique dans
la région du coude entre les hélices H2 et
H3, ainsi qu'une hélice H4 supplémentaire à la
périphérie du motif. De plus, chez K2, la boucle W1
est très courte et se limite aux résidus
P85 et K86 qui forment un coude â de type I entre les brins
S2 et S3. D'après l'analyse DALI,
le domaine structural le plus proche de K2 est le
motif Winged Helix Zá de la protéine ADAR1
(Schwartz et al., 1999). La figure 5.2 représente dans la
même orientation les structures de K2 et de ce domaine, et
illustre bien la forte homologie structurale entre ces deux
protéines.

C
C
N
N
Figure 5.2 :
Représentation du squelette Cá des domaines K2 de KIN17
en rose et Zá de
ADAR1 en bleu (PDB : 1QBJ). L'hélice H4 de K2 n'est
pas représentée.
Un alignement de séquence primaire mené
sur une quarantaine de séquences de KIN17 appartenant à
des espèces eucaryotes met en évidence que 9 des 13
résidus qui forment le coeur hydrophobe de K2 sont
conservés (non montré). Cet alignement fait également
apparaître que 8 des 15 acides aminés qui appartiennent à
la région entre H2 et H3 contenant l'hélice 310 sont
très conservés. Par conséquent, il est raisonnable
d'affirmer que toutes les protéines KIN17 eucaryotes adoptent en
région N-terminale un repliement de type
Winged Helix similaire à celui du domaine K2 de
KIN17 humaine.
Lorsque la caractérisation structurale de la
protéine KIN17 a été entreprise, la
prédiction bio-informatique menée par le
programme SMART suggérait la présence d'un domaine
FF de liaison aux peptides phosphorylés dans la région
50-150 de KIN17. De manière intéressante, la topologie
canonique d'un motif FF de type H1-H2-H2.5-H3 (avec H2.5 une
hélice 310) est très proche de la topologie de la région
N-terminale du domaine K2 (H1-S1-H2-H2.5-H3). Ces 2 domaines sont
constitués d'un tonnelet orthogonal central formé
par les hélices H1, H2, et H3. Nous avons
soumis la région N-terminale de K2 au serveur DALI afin de
rechercher les domaines qui présentent une homologie structurale
avec cette région. Malgré l'absence du double brin â
C-terminal, les résultats obtenus montrent que les domaines les plus
structuralement proches de cette région de K2 sont tous des motifs
Winged- Helix. L'analyse DALI ne fait apparaître aucun domaine
FF comme homologue structural de cette région N-terminale de K2. La
région 51-160 de KIN17 adopte donc un repliement divergent des
domaines FF.
2) Le motif Winged Helix de KIN17 est-il capable
de lier l'ADN
ou l'ARN ?
L'étude préliminaire bio-informatique de la
protéine KIN17, qui a été présentée dans
le premier chapitre, suggère la présence d'un motif
de liaison aux acides nucléiques de type
« doigt de zinc » entre les résidus 28
et 50 en amont du domaine K2. Les chercheurs du Laboratoire de
Génétique de la Radiosensibilité (LGR) du CEA de
Fontenay-aux-Roses ont confirmé l'existence de ce motif en
démontrant sa capacité à lier l'ADN, et notamment
l'ADN courbe, de manière dépendante aux ions Zn2+
(Mazin et al., 1994). Au cours de cette étude, les auteurs ont
également montré qu'il existe un second domaine de liaison
à l'ADN situé dans la région N-terminale de KIN17
entre les résidus 71 et 281. De manière
intéressante, cette région de KIN17 comporte le motif
Winged Helix du domaine K2 (65 à
157). Récemment, de nouvelles investigations
menées par les chercheurs du LGR ont mis en évidence la
capacité de KIN17 à lier l'ARN et notamment l'ARN riche en bases
guanine et uracile (Pinon-Lataillade et al., 2004). Dans l'optique
d'améliorer la connaissance des fonctions précises et des
mécanismes d'action de KIN17, il serait intéressant de
déterminer quelle est l'implication du motif Winged Helix
du domaine K2 dans la liaison à l'ADN et l'ARN. Aussi, d'un
point de vue structural et fonctionnel, ce motif a-t-il la capacité de
lier
l'ADN ou l'ARN ?
2.1) Approche structurale
2.1.1) Reconnaissance de l'ADN par les protéines
à motif Winged Helix
Les principales fonctions des domaines protéiques
à motif Winged Helix sont la reconnaissance et la
fixation de l'ADN. En 1999, près d'une dizaine de structures
de complexe Winged Helix-ADN résolues par
radiocristallographie ou par RMN étaient disponibles dans la
Protein Data Bank. A partir de ces structures, Gajiwala
et Burley ont proposé deux modes de reconnaissance de
l'ADN par les protéines à motif Winged Helix
(Gajiwala & Burley, 2000) :
· Le premier mode dit « classique » est
à ce jour le plus rencontré dans les complexes Winged
Helix-ADN. Il a été découvert pour la
première fois à partir de la structure cristallographique
de la protéine HNF-3 liée à un fragment d'ADN (Clark et
al., 1993). Dans ce modèle, l'hélice de reconnaissance H3 plonge
dans le sillon majeur de l'ADN et établit plusieurs contacts avec
les bases nucléotidiques (contacts spécifiques) et le
squelette phosphate (contacts non spécifiques). La boucle W1
située entre les brins S2 et
S3 participe également à la liaison à
l'ADN en établissant quelques contacts non spécifiques avec
le squelette phosphate et le ribose dans le petit sillon de l'ADN (Figure
5.3). L'hélice H3 constitue donc
l'élément majeur de ce mode de reconnaissance en assurant
la spécificité de l'interaction, alors que la boucle W1 a pour
principale fonction d'augmenter l'affinité de la liaison (Huffman &
Brennan, 2002). Dans certains cas, des résidus de l'hélice H2
ou de la région N-terminale de H1 interagissent également
avec l'ADN et contribuent à améliorer l'affinité de
l'interaction. La plupart des interactions Winged Helix-ADN dans
ce mode d'action impliquent des résidus à chaîne
latérale polaire et notamment chargée positivement. Par
conséquent, les domaines Winged Helix qui adoptent ce mode
de reconnaissance classique présentent une surface
d'interaction
H3-W1 largement positive.
· Le second mode dit « atypique » n'a
été identifié à ce jour qu'à une seule
reprise dans la structure cristalline de la protéine RFX1
liée à un fragment d'ADN (Gajiwala et al.,
2000). Il implique également les éléments H3
et W1 mais leur rôle est inversé. Ainsi, la
spécificité de l'interaction est ici assurée par la boucle
W1, riche en résidus basiques, et
qui établit plusieurs contacts avec les bases du
grand sillon de l'ADN (Figure 5.3). La
surface de l'hélice H3 apparaît comme neutre et
interagit principalement avec le squelette
phosphate du sillon mineur. De manière
intéressante, plusieurs domaines Winged Helix capables de
lier l'ADN comme par exemple ORC2 (Singleton et al., 2004), ou SmtB
(Cook et al., 1998), présentent une surface neutre au niveau de
l'hélice H3 et un amas de résidus basiques dans la boucle W1. Il
est donc fort probable que ces domaines utilisent le mode de reconnaissance
atypique pour lier l'ADN.
« mode classique »
« mode atypique »
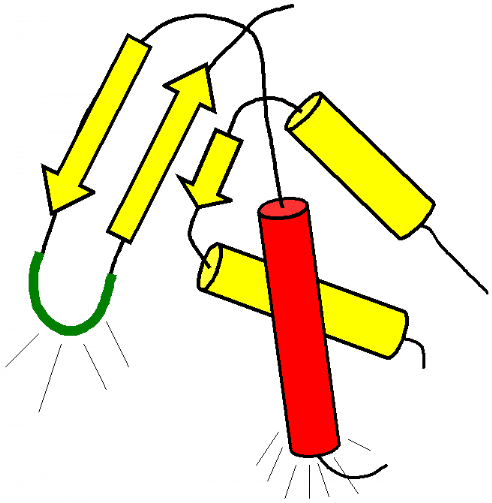
sillon mineur
sillon majeur
sillon mineur
sillon majeur
Figure 5.3 : Illustration des modes
de reconnaissance de l'ADN par les motifs Winged Helix.
L'hélice H3 est représentée en rouge et
la boucle W1 en vert.
2.1.2) Comparaison structurale du domaine K2 avec des
motifs Winged Helix
de liaison à l'ADN
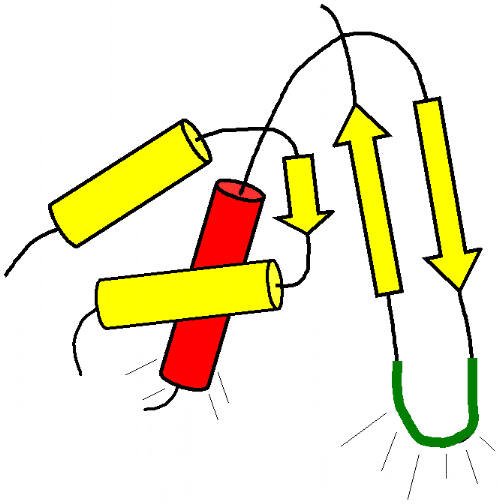
L'analyse de la structure du domaine K2 par l'algorithme
DALI fait apparaître une homologie de structure importante entre
K2 et les protéines ORC2 (Singleton et al., 2004),
PA-Fur (Pohl et al., 2003), TFIIEâ (Okuda et
al., 2000), BlaI (Safo et al., 2005), MecI (Garcia-Castellanos et al.,
2004), et DP2 (Zheng et al., 1999). Ces protéines sont des
régulateurs de la transcription des gènes impliqués
dans des mécanismes variés comme le contrôle de
l'incorporation du fer (PA-Fur), la résistance bactérienne aux
antibiotiques (BlaI et MecI), la régulation du cycle cellulaire
(ORC2 et DP2), et l'initiation de la transcription (TFIIEâ). Pour
toutes ces protéines, le domaine Winged Helix homologue
à K2 est un domaine
de liaison à l'ADN. Parmi ces domaines structuralement
proches, figure le motif Winged Helix
des protéines BlaI, MecI, et DP2 dont la structure en
complexe avec un fragment d'ADN a été
résolue par cristallographie des rayons X. Aussi, il est
à noter que la structure de la protéine
BlaI a été initialement
déterminée par RMN en solution par les chercheurs de
l'IBS Jean- Pierre Ebel de Grenoble (Van Melckebeke et al., 2003). Ces trois
motifs adoptent un mode de reconnaissance de l'ADN de type classique. Nous
avons superposé la structure du domaine K2 avec celle de ces 3
protéines en complexe afin d'évaluer les potentialités du
Winged Helix de
K2 à fixer l'ADN selon ce mode de reconnaissance.
La topologie du domaine Winged Helix de la
protéine MecI liée à un double brin d'ADN de 25
paires de bases est du type H1-H2-T-H3-S1-S2. Le programme DALI a
été utilisé pour identifier les régions
structuralement proches de K2 et MecI. Malgré une absence d'homologie
de séquence entre ces 2 protéines (< 10 %), la
déviation moyenne « rmsd » atteint 2.7 Å sur 66
carbones á. Comme le montre la Figure 5.4, les 2 structures
se superposent de manière quasi parfaite au niveau du feuillet â,
de la boucle W1, et de l'hélice
H2 (en arrière plan). L'hélice H1 du
domaine K2 comporte 2 tours de plus que son homologue de MecI et
son orientation est un peu moins ouverte par rapport au reste de la
structure. La plus grande différence structurale entre ces 2
protéines se situe au niveau de l'hélice de reconnaissance
H3 et de la région du coude entre H2 et H3. En effet, l'hélice
H3
du domaine K2 est d'une part, plus courte que son homologue de
MecI d'un peu moins de 2 tours et d'autre part, son orientation sensiblement
plus ouverte ne lui permet pas de pénétrer le sillon majeur de
la même manière que H3 de MecI. Il apparaît ainsi
en superposant ces 2 structures que la large boucle entre H2 et H3, et
notamment l'hélice 310 H2.5, sont dans une
position beaucoup plus favorable pour interagir avec les bases du
grand sillon de l'ADN.
H1
H1
W1
W1
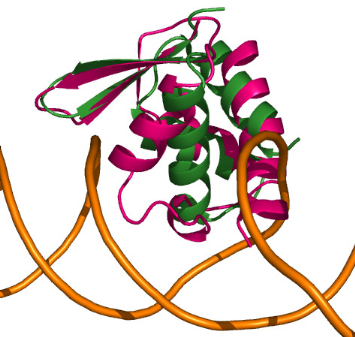
H3 H3
H2 K2 de KIN17
H2 MecI
H2.5
Figure 5.4 : Vue d'ensemble de la
superposition de structure du domaine K2 avec la protéine
MecI en complexe avec un double brin d'ADN de 25 paires de
bases (PDB : 1SAX). Le squelette phosphate de l'ADN est
représenté en orange.
Dans la structure cristallographique du complexe MecI-ADN, 7
résidus de l'hélice de
reconnaissance H3 sont impliqués dans la liaison avec
l'ADN et parmi ceux-ci, les 3 résidus K43, T47, et R51
établissent des contacts majeurs et spécifiques avec 4 bases
nucléotidiques. Ces trois résidus, mis en évidence dans la
Figure 5.5, appartiennent à la région N-terminale de
H3 qui ne se superpose pas avec l'hélice H3 de K2. A la
place de l'arginine R51 de MecI qui forme 2 liaisons hydrogène avec
une guanidine, on trouve dans la même orientation le tryptophane
W72 de K2, d'une part plus volumineux et hydrophobe, et d'autre part qui n'a
pas la capacité à former 2 liaisons hydrogène. La lysine
K43 de MecI qui forme une liaison hydrogène avec une thymine se
superpose quasi parfaitement à l'asparagine N59 de l'hélice
310 de K2. Cette asparagine pourrait également
former cette liaison hydrogène mais ne possède pas la
basicité d'une lysine. Il est d'ailleurs intéressant de noter que
le domaine K2 ne présente quasiment aucun résidu basique dans la
région contenant la large boucle et l'hélice H3. Aussi,
l'orientation des chaînes latérales négatives des
résidus E64 et D68 vers le double
brin d'ADN n'est pas très favorable à l'approche du
squelette phosphate chargé négativement.
H3 W72
D68 H3
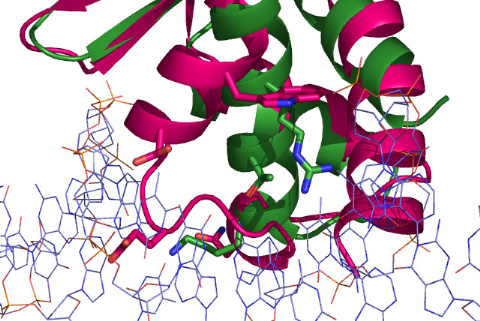
T47
R51
K2 de KIN17
MecI
N59
E64
K43
Figure 5.5 : Comparaison
structurale des hélices H3 de K2 et MecI. Les 2
protéines sont
superposées comme dans la Figure 5.4. Les
chaînes latérales des résidus mis en évidence sont
représentées par des sticks violets pour K2, et verts pour MecI.
Les hétéroatomes N, et O sont différenciés et
sont respectivement représentés par des sticks bleus, et
oranges. Le double brin d'ADN est représenté par des traits
bleus et oranges pour le squelette phosphate.
Nous avons également superposé la structure de K2
avec celle des protéines BlaI et
DP2 en complexe avec un fragment d'ADN. Ces 2 domaines
présentent une forte homologie
de structure avec le domaine K2 : la déviation moyenne
rmsd est de 2.5 Å sur 62 carbones á
pour DP2, et de 2.9 Å sur 67 carbones á
pour BlaI. De manière très analogue à MecI, la
structure de K2 est très proche de celle de BlaI et DP2 au
niveau de l'hélice H2, du feuillet â,
de la boucle W1, et aussi au niveau de l'hélice
H1 pour DP2 (Figure 5.6). La plus grande divergence structurale se situe
au niveau de l'hélice H3 dont la longueur dans K2 (10 résidus)
est plus courte que dans BlaI et DP2 (respectivement
15 et 21 acides aminés). De plus, l'orientation plus ouverte de
l'hélice H3 de K2 ne semble pas très favorable à une
interaction avec les bases du sillon majeur de l'ADN et comme
précédemment, celui-ci apparaît principalement
occupé par la large boucle entre H2 et H3 de K2. Ainsi, les
résidus majeurs de l'hélice H3 de BlaI et DP2 qui
établissent des contacts spécifiques avec les bases de l'ADN
appartiennent à une région qui ne se superpose pas avec
l'hélice H3 de K2, mais plutôt avec
l'hélice 310 H2.5.
W1 H3
H3
H1 H1
H2
W1
K2
H1
H2
H1
H2.5
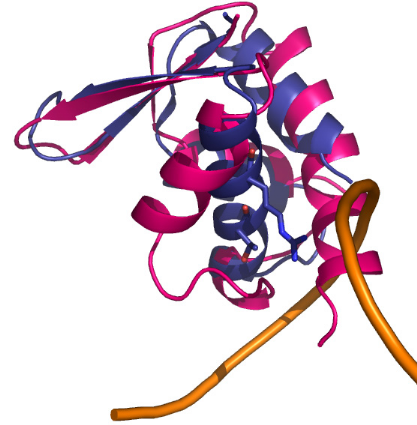
T47
R51
BlaI
DP2
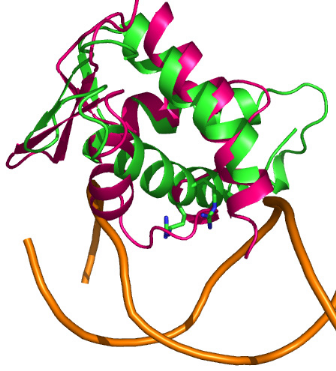
H3
H3
R121
R122
H2.5
Figure 5.6 : Superposition de
structure du domaine K2 avec le motif Winged Helix des
protéines BlaI et DP2 en complexe avec l'ADN. Les
chaînes latérales des résidus de l'hélice
H3 qui interagissent avec les bases du grand sillon
sont représentées par des sticks. Le squelette phosphate de
l'ADN est coloré en orange. A) Superposition de K2 avec BlaI liée
à
un double brin de 25 paires de bases (PDB : 1XSD). Seul un
des deux brins est représenté. B) Superposition de K2 avec la
protéine DP2 du complexe hétérodimère
DP2-E2F4-ADN contenant un fragment de 15 paires de bases (PDB : 1CF7).
La superposition de structure du domaine K2 avec les
motifs Winged Helix des protéines MecI, BlaI, et DP2 fait
donc apparaître des divergences structurales significatives au niveau de
l'hélice de reconnaissance H3. Contrairement à ces 3 motifs, K2
présente une large boucle de 15 résidus entre H2 et H3 (Figure
5.7) dont la présence semble réduire la taille de l'hélice
H3 et ne lui permet pas d'adopter une orientation favorable pour
interagir avec le grand sillon de l'ADN selon le mode de reconnaissance
classique des Winged Helix. Pour de
nombreux motifs Winged Helix, la liaison à
l'ADN s'accompagne d'une déformation ou
d'une réorientation de l'hélice H3 qui
peut atteindre une quinzaine de degrés (Gajiwala & Burley,
2000). Dans le cas du domaine K2, la réorientation de l'hélice H3
dans une position plus favorable nécessiterait une modification majeure
de l'organisation de la boucle et de la position de l'hélice 310 H2.5.
Or, il apparaît, d'après les données RMN, que cette
hélice 310 est stabilisée par un réseau de liaisons
hydrogène et qu'elle incorpore le corps rigide de la
protéine tout autant que la boucle. Par conséquent, la
réorientation de l'hélice H3 pour adopter
le mode de reconnaissance classique nécessiterait une
modification structurale importante du domaine K2 dont l'envergure, à
notre connaissance, n'a jamais été observée dans un
motif Winged Helix. Il est toutefois important de noter que
ce n'est pas la présence même d'une hélice
additionnelle dans la boucle entre H2 et H3 qui compromet l'interaction
potentielle de
H3 avec le sillon majeur. La protéine Genesis, dont la
structure en complexe avec un fragment d'ADN a été résolue
par RMN (Jin et al., 1999), contient une hélice á additionnelle
H4 de 5 résidus entre H2 et H3 (Figure 5.7). Dans la structure, cette
hélice est positionnée à l'extérieur
du sillon majeur et l'hélice de reconnaissance
H3, dont l'orientation et la longueur sont comparables à H3 de
MecI et BlaI, pénètre et interagit avec le sillon majeur
de manière classique. Par conséquent, il s'agit bien de
la position de l'hélice 310 de K2, et non de sa
présence, qui distingue le plus le motif Winged Helix
du domaine K2 du Winged Helix de BlaI, MecI, DP2, et
Genesis. Il est d'ailleurs intéressant de noter que Genesis
n'apparaît pas comme un homologue structural de K2 d'après
l'analyse DALI, et ceci en dépit d'une certaine
homologie de topologie dans la région H2-H3.
BlaI : SANEIVVEIQKYKEVSDKTIRTLITRLYKKEII
DP2 : SYNEVADELVSEFTNSNNHLAADSAYDQKNIRRRVYDALNVLVAMN
MecI : SANNIIEEIQMQKDWSPKTIRTLITRLYKKGFI
Genesis : LSGICEFISNRFPYYREKFPAWQNSIRHNLSLNDC
K2 : HNNIVYNEYISHREHIHMNATQWETLTDFTKWLGREG
Figure 5.7 : Définition
des éléments de structure secondaire entre les hélices H2
et H3 du motif Winged Helix des protéines BlaI, DP2, MecI, Genesis, et
K2 (KIN17). Les résidus de l'hélice H2 sont surlignés en
vert, et de l'hélice H3 en orange. Les hélices additionnelles
de
K2 (H2.5) et Genesis (H4) apparaissent en rouge.
Le calcul des potentiels électrostatiques de surface
met également en évidence des différences majeures
entre le domaine K2 et les motifs Winged Helix structuralement
proches
capables de lier l'ADN. Comme le montre la Figure 5.8,
le potentiel électrostatique de la
surface H3-W1 est largement positif chez les protéines
MecI, BlaI, et DP2. C'est également le
cas pour la protéine Genesis. Cette caractéristique
structurale est typique des protéines à motif
Winged Helix qui adoptent un mode de reconnaissance de
l'ADN de type classique.
A) B)
K2
KIN17 humaine
MecI
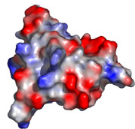
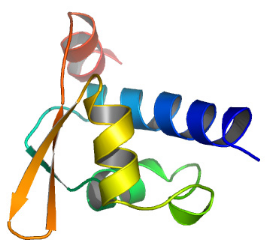
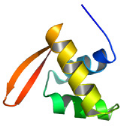

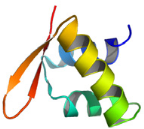

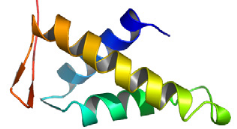
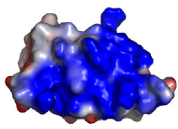
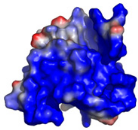
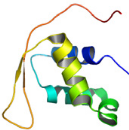
BlaI
DP2
Genesis
Figure 5.8 : Comparaison des
potentiels électrostatiques de surface des protéines
K2
(KIN17), MecI, BlaI, DP2 (du complexe E2F4-DP2) et
Genesis. A) Représentation de la structure de ces protéines.
L'hélice H1 apparaît dans les tons bleus, H2 dans les tons
verts,
H3 dans les tons jaunes, et la boucle W1 et les
brins S2 et S3 dans les tons oranges.
B) Représentation du potentiel électrostatique
de surface calculé avec le programme APBS (Baker et al., 2001) dans la
même orientation que A). Les régions chargées
négativement sont
en rouge et celles chargées positivement en bleu. Les
régions blanches correspondent à des régions peu
chargées ou hydrophobes.
Dans ces protéines, l'hélice H3 et la boucle
W1 contiennent plusieurs résidus lysine et
arginine dont les chaînes latérales
positives sont projetées vers l'extérieur du domaine et
établissent de nombreux contacts électrostatiques avec le
squelette phosphate de l'ADN chargé négativement. Dans le
cas du domaine K2, seuls 2 résidus lysine et arginine sont
dénombrables au niveau de l'hélice H3, ainsi qu'une lysine dans
la boucle W1. Il en résulte une surface électrostatique H3-W1 peu
polaire et très faiblement chargée positivement.
2.1.3) Comparaison avec la protéine
ADAR1
Comme nous l'avons évoqué
précédemment, la protéine qui présente la
plus forte homologie structurale avec K2 est le domaine Zá de ADAR1
humaine. La protéine ADAR1, impliquée dans la régulation
de l'ARN (Herbert et al., 1995), est capable de lier l'ADN de type Z via son
domaine Winged Helix Zá. L'ADN Z est une forme rare de l'ADN
dont la fonction est encore mal connue. Ce polymère de bases
nucléotidiques se structure en une double hélice gauche riche
en bases guanine et cytosine, et dont l'alternance de conformation anti
et syn a pour conséquence la formation d'un sillon
unique et profond. De manière intéressante, les bases
moléculaires de la reconnaissance de l'ADN Z par le domaine Zá
sont très différentes de celles de la reconnaissance de l'ADN B
(forme classique) par les motifs Winged Helix, mais elles impliquent
les mêmes éléments de structure secondaire (Schwartz et
al., 1999). Ainsi, Zá utilise principalement son
hélice de reconnaissance H3 et sa boucle W1 pour établir des
contacts avec le squelette phosphate et le cycle furanose des sucres.
Cependant, cette protéine n'établit aucun contact direct
avec les bases nucléotidiques. Par conséquent, la
reconnaissance de l'ADN Z par le domaine Zá de ADAR1 n'est pas
spécifique
de la séquence mais plutôt de la conformation Z.
Aussi, à l'image des motifs Winged Helix
qui lient l'ADN classique de type B, la majorité des
interactions avec l'ADN Z sont de nature électrostatique et impliquent
des résidus à chaîne latérale chargée
positivement. Il en résulte que les domaines Winged Helix qui
lient l'ADN Z comme ADAR1 présentent un potentiel de surface H3-W1
largement positif (Figure 5.9B), ce qui n'est pas le cas du domaine K2.
La superposition de structure de K2 avec Zá de ADAR1
fait apparaître une très forte homologie de structure entre ces 2
protéines : la déviation moyenne rmsd est de 2.0 Å sur 61
carbones á (Figure 5.9C). Les hélices H1 et H2 sont d'une
longueur quasiment identique et s'alignent presque parfaitement l'une sur
l'autre. Zá contient une boucle W1 un peu plus longue et 2 brins
â S2 et S3 un peu plus courts, mais le nombre de résidus dans la
région S2-
W1-S3 est sensiblement identique dans les 2 domaines (une
quinzaine de résidus). La plus
grande différence structurale concerne une
nouvelle fois la région de l'hélice H3 et de la boucle
entre H2 et H3. L'hélice H3 de K2 est plus courte d'environ un tour et
la plupart des résidus de Zá qui interagissent avec l'ADN
Z appartiennent à une région de H3 qui ne se superpose
pas sur H3 de K2 mais plutôt sur la boucle ou l'hélice 310.
K2
Zá
A)
H1
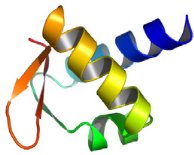
Zá de ADAR1
B)
W1
C)
Y177
K169
H3 H3
K170
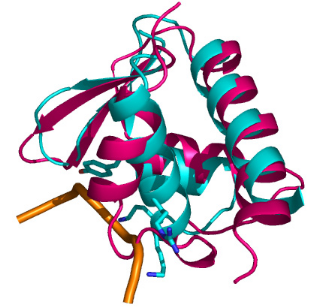
H2
R174
H2.5
Figure 5.9 : Comparaison
structurale du domaine K2 de KIN17 et de Zá de ADAR1
en
complexe avec un fragment d'ADN Z de 6 paires de bases. A)
Structure tridimensionnelle de
Zá. Le code couleur des éléments de
structure secondaire est relatif à la figure
précédente.
B) Surface électrostatique de Zá
calculée avec le programme APBS dans la même orientation que A).
C) Superposition de structure de K2 avec Zá. Les chaînes
latérales des résidus de l'hélice H3 de Zá qui
interagissent avec l'ADN de type Z sont représentées par des
sticks. Le squelette phosphate de l'ADN est coloré en orange. Seul un
brin est représenté.
La protéine ADAR1 humaine comporte un second domaine
Winged Helix (Zâ) situé
en aval de Zá à environ 80 acides aminés
dans la séquence primaire et dont la fonction est inconnue. La
récente résolution de structure de ce domaine par
radiocristallographie (Athanasiadis et al., 2005) montre une très forte
homologie structurale entre Zâ et Zá (rmsd
de 1.3 Å sur 62 résidus). Par conséquent, la
structure de Zâ est également très proche de K2
de KIN17 et les résultats de l'analyse DALI
font apparaître que ce domaine présente la seconde plus
forte homologie structurale avec K2 (rmsd de 2.7 Å sur 67
résidus). De manière intéressante, contrairement à
Zá, le Winged Helix de Zâ n'est pas capable de lier l'ADN
de type Z. En comparant ces 2 structures de manière fine,
Athanasiadis et al., ont mis en évidence des divergences
structurales au niveau de l'hélice de reconnaissance H3 qui
expliquent cette différence fonctionnelle. Ainsi, les 3
résidus conservés K169, R174, et Y177
de Zá qui établissent des contacts majeurs avec
l'ADN ne sont ni présents, ni conservés, chez
Zâ, et sont respectivement remplacés par A327,
A332, et I335 qui n'ont pas la capacité de former de liaison
hydrogène avec leur chaîne latérale. Aussi, les
résidus K169 et R174 de Zá appartiennent à une
région de l'hélice H3 qui ne se superpose pas avec H3 du domaine
K2.
De plus, le potentiel de surface H3-W1 de Zâ est
similaire à celui de K2 et apparaît peu chargé et
plutôt neutre contrairement à Zá qui présente
un potentiel de surface largement positif. Par conséquent, en se
basant sur les relations structure-activité des motifs
Winged Helix de ADAR1, il apparaît que le domaine K2 de KIN17
n'a pas la capacité structurale pour lier l'ADN de type Z selon le mode
de reconnaissance de Zá, et cela en dépit d'une très forte
homologie structurale. Aussi, il n'existe à ce jour aucune donnée
fonctionnelle qui suggère
que la protéine KIN17 est capable de lier l'ADN de type
Z.
2.1.4) Reconnaissance de l'ARN par les protéines
à motif Winged Helix
La publication récente de nouvelles structures de motif
Winged Helix a élargi l'horizon des fonctions adoptées
par ce type de domaine. Ainsi, il a été montré en 2002 par
Selmer et
al., que le domaine de liaison à l'ARN
du facteur d'élongation SelB est un motif Winged Helix
de topologie canonique (Selmer & Su, 2002). Trois
années plus tard, l'institut de Chimie des Substances Naturelles
de Gif-sur-Yvette a caractérisé pour la première
fois les bases moléculaires de la reconnaissance de l'ARN par un motif
Winged Helix en cristallisant
le domaine de liaison à l'ARN de la protéine SelB
avec un fragment de 16 bases (Yoshizawa
et al., 2005). Dans cette structure, plusieurs résidus
des hélices H2, H3, et de la boucle W1 forment une surface
conservée qui interagit de manière quasi exclusive avec les
groupements phosphate de l'ARN chargés négativement. Ce mode de
reconnaissance des acides nucléiques tout à fait unique
implique par conséquent une surface H2-H3-W1 fortement
chargée positivement, ce qui n'est absolument pas le cas du domaine K2
de la protéine KIN17 dont la structure n'apparaît pas homologue
à celle de SelB d'après l'algorithme DALI.
A ce jour, la structure du complexe SelB-ARN est la seule
structure connue d'un motif Winged Helix lié à
l'ARN. Cependant, il existe à notre connaissance au moins 2
autres domaines capables de lier l'ARN qui adoptent un repliement de type
Winged Helix. Il s'agit
du domaine N-terminal LM de la phosphoprotéine La
(Dong et al., 2004) et du domaine
U2AF du facteur d'épissage U2 (Kielkopf et al., 2004). La
topologie du domaine LM, de type
H1-H1'-H1''-B1-H2-H2'-H3-B2-B3, est très
différente de celle des motifs Winged Helix
canoniques, et donc de celle de K2. H1', H1'', et H2' sont des
hélices additionnelles insérées
entre les hélices du tonnelet orthogonal ancestral
formé par H1, H2, et H3. Des expériences de mutagenèse
montrent que des résidus des hélices H1, H1', et H1'' et de la
boucle entre H2 et H2' forment une surface conservée, aromatique, et
hydrophobe susceptible d'interagir avec l'ARN. Il apparaît donc qu'il
existe au moins 2 modes de reconnaissance de l'ARN par des motifs Winged
Helix radicalement différents. La découverte de ces
nouveaux modes de reconnaissance montre la grande versatilité avec
laquelle ce type de repliement est capable de reconnaître les acides
nucléiques. Toutefois, la capacité des domaines Winged
Helix à lier l'ARN reste à ce jour une fonction
atypique de ce genre de motif. Il est donc difficile d'émettre
une hypothèse quant à la capacité structurale du domaine
K2 à lier l'ARN.
2.2) Approche fonctionnelle
Parallèlement à l'étude structurale par RMN,
des études fonctionnelles d'interaction et
de recherche de partenaires biologiques ont été
menées pour les domaines K2 et K3 de KIN17 humaine par Albane le
Maire du Laboratoire de Structure des Protéines. Comme je l'ai
évoqué dans le premier chapitre, le domaine K3 correspond aux 160
premiers résidus de la protéine KIN17 humaine. Il est donc
principalement constitué du domaine K2 (51-160) et du motif
prédit structuré en « doigt de zinc » (28-50).
Deux techniques ont été utilisées pour tester
l'interaction in vitro de K2 et K3 avec l'ADN et l'ARN : il
s'agit de l'hybridation Southwestern et Northwestern.
2.2.1) Interaction avec l'ADN par hybridation
Southwestern
La technique de Southwestern consiste à
incuber des protéines avec un fragment d'ADN radiomarqué
sur une membrane de nitrocellulose afin de révéler les
protéines qui fixent le ligand marqué. Les protéines
d'intérêt sont dans un premier temps séparées sur
un
gel d'acrylamide de type SDS-PAGE, puis
transférées sur une membrane de nitrocellulose. La coloration au
rouge Ponceau de la membrane permet de vérifier la présence et
les quantités des protéines déposées sur le gel. La
membrane de nitrocellulose est ensuite incubée avec une solution
d'hybridation contenant 50 mM de NaCl, 10 mM de Tris-HCl (pH 7.4),
1 mM d'EDTA, du tampon Denhardt 1X, et le fragment d'ADN cible marqué
radioactivement au
phosphore 32P. Après plusieurs lavages de
la membrane, les protéines qui lient la cible
radioactive sont révélées sur plaques
radiosensibles phosphorescentes (phosphoimager).
Pour tester l'interaction de K2 et K3 avec l'ADN, la
cible utilisée est une sonde
d'ADN double brin de 500 nucléotides provenant d'une
origine de réplication reconnue par la protéine KIN17 humaine. La
Figure 5.10 présente les résultats obtenus par Southwestern
de l'interaction de cette sonde avec K2, K3, et KIN17 humaine.
La révélation de la membrane de
nitrocellulose sur plaques radiosensibles fait apparaître un signal
au niveau des bandes protéiques de KIN17 entière et du
domaine K3. Cette expérience confirme donc dans un premier temps
la capacité de la protéine KIN17 humaine à lier
l'ADN cible de 500 paires de bases in vitro. La forte
intensité de la bande correspondant à K3 met en
évidence le rôle important de ce domaine dans la liaison à
cette sonde d'ADN. En revanche, aucun signal de radioactivité n'est
détecté au niveau de la bande protéique relative au
domaine K2. Par conséquent, il semble d'une part, que la région
1-50 de KIN17 humaine contenant le « doigt de zinc » joue un
rôle majeur dans cette liaison à l'ADN
et d'autre part, que le motif Winged Helix de KIN17 n'a
pas la capacité, ou n'est pas suffisant,
pour lier ce fragment d'ADN de manière autonome.

(kDa)
75
50
37
25
20
15
K2 K3 Kin17

·
·

·

32P-ADN
K2 K3 Kin17
« rouge ponceau » « phosphoimager »
Figure 5.10 : Test d'interaction
de K2, K3, et KIN17 avec une sonde d'ADN de 500 paires de bases par
Southwestern. Sur la membrane colorée au rouge ponceau,
les bandes correspondant à K2, K3, et KIN17 sont indiquées par
des points. Les bandes révélées par le phosphoimager sont
mises en évidence par des flèches.
2.2.2) Interaction avec l'ARN par hybridation
Northwestern
Le principe de l'hybridation Northwestern
est tout à fait similaire à celui de
l'hybridation Southwestern et permet de tester l'interaction d'une
protéine avec un fragment d'ARN radiomarqué au phosphore
32P. Les trois protéines KIN17 ont été
séparées sur gel
SDS-PAGE, transférées sur membrane de
nitrocellulose, puis incubées avec une sonde
d'ARN radiomarquée de 1200 nucléotides.
Les résultats de cette expérience
d'hybridation Northwestern sont tout à fait
comparables à ceux obtenus précédemment. La
révélation de la membrane de nitrocellulose
sur plaques photosensibles fait apparaître les
bandes protéiques correspondant à KIN17 entière et
au domaine K3, mais pas à celle de K2 (Figure 5.11). Par
conséquent, ce test d'interaction in vitro montre une
implication de la région 1-50 contenant le motif « doigt de zinc
» dans la liaison à l'ARN. Cette expérience suggère
également que le motif Winged Helix
de KIN17 humaine n'est pas suffisant pour lier l'ARN de
manière autonome dans ces
conditions d'analyse.
Kin K3 K2
Kin K3 K2
·

32P-ARN

·

·
« rouge ponceau »
« phosphoimager »
Figure 5.11 : Test d'interaction
de K2, K3, et KIN17 avec une sonde d'ARN de 1200
nucléotides par Northwestern. Sur la membrane
colorée au rouge ponceau, les bandes correspondant à K2, K3,
et KIN17 (Kin) sont indiquées par des points. Les bandes
révélées par le phosphoimager sont mises en
évidence par des flèches.
3) Le motif Winged Helix du domaine K2
présente une surface ultra conservée
Nous avons utilisé le logiciel CONSURF
(http://consurf.tau.ac.il/) disponible
sur le web afin de déterminer s'il existe une ou plusieurs surfaces
conservées durant l'évolution du domaine K2 de KIN17. A partir
d'un alignement de séquences issues d'organismes différents
et de la structure de K2, CONSURF permet de visualiser
grâce à un code couleur l'état de
conservation phylogénétique des surfaces de la
protéine.
Nous avons vu précédemment que les domaines
à motif Winged Helix capables de lier
l'ADN utilisent leur hélice H3 et leur boucle W1 pour
interagir avec les acides nucléiques, et ceci quel que soit le mode de
reconnaissance adopté ou le type d'ADN reconnu. Aussi, ces domaines
présentent une surface fonctionnelle H3-W1
généralement très conservée qui maintient leur
fonction au cours de l'évolution. De manière
générale, KIN17 est une protéine remarquablement
conservée chez les organismes eucaryotes et notamment au niveau
de sa région N-terminale contenant le motif Winged Helix.
L'analyse de la structure de K2 humaine
par le logiciel CONSURF fait apparaître que les
résidus exposés de l'hélice H3 semblent peu
conservés au vu de l'état de conservation général
du domaine, alors que ceux qui forment la
surface de la boucle W1 et du brin S2 sont relativement bien
conservés (Figure 5.12).
H4 +
C
H1
très conservé
S1
H3
S2
S3 H2
W1
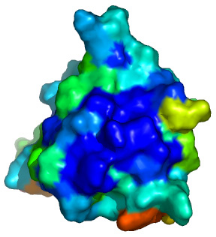
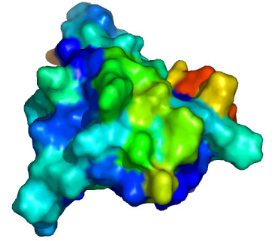
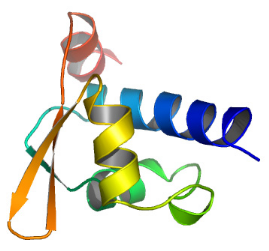
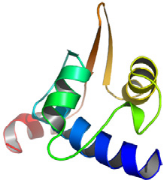
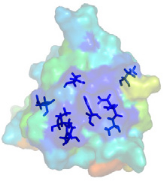

N
H2.5
pression
évolutive
_ non conservé
90°
W1
S3
S1
S2 H3
H2
H4
H2.5
C
N43
E48
I50
H57
T65
N59
A60
H1 N
H55
Figure 5.12 : Mise en
évidence d'une surface ultra conservée du domaine K2 de KIN17.
Les
vues du dessus sont dans la même orientation et
mettent en évidence la surface H3-S2-W1 de K2. De la même
manière, les vues du dessous correspondent à la surface H2-H2.5
(soit une rotation d'environ 90°). Les surfaces
représentées à gauche sont colorées en
fonction de l'état de conservation des résidus au cours de
l'évolution (d'après CONSURF).
De manière intéressante, l'analyse CONSURF met
également en évidence une surface ultra conservée
située sur la face perpendiculaire et adjacente à la
surface S2-W1. Cette surface que nous nommerons H2-H2.5 est formée par
les chaînes latérales de 8 résidus qui appartiennent
à l'hélice H2, à la boucle entre H2 et H3, et à
l'hélice 310 H2.5. Le calcul du
potentiel électrostatique de surface avec le programme
APBS montre que cette face H2-H2.5,
qui comporte 4 résidus hydrophobes (2 histidines,
une isoleucine, et une alanine), est peu polaire et plutôt neutre.
Ce type de surface relativement hydrophobe est tout à fait favorable
à une interaction de type protéine-protéine. Que
signifie d'un point de vue fonctionnel la présence d'une telle
surface ultra conservée ? Le motif Winged Helix de K2 serait-il
capable d'interagir avec un partenaire protéique via cette surface ?
Les domaines à motif Winged Helix sont
principalement connus pour leur capacité à lier et fixer l'ADN.
Cependant, il existe des exemples de motif impliqué dans des
interactions
de type protéine-protéine. Jusqu'au début
des années 2000, les seules interactions protéine-
protéine connues des Winged Helix se limitaient à
des interactions d'homodimérisation ou
d'hétérodimérisation avec d'autres motifs Winged Helix
(Littlefield & Nelson, 1999 ; Zheng
et al., 1999). Ces domaines, qui présentent une
interface de dimérisation, sont également capables de lier
l'ADN et possèdent une surface H3-W1 positive et très
conservée au cours de l'évolution, ce qui n'est pas le cas de
cette surface dans le domaine K2.
A ce jour, il existe à notre connaissance 2
structures de domaine Winged Helix capables de se lier à un
partenaire protéique de nature différente, et qui ne lient pas
l'ADN. Il s'agit du domaine C-terminal de la sous-unité RPA32 de la
protéine de réplication A (Mer et
al., 2000), et du domaine RAP74 de la
sous-unité TFIIF du facteur de transcription TFII (Nguyen et
al., 2003). RPA32 fixe le peptide 73-88 de la protéine UNG2
via une surface négative et hydrophobe composée de
résidus de l'extrémité C-terminale de H3 et des brins
S2
et S3. L'interface d'interaction de RAP74 avec le peptide
944-961 de FCP1 est différente : elle implique des résidus
des hélices H2 et H3 qui forment une surface positive et
hydrophobe. De manière intéressante, RAP74 contient une
hélice á additionnelle de 4 résidus entre H2 et H3.
Cette hélice H2.5 n'est cependant pas impliquée dans
l'interaction avec FCP1. Les bases moléculaires de la
reconnaissance du partenaire protéique sont donc différentes
chez RPA32 et RAP74. Cependant, ces 2 protéines
possèdent plusieurs caractéristiques structurales communes au
domaine K2 : elles présentent une surface H3-W1 peu polaire et peu
conservée, et leur surface d'interaction avec leur partenaire
protéique est conservée et plutôt hydrophobe, ce qui est
également le cas de la surface ultra conservée de
K2 de KIN17.
La surface hydrophobe conservée du motif Winged Helix
de KIN17 pourrait donc être
impliquée dans une interaction avec un partenaire
protéique. Elle pourrait également servir à interagir
avec un autre domaine au sein même de la protéine,
et ainsi contribuer à l'organisation intramoléculaire des
domaines de KIN17. De manière intéressante, le module
prédit en « doigt de zinc » est très proche du motif
Winged Helix dans la séquence primaire de KIN17. Ceci laisse
supposer l'existence d'interactions entre ces 2 domaines.
4) Caractérisation de la position du motif
prédit en « doigt de zinc »
autour du domaine Winged Helix
Dans l'optique d'approfondir notre recherche des fonctions de
KIN17, et notamment
du motif Winged Helix, nous avons abordé
l'étude structurale en solution du domaine K3 (région
1-160). Comme le montre l'alignement de la Figure 5.13, le motif prédit
en « doigt de zinc » par le programme SMART n'est
séparé du motif Winged Helix que par un segment de
taille conservée d'environ 15 résidus dans les
séquences primaires eucaryotes. Avant d'entreprendre à
terme une étude structurale complète du domaine K3 par RMN, nous
avons dans un premier temps voulu caractériser les relations
structurales qui existent entre ces deux modules, et notamment la position
du motif prédit en « doigt de zinc » autour du module
Winged Helix.
|
10
|
20
|
30
|
40
|
50
|
60
|
70
|
80
|
90
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Humain
MGK-SDFLTPKAIANRIKSKGLQKLRWYCQMCQKQCRDENGFKCHCMSESHQRQLLLASENPQQFMDYFSEEFRNDFLELLRRRFGTKRV...
Plante
MGK-NDFLTPKAIANRIKAKGLQKLRWYCQMCQKQCRDENGFKCHCMSESHQRQMQVFGQNPTRVVDGYSEEFEQTFLDLMRRSHRFSRI...
Spore
MGR-AEAGTPKAISNALKSKGLQRLRWYCSACQKQMRDENGFKCHTQSEGHIRQMNVIAMNPGKRIQDFSNQFLRDFISLLRTAHGEKKI...
Ver
MGK-HEKGSSKDLANRTKSKGLQKLKFFCQMCQKQCRDANGFKCHLTSEAHQRQLLLFAENSNSYLRQFSNDFEKNFMQLLRTSYGTKRV...
Neurospore
MPK-AEVGSAKYLANKMKSRGLNRLRWYCQLCEKSCRDENGYKMHCQSPSHTAKALEAGANFKGVQDTFSDQFLKDFIAQLKTSHGEKEI...
Levure
MAD---YDSAKYWSKQGARRGLQKTRYYCQICQRQCKDANGFQSHNKSPSHLRKISQVTAEDAR---RYNIQFEKGFLQLLKQRHGEKWI...
Humain
MGK-SDFLTPKAIANRIKSKGLQKLRWYCQMCQKQCRDENGFKCHCMSESHQRQLLLASENPQQFMDYFSEEFRNDFLELLRRRFGTKRV...
motif prédit C2H2
(ou "doigt de zinc")
région N-terminale du motif Winged
Helix
K2
K3
Figure 5.13 : Alignement de la
région N-terminale de KIN17 réalisé avec les
séquences de
l'Homme, de la plante Arabidopsis Thaliana, du ver
Caenorhabditis elegans, du spore Emericella Nidulans, du neurospore
Neurospora Crassa, et de la levure Saccharomyces Cerevisiae. Les
résidus sont colorés en rouge lorsqu'ils sont conservés,
en vert lorsqu'ils sont fortement similaires, et en bleu lorsqu'ils sont
faiblement conservés. Les 4 résidus cystéine et histidine
ultra conservés et caractéristiques du motif C2H2 prédit
(ou « doigt de zinc ») sont indiquées par des points
noirs.
4.1) Stratégie employée
Par RMN, l'interaction entre deux partenaires peut être
facilement caractérisée à partir
de la connaissance des fréquences de
résonance de l'un des deux partenaires. La méthode
généralement employée consiste à réaliser
une titration du partenaire enrichi en isotope 15N, et dont les
déplacements chimiques 1HN et 15NH sont connus,
par l'autre partenaire non marqué.
Le suivi de l'évolution des pics de
corrélation sur un spectre HSQC 1H-15N
permet alors
d'identifier les résidus dont les noyaux de
groupement amide ont changé d'environnement chimique, et ainsi de
caractériser une surface d'interaction du partenaire marqué
15N avec le partenaire non marqué.
Nous avons adapté le principe de cette méthode,
appelée cartographie des variations
de déplacement chimique, pour déterminer la
position du « doigt de zinc » autour du Winged Helix. Le
domaine K3 (région 1-160) étant constitué de la
totalité des acides aminés de K2 (région 51-160), la
démarche consiste à rechercher les pics de corrélation
1H-15N des résidus
du Winged Helix de K2 sur le spectre HSQC de K3
enregistré dans des conditions identiques
ou très proches. Pour cela, nous avons
enregistré en parallèle une expérience 3D
15N-NOESY- HSQC sur l'échantillon de K3 afin de
distinguer les pics du Winged Helix des pics de la
région 1-50 sur l'HSQC de K3 (par comparaison avec l'expérience
3D 15N-NOESY-HSQC de K2). La comparaison des fréquences de
résonance des noyaux 1HN et 15NH du Winged
Helix permet alors de mettre en évidence l'influence
de la région N-terminale de K3 sur l'environnement
électronique des noyaux amides du Winged Helix. Une
absence totale de modification significative de déplacement chimique
et/ou d'intensité signifierait alors que le
« doigt de zinc » n'adopte aucune position
préférentielle autour du Winged Helix, ou qu'il ne
se positionne pas à sa proximité.
4.2) Préparation de l'échantillon de
protéine K3 simplement marquée 15N
Pour préparer l'échantillon de
protéine K3 simplement marquée 15N, nous avons
eu recours aux mêmes stratégies et méthodologies que celles
utilisées pour produire, purifier, et isoler le domaine K2 (cf. chapitre
2). La région 1-160 de KIN17 humaine a été exprimée
en fusion avec le partenaire ZZ, purifiée sur résine d'amylose,
et le partenaire ZZ a été clivé sur colonne avec la
protéase TEV. Cependant, pour des raisons inhérentes
à la présence du
« doigt de zinc » dans le domaine K3, il
n'a pas été possible d'utiliser les mêmes
compositions de solutions tampons pour purifier la
protéine et préparer l'échantillon RMN.
En effet, la présence de réducteurs de
type DTT ou TCEP, ou d'agent chélateur comme l'EDTA, pourrait
décrocher l'ion Zn2+ lié aux 2 cystéines et 2
histidines du motif C2H2, ou réduire le pont disulfure formé
par les 2 cystéines impliquées dans la chélation
du métal. D'autre part, le phosphate de zinc étant insoluble, il
est préférable d'utiliser un tampon Tris- HCl plutôt
qu'un tampon phosphate qui pourrait favoriser l'agrégation de la
protéine (Vallee & Auld, 1995). La composition finale des
échantillons RMN marqués 15N des protéines
K2 et K3 est indiquée dans le Tableau 5.1. Afin de limiter
les différences de composition entre les 2 échantillons, les
valeurs de pH et de force ionique de l'échantillon K3
ont été choisies identiques à celles de
l'échantillon K2.
|
K2 (C=0.7 mM)
région 51-160 de KIN17 humaine
|
K3 (C=0.2 mM)
région 1-160 de KIN17 humaine
|
|
? 50 mM phosphate (pH 6.0)
? 150 mM NaCl
? 1 mM EDTA
? 1 mM TCEP
|
? 50 mM Tris-HCl (pH 6.0)
? 150 mM NaCl
|
Tableau 5.1 : Composition des
échantillons RMN marqués 15N des protéines K2
et K3.
4.3) Résultats de la cartographie des variations
de déplacement chimique
Nous avons dans un premier temps relevé le nombre de pics
de corrélation 1H-15N sur
le spectre HSQC de K3. Ce nombre de pics est
difficile à comptabiliser en raison de la présence de
recouvrement dans la zone centrale du spectre. On peut toutefois
estimer sa valeur proche de 130 sachant que le nombre de groupements amides
attendu s'élève à 156 (161 résidus dont 4
prolines et une glycine N-terminale). Aussi, près de 90 % des
pics correspondant aux résidus du domaine K2 ont été
retrouvés sur le spectre HSQC de K3. Par déduction, il
apparaît donc que près de la moitié des résonances
1H-15N correspondants aux groupements amides de la
région N-terminale 1-50 de K3 sont absentes sur le spectre. A ce stade
de l'étude structurale, il est difficile d'expliquer la
dégénérescence partielle du signal dans cette
région. Il est toutefois possible que l'extrémité 1-25 en
amont du motif prédit en
« doigt de zinc » soit en échange
conformationnel entre une forme structurée et une forme
déstructurée, ce qui expliquerait l'absence de près de 25
pics de corrélation. D'autre part, la séquence humaine
prédite en « doigt de zinc » comporte 3
cystéines supplémentaires non
conservées et différentes des 2
cystéines ultra conservées qui caractérisent le
motif C2H2.
(Figure 5.13). Ces 3 cystéines pourraient être
responsables de sites d'interaction non
spécifique avec l'ion Zn2+, ce qui induirait un
échange de la position de cet ion entre les 5
cystéines de cette région, accompagné
d'échange conformationnel.
Comme le montre la superposition des spectres HSQC
1H-15N de K2 et K3 (Figure
5.14), la grande majorité des pics relatifs aux
résidus du motif Winged Helix n'ont subi qu'une faible
modification de déplacements chimiques 15N et 1H.
Le domaine K3 comporte donc un motif Winged Helix de repliement
similaire à celui du domaine K2. Toutefois, comme nous nous y
attendions, la présence de la région N-terminale de KIN17,
contenant le
« doigt de zinc », en amont du motif
Winged Helix induit une modification importante de
l'environnement électronique des résidus de
l'extrémité N-terminale 1-18 du domaine K2. En effet, les
changements de déplacements chimiques dans cette région sont tels
que l'expérience
15N-HSQC-NOESY ne permet pas de retrouver les pics de
corrélation 1H-15N correspondants
sur l'HSQC de K3. Cette région 1-18 comporte le segment
1-15 qui sépare le Winged Helix
du « doigt de zinc » potentiel, et les 3 premiers
résidus de l'hélice H1 du Winged Helix.
K2
15N
K3
I50
T61
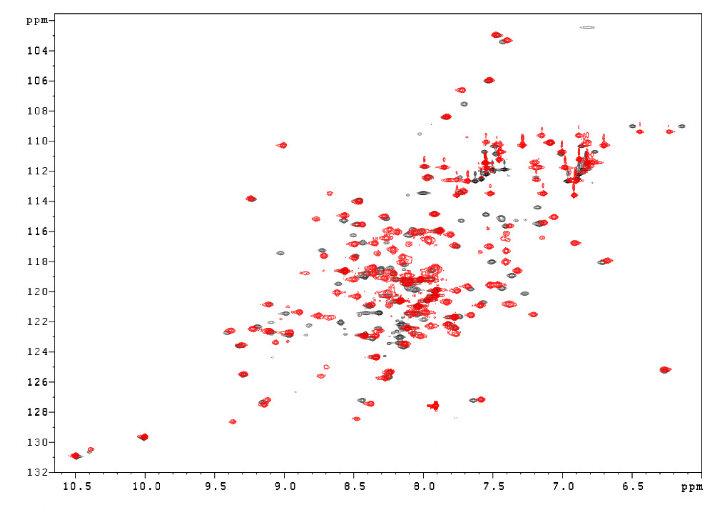


L28
D26
H52
A60
M58
H41
W72å
1H
Figure 5.14 : Superposition
des spectres HSQC 1H-15N de K2 et K3. Les
résidus qui présentent une variation significative de
déplacements chimiques 1H et 15N sont mis
en évidence (?äm > 0.7 ppm). La numérotation des
résidus est relative au domaine K2.
La variation moyenne ?äm de déplacements
chimiques 1H et 15N par résidu a
été
évaluée en utilisant la relation suivante :
?äm = |?äHN| + |?äNH/10|. Comme le montrent les
Figures 5.14 et 5.15, des valeurs significatives de ?äm
(> 0.07 ppm) sont observées au niveau
de résidus de la boucle H2-H3 (I50, H52, et T61), et de
l'hélice 310 (M58 et A60). Ces valeurs
de ?äm indiquent une modification de l'environnement
chimique des groupements amides de
ces résidus induite par la proximité avec la
région N-terminale de KIN17 contenant le motif prédit en «
doigt de zinc ». Des modifications d'intensité de pic sont
également observées au niveau de la boucle H2-H3. Ainsi, les pics
de corrélation 1H-15N correspondant aux
résidus E54, H55, I56, H57, et N59, et qui appartiennent à des
zones dégagées du spectre HSQC de
K2, n'ont pas été retrouvées sur le
spectre HSQC de K3. Cette dégénérescence du signal
pourrait s'expliquer par une mobilité du motif « doigt de
zinc » qui induirait un équilibre conformationnel (à une
vitesse d'échange intermédiaire) entre une forme liée,
où ces 5 résidus seraient en interaction avec « le doigt de
zinc », et une forme libre où ils ne le seraient pas. Les
résidus dont le pic de corrélation n'a pas été
retrouvé (hors extrémité 1-18) sont imputés
d'une valeur de ?äm de 0.1 ppm.
0,25
0,2
?äm = |?äHN| + |?äNH/10|
pic disparu : pénalité de 0.1 ppm
?äm ppm
0,15
0,1
0,05
0
0 5 10 15 20 25 30 35 40 45 50 55 60 65 70 75 80 85 90 95 100
105 110
H1 S1 H2 310 H3 S2 S3 H4
Figure 5.15 : Graphique des
variations moyennes ?äm des déplacements chimiques 1H
et 15N
des résidus du motif Winged Helix. La
numérotation des résidus est relative au domaine K2.
Les résidus de la boucle H2-H3 affectés par la
présence de la région N-terminale de
KIN17 contenant le doigt de zinc forment une surface qui est mise
en évidence sur la Figure
5.16. De manière intéressante, cette surface
se superpose parfaitement avec la surface hydrophobe ultra
conservée que nous avons mise en évidence
précédemment. Le motif prédit
en « doigt de zinc » adopte donc une position
préférentielle au niveau de la surface ultra
conservée H2-H3 du motif Winged Helix.
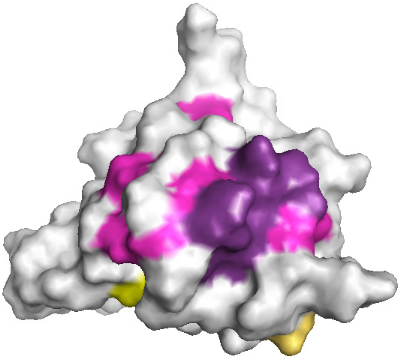
A)
W1
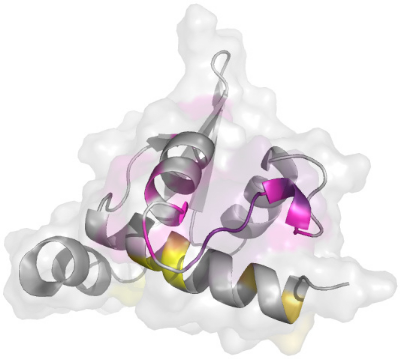
H2
H3
310
H4 H1
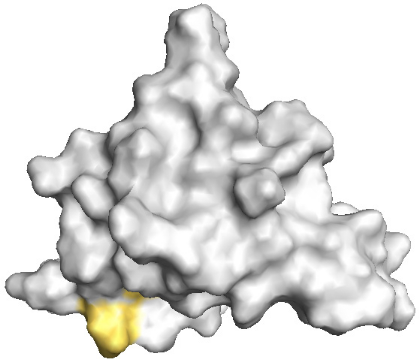
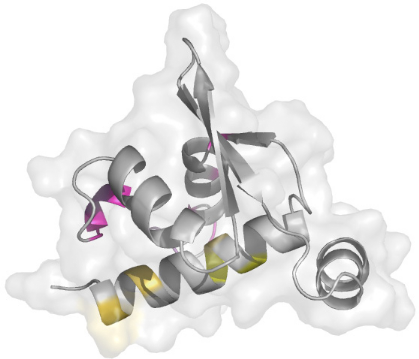
B) W1
H3 H2
H4
310
H1
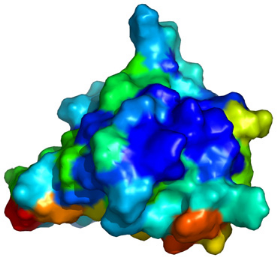
C)
+ très
conservé
pression
évolutive
_ non conservé
Figure 5.16 : Mise en
évidence d'une surface d'interaction du motif Winged Helix avec
la
région N-terminale de KIN17 contenant le module
prédit en « doigt de zinc ».
A) Représentation de la face avant H2-H3.
Les résidus dont la modification moyenne de déplacements
chimiques ?äm est significative (?äm > 0.07 ppm) sont
colorés en violet (hors hélice H1). Les résidus dont le
pic de corrélation n'a pas été retrouvé sont
colorés en rose (hors hélice H1). Les résidus de
l'hélice H1 dont la valeur de ?äm est significative sont
colorés en jaune. B) Représentation de la face arrière
opposée à la surface H2-H3. Le code couleur est identique
à celui utilisé en A). C) Mise en évidence de la surface
ultra conservée
H2-H3 du motif Winged Helix. Les surfaces sont
colorées en fonction de l'état de conservation des
résidus au cours de l'évolution (d'après CONSURF).
Par ailleurs, des modifications significatives de
?äm apparaissent également au niveau
des résidus D26, L28, et E29 de l'hélice H1. Ces 3
résidus appartiennent à une surface située
à l'opposé de la surface ultra conservée
(Figure 5.16). De plus, dans la structure du domaine K2, les protons amides des
résidus D26 et L28 sont protégés du solvant et
orientés vers le coeur hydrophobe. Par conséquent, nous
proposons que la modification significative de déplacements
chimiques de ces 3 résidus soit plutôt due à une
modification structurale de l'hélice H1 (orientation ou longueur),
ou des résidus qui la précédent qui sont
déstructurés dans la structure du domaine K2. La
structuration des 15 résidus qui séparent le module
« doigt de zinc » du motif Winged Helix
pourrait également provoquer une modification ou une rupture
partielle du réseau de liaisons hydrogènes du Winged
Helix, ce qui expliquerait
les modifications significatives de ?äm
enregistrées.
Chapitre 6 : Conclusions et perspectives
CHAPITRE 6
Conclusions & Perspectives
L'objectif du travail présenté dans la seconde
partie de ce manuscrit était d'améliorer
la connaissance des fonctions, des partenaires biologiques, et
des mécanismes d'action de la protéine KIN17 humaine en
s'intéressant plus particulièrement à la région
51-160. Pour cela, nous avons abordé une approche structurale qui
consiste à émettre des hypothèses sur les fonctions
potentielles adoptées par cette région à partir de
la connaissance de sa structure. Nous avons montré par
Résonance Magnétique Nucléaire et Modélisation
Moléculaire que la région 51-160 de la protéine KIN17
humaine adopte un repliement de type Winged Helix de topologie quasi
canonique. La détermination de la structure de ce domaine
réfute donc la prédiction des logiciels bio-informatiques de
détections de domaines qui suggèrent l'existence d'un domaine
FF de liaison à des peptides phosphorylés dans la
région 50-150 de KIN17 humaine. Les domaines à motif
Winged Helix sont principalement connus pour leur capacité
à lier l'ADN et de ce fait, ils sont souvent retrouvés
chez des facteurs de transcription eucaryotes ou procaryotes. Ce simple
constat constitue un premier argument structural qui
supporte l'implication de KIN17 dans la régulation et la
maintenance de l'ADN.
? Interaction avec l'ADN ?
Nous avons comparé la structure du domaine K2 avec celle
de motifs Winged Helix
structuralement proches afin d'évaluer les
potentialités de K2 à lier l'ADN. A ce jour, il existe
2 modes de reconnaissance de l'ADN par les domaines à
motif Winged Helix dont les bases moléculaires sont
parfaitement connues. La comparaison structurale du domaine K2 avec ces motifs
fait apparaître des divergences significatives qui mettent en cause la
capacité de K2 à lier l'ADN selon ces modes de reconnaissance
:
- Les motifs Winged Helix qui adoptent le mode
de reconnaissance classique utilisent principalement leur hélice H3
pour pénétrer le sillon majeur et interagir avec le squelette
et les bases de l'ADN. En superposant K2 avec plusieurs de ces
motifs, nous avons mis
en évidence des différences structurales au
niveau de l'hélice H3 qui apparaît plus courte chez K2 et dont
l'orientation, plus ouverte par rapport au reste du domaine, n'est
pas favorable à une interaction avec le grand sillon de
l'ADN selon le mode de reconnaissance classique. La présence
et la position d'une large boucle rigide et très
conservée, et d'une hélice 310 atypique, entre H2 et H3
sont responsables de cette différence structurale et
différencient le motif Winged Helix de K2 des
Winged Helix
classiques de liaison à l'ADN.
- L'analyse du potentiel électrostatique de surface
a également mis en évidence des
différences structurales notables : tous les motifs
Winged Helix qui lient l'ADN de type B
ou Z selon le mode de reconnaissance classique
présentent un potentiel de surface H3-W1 largement positif alors que
cette surface apparaît peu polaire et faiblement chargée
positivement chez K2.
- Cette surface fonctionnelle de liaison à l'ADN H3-W1
est fortement conservée chez les motifs Winged Helix de liaison
à l'ADN, ce qui n'est pas le cas chez K2 où nous avons
montré que l'hélice H3 est peu conservée au vu de
l'état de conservation générale du domaine K2.
- Le seul motif Winged Helix connu à ce
jour qui adopte un mode de reconnaissance atypique utilise sa boucle W1
qui contient plusieurs résidus basiques pour interagir avec
le grand sillon de l'ADN. Chez K2, cette boucle ne contient
que 2 résidus qui forment un coude â de type I et les surfaces
formées par les résidus de W1 et des brins â S2 et S3 sont
peu polaires et plutôt hydrophobes.
L'ensemble de ces observations suggère que le motif
Winged Helix de la région 51-160 de KIN17 humaine n'est pas
capable de lier l'ADN selon les modes de reconnaissance connus des Winged
Helix. Les premiers tests d'interaction in vitro que nous
avons menés par Southwestern corroborent nos conclusions
structurales et montrent que cette région de KIN17 n'est pas suffisante
pour lier l'ADN de manière autonome.
? Interaction avec l'ARN ?
Les données récentes de la littérature
ont mis en évidence la capacité de 3 motifs
Winged Helix à fixer l'ARN. Toutefois, les bases
moléculaires de cette reconnaissance n'ont
été identifiées à ce jour
qu'à une seule reprise chez le facteur d'élongation SelB.
Celles-ci reposent principalement sur une interaction avec l'ARN via
une surface formée par des résidus des hélices H2 et
H3 particulièrement basiques. Le motif Winged Helix de la
région
51-160 de KIN17 ne présente pas cette
caractéristique structurale et par conséquent, ne
possède certainement pas la capacité à lier l'ARN
selon ce mode de reconnaissance. Ces données structurales sont
toutefois insuffisantes pour conclure quant à la
potentialité du
Winged Helix de KIN17 à interagir avec l'ARN.
D'un point de vue fonctionnel, nous avons
montré par hybridation Northwestern que ce
motif n'est pas suffisant pour lier une sonde
d'ARN de 1200 nucléotides reconnue par KIN17 humaine.
? Interaction de type protéine-protéine
?
L'utilisation du programme CONSURF nous a
permis de mettre en évidence la présence d'une surface
ultra conservée formée par les chaînes
latérales de 8 résidus qui appartiennent à
l'hélice H2, à la boucle entre H2 et H3, et à
l'hélice 310 H2.5. Cette surface plutôt neutre et relativement
hydrophobe est située sur la face adjacente à la surface H3-W1.
Nous avons trouvé dans la littérature quelques motifs Winged
Helix qui possèdent une telle surface hydrophobe et
conservée située sur une face différente de H3-W1. Dans la
plupart des cas, ce type de surface constitue une interface autonome
de dimérisation qui permet une liaison à l'ADN sous forme
dimérique. Nos résultats ont montré d'une part, que le
domaine
K2 se trouve sous forme monomérique en solution et d'autre
part, qu'il n'est pas capable de lier l'ADN selon les modes de reconnaissance
connus. Par conséquent, il est peu probable que
la surface ultra conservée de K2 soit impliquée
dans un mécanisme de dimérisation autonome.
La récente résolution de structure des domaines
RPA32 et RAP74 à motif Winged Helix a mis
en évidence l'implication de ce type de motif dans des
interactions de type protéine-protéine avec un partenaire
protéique de nature différente. Ces 2 domaines ne sont pas des
homologues structuraux de K2 mais ils présentent des
caractéristiques structurales communes : ils ne lient pas l'ADN, leur
surface H3-W1 est peu conservée, et ils possèdent une surface
hydrophobe très conservée qui interagit avec leur partenaire
respectif. Sur la base de ces similitudes, il est apparu possible que la
surface ultra conservée du motif Winged Helix de la
région 51-160 de KIN17 soit impliquée dans des interactions de
type protéine-protéine.
Dans l'optique d'améliorer la connaissance des
fonctions du motif Winged Helix de KIN17, l'étude
structurale en solution de la région 1-160 (domaine K3) de la
protéine humaine a été initiée par RMN. Les
prédictions bio-informatiques préliminaires que nous avons
réalisées suggèrent la présence d'un motif de
liaison aux acides nucléiques de type
« doigt de zinc » en amont du module Winged Helix
au niveau de la région 28-50 de KIN17
humaine. Les premiers tests d'interaction in vitro
menés par Nortwestern et Southwestern sur
le domaine K3 mettent en évidence le rôle majeur
de la région 1-50 dans la liaison de KIN17 humaine à l'ADN
et l'ARN. Nous avons montré par cartographie des
déplacements chimiques sur HSQC 1H-15N
l'existence de relations structurales entre le module Winged
Helix et la région N-terminale 1-50 contenant le
motif « doigt de zinc ». Cette étude révèle
ainsi que le motif « doigt de zinc » adopte
une position préférentielle autour du module Winged
Helix au niveau de sa surface hydrophobe ultra conservée.
Cette surface est donc impliquée dans des interactions de type
protéine-protéine intra-moléculaires.
En conclusion, notre approche structurale n'a pas abouti
à la caractérisation de la fonction du domaine Winged
Helix de KIN17 humaine, mais, dans un cadre plus général,
elle
a permis de contribuer à l'amélioration de
la connaissance des fonctions et des partenaires biologiques de la
protéine KIN17. Ainsi, nous avons montré d'une part, que
ce domaine Winged Helix n'est pas capable de lier l'ADN ou l'ARN de
manière autonome, et d'autre part, qu'il entretient des relations
structurales avec le module « doigt de zinc » de liaison à
l'ADN
et l'ARN de la région N-terminale de KIN17.
Sur la base de ces conclusions, il serait à présent
intéressant de déterminer le rôle exact
du motif Winged Helix dans la liaison à l'ADN et
l'ARN du domaine K3. La caractérisation structurale et dynamique du
domaine K3 par RMN nous permettrait de savoir si ce domaine
est composé d'une ou de deux unités de repliement.
En effet, il est possible que le module
« doigt de zinc » ne soit pas capable de se replier de
manière autonome. La fonction du motif
« Winged Helix » pourrait donc être
essentiellement structurale et consisterait à favoriser et stabiliser
le repliement biologiquement actif du module « doigt de zinc
». Le rôle exact du motif Winged Helix dans la
liaison à l'ADN et l'ARN pourrait être rapidement mis en
évidence par RMN en réalisant une titration du domaine K3 par des
acides nucléiques après avoir attribué les raies de
résonance de la chaîne principale de K3. Cependant, une
étude par RMN de ce domaine nécessiterait dans un premier
temps une optimisation des conditions d'analyse afin de pallier la
dégénérescence du signal observée dans la
région N-terminale contenant le « doigt de zinc »,
probablement due à de l'échange conformationnel. Dans le cas
où cette optimisation ne serait pas suffisante,
la mutation d'une ou plusieurs cystéines non
caractéristique(s) du motif C2H2 pourrait être une
solution afin de limiter cet échange conformationnel. A l'issue de
cette étude structurale, la mise en évidence d'une interaction
conjointe du module « doigt de zinc » et du motif Winged Helix
avec l'ADN ou l'ARN serait considérable d'un point de vue
structural. Cela signifierait que ces 2 modules constituent un
nouveau repliement de domaine de liaison aux acides
nucléiques. A notre connaissance, il
n'existe en effet aucune structure tridimensionnelle de
protéine qui présente un tel domaine
formé par un motif Winged Helix et un module
« doigt de zinc ».
D'une manière plus globale, l'étude structurale de
KIN17 pourrait être poursuivie en recherchant les interactions du domaine
K3 avec les autres domaines de la protéine. La région
C-terminale 268-393 de KIN17 humaine vient d'être
résolue au LSP par cristallographie des rayons X. Ce domaine contient un
double motif SH3 de liaison à l'ARN. La reconstitution de
la structure entière de KIN17 à partir de la
structure de ses domaines structuraux pourrait être
réalisée à partir d'une analyse SAXS (Small
Angle X-ray Scaterring) qui permet de reconstituer l'enveloppe
globale de la protéine.
Enfin, au-delà de l'approche structurale, les
chercheurs du LSP poursuivent actuellement la recherche des partenaires
protéiques des domaines de KIN17 par des études biochimiques de
type « double hybride » ou pull down réalisées
sur des extraits nucléaires. En
cas de résultat favorable pour les domaines K2
et K3, la RMN sera sans aucun doute la méthode de choix pour
caractériser les bases moléculaires de l'interaction de ces
domaines avec leur(s) partenaire(s), et ainsi améliorer la
connaissance des fonctions précises et des
modes d'action de la protéine KIN17 dans le noyau de la
cellule.
Annexe : Criblage des conditions d'expression des
protéines PROentier, PROcatal, PROter, et PROinser
ANNEXE
Criblage des conditions d'expression des
protéines PROentier, PROcatal, PROter, et
PROinser
1) Matériels et Méthodes
1.1) Construction des plasmides d'expression par
recombinaison homologue
1.1.1) Stratégie employée
Commercialisée par la société
Invitrogen, la technologie Gateway s`appuie sur les
propriétés de recombinaison homologue du
bactériophage lambda au niveau des sites spécifiques
att, qu'il utilise naturellement pour s'intégrer dans le
génome d'E. coli (Landy,
1989). Ce mécanisme naturel a été
détourné pour en faire un système de recombinaison
in vitro simple et efficace. Le système de clonage Gateway
présente de nombreux avantages et permet notamment de
s'affranchir des étapes de restriction-ligation obligatoires lors
d'un clonage classique. L'insertion d'un insert d'ADN est spécifique et
directionnelle (conservation du cadre de lecture), et le moyen de
sélection est rapide. Il est alors facile d'obtenir
différents plasmides d'expression à partir d'un seul plasmide
d'entrée.
Le principe de la méthodologie de construction des
différents vecteurs d'expression à partir de la technique
Gateway est représenté dans la Figure A1. La
première étape consiste à adjoindre aux gènes
encodant les domaines PRODH les 2 sites de recombinaison spécifique,
attB1 en amont, et attB2 en aval du gène, par PCR
d'assemblage. Un site de clivage reconnu
par la protéase TEV (Tobacco Etch Virus)
est également incorporé en amont du gène
d'intérêt. Les amplicons de PCR, flanqués des sites
attB, sont alors clonés dans le vecteur donneur pDONR221
(Invitrogen) possédant les sites attP1 et attP2
par réaction de BP clonase. Ce vecteur donneur contient
également un gène de résistance à la kanamycine
(KmR), ainsi qu'un second au chloramphénicol (CmR)
inséré entre les 2 sites attP. Les enzymes de la
BP clonase reconnaissent de manière
spécifique les sites attB et attP, ce qui
conduit à l'insertion du gène d'intérêt dans le
vecteur donneur. Il se forme alors par cette réaction un vecteur
d'entrée possédant le gène de résistance
à la kanamycine (KmR), les sites de recombinaison
homologue AttL1 et AttL2, et le gène
d'intérêt.
Le vecteur d'entrée est utilisé pour
obtenir l'ensemble des vecteurs d'expression à partir de
vecteurs pDEST, qui contiennent un gène encodant un des 6 partenaires de
fusion,
un gène de résistance à l'ampicilline
(AmpR), et les sites de recombinaison attR1 et
attR2. Le
gène d'intérêt est «
transféré » du vecteur d'entrée au
vecteur d'expression de manière unidirectionnelle
grâce aux enzymes de la LR clonase qui reconnaissent les sites attL
et attR
de manière spécifique. Au final, ces
plasmides d'expression comportent, un gène de
résistance à l'ampicilline (AmpR), une
étiquette 6xHis, un gène encodant un des 6 partenaires
de fusion, le gène d'intérêt, et un site de
clivage par la TEV inséré entre le partenaire et le gène
d'intérêt.
Gène
PCR
TEV Gène
CmR
+
attP1 attP2
pDONR
221
attB1 attB2
KmR
BP clonase
Gène
attL1 attL2
Vecteur d'entrée
KmR
LR clonase




CmR
attR1 attR2
pDEST
Gène
attB1 attB2
Vecteur d'expression
AmpR
AmpR
Figure A1 : Résumé
des principales étapes de la construction des vecteurs
d'expression à
partir de la technique de clonage par recombinaison homologue
Gateway.
1.1.2) PCR d'assemblage
Outre l'amplification des gènes
d'intérêt, la PCR d'assemblage a pour objectif d'adjoindre,
un site de reconnaissance AttB1 et un site de clivage
reconnu par la TEV en amont du gène, et un site de reconnaissance
AttB2 et une étiquette Tag-S en aval du gène. Le Tag-S
encode un fragment protéique qui permet de détecter
l'expression des protéines de fusion par un test de
fluorescence. Dans le cas des domaines PRODH, ce test, qui
était en cours de mise au point, n'a pas été
utilisé et l'expression des protéines
hétérologues a été uniquement analysée sur
gel SDS-PAGE.
L'introduction de longues séquences de nucléotides
non spécifiques du gène en amont
et en aval des gènes d'intérêt
(respectivement 57 et 60 bp) nécessite de réaliser 2
réactions de PCR successives. La première PCR (PCR1) fait
intervenir une amorce sens spécifique du gène contenant le
site de clivage par la TEV et une partie du site attB1, ainsi qu'une
amorce anti-sens complémentaire à la fin du gène
comportant l'étiquette Tag-S. L'ensemble des amorces
utilisées pour amplifier les gènes encodant les domaines PRODH
est reporté dans le Tableau A1.
|
PCR
|
Amorces
|
Séquences
|
|
PCR1
|
S
PROentier AS
|
5'-GGCTTCGAGAATCTTTATTTTCAGGGCGGACATTTTGTAGCCGGGGAG-3'
5'-TTAGCTGTCCATGTGTTGGCGTTCGAATTTAGCAGCAGCGGTTTCTTTCTAGGC
AGGGCGATGGAAGAGGTT-3'
|
|
S
PROcatal AS
|
5'-GGCTTCGAGAATCTTTATTTTCAGGGCGGACATTTTGTAGCCGGGGAG-3'
5'-TTAGCTGTCCATGTGTTGGCGTTCGAATTTAGCAGCAGCGGTTTCTTTCTAGGC
AGGGCGATGGAAGAGGTT-3'
|
|
S
Proter AS
|
5'-GGCTTCGAGAATCTTTATTTTCAGGGCGGAGGAGGGTCGGCAACGGCAGTG-3'
5'-TTAGCTGTCCATGTGTTGGCGTTCGAATTTAGCAGCAGCGGTTTCTTTCTAGGA
CTCGTGGTCTTCCCCGGC-3'
|
|
S
Proinser AS
|
5'-GGCTTCGAGAATCTTTATTTTCAGGGCGGGAGACCACAGTTTCTGCTGCAG-3'
5'-TTAGCTGTCCATGTGTTGGCGTTCGAATTTAGCAGCAGCGGTTTCTTTCTACTC
CTCCTCCTCAGTGAACCCGGA-3'
|
|
PCR2
|
AttB1-TEV-S
|
5'-GGGGGGGGGACAAGTTTGTACAAAAAAGCAGGCTTCGAGAATCTTTATTTTCAG
GGC-3'
|
|
AttB2-TagS-AS
|
5'-GGGGGGGGGACCACTTTGTACAAGAAAGCTGGGTCCTTAACTGTCCATGTGCTG
GCGTTCGAA-3'
|
Tableau A1 : Amorces sens (S) et
anti-sens (AS) utilisées pour les PCR d'assemblage. Les
oligonucléotides qui apparaissent en orange
correspondent au site de recombinaison AttB1,
en bleu au site de clivage par la TEV, en violet
au Tag-S, et en rouge au site de reconnaissance AttB2.
Les mélanges réactionnels se composent de
12 ng de vecteur de départ contenant le gène
d'intérêt, 0.1 mM de dNTP, 0.4 uM d'amorce sens, 0.4 uM d'amorce
anti-sens, 4.5 unités de polymérase, et 5uL de tampon Pfu
10X dans un volume final de 50 uL complété avec de l'eau
milliQ. L'enzyme utilisée est la Pfu DNA polymerase
commercialisée par Promega. Ces mélanges sont introduits
dans des barrettes de 16 tubes PCR de 250 uL (Promega) et traités en
parallèle avec des pipettes multicanaux. Les conditions de PCR
utilisées sont : un cycle de dénaturation thermique (2 min
à 94°C), suivi de 30 cycles de PCR (45 sec à 94°C, 45
sec à
50°C, et 4 min 30 à 72°C), puis 10 min
d'élongation finale à 72°C. Les produits de PCR1 sont
purifiés sur gel avec le kit QIAquick purification (Qiagen), et
quantifiés sur gel d'agarose.
La seconde PCR (PCR2) utilise une amorce sens non
spécifique du gène qui adjoint la région
N-terminale du site attB1 en amont du site de
clivage par la TEV. L'amorce anti-sens est également non
spécifique du gène et permet d'insérer le site de
reconnaissance attB2 en aval
du Tag-S. La composition des mélanges réactionnels
et les conditions de PCR sont identiques
à celles utilisées pour la PCR1. Environ 5 ng de
produit de PCR1 purifié servent de matrice.
La purification et la quantification des produits de PCR2 se font
de la même manière que pour
la PCR1.
1.1.3) Construction des vecteurs
d'entrée
Les produits de PCR2, contenant les gènes
d'intérêt flanqués des sites AttB, sont
clonés dans le vecteur donneur pDONR 221 par réaction de BP
clonase. Les réactifs utilisés, ainsi que le protocole
original, sont fournis par Invitrogen. Le mélange
réactionnel est composé de 1 uL de Clonase Reaction
Buffer 5X, 150 ng de vecteur pDONR 221 (1 uL),
1 uL de tampon Tris-EDTA (Tris 10mM à pH 8.0 et EDTA 1mM),
1 uL de produit de PCR2,
et 1 uL de mélange enzymatique BP clonase. L'ensemble est
incubé 3 heures à 25°C, puis traité à la
protéinase K (0.4 ug/uL) pendant 10 min à 37°C afin de
stopper la réaction. 50 uL
de bactéries thermocompétentes DH5á
sont transformées par le produit de réaction BP clonase
par choc thermique à 42°C pendant 45 secondes. Le produit
de transformation est étalé sur boîte LB/agar contenant
50 ug/mL de kanamycine, puis incubé à 37°C pendant toute une
nuit. Le lendemain, une dizaine de colonies sont repiquées sur milieu
LB/agar/kanamycine, puis une PCR sur colonie est réalisée sur
chacun des clones en utilisant une amorce sens (M13) s'hybridant sur le
plasmide, et une amorce anti-sens s'hybridant sur le gène
d'intérêt. Les colonies, conduisant après PCR à une
bande unique dont la taille apparente correspond à la taille
moléculaire attendue, sont mises en culture dans du LB à
37°C. Les vecteurs d'entrée sont alors extraits et
purifiés avec le kit Wizard Plus Minipreps DNA Purification
System (Promega). Les produits de ces minipréparations
sont finalement séquencés sur appareil 310 Genetic
Analyser (ABI Prism, Applied Biosystems).
1.1.4) Construction des vecteurs
d'expression
Les 7 plasmides de type pDEST possèdent un gène
encodant un partenaire de fusion
en aval de l'étiquette 6xHis, à l'exception du
vecteur pDEST-17 (Invitrogen) qui ne contient pas de partenaire. La formation
des vecteurs d'expression est réalisée en mélangeant 1.8
uL
de vecteur d'entrée (300 ng), 1.2 uL de chacun
des vecteurs pDEST (100 à 300 ng), 1 uL de
Clonase Reaction Buffer 5X, et 1 uL de mélange
enzymatique LR clonase. L'incubation dure
5 heures à température ambiante, puis
la protéinase K est ajoutée (0.4 ug/uL). La
transformation des bactéries DH5á thermocompétentes est
alors menée comme
précédemment par choc thermique à 42°C
pendant 45 secondes. L'analyse des transformants
se fait d'une part, par PCR sur colonie en utilisant une
amorce sens spécifique du promoteur T7, et une amorce anti-sens
spécifique du site AttB2. D'autre part, un repiquage sur milieu
LB/agarose-kanamycine, -chloramphénicol, et -ampicilline, permet de
vérifier le profil de résistance des vecteurs d'expression.
Seules les colonies qui présentent une résistance unique
à l'ampicilline sont sélectionnées puis
stockées à -80°C avec du glycérol.
1.2) Criblage des conditions d'expression en
microplaque
Le criblage des conditions d'expression des domaines
PRODH sur microplaques 96 puits (Falcon) a été
réalisé en suivant les protocoles mis au point par les chercheurs
du LMP dans le cadre de la plate-forme 3PM (une microplaque par souche et par
température).
1.2.1) Transformation des souches d'E. coli et
préparation des précultures
Pour chaque puits, 50 uL de bactéries
thermocompétentes sont transformées par 20 ng
de vecteur d'expression par choc thermique
à 42°C pendant 45 secondes. Après ajout de
200 uL de milieu SOC, les microplaques sont incubées
pendant 1 heure à 37°C. Chaque puits
est alors doté des antibiotiques appropriés
(Ampicilline à 100 ug/mL pour la sélection des plasmides
d'expression, et chloramphénicol à 35 ug/mL uniquement pour la
souche Rosetta pLysS), et les plaques sont incubées sur la nuit à
37°C sous une agitation de 250 rpm. On peut ainsi noter que les cultures
de transformation servent également de préculture.
1.2.2) Cultures d'expression
Les cultures d'expression, contenant 240 uL de LB et les
antibiotiques appropriés, sont ensemencées avec un volume de
préculture de manière à obtenir une DO600 initiale de
0.05 (utilisation du lecteur de microplaques ELX 800
Universal Microplate Reader de la
société Bio-Teck Instruments). La croissance est
maintenue sous agitation (1000 rpm) à 37°C jusqu'à une DO600
de 1.2 pour une induction à 20°C, et de 0.4 à 0.6 pour une
induction à 37°C (0.4 pour les BL21 Star, 0.5 pour les C41, et 0.6
pour les Rosetta). Les souches C41 et Rosetta sont induites avec 1mM d'IPTG, et
la souche BL21 Star avec 0.5 mM d'IPTG. L'induction
est alors prolongée pendant 3 heures à 37°C,
ou sur la nuit à 20°C sous une agitation de 1000
rpm. La croissance bactérienne est alors stoppée
par centrifugation à 2830xg et à 4°C pendant
20 minutes.
1.2.3) Lyse des bactéries et séparation des
fractions soluble et insoluble
Les culots bactériens sont lysés par reprise dans
50 uL de tampon de lyse contenant,
100 mM de Tris-HCl (pH 8.0), 150 mM de NaCl, 0.1 % de
Triton X-100, 1 uM de phosphoramidon, et 1 mM de PMSF. 0.04 ug/mL de
lysozyme (Sigma) sont ensuite ajoutés,
et les microplaques sont mises à incuber sous agitation
modérée à 4°C pendant 1 heure. La lyse de l'ADN est
initiée en ajoutant 0.125 U/uL de benzonase (Merck) et 10 mM de MgCl2.
Les microplaques sont alors incubées à température
ambiante pendant 10 minutes sous légère agitation. Les fractions
soluble et insoluble sont séparées par centrifugation à
2830xg pendant
1 heure 30 à 4°C. L'expression des
protéines de fusion est analysée sur gel SDS-PAGE.
10 uL de chaque échantillon sont dilués dans du
« bleu de charge » réducteur 2X [Tris-Hcl
62.5 mM pH 6.8, bleu de bromophénol 0.02 % (w/v), SDS 2
% (v/v), â-mercaptoéthanol 280 mM, glycérol 20 %
(v/v)], avant de subir une dénaturation thermique à
100°C pendant 5 minutes. 10 uL sont finalement déposés sur
gel de polyacrylamide (8 à 12 %).
2) Résultats
Comme je l'ai exposé dans le chapitre 4 de la
première partie de ce manuscrit (cf. § 2.2.2), les domaines
PROcatal et PROentier ont été exprimés dans 28
conditions expérimentales différentes (7 partenaires de
fusion, 2 souches d'expression, et 2 températures), contre 42
conditions pour les domaines PROter et PROinser (7 partenaires, 3 souches, et 2
températures). Pour chaque condition testée, les fractions
contenant les protéines solubles et insolubles ont été
déposées sur gel SDS-PAGE. L'analyse de l'expression des
protéines de fusion à partir de l'interprétation
des profils électrophorétiques des gels SDS- PAGE est
reportée, domaine par domaine, dans un tableau récapitulatif
(Figures A2 à A5).
2.1) Le domaine PROcatal
Que ce soit en souche BL21 Star ou Rosetta, les
protéines de fusion PROcatal sont globalement bien exprimées, et
ceci avec l'ensemble des partenaires de fusion (Figure A2). Seule la
construction avec Gb1 conduit à une expression faible, voire nulle en
souche Rosetta
et à 20°C. Cependant, ces bons résultats en
terme d'expression ne sont pas associés à de bons profils de
solubilité. En effet, on constate que la majorité des
protéines hétérologues PROcatal sont exprimées dans
la fraction insoluble. C'est le cas notamment pour les constructions avec
ZZ et la GST, ainsi que pour les fusions Gb1, Trx, et
His, qui conduisent toutefois dans
certaines conditions à une expression soluble très
minoritaire.
PM
(kDa)
250
Gb1
S I
ZZ GST
S I S I
Trx
S I
MBP
S I
His
S I
NusA
S I
PM
(kDa)
250
Gb1
S I
ZZ GST
S I S I
Trx
S I
MBP
S I
His
S I
NusA
S I
150
100
75
50
37
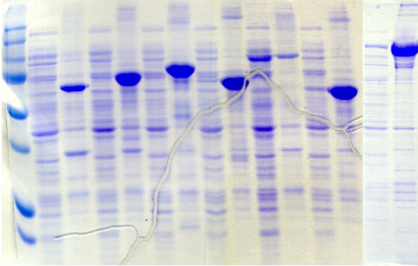
·
·
·
·
·
·
· 150
100
75
50
37
·
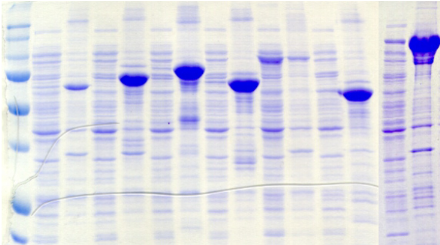
·
·
·
·
·
·
25
20
BL21 Star (37°C)
25
20
BL21 star (20°C)
PM
(kDa)
250
150
100
75
Gb1
S I
ZZ GST
S I S I
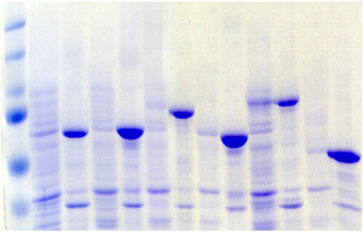
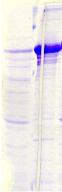
·
Trx
S I
MBP
S I
·
His
S I
NusA
S I
·
(kDa)
250
150
100
PM Gb1
S I
ZZ GST
S I S I
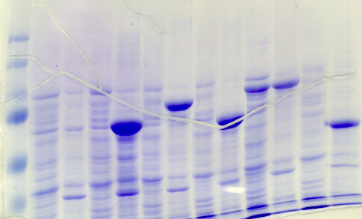
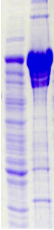
·
Trx
S I
MBP
S I
·
His
S I
NusA
S I
·
·
·
·
·
50
75
·
·
·
·
50
37
Rosetta (37°C)
37
Rosetta (20°C)
PROcatal
Gb1 ZZ GST Trx MBP His NusA
S I S I S I S I S I S I S I
BL21 Star
37°C - ++(+) - +++(+) - +++(+) +
+++(+) +(+) + - +++(+) +(+) ++++
BL21 Star
20°C + ++ - ++++ - ++++ + ++++ +(+) +
- ++++ +(+) ++++
Rosetta
37°C - ++ - +++ +/- ++(+) + +++(+) +
++ + +++ +(+) +++(+)
Rosetta
20°C +/- +/- - ++++ - +++ - +++(+)
++(+) ++ - ++(+) ++ ++++
Figure A2 : Analyse de l'expression
et de la solubilité des protéines de fusion PROcatal dans
28 conditions expérimentales
différentes.
Les points rouges sur les gels SDS-PAGE
correspondent aux masses attendues des protéines de fusion.
Après dépôt des fractions soluble (S) et insoluble
(I), chaque bande de surexpression est analysée, et une valeur
semi-quantitative est associée à son intensité, de
faible (+) à très forte (++++). Les fractions non
lisibles sont annotées du sigle NL, et les fractions se
confondant aux protéines intrinsèques à E. coli et
constituant un doute, sont associées au sigle +/-. Les
notations surlignées en couleur mettent en évidence les
meilleures conditions d'expression et de solubilité : couleur
orange pour la meilleure condition, et couleur jaune pour la deuxième
meilleure condition.
De manière plus intéressante, on retrouve ce profil
d'expression pour la construction
PROcatal fusionnée avec le partenaire NusA.
L'expression dans la fraction insoluble y est très intense, mais
contrairement aux autres protéines de fusion, le taux d'expression de
protéines solubles est significatif, et notamment en souche Rosetta
cultivée à 20°C.
Le meilleur résultat en terme d'expression de
protéines solubles PROcatal est obtenu avec le partenaire MBP en
souche Rosetta cultivée à 20°C. La combinaison de
ces trois conditions permet en effet d'obtenir une bonne expression avec un
taux de protéines solubles tout à fait satisfaisant.
Au final, il apparaît que la nature du partenaire de
fusion joue un rôle primordial sur la solubilité des
protéines hétérologues PROcatal. L'influence de la souche
bactérienne et de la température d'expression semble à
première vue moins prononcée, mais elle est flagrante
lorsque PROcatal est exprimée en fusion avec NusA et MBP.
Ainsi, l'association souche Rosetta-température d'expression de
20°C se démarque nettement des autres conditions dans
la mesure où elle permet d'améliorer
sensiblement le profil de solubilité des constructions avec NusA
et MBP.
2.2) La protéine PROentier
PROentier est constitué à 86 % des acides
aminés du domaine PROcatal et de manière intéressante, on
constate que le profil d'expression des protéines de fusion
PROentier est similaire à celui des protéines
hétérologues PROcatal, voire identique dans certaines
conditions. Ainsi, le meilleur résultat, en termes d'expression et de
solubilité, est obtenu avec
le partenaire MBP en souche Rosetta cultivée à
20°C.
Comme le met en évidence la Figure A3, la nature
du partenaire de fusion semble jouer un rôle essentiel sur
l'expression et la solubilité des protéines
hétérologues PROentier :
les constructions avec His, Gb1, ZZ, Trx, et GST conduisent
à une expression quasi exclusive
de protéines insolubles ; et même si pour Trx, on
retrouve systématiquement une expression très faible dans la
fraction soluble, seules les constructions avec MBP et NusA permettent, dans
certains cas, d'observer une expression suffisante de protéines
solubles.
(kDa)
250
150
100
Gb1
S I
ZZ GST
S I S I
Trx
S I
MBP
S I
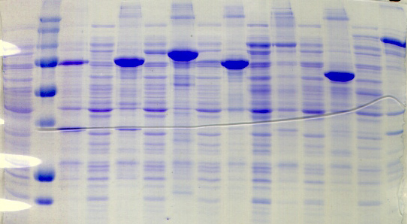
·
His
S I
NusA
S I
·
(kDa)
250
150
100
Gb1
S I
ZZ GST
S I S I
Trx
S I
MBP
S I
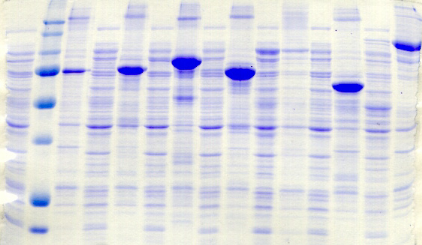
·
His
S I
NusA
S I
·
75
·
·
·
·
·
50
37
75
·
·
·
·
·
50
37
25
20
BL21 Star (37°C)
25
20
BL21 Star (20°C)

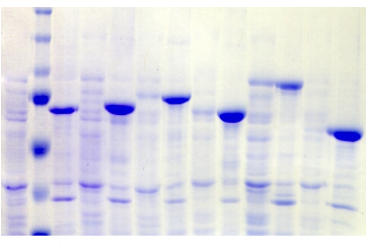
(kDa)
250
150
100
Gb1
S I
ZZ GST
S I S I
Trx
Rosetta (37°C)
S I
MBP
S I
·
His
S I
NusA
S I
·
(kDa)
250
150
100
Gb1
S I
ZZ GST
S I S I
Trx
S I
MBP
S I
His
S I
NusA
S I
·
75
·
·
·
·
·
50
·
·
75
·
·
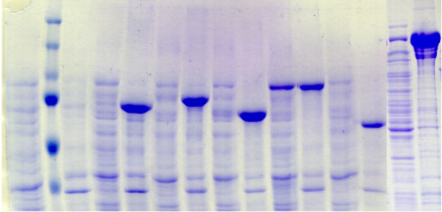
·
·
50
37
37
25
Rosetta (20°C)
PROentier
Gb1 ZZ GST Trx MBP His NusA
S I S I S I S I S I S I S I
BL21 Star
37°C - ++(+) - +++(+) + +++(+) +
+++(+) + + - +++(+) + +++
BL21 Star
20°C - +(+) + ++(+) +/- ++++ + ++++ +
+ - +++(+) + +++(+)
Rosetta
37°C - ++ +/- +++ - ++(+) + +++(+) +
++ + +++ + +++(+)
Rosetta
20°C +/- +/- - +++(+) - +++ + +++ ++
++ - ++ +(+) ++++
Figure A3 : Analyse de
l'expression et de la solubilité des protéines de fusion
PROentier
dans 28 conditions expérimentales
différentes.
Comme précédemment, les résultats du
criblage des conditions d'expression des
protéines de fusion PROentier ne mettent en
évidence aucun effet direct de la souche bactérienne ou de
la température d'expression sur l'expression et la solubilité des
protéines de fusion recombinantes. Cependant, on retrouve pour les
constructions avec MBP et NusA, un effet souche couplé à un effet
température qui met une nouvelle fois en valeur le pouvoir de
solubilisation de la combinaison souche Rosetta-température d'expression
de 20°C.
2.3) Le domaine PROter
L'expression des protéines de fusion PROter n'a
été initialement testée qu'en souches BL21 Star et
Rosetta. Comme le montrent les gels SDS-PAGE et le tableau d'analyse de la
Figure A4, la majorité de ces protéines
hétérologues ne s'expriment pas dans ces deux souches ; ou
l'expression est si faible qu'elle n'est pas décelable sur gel SDS-PAGE.
C'est le
cas notamment des constructions avec Gb1, Trx, MBP, et His.
Pour PROter en fusion avec le partenaire ZZ, une expression faible
est observable dans la fraction insoluble, mais uniquement en
souche BL21 Star. Seule la construction avec GST s'exprime dans les deux
souches. Cette expression est exclusivement insoluble, sauf en BL21 Star
cultivée à 20°C où une bande de faible
intensité apparaît dans la fraction soluble.
En ce qui concerne la construction avec NusA, nous avons
rencontré des difficultés lors de l'étape de
transformation des BL21 Star par le plasmide d'expression. En effet,
malgré plusieurs essais, nous n'avons pas été en mesure de
transformer cette souche par notre vecteur d'intérêt. Ce
problème ne s'est pas posé avec la souche Rosetta, et
contrairement aux autres protéines de fusion, les taux d'expression
obtenus à 37°C et 20°C avec NusA sont significatifs dans les
fractions soluble et insoluble.
De manière intéressante, nous avons
constaté que la transformation des souches par les vecteurs d'expression
PROter induit une diminution importante de la croissance
bactérienne après induction par l'IPTG. Cette baisse de la
vitesse de croissance associée à une expression réduite
de la plupart des protéines de fusion est tout à fait
caractéristique de protéines hétérogènes
toxiques. Par conséquent, il nous est apparu nécessaire de tester
l'expression des protéines de fusion PROter en souche C41 qui
est adaptée à la production de ce type de
protéines.
(kDa)
100
75
50
37
25
20
Gb1
S I
·
ZZ GST
S I S I
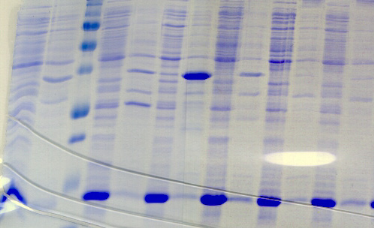
·
·
Trx
S I
·
MBP
S I
·
His
S I
(kDa)
100
75
50
37
25
20
Gb1
S I
·
ZZ GST
S I S I
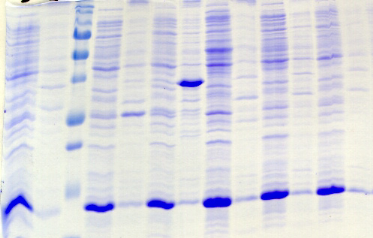
·
·
Trx
S I
·
MBP
S I
·
His
S I
15
·
BL21 Star (37°C)
15
·
BL21 Star (20°C)
(kDa)
100
75
50
37
25
20
Gb1
S I
·
ZZ GST
S I S I
·
·
Trx
S I
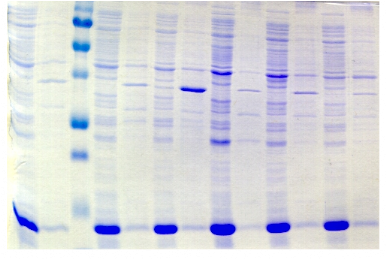

·
MBP
S I
·
His
S I
NusA
S I
·
(kDa)
150
100
75
50
37
25
20
Gb1
S I
·
ZZ GST
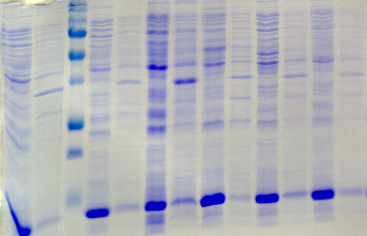
S I S I
·
·
Trx
S I
·
MBP
S I
·
His
S I
NusA
S I
·
15
·
Rosetta (37°C)
15
·
Rosetta (20°C)
|
Gb1
|
ZZ
|
GST
|
Trx
|
MBP
|
His
|
NusA
|
|
S I
|
S I
|
S I
|
S I
|
S I
|
S I
|
S I
|
|
|
|
|
·
|
|
·
|
(kDa)
150
100
75
50
37
·
25
·
·
20
·
(kDa)
150
100
75
50
37
25
20
Gb1
S I
·
ZZ GST
S I S I
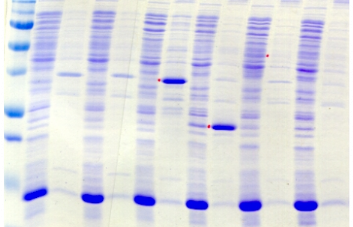
·
·
Trx
S I
·
MBP
S I
·
His
S I
NusA
S I
·
15 15
·
10 10
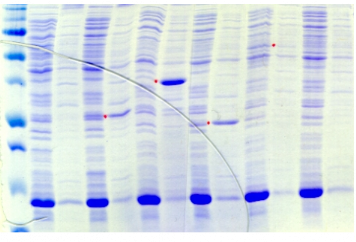

C41 (37°C)
·
C41 (20°C)
|
S
|
I
|
S
|
I
|
S
|
I
|
S
|
I
|
S
|
I
|
S
|
I
|
S
|
I
|
|
-
|
-
|
+/-
|
+
|
+/-
|
+++
|
-
|
-
|
-
|
-
|
-
|
-
|
ND
|
ND
|
|
-
|
-
|
+/-
|
+
|
+
|
++
|
-
|
-
|
-
|
-
|
-
|
-
|
ND
|
ND
|
|
-
|
-
|
-
|
-
|
+/-
|
++(+)
|
-
|
-
|
-
|
-
|
-
|
-
|
+
|
+(+)
|
|
-
|
-
|
-
|
-
|
-
|
+(+)
|
-
|
-
|
-
|
-
|
-
|
-
|
++
|
++
|
|
-
|
-
|
+/-
|
+
|
+
|
++(+)
|
+/-
|
++
|
+
|
+/-
|
-
|
-
|
+(+)
|
++(+)
|
|
-
|
-
|
-
|
-
|
+/-
|
++(+)
|
+
|
++(+)
|
+/-
|
-
|
-
|
-
|
+(+)
|
+(+)
|
PROter
Gb1 ZZ GST Trx MBP His NusA
BL21 Star
37°C
BL21 Star
20°C
Rosetta
37°C
Rosetta
20°C
C41
37°C
C41
20°C
Figure A4 : Analyse de
l'expression et de la solubilité des protéines de fusion
PROter dans 42 conditions expérimentales différentes.
Comme le met en évidence la Figure A4, on constate que la
souche C41 apporte une
amélioration significative des taux d'expression des
protéines hétérologues PROter. Ainsi, alors qu'en
souches BL21 Star et Rosetta l'expression des constructions Trx et MBP
n'était pas détectable, des bandes de surexpression
sont observables en C41 pour ces deux constructions, et notamment
pour le partenaire Trx où l'effet souche est assez spectaculaire.
En revanche, la production de PROter en fusion avec Gb1 et His
n'est toujours pas détectable
en souche C41. En ce qui concerne les constructions avec ZZ et
GST, les profils d'expression obtenus en souche C41 sont comparables à
ceux obtenus avec les deux autres souches.
Au vu de l'ensemble de ces résultats, il apparaît
que la nature du partenaire de fusion
et de la souche bactérienne est déterminante
pour l'expression et la solubilité des protéines
hétérologues PROter. En effet, seule la construction avec
NusA permet d'obtenir une expression suffisante de protéines
solubles en souches Rosetta ou C41. L'influence de la
température semble plus limitée, mais comme observé
précédemment, l'association souche Rosetta-température
de 20°C combinée à un partenaire de fusion efficace
conduit aux meilleurs taux d'expression de protéine soluble.
2.4) Le domaine PROinser
Les protéines hétérologues PROinser
présentent un profil d'expression qui ressemble davantage à
celui des protéines de fusion PROcatal et PROentier,
plutôt qu'à celui des protéines PROter. Ainsi, le
meilleur résultat est obtenu avec le partenaire MBP en souche
Rosetta cultivée à 20°C avec un taux d'expression de
protéines solubles tout à fait satisfaisant.
Comme le montre la Figure A5, les niveaux d'expression obtenus
sont très élevés pour l'ensemble des partenaires de
fusion, à l'exception de la construction avec Gb1, qui est peu ou non
exprimée. Cependant, cette production est majoritairement insoluble pour
la plupart des partenaires de fusion : c'est le cas notamment pour His, GST,
Trx, et ZZ. Aussi, dans le cas
de GST et Trx, une faible expression est presque
systématiquement observable dans la fraction soluble. A l'instar des
3 autres domaines PRODH, seules les constructions avec MBP
et NusA conduisent à une production significative dans la
fraction soluble.
(kDa)
100
75
50
37
Gb1
S I
ZZ GST
S I S I
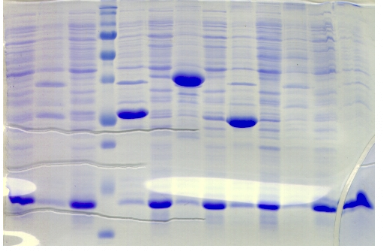

·
Trx
S I
MBP
S I
·
His
S I
NusA
S I
·
(kDa)
100
75
50
37
Gb1
S I
ZZ GST
S I S I

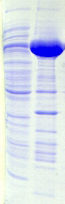
·
Trx
S I
MBP
S I
·
His
S I
NusA
Annexe : Criblage des conditions d'expression des
protéines PROentier, PROcatal, PROter, et PROinser
S I
·
25
·
·
·
20
25
·
·
·
20
15
·
10
BL21 Star (37°C)
15
·
10
BL21 Star (20°C)
(kDa)
100
75
50
37
Gb1
S I
ZZ GST
S I S I
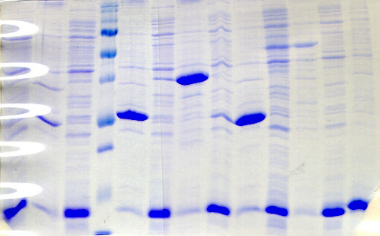
·
Trx
S I
MBP
S I
·
His
S I
NusA
S I

·
(kDa)
100
75
50
37
Gb1 ZZ
S I S I
GST
S I
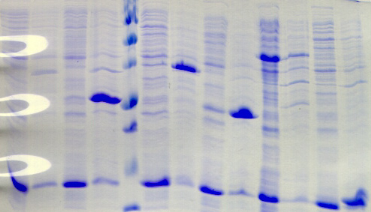
·
Trx
S I
MBP
S I
·
His
S I
NusA
S I

·
25
·
·
·
20
25
·
·
·
20
15
·
10
Rosetta (37°C)
15
10
·
Rosetta (20°C)
(kDa)
100
75
50
37
Gb1
S I
ZZ GST
S I S I
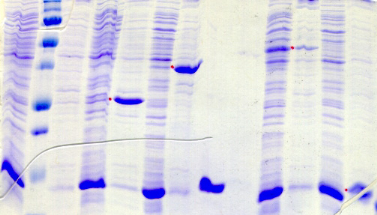
·
Trx
S I
MBP
S I
·
His
S I
NusA
S I
·
(kDa)

100
75
50
37
Gb1
S I
ZZ GST
S I S I
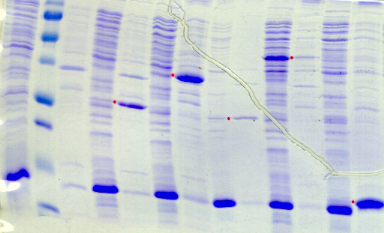
·
Trx
S I
MBP
S I
·
His
S I
NusA
S I
·
25
·
·
·
20
25
·
·
20
·
15
15
· 10
10
C41 (37°C)
·
C41 (20°C)
PROinser
BL21 Star
Gb1 ZZ GST Trx MBP His NusA
S I S I S I S I S I S I S I
37°C - +/- +/- +++(+) + ++++ + ++++
+ + +/- +++ +(+) +++(+)
BL21 Star
20°C - - - +++ + ++++ + +++ + + +/-
++(+) +(+) ++++
Rosetta
37°C - + - +++ + +++(+) + +++(+)
+(+) +(+) +/- ++(+) +(+) +++(+)
Rosetta
20°C - - +/- +++ + +++ + +++ ++(+) +
+/- +++ +(+) ++++
C41
37°C - - +/- +++ +/- +++ ND ND ++ +(+)
- +++ +/- +++
C41
20°C - - + ++(+) + +++ + + ++ + -
+++(+) +(+) +++(+)
Figure A5 : Analyse de
l'expression et de la solubilité des protéines de fusion
PROinser dans 42 conditions expérimentales différentes.
Annexe : Criblage des conditions d'expression des
protéines PROentier, PROcatal, PROter, et PROinser
L'analyse du criblage des conditions d'expression de PROinser met
donc une nouvelle
fois en évidence le rôle essentiel du
partenaire de fusion sur la solubilité des protéines
hétérologues. Aussi, contrairement aux résultats obtenus
avec les autres domaines PRODH, il
est plus difficile de mettre en évidence un effet
direct de la souche bactérienne ou de la température sur
l'expression et la solubilité des protéines de fusion PROinser.
Dans le cas de
la construction avec MBP, on constate toutefois que la
souche Rosetta donne de meilleurs résultats que les souches
BL21 Star et C41. Par ailleurs, même si un effet solubilisant
de l'association souche Rosetta-température de 20°C est
observé pour la construction avec MBP,
ce dernier ne se retrouve pas pour la construction avec NusA.
Références bibliographiques
Références Bibliographiques
Adams, E. & Frank, L. (1980) Metabolism of proline
and the hydroxyprolines. Annu Rev
Biochem, 49, 1005-1061.
Allen, M., Friedler, A., Schon, O. & Bycroft, M. (2002) The
structure of an FF domain from human HYPA/FBP11. J Mol Biol,
323, 411-416.
Ames, B.N., Shigenaga, M.K. & Gold, L.S. (1993) DNA
lesions, inducible DNA repair, and cell division: three key factors in
mutagenesis and carcinogenesis. Environ Health Perspect,
101 Suppl 5, 35-44.
Angulo, J.F., Rouer, E., Mazin, A., Mattei, M.G., Tissier, A.,
Horellou, P., Benarous, R. & Devoret, R. (1991) Identification and
expression of the cDNA of KIN17, a zinc-finger gene located on mouse chromosome
2, encoding a new DNA-binding protein. Nucleic Acids Res,
19, 5117-5123.
Arfken, G. (1985) The method of Steepest Descents.
Mathematical Methods for Physicists,
3rd ed., Orlando, FL: Academic Press,
428-436.
Athanasiadis, A., Placido, D., Maas, S., Brown, B.A., 2nd,
Lowenhaupt, K. & Rich, A. (2005) The crystal structure of the Zbeta domain
of the RNA-editing enzyme ADAR1 reveals distinct conserved surfaces among
Z-domains. J Mol Biol, 351, 496-507.
Baker, N.A., Sept, D., Joseph, S., Holst, M.J. &
McCammon, J.A. (2001) Electrostatics of nanosystems : application to
microtubules and the ribosome. Proc Natl Acad Sci USA,
98, 10037-10041.
Bannai, H., Inenaga, S., Shinohara, A., Takeda, M.
& Miyano, S. (2002) A string pattern regression algorithm and its
application to pattern discovery in long introns. Genome Inform Ser
Workshop Genome Inform, 13, 3-11.
Becker, D.F. & Thomas, E.A. (2001) Redox properties of the
PutA protein from Escherichia coli and the influence of the flavin
redox state on PutA-DNA interactions. Biochemistry,
40, 4714-4721.
Biard, D.S., Miccoli, L., Despras, E., Frobert, Y.,
Creminon, C. & Angulo, J.F. (2002) Ionizing radiation triggers
chromatin-bound kin17 complex formation in human cells.
J Biol Chem, 277, 19156-19165.
Blum, A. & Ebercon, A. (1976) Genotypic responses in
sorghum to drought stress. Free proline accumulation and drought
resistance. Crop Sci, 16, 428-431.
Bohm, S., Frishman, D. & Mewes, H.W. (1997) Variations of the
C2H2 zinc finger motif in
the yeast genome and classification of yeast zinc finger
proteins. Nucleic Acids Res,
25, 2464-2469.
Bondy, S.C. & Naderi, S. (1994) Contribution of
hepatic cytochrome P450 systems to the generation of reactive oxygen
species. Biochem Pharmacol, 48, 155-159.
Braud, S., Moutiez, M., Belin, P., Abello, N., Drevet,
P., Zinn-Justin, S., Courcon, M., Masson, C., Dassa, J., Charbonnier,
J.B., Boulain, J.C., Menez, A., Genet, R. & Gondry, M. (2005) Dual
expression system suitable for high-throughput fluorescence- based screening
and production of soluble proteins. J Proteome Res,
4, 2137-2147.
Brennan, R.G. & Matthews, B.W. (1989) The
helix-turn-helix DNA binding motif. J Biol
Chem, 264, 1903-1906.
Brooks, B.R., Bruccoleri, R.E., Olafson, B.D., States, D.J.,
Swaminathan, S. & Karplys, M. (1983) CHARMM: a program for
macromolecular energy, minimization, and dynamics calculations. J
Comput Chem, 4, 187-217.
Brosemer, R.W. & Veerabhadrappa, P.S. (1965) Pathway of
proline oxidation in insect flight muscle. Biochim Biophys Acta,
110, 102-112.
Brown, E.D. & Wood, J.M. (1992) Redesigned
purification yields a fully functional PutA
protein dimer from Escherichia coli. J Biol Chem,
267, 13086-13092.
Brünger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros,
P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu,
N.S., Read, R.J., Rice, L.M., Simonson,
T. & Warren, G.L. (1998) Crystallography & NMR system: A
new software suite for macromolecular structure determination. Acta
Crystallogr D Biol Crystallogr, 54,
905-921.
Brünger, A.T. (1992). X-PLOR Manual Version
3.1, Yale University New Haven, Connecticut.
Brunner, G. & Neupert, W. (1969) Localisation of
proline oxidase and Delta-pyrroline-5- carboxylic acid dehydrogenase in rat
liver. FEBS Lett, 3, 283-286.
Campbell, H.D., Webb, G.C. & Young, I.G. (1997) A human
homologue of the Drosophila melanogaster sluggish-A (proline oxidase) gene maps
to 22q11.2, and is a candidate gene for type-I hyperprolinaemia. Hum
Genet, 101, 69-74.
Carlier, L., le Maire, A., Braud, S., Masson, C., Gondry, M.,
Zinn-Justin, S., Guilhaudis, L., Milazzo, I., Davoust, D., Gilquin, B.
& Couprie, J. (2006) NMR Assignment of Region 51-160 of Human KIN17,
a DNA and RNA-binding Protein. J Biomol NMR.
Cavanagh, J., Fairbrother, W.J., Palmer III, A.G. & Skelton,
N.J. (1996). NMR Spectroscopy.
Principles and Practice, London 1996, Academic
Press.
Chakravarti, A. (2002) A compelling genetic hypothesis for a
complex disease: PRODH2/DGCR6 variation leads to schizophrenia susceptibility.
Proc Natl Acad Sci
U S A, 99, 4755-4756.
Chen, C.C., Tsuchiya, T., Yamane, Y., Wood, J.M. & Wilson,
T.H. (1985) Na+ (Li+)-proline cotransport in Escherichia coli. J Membr
Biol, 84, 157-164.
Clark, E.D. (2001) Protein refolding for industrial processes.
Curr Opin Biotechnol, 12, 202-
207.
Clark, K.L., Halay, E.D., Lai, E. & Burley, S.K.
(1993) Co-crystal structure of the HNF-
3/fork head DNA-recognition motif resembles histone H5.
Nature, 364, 412-420.
Claros, M.G. & Vincens, P. (1996) Computational method
to predict mitochondrially imported proteins and their targeting sequences.
Eur J Biochem, 241, 779-786.
Clore, G.M. & Gronenborn, A.M. (1991) Two-, three-, and four-
dimensional NMR methods
for obtaining larger and more precise three-dimensional
structures of proteins in solution. Annu Rev Biophys Biophys Chem,
20, 29-63.
Cook, W.J., Kar, S.R., Taylor, K.B. & Hall,
L.M. (1998) Crystal structure of the cyanobacterial
metallothionein repressor SmtB: a model for metalloregulatory
proteins. J Mol Biol, 275, 337-346.
Cornilescu, G., Delaglio, F. & Bax, A. (1999) Protein
backbone angle restraints from searching a database for chemical shift and
sequence homology. J Biomol NMR, 13,
289-302.
De Silva, R.S., Kovacikova, G., Lin, W., Taylor, R.K.,
Skorupski, K. & Kull, F.J. (2005) Crystal structure of the virulence
gene activator AphA from Vibrio cholerae reveals it
is a novel member of the winged helix transcription factor
superfamily. J Biol Chem,
280, 13779-13783.
Dillon, E.L., Knabe, D.A. & Wu, G. (1999) Lactate inhibits
citrulline and arginine synthesis from proline in pig enterocytes. Am J
Physiol, 276, G1079-1086.
Dong, G., Chakshusmathi, G., Wolin, S.L. & Reinisch, K.M.
(2004) Structure of the La motif:
a winged helix domain mediates RNA binding via a conserved
aromatic patch. Embo
J, 23, 1000-1007.
Efron, M.L. (1965) Familial Hyperprolinemia. Report of
a Second Case, Associated with Congenital Renal Malformations, Hereditary
Hematuria and Mild Mental Retardation, with Demonstration of an Enzyme Defect.
N Engl J Med, 272, 1243-1254.
Elantak, L., Ansaldi, M., Guerlesquin, F., Mejean, V.
& Morelli, X. (2005) Structural and genetic analyses reveal a key role
in prophage excision for the TorI response regulator inhibitor. J Biol
Chem, 280, 36802-36808.
Emanuelsson, O., Nielsen, H., Brunak, S. & von
Heijne, G. (2000) Predicting subcellular localization of proteins based on
their N-terminal amino acid sequence. J Mol Biol,
300, 1005-1016.
Emanuelsson, O., von Heijne, G. & Schneider, G.
(2001) Analysis and prediction of mitochondrial targeting peptides.
Methods Cell Biol, 65, 175-187.
Englander, S.W., Downer, N.W. & Teitelbaum, H. (1972)
Hydrogen exchange. Annu Rev
Biochem, 41, 903-924.
Erecinska, M. (1965) Ubiquinone in proline oxidation. Arch
Int Pharmacodyn Ther, 158, 209-
215.
Fan, J.B., Ma, J., Zhang, C.S., Tang, J.X., Gu, N.F., Feng, G.Y.,
St Clair, D. & He, L. (2003)
A family-based association study of T1945C
polymorphism in the proline dehydrogenase gene and schizophrenia in the
Chinese population. Neurosci Lett, 338,
252-254.
Farrow, N.A., Zhang, O., Forman-Kay, J.D. & Kay, L.E. (1994)
A heteronuclear correlation
experiment for simultaneous determination of 15N
longitudinal decay and chemical exchange rates of systems in slow
equilibrium. J Biomol NMR, 4, 727-734.
Finn, R.D., Mistry, J., Schuster-Bockler, B.,
Griffiths-Jones, S., Hollich, V., Lassmann, T., Moxon, S., Marshall, M.,
Khanna, A., Durbin, R., Eddy, S.R., Sonnhammer, E.L. & Bateman, A. (2006)
Pfam: clans, web tools and services. Nucleic Acids Res,
34, D247-
251.
Friedberg, E.C. (2001) How nucleotide excision repair
protects against cancer. Nat Rev
Cancer, 1, 22-33.
Frishman, D. & Argos, P. (1995) Knowledge-based protein
secondary structure assignment.
Proteins, 23, 566-579.
Gajiwala, K.S. & Burley, S.K. (2000) Winged helix proteins.
Curr Opin Struct Biol, 10, 110-
116.
Gajiwala, K.S., Chen, H., Cornille, F., Roques, B.P.,
Reith, W., Mach, B. & Burley, S.K. (2000) Structure of the
winged-helix protein hRFX1 reveals a new mode of DNA binding.
Nature, 403, 916-921.
Garcia-Castellanos, R., Mallorqui-Fernandez, G., Marrero,
A., Potempa, J., Coll, M. & Gomis-Ruth, F.X. (2004) On the
transcriptional regulation of methicillin resistance: MecI repressor in
complex with its operator. J Biol Chem, 279,
17888-17896.
Georgiou, G. & Valax, P. (1999) Isolating inclusion bodies
from bacteria. Methods Enzymol,
309, 48-58.
Gogos, J.A., Santha, M., Takacs, Z., Beck, K.D.,
Luine, V., Lucas, L.R., Nadler, J.V. & Karayiorgou, M. (1999)
The gene encoding proline dehydrogenase modulates sensorimotor gating
in mice. Nat Genet, 21, 434-439.
Grzesiek, S. & Bax, A. (1993) Amino acid type determination
in the sequential assignment procedure of uniformly 13C/15N-enriched proteins.
J Biomol NMR, 3, 185-204.
Gu, D., Zhou, Y., Kallhoff, V., Baban, B., Tanner, J.J. &
Becker, D.F. (2004) Identification and characterization of the
DNA-binding domain of the multifunctional PutA flavoenzyme. J
Biol Chem, 279, 31171-31176.
Hagedorn, C.H. & Phang, J.M. (1983) Transfer of reducing
equivalents into mitochondria by
the interconversions of proline and delta
1-pyrroline-5-carboxylate. Arch Biochem
Biophys, 225, 95-101.
Hagedorn, C.H. & Phang, J.M. (1986) Catalytic
transfer of hydride ions from NADPH to oxygen by the interconversions
of proline and delta 1-pyrroline-5-carboxylate. Arch Biochem
Biophys, 248, 166-174.
Hagedorn, C.H., Yeh, G.C. & Phang, J.M. (1982)
Transfer of 1-pyrroline-5-carboxylate as oxidizing potential from
hepatocytes to erythrocytes. Biochem J, 202,
31-39.
Hammarstrom, M., Hellgren, N., van Den Berg, S., Berglund, H.
& Hard, T. (2002) Rapid
screening for improved solubility of small human proteins
produced as fusion proteins
in Escherichia coli. Protein Sci, 11,
313-321.
Hayward, D.C., Delaney, S.J., Campbell, H.D., Ghysen,
A., Benzer, S., Kasprzak, A.B., Cotsell, J.N., Young, I.G. & Miklos,
G.L. (1993) The sluggish-A gene of Drosophila melanogaster is expressed in
the nervous system and encodes proline oxidase, a mitochondrial enzyme
involved in glutamate biosynthesis. Proc Natl Acad Sci U S A,
90, 2979-2983.
Herbert, A., Lowenhaupt, K., Spitzner, J. & Rich, A. (1995)
Chicken double-stranded RNA
adenosine deaminase has apparent specificity for Z-DNA. Proc
Natl Acad Sci U S A,
92, 7550-7554.
Hoeijmakers, J.H. (2001) Genome maintenance mechanisms for
preventing cancer. Nature,
411, 366-374.
Hoffmann, M.H. & Vadstrup, S. (2000) [DiGeorge syndrome.
Velocardiofacial syndrome/chromosome 22q11 deletion syndrome]. Ugeskr
Laeger, 162, 2736-2739.
Holden, J.S. (1973) Free amino acid levels in the cockroach,
Periplaneta americana. J Physiol,
232, 61P-62P.
Holm, L. & Sander, C. (1993) Protein structure
comparison by alignment of distance matrices. J Mol Biol,
233, 123-138.
Hong-qi, Z., Croes, A.F. & Linskens, H.F. (1982) Protein
synthesis in germinating pollen of
Petunia: role of proline. Planta, 154,
199-203
Huffman, J.L. & Brennan, R.G. (2002) Prokaryotic
transcription regulators: more than just the helix-turn-helix motif. Curr
Opin Struct Biol, 12, 98-106.
Humbertclaude, V., Rivier, F., Roubertie, A., Echenne, B.,
Bellet, H., Vallat, C. & Morin, D. (2001) Is hyperprolinemia type I
actually a benign trait? Report of a case with severe neurologic involvement
and vigabatrin intolerance. J Child Neurol, 16,
622-623.
Hutchinson, E.G. & Thornton, J.M. (1994) A revised set of
potentials for beta-turn formation
in proteins. Protein Sci, 3,
2207-2216.
Hutchinson, F. (1985) Chemical changes induced in DNA by ionizing
radiation. Prog Nucleic
Acid Res Mol Biol, 32, 115-154.
Jacquet, H., Berthelot, J., Bonnemains, C., Simard, G.,
Saugier-Veber, P., Raux, G., Campion,
D., Bonneau, D. & Frebourg, T. (2003) The severe form of type
I hyperprolinaemia results from homozygous inactivation of the PRODH gene.
J Med Genet, 40, e7.
Jacquet, H., Raux, G., Thibaut, F., Hecketsweiler, B.,
Houy, E., Demilly, C., Haouzir, S., Allio, G., Fouldrin, G., Drouin,
V., Bou, J., Petit, M., Campion, D. & Frebourg, T. (2002) PRODH
mutations and hyperprolinemia in a subset of schizophrenic patients. Hum
Mol Genet, 11, 2243-2249.
Jansson, M., Li, Y.C., Jendeberg, L., Anderson, S., Montelione,
B.T. & Nilsson, B. (1996)
High-level production of uniformly 15N- and
13C-enriched fusion proteins in
Escherichia coli. J Biomol NMR, 7,
131-141.
Jeggo, P.A. (1998) DNA breakage and repair. Adv Genet,
38, 185-218.
Jin, C., Marsden, I., Chen, X. & Liao, X. (1999)
Dynamic DNA contacts observed in the
NMR structure of winged helix protein-DNA complex. J Mol
Biol, 289, 683-690.
Johnson, A.B. & Strecker, H.J. (1962) The interconversion of
glutamic acid and proline. IV.
The oxidation of proline by rat liver mitochondria. J Biol
Chem, 237, 1876-1882.
Jonasson, P., Liljeqvist, S., Nygren, P.A. & Stahl,
S. (2002) Genetic design for facilitated production and recovery of
recombinant proteins in Escherichia coli. Biotechnol Appl Biochem,
35, 91-105.
Kanelis, V., Forman-Kay, J.D. & Kay, L.E. (2001)
Multidimensional NMR methods for protein structure determination. IUBMB
Life, 52, 291-302.
Kannouche, P., Mauffrey, P., Pinon-Lataillade, G., Mattei, M.G.,
Sarasin, A., Daya-Grosjean,
L. & Angulo, J.F. (2000) Molecular cloning and
characterization of the human KIN17 cDNA encoding a component of the
UVC response that is conserved among metazoans.
Carcinogenesis, 21, 1701-1710.
Kannouche, P., Pinon-Lataillade, G., Mauffrey, P., Faucher,
C., Biard, D.S. & Angulo, J.F. (1997) Overexpression of kin17 protein forms
intranuclear foci in mammalian cells. Biochimie, 79,
599-606.
Kannouche, P., Pinon-Lataillade, G., Tissier, A.,
Chevalier-Lagente, O., Sarasin, A., Mezzina,
M. & Angulo, J.F. (1998) The nuclear concentration of
kin17, a mouse protein that binds to curved DNA, increases during cell
proliferation and after UV irradiation. Carcinogenesis,
19, 781-789.
Kapust, R.B. & Waugh, D.S. (1999) Escherichia coli
maltose-binding protein is uncommonly effective at promoting the solubility of
polypeptides to which it is fused. Protein Sci,
8, 1668-1674.
Karplus, M. & Grant, D.M. (1959) A Criterion for
Orbital Hybridization and Charge
Distribution in Chemical Bonds. Proc Natl Acad Sci U S
A, 45, 1269-1273.
Kay, L.E., Torchia, D.A. & Bax, A. (1989) Backbone
dynamics of protein as studied by 15N inverse detected heteronuclear NMR
spectroscopy : application to staphylococcal nuclease.
Biochemistry, 28, 8972-8979.
Kay, L.E., Xu, G.Y., Singer, A.U., Muhandiram, D.R.
& Forman-Kay, J.D. (1993) A Gradient-Enhanced HCCH-TOCSY Experiment for
Recording Side-Chain 1H and 13C Correlations in H2O
Samples of Proteins. J Magn Reson B, 101, 333-337
Khanna, K.K. & Jackson, S.P. (2001) DNA double-strand
breaks: signaling, repair and the cancer connection. Nat Genet,
27, 247-254.
Kielkopf, C.L., Lucke, S. & Green, M.R. (2004) U2AF homology
motifs: protein recognition
in the RRM world. Genes Dev, 18,
1513-1526.
Kowaloff, E.M., Phang, J.M., Granger, A.S. & Downing, S.J.
(1977) Regulation of proline oxidase activity by lactate. Proc Natl Acad
Sci U S A, 74, 5368-5371.
Kramar, R. (1967) Studies on the proline oxidase complex.
Enzymologia, 33, 33-37.
Kurumizaka, H., Aihara, H., Ikawa, S., Kashima, T., Bazemore,
L.R., Kawasaki, K., Sarai,
A., Radding, C.M. & Shibata, T. (1996) A possible role of the
C-terminal domain of
the RecA protein. A gateway model for double-stranded DNA
binding. J Biol Chem,
271, 33515-33524.
Kyrpides, N.C., Woese, C.R. & Ouzounis, C.A. (1996)
KOW: a novel motif linking a bacterial transcription factor with
ribosomal proteins. Trends Biochem Sci, 21,
425-
426.
Landy, A. (1989) Dynamic, structural, and regulatory
aspects of lambda site-specific recombination. Annu Rev
Biochem, 58, 913-949.
Laskowski, R.A., Rullmannn, J.A., MacArthur, M.W., Kaptein, R.
& Thornton, J.M. (1996) AQUA and PROCHECK-NMR: programs for
checking the quality of protein structures solved by NMR. J Biomol
NMR, 8, 477-486.
Lee, Y.H., Nadaraia, S., Gu, D., Becker, D.F. & Tanner, J.J.
(2003) Structure of the proline dehydrogenase domain of the multifunctional
PutA flavoprotein. Nat Struct Biol, 10,
109-114.
Lindahl, T. (1974) An N-glycosidase from Escherichia coli
that releases free uracil from
DNA containing deaminated cytosine residues. Proc Natl Acad
Sci U S A, 71, 3649-
3653.
Lindahl, T. (1993) Instability and decay of the primary structure
of DNA. Nature, 362, 709-
715.
Littlefield, O. & Nelson, H.C. (1999) A new use for the
'wing' of the 'winged' helix-turn-helix motif in the HSF-DNA cocrystal. Nat
Struct Biol, 6, 464-470.
Liu, H., Heath, S.C., Sobin, C., Roos, J.L., Galke,
B.L., Blundell, M.L., Lenane, M., Robertson, B., Wijsman, E.M., Rapoport,
J.L., Gogos, J.A. & Karayiorgou, M. (2002) Genetic variation at the
22q11 PRODH2/DGCR6 locus presents an unusual pattern and increases
susceptibility to schizophrenia. Proc Natl Acad Sci U S A,
99, 3717-
3722.
Logan, T.M., Olejniczak, E.T., Xu, R.X. & Fesik,
S.W. (1993) A general method for assigning NMR spectra of denatured
proteins using 3D HC(CO)NH-TOCSY triple resonance experiments. J Biomol
NMR, 3, 225-231.
Lopez, P.J., Marchand, I., Joyce, S.A. & Dreyfus, M.
(1999) The C-terminal half of RNase E, which organizes the Escherichia coli
degradosome, participates in mRNA degradation but not rRNA processing in vivo.
Mol Microbiol, 33, 188-199.
Majumdar, A., Wang, H., Morsauher, R.C. &
Zuiderweg E.R.P. (1993) Sensitivity
improvement in 2D and 3D HCCH spctroscopy using
heteronuclear cross- polarization. J Biomol NMR, 3,
387-397.
Maniatis, T., Fritsch, E.F. & Sambrook, J. (1982).
Molecular cloning, a laboratory manual,
Cold Spring Harbor: Cold Spring Harbor
Laboratory.
Marion, D., Kay, L.E., Sparks, S.W., Torchia, D.A. &
Bax, A. (1989) Three-dimensional heteronuclear NMR of 15N labeled proteins.
J Am Chem Soc, 111, 1515-1517.
Marsden, I., Chen, Y., Jin, C. & Liao, X. (1997) Evidence
that the DNA binding specificity of winged helix proteins is mediated by a
structural change in the amino acid sequence adjacent to the principal DNA
binding helix. Biochemistry, 36, 13248-13255.
Marti, T.M., Kunz, C. & Fleck, O. (2002) DNA
mismatch repair and mutation avoidance pathways. J Cell Physiol,
191, 28-41.
Masson, C., Menaa, F., Pinon-Lataillade, G., Frobert, Y.,
Chevillard, S., Radicella, J.P., Sarasin, A. & Angulo, J.F. (2003)
Global genome repair is required to activate KIN17,
a UVC-responsive gene involved in DNA replication. Proc Natl
Acad Sci U S A, 100,
616-621.
Maynard, T.M., Haskell, G.T., Peters, A.Z., Sikich, L.,
Lieberman, J.A. & LaMantia, A.S. (2003) A comprehensive analysis
of 22q11 gene expression in the developing and adult brain. Proc
Natl Acad Sci U S A, 100, 14433-14438.
Mazin, A., Timchenko, T., Menissier-de Murcia, J., Schreiber, V.,
Angulo, J.F., de Murcia, G.
& Devoret, R. (1994) Kin17, a mouse nuclear
zinc finger protein that binds preferentially to curved DNA.
Nucleic Acids Res, 22, 4335-4341.
McGuffin, P., Asherson, P., Owen, M. & Farmer, A. (1994) The
strength of the genetic effect.
Is there room for an environmental influence in the aetiology of
schizophrenia? Br J Psychiatry, 164, 593-599.
Menzel, R. & Roth, J. (1981) Purification of the
putA gene product. A bifunctional membrane-bound protein from Salmonella
typhimurium responsible for the two-step oxidation of proline to glutamate.
J Biol Chem, 256, 9755-9761.
Mer, G., Bochkarev, A., Gupta, R., Bochkareva, E., Frappier, L.,
Ingles, C.J., Edwards, A.M.
& Chazin, W.J. (2000) Structural basis for the
recognition of DNA repair proteins
UNG2, XPA, and RAD52 by replication factor RPA. Cell,
103, 449-456.
Miccoli, L., Biard, D.S., Creminon, C. & Angulo, J.F.
(2002) Human kin17 protein directly interacts with the simian virus 40
large T antigen and inhibits DNA replication. Cancer Res,
62, 5425-5435.
Miccoli, L., Frouin, I., Novac, O., Di Paola, D., Harper, F.,
Zannis-Hadjopoulos, M., Maga,
G., Biard, D.S. & Angulo, J.F. (2005) The human
stress-activated protein kin17 belongs to the multiprotein DNA
replication complex and associates in vivo with mammalian replication
origins. Mol Cell Biol, 25, 3814-3830.
Murphy, K.C. (2002) Schizophrenia and velo-cardio-facial
syndrome. Lancet, 359, 426-430.
Murphy, K.C., Jones, L.A. & Owen, M.J. (1999) High rates of
schizophrenia in adults with
velo-cardio-facial syndrome. Arch Gen Psychiatry,
56, 940-945.
Nakai, K. & Horton, P. (1999) PSORT: a program for detecting
sorting signals in proteins and predicting their subcellular localization.
Trends Biochem Sci, 24, 34-36.
Nguyen, B.D., Abbott, K.L., Potempa, K., Kobor, M.S.,
Archambault, J., Greenblatt, J., Legault, P. & Omichinski, J.G.
(2003) NMR structure of a complex containing the TFIIF subunit RAP74
and the RNA polymerase II carboxyl-terminal domain phosphatase
FCP1. Proc Natl Acad Sci U S A, 100, 5688-5693.
Nielsen, H., Engelbrecht, J., Brunak, S. & von Heijne, G.
(1997) Identification of prokaryotic and eukaryotic signal peptides and
prediction of their cleavage sites. Protein Eng,
10,
1-6.
Nilges, M. (1995) Calculation of protein structures with
ambiguous distance restraints.
Automated assignment of ambiguous NOE crosspeaks and disulphide
connectivities. J Mol Biol, 245, 645-660.
Nilges, M., Macias, M.J., O'Donoghue, S.I. &
Oschkinat, H. (1997) Automated NOESY
interpretation with ambiguous distance restraints: the refined
NMR solution structure
of the pleckstrin homology domain from beta-spectrin. J Mol
Biol, 269, 408-422.
Nishikawa, J., Amano, M., Fukue, Y., Tanaka, S., Kishi, H.,
Hirota, Y., Yoda, K. & Ohyama,
T. (2003) Left-handedly curved DNA regulates accessibility to
cis-DNA elements in chromatin. Nucleic Acids Res, 31,
6651-6662.
Okuda, M., Watanabe, Y., Okamura, H., Hanaoka, F., Ohkuma, Y.
& Nishimura, Y. (2000) Structure of the central core domain of TFIIEbeta
with a novel double-stranded DNA- binding surface. Embo J,
19, 1346-1356.
Ostrovsky de Spicer, P., O'Brien, K. & Maloy, S. (1991)
Regulation of proline utilization in
Salmonella typhimurium: a membrane-associated dehydrogenase binds
DNA in vitro.
J Bacteriol, 173, 211-219.
Ostrovsky, P.C. & Maloy, S. (1995) Protein
phosphorylation on serine, threonine, and tyrosine residues modulates
membrane-protein interactions and transcriptional regulation in
Salmonella typhimurium. Genes Dev, 9, 2034-2041.
Pardi, A., Billeter, M. & Wuthrich, K. (1984) Calibration
of the angular dependence of the amide proton-C alpha proton coupling
constants, 3JHN alpha, in a globular protein. Use of 3JHN alpha for
identification of helical secondary structure. J Mol Biol,
180,
741-751.
Pervushin, K., Riek, R., Wider, G. & Wuthrich, K. (1997)
Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling
and chemical shift anisotropy indicates an avenue to NMR structures of
very large biological macromolecules in solution. Proc Natl Acad Sci U S
A, 94, 12366-12371.
Pfanner, N. & Neupert, W. (1990) The mitochondrial
protein import apparatus. Annu Rev
Biochem, 59, 331-353.
Phang, J.M. (1985) The regulatory functions of proline and
pyrroline-5-carboxylic acid. Curr
Top Cell Regul, 25, 91-132.
Pinon-Lataillade, G., Masson, C., Bernardino-Sgherri, J.,
Henriot, V., Mauffrey, P., Frobert,
Y., Araneda, S. & Angulo, J.F. (2004) KIN17 encodes an
RNA-binding protein and is expressed during mouse spermatogenesis. J Cell
Sci, 117, 3691-3702.
Pohl, E., Haller, J.C., Mijovilovich, A., Meyer-Klaucke,
W., Garman, E. & Vasil, M.L. (2003) Architecture of a protein
central to iron homeostasis: crystal structure and spectroscopic analysis
of the ferric uptake regulator. Mol Microbiol, 47,
903-915.
Ponting, C.P. (2002) Novel domains and orthologues of eukaryotic
transcription elongation factors. Nucleic Acids Res,
30, 3643-3652.
Powell, M.J.D. (1977) Restart procedures of the conjugate
gradient method. Math Program,
2, 241-254.
Ramachandran, G.N., Chandrasekaran, R. & Kopple,
K.D. (1971) Variation of the NH-C alpha-H coupling constant with
dihedral angle in the NMR spectra of peptides. Biopolymers,
10, 2113-2131.
Rappsilber, J., Ryder, U., Lamond, A.I. & Mann, M. (2002)
Large-scale proteomic analysis of the human spliceosome. Genome Res,
12, 1231-1245.
Redfield, A.G., McIntosh, L.P. & Dahlquist, F.W. (1989) Use
of 13C and 15N isotope labels
for proton nuclear magnetic resonance and nuclear Overhauser
effect. Structural and dynamic studies of larger proteins and nucleic acids.
Arch Biol Med Exp (Santiago),
22, 129-137.
Reilly, D. & Fairbrother, W.J. (1994) A novel isotope
labeling protocol for bacterially expressed proteins. J Biomol
NMR, 4, 459-462.
Safo, M.K., Zhao, Q., Ko, T.P., Musayev, F.N., Robinson, H.,
Scarsdale, N., Wang, A.H. & Archer, G.L. (2005) Crystal structures of
the BlaI repressor from Staphylococcus aureus and its complex with DNA:
insights into transcriptional regulation of the bla and mec operons. J
Bacteriol, 187, 1833-1844.
Sancar, A., Lindsey-Boltz, L.A., Unsal-Kacmaz, K. & Linn, S.
(2004) Molecular mechanisms
of mammalian DNA repair and the DNA damage checkpoints. Annu
Rev Biochem, 73,
39-85.
Sattler, M., Schleucher, J. & Griesinger, C. (1999)
Heteronuclear multidimensional NMR experiments for the structure
determination of proteins in solution. Prog Nucl Mag Res Sp,
34, 93-158.
Savarin, P., Zinn-Justin, S. & Gilquin, B. (2001)
Variability in automated assignment of NOESY spectra and
three-dimensional structure determination: a test case on three small
disulfide-bonded proteins. J Biomol NMR, 19,
49-62.
Schultz, J., Milpetz, F., Bork, P. & Ponting, C.P.
(1998) SMART, a simple modular architecture research tool: identification
of signaling domains. Proc Natl Acad Sci U S
A, 95, 5857-5864.
Schwartz, T., Rould, M.A., Lowenhaupt, K., Herbert, A. &
Rich, A. (1999) Crystal structure
of the Zalpha domain of the human editing enzyme ADAR1 bound to
left-handed Z- DNA. Science, 284, 1841-1845.
Selenko, P., Sprangers, R., Stier, G., Buhler, D., Fischer, U.
& Sattler, M. (2001) SMN tudor domain structure and its interaction with
the Sm proteins. Nat Struct Biol, 8, 27-31.
Selmer, M. & Su, X.D. (2002) Crystal structure of an
mRNA-binding fragment of Moorella thermoacetica elongation factor SelB.
Embo J, 21, 4145-4153.
Singleton, M.R., Morales, R., Grainge, I., Cook, N.,
Isupov, M.N. & Wigley, D.B. (2004) Conformational changes induced by
nucleotide binding in Cdc6/ORC from Aeropyrum pernix. J Mol Biol,
343, 547-557.
Small, I., Peeters, N., Legeai, F. & Lurin, C. (2004)
Predotar: A tool for rapidly screening proteomes for N-terminal targeting
sequences. Proteomics, 4, 1581-1590.
Smith, L.J., Bolin, K.A., Schwalbe, H., MacArthur, M.W.,
Thornton, J.M. & Dobson, C.M. (1996) Analysis of main chain torsion angles
in proteins: prediction of NMR coupling constants for native and random coil
conformations. J Mol Biol, 255, 494-506.
Strecker, H.J. (1960) The interconversion of glutamic acid
and proline. II. The preparation and properties of delta
1-pyrroline-5-carboxylic acid. J Biol Chem, 235,
2045-2050.
Surber, M.W. & Maloy, S. (1999) Regulation of flavin
dehydrogenase compartmentalization: requirements for PutA-membrane
association in Salmonella typhimurium. Biochim Biophys Acta,
1421, 5-18.
Szostak, J.W., Orr-Weaver, T.L., Rothstein, R.J. &
Stahl, F.W. (1983) The double-strand- break repair model for recombination.
Cell, 33, 25-35.
Timchenko, T., Bailone, A. & Devoret, R. (1996) Btcd, a mouse
protein that binds to curved
DNA, can substitute in Escherichia coli for H-NS, a bacterial
nucleoid protein. Embo
J, 15, 3986-3992.
Tissier, A., Kannouche, P., Biard, D.S., Timchenko, T., Mazin,
A., Araneda, S., Allemand, I., Mauffrey, P., Frelat, G. & Angulo, J.F.
(1995) The mouse Kin-17 gene codes for a new protein involved in DNA
transactions and is akin to the bacterial RecA protein. Biochimie,
77, 854-860.
Tugarinov, V., Hwang, P.M. & Kay, L.E. (2004) Nuclear
magnetic resonance spectroscopy of high-molecular-weight proteins. Annu Rev
Biochem, 73, 107-146.
Tusnady, G.E. & Simon, I. (1998) Principles governing amino
acid composition of integral membrane proteins: application to topology
prediction. J Mol Biol, 283, 489-506.
Vallee, B.L. & Auld, D.S. (1995) Zinc metallochemistry in
biochemistry. Exs, 73, 259-277.
Van Melckebeke, H., Vreuls, C., Gans, P., Filée,
P., Llabres, G., Joris, B. & Simorre, J.P. (2003) Solution
structural study of BlaI: implications for the repression of genes
involved in â-Lactam antibiotic resistance. J Mol Biol,
333, 711-720.
Venyaminov, S.Y. & Yang, J.T. (1996) Circular Dichroism and
the Conformational Analysis
of Biomolecules. Plenum Press, New York,
1996.
Verbruggen, N., Hua, X.J., May, M. & Van Montagu,
M. (1996) Environmental and developmental signals modulate proline
homeostasis: evidence for a negative transcriptional regulator.
Proc Natl Acad Sci U S A, 93, 8787-8791.
Verlet, L. (1967) Computer experiments on classical fluids. Part
I. Phys Rev, 159, 98-103. Vincentelli, R.,
Bignon, C., Gruez, A., Canaan, S., Sulzenbacher, G., Tegoni,
M.,
Campanacci, V. & Cambillau, C. (2003) Medium-scale structural
genomics: strategies for protein expression and crystallization. Acc Chem
Res, 36, 165-172.
Vuister, G.W. & Bax, A. (1993) Quantitative J
correlation: a new approach for measuring homonuclear three-bond
J(HNHá) coupling constants in 15N-enriched proteins.
J Am Chem Soc, 115, 7772-7777.
Wakasugi, M. & Sancar, A. (1999) Order of assembly
of human DNA repair excision nuclease. J Biol Chem,
274, 18759-18768.
Wang, S.S. & Brandriss, M.C. (1986) Proline
utilization in Saccharomyces cerevisiae:
analysis of the cloned PUT1 gene. Mol Cell Biol,
6, 2638-2645.
Williams, D.C., Jr., Cai, M., Suh, J.Y., Peterkofsky, A. &
Clore, G.M. (2005) Solution NMR structure of the 48-kDa IIAMannose-HPr
complex of the Escherichia coli mannose phosphotransferase system. J
Biol Chem, 280, 20775-20784.
Williams, H.J., Williams, N., Spurlock, G., Norton, N.,
Zammit, S., Kirov, G., Owen, M.J. & O'Donovan, M.C. (2003) Detailed
analysis of PRODH and PsPRODH reveals no association with schizophrenia.
Am J Med Genet B Neuropsychiatr Genet, 120, 42-46.
Wishart, D.S. & Sykes, B.D. (1994) The 13C chemical-shift
index: a simple method for the identification of protein secondary structure
using 13C chemical-shift data. J Biomol NMR, 4,
171-180.
Wishart, D.S., Sykes, B.D. & Richards, F.M. (1992)
The chemical shift index: a fast and simple method for the assignment
of protein secondary structure through NMR spectroscopy.
Biochemistry, 31, 1647-1651.
Wojciak, J.M., Iwahara, J. & Clubb, R.T. (2001) The Mu
repressor-DNA complex contains an immobilized 'wing' within the minor groove.
Nat Struct Biol, 8, 84-90.
Wood, J.M. (1987) Membrane association of proline
dehydrogenase in Escherichia coli is redox dependent. Proc Natl Acad
Sci U S A, 84, 373-377.
Wüthrich, K. (1986). NMR of Proteins and Nucleic
Acids, New York, John Wiley & Sons.
Xu, R., Ayers, B., Cowburn, D. & Muir,
T.W. (1999) Chemical ligation of folded recombinant proteins:
segmental isotopic labeling of domains for NMR studies. Proc Natl Acad Sci
U S A, 96, 388-393.
Yang, J.T., Wu, C.S. & Martinez, H.M. (1986)
Calculation of protein conformation from
circular dichroism. Methods Enzymol,
130, 208-269.
Yeldandi, A.V., Rao, M.S. & Reddy, J.K. (2000)
Hydrogen peroxide generation in peroxisome proliferator-induced
oncogenesis. Mutat Res, 448, 159-177.
Yoshizawa, S., Rasubala, L., Ose, T., Kohda, D., Fourmy,
D. & Maenaka, K. (2005) Structural basis for mRNA recognition by
elongation factor SelB. Nat Struct Mol Biol,
12, 198-203.
Zheng, N., Fraenkel, E., Pabo, C.O. & Pavletich, N.P.
(1999) Structural basis of DNA
recognition by the heterodimeric cell cycle transcription factor
E2F-DP. Genes Dev,
13, 666-674.
Zhu, W. & Becker, D.F. (2003) Flavin redox state
triggers conformational changes in the
PutA protein from Escherichia coli. Biochemistry,
42, 5469-5477.
Zimmerman, D.E., Kulikowski, C.A., Huang, Y., Feng, W.,
Tashiro, M., Shimotakahara, S., Chien, C., Powers, R. & Montelione, G.T.
(1997) Automated analysis of protein NMR assignments using methods from
artificial intelligence. J Mol Biol, 269, 592-610.
Résumé
Le maintien de l'intégrité du patrimoine
génétique est essentiel à la survie des organismes
vivants. Face aux nombreuses sources de stress génotoxiques qui
induisent des dommages de l'ADN,
les cellules ont mis en place des mécanismes complexes
capables de détecter et réparer ces lésions. Parmi ces
sources, figurent les rayonnements ultraviolets (UV) contenus dans la
lumière solaire, qui modifient la structure de l'ADN et peuvent conduire
à l'introduction de mutations. Chez l'homme, la grande majorité
des dommages de l'ADN produits par les rayonnements est éliminée
par le système NER (Nucleotide Excision Repair), un
système capable d'exciser les nucléotides
lésés et de les remplacer. Une déficience de cette voie
de réparation peut mener à l'apoptose (mort programmée des
cellules), et augmente la susceptibilité de développer des
maladies graves telles que le cancer. Le système NER met en
oeuvre de nombreuses protéines impliquées dans des
mécanismes variés tels que
la détection, la signalisation, ou la
réparation de l'ADN. La protéine eucaryote KIN17,
récemment découverte dans le noyau de la cellule humaine,
semble appartenir à ce système de réparation.
Cependant, son rôle précis dans la réponse aux dommages de
l'ADN reste à ce jour inconnu. C'est pourquoi, nous avons entrepris une
caractérisation structurale et fonctionnelle de la région 51-160
de
la protéine KIN17 humaine (domaine K2) afin
d'améliorer la connaissance de ses fonctions, de ses partenaires
biologiques, et de ses modes de fonctionnement.
La première partie de ce manuscrit est consacrée
à la préparation de l'échantillon de protéine
en vue d'une analyse structurale par Résonance
Magnétique Nucléaire (RMN). Le travail a dans un premier temps
consisté à choisir et optimiser le système d'expression de
domaines structuraux d'une autre protéine : la proline
déshydrogénase PRODH.
Dans un second temps, la meilleure stratégie de
préparation de l'échantillon a été appliquée
à
la protéine KIN17 pour produire, puis
résoudre la structure tridimensionnelle du domaine K2 par RMN et
Modélisation Moléculaire. Nous avons montré que la
région 51-160 de la protéine KIN17 humaine adopte un repliement
caractéristique de la famille structurale « Winged Helix
» des protéines
de liaison aux acides nucléiques. Cependant,
l'analyse des détails structuraux du domaine K2, la comparaison
avec des protéines de fonction connue de la même classe
structurale, et des études électrophorétiques,
révèlent l'incapacité de ce domaine à lier l'ADN et
l'ARN de manière autonome.
En revanche, nous avons mis en évidence, par
une étude RMN complémentaire, l'existence d'une surface
ultra conservée impliquée dans des interactions de type
protéine-protéine intra-moléculaires entre le motif
« Winged Helix » de la région 51-160 et la
région N-terminale 1-50 de KIN17 humaine.
| 

