|
???ü?? L4IÂ?? J
??¾ü?? ???iÈ?? Þ?ß?

BADJI MOKHTAR- ANNABA ÄÂ ¾?û - J
¾ÈØ? ?о Äü?
¾Ð
UNIVERSITY
UNIVERSITE BADJI MOKHTAR Année 2007
ANNABA
Faculté des Sciences
Département de
Chimie
MEMOIRE
Présenté pour l'obtention du diplôme de
Magister
Option : Synthèse et Biocatalyse
Organique
THEME

|
|
|
|
|
|
|
|
|
|
|
SYNTHESE DES ISOXAZOLIDINES PAR
REACTION DIPOLAIRE-1 ,3 ENTRE
NITRONES ET OLEFINES
|
|
Par :
Mr Abdelmalek KHORIEF
NACEREDDINE
Devant le jury :
Président : Mr. F. Ismail M.C
Université de Annaba
Rapporteur Mr. A.Boukhari M.C Université
de Annaba
Examinateur : Mr. F. Halaimia M.C
Université de Annaba
Examinateur : Mr. A. Djilani M.C
Université de Annaba
Dédicaces
A mes parents, mes Frères, stxurs et ma
fiancée
A toute ma famille
Et à tous mes
amis
Je dédie ce travail.
Remerciements
Ce travail a été réalisé au
Laboratoire de Synthèse et Biocatalyse Organique(LSBO) du
Département de Chimie de l'Université de Annaba.
Je suis particulièrement heureux de
témoigner ma profonde gratitude à monsieur A. Boukhari
Maître de conférences à l'université de Annaba mon
directeur de mémoire, pour m'avoir suivi tout au long de mon
apprentissage au laboratoire, pour m'avoir fait découvrir la chimie des
composés hétérocycliques, pour son soutien et son aide
précieuse.
J'adresse mes très vifs remerciements à
Monsieur F. Ismail Maître de conférences au Département de
Génie de Procédés à l'Université de Annaba
de m'avoir fait l'honneur d'accepter la présidence du jury.
Je remercie vivement monsieur F. Halaimia Maître de
conférence à l'Université de Annaba,
qui me fait l'honneur d'examiner ce travail et de faire
partie du jury de ce mémoire.
Mes remerciements vont aussi à monsieur A. Djilani
Maître de conférence à l'Université
d'Annaba d'avoir bien voulu accepter de juger ce travail et
être membre du jury de ce mémoire. Que tous les membres de la
promotion biocatalyse et surtout du labo 8.
Mes remerciements vont également aux personnels
techniques et administratifs du département de chimie.
Je ne saurais oublier ma famille proche et mes amis pour
l'intérêt qu'ils ont toujours porter à mon travail.
Résumé :
Mots clé : nitrones, isoxazolidines,
cycloaddition dipolaire-1,3
Dans la première partie de ce mémoire nous avons
réalisé la synthèse des différents nitrones par
plusieurs procédés, et à partir de différents
aldéhydes aromatiques et aliphatiques pour l'étude de la
stabilité et la réactivité de ces nitrones en fonction de
leurs structures.
Dans la deuxième partie nous avons effectué la
synthèse de quelques isoxazolidines utilisant la réaction de
cycloaddition dipolaire-1,3, pour cela on a engagé la
a-phényl-N-méthylnitrone avec l'alcool allylique,
l'anhydride malièque et en fin le parafluorostyrène
respectivement. Ces réactions donnent un mélange de deux
diastérioisomères et deux régioisomères, le but est
l'étude de la sélectivité en fonction de la nature des
substituants portés par l'alcène.
Abstract:
Key words: nitrones, isoxazolidines, 1,
3-dipolar cycloaddition.
In the first part of this Work we have synthesis different
nitrones with many proceeds, starting from aliphatic and aromatic
aldéhydes. In order to study the reactivity and the stability of those
nitrones that is function of his structures.
In the second part we have synthesis some isoxazolidines using
the 1.3-dipolar cycloaddition, so we have engaged the
a-phényl-N-méthylnitrone with allylic alcohol, maleic
anhydride and parafluorurestyrène respectively. Those reactions give a
mixture of two diasterioisomers and two of régioisomers. The goal of our
work consist the study of the selectivity depending on the nature of the
substituants in the olefins.
|
:
|
ki2&101
|
|
. ykill 401:L.
|
-
|
3 . 1
|
":,Liil_N.11
|
rtithVI ,a1.49... 9..)41
'&1;10 :
|
411.111 c.i'LLLCII
|
,A41:11,11 3 Aultc,
`J. 1.1Ablii cid:I-N.A.' & A 1:01.11 3
cCulca &lc. JI.A..:A.,,' 14 `J*1.1..1)41:
&lc. 104.1&1. LIAi sJS:.11 63.4 4).4
Jill .,..)31 ci
. .144-.-.
3-1V.1.1 cL3.1-iiii:1-11:
1 b3A A71.1.1-zi 9 JU?È")
4...AJ
·11
Ialc.li 1413 dill 401t 3.1
.....1 :411-41 ci-.41-1
,L3* U1.4. 3..)-S3..):1.71
c.);...-1...L3411:u..1-1Ai
criM s.,11 cri
|
.4219:111 Lsic.
WA"9..)91..)14 U ...*1 9
4 41...).litil 9 4211.'1"
L39-N111 Q (:).9j:1-1. LL113
|
N- J:L.ii
|
-
|
a
|
LI ja, 4).4 A.134_N..411 4:JI21a,,,,All "
4..41.Z1V.1.1A411.iIIVI ;L,,I. j. 3..11 11%
4-)A ,_;41 ,4:J*14SL4.411
4)..3.14.1&. .1,..:1 4:Zt.clii:111 b3.4
All
Abréviations
Groupements chimiques et
molécules.
Aux : Auxiliaire chirale
Boc : Tert butoxycarbonyl [-CO2CMe3]
Bn : Benzyle [PhCH2]
DBU:
1,8-Diazabicyclo[5,4,0]undec-7-ène
Et: Ethyle
i-Pr: Isopropyle
m-CPBA: Acide métachloroperbenzoique
Me: méthyle
MTO: méthyltrioxorhenium
NBS: N-Bromosuccinimide NIS:
N-Iodosuccinimide Ph: Phényle
TFA: Acide trifluoroacétique
THF: Tétrahydrofurane
TM: Tamis moléculaire
TMS: Triméthylsilyle
Tf: Trifluorométhanesulfonyle
[-SO2CF3]
t-Bu: tertbutyle
TBS (TBDMS) : t-Butyldiméthylsilyle
Autres Abréviations
Arom : Aromatique Atm :
Atmosphère
: Angström
Cat : Catalytique
Cm : Centimètre
|
CCM : Chromatographie sur Couche Mince °C : Degré
Celsius
o : Déplacement chimique
A : Chauffage
eq : équivalents
ed : excès diasterioisomérique
g : gramme
GC : Chromatographie en phase Gazeuse
h : heures
Hz : Hertz
IR : Infra Rouge
IE : Impact Electronique
J : Constante de couplage en RMN
M : Masse molaire
min : minute
MHz : Méga Hertz
mg : milligramme
ml : millilitre
m/z : Unité de masse par charge élémentaire
Nu : Nucléophile
v : Nombres d'ondes
ppm : parties par million
Pf : Point de fusion
Rdt : Rendement
Rf : Rapport frontale
RMN : Résonance Magnétique et Nucléaire SM :
Spectroscopie de Masse
TA : Température Ambiante
tr: temps de rétention
|
SOMMAIRE
Pages
Introduction générale .1
Références .2
Partie I. Synthèse des nitrones
Chapitre I. Rappels bibliographique
Introduction 3
1.1. Apartir de l'hydroxylamine-N-substitué
1.1.1 Réaction de condensation
a. Sur un aldéhyde ...4
b. sur un composé carbonylé 5
1.1.2.
Réaction d'oxydation
a. Par l'hypochlorite de sodium (eau de javel) 5
b. Par l'oxyde de mercure 6
c. Par le dioxyde de manganèse .6
d. Par le chlorure de sulfinimidoyl en présence de DBU
7
e. Traitement par le TFA .7
1.2. A partir d'oximes
a. Thermolyse d'oxime .8
b. Condensation entre un oxime et un composé
carbonylé 8
c. Réaction avec un alcène .9
d. Alkylation des oximes 9
e. Alkylation des oximes aromatiques 10
f. a partir d'oxime et divenylsulfone 10
g. Réaction intramoléculaire d'oxime et un
alcène 11
1.3. A partir de composés nitrosés
a. Réaction de cétone avec le
1-chloronitrosocyclohéxane 11
b. Réaction avec un méthylène activé
.12
1.4. A partir de nitroarènes 13
1.5. Apartir d'amine secondaire 13
1.6. A partir d'imines 14
1.7. A partir des oxaziranes ..16
|
1. Synthèse de nitrones par la condensation entre un
aldéhyde et l'hydrochlorure de
|
|
|
la N-lméthylhydroxylamine
|
18
|
|
a. Réaction avec le propanaldéhyde
|
19
|
|
b. Réaction avec le hexanaldéhyde
|
20
|
c. Réaction avec le benzaldéhyde
|
20
|
d. Réaction avec le 4-nitrobenzaldéhyde
|
20
|
e. Réaction avec l'anisaldéhyde
|
|
.21
|
|
|
2. Synthèse de a,N-diphénylnitrone
|
22
|
|
3. Synthèse de
a-phényl-N-(4-hydroxy)phénylnitrone
|
23
|
|
Conclusion
|
...25
|
|
Partie Expérimentale
|
.26
|
|
Références
|
32
|
|
Annexe
|
.34
|
|
Partie II Synthèse des isoxazolidines
|
|
|
Chapitre I Rappels bibliographiques
|
|
|
Introduction
|
42
|
|
1. A partir de nitrones
|
|
|
1.a Réaction avec les alcènes
|
43
|
|
1.b Réaction avec l'énolate de lithium
|
43
|
|
1.c Réaction avec le réactif de réformasky
|
.44
|
|
1.d Réaction avec l'allyl triméthylsilane
|
45
|
|
1.d Réaction avec l'allyltributylstannane
|
45
|
|
1.e Réaction avec le silylénoléther
|
.46
|
|
1.f. Réaction avec triméthylsiloxy furane
|
47
|
|
2.A partir d'oxime et un alcène
|
.48
|
|
3.A partir de nitronate et un alcène
|
.49
|
4. Apartir d'hydroxylaminey-ö insaturée
|
50
|
5. A partir de nitroalcène et l'allylamine
|
50
|
6. Réaction de plusieurs composés
sumiltanèment
|
.52
|
7. A partir de cyclisation électrophile de
dérivées de O-homoallylhydroxylamine
|
|
53
|
|
Réaction avec l'alcool allylique
54
|
2. Réaction avec l'anhydride maléique
|
61
|
3. Réaction avec le para fluorostyrène
|
|
.62
|
|
|
Conclusion
|
64
|
|
Partie expérimentale
|
.65
|
|
Références
|
69
|
|
Annexe
|
.71
|
|
Conclusion générale
|
.77
|
Introduction Générale
|
INTRODUCTION GENERALE
La série hétérocyclique constitue un
vaste domaine de la chimie organique, approximativement deux
tiers des publications en chimie concernent de prés ou de loin ces
composés. Un très grand nombre de substances naturelles et par
conséquent de médicaments sont à base
d'hétérocycles.
Les hétéro atomes les plus courants sont
l'oxygène, l'azote et le soufre, et les cycles les plus stables sont,
comme dans le cas des hydrocarbures, ceux qui comportent cinq ou six
chaînons.
Nous nous sommes intéressés aux composés
hétérocycliques à la fois oxygénés et
azotés, en l'occurrence les 1,2-oxazolidines ou isoxazolidines. En effet
ces produits sont connus pour leur activité
anti-bactérienne1. En ce qui concerne leur
synthèse, notre choix s'est porté sur la réaction de
cycloaddition dipolaire-1,3 qui est une méthode extrêmement
efficace pour la création des structures hétérocycliques
complexes. Ce type de réaction qui met en jeu les
nitrones2,3,4 et les alcènes, aboutit aux
isoxazolidines (schéma).
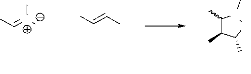
N O +
3
2
N
O
1
4
5
- Schéma -
Pour la réalisation de ce projet, nous avons
commencé par la préparation des nitrones qui englobe donc la
première partie de notre mémoire, à savoir, bibliographie,
résultats et discussion, et enfin étude expérimentale.
La deuxième partie est consacrée à la
synthèse des isoxazolidines, au développement de la
bibliographie, des résultats et discussion, ainsi qu'à
l'étude expérimentale.
Les deux parties sont dépendantes l'une de l'autre.
Nous terminerons notre travail par la conclusion générale. Afin
de confirmer nos résultats, et compléter notre étude, une
annexe est conçue, nous avons effectué des analyses spectrales
(RMN, IR, Masse ...) pour l'identification des différents produits
préparés.
|
Références:
[1] D.P.A Iannazzo,V.Pistara, A.Rescifina and R.Romeo.
Tetrahedron, 2002, 58, 581.
[2] R. Huisgen, Angew.Chem.Int.Ed.Engl,
1963, 2, 565.
[3] D.S.C. Blank, R.F. Crozier and V.C.Davis,
Synthesis, 1975, 205.
[4] K. Hensley, J..M. Carney, C.A. Stewart, T.Tabafabaie, Q.Pye
and R.A.Floyd, Int. Rev. Neurobiol, 1999, 40, 299.
|
Partie I
Synthèse des Nitrones
|
Chapitre I
Rappels Bibliographiques
|
Introduction
Les nitrones sont des intermédiaires
synthétiques importants qui ont servi d'une manière extensive en
chimie organique1,2,3,4. Quelques nitrones ont
été utilisées pour le piégeage et l'identification
des radicaux libres5,6, particulièrement
dans les études biologiques7. De nos jours,
les nitrones ne représentent pas seulement un outil largement
utilisé pour la détection des radicaux libres, mais aussi comme
agents chimiothérapiques prometteurs au niveau cérébral et
autres pathologies8. Elles peuvent réagir de
façon dipolaire 1,3 avec une grande variété de
dipôlarophiles pour donner différents produits. Une grande
application synthétique de nitrone est leur capacité
d'utilisation comme dipôle 1,3 dans les réactions de cycloaddition
dipolaire-1,3 avec les oléfines pour la préparation des
isoxazolidines9,10,11.
Le nom de nitrone est la contraction de nitrogène et
cétone. Ce terme a été proposé par
Pffeifer12 en 1916 pour les composés
possédant le groupe imine-N-oxide par analogie avec les cétones
(schéma 1). L'analogie se résume à l'effet
mésomère où ils sont prédominés dans les
deux classes de ces groupes faisant le groupe nitrone ou
azométhine-N-oxide, et considérer comme une extension de la
fonction carbonyle.
N

R
X

Y
X O
Y
O
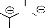
R
X
Y

O

N
X
Y
O

- Schéma 1 -
Nous présenterons tout d'abord un rappel
bibliographique sur les préparations des nitrones existant dans la
littérature et ensuite l'approche expérimentale dérivant
directement des résultats obtenus.
Nous avons envisagé de préparer des nitrones
à partir de la réaction de condensation entre des
aldéhydes aromatiques et aliphatiques et des hydroxylamines dans le but
de prévoir leurs stabilité et réactivité
réciproques. Nous avons également projeté d'étudier
l'effet électronique des groupements donneurs et accepteurs
d'électrons placés sur le cycle aromatique de l'aldéhyde,
qui
pourraient influer sur la stabilité des nitrones. Ces
composés sont synthétisés en vue de préparer des
isoxazolidines dont le travail sera développé dans la
deuxième partie du mémoire.
I- Rappels bibliographiques :
Nous notons que dans la littérature, les nitrones sont
généralement préparées à partir des
hydroxylamines-N-substituées. Ces dernières subissent
différentes réactions, que nous développerons ci-dessous.
Cependant, la synthèse de ces composés utilise aussi d'autres
méthodes plus intéressantes.
I. 1 A partir de l'hydroxylamine-N-substitué
:
I. 1.1 Réaction de condensation.
a. Sur un aldéhyde.
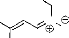
R
O RCH2NHOH,HCl
Ph
N O
Ph H NEt3,toluène, reflux, 1 h Ph
R=H, Rd =79% R=Ph, Rd =71%
Les nitrones sont facilement disponibles à partir de la
réaction entre les aldéhydes et les
hydroxylamines-N-substituées 13, 14,15. En effet, le
phénylcinnamaldéhyde réagit avec la N-méthyl et la
N-benzyl hydroxylamine (schéma 2) pour donner des produits stables
(stabilité qui est attribuée à son énergie de
résonance) et identifiables par spectroscopie de masse et par son
analyse élémentaire. Il faut noter que l'hydroxylamine est
utilisée sous forme de sel.
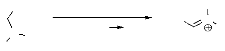
b. Sur un composé carbonylé.
On peut accéder aux nitrones en utilisant la
réaction de condensation entre un aldéhyde et la
N-Phénylhydroxylamine. La réaction s'effectue à reflux
d'éthanol et pendant un temps relativement court. Les rendements se
situent entre 50 et 90 % (schéma 3)16.

O
R O
H
+ PhNHOH
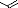
EtOH
reflux, 2h
R
H
N
Ph
50 - 90%
- Schéma 3 -
R
N
R1
OH
CH2Cl2 , 0°C
2-28h
NaClO(5%)
TA
R1
50-85%
N
R
O
1.1.2 Réactions d'oxydation.
a. Par l'hypochlorite de sodium (eau de javel).
Le traitement de l'hydroxylamine par l'agent oxydant, à
savoir l'hypochlorite de sodium (NaClO) à basse température
(0°C) et progressivement à température ambiante, aboutit
à la nitrone recherchée (schéma 4). Les produits obtenus
à la fin de la réaction sont purifiés par chromatographie
sur colonne et les rendements sont bons (50 -85 %) 17,18.
Par l'oxyde de mercure.
Plusieurs méthodes ont été
employées pour l'oxydation des N,N-dialkylhydroxylamines pour
obtenir les nitrones correspondants, et le plus couramment utilisé est
l'oxyde de mercure 19. C'est un oxydant doux et
sélectif de cette classe de composés, mais son utilisation d'une
manière quantitative, afin d'assurer une oxydation complète,
s'avère très toxique (schéma 5).

HgO
CH2Cl2, 2h
N OH
N
O

90%
- Schéma 5 -
c. Par le dioxyde de manganèse.
Cicchi et al20,21 ont
trouvé une méthode d'oxydation non toxique et efficace avec le
réactif de Bleach (MnO2). Elle est plutôt choisie à la
place de l'oxyde de mercure car l'oxyde de manganèse (Mn II) est connu
comme un oxydant pour la déshydrogénation des alcools et des
amines activées. Ce type de réaction donne des nitrones avec
d'excellents rendements (schéma 6).

R1
R2
N R2
O
CH2Cl2 , TA
MnO2 (1,5eq) 90%
R1 N
OH
85-95 %
d. Par le chlorure de Sulfinimidoyle en présence
de DBU
Les hydroxylamines N,N-disubstituées sont
généralement oxydées en leurs nitrones correspondantes
à très basse température (- 78 °C) par le chlorure de
sulfinimidoyle et le DBU dans dichlorométhane. Il est noté que
l'oxydation doit s'effectuer via l'intermédiaire qui sera ensuite
converti en nitrone après élimination du
N-tertiobutylphénylsulfenamide lors du transfert de proton via
l'état de transition cyclique à six chaînons (schéma
7)22.

Ph
OH S Ntbu
Cl
Ph N
DBU( 2,0 eq), CH2Cl2
-78C°, 15 min 97%

(1,5eq)
O
Ph N

tbu
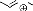
- Schéma 7 -
Le mécanisme d'obtention de la nitrone est montré
dans le schéma 8.
R

R'
N
H
R
N
OH
DBU O
N R'
S
Ph
S
Ph NH
tBu tBu
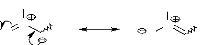
O
O
N R'
R
R
N R'
- Schéma 8 -
e. Traitement par le TFA :
Le traitement du
N,O-bis(tert-butoxycarbonyl)hydroxylamineglycinate d'éthyle
avec le TFA23,24, suivi par la condensation avec le
pyruvate donne les nitrones attendues (schéma 9).
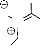
OBoc TFA , CH2Cl2 OHH TFA
EtO2C N H


N
EtO2C
Boc

Me
CH2Cl2 ,NaHCO3 ,MgSO4
O
N
COR

- Schéma 9 -

COR
EtO2C
85 -90%
Me O
1. 2. A partir d'oximes.
1.2. a- thermolyse d'oxime :
La thermolyse25 des oximes aboutit
aux nitrones correspondantes. La réaction s'effectue à reflux du
toluène et donne de bons rendements (schéma10).
PhCH3
NOH
°
reflux, 4A
N
H
O
N
N
81%
- Schéma 10 -
1.2. b-condensation entre une oxime et un composé
carbonylé :
Vassela et al26,27 ont
développé un autre type de nitrones, dérivées de
glucosides, en l'occurrence le D-mannose. Celui-ci, partiellement
protégé, réagit avec le formaldéhyde ou
l'acétone pour conduire aux nitrones recherchées (schéma
11).
O OH
O
O
R O
O
N
R
R
R= H, CH3
- Schéma 11 -
HO
O
N
O OH
O
O
R O
°
TM 4A
1. 2. c. Réaction avec un alcène
:
Les oximes réagissent rapidement avec les
alcènes quand elles sont traitées par l'iode ou le
N-bromo-succinimide, dans le dichlorométhane et à
température ambiante. Elles fournissent, via l'attaque de l'atome
d'azote sur l'intermédiaire ion halonium, des sels de nitrone
correspondantes. Les rendements faibles de ces réactions sont dus
à la stéréochimie de l'oxime (E/Z) en
général c'est l'isomère trans qui subit l'addition
(schéma 12)28,29.

I2, CH2Cl2
+ R N
OH
-5 TA
I
HO
R
I
N H
50%
- Schéma 12 -
1.2.d. Alkylation des oximes:
L'alkylation30,31 de ce type de
composés donne généralement des mélanges
d'oxime-ethers et nitrones. L'isomère formé et leur
prépondérance dans le mélange dépendent de la
nature de l'oxime, de l'agent alkylant et des conditions expérimentales.
Par conséquent cette méthode ne conduit pas à de bons
rendements (schéma 13).

R2 N
R1
R2 N
OH + R-X
R1
R
R1
N
R2
O
OR
+
N OH
+ S O toluène
O
reflux O
Ph
O
N
O
O
O
S
Ph
O
O
- Schéma 13 -
1.2. e.Alkylation des oximes aromatiques :
L'alkylation des aldoximes aromatiques aboutit à la
formation des nitrones correspondantes (schéma
14)32, tandis que l'alkylation des cétoximes
aromatiques donne un mélange dont le produit majoritaire est issu de
l'O-alkylation (éther oxime).

87%
- Schéma 14 -

+
Br
O
OEt
H
O Na
EtOH
N OH
O
O
N
O OEt
1.2. e. A partir d'oxime et du
divenylsulfone.
Frederickson et al33 ont
développé une nouvelle approche de synthèse de
N-alkenylnitrone. Celle-ci a été formée par
attaque de l'atome d'azote de l'oxime sur le divenylsulfone (schéma 15).
La réaction a lieu à reflux du toluène.

- Schéma 15 -
Aux O
O
N

Aux O O
O
1) NaN(SiMe3)2
2) Cl
NO
4) H3O
Aux =
NH
S O O
1.2. f. Réaction intramoléculaire d'oxime
et un alcène:
Grigg et al34 ont
préparé la nitrone cyclique par la réaction de cyclisation
intramoléculaire catalysé par PdCl2 de l'alkenyloxime avec un
rendement de 81 % (schéma 16)

N
OH
THF,reflux N O
Pd Cl2
81%
- Schéma 16 -
1.3. A partir de composés nitrosés :
1.3. a. Réaction de cétone et du
1-chloro-1-nitrosocyclohexane
Oppelzer et al35,36,37 ont
réussi à synthétiser la nitrone cyclique qui est un
intermédiaire pour la synthèse du (-)allosedamine, un agent
potentiel dans le traitement de SIDA ( syndrome immunodéficitaire
acquis). Ces auteurs ont utilisé comme catalyseur le composé
chiral le plus connu. La nitrone obtenue, après attaque de
l'énolate qui est généré par traitement basique sur
le 1-chloro-1- nitrosocyclohexane, est énantiomériquement pure.
L'addition de l'acide chlorhydrique conduit à l'hydroxylamine et
à l'aldéhyde dont la condensation entre eux donne la nitrone
cyclique (schéma 17).
NaN(SiMe3)2
Aux O O
Aux
O
Aux
O
O
N
H
OH
N
O
O O
O
O
O
H
Aux O O
H
H
O
Aux
Aux
O
O
NHOH O N
Aux O
N
O
O O
O
O O
N
H
H
H
Cl
O
Aux
Aux
O
N
OH
O H
N
O
O
Cl
O
O
O
Le mécanisme d'obtention de la nitrone est montré
dans le schéma 18.

- Schéma 18 -
1.3. b. A partir de méthylène activé
et de composé nitrosé :
Les composés qui possèdent un
méthylène activé, par un bon groupe partant, réagit
en présence d'une base avec les composés aromatiques
nitrosés, pour donner les nitrones Narylsubstituées. Ce
procédé est plus connu sous le nom de réaction de
Kröhnke (schéma 19)38.
NaOH 2N
N
Pyridine anhydre
N
ON
N
NH
N
N
N
N
N N H
+
O
N
N
94%

N
N
N
N
N NaOH HN N N
O
N
N
HN
O
N
N
N
N
N
N
N
NH
NH
ON
N
N
N
N
- Schéma 20 -
1.4. A partir de nitroarénes:
La réaction des aldéhydes avec les
nitroarénes s'effectue en milieu aqueux et en présence de l'agent
réducteur le zinc. Elle se déroule en deux étapes
simultanément, à savoir, la réduction de la fonction nitro
en hydroxylamine et l'attaque de cette dernière sur l'aldéhyde.
Nous obtenons la nitrone correspondante avec des rendements
acceptables39 (schéma 21).

O
O O O NH4Cl ,Zn
+ N
Ar
N
H
R
Ar
R
H
EtOH , H2O 16 h
12 - 65%
- Schéma 21-
1.5. A partir d'amine secondaire :
a. L'oxydation d'amine secondaire utilisant le
complexe (MTO/Urée, H2O) 40 a montré
son efficacité et sa non-toxicité pour la préparation des
nitrones a partir des amines secondaires.
Les rendements sont excellents (schéma 22).
R
R1 N
H
CH3ReO3 (0.5%)

urée, H2O2,CH3OH, 2.5h
R
R1 N
O

50 - 91%
- Schéma 22 -
b. L'oxydation des produits organiques,
telles que les amines, par l'oxygène41 en
présence du perchlorate de 5-éthyl-3-méthyllumiflavinium
comme catalyseur et l'hydrazine monohydraté conduit aux nitrones avec de
très bons rendements (schéma 23).

O2
(1 atm)
CF3CH2OH
, 60°C, 3h
flavin 5mol
1.1eq hydrazine/eau
NH
N
O
85 %
flavin:
N
N
O
O
ClO
N
N
- Schéma 23 -
1.6. A partir d'imines.
a. Larson et al42
ont développé une méthode de synthèse des nitrones
cyciques à partir des imines correspondantes. Ces composés ont
une activité anti-stroke (endommagement cellulaire) potentiel. La
procédure consiste en la réduction de l'imine en amine avec le
borohydrure de sodium, suivie de l'oxydation par l'eau
oxygénée. Le résultat de la réaction est la
formation de la nitrone cyclique (schéma 24)
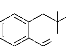
R1
R2
N
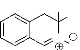
R1
N
R2
O
1)NaBH4
2) H2O2 , Cat NaWO4
- Schéma 24 -
b. Le couple urée/eau
oxygéné43 est stable peu
coûteux et facilement manipulable. Ce réactif est utilisé
dans l'oxydation efficace des différentes molécules organiques
(schéma 25).
c.
87%
-schéma 25-
N
85°C, 25min
N O
O
H2N NH2
, HOOH
L'oxydation par l'acide
méta-chloroperbenzoïque44 de la
8-hydroxyquinolèine, en présence du 1,2dibromoéthane et du
dichlorométhane, en phase fluorée (C6F14) fournit une
nitrone stable (schéma 26).
CH2Cl2, C6F14
BrCH2CH2Br
1,5eq. m-CPBA, TA, 72h
N
O
OH
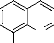
N
OH
- Schéma 26 -
d. La N-oxydation, des imines
aromatiques, donne directement la fonction nitrone. La réaction a lieu
généralement par l'intermédiaire de peracides
(schéma 27)45.


O
N
H
oxone
KHCO3
(CH3)2CO ou CH3CN
2H2O
O
O
N
H
97%
Oxone : 2KHSO5.KHSO4.K2SO4
- Schéma 27 -
e. Oxydation par perborate de sodium.
Le Perborate de sodium est un réactif très efficace
pour l'oxydation des imines conjugués en leur N-Oxydes
(schéma 30)46.
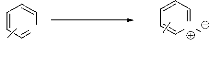
60-70%
1.1eq SPB
CH3CO2H 40°C, 3-4 h
N
R O
N
R
- Schéma 30 - Les résultats obtenus sont
illustrés dans le tableau suivant. Tableau
|
Produits
|
O
N
|
O
N
HO
|
N O
NC
|
|
Rdt (%)
|
75
|
73
|
60
|
1.1.7. A partir des oxaziranes:
La réaction entre les imines et l'eau
oxygénée a été employée pour la
synthèse de grandes variétés de dérivés de
3-aryloxazirane. Le réarrangement thermique des dérivés
de3-aryloxazirane donne les nitrones correspondantes avec des rendements qui
varient entre 50 et 100 %. L'isomérisation thermique des
oxaziranes47,48 autres que les 3-aryloxaziranes ne
donnent pas des nitrones, mais d'autres produits, généralement
des amides (schéma 31).

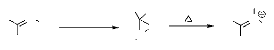
R N
Ar
R1 H2O2
R Ar
O
N
R1
O
R
Ar
N
R1
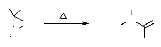
R
R
R R N O
R1
N O
R1
- Schéma 31-
Résultats
Comme nous l'avons mentionné dans la première
partie, en ce qui concerne la préparation des nitrones, les
méthodes de condensation entre les composés carbonylés et
les hydroxylamine-Nsubstituées sont les plus souvent
employées pour la synthèse de ces
produits13,14,15.
Nous avons pu préparer quelques nitrones à
partir de différents composés de départ tels que les
aldéhydes (aliphatiques, et aromatiques para substitués, et non
substitués) et les hydroxylaminesN- Substituées. En
plus, nous avons utilisé différents procédés de
synthèse en tenant compte de la structure des produits de
départ.
Les hydroxylamines utilisées sont les suivantes :
CH3NHOH , HCl PhNHOH
1 2
Nous avons également synthétisé une nitrone
9 à partir de la condensation entre le parahydroxynitro
benzène et le benzaldéhyde.
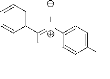
O
N
H
OH
9
1. Synthèse de nitrones à partir de la
condensation entre un aldéhyde et l'hydrochlorure
de-N-méthylhydroxylamine.
Cette réaction nous a permis d'aboutir aux
différentes nitrones aliphatiques et aromatiques par des manipulations
relativement simples. En effet, nous procédons à l'addition d'une
base en l'occurrence la triméthylamine, qui a pour rôle de
libérer l'hydroxylamine, dans le but de former le sel correspondant, que
nous séparons ensuite par filtration sous vide. Les molécules
d'eau générées au cours de la réaction ont
été éliminées par le Dean Stark pour éviter
la réaction inverse.
a. Réaction avec le propanaldéhyde.
La réaction de l'hydrochlorure de
N-méthylhydroxylamine 1 avec le
propanaldéhyde à reflux du toluène pendant une trois
heures du temps donne la nitrone 3 (schéma 1). Le
rendement est estimé à 71 % (d'après le brut), ce produit
est instable et décompose par le contacte avec des solvants polaires ou
portiques, nous pensons que ce ci est dû à l'absence de
conjugaison. Donc on a pas pu l'identifié avec les méthodes
spectroscopiques usuelles.

H3CH2C
H
O + 1
NEt3

toluène,reflux 3h
H3CH2C
H
N
+ HNEt3,Cl + H2O
CH3
3
- Schéma 1-
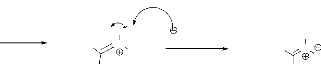
Le mécanisme réactionnel s'effectue selon le
schéma suivant :

CH3NHOH,HCl + NEt3 CH3NHOH + HNEt3 ,Cl
O
H3CH2C H
H H
- Schéma 2 -
H
O
OH
CH3
-H2O
H3CH2C N CH3
H3CH2C N O
CH3NHOH O
H3CH2C N
H
OH H3CH2C N
CH3
OH
CH3
Le doublet de l'atome d'azote, de l'hydroxylamine,
formée à partir de l'hydrochlorure d'hydroxylamine de
départ, attaque le carbone du carbonyle. Après élimination
d'une molécule d'eau, nous obtenons la nitrone 3
(schéma 2).
b. Réaction avec le n-hexanaldéhyde.
La réaction du n-hexanaldehyde avec l'hydroxylamine
1 fournit la nitrone 4 avec un rendement de
52% (schéma 3). Ce produit est également instable pour les
mêmes raisons que le composé 3.

O
H3CH2CH2CH2CH2C
O + 1
NEt3
toluène,reflux 3h
H3CH2CH2CH2CH2C
N
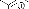
CH3
H
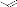
H
4
- Schéma 3 -
c. Réaction avec le benzaldéhyde.
La réaction entre l'hydroxylamine 1 et
le benzaldéhyde se déroule selon le même
procédé. Nous obtenons la nitrone 5 avec un bon
rendement 67% (schéma 4). Ce produit est purifié par
recristallisation dans l'éther diéthylique. Celui-ci a
été identifié grâce à ses
caractéristiques spectrales (IR, RMN
1H et SM).
O H
NEt3
+ 1
toluène,reflux 2h
O
CH3
N
5
H
- Schéma 4 -
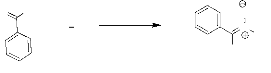
d. Réaction avec le 4-nitrobenzaldéhyde.
L'action de l'hydroxylamine 1 sur le
4-nitrobenzaldéhyde à reflux du toluène, et pendant 12
heures a permis d'isoler la nitrone 6 (schéma 5). Le
rendement est 12% après purification par chromatographie sur colonne de
silice, en utilisant l'acétate d'éthyle et le chlorure de
méthylène (8/2) comme éluant.
O H
NO2
+ 1
O
12h
H
6
N
toluène,reflux
CH3
O2N
NEt3
- Schéma 5 -
e. Réaction avec l'anisaldéhyde.
Cette réaction a été conduite
après deux heures de temps pour aboutir à la formation de la
nitrone 7 (schéma 6), qui a été
isolée par chromatographie sur colonne de silice (éluant :
l'acétate d'éthyle / chlorure de méthylène
(1/1).
- Schéma 6 -
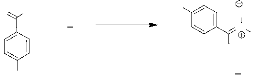
NEt3
+ 1
toluène,reflux 2h
O H
OMe
MeO
O
CH3
N
7
H
La spectroscopie infrarouge confirme tout à fait nos
résultats. En effet, les spectres montrent clairement la présence
de bandes d'absorption à (1600-1700 cm-1) qui
représentent ce type de composés (vibration de valence de la
fonction nitrone). En plus, la bande caractéristique de vibration de
valence de la liaison N-O apparaît aux environs de 1000-1200
cm-1.
La spectroscopie RMN 1H montre la présence
d'un pic caractéristique du proton oléfinique (HC =
N) qui appartient aux nitrones issues de la condensation avec les
aldéhydes. Celui-ci apparaît dans la région des champs
faibles, et ceci est dû à l'effet attracteur de l'atome
d'oxygène et de la conjugaison avec le noyau aromatique. Dans le cas de
la a-phényl-N-méthyl-nitrone 5, ce
proton est couplé avec les hydrogènes du groupement
méthyle porté par l'atome d'azote.
La spectroscopie de masse confirme la structure des
composés préparés grâce à l'apparition des
pics d'ions moléculaires. Pour
l'a-phényl-N-méthyl-nitrone 5 le pic
moléculaire apparaît à m/z= 135 avec une abondance de 70
%.
Le chromatogramme indique la pureté de ces produits ainsi
que l'existence de l'isomérie trans et cis à haute
température, pour le composé 5, les deux pics
apparaissent à 10.90 et 10.96 min.
2. Synthèse de l'a, N-diphénylnitrone 8
à partir de la N-phénylhydroxylamine 2 et du
benzaldéhyde.
La a,N-diphénylnitrone 8 a
été synthétisée en utilisant la condensation du
benzaldehyde16 avec
l'hydroxylamine 2. Cette dernière a été
préparée au niveau de notre laboratoire à partir du
benzène comme produit de départ, selon l'enchaînement
réactionnel décrit dans le schéma 7. La réaction de
substitution électrophile, sur le noyau benzénique, a
été réalisée en prenant de multiples
précautions du fait de la toxicité du benzène et de
l'emploi des acides forts. Le nitrobenzène obtenu a été
ensuite réduit en N-phénylhydroxylamine
2. Le rendement obtenu après recristallisation est de
67 %.
2
8
- Schéma 7 -

HNO3 H2SO4
NO2
Zn / NH4Cl
H2O
NHOH O
EtOH,TA,24h
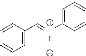
N
O
La condensation de l'hydroxylamine 2 avec le
benzaldéhyde conduit finalement à la formation de la nitrone
8 avec un rendement satisfaisant (76%).
Le mécanisme réactionnel de la condensation est
détaillé dans le schéma suivant :
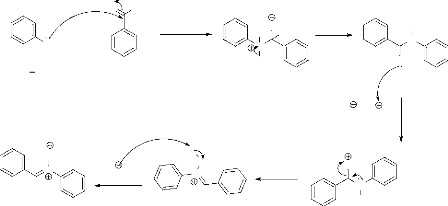
O H
EtOH
OH
N OH
NHOH
2
EtO
, H
H
O
O
N
EtO
N
O
OH H
N
H
-H2O
OH2
N OH
8
- Schéma 8 -
La spectroscopie IR a permis de justifier la présence des
bandes caractéristiques de la nitrone, à savoir 1570
cm-1 qui est la bande de vibration de valence de C=N et 1210 celle
de N-O.
Le spectre RMN 1H de la nitrone 8
montre que le pic du proton, de la fonction imine, apparaît sous
forme d'un singulier à 7,90 ppm, ce qui confirme bien la structure de
notre produit.
En spectroscopie de masse ce produit est
caractérisé par l'apparition du pic d'ion moléculaire
à m/z =197.
3. Synthèse de
a-phényl-N-(4-hydroxy)-phényl-nitrone 9 par réaction entre
le benzaldéhyde et le para-hydroxynitrobenzène :
Cette réaction nous a permis d'obtenir des structures
très complexes de nitrones39 à partir
de produits commerciaux simples. Nous avons pu réaliser la
synthèse de la a-phényl-N-4- hydroxyphénylnitrone
9 en utilisant le benzaldéhyde et le
4-hydroxynitrobenzène. Cette nitrone
possède un substituant hydroxyle situé en
position para dans le noyau benzénique porté par l'atome d'azote.
C'est par conséquent une réaction clé pour la
préparation de ce genre de composés, qui est la condensation
entre les nitroarènes et les composés
carbonylés39, donc c'est la raison pour
laquelle nous avons employé cette méthode pour l'obtention de la
nitrone 9. La 4- hydroxyphénylhydroxylamine est
préparée, à partir de la réduction du
4-hydroxynitrobenzène, in situ, que nous faisons,ensuite,
réagir avec le benzaldéhyde pour aboutir à la nitrone
attendue 9 (Schéma 9).
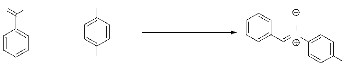
NH4Cl, EtOH
N
H2O,Zn,16h, 0 - 25C°
O
OH
+
O H
NO2
OH
9
- Schéma 9 -
Le produit 9 a été
identifié par spectroscopie infrarouge. L'apparition de la bande
d'absorption caractéristique de OH à 3300 cm-1, et les
autres bandes caractéristiques de la fonction nitrone (1680
cm-1 et 1100 cm-1) confirment d'une manière claire
la structure du produit 9.
CONCLUSION
La condensation entre les aldéhydes aliphatiques et
aromatiques para substitués, en l'occurrence le benzaldéhyde, le
paranitrobenzaldéhyde, anisaldéhyde et propanaldéhyde,
nous fournit les nitrones correspondantes avec des rendements variés en
fonction de la structure de ces derniers et aussi de par la nature des
groupements portés par le noyau benzénique de
l'aldéhyde.
Les nitrones préparées à partir des
aldéhydes aliphatiques sont instables et se décomposent en leurs
produits de départ, pendant la purification ou au contact avec des
solvants polaires comme l'éthanol. Tandis que les nitrones aromatiques
sont généralement plus stables et cristallisent dans
l'éthanol ou l'éther diéthylique.
L'introduction des substituants donneurs d'électrons
tel que le groupement méthoxy, joue un rôle important dans la
formation des nitrones correspondantes, où l'on obtient de bons
rendements (de l'ordre de 80%). Par contre les substituants attracteurs
d'électrons, comme le groupement nitro, défavorisent la formation
de nitrones, et l'on constate l'obtention de rendements faibles (environ
12%).
Les nitrones aromatiques porteurs du groupement aryles sur
l'atome d'azote sont plus stables et ceci est dû à la forte
conjugaison qui existe dans la molécule.
TECHNIQUES GENERALES
Sauf indication contraire, les analyses ont été
conduites dans les conditions générales indiquées
cidessous.
Les spectres IR ont été enregistrés sur
le spectrophotomètre infrarouge SHIMADZU- FTIR 8400S, avec des cellules
en chlorure de sodium à épaisseur fixe pour les solutions
liquides en film. Pour les solides, les pastilles sont préparées
(2-4mg) de l'échantillon dans 200-225mg de KBr. La position des bandes
caractéristiques est donnée en cm-1, les lettres F, m,
et f placés après ces derniers signifient respectivement forte,
moyenne, faible.
Les spectres de masse ont été enregistrés
avec un spectromètre de masse couplé à un appareil de
chromatographie en phase gazeuse équipé d'une colonne capillaire
de polarité moyenne de type (25m FS-OV-1701-CB-0.25 CS-25292-82). On
utilise l'ionisation par impact électronique (70ev), les
intensités relatives sont indiquées entre parenthèses, le
chiffre 100 est attribué au pic de base.
Les spectres de Résonance Magnétique
Nucléaire du proton et de carbone 13 (RMN 1H et RMN
13C) ont été enregistrés sur un appareil BRUKER
AC 200 à 250 MHz. Les spectres ont été effectués
dans le CDCl3. Les déplacements chimiques (ö) sont exprimés
en partie par million (ppm) par rapport au tétraméthylsilane
(TMS) pris comme référence interne. Pour la description des
signaux, nous utiliserons les abréviations suivantes:
S : singulet, d : doublet, t : triplet, q : quadruplet, m :
multiplet,
M: massif, dd : doublet dédoublé.
Les points de fusion (Pf) ont été
déterminés en tube capillaire au moyen de l'appareil BUCHI
electrothermal 9100.
Chromatographie :
Les chromatographies sur couche mince (ccm) ont
été réalisées sur des plaques de gel de silice sur
aluminium 60 F 254 SDS, les ccm sont observées en lumière
ultraviolette à 254 nm ou trempées dans un
révélateur d'iode (SiO2 +I2).
Les chromatographies sur colonne ont été
effectuées sur gel de silice SDS.
Solvants et réactifs :
Le benzène et le toluène sont distillés et
stockés sur tamis moléculaire 4 Å.
Les réactifs ont généralement
été utilisés sans purification
supplémentaire.
I. Synthèse de nitrones par réaction de
condensation entre un composé carbonylé et
l'hydrochlorure de
N-méthyl-hydroxyl-amine.
1. Mode opératoire général
:
Dans un bicol de 250 ml et sous agitation magnétique,
on introduit une solution d'aldéhyde dans le toluène ou le
benzène anhydre. On additionne de l'hydrochlorure de
Nméthylhydroxylamine (CH3NHOH, HCl), dissous dans les
mêmes solvants, puis on ajoute goutte à goutte à l'aide
d'une ampoule à brome la triéthylamine (NEt3). Le mélange
réactionnel est porté à reflux du solvant. On se sert du
Dean Stark pour évacuer les molécules d'eaux
générées au cours de la réaction. Le reflux est
d'une durée déterminée pour chaque cas (l'évolution
de la réaction peut être suivie par des tests de chromatographie
sur couche mince). Une fois la réaction terminée, le ballon est
plongé dans un bain de glace, on filtre sous vide le
précipité, puis on évapore le solvant sous pression
réduite. Le produit brut, ainsi obtenu, est purifié par
recristallisation ou par chromatographie sur colonne.
2.
Détail des expériences :
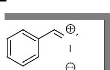
a-phényl-N-methylnitrone 5
Me
N
O
C8H9NO
M =135 g/mole
La réaction est réalisée sur 1.06 g
(0.01mol) de benzaldéhyde dissous dans 20ml de toluène et 0.835 g
(0.01mol) de 1 en solution dans 20ml du toluène et de 1.4 ml (0.01 mol)
de triéthylamine. Temps de réaction 2 heures à reflux du
solvant. La purification par recristallisation permet d'isoler 900 mg
(0.0066mol, 67%) de 5.
Rdt =67%
Aspect : cristaux (recristallise dans
l'éther diéthylique)
Rf: 0.27 (Et2O/ CH2Cl2)
Pf : 80 - 81°C
tr: 10.90 min (80°C
+10°C/min)
IR (KBr) : 3055 (m) VC-H, 2993 (m) VC-H, 1589
(f) VC=N, 1558 (f) v C=C, 1080 (m) v N-C, 1296 (m)
VNO.
SM: [M.] +=135 (70), 134
(100), 118(20), 107(25), 89 (30), 77(50), 65(30), 44(73).
RMN 1H (CDCl3): ö = 3.75 (s, 3H,
CH3); 7.28-7.38 (m, 5H,C6H5), 8.15 (q, 1H, J=3.22 Hz, CH=N).
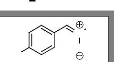
a-(4-nitro)-phényl-N-méthyl-nitrone
6
Me
N
O
O2N
C8H8N2O3
M =180 g/mole
La réaction est effectuée à partir de
1.51 g (0.01 mol) de 4-nitrobenzaldéhyde en solution dans 20 ml de
toluène et de 0.835 g (0.01 mol) de 1 dissous dans 20 ml de
toluène et de 1.4 ml de triéthylamine en solution dans 20 ml de
toluène. Après purification par chromatographie sur colonne de
silice (10 g), nous avons isolé 200 mg (0.0011 mol, 11 %) de 6.
Rd =11 %
Aspect : poudre rouge brique
Rf : 0.48 (Et2O/ CH2Cl2 (1/1)). Pf : 199 -200
°C
IR (KBr) 2900 (m) VC-H ; 1680 (f) VC=N ; 1600
(m) VC=C ; 1350 (f) vC-N ; 1250 (m) VN-O 3400 (L) due à la
présence d'une quantité d'eau dans la pastille de KBr.
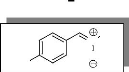
a-(4-méthoxy)-phényl-N-méthylnitrone
7
Me
N
O
MeO
C9H11NO2
M =165 g/mole
2.27 g (0.02 mol ) d'anisaldéhyde
(4-méthoxybenzaldéhyde) est dissous dans 30 ml du benzène,
on additionne 1.67 g (0.02 mol) de 1 en solution de 30 ml de benzène,
puis on ajoute, goutte à goutte, 2.8 ml (0.02 mol) de
triéthylamine dans 20 ml du même solvant. Après
purification du brut par chromatographie sur colonne on obtient 2.40 g (0.0145
mol, 73%) de la nitrone 7.
Rd =73 %
Aspect: huile jaune
Rf: 0.20 (Et2O/ CH2Cl2).
Tr: 9.71min (80C°
+10C°/min)
IR (CH2Cl2): 2900 (m) VC-H ; 1750 (m) VC=N ;
1600 (f) VC=C ; 1420 (m) VC-N ; 1280 (m) VN-O .
a,N-diphénylnitrone 8
C13H11NO
M =197 g/mole
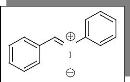
N
O
Mode opératoire :
Dans un bicol de 250ml, on introduit 0,33 g (0,30 mmol) de la
N-phénylhydroxylamine 2 que l'on dissous dans
30 ml d'éthanol absolue, à une température comprise entre
40 - 60 °C, puis on ajoute 0,30 ml (2,94 mmol) de benzaldéhyde
anhydre. On agite à température ambiante pendant 24 heures.
Après concentration du mélange, la recristallisation dans
l'éthanol donne 0.45 g (0.22 mmol, 76%) de 8.
Rd =76 %
Aspect : cristaux (recristallise dans
l'éthanol) Rf: 0.30 (Et2O/ CH2Cl2, 8/2).
tr: 27min
(80C°+10C°/min)
Pf : 112-113C°
IR (KBr) : 2990 (f) VC-Harom ; 1570 (m) VC=N ;
1550 (m) VC=C ; 1470 (m) VC-N ; 1400 (m) VN-O .
SM: [M.] + =197 (20); 182(5); 169(20); 141(15);
115(10); 105(20); 91(25); 77(100); 65(30); 51(40); 41(05).
RMN 1H(CDCl3) : 7.38-7.68 (m, 4H) ;
7.70-7.77 (m, 2H); 7.90(s, 1H); 8.20 (d, 1H, J=2.9Hz); 8.33 (d, 1H, J=2.38) ;
8.37-8.50 (m, 2H).
Mode opératoire de la réaction de
nitration du benzène :
Dans un bicol de 100 ml équipé d'un barreau
magnétique, d'une ampoule à brome et d'un
réfrigérant, on introduit 20 ml d'acide nitrique concentré
et 10 ml d'acide sulfurique concentré. On laisse refroidir à
l'aide d'un bain de glace fondante et on fait tomber goutte à goutte
dans l'ampoule à brome, 20ml de benzène anhydre. Il faut
régler le goutte à goutte de telle sorte que la couleur brune,
crée par une goutte, disparaisse avant d'ajouter la goutte suivante.
Lorsque l'addition est complète, on agite encore
pendant 10 minutes avant de transférer le mélange
réactionnel dans une ampoule à décanter et
récupérer la phase organique. On la reprend dans 20ml
d'éther et on sèche sur du sulfate de magnésium.
L'évaporation du solvant sous pression réduite donne 15 ml de
nitrobenzène.
Mode opératoire de la réduction du
nitrobenzène en N-phénylhydroxylamine :
Dans un bicol de 250 ml on introduit 2,32 g (0,043 mol) de
chlorure d'ammonium, 50ml d'eau distillée et 47,24 g (39,31ml, 0,31 mol)
de nitrobenzène. Après agitation du mélange on additionne
lentement 5,46 g (0,083 mol) de zinc en solution dans 20 ml d'eau
distillée, pendant 15 minutes. Après augmentation de la
température jusqu'à 60 - 65 °C, on agite encore le
mélange jusqu'à l'abaissement de la température qui
signale la fin de la réduction. On filtre le mélange
réactionnel pour enlever l'oxyde de zinc (patte blanche), puis on lave
le filtrat avec 50 ml d'eau distillée. On transvase le filtrat dans un
Becher de 200 ml qu'on sature par du sel (NaCl). Le refroidissement dans un
bain de glace pendant une heure du temps donne le maximum de recristallisation
de l'hydroxylamine attendue. Cette dernière contient un petite
quantité de sel, la recristallisation dans un solvant organique tel que
l'éther donne 0.67 g (0,61 mol, 51%) d'hydroxylamine
2.
Synthèse de a-phényl-N-(4-hydroxy)
phénylnitrone 9.
C13H11NO2
M=213 g/mole
N
O
OH
Rd = 83%
Aspect : cristaux (recristallise après
chromatographie)
Rf: 0.46 (Et2O/CH2Cl2 1/1)
Pf: 170 °C
IR (KBr): 3350(L) vO-H ; 3000 (f) VC-Harom ;
1680 (m) VC=N ; 1580 (m) VC=C ; 1320 (m) vC-N ; 1100 (m) VN-O
Mode opératoire :
Dans un bicol de 100ml on introduit 190 mg (1.36 mmole) de
para-hydroxynitrobenzène dissous dans 2.6 ml d'éthanol, puis on
ajoute 90.4 mg de NH4Cl dissous dans 3 ml d'eau distillée. Après
agitation, on additionne 138 mg (1.30 mmol) de benzaldéhyde. Le
mélange est refroidi jusqu'à 0°C, puis on ajoute goutte
à goutte 170 mg (0.21 mmol) de Zn en solution dans 100 ml
d'éthanol. On agite pendant 12 heures. Après filtration sous
vide, le filtrat est lavé par deux fois 20 ml de dichlorométhane.
La phase organique est ensuite séchée sur sulfate de sodium
(Na2SO4). Après avoir filtré, la solution est alors
évaporée sous vide. Le produit brut obtenu est purifié par
chromatographie sur colonne de silice (10 g). On recueille 242 mg (1.136 mmol,
83%) de produit 9 sous forme de cristaux.
Bibliographie
|
|
Références :
[1] S. Cicchi, M. Corsi and A. Goti,
J. Org. Chem, 64, 7243
(1999).
[2] R.W. Murray and M. Singh,
J.Org. Chem, 55, 2954,
(1990).
[3] W. W. Zajac. J., T. R. Walters and M. G. Darey,
J.Org. Chem, 53, 5856
(1988).
[4] S-I. Murahashi, T. Shiota and Y. Imada, Org. synth,
70, 265 (1991).
[5] R. H. D. Barton and J. M. Beaton, J. Am. Chem. Soc,
82, 2641 (1960).
[6] M, Iwamura,.; S. Futibe; Matukura, T.; Sano, M. Chem.
Lett. 1983, 1023.
[7] Thomas. L.F., Marc-Bowen. D., Janowick.A, J. Med.
Chem. 1996, 39, 4988-4996.
[8] K. Hensley, J.M. Carney, C.A. Stewart, T.Tabafabaie, Q.Pye
and R.A.Floyd, Int. Rev. Neurobiol, 40, 299
(1999).
[9] P. C. Dennis, Advances in cycloaddition, Vol 3,
1993.
[10] Tufariello, J. J.In 1,3-Dipolar Cycloaddition
Chemestry ; Padwa, A., Ed.;WileyInterscience; New-York,
1984; pp 83-168.
[11] Balasubramanian, N.; Org. Prep.Proced. Int.
1985, 17, 23.
[12] P. Pfeiffer, Annalen, 1916, 411,
72.
[13] P. W. Grounwater, M. N. Yerges, I. Fejes, D. E. Hibbs, D.
Bendell, R. J. Anderson, A.
Mc Killop, T. Sharif, W. Zhanga, ARKIVOC,
2000, 1, 684-697.
[14] P. N. Confalone and E. M. Huie, Org. React.
1988, 36, 1.
[15] K. B. G. Nitrile oxides, Nitrones and Nitronates in
Organic Synthesis, H. Feuer. Ed.,
VCH Publishers, New-York, 1988.
[16] Sandler. S. R.; Karo. W. Organic Functionnal
GroupPreparation 2nd ed, Academic press,
San Diego, Vol 3, pp 351-376 (1989).
[17] Döpp, D.; Döpp, H. In Houben-Weyl Methoden
der Organishen Chemie, Vol. E14 b/ part.
2; Klamann, D.; Hagemann, H.; Eds.; Gearg Thieme Verlag :
Stuttgart, 1990.
[18] Cicchi, S. Corsi. M,
Goti. A. J.Org. Chem
.1999, 64, 7243-7245.
[19] Goti. A.; Desarlo. F.; Rmani. M. Tetrahedron Lett
1994, 35, 6571-6574.
[20] Paquette, L. A.; Heidelbaugh, T.
M.Org. Synth.
1996, 73, 44 -49.
[21] Cicchi, S.;Marradi, Gotti, A.; Brandi. A. Tetrahedron
Letters. 42 (2001) 6503-6505.
[22] J. Matsuo, T. Shibata,
H. Kitagawa. Archi.
Org. Chem. 2001. 58.
[23] Chiacchio, U.; Piperno, A.; Rescifina, A.; Romeo, G.;
Uccella, N. Tetrahedron 1998,
54, 5695.
[24] Dondoni, A.; Merino, P.; Franco, S.; Merchan, F.; Tejero,
T.; Junquero, F. Synth. Commun. 1994, 24, 2573.
|
[25] Hassner, A.; Singh, S.; Sharma, R.; Maurya, R.
Tetrahedron, 1993, 49, 2317-2324.
[26] Vassela, A. Helv. Chim.Acta 1977,
60, 1273.
[27] Huber, R.; Vassella, A. Tetrahedron
1990, 46, 33
[28] Dondas, H.A.; Grigg, R.;Hadjisoteriou, H.;Markandu, J,
Kennwell. P.; Thomton-Pett, M. Tetrahedron 2001, 57,
1119-1128.
[29] Dondas, H.A.; Grigg, R.;Hadjisoteriou, H.;Markandu, J.;
Thomas, W. A.; Kennwell. P.Tetrahedron 2000, 56,
10087-10096
[30] Brady. O. L.; Goldsteur. R.F. J. Chem. Soc.
1926, 2403
[31] Brady. O. L.; Chokshi. N. M. J. Chem.
Soc.1929, 2271.
[32] Miki, T.; Goto.; Kawakita, K.; Okutani, T. Chem. Pharm.
Bull., 1986, 34, 3202.
[33] Frederickson, M.; Grigg, R.; Rankovic, Z.; Thornthon-Pett,
M.; Redpath, J.; Corssley, R. Tetrahedron 1995, 51,
6835.
[34] Frederickson, M.; Grigg, R.; Redpath, J.; Thornthon-Pett,
M. Tetrahedron 1994, 50, 5495.
[35] Oppelzer, W.; Deerberg, J.; Tamura, O. Helv. Chim. Acta
1994, 34, 3939.
[36] Kim, B. H.; Curron, D. P. Tetrahedron
1993, 43, 1969.
[37] Oppelzer, W. Tetrahedron 1987,
43, 1969.
[38] Giner-Sonolla, A.; Zimmerman, I.; Bendich, A.J. Am.
Chem. Pharm. Soc., 1959, 81, 2515.
[39] West, P. R.; Davis, G. C.
J. Org. Chem.
1989, 54, 5176-5180.
[40] Goti. A.; Cardona,
G. Soldaini. Org.
synth, Vol. 81, p 204 (2005).
[41] Y. Imada, H. Lida, S. Ono, S. I. Murahaschi, J.Am.
Chem.Soc, 2003, 125, 2868-2869.
[42] Larsen, R. D.; Reamer, R. A.; Carley, E. G.
J. Org. Chem.
1991. 56. 6034-6038.
[43] R. S. Varma, K. P. Naicker, Org. Lett,
1999, 1, 189-191.
[44] N. K. Jana, J.G. Verkade, Org. Lett.,
2003, 5, 3787-3790.
[45] Hajipur, A. R.; Pyne, S. G. J. Chem. Res. Synop,
1992, 11, 388.
[46] A. Mc Killop, D. Kemp, Tetrahedron,
1989, 45, 3299-3306.
[47] Emmons, W.D., J. Am. Chem. Soc., 79, 5739
(1957).
[48] Hawthorne, M. F., and Strahm, R. D.,
J. Org. Chem., 22, 1263
(1957).
|
Annexe
|

g tt-evai
2 .221 I
H2 2048_0 2044.01 040.39 2136..0 2.1134 .24 2 0V2. 23 213.68
1847.ti6 160.06 103'9.4
----..., 14131.16
183b.S3 1833,Ba 1033.2b li30.5 le3C.A0 1423.0
W..5-,a 3821.7E

s-6-3
PS
g».24
1 964
934.24



co
1,11
...1210.1.99 2104,35 2101.86
_7-'49#3.146 1949,95 1944.09 1141.24 1939.02
111111
-----1991.06
1912.99 1971.21 1870.68 1965,03 1961.24 1965.70 1E64.54
1E62.31 1659.98 1923.2?

L
27.73 10.15 10.64 10.26 9,91 5.54 1.70 .6.01
4.36
41,2,7QEa1WW22Z FARM-2MR2NtilgMFAVW Agq
pRk i
Y8 P,
-
9
Et2:::!2.414 r
aiXi xi o 2 g AiWit UU
tv1N
I I I Iliiillii 1 'I i . . .. . . ,
II
6 oP XI 0.1-PocTs
TIT I rrin PI
r...)
r
ji \Ufs $ i
01
1
1.4 boat
osu
-1)46, 11
I
it
if
r
(1\
u
1,..
.. .
,..:
,1
,
Partie II
Synthèse des Isoxazolidines
|
Chapitre I
Rappels Bibliographiques
|
Introduction
Les isoxazolidines sont des hétérocycles
à cinq chaînons saturés et dont les hétéro
atomes sont l'oxygène et l'azote qui sont situés en position
adjacente. Ce sont des intermédiaires synthétiques très
importants pour la synthèse d'une grande variété de
produits naturels et de molécules à intérêt
biologique, en particulier les alcaloïdes, les aminoacides, les
aminosucres, et les f3-lactames1,2,3. Il y a un
intérêt croissant dans la synthèse d'analogues
nucléosides et leur incorporation dans les séquences ADN, pour la
recherche de nouveaux agents anti-viraux et pour l'étude des
interactions ligand ADN et protéine-ADN4,5.
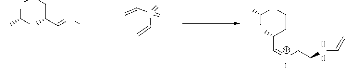
Récemment, certains auteurs6,7,8
ont été intéressés par la synthèse
de N,O-psiconucléosides, une classe d'analogues
nucléosides, dans laquelle une unité de sucre a été
changé en un système isoxazolidinique (figure 1).
HO
H
N
OH
Me O
Me
H CH2OTBDPS
N
Me O
CO2Et
B
N
O
B :bases azotées
- Figure 1-
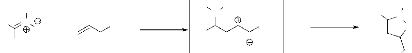
Les cycloadditions dipolaires-1,3 entre les nitrones et les
alcènes sont les plus utilisées et les plus commodes pour la
préparation de dérivés isoxazolidiniques, lesquelles sont
facilement converties en 1,3-aminoalcool sous les conditions douces de
réduction, avec rétention de configuration des centres chiraux
(figure 2).
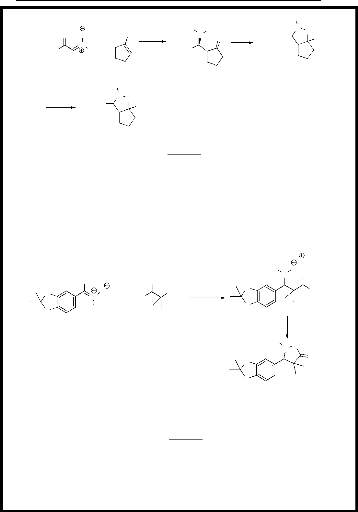
N O réduction
NHR OH
- Figure2 -
La plupart des réactions de réduction avec
ouverture de cycle sont des hydrogénations catalysées par le
palladium ou le nickel de Raney, ou le traitement des isoxazolidines par le
zinc et l'acide, mais une variété d'autres méthodes sont
aussi accessibles9,10. Aussi, avant de
présenter nos résultats, nous développerons
dans les paragraphes suivants les principales études décrites
dans la littérature.
I. Rappel bibliographique.
1. A partir de nitrones.
1-a. Réaction avec les alcènes
Les nitrones possèdent une structure dipolaire -1,3
peut être exploitée dans les réactions de cycloaddition du
même type11. La réaction de
cycloadditoion dipolaire-1,3 des nitrones avec les alcènes engendrant
les isoxazolidines, (schéma 1) est une réaction fondamentale en
chimie organique. La littérature y est largement disponible.
Balasubramanian2 a rassemblé, les travaux
effectués sur les isoxazolidines obtenues par réaction de
cycloaddition pour la synthèse des produits naturels.
Frederickson3 a mené la synthèse des
isoxazolidines optiquement actifs 3. En effet, dans cette
réaction trois centres asymétriques peuvent être
crées. La stéréochimie relative du centre C4 et C5 sont
toujours contrôlés par les substituants porté par
l'alcène.
Toluène reflux, 3h
N
O
+
N
O
- Schéma 1-
1. b. Réaction avec l'énolate de
lithium.
L'énolate de cyclopentanone lithium
généré in situ à partir de tri méthyle
silyle enol éther correspondant, en présence de méthyle
lithium, a été additionné à la nitrone. Nous
obtenons un produit qui va être soumis à une cyclisation
intramoléculaire d'une manière spontanée pour donner le
produit recherché à savoir l'isoxazolidine après
confirmation par le piégeage du produit par un agent silylant (le
chlorure de triméthylsilyle) (schéma 2)12.
Me
Me
1. c. Réactif de reformasky .
TMSCl
MeO2C
OTMS
N
O
- Schéma 2 -
L'addition du réactif de reformasky sur une nitrone,
suivie par une lactonisation spontanée, conduit directement au produit
qui est l'isoxazolidin-5-one (schéma
3)13.
ZnBr
Me
O
N
O
- Schéma 3 -
Me OLi NO
MeO2C OLi
MeO2C
OLi N O
O
+
N
Me
O
MeO
O
ZnBr
O
OEt
N
O
O
O
O
Rd = 52%
Me
H
O
+
EtO
O
N
Me
O
O
1. d.
80 - 90 %
- Schéma 5 -
Me OTMS
TMSOTf N
+ Sn(Bu)3
O
R
H
N R1
45-85%
R1 I
NIS N O
R
Réaction avec l'allyl trimétyl
silane.
Selon Wuts et Jung14 l'addition
d'allyltriméthylsilane sur les nitrones cycliques conjuguées, en
présence de TMSOTf donne l'hydroxylamine O-silylée
homoallylique (schéma 4). Tandis qu'avec les nitrones acycliques non
conjugues il se forme le
5-silylméthylisoxazolidine15. Cette
réaction passe par l'intermédiaire carbenium dont la durée
de vie est courte.
TMS TMSOTf
+
Me OTMS N
R
TMS
TfO
Me
R
N
O
Me
R
H
N O
TMS
R = tBu
48%
Me OTMS N
R
R = Ph
- Schéma 4 -
1. e. Réaction avec l'allyltributyl stannane
.
Le passage de l' allyltriméthylsilanne à
l'allyltributylstannane qui est beaucoup plus réactif, donne avec
succès le O-silyl hydroxylamine avec de bons rendements, et en
présence de TMSOTf. A partir de ces résultats ces
auteurs16 ont pu développer la
synthèse des 5- iodométhyl-isoxazolidines par simple
piégeage du mélange réactionnel avec le
Niodosuccinimide (NIS) (schéma 5) en une seule étape.
1.f. Réaction avec le silylènol
éther.
l'addition des silyle énol éthers
d'acétaldéhyde, d'acétone et d'acétophénone
sur des nitrones en présence de TMSOTf (1équi.) à 0°
C donne après 24 heures les 5- silyloxyisoxazolidines correspondants
avec de très bons rendements17,18.
L'utilisation d'une quantité catalytique de TMSOTf est possible, mais la
lenteur de la réaction nécessite une bonne conversion, car sous
ces conditions une partie du produit subit une élimination du silanol en
2,3- dihydro-isoxazole. Il a été prouvé que la
réaction procéde via la formation d'un ion
Osilyloxyiminium, selon le mécanisme décrit dans le
schéma 6
(1)
(2)
R2
OSi
TfO
R
H
OTMS
N R1
TfO
R1 OTMS
N
R
OSi
R2
O
R
H
N R1
TMSOTf
TfO
TfO
TMS
R1 O
OSi
R1
O
H2O N
R2
N O
R2
Si
N O
R1
TMS
OSi
R2
R
R
R
75-95%
- Schéma 6 - Les résultats obtenus sont
illustrés dans le tableau I
Tableau I
|
Nitrone
|
R
|
R1
|
R2
|
Si
|
Isoxazolidine
|
|
|
|
|
|
Rd(%)
cis/trans
|
|
1
|
Et
|
Bn
|
H
|
TBS
|
92 70/30
|
|
2
|
iPr
|
Bn
|
H
|
TBS
|
95 38/62
|
|
3
|
Et
|
Bn
|
Me
|
TMS
|
91 35/65
|
|
4
|
iPr
|
Bn
|
Ph
|
TMS
|
74 55/45
|
Puisque l'équilibre entre les ions oxoniums (1) et (2)
subsiste, Dhavale et al18 ont
développé un procédé, où, dans un premier
temps le TMSOTf provoque la réaction entre le silyl énol
éther et la nitrone, ensuite l'addition, à haute
température, du nucléophile silylé (cyanure de
triméthylsilyle ou allyltriméthylsilane) sur l'acétal pour
conduire finalement à l'isoxazolidine (schéma 7).
O
OTBS
60°C
TMSOTf -10°C
Ph
H
N
Me
Nu -TMS
Me
N O
Nu
Ph
Nu =allyl, Rd =44% Nu =CN, Rd =53%
- Schéma 7 -
1. g. Réaction avec le triméthyl
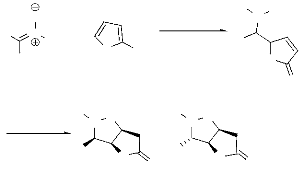
silyloxy-furane.
La réaction du 2-tri méthyl-silyloxy-furane avec
les nitrones qui est un cas particulier du silylcetène-acétal
conjugué, procure une voie complètement
régéosélective. En outre, le produit de cette
réaction qui est formé en premier lieu, souvent cyclisé
intramoléculaire il se forme le tetrahydrofuro[2.3.0]isoxazol-5(2H)one,
au contact du gel de silice pendant l'étape de purification par
chromatographie. Le butenolide peut être forcé à la
cyclisation en isoxazolidine par utilisation du fluorure de
tétrabutylammonium (Bu4NF) 19.
La stéréochimie de cette réaction est
contrôlée par la nature des deux substituants a situés sur
la nitrone. Quand le substituant placé en a est un cycle aromatique nous
obtenons principalement un produit syn, tandis qu'avec l'utilisation des
a-alkyl-N-benzylnitrones, le produit majoritaire est l'anti
(schéma 8)20.
O
SiO2 ou
Bu4NF
R1
N
O
TMSOTf
+
R
H
R
O OTMS
OTMS
O
N R1
R1
N O
O O
R
syn
R1
N O
R
O anti
O
-Schéma 8-
Les résultats obtenus sont illustrés dans le
tableau II
|
Nitrone
|
R
|
R1
|
Anti
(%)
|
Syn
(%)
|
Anti /
syn
|
|
1
|
Ph
|
Me
|
15
|
81
|
16/84
|
|
2
|
Ph
|
2-
|
3
|
81
|
4/96
|
|
3
|
iPr
|
Thienyl
|
70
|
10
|
88/12
|
|
|
Bn
|
|
|
|
-Tableau II-
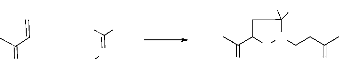
2. A partir d'oxime et d'alcène.
Un travail de recherche
bibliographique21,22 a révélé
que lorsque le formaldoxime réagit avec l'acrylate de méthyle,
nous obtenons un mélange d'isoxazolidines dont l'une (la
N-substituée) est majoritaire par rapport à l'autre. Par
contre la réaction de la benzophénone oxime avec la
méthyle vinyle cétone donne l'isoxazolidine
N-substituée (schéma 9). Ces produits sont formés
à partir de l'addition de Michaël de l'oxime sur l'oléfine
consécutive à la réaction dipolaire-1,3.
Ce mécanisme est corroboré par le chauffage
à 80 - 90°C, pendant quelques heures et l'utilisation de deux moles
d'acrylate de méthyle, ou acrylonitrile ou méthyle vinyle
cétone et une mole d'oxime. Il faut noter que l'emploi du
1-hexène n'aboutit à aucune réaction
23.
H3COOC
+ N
HO
H3COOC O
COOCH3 +
H3COOC O

N



NH
majoritaire minoritaire
Ph
HO O
+
Ph
N
O
Ph
Ph
O N
O
-Schéma 9 -
3. A partir de nitronate et d'alcène.
Les alkyl et silyl nitronates ou les N-alkoxy et
N-silyloxy nitrones peuvent réagir avec les alcènes par
réaction de cycloaddition dipolaire-1,3 pour obtenir la
N-alkoxy/Nsilyloxyisoxazolidine (schéma
10)24. Les produits obtenus à partir de
nitronates acycliques qui possèdent un proton en position 3 de
l'isoxazolidine, sont facilement transformés en 2- isoxazoline par
élimination du groupe alkoxy ou silyloxy selon un traitement acide ou
par simple chauffage.
OR
OR
N O
+
N
O
N
O
R= silyl, alkyl isoxazolidine 2-isoxazoline
- Schéma 10 -
4.
A partir d'hydroxylamine y-ö
insaturée.
Les hydroxylamines y-ö insaturées ont
été synthétisées à partir de l'addition du
réactif de grignard allylique sur une nitrone. L'avantage de ces
composés est leur utilisation dans les réactions
d'iodocyclisation des dérivés
O-silylés25,26. Ces derniers
peuvent être transformés en 5-iodométhylisoxazolidine dont
l'ouverture, par un traitement acide, est possible pour aboutir à la
chaîne ouverte que est la 1,3-amino alcool (schéma
11)27.
5.
R1
TMSO R1
N
HO R1
N
Nu
N
I-I
TMSCl
R
O
I
R
R
R1
N
O
H
NH
OH
R
R1
Nu
R
Nu
- Schéma 11 -
A partir de nitroalcène et
d'allylamine.
a. Hassner et
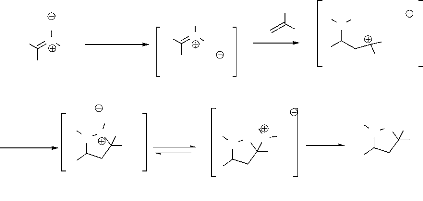
al28,29 ont découvert la réaction de
cycloaddition intramoléculaire de silylnitonate obtenu à partir
de l'addition de Michael de l'allylamine sur le nitroalcène, suivie par
le piégeage au moyen du chlorure de tri méthyle silane (TMSCl)
(schéma 13). La nitronate de silyle obtenue, réagit de
façon dipolaire-1,3 et intramoléculairement pour former
l'isoxazolidine correspondante, et en un seul
diastéréoisomère (schéma 12).
O
H
R1
R2 H
N
N
OSiMe3
-Schéma 12-
TMSCl
H
R1
N
N
+
NO2
R2
NEt3
OSiMe3
R1 N
O
R2
R1
R2
N
H
NO2
ON O
MeO2C Me
H
H
OAux
- Schéma 13 -
Me
CO2Me
+ OAux
H
Me
O O
N
OAux
TiCl2
O O
N
CO2Me
b. Denmark et
al30 ont pris un exemple typique, à savoir
la réaction catalysée par un acide de Lewis, entre
l'alcène possédant un auxiliaire chiral et un nitroalcène
selon un mécanisme d'hétéro Diels-Alder. Il se forme
l'intermédiaire nitronate de cycloalkyl chiral. Celui-ci donne,
après réaction de cycloaddition dipolaire-1,3, le produit
isoxazolidinique (schéma 13)31.
6. Réaction de plusieurs réactifs
simultanément.
Récemment l'hydroxylamine chirale a été
introduite comme un bon réactif pour la réaction de cycloaddition
dipolaire-1,3. Celle-ci conduit après action avec un mélange
d'aldéhyde et d'alcool allylique, à l'isoxazolidine
correspondante, avec un bon rendement32. La
diastéréosélectivité a été grandement
avantagée par l'utilisation de MgBr2 anhydre en engendrant le complexe
magnésium portant l'alcool allylique et la nitrone formée in situ
grâce à la coordination des atomes d'oxygène (schéma
14).
OH
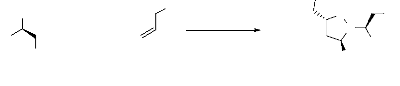
OH
+ RCHO +
OH
NHOH
Ph
MgBr2/iPrOH
THF
OH
O
N
Ph
90%
R
-Schéma 14-
Le mécanisme réactionnel de l'obtention de
l'isoxazolidine dans le schéma 15.
Br2
Mg
OH
NHOH
Ph
OH
R O
H
+
MgBr2
HO
O
N
Ph
H
OH
O
N
Ph
R
OH
Ph
-Schéma 15-
7. A partir de la cycisation électrophile de
dérivés O-homoallyl-hydroxylamine.
Les cyclisations électrophiles, dérivées
de différents O-homoallyl hydroxylamine, ont été
étudiées. En effet les O-homoallyl hydroxylamines non
protégées subissent une cyclisation pour donner des isoxazolines.
Cette nouvelle synthèse permet une cyclisation électrophilique
pour donner la formation des isoxazolidines.
La cyclisation peut être conduite par différents
agents électrophiles (tBuOCl, PhSeBr, NBS et NIS). L'oxydation
ne peut pas avoir lieu si les O-homoallyl hydroxylamines de
départ sont Sulfonées. La cyclisation électrophile fournit
des isoxazolidines-N-sulfonées avec une sélectivité
cis élevée (plus de 7:1). La cyclisation
électrophile d'O-homoallyl hydroxylamines
N-acylées donne les isoxazolines ou les isoxazolidines selon
les conditions de réaction (réactifs). La cyclisation en
milieu t-BuOCl aboutit aux isoxazolines via la cyclisation oxydante,
tandis que le NIS permet d'obtenir le produit de cyclisation 5-exo avec une
haute stéréo sélectivité (cis : trans. =13 :1)
(schéma 16)33.
O
O
O N Cl
Ph
tBuOCl
O NH
NIS
O N
I
Ph
Ph
69% 82%
e.d =12.5:1
-Schéma 16-
Résultats
La littérature nous rapporte que dans la
majorité des réactions 1.3-dipolaires entre les nitrones et les
oléfines nous avons la formation d'un mélange de
régioisomères et de diastérioisomères
(schéma 1), Nous avons opté pour la synthèse des
isoxazolidines en utilisant la réaction de cycloaddition dipolaire-1,3
entre l'a-phényl-N-méthylnitrone 1,
préparée à partir de la réaction de condensation
entre le benzaldéhyde et la CH3NHOH,HCl ,avec les alcènes
2, 3, et 4 (figure 1).
O
OH
F
H
O O O
N CH3
1 2 3 4
- Figure 1 -
Le but de ce travail est d'étudier la
réactivité, la régiosélectivité et la
diastéréosélectivité de cette réaction en
fonction de la nature des substituants portés par l'alcène
utilisé.
R1 H3C
N
benzène
reflux
R2
R2
O
R1
H3C
N
O
R2
+
R1
H
O
N
+
CH3
- Schéma 1 -
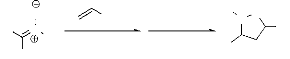
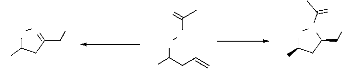
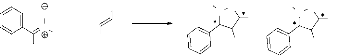
a. Réaction avec l'alcool allylique 2.
L'alcool allylique 2 est un alcène
monosubstitué portant le groupement hydroxyle en position allylique, ce
qui entraîne un effet inductif attracteur, par conséquent
l'alcène devient polarisée. Nous avons obtenu deux
régioisomères 5 et 6, dont
chaque régioisomère contient deux diastérioisomères
(5a, 5b) et
(6a, 6b). L'obtention du
régioisomère 5 est expliquée par l'effet
stérique du groupement phényle porté par la nitrone
1 et le groupement hydroxyle de l'alcène
2. En effet, le rapprochement de l'alcène et de la
nitrone s'effectue par l'état de plus basse énergie (minimisation
des interactions des substituants). Les diastérioisomères sont
obtenus en tenant
compte de l'isomérie de la nitrone (schéma 3).
Ces produits ont le même Rf ce qui n'a pas permis leur séparation
par chromatographie sur colonne. On a pu identifier ces produits uniquement par
RMN H1 et RMN C13.
La réaction s'effectue selon le schéma
réactionnel suivant :
5a, 5b 5a,
5b
O
*
+
1
2
*
benzène
reflux, 24h
H3C
N
HO
H3C
N
O*
*
+
OH
- Schéma 2 -
3b
- Schéma 3 -
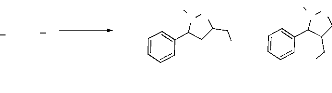
P
P
P
P
H
Hb H
OH
Ha
Hb H
OH
Ha
CH3 N O
H
N O
CH3
H3C
H3C
3a
trans
N O
H HaHb
N O
cis
H HaHb
H
H
OH
OH
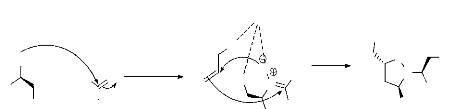
Le mécanisme d'obtention de ces produits est le suivant
:
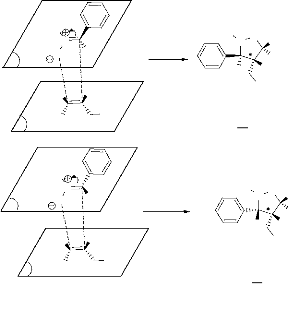
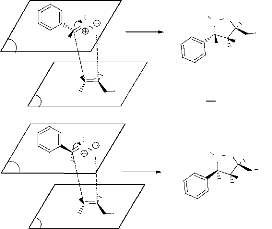
H3C
N
O
O H
N
*
P
Hb
Ha
H3C
H
O
Hb
Ha
P
H3C
N
N
*
O H
OH
trans
4a
P
Hb H
OH
Ha
H3C
H
cis
4b
H
OH
P
Hb Ha
H
OH
- Schéma 4 -
H
Le spectre infra rouge montre :
- une bande à 3371 cm-1, due à la
vibration de valence de la liaison O-H - la bande de vibration de valence de la
liaison C=Carom à 1604 cm-1
- la bande de vibration de valence de C-O à 1275
cm-1 - la bande de vibration de valence de C-N à 1180
cm-1
- et la bande de vibration de valence de N-O à 1033
cm-1.
Dans l'analyse par spectroscopie de masse, le spectre de masse
n'indique pas la présence du pic de l'ion moléculaire mais des
fragments d'ions qui confirment la structure de la molécule.

Le schéma suivant montre l'explication de quelques
fragments de régioisomère 5 et
6.
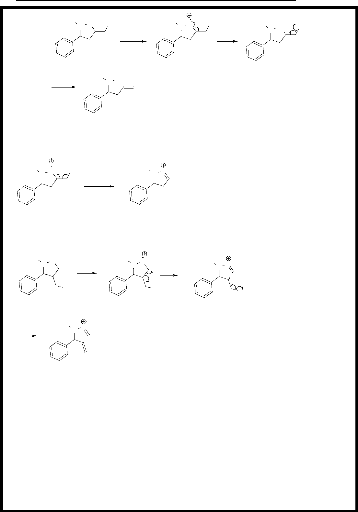
- .OH
H3C
N O
m/z =176
H3C N O OH
H3C N O OH
H3C N O OH
-e
m/z =162
- CH2OH
H3C
N O
H3C N O OH
OH
OH
H3C N O
-e
H3C
N O
H3C
N O
m/z = 176
H3C
N O
OH
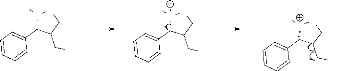
H3CN O
-é
H3C
N O
OH
OH
H3C
N O
OH

H3C
N O
m/z =162
- Schéma 5 -
L'analyse par spectroscopie RMN H1
précise d'une manière évidente, l'existence de deux
régioisomères 5 et 6. Quant
à l'identification de chaque diastérioisomère
(5a, 5b, 6a et
6b) par la RMN 1H,
ceci s'est avéré très difficile, ce qui a
nécessité l'emploi d'autres méthodes spectroscopiques plus
développées*.
a. le Régioisomère 5 :
S,2.5
dd,7.28
dd,7.28
d,7.25
H
H
d,7.2
H
H
dd,4.4
H3C m,4.2
N O H OH
H Hdd,3.6
Hdd,3.7
d,7.2
H
H
Hm,1.12
m, 1.2
S, 2.12
5
Les protons des groupements méthylènes sur le C4
voient leurs signaux apparaître à 1.12 et 1.20 ppm sous la forme
de multiplet vers la région des champs forts. Le proton situé sur
le carbone C5 résonne sous forme de multiplet à 4.2 ppm. Ce
phénomène de déblindage est du à l'effet attracteur
des deux atomes d'oxygène voisins. Le signal du proton placé sur
le carbone 3 apparaît sous la forme de doublet de doublets à 4.4
ppm. Ceci est dû à la présence, sur le carbone voisin, de
l'atome d'azote. Les deux protons de CH2OH apparaissent
à 3.6 et 3.7 ppm, chacun sous la forme
de doublet de doublets et le proton de l'hydroxyle
résonne à 2.12 ppm (singulier). Les hydrogènes du
groupement méthyle ont leur signal à 2.5 ppm sous la forme d'un
singulier. Les protons du groupement phényle apparaissent comme suit
:
- Les protons en position ortho à 7.2 ppm.
- Les protons en position méta à 7.28 ppm.
- Les protons en position para à 7.25 ppm.
b. le Régioisomère 6 :
dd,7.22
S,2.5
H3C
H
d,7.40
dd,2.6
H H
2 dd,2.7
dd,2.7
H
N O
d,7.4
dd,3.5
H
H
Hdd.2.6
H
OH
S,2.12
H
m,2.
H
dd,7.22
H
dd,7
6
Les protons du groupe CH2OH et celles
situés sur C5 sont équivalents et résonnent à 2.6
et 2.7ppm sous la forme d'un doublet de doublets. Le proton en C4 voit son
signal apparaître à 2.2ppm. Le pic du proton, appartenant au C3,
apparaît à 3.5ppm sous la forme d'un doublet de doublet, le proton
de OH à 2.12ppm et les protons de CH3 à 2.5 ppm sous la forme
d'un singulier. Les protons du phényle apparaissent comme suit :
- Les protons en ortho à 7.40 ppm
- Les protons en méta à 7.22 ppm
- Les protons en para à 7.00 ppm.
L'analyse par spectroscopie de RMN 13C
indique aussi l'existence de ces régioisomères 5
et
6.
a.
Le Régioisomère 5 :
44.5
H3C
N O
65
129
128
138
60
74
35
OH
127
127
129
5
Le carbone C3 résonne à 60 ppm du à
l'effet de l'atome d'azote, et ce ci aussi pour C5 et CH2OH,
le C4 résonne à champs fort (plus loin par rapport aux autres
carbones) ; le CH3 résonne à 44.5ppm. Les
carbones du groupement phényle résonnent entre 127 et 129ppm, le
carbone 1résonne à champ faible (138ppm).
b. Le Régioisomère 6 :
44.5
H3C
N O
127
40
135
74
126
OH
124
76
127
6
77
124
Le carbone en position 3 résonne à champ fort
(pas d'effet électronique d'héteroatomes) les deux carbones
CH2OHet C5 résonnent dans le même champ (ils sont
presque équivalents) ; les carbones du noyau aromatique résonnent
entre 124 et 127ppm.
2. Réaction avec l'anhydride maléique
3
La réaction entre la nitrone 1 et
l'alcène 3, fournie un mélange de deux
diastérioisomères (cis et trans), un est sous forme de poudre
blanche après traitement par chlorure du méthylène, et
l'autre sous forme d'huile rouge. La réaction se réalisée
selon le schéma suivant :
, 16h
reflux
benzène
1 + 3
H3C
O
O
O
N
O
7 + 8
- Schéma 6 -
Le mécanisme de l'obtention des deux
diastérioisomères 7 et 8 est le
suivant :
P
P
O
H
H H
O O
N O
CH3
Me
H
O
* *
*
H
H
O
O
N
O
cis
7
- Schéma 7 -
P
H H
O O
O
trans
8
P
CH3 N O
H
Me O H
O
O
O
N
*
H
*
*
H
En spectroscopie infra rouge les deux
diastérioisomères sont similaires. Ils montrent une bande
d'absorption à 1800 cm-1 dûe à la vibration de
valence de C=O ; 1640 cm-1 vibration de valence de C=C
arom; 1210 cm-1 vibration de valence de C-O ; 1150
cm-1 vibration de valence de C-N et 1080 cm-1 vibration
de valence de N-O.
Ces produits sont instables lors de l'analyse par GCMS ils
donnent leur produits de départs.
3. Réaction avec le parafluorostyrène 4
:
L'alcène 4 porte un groupement aryle
(4-fluorophényl), le fluore dans ce cas est un donneur d'électron
ce qui donne un alcène fortement polarisé. Nous avons obtenu un
produit sous forme d'huile après purification sur colonne de silice. La
réaction avec 1 donne un mélange de deux
diastérioisomères 9 et
10.
La réaction s'effectue selon le schéma suivant :

1 + 4
9 + 10
- Schéma 8 -
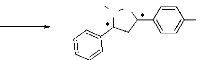
benzène reflux, 24h
F
N
Me
O
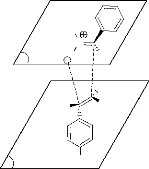
H3C
N
P
P
O
H
F
H
H
H
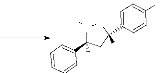
F
H3C
N O
H
H
trans
9
Le mécanisme de l'obtention de deux
diastérioisomères est le suivant :
- Schéma 9 -
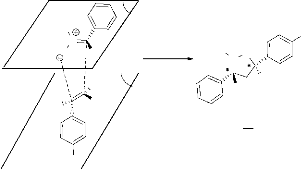
F
H3C
N
O
H
H
cis
10
H3C
O
H
F
N
H
H
H
P
P
Le spectre infra rouge montre :
- une bande à 3000 cm-1, due à la
vibration de valence de la liaison C-Harom - la bande de vibration de valence
de la liaison C=Carom à 1600 cm-1
- la bande de vibration de valence de C-O à 1210
cm-1 - la bande de vibration de valence de C-N à 1250
cm-1
- et la bande de vibration de valence de N-O à 1145
cm-1.
Dans l'analyse par spectroscopie de masse, le spectre de masse
indique la présence du pic de l'ion moléculaire à m/z
=257.
CONCLUSION
La condensation entre les aldéhydes aliphatiques et
aromatiques para substitués, en l'occurrence le benzaldéhyde, le
para-nitro-benzaldéhyde, anisaldéhyde et propanaldéhyde,
nous fournit les nitrones correspondantes avec des rendements variés en
fonction de la structure de ces derniers et aussi de par la nature des
groupements portés par le noyau benzénique de
l'aldéhyde.
Les nitrones préparées à partir des
aldéhydes aliphatiques sont instables et se décomposent en leurs
produits de départ, pendant la purification ou au contact avec des
solvants polaires comme l'éthanol. Tandis que les nitrones aromatiques
sont généralement plus stables et cristallisent dans
l'éthanol ou l'éther di-éthylique.
L'introduction de substituants donneurs d'électrons tel
que le groupement méthoxy, joue un rôle important dans la
formation des nitrones correspondantes, où l'on obtient de bons
rendements (de l'ordre de 80%). Par contre les substituants attracteurs
d'électrons, comme le groupement nitro, défavorisent la formation
de nitrones, et l'on constate l'obtention de rendements faibles (environ
12%).
Les nitrones porteurs du groupement phényle sur l'atome
d'azote sont plus stables et ceci est dû à la forte conjugaison
qui existe dans la molécule.
Mode opératoire général
:
Dans un ballon de 250 ml on introduit la nitrone 1
et l'alcène avec des quantités
équimoléculaires. Le mélange est dissous dans le
benzène ou le toluène et qu'on porte à reflux du solvant.
L'évolution de la réaction est contrôlée par ccm.
Après évaporation du solvant le produit est purifié par
chromatographie sur colonne de silice.
1. Réaction entre 1 et 2.
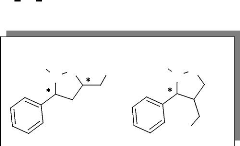
H3C
N
O OH
+
H3C
N
HO
O
*
5, 6
C11H15NO2
M = 193 g/mole
La réaction est réalisée à partir
de 0,56 g (4,148 mmol) de nitrone 1 et 0,24 g (4,148 mmol,
0.281 ml) de l'alcène 2 en solution dans le
benzène. La réaction a lieu à reflux de ce solvant et
pendant 24 heures. Après purification du mélange
réactionnel on isole 0,57 g (2,95 mmol, 72%) des produits 5
et 6.
Rdt =72 %
Aspect: huile orange
Rf = 0.69 (Et2O / CH2Cl2(8/2))
tr= 9.02 min (80°C +10°C/min)
IR: 3350 (L) VO-H ; 3000(f) VC-Harom ; 2900 (f)
VC-Haliph ; 1600 (m) VC=Carom ; 1410 (m) VC-O ; 1250 (m) vC-N ; 1180 (m) vN-O
.
SM: 193 (0); 176 (35); 162 (10); 146(15);
132(5); 118(5); 105(20); 100(100); 91(20); 72(20); 56(20); 42(18); 41(5).
RMN pour les deux régioisomères
qui diffèrent de par ces analyses.
a. le régioisomère 5






RMN H1 (CDCl3) : 1,12 (m,
2H, C4,); 2(s, OH); 2,5(s,
CH3); 4,2 (m, H, C5); 3,56 (m,
CH2OH); 4,1 (m, H, C3).
RMN C13 (CDCl3): 35 (C4) ; 44.5
(CH3) ; 60 (C3) ; 65(CH2OH) ; 74 (C5) ; les
carbones du phényle apparaît comme suit: C1 (138); C2 et C6 (127)
; C3 et C4 (129) ; C4 (128).
b. le régioisomère 6
RMN H1 (CDCl3) : 2,2 (m,
H en C4,); 2(s, OH); 2,5(s,
CH3); 2,75 (m, 2H en C5 et
CH2OH); 3,5 (m, H en C3).
Les protons de phényle apparaît comme suit :
Ortho (7,40) ; méta (7,22) ; para (7,00)
RMN C13 (CDCl3): 40 (C4); 44.5
(CH3); 74 (C3); 77 (C5); 76 (CH2OH) Les
carbones du phényle apparaît comme suit:
C1 (135); C2 et C6 (127) ; C3 et C4 (124) ; C4 (126).
2. Réaction entre 1 et 3
H3C
N O
O
O
O

7 + 8
M = 233 g/mole
C12H11NO4
La réaction s'effectue à partir de 0,135 g (1 mmol)
de nitrone 1 et 0,098 g (1 mmol) de l'alcène
3, en solution dans 40 ml du benzène anhydre, à
reflux pendant 16 heures. Après traitement
par le chlorure du méthylène on isole 0,134 g
(0,575 mmol) de poudre blanche (80%), et 0,034 g (0,146mmol) d'huile rouge
(20%). Le rendement total est de l'ordre de 72 %.
H3C
H
N O
H
H
O
O
O
7
Trans (exo)
Aspect : Poudre blanche
Rf = 0.26 (Et2O / CH2Cl2 (8/2))
Pf : 119 -120 °C.
IR (KBr) : 2960 (f) VC-Harom ; 1720 (m) VC=O,
1600 (m) VC=Carom ; 1210 (m) VC-O ; 1150 (m) VC-N ; 1049 (m) VN-O.
H3C
H
N O
H
H
O
O

O
8
Cis (endo)
Aspect : Huile rouge.
Rf = 0.72 (Et2O / CH2Cl2 (8/2))
IR (KBr) : 2940 (f) VC-Harom ; 1800 (m) VC=O ,
1640 (m) VC=Carom ; 1210 (m) VC-O ; 1150 (m) VC-N ; 1080 (m) VN-O .
3. Réaction entre 1 et 4.

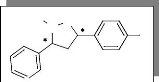
H3C
N
O
F
9 , 10
C16H16NOF
M = 257 g/mole
A 500 mg (3,7 mmol) de 1, dissous dans 20 ml
de benzène, on ajoute 3,85 mg (0,44 ml, 3,7 mmol) de 4
dissous dans la même quantité de benzène anhydre,
et porté à reflux pendant 24 heures. Après purification du
brut on a obtenu 575 mg (2,23 mmol, 60%) du produit
9.
Rdt = 65%
Aspect: huile jaune.
Rf =0,77 (Et2O / CH2Cl2 (8/2)).
IR (CH2Cl2) : 3000 (f) VC-Harom ; 1600 (m) VC=C,
1 ; 1210 (m) VC-O ; 1250 (m) VC-N ; 1145 (m) VN-O.
SM : 259 (2) ; 257 (10) ; 211(45) ; 196 (5) ;
134 (100) ; 115 (20) ; 106 (10) ; 91 (15) ; 77 (20) ; 65 (10) ; 41 (10).
tr: 17,4 min (80°C +
10°C/min).
Bibliographie
|
|
Références :
[1] Tufariello, J. J. Acc. Chem. Res.,
1979, 12, 396-403
[2] Balusubramanian, N.; Org. Prep. Proced. Int.
1985, 17, 23
[3] Frederichson, M. Tetrahedron 1997,
53, 403]
[4] Gearing. B. k.Ph. D. Dissertation, Cornell
University, 1995.
[5] Hastam, E. Shikimiv Acid Metabolism and Metabolites,
Jhon Wiley &Sons ; New York, 1993.
[6] Chiacchio, U.; Corsano, A.. Iannazzo, D.; Rescifina, A.;
Piperno, A . Romeo, G. Tetrahedron Lett. 2001, 42,
1777-1780
[7] Chiacchio, U.; Corsano, A.; Pistarà, V.; Iannazzo,
D.; Piperno, A.; Romeo, G.; Romeo, R.; Grassi, G.; Eur. J. Org.Chem.
2002, 1206-1212
[8] Iannazzo, D.; Piperno, A.; Pistarà, V.; Rescifina,
A.; Romeo, R. Tetrahedron 2002, 58, 581-587.
[9] Smith, C. J.; Holmes, A. B.; Press, N. J. Chem.
Commun., 2002, 1214-1215
[10] Tufariello, J. J. In 1,3-Dipolar Cycloaddition
Chemistry, Padwa, A., Ed.; Wiley: New York, 1984; Vol. 2,
p83.
[11] Tufariello, J. J. In 1,3-Dipolar Cycloaddition
Chemistry, Padwa, A., Ed.; Wiley; Interscience: New York.
1994; pp83-188
[12] Schlecht, M. F. J. Chem. Soc. Chem. Commun.
1985, 1293
[13] Stamm, H.; Steudle, H. Tetrahedron
1979, 35, 647.
[14] Wuts, P. G. M.; Jung, Y, w,
j. Org. Chem.
1988, 53, 1957
[15] Dhavale, D, D,.; Trombini, C.
Hétérocycles 1992, 34, 2253
[16] Gianotti, M.; Lombardo .; M.; Trombini, C. Tetrahedron
Lett 1998, 39, 1643
[17] Dhavale, D, D,.; Trombini, C. J. Chem. Soc., Chem.
Commun. 1992, 1268
[18] Camiletti, C.; Dhavale, D, D.; Gentilucci, L; Trombini, C.
J. Chem. Soc., Perkin Trans. 1, 1993, 3157
[19] Camiletti, C.; Poletti, L.; Trombini, C.
j. Org. Chem..
1994, 59, 6843
[20] Lombardo, M.; Trombini, C. Tetrahedron
2000, 56, 323
[21] A. Dalgard. K. E. Larsen, K. B. G. Torssel. Acta
Chemica. B 38 (1984)
[22] Ochiai, M., Obayashi, M.and Morita, K. Tetrahedron
23(1967) 2641
[23] Lablanche-Combier, A.; Villaume, M. et Jaquesy, R.
Tetrahedron Lett (1967) 4956
[24] Torssel, K. B. G, Nitriles Oxides, Nitrones and
Nitronates in Organic Synthesis; VCH: Weinheim, 1988
[25] Manwni, F.; Piazza, M. G.; Trombini, C.
J. Org. Chem
1991, 56, 4246
|
[26] Fwmana, A.; Lombardo, M.; Trombini, C.J. Org. Chem
1997, 62, 5623.
[27] Dhavale, D. D.; Gentilucci, L.; Piazza, M. G.; Trombini, C.
Liebigs. Ann. Chem. 1992, 1289
[28] Dehaen, W.; Hassner, A. Tetrahedron Lett.
1990, 31, 743.
[29] Gottlieb, L.; Hassner, A.
j. Org. Chem.
1995, 60, 3759.
[30] Denmark, S. E.; Thorarensen, A. Chem. Rev.
1996, 96, 137.
[31] Denmark, S. E.; Senanayake, C. B. W. Tetrahedron
1990, 46, 4857
[32] Hanselmann, R.; Zhon, J. P.; Confalone, P. N.
J.Org. Chem.
2003, 68, 8739- 8741
[33] B. Janza, A. Studer, Synthesis,
2002, 2117-2123.
|
Annexe
|
LOO:
°3642 23 --
F
pt
h7884',
= o ro
·
711
1 3371,34 -- 04
ALL
|
Q
2885,613
cc
0
0
C
C
|
3201,81 -- -
2818,17
|
.
0
141*
41
0
5
r.,
01
40
|
|
2830,02 -- --
2306.71
|
NJ
0
07
m 10406
e-§
37
|
157301
|
|
|
1303,78
1033,77
|
144208 _
|
|
|
848
894131 -
84001
4 80
15
· 124 31 -
7 C. 11 17
ndlAWI :HOE: uolo7d azqoads

;41
h 1
|
|
|
|
|
|
|
|
\----........9914:.
94.34 ----____\
\-....._ 91 li . 62
92933,
138 e2, 5 :
40 61
:9E; 141
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O

:84.54
6;23
648.89 644.17 576.01 570.21 566.32 563,75 563.48 54-0.08 543
,
361.26 348 b-1 301,22 301.11 295-1.4
CL,
7.48
4.?1
· - -

· .1 A 4,,
.7;
r,4
a
rF
a
a
r.4
z
a
0
rr
a

N. I
J
147
N w
41,1E
1'
1J
-- -- -- --

2
(11
0
0
0
0
P.
N
0
0
3
C
0
0
0»
a
a
- 0
PPG
--
a,
Ca
9 Zama ilOSE T

fl

|
139.226
136,301
1136.314
1131.502
1132.RE 129.750 29.500 128.998 128.775 128.65'9 128.327
129.079 127.927 127.722 127.293 12E1.977 126.728 126.549 77.705 77.490 77.195
77.105 76.641S 73.981 73.094 61 1.751 ---- 63.012 60.351;
|
|
|
|
|
|
·
|
|
42.924
40.987
15,142
-- 0.230
rS7
g 741,'
, F -
·
'42
E
- .... ,, _A ,
...
· .--.
· 9 u0'4. 0
0 0 -
· 61: 4.--4 L-. 9 1., 1...,
Ot ., %I r+ UN I c. l...7 ,--..«-- .-
· a a , ,
.2. ... r.,
· a
· .
· N
· 0 9 . 0, , , :, CO
47. a R
NB ...--.. ._.. ,..a . ._. ,.. , IT -4 I, -.A 0 0
· CO
-, NO
·
,J. VD ary 9 0 0 U Z IA g '-. ' ' " '-'' ' ' '. ''' '
' -.'
r., ..... 0 9 W GO en al a. ,-+a ,.., -.., ..., ,a,
e..., +i...., 1,r, ,,I
... ,-
· . . ,-
· G t. 9
4. 0 9
0
Wrg WE Z 0 = r,, .R; 4gC 6Iti ..g ffi'.4"'r..g
g?.7Z7
3 3 ' n "RR' " f-1 ^ VA '
q i
|
0
|
1.4
0
|
|
|
|
|
304223-
|
|
503
|
2E7760-- -
·
|
|
|
24 .73
|
2822.72
2407 00 -
|
2/15.77
1
·Z.
0
O
Mt as
0
·
CONCLUSION GENERALE
Dans la première partie de ce travail, les
résultats que nous avons obtenus au cours de nos travaux sont
globalement positifs. En effet sur le plan de la synthèse, nous avons
accédé à des nitrones par la réaction de
condensation entre les aldéhydes et les
hydroxylamines-N-substituées. Sur le plan de l'analyse
spectrale nous avons pu identifié les produits préparés
par les méthodes spectroscopiques usuelles.
Dans la deuxième partie de ce travail, on s'est
intéressé à la synthèse des isoxazolidines à
partir des nitrones initialement préparées. Nous pouvons dire que
la réaction de cycloaddition dipolaire-1,3, entre ces composés
préalablement synthétisés d'une part, et les
oléfines d'autre part, est une méthode très efficace pour
l'obtention des isoxazolidines chirales et racémiques.
L'efficacité et la régiosélectivité de cette
réaction sont généralement dépendants des facteurs
stérique et électronique des substrats de départ.
La réaction de
l'a-phényl-N-méthylnitrone avec l'alcool allylique n'est
pas régiosélective, où elle donne un mélange de
deux régioisomères et deux diastérioisomères ceci
est dû à l'effet stérique du groupement phényle
porté par la nitrone, et du groupement hydroxy de l'alcène.
Tandis que pour le para fluorostyrène la réaction est
régiosélective du fait de l'influence des deux facteurs, à
savoir l'effet électronique de l'atome du fluore en position para, dans
le styrène, et l'effet stérique du groupement aryle porté
par cette alcène.
La réaction de l'
a-phényl-N-méthylnitrone avec l'anhydride
maléique donne un seul régioisomère qui est dû
essentiellement à l'alcène symétrique.
Dans toutes ces réactions on a obtenu un mélange
de deux diastérioisomères endo et exo (à savoir cis et
trans), La régiosélectivité et la
diastériosélectivité de cette réaction sont
déterminés à partir de l'analyse spectrale des
produits.
Dans le prochain travail nous essayerons de synthétiser
des isoxazolidines optiquement actifs par l'introduction du groupement chiral
sur les substrats ou l'utilisation de catalyseurs chiraux, et par de la
approfondir nos connaissances sur la synthèse asymétrique.
|
|


