La cytogénétique et classification des hémopathies malignes( Télécharger le fichier original )par Achouria BOURIACH Universdité d'Oran - Algérie - Diplôme des études supérieures en génétique 2012 |

I.1.2.2.2 La maladie de h odgkin :C'est une maladie qui se rapproche des lymphomes, mais qui en diffère par de nombreux aspects, cliniques pronostiques et thérapeutiques La maladie de Hodgkin ou lymphome de Hodgkin est un type de lymphome caractérisé par la présence de grosses cellules atypiques, (Haluska et al., 1987; Hecht et al., 2000) les cellules de Reed-Sternberg. C'est un cancer en très forte et rapide augmentation, notamment chez les femmes (croissance de + 6 % par an). En France, c'est le troisième cancer le plus fréquent chez les femmes, ce qui laisse supposer des causes environnementales (Deininger et al., 2000; Maru 2001) 1.1.1.3 Toxicologie : On connait plusieurs facteurs environnementaux de risques L'exposition aux solvants . Selon une étude américaine de 2008 ayant porté sur 1 318 femmes salariées du Connecticut (dont 601 atteintes d'un lymphome de 1996 à 2000), l'exposition des femmes aux solvants chlorés provoque une hausse de 40 % du risque de Lymphome non Hodgkinien (LNH) et de plus de 100 % dans le cas du tétrachlorométhane. Être exposé à tout solvant organique augmente le risque de développer un LNH selon cette étude. L'exposition aux pesticides, chez les agriculteurs en particulier : Une étude de 2009, portant sur la population masculine française, a montré que l'incidence des lymphomes était deux à trois fois plus élevée parmi les agriculteurs. L'exposition aux antidépresseurs tricycliques : Une étude, étudiant 2768 cas et 22177 témoins, montre une augmentation de 1,2 fois le risque en cas d'utilisation prolongée. 1.1.3 Predispositions genetiques ou caractere mutagene des pesticides :En cherchant des biomarqueurs prédictifs de lymphomes chez des personnes exposées aux pesticides, à partir de la base de donnée Agrican de l'INSERM, des chercheurs marseillais ont trouvé dans le sang de participants à l'étude Agrican des cellules anormales qui semblent être les précurseurs des cellules tumorales constituant les lymphomes folliculaires. Selon Bertrand Nadel, ces biomarqueurs témoignent d'un lien moléculaire entre l'exposition des agriculteurs aux pesticides, une anomalie génétique" et "la prolifération de ces cellules, qui sont des précurseurs de cancer", et "cet effet est fonction de la dose et du temps d'exposition. Cette anomalie génétique ferait qu'un fragment du chromosome 14 s'en détacherait pour aller activer un oncogène situé sur le chromosome 18. L'expression de cet oncogène n'étant plus inhibée, des cellules qui auraient dû mourir vont proliférer (Rowley 1999). Les personnes plus exposées aux pesticides étant plus nombreuses que la moyenne à présenter dans leurs lymphocytes sanguins cette anomalie génétique, il semble que les pesticides puissent être responsables de cassures et mutation délétères de l'ADN. D'autres effets sont associés à cette anomalie, dont une instabilité générale du génome : deux gènes sont exprimés en même temps alors que normalement ils ne le sont pas, ce qui permet aux cellules anormales de résister aux mécanismes de mort cellulaire programmée ajoute Bertrand Nadel (respectivement Bai et al., 1998; Hernandez et al., 1999) . Environ 50% de la population française porte la translocation, soit environ 30 millions de Français, mais moins d'une personne sur 17 000 déclare ce type de cancer). Chez les agriculteurs ou les individus exposés aux pesticides, le sang présente « 100 à 1000 fois plus de cellules "transloquées" » que la moyenne. 1.1.4 Le myélome multiple ou maladie de kahler :C'est une maladie qui se rapproche beaucoup des lymphomes de bas grade. Comme pour la maladie de Hodgkin, le myélome multiple présente une évolution particulière et une approche thérapeutique différente. 1.1.5 Epidémiologie :Mortalité ajustée à l'âge (avec ajustement standardisé) par lymphomes et myélomes multiples pour 100,000 habitants en 2004. Pris conjointement, les lymphomes représentent 5.3% de tous les cancers (hors cancer des cellules basales de la peau) aux Etats-Unis et 55.6% de tous les cancers du sang. Selon le National Institute of Heath, les lymphomes représentent environ cinq pour cent de tous les cas de cancer aux Etats-Unis, et le lymphome de Hodgkin en particulier intervient dans moins de un pour cent des cas. Etant donné que l'ensemble du système lymphatique fait partie du système immunitaire du corps, les patients chez qui le système immunitaire est affaibli, comme par exemple dans le cas d'une infection par le VIH ou par l'usage de certaines drogues ou médicaments sont également exposés à un risque accru de développer un lymphome. Une étude menée sur des enfants atteints de graves pathologies immunologiques (leucémie lymphoblastique aigüe, lymphome) a démontré leurs graves carences en zinc ainsi qu'en magnésium mais n'en donne pas les causes. 1.1.6 Les signes et symptomes du lymphome :Les signes et symptômes sont tous les troubles, observés et signalés par le patient, qui peuvent avoir un lien avec la maladie. Les signes sont des états anatomiques ou physiologiques anormaux. Des ganglions enflés, douloureux ou pas, sont souvent le signe le plus fréquent d'un lymphome. Généralement, les ganglions sont enflés au niveau du cou ou des aisselles, mais beaucoup de patients peuvent aussi avoir des ganglions qui enflent à d'autres parties du corps et qui entraînent différents symptômes. Ainsi, des ganglions enflés au niveau de l'aine peuvent provoquer des jambes lourdes et des chevilles gonflées, alors que des ganglions enflés au niveau de l'abdomen peuvent provoquer une gêne abdominale, des maux de dos ou des ballonnements. Les signes d'un lymphome extra-ganglionnaire peuvent varier selon la partie du corps où se développe la tumeur (Okuda et al., 1996; Voss et al., 2000). C'est pourquoi, un lymphome présent dans l'estomac peut parfois provoquer des symptômes semblables à ceux d'un ulcère, tels que des douleurs et des saignements internes. Plus rarement, il arrive que certains patients atteints d'un lymphome n'aient pas de ganglions enflés (Lengauer et al., 1998; Ponder 2001). Les symptômes sont les phénomènes inhabituels observés par le patient indiquant la présence possible de la maladie. Les symptômes d'un lymphome sont des frissons, une fièvre, des sueurs profuses (souvent nocturnes), une perte de poids inexpliquée, une baisse d'énergie, des démangeaisons ou d'autres symptômes provoqués par des ganglions enflés. Ces symptômes ne sont pas spécifiques et la plupart des personnes qui les ressentent ne sont pas atteintes d'un lymphome. Toutefois, dans le cas de symptômes persistants, il important de consulter un médecin pour s'assurer qu'il n'y a pas présence d'un lymphome. Les maladies graves ne disparaissent pas du jour au lendemain ; elles persistent. 1.1.7 Diagnostique biologique : I.1.7.1 Examens biologiques :L'hémogramme recherche une leucocytose, parfois marquée dans les formes évolutives, avec souvent anémie normochrome. Une anemie hemolytique avec test de Coombs negatif est plus rare. Une accélération de la vitesse de sédimentation érythrocytaire (VS) est souvent présente : elle est utilisée comme facteur de stratification pronostique dans de nombreux protocoles européens. Une élévation des phosphatases alcalines (en l'absence d'atteinte osseuse), ou une hypo albuminémie sont aussi des critères d'évolutivité. egiagotertique eteieloo0:eation deo détutpatideo tiralipteo
I.1.7.2 Autres examens : 1.1.7.2.1 Examens d'imagerie : Généralement, les médecins prescrivent des examens qui vont leur fournir des clichés de l'intérieur de votre corps. La plupart des ces examens sont sans douleur et sont réalisés sans anesthésie. Plusieurs techniques d'imagerie médicale peuvent être nécessaires pour effectuer le meilleur diagnostic de votre cancer. Parmi ces examens, il y a notamment : La radiographie, le scanner, l'IRM (Imagerie par Résonance Magnétique), les TEP (Tomographie par Emission de Positrons (McWhirter et al., 1993; Golub et al., 1996) 1.1.7.2.2 Chirurgie diagn ostique :Une laparotomie avec splénectomie a été largement utilisée pour préciser l'extension sous diaphragmatique. L'avènement de la TDM et surtout l'utilisation très large de la chimiothérapie, capable de stériliser de petites les ions sous diaphragmatiques non repérées par l'imagerie, ont permis de renoncer a ce type d'exploration 1.1.7.2.3 Les examens sanguins :Les examens sanguins permettent de déterminer si les différentes cellules sanguines examinées à l'aide d'un microscope sont normales en termes de nombre et d'aspect. Parmi ces cellules, on retrouve les globules rouges, les globules blancs et les plaquettes. Des anomalies observées au niveau des cellules sanguines anormales sont parfois les premiers signes de lymphome. 1.1.7.2.4 Examen de la malle osseuse :La moelle osseuse est la matière spongieuse que l'on trouve à l'intérieur des os. Elle contient des cellules immatures, appelées cellules souches, qui se développent en trois sortes de cellules : les globules rouges dont la fonction est de distribuer l'oxygène dans toutes les parties du corps et d'évacuer le gaz carbonique ; les globules blancs qui protègent l'organisme des infections ; et les plaquettes qui jouent un rôle dans la coagulation du sang. Le lymphome peut s'étendre ou commencer dans la moelle osseuse. C'est pourquoi les médecins tiennent à examiner des échantillons de la moelle osseuse pour vérifier s'il y a présence d'un cancer. Après application d'une anesthésie locale, une "carotte" de moelle osseuse de 15 mm de long sur 2 mm de large environ est prélevée dans l'os du bassin. Le procédé peut être douloureux au moment où la moelle osseuse est aspirée. Si le patient le souhaite, il peut demander au médecin ou aux infirmiers de lui administrer une prémédication calmante.
1.1.8 Evolution et complication :
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
||
|
|
|
||
|
|
|
||
|
|
|
||
|
|
|||
La nouvelle classification OMS des myélodysplasies définit également 5 catégories, différentes de celles du FAB :
· l'anémie réfractaire (AR) sans ou avec sidéroblastes en couronne (ARS),
· la cytopénie réfractaire ou syndrome myélodysplasique avec myélodysplasie touchant plusieurs lignées (CRMD),
· le syndrome 5q-,
· l'anémie réfractaire avec excès de blastes (AREB),
· les SMD inclassables
·
Tableau III : Classification OMS (1999); Remplace la classification FAB.
http://atlasgeneticsoncology.org/Educ/Hempat_t.html
|
|
|
|
|
|
|
|
||
|
|
|
||
|
|
|
||
|
|
|
||
|
|
|
||
|
||||
Les syndromes myélodysplasiques, longtemps appelés « anémies réfractaires » (aux traitements vitaminiques), sont des hémopathies clonales (ce qui signifie qu'elles dérivent toutes d'un même précurseur hématopoïétique anormal). Les cellules provenant de ce précurseur présentent un trouble de la maturation qui les fait mourir au sein même de la moelle (avortement intramédullaire).
Les conséquences cliniques sont les cytopénies périphériques et les troubles morphologiques des précurseurs myéloides (dysmyélopoïèse).
Les syndromes myélodysplasiques sont les plus fréquents des états préleucémiques. Ils prédominent chez le sujet âgé avec une prédominance chez l'homme.
Il n'y a pas d'étiologie connue mais seulement des facteurs favorisants : facteurs familiaux, facteurs toxicologiques (exposition benzène, chimiothérapie ou radiothérapie.
La clinique est dominée par les conséquences liées aux cytopénies : syndrome hémorragique par thrombopénie, infections sévères et répétées par neutropénie, syndrome anémique. Dans de rares cas, une splénomégalie modérée est notée, en rapport avec une leucémie myélomonocytaire chronique (LMMC), variété de syndrome myélodysplasique.
Ici aussi, c'est l'association des cytopénies, en nombre et d'importance variable, qui oriente le diagnostic :
· L'anémie est macrocytaire arégénérative
· La leucopénie avec neutropénie (deux fois sur trois) est parfois accompagnée d'une monocytose dans le cas d'une LMMC.
· La thrombopénie
Un petit pourcentage de blastes circulants et une hypogranulation des polynucléaires neutrophiles sont très évocateurs du diagnostic.
L'étude cytologique met en évidence la dysmyélopoïèse et/ou un excès de blastes médullaires:
· La richesse médullaire est le plus souvent augmentée (production inefficace).
· La lignée rouge est souvent hyperplasique. La dysérythropoïèse peut consister en une mégaloblastose, des anomalies d'hémoglobinisation, des anomalies nucléaires (multinucléarité, caryoréxie). La coloration de Perls met en évidence des sidéroblastes en couronnes dans l'ARS.
· La lignée granuleuse est souvent hyperplasique dans les formes avec excès de blastes. Une dysgranulopoïèse peut être représentée par une diminution ou une disparition des grains neutrophiles intracytoplasmiques et/ou des troubles de la segmentation des noyaux.
· La lignée plaquettaire peut montrer des micros mégacaryocytes (mégacaryocytes de petite taille mononuclées), des mégacaryocytes mononuclées (évocateur d'une anomalie cytogénétique 5q-), ou de grands mégacaryocytes à noyaux séparés et arrondis. Le nombre de ces mégacaryocytes est très variable.
L'étude cytogénétique de la moelle est complémentaire de l'analyse cytologique.
1.1.10.6.~ Autres examens :
Ils sont inutiles au diagnostic mais certains sont utiles à l'estimation du pronostic.
· Sur la biopsie osseuse, la dysmyélopoïèse est mieux visible. Une fibrose réticulinique se voit parfois. La localisation anormale de précurseurs au sein des espaces médullaires a une certaine valeur pronostique. Enfin, la biopsie peut être utile pour faire la différence avec une aplasie médullaire dans le cas où la moelle du syndrome myélodysplasique montrerait une moelle pauvre.
· Si l'exploration isotopique de l'érythropoïèse au Fe59 est pratiquée, elle montre une forte captation médullaire mais une faible incorporation globulaire.
· L'étude cytogénétique montre des anomalies clonales dans la moitié des cas : délétion 5q, délétion 20q, monosomie 7 ou délétion 7q, trisomie 8. Elle a une valeur pronostique importante.
La classification franco-américano-britannique (FAB) basée sur le pourcentage de blastes sanguins et médullaires, le pourcentage de sidéroblastes en couronne de la moelle et la
monocytose sanguine permet de distinguer les différents types de syndrome myélodysplasique.
1. L'anémie réfractaire sidéroblastique idiopathique (ARSI).
Elle s'accompagne d'une anémie isolée et d'une sidéroblastose médullaire > 15 %.
2. L'anémie réfractaire avec excès de blastes (AREB) et l'AREB en transformation (AREB-t). Elle se manifeste par une pancytopénie sévère. La blastose sanguine est < 5 % et la blastose médullaire est comprise entre 6 et 20 %.
L'AREB-t répond aux critères de l'AREB à l'exception d'une des caractéristiques suivantes : blastes circulants > 5 % ou blastes médullaires entre 20 et 30 % ou présence de corps d'Auer dans les blastes.
3. La leucémie myélo-monocytaire chronique (LMMC).
Elle est caractérisée par une monocytose > 1 000 par millimètres cubes et une splénomégalie. Il s'agirait d'une forme frontière avec la leucémie myéloïde chronique (LMC).
4. Cytopénie réfractaire simple.
Elle n'est définie que par des critères négatifs : c'est elle qui peut poser des problèmes de diagnostic.
Les anémies sidéroblastique peuvent rarement être secondaires à des toxiques (isoniazide...) ou héréditaires liées à l'X.
Les AREB-t ne posent pas de problème de diagnostic différentiel. Elles doivent cependant être distinguées des leucémies aigües myéloïdes où la blastose est par convention > 30 %.
La LMMC pose le problème du diagnostic différentiel d'une monocytose. Dans un premier temps, une monocytose réactionnelle post-infectieuse doit être éliminée : toute monocytose durable est à priori leucémique. Dans un 2e temps, une LMC doit être éliminée par l'absence de chromosome Philadelphie.
Le problème principal reste donc les cas de cytopénies simples où il faudra éliminer :
· Une carence vitaminique où des cytopénies peuvent accompagner l'anémie. Les dosages vitaminiques permettent de trancher.
· L'anémie macrocytaire de la cirrhose éthylique où la dysmyélopoïèse médullaire ne touche que la lignée rouge.
· Une neutropénie isolée doit faire discuter des étiologies médicamenteuses, infectieuses ou auto-immunes.
· Une thrombopénie isolée doit éliminer une thrombopénie périphérique auto-immune. Dans les cas difficiles, la mise en évidence d'une anomalie cytogénétique clonale, d'une poussée anormale de précurseurs ou une étude de l'érythropoïèse par le radiofer permettent de trancher.
Les complications infectieuses sont la cause du décès dans 50 % des cas. Elles se voient principalement quand la neutropénie est < 500/mm3, un déficit fonctionnel des polynucléaires neutrophiles aggravant le risque.
Le syndrome hémorragique est la cause de la mort dans 20 % des cas. Ici aussi, une thrombopathie aggrave la symptomatologie.
·Les complications infectieuses et les hémorragies sont surtout le fait des AREB-t. L'anémie récidivante expose à l'hémochromatose transfusionnelle.
Le risque global de transformation est de 30 % mais dépend aussi du type de syndrome myélodysplasique.
La transformation peut être brutale ou progressive avec augmentation de la blastose jusqu'à 30 % sur différents prélèvements. La leucémie myéloïde aiguë secondaire à un syndrome myélodysplasique est de très mauvais pronostic car elle ne répond pas aux traitements usuels.
1.1.10.83 Complications de la LMMC .
La LMMC peut évoluer vers un syndrome myéloprolifératif ou donner des épanchements spécifiques des séreuses et des localisations cutanées.
Tout épisode fébrile chez un neutropénique justifie la prescription d'une antibiothérapie à large spectre couvrant les bacilles Gram négatifs puis les staphylocoques et enfin les levures. Un syndrome hémorragique lié à une thrombopénie doit être traité par des concentrés plaquettaires.
L'anémie nécessite des transfusions répétées de concentrés érythrocytaires, pouvant aboutir à une surcharge en fer (hémochromatose transfusionnelle). La prévention de l'hémochromatose transfusionnelle repose sur l'administration de desférioxamine (Desféral) en sous-cutanée.
Le traitement spécifique efficace, l'allogreffe de la moelle, se heurte à l'âge souvent élevé des patients. De très nombreux sujets âgés tolérant parfaitement leur maladie n'ont besoin que d'un traitement symptomatique et d'une surveillance.
Il s'agit d'une nouvelle classe de chimiothérapie basée sur l'épigénétique. Il existe deux molécules qui ont été testées : La décitabine et l'azacitidine. Seule l'azacitidine a montré un avantage significatif en survie comparée aux traitements conventionnels (transfusions, cytarabine faible dose et chimio standard.
Elle comprend numération-formule-plaquettes et myélogramme réguliers. Le premier bilan doit se faire à la sixième cure seulement. Parfois la première réponse ne peut intervenir qu'au 9ème cycle. La réponse se manifeste souvent sur la lignée érythrocytaire. Il suffit qu'une lignée réponde pour que les autres suivent généralement. Il faut stopper le traitement qu'en cas de progression de la maladie, d'effet secondaire de stade élevé compromettant le pronostic vital du malade ou sa qualité de vie.
Un syndrome myéloprolifératif est une maladie caractérisée par une production anormale, d'allure cancéreuse, de certains types de
Il s'agit d'une prolifération clonale des cellules
conservent une capacité de différenciation à l'inverse des leucémies aiguës. Il peut éventuellement évoluer en syndrome myélodysplasique
%c3%
La cancérogenèse chez l'homme est un processus "multi
multiplicité des événements génétiques pouvant conduire à la maladie. Ces altérations génétiques induisent une transformation progressive d'une cellule normale en une cellul dérivée maligne. Les cellules cancéreuses ont des défauts dans leur programme de régulation qui gouverne la prolifération des cellules normales et l'homéostasie.
Le phénotype des cellules malignes es
l'immortalité, la tumorigénicité et l'instabilité génomique (Tlsty et al, 1992). L'instabilite du génome est un événement précoce important dans la progression tumorale : elle se manifeste par l'apparition de mutations ponctuelles et/ou de réarrangements de régions plus ou moins grandes du génome. Elle aboutit à la perte de la ségréga
à la production de cellules aneuploïdes (perte ou gain d'un chromosome comme la trisomie 21 ou la monosomie X) et hétéroploïdes (copies supplémentaires de tous les chromosomes comme la triploïdie 3n et la tétraploïdi
egiagotertique eteieloo0:eation deo détutpatideo tiralipteo
Les cellules malignes présentent des anomalies acquises et parfois des anomalies constitutionnelles qui participent à la cancérogenèse. Les anomalies acquises sont présentes dans les cellules tumorales et absentes des tissus qui ne sont pas impliqués dans le phénotype malin. Ce sont des anomalies clonales apparues dans une seule cellule et présentes, en majorité, dans toutes les cellules de la tumeur considérée. Dans certains cas il peut y avoir perte du clone initial et donc perte des anomalies dans les cellules de la tumeur. La cellule portant l'anomalie présente des modifications génétiques lui conférant un avantage prolifératif.
Trois types d'anomalies chromosomiques sont retrouvés dans les cellules cancéreuses (Holliday 1989) :
· le "bruit chromosomique", c'est-à-dire les anomalies chromosomiques dues au hasard, sans liaison ni conséquence directe avec le processus malin.
· Les anomalies primaires, essentielles dans l'apparition des cellules tumorales.
· Les anomalies secondaires, apparues au cours de l'évolution de la tumeur et importantes dans sa progression.
· Les anomalies chromosomiques rencontrées dans les cancers peuvent être :
· Des anomalies de nombre : perte et/ou gain d'un ou plusieurs chromosomes ce qui entraîne une modification de la quantité d'ADN totale de la cellule.
· Des anomalies de structure : elles impliquent des cassures suivies de réarrangements aberrants. Elles peuvent être inter- ou intra-chromosomiques. On distingue les anomalies équilibrées (c'est-à-dire sans perte de matériel chromosomique), comme les translocations réciproques, les inversions et les anomalies déséquilibrées, comme les délétions, les duplications, les insertions et les isochromosomes (chromosomes dicentriques et en anneau). Les translocations réciproques associées à une délétion au point de cassure sont également des anomalies déséquilibrées.
Ces deux types d'aberrations chromosomiques peuvent coexister dans la même cellule cancéreuse et reflètent un caryotype complexe. Je ne détaillerai par la suite que les translocations réciproques car mon travail de thèse a porté plus particulièrement sur ce type d'anomalies équilibrées.
I.1.13.4.1 Les translocations chrom osomiques dans les cancers en générales :
egiagotertique et laid/ Caton deo détutpatideo tiralipteo
La découverte des translocations chromosomiques a transformé notre compréhension des mécanismes génétiques impliqués dans la leucémogenèse. Les translocations réciproques ont été identifiées dans les hémopathies malignes, les sarcomes et quelques tumeurs bénignes. Boveri en 1914 a été le premier à énoncer le concept suivant : un cancer dérive d'une seule cellule anormale. Il propose que des aberrations chromosomiques peuvent être la cause directe d'un cancer (pour revue Balmain 2001). Cette notion a été alors mise en évidence par la découverte du chromosome Philadelphie par Nowell et Hungerford en 1960 qui montrent pour la première fois qu'une anomalie chromosomique spécifique est liée à un type particulier de leucémie humaine, la leucémie myéloïde chronique. D'autres exemples confirment la notion que certaines anomalies chromosomiques sont spécifiques d'une tumeur maligne donnée. Dans les hémopathies malignes, la translocation t (15;17) (q21;q11-22) est associée à la leucémie aiguë promyélocytaire avec le gène de fusion PML-RARá (Kakizuka et al., 1991). Dans les tumeurs solides, la translocation t (11;22) (q24;q12) est retrouvée dans 85% des tumeurs d'Ewing et aboutit à la formation du gène de fusion EWS-FLI1 (Desmaze et al., 1997).
Dans de nombreux cancers, la présence de translocation spécifique est donc souvent importante pour le diagnostic et le pronostic et cette information à un impact direct sur la thérapie. De nouvelles translocations chromosomiques récurrentes sont sans cesse identifiées, il est donc difficile d'établir une liste exhaustive des aberrations chromosomiques dans les cancers. Un catalogue des aberrations chromosomiques mis en place par (Mitelman et al., (2001) est disponible sous forme de data base mais elle est encore incomplète. La découverte de ces anomalies chromosomiques conduit à l'identification de nouveaux gènes qui pourraient jouer un rôle dans la leucémogenèse.
Les cassures peuvent être dues au hasard et la probabilité d'apparition est théoriquement la même dans n'importe quel point du génome. Des variations peuvent être causées par la structure chromatinienne (des piralisation, ADN-Z), par des sites spécifiques de coupure des DNA topoisomérase II, par des sites hypersensibles à la DNAse I ou par des séquences Alu répétées (Strissel et al., 1998).
Ces cassures sont associées à deux processus distincts : un processus de dérégulation de l'expression des gènes qui aboutit à la production d'une protéine normale en excès. Un processus de sélection qui génère des protéines de fusion anormales et qui ne favorise que les produits de fusion conférant un avantage prolifératif.
Les translocations équilibrées sont plus fréquemment étudiées dans les hémopathies malignes que dans les tumeurs solides. Les cellules hématopoïétiques malignes représentent un matériel de choix par leur facilité de culture. Au contraire, l'extension de l'analyse cytogénétique aux tumeurs solides s'est avérée beaucoup plus délicate en raison de la difficulté de leur mise en culture, de leur indice mitotique souvent faible, et fréquemment, du nombre et de la complexité de leurs anomalies chromosomiques. Les anomalies de structure équilibrées sont bien caractérisées dans les tumeurs d'origine mésenchymateuse (les sarcomes) et ont été plus rarement décrites dans les tumeurs d'origine épithéliale (les carcinomes) qui sont les plus retrouvées chez l'homme. Je ne décrirai par la suite que les translocations retrouvées dans les hémopathies malignes.
Après une brève description des hémopathies malignes, je décrirai les gènes fréquemment réarrangés dans ces tumeurs et les conséquences que peuvent avoir les translocations réciproques.
La dérégulation de l'homéostasie hématopoïétique peut être à l'origine des hémopathies malignes. Ce dysfonctionnement peut être consécutif à une expression inadaptée ou à des altérations structurales de certaines protéines suite à des mutations ponctuelles ou à des réarrangements chromosomiques. Les facteurs de croissance et leurs récepteurs, impliqués dans la régulation de l'hématopoïèse normale, sont fréquemment altérés dans les hémopathies malignes. La dérégulation de l'hématopoïèse normale peut être également due à une perte du contrôle de l'activité de facteurs participant à la régulation négative de la croissance normale des cellules hématopoïétiques.
Les leucémies humaines sont classées en fonction de leur degré de sévérité et du stade de maturation représenté. Quatre groupes peuvent être distingués :
· Les leucémies aiguës (LA).
· Les lymphomes.
· Les syndromes myéloprolifératifs (SMP).
· Les syndromes myélodysplasiques (SMD).
Les syndromes myélodysplasiques sont le résultat d'anomalies qualitatives et quantitatives du fonctionnement médullaire se traduisant par des anomalies de maturation de la cellule souche et par des degrés divers d'hématopoïèse inefficace. Un trouble de la production des
globules rouges, des polynucléaires et des plaquettes est observé, entraînant habituellement une cytopénie périphérique malgré une moelle hypercellulaire.
Les syndromes myéloprolifératifs (Dickstein et al., 1995; Albitar et al., 2000) sont des manifestations d'une anomalie clonale de la cellule souche hématopoïétique multipotente. Cette anomalie confère à la cellule souche un avantage prolifératif avec l'émergence d'un clone pathologique. Il se produirait dans un deuxième temps une lésion spécifique à chaque type de SMP, entraînant l'émergence de cellules progénitrices anormales. Contrairement aux leucémies aiguës, les SMP sont chroniques c'est-à-dire que les éléments myéloïdes gardent un potentiel de différenciation terminale normal ou quasi-normal. L'évolution des SMP se fait, après une période plus ou moins longue, vers la transformation en leucémie aiguë et/ou vers l'apparition de myélofibrose (fibrose médullaire sous forme réticulinique ou collagénique). Les SMP s'associent généralement à une reprise d'activité d'organes de l'hématopoïèse embryonnaire (rate et foie) (hépato splénomégalie fréquemment associée).
Les SMP regroupent la leucémie myéloïde chronique (LMC), la polyglobulie de Vaquez (PV), la thrombocytopémie essentielle (TE) et la myélofibrose idiopathique (MFI) (Figure6). Les cellules souches semblent être touchées à différentes étapes de leur différenciation par des facteurs
dont l'origine reste inconnue, mais qui induirait une prolifération chronique maligne de la lignée granulocytaire (dans le cas de la LMC), de la lignée érythrocytaire (dans le cas de la PV) ou de la lignée plaquettaire (pour la TE). La fibrose médullaire précoce ou myélofibrose avec métaplasie myéloïde, comme les autres SMP, est due à une transformation néoplasique de la cellule souche multipotente. Cependant, contrairement aux autres SMP chroniques, la caractéristique de cette affection est une myélofibrose qui étouffe l'hématopoïèse médullaire,
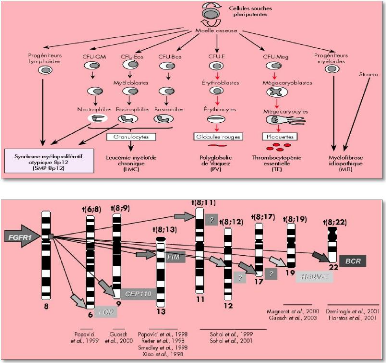
atifs, et au moins huit r&
Le clonage des points de cassure des translocations chromosomiques a permis l'identification de différentes classes d'oncogènes putatifs. Les oncogenes sont des genes cellulaires appelés proto-oncogène devenus, suite à des modifications qualitatives ou quantitatives, des gènes "transformant
mutations du type gain de fonction se manifestent à
l'état hétérozygote. Ils peuvent
être
réarrangés par translocations réciproques ou
par inversions. Ces oncogènes in des facteurs
de croissance, des
récepteurs aux facteurs de croissance, des protéines de la
transduction du
signal, des activateurs de la transcription et des protéines kinase. Les translocations ainsi créées activent ces gènes qui sont impliqués dans le contrôle de la prolifération et de la différenciation cellulaire.
Des études moléculaires des translocations ont permis de mettre en évidence deux mécanismes de leucémogenèse : la dérégulation de l'expression de gènes et la formation d'un gène de fusion. Chaque mécanisme, décrit ci-dessous, sera illustré par des exemples.
Dans le cas de la dérégulation de l'expression de gènes, le point de cassure est situé le plus souvent en dehors de la partie codante du gène transloquées. Suite à la translocation, le gène est mis sous le contrôle des séquences régulatrices (enhancer et promoteur) d'un autre gène.
Ces translocations juxtaposent un promoteur fort, comme les loci d'immunoglobuline ou du TCR, à un gène régulateur de la différenciation ou de la survie cellulaire. Elles conduisent à la surexpression d'un gène normal. Ces remaniements sont spécifiques des proliférations lymphoïdes malignes et en particulier les leucémies aiguës T (les gènes des immunoglobulines Ig ou des récepteurs des lymphocytes T y sont impliqués) et les lymphomes différenciés.
Les gènes affectés par ces translocations peuvent coder des facteurs de transcription comme MYC (Cleary 1991; Look 1997). Les prototypes de ces translocations sont la t (8;14), t (2;8) et t(8;22) du lymphome de Burkitt qui activent l'expression du gène MYC localisé en 8q24, sous le contrôle de différentes régions enhancer des immunoglobulines et aboutissent à la formation des fusions respectivement IgH-MYC, Igê-MYC et Igë-MYC. MYC est une protéine proto-oncogène à fonction de facteur de transcription. Il appartient à une famille de protéine contenant des motifs helix-loop-helix (HLH) qui permettent la fixation à l'ADN et une dimérisation avec des membres hétérologues de cette famille. Ces réarrangements engendrent la surexpression du gène MYC qui est transcriptionnellement silencieux à l'état normal.
Le gène BCL2, localisé en 18q21, peut être associé avec le gène des chaînes lourdes des immunoglobulines dans des lymphomes folliculaires ayant la t (14;18). BCL2 code pour une protéine mitochondriale impliquée dans la régulation de l'apoptose. Ce gène peut également être associé avec les gènes des chaînes légères des immunoglobulines Igê et ë dans respectivement les translocations t (2;18) et t(18;22), présentes dans diverses proliférations lymphoïdes de la lignée B (Willis et al., 2000).
Les facteurs de croissance, impliqués dans la régulation de l'hématopoïèse normale, sont fréquemment altérés dans les hémopathies malignes. L'interleukine 3 (IL-3) par exemple est
surexprimée dans un sous type de leucémies aiguës pré-lymphocytaires B avec éosinophilie suite à une translocation chromosomique t (5;14). Dans cette translocation, l'IL-3 est mis sous le contrôle des séquences régulatrices des chaînes lourdes des immunoglobulines.
Les translocations aboutissant à la formation d'un gène de fusion, sont généralement retrouvées dans les leucémies myéloïdes et les leucémies lymphoïdes B. Deux gènes de fusion résultent de ces translocations équilibrées, ce qui conduit à l'expression d'une ou deux protéines chimères possédant des éléments de séquence des deux protéines impliquées (comme par exemple les protéines BCR-ABL et ABL-BCR résultant de la translocation t (9;22)). Le plus souvent, une seule des deux protéines de fusion a un rôle évident dans le processus leucémogènes. Cette protéine de fusion présente un gain et/ou un effet dominant négatif sur la fonction des protéines parentales.
De nombreux gènes, incluant les gènes codant pour des régulateurs du cycle cellulaire, de la prolifération et de la différenciation cellulaire, sont impliqués dans le mécanisme de transformation maligne suite à des translocations réciproques. Parmi eux, deux grandes classes peuvent être décrites : les gènes codant pour des facteurs de transcription ayant des motifs de liaison à l'ADN et les gènes codant pour des protéines à activité tyrosine kinase. Les protéines de fusion qui résultent de ces translocations ont un rôle crucial dans la leucémogenèse.
Les protéines de fusion créées suite aux translocations chromosomiques, peuvent agir différemment sur les cellules hématopoïétiques. Les facteurs de transcription chimères perturbent les voies normales de différenciation et les protéines tyrosine kinase activées confèrent un potentiel prolifératif et/ou anti-apoptotique aux cellules hématopoïétiques.
Les facteurs de transcription, réarrangés dans les leucémies humaines, jouent un rôle important dans l'hématopoïèse normale. Les facteurs de transcription impliqués dans ces leucémies incluent CBF (Core Binding Factor)/AML, RARá (Retinoic Acide Receptor á), les membres de la famille HOX et les membres de la famille Ets (Voir pour revue Dash et al., 2001).
Les protéines de fusion impliquant les facteurs de transcription, peuvent être regroupées en deux classes suivant leur structure : celles formées d'un domaine d'oligomérisation et d'un domaine d'activation transrationnel et celles composées d'un domaine d'activation
transrationnel fusionné à un domaine de liaison à l'ADN. Ces deux catégories de protéines de fusion seront décrites et illustrées par des exemples.
La conséquence de ces produits de fusion est la modification de la spécificité de liaison des facteurs de transcription. Le facteur de transcription chimérique agit comme un dominant négatif en inhibant la fonction du partenaire normal (Hiebert et al., 2001). Ce cas de figure est illustré par les exemples suivants :
Le gène TEL/ETV6 (Translocated ETS Leukemia) est un gène de la famille ETS qui code pour des régulateurs de la transcription (Figure7). La protéine TEL est composée d'un domaine d'oligomérisation en N-terminal et d'un domaine ETS de liaison à l'ADN. Dans la translocation réciproque t (12;21)(p13;q22) associée à la leucémie aiguë lymphoblastique pré-B, et dans la translocation t(1;12)(q21;p13) associée à la leucémie aiguë myéloïde, deux protéines de fusion sont formées : respectivement TEL-AML1 et TEL-ARNT.
TEL-AML1 (Romana et al., 1995; Golub et al., 1995) : le gène AML1 (CBFA2) code pour la sous-unité á du "Core Binding Factor" (CBF). Ce complexe a été initialement identifié comme un facteur de transcription liant une région régulatrice dans les "enhancers" de virus leucémogènes murins. Ces facteurs sont caractérisés par la conservation d'un domaine de 128 résidus, le domaine RUNT. Ce domaine est responsable de leur liaison spécifique à l'ADN, de leur localisation nucléaire et de leur interaction avec CBFâ. Des expériences de transfection transitoire ont montré qu'AML1 augmente l'activité de l'enhancer du TCRâ. La protéine de fusion TEL-AML1 comporte les domaines d'oligomérisation de TEL et le domaine RUNT d'AML1. Ont montré que la protéine de fusion TEL-AML1 inhibe l'activité d'un gène indicateur placé sous le contrôle de l'enhancer du TCRâ et interfère avec l'activation de cet enhancer par AML1. Ainsi la protéine TEL-AML1 a un effet dominant négatif sur les propriétés normales d'AML1 et cette activité de répression est dépendante de l'intégrité du domaine HLH de TEL (Hiebert et al., 1996). Récemment, il a été montré que la protéine TEL-AML1 interagit avec le corépresseur SIN3A via le domaine HLH de TEL et via un domaine de 29 acides aminés en aval du domaine RUNT de AML1 (Fenrick et al., 1999).
TEL-ARNT: le gène ARNT
(Aryle hydrocarboné Receptor Nuclear Translocator) code
pour un
facteur de transcription de la famille bHLH-PAS impliqué dans
la réponse à l'hypoxie et la
réponse aux hydrocarbures
aromatiques. La fusion TEL-ARNT a un domaine d'oligomérisation
fonctionnel et la conséquence de cette fusion pourrait être de convertir ARNT en un répresseur transcriptionnel.
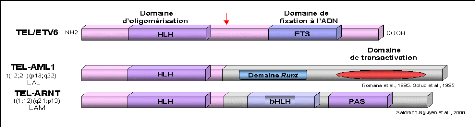
Figure 7:Les fusions de TEL avec les
régulateurs transcriptionnes.
http://www.imgt.org/IMGTeducation/Tutorials/index.php?article=Cancer&chapter=ChromosomalTranslocation&lang=FR&nbr=
2
La flèche rouge positionne le point de cassure hydrocarbon receptor nuclear translocator. bHLH: Region basique «Helix-
loop-Helix».Domaine. Runt: domaine de liaison a l'AND. PAS:per ARNt sim domaine.
Dans les translocations réciproques t(8;21)(q22;q22) et t(3;21)(q26;q22) associées à une leucémie aiguë myéloïde, deux protéines de fusion sont formées, respectivement AML1-ET0 et AML1-EVI-1 (Miyoshi et al., 1993; Mitani et al., 1994), (Figure8).
Les protéines de fusion conservent le domaine RUNT de AML1 fusionné à des domaines contenant des motifs en doigts de zinc. Ces protéines engendrent une accumulation nucléaire plus importante de la protéine CBFâ que celle déterminée par la protéine AML1 sauvage (Tanaka et al., 1998). La répression de la fonction de la protéine AML1 sauvage par les protéines de fusion AML1-ETO et AML1-EVI-1, pourrait être un des mécanismes engendrant la leucémogenèse.
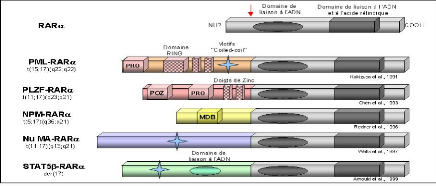
La leucémie aiguë promyélocytaire est associée à différentes translocations chromosomiques impliquant toujours le gène codant pour le récepteur nucléaire à l'acide rétinoïque, RARá (Figure9). Dans toutes les translocations décrites à ce jour, les protéines de fusion conservent le domaine de liaison à l'ADN et à l'acide rétinoïque de RARá. La translocation t (15;17), la plus fréquente, conduit à la formation de la protéine de fusion PML-RARá qui
Contient des motifs d'oligomérisation dans sa partie N-terminale. Trois autres translocations,
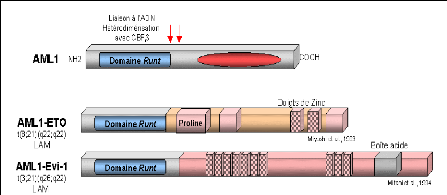
Figure 8: Les fusions AML 1 avec les régulateurs transcriptionnels.
http://www.imgt.org/IMGTeducation/Tutorials/index.php?article=Cancer&chapter=ChromosomalTr
anslocation&lang=FR&nbr=3
Les flèches rouges positionnent les points de cassure.
ETO: Eight Twenty one.Est aussi appelé MTG8: Myeloid Translocation gene on 8. ETO contient trios regions riches en proline retrouvées dans les domains d'activation de la transcription.
Evi-1: Ecotropic viral integration site 1.
ETO et Evi-1 sont des proteins considérées comme des facteurs de transcription car ells contiennent des motifs de type doigt de zinc de liaison à l'ADN.
impliquant toujours RARá, ont été décrites chez de rares patients atteint de leucémie aiguë promyélocytaire : la translocation t(11;17) implique le gène PLZF qui code pour un répresseur transcriptionnel possédant des motifs de dimérisation, la translocation t(5;17) implique le gène NPM codant pour la nucléophosmine et la translocation variante t(11;17) implique le gène NuMA dont la protéine est associée à la matrice nucléaire comme PML et NPM (Wells et al., 1997). Enfin un chromosome dérivatif der(17) implique STAT5â qui code pour un facteur de transcription activé par certaines cytokines.
Les protéines de fusion X-RARá se comportent comme des répresseurs transcriptionnels en se liant avec une plus grande affinité que RARá aux co-répresseurs. Ceci a pour conséquence le blocage de la différenciation (He et al., 1998).
Des gènes codant pour des cofacteurs transcriptionnels sont souvent dérégulés dans les leucémies. Le gène PBX1, codant pour des protéines à homéodomaines, est réarrangé avec le facteur de transcription E2A dans la t(1;19) associée à une leucémie aiguë lymphoblastique pré-B
egtagoterfique eteieloo0:eation deo détutpatideo tiraiipteo
(Kamps et al., 1990). Un autre exemple est la translocation t (7;11) associée à une leucémie
aiguë myéloïde, qui fusionne le gène NUP98 (codant pour une nucléoporine, un composant du
complexe du pore nucléaire qui permet le transport bi-directionnel des protéines et de l'ARN entre le noyau et le cytoplasme) à un gène HOXA9 codant pour une protéine à homéodomaines (Borrow et al., 1996).
|
Figure 9:Les fusions de RARá avec les
régulateurs transcriptionnels dans la leucémie
aigue http://www.imgt.org/IMGTeducation/Tutorials/index.php?article=Cancer&chapter=ChromosomalTranslocation&lang=FR&nbr=4 La fléche rouge positionne le point de cassure. RARá : Retinouc Acid Receptor á. PML : promyelocytic Leukemia.PLZF :Promyelocytic Leukemia Zinc Finger.NPM : Nucleophosmin.Nu MA :Nuclear Mitotic Aparatus. PRO :motifs riches en proline. RNG finger : domaine contenant des motifs en doigts de zinc. POZ :domaine dedimérisation.MDB : domaine de liaison aux microtubules. PML et PLZF sont des protéines qui contiennent des motifs de type doigt de Zinc de liaison à l'ADN .Elles peuvent s'hétérodimériser et co-localiser dans le noyau et dans des structures appelées corps nucléaires. NPM est une protéine nucléolaire.Nu MA est nucléaire et joue un rôle dans la coordination de la mitose. |
Les protéines de fusion résultantes E2A-PBX1 et NUP98-HOXA9, conservent le domaine d'activation transcriptionnel fusionné à un domaine de liaison à l'ADN. Ces deux protéines de fusion sont capables de transformer des fibroblastes NIH3T3 en culture (et de bloquer la diffenciation en immortalisant des myéloblastes dans le cas de E2A-PBX1) probablement en activant la transcription des gènes cibles de PBX1 et HOXA9 (respectivement Monica et al., 1994; Kamps et al., 1996 et Kasper et al., 1999). Dans le cas d'E2A-PBX1, les propriétés transformantes de la protéine de fusion dépendent de la partie amino-terminale d'E2A, responsable de l'activation de la transcription.
Les régulateurs de la transcription, tels que CBP/p300 (Creb Binding Protein) et MLL (Mixed Lineage Leukaemia) sont également associés à des translocations dans les leucémies humaines, ce qui suggère un rôle critique joué par ces protéines dans la leucémogenèse.
1.1.14.7.1 MOZ et les leucimies aigtds myeloieles :
Le gène MOZ (Monocytic leukemia zinc finger protein) qui code pour une protéine ayant une activité acétyltransférase (Borrow et al., 1996; Champagne et al., 2001), est réarrangé avec des gènes codant pour des coactivateurs transcriptionnels tels que CBP (CREB binding protein), p300 (adenovirus E1A-associated 300-kD protein) et TIF2 (Transcriptional intermediary factor 2) (Figure 10). L'acétylation posttraductionnelle des histones est une des caractéristiques de la chromatine transcriptionnellement active. La protéine MOZ possède, entre autres, des motifs en doigt de zinc atypiques retrouvés dans des protéines de levures SAS2, YBF2/SAS3 (something about silencing) et dans la protéine humaine TIP (HIV TAT interactive protein) (Reifsnyder et al., 1996), un domaine histone acétyltransférase (HAT), un domaine acide et dans sa partie Cterminale un domaine riche en sérine-méthionine.
MOZ est réarrangé avec CBP dans la translocation t(8;16)(p11;p13) (Borrow et al., 1996) et dans une translocation cryptique t(8;16) associée à une inversion péricentrique inv(8)(p11;q24) (Chaffanet et al., 1999). Dans la translocation t(8;22)(p11;q13) MOZ est réarrangé avec p300 (Chaffanet et al., 2000; Kitabayashi et al., 2001). Il est intéressant de noter que dans les translocations t(8;16) et t(8;22), les protéines de fusion conservent toute la partie N-terminale de MOZ contenant les motifs de type doigt de zinc et le domaine histone acétyltransférase en phase avec une grande partie de CBP et p300 qui présentent une structure très similaire (des domaines riches en cystéine- histidine et un domaine CREB de fixation à l'AMP cyclique) (Figure 10). CBP et p300 régulent la transcription par remodelage de la chromatine en acétylant les histones des nucléosomes. Une altération fonctionnelle de l'activité spécifique de ces deux protéines, suite aux translocations chromosomiques décrites, doit être un événement critique dans la leucémogenèse. La découverte des réarrangements des gènes MLL et CBP dans la translocation t(11;16)(q23;p13) associée à des syndromes myélodysplasiques et des leucémies aiguës postthérapeutiques, laisse supposer une importante participation du gène CBP, peut-être par un mécanisme "perte de fonction", dans la transformation maligne associée aux translocations t(8;16) et t(11;16).
Enfin, MOZ est réarrangé avec TIF2, un coactivateur des récepteurs nucléaires dans
l'inversion péricentrique inv(8) (p11;q13). La protéine de fusion MOZ-TIF2 retient les domaines de fixation à l'ADN, les motifs de type doigt de zinc et le domaine HAT de MOZ liés aux domaines d'activation de TIF2 et aux domaines d'interaction avec CBP.
Dans les translocations impliquant les protéines à activité tyrosine kinase, la partie 5' du gène codant pour une protéine à activité tyrosine kinase, est remplacée par des séquences provenant d'un autre gène (Rodrigues et al., 1994). Les protéines de fusion sont composées de domaines d'oligomérisation et d'un domaine catalytique (Figure 11 et Figure 12). La (Figure 11) décrit cinq différentes protéines, retrouvées dans des hémopathies malignes, conservant le
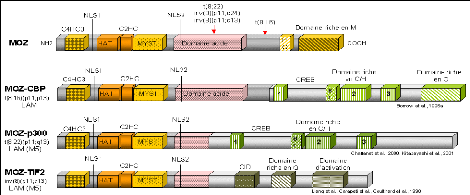
Figure 10: Structure de la protéine
sauvage MOZ et des protéines de fusion MOZ-CBP, MOZ-p300
et
MOZ-TIF2.
http://www.imgt.org/IMGTeducation/Tutorials/index.php?article=Cancer&chapter=ChromosomalTranslocation&la
ng=FR&nbr=5
Les fléches rouges positionnent les points de cassre.MOZ : Monocytic leukemia Zinc finger protein.CBP : CREB binding protein. p300 : adenovirus E1A-associated 300KD protein.TIF2:Transcriptional intermediary factor 2.C4Hc3 et HC: motifs de type doigt de zinc.
NLS1,2 : Séquences de localisation nucléaire.HAT : domaine histone acétyltransférase.Dmaine MYST : domaine d'homologie présent dans les protéines MOZ ,YBF2,SAS2,et TIP .PQ : domaine riche en proline-glutamine. Domaine riche en M :méthionine. Domaines1,2,3 riches en C/H : cystéine/histidine.CREB :domaine de liaison à l'AMP cyclique .Br : bromodomaine.CID :CBP interaction domain
domaine d'oligomérisation en NH2 terminale de TEL/ETV6 fusionné au domaine catalytique de différentes protéines à activité tyrosine kinase. Dans ce cas, l'expression des gènes de fusion est sous le contrôle du promoteur de TEL, dont le profil d'activité et la régulation sont différents de ceux des gènes codant pour les protéines à activité tyrosine kinase.
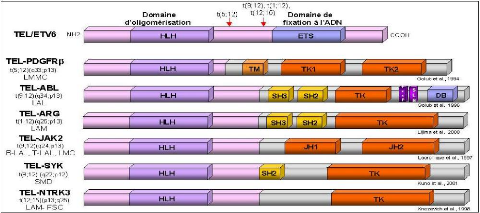
Figure 11 : Les fusions de TEL avec les protéines tyrosine Kinases.
Les fléche rouges positionnent les points de cassure.HLH : domaine d'oligomérisation `'Helix-Loop-Helix''.TM : domaine transmembranaire.TK : domaine tyrosine Kinase.SH : Src Homology : domaine d'homologie semblable à ceux de l'oncogéne SRC .SLN séquence de localisation nucléaire.DB : domaine de liaison à l'ADN.PxxP : région riche en proline. JH JAK Homology region.JH1 : domaine catalytique.JH2 : pseudo-domaine catalytique.TEL : Translocation Ets Leukemia.PDGFR : Platelet-Derived Growth Factor receptor.ABL : Abelson.ARG : ABL-Related Gene .JAK:janus family of tyrosine kinases.NTRK3: neurotrophin-3 receptor.LMMC: Leucémie myélomonocytaire chronique.LAL: Leucémie aigue lymphocytaire.LAM: Leucémie aigue myéloide. LMC :Leucémie myéloide chronique. SMD : Syndrome myélodisplasique.FSC : Fibrosarcome congénital.
ABL est une protéine tyrosine kinase composée d'une partie N-terminale possédant des domaines d'homologies SH semblables à ceux de l'oncogène SRC, impliquée dans la transmission du signal, et une partie C-terminale de liaison à l'ADN (Smith et al., 2002). Dans la leucémie aiguë lymphoblastique avec la translocation t(9;12)(q34;p13) ABL est fusionné au domaine HLH de TEL (Figure 11). Dans diverses leucémies avec la translocation t(9;22), ABL est fusionné au domaine d'oligomérisation de BCR (Figure
12).
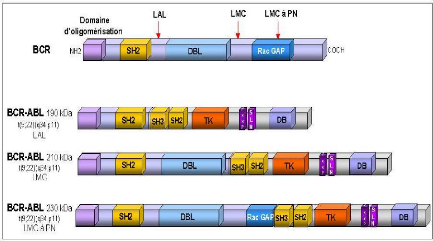
En fonction de l'endroit où se produit le point de cassure sur le gène BCR trois gènes de fusion sont crées (Figure 12). Le gène de fusion qui code pour une protéine BCR-ABL de 190 kDa est retrouvé en majorité dans la leucémie aiguë lymphoblastique (Melo 1996). Il est également associé dans très peu de cas, à des leucémies myéloïdes chroniques atypiques avec monocytose (Melo et al., 1994), des leucémies aiguës myéloïdes et des lymphomes. Le deuxième gène de fusion crée code pour une protéine BCR-ABL plus grande, de 210 kDa, caractéristique de la leucémie myéloïde chronique. Cette protéine de 210 kDa est également retrouvée dans des leucémies aiguës lymphoblastiques, plus rarement dans des leucémies aiguës myéloïdes et des myélomes. La protéine p210 BCR-ABL contient en plus un domaine important DBL-like d'échange
GDP/GTP (domaine d'homologie à la protéine DBL) qui pourrait expliquer les différences cliniques observées entre les différentes protéines BCR-ABL. Enfin une troisième forme BCR-ABL de 230 kDa a été observée en majorité dans les leucémies chroniques à polynucléaires neutrophiles et dans quelques cas de leucémie myéloïde chronique. La protéine p230 BCR-ABL contient pratiquement toute la protéine BCR avec les deux tiers du domaine C-terminal GAP à activité GTPase. Cette différence de structure pourrait expliquer la maturation granulocytaire normale dans ces formes particulières de leucémie myéloïde chronique.
La caractéristique commune entre les protéines de fusion TEL-ABL et BCR-ABL est la présence d'un domaine d'oligomérisation en N-terminal fusionné au domaine tyrosine kinase de ABL. Le domaine d'oligomérisation est crucial pour les propriétés transformantes des protéines de fusion. En effet, suite à leur oligomérisation, il y a activation constitutive du domaine kinase d'ABL et stimulation des voies de transduction similaires.
|
Figure 12: Structure de la protéine BCR et des
protéines de fusion à
http://www.imgt.org/IMGTeducation/Tutorials/index.php?article=Cancer&chapter=ChromosomalTransloc Les fléches rouges positionnent les points de cassure .BCR : Breakpoint Cluster region. DBL : domaine d'homologie avec la Partie qui présente une zone de facteur d'échanges GTP/GDP. Rac GAP : domaine à activité GTPase pour les protéines Rac. DB : domaine de liaison à l'ADN. SLN : séquence de localisation nucléaire.SH : Src homology domain.PxxP : région riche en proline.LAL : Leucémie aigue lymphocyaire.LMC : Leucémie myéloide chronique.PN : Polynucléaires Neutrophiles. |
Les gènes codant pour des récepteurs à activité tyrosine kinase sont fréquemment réarrangés suite à des translocations réciproques (Schlessinger et al., 1992). C'est le cas du récepteur au PDGF (Platelet derived Growth Factor) PDGFRâ dans la leucémie chronique myélomonocytaire avec la translocation t(5;12) (Golub et al., 1994), du récepteur à la neurotrophine 3, NTRK3 (ou TRKC) dans le fibrosarcome congénital et la leucémie aiguë myéloïde associé à la translocation t(12;15)(Figure 11), et du récepteur ALK (anaplastic lymphoma kinase), dérégulé suite à la translocation t(2;5) associé à un lymphome nonhodgkinien (Morris et al., 1994; Hernandez et al., 1999).
Dans les protéines de fusion ainsi créées, il y a perte des séquences codant pour les domaines extracellulaire et transmembranaire des RTKs, domaines responsables respectivement de la fixation du ligand et de leur localisation au niveau de la membrane, et il y a conservation des séquences codant pour le domaine catalytique. Les nouvelles séquences juxtaposées en 5' favorisent la dimérisation, comme dans les fusions TEL-PDGFRâ et TEL-NTRK3 où la partie du gène TEL code pour une région "Helix-Loop-Helix" (HLH) (Figure 11). Ceci est également retrouvé dans les fusions NPM-ALK et TFG-ALK où le domaine NH2 terminal contenant des hélices hydrophobes ou motifs "Coiled-Coil" est responsable de la dimérisation et de l'activation constitutive de la kinase. Les conséquences de ces réarrangements sont importantes pour la fonction du RTK.
Le développement de la génétique médicale au cours des trente dernières années et son implantation dans la pratique médicale ont précédé l'explosion récente de la génétique moléculaire. Deux lecteurs ont principalement contribué à la naissance de cette nouvelle discipline médicale : l'augmentation relative de la pathologie malformative comme cause de morbidité et mortalité infantiles, consécutive à la maîtrise de la pathologie infectieuse, et la possibilité de contrôle des naissances qui compagne d'une exigence de qualité des couples envers ces enfants désirés et programmés, i conseil génétique a pour but d'évaluer le risque de survenue ou de récurrence d'une maladie ou une malformation dans la descendance d'un couple, de proposer à celui-ci les différentes solutions s'offrent à lui pour avoir des enfants normaux et de l'aider dans sa décision. C'est une démarche Jicale originale dans le sens où elle s'adresse le plus souvent à un couple et non à un individu, /elle concerne une tierce personne, l'enfant à venir, qu'elle n'aboutit le plus souvent à aucune thérautique et que la prévention repose sur l'interruption de grossesse.
Cartographie du génome humain et l'identification des gènes à l'origine de la plupart des maladies génétiques fréquentes ont considérablement modifié la pratique du conseil génétique. Il faut actuelle-sont tenir compte non seulement de la disponibilité des tests moléculaires mais aussi des contraintes aosées par la législation entourant la pratique de ces tests.
La cytogénétique humaine (ou génétique chromosomique) est une discipline récente dont l'essor date de 1956 avec la détermination du nombre exact de chromosomes chez l'homme.
Ce dénombrement a été rendu possible pas l'introduction d'un choc hypotonique parmi les différentes étapes de préparation, qui a permis d'obtenir un étalement correct des chromosomes.
En 1970, l'introduction des techniques de marquage en bandes des chromosomes a grandement amélioré la résolution et la sensibilité de l'analyse cytogénétique.
Les techniques classique de caryotypage offrent une vue d'ensemble du génome mais avec une faible résolution (une sous-bande chromosomique correspond au mieux à environ 2Mb sur un caryotype en haute résolution), l'hybridation in situ apport une résolution de l'ordre de quelques Kb(ou même moins si l'hybridation est réalisée sur une fibre d'ADN étiré), ce qui comble le fossé analytique entre cytogénétique et moléculaire.
* La première période (1819-1952) ou "âge des Ténèbres de la Cytogénétique Humaine" débute par le travail d'un cytologiste D.von Hanseman qui en 1819, dénombre le premier 18.24 et plus de 40 chromosomes dans un tissu humain normal. Les comptages des chromosomes étaient difficiles par la superposition des images et la perte d'un ou de plusieurs chromosomes. En 1912, Winiwarter améliore cette technique en prenant des biopsies de testicules prélevés chirurgicalement, immédiatement fixées et coupées à un^ épaisseur de 5 à 7,5 mm qui permettent de garder intact l'ensemble de la garniture chromosomique. Painter conclut en 1923 à l'existence d'un déterminisme du sexe liée à la formule XX pour le sexe féminin ou XY pour les individus de sexe masculin.
* La deuxième période (1952-1959) de la cytogénétique débute avec la découverte hasardeuse, en 1952, de Hsu, du prétraitement par le choc hypotonique technique qui va favoriser le gonflement cellulaire, la dispersion des chromosomes et permet une meilleure individualisation de chaqu'un deux. A l'aide de cette technique, deux cytologistes Albert Levan et Joe Hin Tjio, en 1956, déterminent le nombre chromosomique dans les cellules somatiques humaines qui est de 46.
* La troisième période (1959-1969) commence par la publication le 26 janvier 1959 par le Lejeune, Gautier et Turpin dans les comptes-rendus de l'Académie des Sciences (Paris) de la présence chez neuf enfants mongoliens d'un petit chromosome acrocentrique surnuméraire.
Depuis cette date, un effort collectif des cytogénéticiens et des cliniciens pour rechercher dans les anomalies congénitales une éventuelle étiologie chromosomique. Rapidement, différentes anomalies des nombre et de structure chromosomiques sont décrites.
Lejeune et Coll. (1963) ainsi que Carr (1963) établirent que des anomalies Chromosomiques peuvent être à 1' origine d'avortements spontanés. Les doiées cumulatives montrent que les anomalies chromosomiques trouvées par les cytogénéticiens des plantes et des insectes surviennent spontanément dans les populations humaines avec une fréquence importante donc, on est arrivé à la conclusion que les chromosomes sont très instables, ce qui d'un autre coté à permis l'énorme essor de cette discipline.
*La quatrième période de la cytogénétique (après 1969) est cette du Banding chromosomique. La première technique de Banding est due à Caspersson (1968-1970), qui utilise la moutarde de quinacrine pour individualiser chaque chromosome par l'apparition de bandes caractéristiques: les bandes Q. la découverte des bandes chromosomiques va améliorer considérablement les potentialités d'analyses comme la localisation de points de cassure,
caractérisation des centromères et la nature hétérochromatique de certains segments chromosomique qui n'était pas évidant auparavant. D'autres chercheurs sont rapidement montés au créneau et on observa: le Banding G (Seabright, 1971), le Banding R (Dutrillaux et Lejeune, 1971), le Banding C (Summer et al. 1971), le Banding NOR (Howell et al. 1975).
Les techniques de "haute résolution", mises au point par Yunis en 1976, permettent encore d'améliorer la définition cytogénétique des chromosomes car leur résolution est telle qu'elle permet la détection des microdélétions et microduplications. Par conséquent, cette technologie fine rapprocha les résultats de la cytogénétique de ceux de la génétique formelle.
La cellule Procaryote représentée notamment par les bactéries est constituée d'une
membrane cytoplasmique (associée à une paroi) délimitant le cytoplasme et d'un chromosome circulaire unique directement en contact avec celui-ci.
La cellule Eucaryote possède un noyau pourvu de chromatine et séparé du cytoplasme par une enveloppe nucléaire.
Deux types de chromatine (l'hétérochromatine condensée et l'euchromatine dispersée) forment les chromosomes. Dans cet exposé, nous allons étudier uniquement les chromosomes des cellules humaines qui sont des cellules Eucaryotes.
Les chromosomes sont constitués d'acide désoxyribonucléique (ADN) associé à des
protéines histones. L'ADN, porteur de l'information génétique est un polynucléotide (entité d'un grand nombre de nucléotides) et chaque nucléotide contient trois composants : une base hétérocyclique azotée (bases puriques : Adénine, Guanine ; bases pyrimidiques : Cytosine, thymine), un pentose et une molécule d'acide phosphorique.
La molécule d'ADN s'organise en double hélice stabilisée grâce à la complémentarité entre les bases Guanine (G) Cytosine (C) et les bases Adénine (A) Thymine (T).Les gènes sont des segments définis de la molécule d'ADN et correspondent à des séquences polynucléotidiques particulières.
Il faut également mentionner que l'ADN de l'euchromatine est accessible à la transcription tandis que l'ADN de l'hétérochromatine n'est pas « actif ».
Les protéines histones se rassemblent pour former des nucléosomes (ensemble de huit
histones). L'ADN s'enroule autour des nucléosomes aboutissant à un nucléofilament en « collier
de perles ». Un degré de compaction supplementaire defini un filament de chromatine de 30nm (ou solénoide), c'est à dire la structure primaire du chromosome.
Cette structure déployée et linéaire peut se modifier au cours de la vie de la cellule qui est réglée par le cycle cellulaire. Le cycle cellulaire commence à la naissance de chaque cellule (issue de la division ou mitose de la cellule mère), se poursuit au cours de la croissance cellulaire (ou interphase) et se termine par une nouvelle mitose donnant naissance a une cellule fille ou par une élimination de la cellule.
La mitose comprend cinq phases :
télophase. Elle abouti à une répartition de l'ADN de la cellule mère en les deux cellules filles. Au cours de la mitose, le chromosome se condense progressivement, la fibre chromosomique linéaire se replie sur elle même pour former une structure plus compacte.
Ainsi, c'est au moment de la métaphase que le chromosome est au maximum de sa condensation et qu'il est le facilement observable. Il est bien differencie, constitue de deux chromatides reliées entre elles au niveau du centromere. Le centromere ou constriction primaire est un point de repère cytologique, permettant de diviser les chromosomes en deux bras, un bras court (symbolysé p) et un bras long (symbolysé q)
des chromosomes métaphasiques le caryotype est réalisé.
Il existe des colorants qui permettent de visualiser la chromatine grâce à leur affinité pour
et/ou les protéines qui lui sont associées. Le plus couramment utilise est le Giemsa qui est une association de trois colorants de base et qui donne une coloration rose violacée de la chromatine en lumière visible. (figure 13)
Il existe également des colorants fluorescents qui
permettent de visualiser spécifiquement l'ADN
car ils s'intercalent
entre les bases de la double hélice. C'est le cas de la moutarde de
quinacrine
(figure 13 du DAPI (4',6-diamino ou de
I'lodure de Propidium. (Figure 13)
Figure 13 : métaphase coloré aux colorations visualiser du chromosome du gauche a droit Moutarde de quinacrine et l'iodure de propidium.
Comme on l'a vu plus haut, les chromosomes ne sont visibles que pendant une courte
période du cycle cellulaire, lors de la division cellulaire (mitose ou méiose). Toutes les techniques cytogénétiques visent donc à obtenir un maximum de cellules bloquées à ce stade.
Pour cela, il est nécessaire d'avoir des cellules en phase de multiplication active, soit spontanément (cas des villosités choriales ou de certaines cellules tumorales) soit par une culture préalable le plus souvent (fibroblastes, tout type cellulaire capable de se diviser) parfois associée à une stimulation (lymphocytes sanguins). La durée de cette culture est variable en fonction du type cellulaire considéré et de la quantité de matériel biologique disponible au départ.
L'étape suivante consiste à bloquer les cellules en métaphase afin de pouvoir observer les chromosomes. Pour cela, on utilise un poison du fuseau de division (classiquement c'est la Colchicine qui est utilisée ou son équivalent synthétique la Colcémide) qui empèche la progression de la mitose vers l'anaphase
Les cellules sont alors plongées dans une solution hypotonique ce qui entraîne leur gonflement. Cette étape est indispensable à l'obtention d'un étalement correct des chromosomes.
Enfin, la dernière étape consiste en une fixation par un mélange d'alcool et d'acide acétique. La préparation est alors étalée en laissant tomber une goutte de la suspension cellulaire sur une lame.
Cas particulier : certains types cellulaires comme les fibroblastes adhèrent au support lors de la culture. On peut obtenir des métaphases à partir de ces cellules sans les détacher de leur support, toutes les étapes précédentes étant réalisées directement sur la surface de culture (sauf l'étalement qui est bien sûr inutile dans ce cas).
Lorsque l'on colore des préparations chromosomiques avec du Giemsa, les chromosomes prennent un aspect rose violacé à peu près homogène sur toute leur longueur. On ne peut donc les distinguer les uns des autres que par leur taille et leur forme. Cependant, ces critères sont
insuffisants pour assurer la reconnaissance et l'interprétation correcte des anomalies chromosomiques.
Pour reconnaître spécifiquement chaque paire chromosomique, on utilise donc des techniques de marquage particulières qui permettent d'obtenir une coloration inhomogène des chromosomes par le Giemsa et l'apparition de bandes. C'est la succession de bandes sombres et claires le long d'un chromosome, identique chez tous les individus pour un chromosome donné,

Figure 14 : Les techniques de
marquage en bandes des chromosomes (banding) du gauche à droit
les
bandes G, R et G-R au même temps.
http://cvirtuel.cochin.univ-paris5.fr/cytogen/1-2_fichiers/GTG_L.jpg
qui en permet l'identification précise, selon le même principe qu'un code à barres.
Il existe deux principales techniques de marquage en bandes des chromosomes (banding), utilisées en routine :
Les bandes G, obtenues après traitement des chromosomes par la trypsine (figure 14). Les bandes R obtenues par un traitement à la chaleur. (Figure 14).
Dans les deux cas, les bandes ne deviennent visibles qu'après une coloration avec le Giemsa. Ces deux techniques donnent un marquage réciproque, c'est-à-dire que là où l'on obtient une bande sombre avec l'une des deux techniques, on observe une bande claire avec l'autre (figure15).
D'autres techniques de marquage complémentaires existent qui permettent d'analyser certaines régions particulières du génome :
Bandes C : cette coloration par le Sulfate de Baryium permet de mettre en évidence l' hétérochromatine constitutive, qui correspond à des régions non codantes du génome comme les régions centromériques. (figure15).

Figure 15 : Autres techniques de
marquage complémentaires bandes C a gauche et NOR a
droit.
http://cvirtuel.cochin.univ-paris5.fr/cytogen/1-2_fichiers/modeleGTG_RHG_L.jpg
NOR : cette technique consiste en un dépôt de nitrate d'argent qui met en évidence les
organisateurs nucléolaires. Ces structures correspondent aux régions du génome contenant les gènes qui codent pour les ribosomes.
Les chromosomes métaphasiques sont constitués d'un bras court (noté p) et d'un bras long (noté q), reliés entre eux par le centromère qui correspond à un étranglement situé à un niveau variable du chromosome et qui sert de point d'attache au fuseau de division pendant la division cellulaire.
Plusieurs critères vont permettre de reconnaître et de classer les chromosomes :
*La taille Par convention, les chromosomes sont classés du plus grand au plus petit chromosome
. *l'index centromérique, c'est-à-dire le rapport entre la taille du bras court et la taille totale du Cet index permet de reconnaître trois familles de chromosomes : (figure 16).

n de l'indice
ol_L.jpg
- Les chromosomes métacentriques dont les deux bras ont une taille à peu près équivalente.
- Les chromosomes submétacentriques qui ont un bras franchement plus petit que le bras long Les
chromosomes acrocentriques dont le bras court est quasi inexistant (on ne trouve sur ces bras courts que les gènes codant pour les ribosomes; ces gènes étant présents à plusieurs centaines d'exe
mplaires double génome, la perte du bras court d'un chromosome acrocentrique n'a pas de conséquence clinique)
*les bandes chromosomiques,qui sont caractéristiques de chacune des paires.
Le nombre de bandes visibles est variable d'une mitose à l'autre et dépend du niveau de condensation du chromosome. Plus les chromosomes sont condensés, moins on peut observer de bandes et moins l'analyse permet de dépister des anomalies de petite taille. Le nombre de bandes par lot haploïde (c'est-à-
dire pour 23 chromosomes) permet de définir la résolution de l'analyse cytogénétique ; un caryotype standard a une résolution de 300 à 550 bandes ; certaines techniqu
es dites de haute résolution permettent d'augmenter le nombre de bandes visualisées en bloquant les chromosomes au tout début de
leur condensation : on peut ainsi obtenir 800 ou même 1000 bandes par lot haploïde. Ces techniques de haute résolution sont de réalisation et d'interprétations plus délicates que le caryotype standard, mais permettent la mise en évidence d'anomalies de taille beaucoup
plus réduite (figure 17). hromosomique de
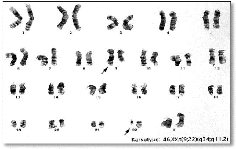
X t (9;22)(q34;q11.2)
egiagotertique eteieloo0:eation deo détutpatideo tiralipteo
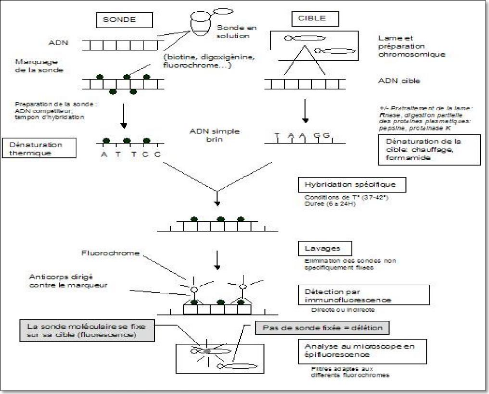
Le principe repose sur l'utilisation d'une sonde moléculaire, c'est-à-dire une petite séquence d'ADN (ou d'ARN) dont l'emplacement normal est connu dans le génome et qui est marquée chimiquement de façon à pouvoir être repérée par la suite. Cette sonde est mise en contact avec les chromosomes d'une mitose (ou de noyaux interphasiques) et va s'hybrider (se fixer) spécifiquement au niveau de sa séquence complémentaire. On peut alors visualiser la sonde au microscope dont l'emplacement identifie précisément la région chromosomique dont elle est complémentaire.
Les sondes sont marquées soit avec une molécule fluorescente, soit avec une haptène (molécule qui peut être reconnue par un anticorps). Dans le premier cas, la sonde est directement visible au microscope à fluorescence, tandis que dans le second, une étape supplémentaire de révélation avec un anticorps fluorescent est nécessaire.
11.3.2.2 Diff~rents types de sondes sont utilisies : 11.3.2.2.1 Sondes centromeriques :
Elles s'hybrident au niveau des centromères des chromosomes. Les séquences dont elles sont complémentaires sont naturellement présentes en un grand nombre d'exemplaires au niveau des centromères ; le signal obtenu est donc en général intense car la sonde s'hybride sur chacune des séquences complémentaires présentes. Ces sondes sont surtout utiles pour dénombrer les chromosomes, aussi bien en métaphase qu'en interphase et pour identifier l'origine de chromosomes marqueurs.
11.3.2.2.2 Sondes de peinture chromosomique :
Elles sont constituées d'un ensemble de sondes de petite taille qui couvrent l'ensemble du chromosome. Ces sondes sont obtenues après isolement et marquage de l'ADN d'un chromosome ; leur réalisation ne nécessite pas de connaître la séquence de cet ADN. Après hybridation, on observe un marquage de tout le chromosome. Il existe également des peintures spécifiques d'un bras ou même de quelques bandes chromosomiques. Ces sondes sont très utiles pour interpréter certaines translocations complexes, mettre en évidence des échanges de petite taille, identifier précisément l'origine d'un fragment non identifié.
11.3.2.2.3 Sondes locus specifique :
Comme leur nom l'indique, ces sondes de petite taille permettent d'identifier une région très précise du génome. Elles sont obtenues par marquage de l'ADN cloné dans différents
egiagotertique et laid/ Caton deo détutpatideo tiralipteo
vecteurs (plasmides, cosmides, YACS, BACs...). Leur intérêt principal réside dans la mise en évidence rapide de remaniements impliquant une région chromosomique précise (microdélétions, translocations, inversions ...)
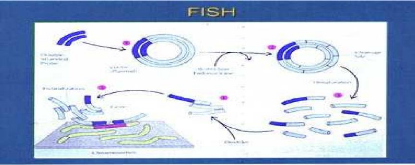
Figure 18 : Principales applications de l'hybridation in situ M.jeaunepier et al.
GENETIQUE MEDICAL ; ISBN2 29400812X ;; 2004.
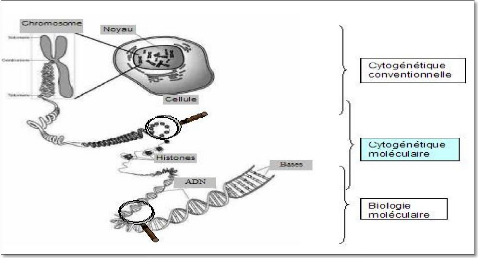
La cytogénétique moléculaire est née de la rencontre de la biologie moléculaire et de la cytogénét La cytogénétique moléculaire est née de la rencontre de la biologie moléculaire et de la cytogénétique conventionnelle, elle se situe à une échelle de résolution intermédiaire entre ces deux disciplines (figure 19). Cet outil a permis de couvrir en partie un champ d'études inexploité jusque là, en comblant l'intervalle qui existe d'une part entre la biologie moléculaire (quelques centaines de Kb) et d'autre part le pouvoir de résolution de la cytogénétique (environ 2 à 5 Mb) (Blume-Jensen et al., 2001). La cytogénétique moléculaire d'apparition relativement récente dont l'outil principal est l'Hybridation In Situ en Fluorescence (FISH) sur préparation chromosomique a révolutionné l'approche traditionnelle de la cytogénétique. Le pouvoir de résolution de la FISH (dans certaines conditions moins de 1Kb) permet une analyse fine de la structure des chromosomes. Les applications en sont multi ples tant en recherche (cartographie physique, hybridation croisée que conventionnelle, elle se situe à une échelle de résolution intermédiaire entre ces deux disciplines (figure 19). Cet outil a permis de couvrir en partie un champ d'études inexploité jusque là, en comblant l'intervalle qui existe d'une part entre la biologie moléculaire (quelques centaines de Kb) et d'autre part le pouvoir de résolution de la cytogénétique (environ 2 à 5 Mb). La cytogénétique moléculaire d'apparition relativement récente dont l'outil principal est l'Hybridation In Situ en Fluorescence (FISH) sur préparation chromosomique a révolutionné l'approche traditionnelle de la cytogénétique. Le pouvoir de résolution de la FISH (dans certaines conditions moins de 1Kb) permet une analyse fine de la structure des chromosomes. Les applications en sont multiples tant en recherche (cartographie physique, hybridation croisée inter-espèce...) qu'en diagnostic (caractérisation ou détection d'un remaniement chromosomique de petite taille...).
Les laboratoires de cytogénétique utilisent actuellement de manière courante ces nouvelles méthodes devenues indispensables en complément de la cytogénétique classique pour les caryotypes constitutionnels (pré et postnatals) et sur tumeurs pour les anomalies chromosomiques acquises.
~~d~g~~~~~~ae el ~~~~~~~~caI~n ~~ ñ6~~pa~~~~ ~~~~çne~
mique sont indiquées chromosome
omes homologues.
2004.
Le principe général de la FISH est illustré par la (figure 20).
Les techniques d'hybridation in situ sont fondées sur la propriété de réassociation spécifique des acides nucléiques. Une sonde dénaturée (ADN simple brin marqué) en solution peut s'hybrider spécifiquement avec sa séquence cible (préparation chromosomique dénaturée) grâce à la complémentarité des bases nucléotidiques. La sonde s'apparie par des liaisons hydrogène établies selon les critères de Watson et Crick. Les hybrides infidèles et les molécules de sonde non hybridées sont éliminés par l
avages, puis les hybrides spécifiques sont révélés, en général par immunodétection et enfin l'observation s'effectue grâce à un microscope à épifluorescence. L'introduction de marqueurs
non radioactifs a permis de s'affranchir de la lourdeur des techniques utilisant des radio-isotopes réservés aux laboratoires de recherche. De nombreuses améliorations ont permis d'obtenir une qualité et une reproductibilité remarquable (sondes clonées, amélioration des solutions d'hybridation....). Son utilisation pratique est relativement facile et rapide.
De nombreuses sondes sont actuellement disponibles et commercialisées, elles sont principalement de 4 types : (figure 22
- peinture chromosomique (spécifique d'un chromosome entier ; figure A et E),
- locus spécifique (correspondant à un petit fragment chromosomique ; figures B et F)
- télomérique (figure C),
- centromérique (figure D)
En dehors d'applications particulières telle que l'HCG dont la sonde correspond à un génome entier, l'ADN est obtenu après clonage dans un vecteur ou par synthèse in vitro (tableau IV). Une méthode permet par diverses approches d'obtenir un caryotype "en couleur" grâce à l'utilisation de l'informatique et du traitement d'image : chaque chromosome est repéré par une
combinaison spécifique de fluorochromes et donc une couleur qui lui est spécifique (Multi-FISH, Planche I, Figure E).
Tableau IV : différents types de sondes avec
indication des tailles maximales
M.jeaunepier et al. GENETIQUE
MEDICAL ; ISBN2 29400812X ; 2004.

11.3.2.3.3 Marquage des sondes :
Pour permettre leur détection spécifique les sondes sont marquées soit par des procédés
enzymatiques (figure 21) par introduction d'haptène (biotine, digoxigénine), soit par procédés chimiques. Actuellement de nombreux fournisseurs proposent des sondes directement marquées par des fluorochromes modifiés.
egiagotertique et laid/ Caton deo détutpatideo tiralipteo
Elle est habituellement identique à la cytogénétique classique : préparation de lames de chromosomes en métaphase ou prométaphase. Des préparations spécifiques particulières sont parfois employées : études en interphase, chromatine étirée, peignage moléculaire de l'ADN, coupes tissulaires...
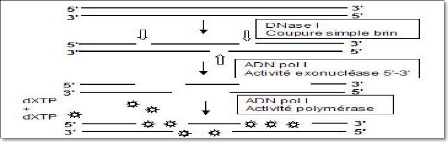
.oupures : actionvité exonucléasique arqués (dxtp).
; 2004.
11.3.1.3.5 Detection des sondes :
- Détection par immunofluorescence :
Les sondes marquées par des haptènes sont révélées par immunoréaction grâce à des anticorps spécifiques couplés à des fluorochromes. Le montage peut comporter un seul anticorps (immunodétection directe, (
figure 20) ou plusieurs anticorps superposés pour amplifier le signal ( immunodétection indirecte). La difficulté est ici d'obtenir un rapport signal/bruit satisfaisant avec un minimum de bruit de fond (Knezevich et al., 1998).
- Détection directe :
Les sondes étant directement fluorescentes, la détection ne nécessite pas d'amplification du
signal ; son utilisation en est donc plus facile, plus rapide et moins parasitée par le bruit de fond 11.3.2.3.6 Analyse au microscope
~
La FISH nécessite un microscope à épifluorescence équipé de filtres appropriés aux nombreux fluorochromes disponibles : isothiocyanate de fluorescéine (vert-jaune), rouge Texas, rhod amine (rouge), coumarine (bleu), etc.. ). Les molécules émettent une lumière lorsqu'elles sont excitées par une longueur d'onde appropriée. Des filtres sont disponibles avec plusieurs bandes passantes permettant de détecter plusieurs couleurs durant la même observation (figure 22) , F1: double bande passante, figure F2 : triple bande). L'observation de couleurs difficilement perceptibles par l'oeil humain (cyanine, rouge sombre) ou la distinction de combinaisons
nombreuses couleurs (Multi-FISH, figure 22 E) bénéficient indéniablement de l'avantage des caméras numériques de capture couplées au microscope. Il en est de même des logiciels adaptés au traitement de l'image.
11.3.2.3.7 Application clinique :
La cytogénétique moléculaire intéresse tous les domaines de la cytogénétique classique : constitutionnelle : post et prénatale ainsi que récemment le diagnostic préimplantatoire ; analyse des anomalies acquises : onco-hématologie, tumeurs solides (tableau V). Devant les différentes possibilités techniques offertes par la cytogénétique, le prescripteur peut être dérouté dans sa démarche diagnostique : caryotype conventionnel standard ou en haute Résolution, sur fibroblastes, recherche de la fragilité de l'X, FISH avec recherche de microdélétions, association d'une étude en biologie moléculaire, Multi-FISH, étude
Tableau V : Principales indications de la FISH
Cytogénétique. (Le diagnostic
préimplantoire n'est pas
indiqué dans ce dernier)
M.jeaunepier et al. GENETIQUE
MEDICAL ;ISBN2 29400812X ;; 2004.

"pantélomèrique"... sont autant d'analyses disponibles : un minimum de connaissances est nécessaire aux médecins impliqués notamment dans le cadre diagnostique du retard mental. Cette connaissance associée à une réflexion clinique appropriée, permet de prescrire à bon escient en collaboration avec le cytogénéticien les examens adaptés.
La caractérisation des anomalies fait appel aux mêmes techniques évoquées précédemment. L'étude des anomalies chromosomiques acquises sur tumeurs à largement bénéficiée du développement de la cytogénétique moléculaire, d'une part les aberrations sont souvent
nombreuses et complexes, d'autre part les préparations chromosomiques sont parfois de qualités insuffisantes ne permettant pas une analyse complète avec les techniques conventionnelles. Par
ailleurs, les hémopathies malignes
remaniement chromosomique spécifique (translocation, inversion) facilement détectable par FISH. Les anomalies détectées
diagnostique, pronostique et parfois dans le suivi du traitement pour en contrôler l'efficacité.
La FISH est un complément précieux du fait de sa précision et de sa facilité. Par ailleurs, du
Point de vue fondamental, le ou les gènes jouant un rôle dans le processus évolutif de la cancérogenèse.Sondes localisées aux points de cassure et révélées par des fluorochromes différents (FISH bicolore) sur noyaux. Par ailleurs, la FISH
surveillance de greffes de moelle avec donneur de sexe différent par recherche de chimérisme XX/XY. La complexité des remaniements chromosomiques rencontrés dans les tumeurs solides et hématopoïétiques, et l'intérêt du point
la cancérogenèse ont amené les cytogénéticiens spécialisés dans ce domaine à
intensive les nouveaux outils basés sur le principe de la FISH et qui permettent une étude globale du génome étudié : Multi-FISH, CGH, biopuces à ADN

egiagotertique et laid/ Caton deo détutpatideo tiralipteo
L'incidence en Algérie de différentes hémopathies a été pendant de nombreuses années impossibles à estimer en raison du nombre insuffisant de structures spécialisées et de l'étendue du pays. Actuellement de nombreux services se sont développés au niveau du territoire national, dont 12 de statut hospitalo-universitaire harmonieusement réparties au nord du pays, ce qui a permis un meilleur accès des patients au diagnostic et au traitement et par la même une meilleure connaissance épidémiologique.
L'incidence spécifique globale (patients âgés de plus de 14 ans) de la leucémie myéloide chronique (LMC) est de 0.41/100.000ht, elle a doublé entre 1994 et 2004 passant de 0,19 à 0,4 elle reste cependant très en deca de l'incidence observée dans les pays occidentaux ou elle se situ 1,4 et 2,2 par contre l'âge médian des patients n'est pas différent il est de 44 ans et entre 30 et 50 ans dans les pays occidentaux. Ainsi peut penser que l'incidence plus faible dans notre pays n'est pas en rapport avec la pyramide des âges comme celle peut être évoqué pour les LNH ganglionnaires de l'adulte, le MN et les LAM.la connaissance de l'incidence de la LMC est primordiale car si depuis fin 2007 l'indication de l'allogreffe qui représentait avant cette date le traitement à visée curatrice le plus utilisé dans notre pratique, actuellement il a été remplacé par l'utilisation des inhibiteurs de la tyrosine kinase essentiellement l'imatinib.
Cependant le cout élevé de ce traitement va s'accroitre au cours des années à venir en raison de son efficacité sur le contrôle de la maladie mais non sa guérison, nécessitant la poursuite du traitement avec une augmentation exponentielle des quantités nécessaires, de plus il nécessite à terme pour mesurer son efficacité l'amélioration des moyens de surveillance biologique basé sur la cytogénétique et la biologie moléculaire.
Le fait important qui ressort de l'étude des aplasies médullaires (AM) dans notre pays est la prédominance nette des patients âge médian est de 26 ans avec deux tiers des patients âge de moins de 30 ans, cette répartition est observée en Asie ou cependant l'incidence globale est plus élevée allant de 3,9 à 7,4/100.000 ht alors qu'elle n'est que 0,30 dans noyer pays, en France l'incidence et de 1,4/100.000 ht avec une répartition bimodale un second pic étant observé après 50 ans non retrouvé en Algérie. Une étude prospective cas-témoins réalisée en Thaïlande a montré que le risque de survenue d'AM est corrélé avec un nombre faible d'années d'étude et inversement corrélé avec les revenus mensuels. Par ailleurs 86% de nos patients présentant une forme sévère ce qui pose un problème de prise en charge thérapeutique basée sur la greffe
~~d~g~~~~~~ae el ~~~~~~~~caI~n ~~ ñ6~~pa~~~~ ~~~~çne~
allogénique entre en raison de l'âge de nos patients et du taux élevé de compatibilité HLA, ce qui potentiellement aurait représenté en 2004 environ 50 patients atteints d'AM acquise pouvant bénéficier d'une greffe alors que seulement 17 ont été greffés ou à défaut par la traitement immunosuppresseur incluant au mieux l'utilisation du sérum anti-lymphocytaire associé à la Ciclosporine par ailleurs en ce qui concerne les formes congénitales dominées par l'anémie de fanconi retrouvée dans 13% des cas, il est inférieur à celui observé dans la littérature, or en raison du taux de consanguinité dans notre pays, il devrait être plus élevé, d'où l'importance de pratiquer un caryotype chez tout sujet présentant une AM avant l'âge de 20 ans en raison des formes à révélation tardive.
Ces études bien que non exhaustives ont permis d'avoir une approche de l'incidence dans notre pays de cinq hémopathies parmi les plus importants et de pouvoir les comparer avec les autres pays. Leur incidence est dans l'ensemble plus faible que dans les pays occidentaux ce qui peut être expliqué par la pyramide des âges pour les LNH ganglionnaires, le MM, les AM mais pas pour la LMC. L'un des facteurs évidents est sous estimation des cas du au biais du recueil des donnée, mais aussi d'autres facteurs notamment géographiques et environnementaux seraient intéressants de recherche par des études épidémiologiques prospectives, non seulement algériennes mais aussi maghrébine. Pas ailleurs ces études ont surtout le mérite de pouvoir quantifier les moyens thérapeutiques actuels et à venir d'une part celui des thérapeutiques ciblées : inhibiteur des tyrosines Kinase essentiellement imatinib et des anticorps monoclonaux représenté par le rituximab et d'autre part la nécessité évidente de créer des unités de greffe tant pour l'allogreffe que pour l'autogreffe.
en effet la capacité actuelle de l'unique centre qui est de 150 greffe en moyenne par an dont 70% d'allogreffe et 30% d'autogreffe ne peut répondre aux demandes telles qu'elles ont été évaluées pour les années 2004 et 2005 et qui à ce jour ont. De plus elles permettent de mettre l'augment. De plus elles permettent de mettre l'accent sur la nécessité de développer des plateaux techniques répartis au centre, à l'est et à l'ouest du pays, ce qui permettrait l'amélioration des diagnostics et du suivi des patients notamment la généralisation des colorations cytchnimiques, le développement de l'immunomarquage, de la cytogénétique constitutionnelle et hématologique et de la biologie moléculaire.
I".5 Approche épidémiologiques des leucémies aigues myéloides en
Les leucémies aigues myéloides (LAM) sont un groupe hétérogènes d'hémopathies malignes caractérises par l'expansion clonale au
Blastes( appartenant à la lignée myéloide( granulocytaire, monocytaire, érythroblastique ou mégacaryocytaire) donnant lieu à différents
cellule souche leucémique.
En Algérie, en l'absence d'un registre national
disponibles. L'objectif de travail, initié par le bureau de la société Algérienne d'Hématologie et de transfusion sanguine (SAHTS), est d'étudier le profil épidémiologique des LAM et leurs moyens diagnostiques.
1".5.1 Répartition selon le sexe
On notre une prédominance des LAL pour le sexe masculin, le sex ratio est 1,68 Redhouane et al ; 2004).
Le sexe ratio sera plus élevé pour la tranche d'
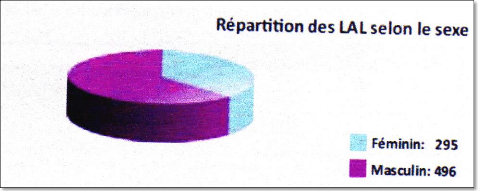
Etude de la fratrie :
Tableau VI : Le taux médian de frères et soeurs est 5.5, avec des extrêmes allant de 0 à 14
|
Fratrie |
3 à 3 |
4 à 6 |
7 à 10 |
> à 10 |
NP |
Total |
|
Nombre % |
33 |
140 |
131 |
29 |
458 |
791 |
|
4.2 |
17.6 |
16.5 |
3.5 |
58 |
100 |
11.5.2 Profession :
Les professions en rapport avec le bâtiment et les produit agricoles semblent plus élevées ceci
mérite d'être vérifier par une étude prospective (Tableau VII)
Tableau VII: répartition selon la profession
|
Profession Non précisé |
Nombre 216 |
|
Sans profession |
321 |
|
Domaine agricole |
42 |
|
Domaine bâtiment |
32 |
|
Corps constitué |
29 |
|
Administration |
16 |
|
Universitaires |
19 |
|
Etudiants, Lycéens.. |
20 |
|
Commerçants |
32 |
|
Chauffeurs |
17 |
|
Autres |
47 |
|
Total |
791 |
11.5.3 Diagnostic :
Etude cytchnimiques : Le diagnostic de LAL a été posé après une étude cytchnimique utilisant la
coloration des péroxydases ou de noir soudan, dans 64% des cas, dans environ 01 cas sur 03 le diagnostic a été porté sur une cytologie seulement (tableau VIII)
Tableau VIII: Fréquence de l'étude cytchnimique.
|
Nombre |
% |
|
|
Non précisé |
67 |
4.4 |
|
Faite |
512 |
|
|
64 |
||
|
Non faite |
250 |
31.6 |
|
Totale |
791 |
100 |
La cytométrie en flux été effectuée dans 79 cas (10%) (Tableau IX).
Tableau IX: Fréquence de l'immunophénotypage.
|
Nombre |
% |
|
|
Non précise |
7 |
01 |
|
Faite |
79 |
10 |
|
Non faite |
705 |
89 |
|
Total |
791 |
100 |
On note une augmentation croissante de l'incidence chez l'adulte qui passe de 0,27/100.000 ha
en 1994 à 0,48 en 2003 (tableau X).
Tableau X: Etude de l'incidence des LAL de l'adulte.
|
1994 |
1995 |
1996 |
1997 |
1998 |
1999 |
2000 |
2001 |
2002 |
2003 |
Total |
|
|
CPMC |
9 |
16 |
16 |
12 |
19 |
11 |
9 |
8 |
23 |
11 |
134 |
|
Setif |
17 |
13 |
12 |
6 |
13 |
11 |
11 |
15 |
11 |
20 |
129 |
|
Constantine |
3 |
10 |
8 |
9 |
14 |
6 |
10 |
14 |
18 |
12 |
104 |
|
Oran |
8 |
11 |
12 |
9 |
10 |
13 |
11 |
9 |
10 |
3 |
97 |
|
Blida |
4 |
4 |
5 |
3 |
7 |
8 |
11 |
8 |
15 |
65 |
|
|
Tizi ouzou |
4 |
1 |
1 |
6 |
7 |
6 |
7 |
4 |
7 |
9 |
52 |
|
Annaba |
2 |
2 |
1 |
1 |
4 |
9 |
10 |
5 |
13 |
47 |
|
|
B messous |
4 |
4 |
2 |
2 |
10 |
4 |
3 |
4 |
8 |
6 |
47 |
|
Tlemcen |
1 |
6 |
6 |
2 |
7 |
4 |
7 |
3 |
1 |
37 |
|
|
SBA |
2 |
2 |
1 |
4 |
2 |
5 |
5 |
1 |
6 |
7 |
35 |
|
HCA |
1 |
2 |
2 |
4 |
4 |
8 |
3 |
1 |
3 |
2 |
30 |
|
Batna |
2 |
3 |
1 |
1 |
1 |
1 |
1 |
5 |
14 |
||
|
Total |
52 |
65 |
68 |
66 |
85 |
83 |
81 |
85 |
103 |
104 |
791 |
|
Pop en |
18.5 |
19 |
19.4 |
19.8 |
20.2 |
20.2 |
20.6 |
20.8 |
21.2 |
21.5 |
|
|
106> 15 ans |
|||||||||||
|
Incidence |
0.27 |
0.59 |
0.35 |
0.33 |
0.42 |
0.42 |
0.39 |
0.40 |
0.45 |
0.48 |
|
|
Adulte |
|||||||||||
HS Approche epidemiologique de la leucemie myeloide
chronique en
Algerie :
Les objectifs principaux de cette étude sont les suivant :
· Connaitre la prévalence et l'incidence de la LMC en Algérie.
· Etablir la répartition de cette affection selon les différentes régions du pays
· Etablir la répartition selon les caractéristiques des patients (âge, sexe, lieu de résidence et profession).
· Evaluer les chances de trouver un donneur apparenté HLA compatible, les patients atteints LMC.
> Prévalence :
La prévalence absolue de la LMC en Algérie en 2004 est de 472 cas, sa prévalence relative est de 1,8cas/ 100.000 habitants par année.
egiagotertique et eiao,Wication deo détutpatideo tiralipteo
> Incidence :
La répartition des nouveaux cas de LMC par année sur la période allant de 1994 à 2004 est de 53 cas en 1994, 69 cas en 1995, 85 cas en 1996, 68 cas en 1997, 89 cas en 1998, 74 cas en 1999, 78 cas en 2000, 99cas en 2001, 118cas en 2002, 105cas en 2003, 130 cas en 2004. Nous avons en moyenne 88 nouveaux cas LMC par année. Le taux d'incidence annuel est en progression
Des patients présentant des hémopathies malignes hospitalisés au service d'ophtalmologie Clinique HAMMOU BOUTLELIS au laboratoire de cytogénétique ont fait l'objet d'une étude cytogénétique sur prélèvement du sang périphérique et sur la moelle osseuse.
L'analyse chromosomique nécessite la préparation de lames riches en étalement chromosomiques analysables, ce qui est réalisé en trois étapes :
- Mise en culture de lymphocytes stimulés par un agent mitogène ou culture primaire de Fibroblastes.
- Accumulation de cellules en métaphase ou en pro métaphase, stade du cycle cellulaire auquel les chromosomes sont les mieux individualisés.
- Récolte des cellules et préparation des étalements chromosomiques.
Le sang, facile à prélever, est la source de cellules la plus utilisée en cytogénétique humaine.
Chez les mammifères, les lymphocytes B et T sont les seules cellules du sang susceptibles d'être transformées en cellules actives et proliférantes.
Cette transformation lymphoblastiques induite in vivo par les antigènes peut être stimulée in vitro par des composés tels que les lectines.
I l existe plusieurs types de lectines (Sharon et Lis, 1989), dont les plus couramment Employées pour la transformation lymphoblastiques sont la phytohémagglutinine (PHA), la Concanavaline A (Con A) et le pokeweed mitogène (PWM). Leurs effets mitogènes Diffèrent :
Ainsi la PHA et la Con A activent de préférence les lymphocytes T alors que le PWM active Les lymphocytes T et surtout B.
Le sang est prélevé à une veine facilement accessible dans une seringue contenant de
de sodium. Pour chaque échantillon de sang, différents milieux nutritifs sont utilises pour augmenter la probabilité d'obtenir des lames de bonne qualite. Le sang preleve (0,5m1) est ensemencé dans un volume final de 8,5 ml d'un mélange composé d'un milieu de
1640 auquel sont ajoutés 10 à 20 % de sérum de veau foetal (SVF) et un mitogene (PHA) à la concentration finale de 10ug/I
Les cultures sont incubées dans des tubes fermés à 37°C pendant 3 jours
D'une manière générale, de meilleurs résultats (préparations riches en étalements chromosomiques analysables) sont obtenus avec la mise en culture de l
Chez certains, les résultats ne sont pas reproductibles d'un essai à l'autre et l'indice mitotique souvent faible (figure 24).
Le manque de reproductibilité est probablement dû au fait que le nombre de lymphocytes au
moment du prélèvement et leur stimulation potentielle par les mitogène varient beaucoup en
egiagotertique eteieloo0:eation deo détutpatideo tiralipteo
fonction de l'état sanitaire du patient. L'indice mitotique dépend du nombre de lymphocytes qui peuvent être transformés spécifiquement par l'agent mitogène présent.
111.4 Preparation des italements chrom osomiques :
Pour obtenir des préparations chromosomiques, i l faut récolter les cellules au stade de la Mitose, les gonfler par un traitement particulier pour avoir une bonne dispersion des Chromosomes, les fixer et les étaler sur lames (figure 25).
111.4.1 Accumulation des cellules mitotiques :
Une solution de colchicine 1 X est ajoutée au milieu de culture à une concentration finale de 0,04 ug/ml, 90 mn avant la récolte des cellules. Les flacons sont remis à l'étuve à 37 °C.
La suspension cellulaire est centrifugée pendant 5 mn à 1400 rpm. le culot est remis en
Suspension délicatement dans 9 ml d'un mélange hypotonique préchauffé à 37 °C pendant 20 Mn (Figure 25 A).
Une préfixation est réalisée en ajoutant 1ml de fixateur (3vol. d'éthanol- 1 vol. d'acide Acétique)
directement dans le milieu hypotonique pendant 5 mn.
La suspension cellulaire est centrifugée 5mn à 1400 rpm et le culot est repris d'abord dans 0,5 ml de fixateur pour bien homogénéiser la suspension, puis environ 6ml sont ajoutés. Cette première
fixation dure 30 min à +4 °C. Après une nouvelle centrifugation pendant 5 à 1400 rpm, le culot est repris dans 6ml de fixateur et les tubes sont laissés à température ambiante pendant 5 mn afin de réaliser une 2 e m e fixation (Figure 25 B).
Après fixation et centrifugation, les cellules sont remises en suspension dans un volume
Minimum de fixateur. La suspension cellulaires peut être étalé soit sur lame humides, soit sur Lames sèches (Figure 25 C).
La solution de coloration est préparée extemporanément en mélangeant (Figure 25 D):
- Solution tampon Sorensen pH 6, 8 : 3ml.
- Colorant de Giemsa R : 3 m l.
- Eau déminéralisée : 94 ml.
Les lames sont incubées durant 10 mn dans la solution à température ambiante, rincées Abondamment à l'eau courante, puis à l'eau déminéralisée. Elles sont ensuite séchées à l'air. Les préparations ainsi colorées sont observées au microscope et muni D'une caméra.

ujet présentant un ue (Gr X100).

plaque métaphasique
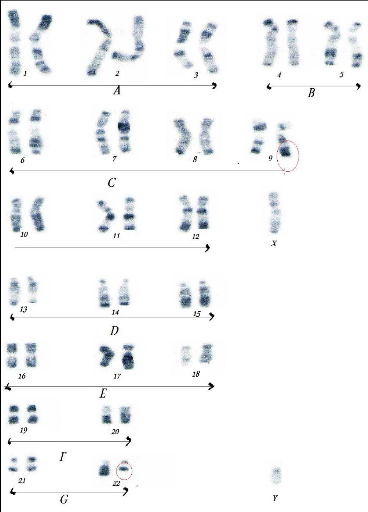
Figure 28: caryotype 46 XY t (9;22Xq34;q11).
Discussion
Le chromosome Philadelphie (ou de Philadelphie, PH1 ou translocation de Philadelphie) est une anomalie chromosomique spécifique qui est associée à la leucémie myéloide chronique (LMC°. ceci est le résultat d'une translocation réciproque, un échange de matériel génétique, entre les chromosomes 9 et 22 [nommée t (9 ; 22) (q34 ; q11)].
Cette anomalie a été décrite en 1960 par PETER NOWELL (university of pennsylvania school
of medicine) et DAVID HUNGERFORD (Fox chase cancer center's institute for cancer Research).
En 1973, Janet D. Rowley de l'université de Chicago identifia les mécanismes aboutissant au chromosome de Philadelphie.
La présence de cette translocation est désormais utilisée comme test de confirmation de LMC, car 95% des personnes ayant développé la maladie sont porteuses de cette anomalie chromosomique. Les 5% restant présentent d'autres translocations impliquant d'autres chromosomes.
Les techniques modernes de cytogénétique (caryotype, FISH) permettent désormais d'examiner avec précision les anomalies génétiques au niveau cellulaire.
Cependant, la présence du chromosome de Philadelphie n'est pas suffisante pour diagnostiquer une LMC car cette anomalie est également présente chez certaines personnes développant une leucémie lymphoide aigue (LLA, 25-30% chez l'adulte et 2-10% chez les enfants) et plus rarement une leucémie myéloide aigue (LMA) Comme dit précédemment, le défaut du chromosome de Philadelphie est une translocation. Un segment de chromosomes 9 et 22 s'inter-change et prenne la place l'un de l'autre.
Ceci résulte en la fusion d'une part du gène BCR (break point cluster region) du chromosome 22 avec une partie du gène ABL (Abelson) du chromosome 9. La protéine issue de ce gène bcr-abl est une protéine de 185 ou 210 KDa ayant une activité de tyrosine kinase.
Elle interagit notamment avec une sous-unité du récepteur à l'interleukine-3(IL-3).
La protéine est constitutivement active et ne requiert aucune activation par d'autres protéines cellulaires. Ceci pour conséquence que de nombreuses protéines et enzymes contrôlant le cycle cellulaire sont activées par bcr-abl, ceci accélérant fortement la vitesse de division cellulaire.
De plus, bcr-abl inhibe la réparation de l'ADN, causant une forte instabilité génomique et induisant considérablement la crise blastique de la LMC.
Les hémopathies malignes constituent en groupe de tumeurs qui se posent dans la transformation maligne de la moelle osseuse des cellules dérivées.
La grande diversité vue dans ce groupe de troubles est le reflet de la complexité d'hématopoïèse normale et le système immunitaire.
Syndromes myéloprolifératifs sont actuellement classés selon la classification FAB.
La leucémie aigue myéloide compose de très tôt sur les cellules souches et des cellules progénitrices et sont classées selon le témoignage de la différenciation lignée.
Le syndrome myélodysplasique est un trouble de cellules souches dans lesquelles, il ya une défaillance de non-hématopoïèses conduisant à une pancytopénie.
Dans de nombreux cas de myélodysplasie subissent la progression à la leucémie aigue.
Dans les troubles myéloprolifératifs chroniques, il y a surproduction d'un ou plusieurs types de cellules du sang périphérique.
Les translocations réciproques du chromosome ph1 ou bien t (9 ; 22) (q34 .1 ; q11.21) peuvent être détecté par plusieurs techniques or les méthodes révélant les bandes R ; par digestion enzymatique ou bien par dénaturation thermique.
L'amélioration des techniques telles que la biologie moléculaire, l'hybridation in situ (FISH) et la PCR peuvent augmenter le taux de détection du ph1. Enfin, cette étude montre l'importance d'installer les techniques de cytogénétiques pour des fins de diagnostic différentiel positif et le suivi du traitement.
egiagotertique et laid/ Caton deo détutpatideo tiralipteo
Abruzzo L.V., Jaffe E.S., Cotelingam J.D., Whang-Peng J., Del Luca V., Medeiros L.J et Am. J. Surg. T-cell lymphoblastic lymphoma with eosinophilia associated with subsequent myeloid
malignancy. Pathol. 16: 236-245. 1992.
Adams J.M., Cory S., Life-or-death decisions by the Bcl-2 protein family. Trends. Biochem. Sci. 26:
61-66. 2001.
acutemyelogenous leukemia with anormal karyotype. n. engl. j . med., 2005, 352, 254-266.
Adnane J., Gaudray P., Dionne C.A., Crumley G., Jaye M., Schlessinger J., Jeanteur P., Birnbaum
Ahmed Nacer Redhouane et Pr Hamladji Rose Marie, Pr Belhani Meriem, Pr Ardjoun Fatma
Zohra, Pr Abad Mohand Tayeb, Pr Touhami Hadj, Pr Ait Ali Hocine, Pr Zouaoui Zahia, Pr Bekadja
Mohamed Amine, Pr Ahmed Nacer Redhouane, Pr Boudjerra Nadia, Pr
Sidi Mansour Nourredine,
Pr Hamladji Rose Marie, Pr Belhani Meriem, Pr
Ardjoun Fatma Zohra, Pr Abad Mohand ,Tayeb,Pr
Touhami Hadj,Pr Ait Ali Hocine, Pr Zouaoui Zahia, Pr Bekadja Mohamed Amine, Pr Ahmed Nacer
Redhouane,Pr Boudjerra Nadia,Pr Sidi Mansour Nourredine,Pr Ainas Lahlou,Pr Mesli Naima,Pr Hamdi Salima, Pr Saidi Mahdia et Pr Benakli Malek ;;truc de approches épidémiologique en
algérie ;;2009.
BEK and FLG, two receptors to members of the FGF family, are amplified in subsets of human breast cancers. Oncogene. 6: 659-663; 1991.
Bai R-Y., Dieter P., Peschel C., Morris S.W., and Duyster J., Nucleophosmin-anaplastic lymphoma
kinase of large-cell anaplastic lymphoma is a constitutively active tyrosine kinase that utilizes phospholipase C-ã to mediate its mitogenicity. Mol. Cell. Biol. 18: 6951-6961; 1998.
Bai R-Y., Ouyang T., Miething C., Morris S.W., Peschel C., Duyster J., Nucleophosmin-anaplastic lymphoma kinase associated with anaplastic large-cell lymphoma activates the
phosphatidylinositol 3-kinase/Akt antiapoptotic signaling pathway. Blood. 96: 4319-4327;2000. BELL RJ. lntroductory Fourier transforms spectroscopy. Londres: académie press, 1972. BENNETT, CATOVSKY, DANIEL, et- al (The french-american-british (fab) coopérative
BENNETT, CATOVSKY, DANIEL, et al FRENCH-AMERICAN-BRITISH (FAB) co-operative
Brown D., Kogan S., Lagasse E., Weissman I., Alcalay M., Pelicci P.G., Atwater S., Bishop J.M., PMLRARá transgene initiates murine acute promyelocytic leukemia Proc. Natl. Acad. Sci. 94:
2551-2556; 1997.
CAVE H, VAN DER WERFF TEN BOSCH J . , SUCIU S., GULDAL C , WATERKEYN C ,
CHUDOBA L PLESCH A, LORCH T, et aL high resolution multicolor-banding: a new technique Cleary ML. Oncogenic conversion of transcription factors by chromosomal translocations. Cell. 66: 619-622; 1991.
CREMER T, LANDEGENT J , BRUCKNER
A, et ai. détection of chromosome aberration
in the
Deininger M.W.N., Goldman J.M., and Melo J.V., the
molecular biology of chronic myeloid
leukemia. Blood. 96: 3343-3356;; 2000.
Desmaze C., Brizard F., Turc-Carel C., Melot T., Delattre O., Thomas G., Aurias A., Multiple
chromosomal mechanisms generate an EWS/FLI1 or EWS/ERG fusion gene in Ewing tumors.
Cancer Genet. Cytogenet. 97: 12-19;1997.
F L A N D R I N MDS SCORING system / système de score pour les myélodysplasies. Tutorial FALINI B., MECUCCI C., TIACCI E . , ALCALAY M ROSATI R., PASQUALUCCI L . , LA
FE1NBERG P, VOGELSTEJLN B. A technique for radio Jabeling DNA restriction enzyme fragments
FEINGOED J , F E L EO E S M, SOLIGNAC M. Principes de génétique humaine, paris : Hermann, Fenrick R., Amann J.M., Lutterbach B., Wang L., Westendorf J.J., Downing J.R., Hiebert S.W
Both TEL and AML-1 contribute repression domains to the t(12;21) fusion protein. Mol. Cell. Biol. 19: 6566-6574., 1999.
for refined fish analysis of human chromosomes. C'ytogenetic cell genêt 1999; 84: 156-60. group) the chronic myeloid leukaemias: guidelines for distinguishing chronic granulocytic, groupproposals for the classification of the acute leukaemias br j haematol 33 1976 451-458
He L.Z., Guidez F., Tribioli C., Peruzzi D., Ruthard M., Zelent A., Pandolfi P.P Distinct interactions
of PML-RARá and PLZF-RARá with co-repressors determine differential responses to RA in APL. Nature Genet. 18: 126-135., 1998.
Heim S., Mitelman F. Cancer Cytogenetics. Chromosomal and Molecular Genetic Aberrations of Tumor Cells. Second edition. Wiley-Liss. http://www.infobiogen.fr/services/chromosomes cancer Hernandez L., Pinyol M., Hernadez S., Bea S., Pulford K., Rosenwald A., Lamant L., Falini B., Ott G.,
Mason D.Y., Delsol G., and Campo E;TRK-Fused Gene (TFG) is a new partner of ALK in anaplastic large cell lymphoma producing two structurally different TFG-ALK translocations; Blood. 94: 3265-3268; 1999.
human interphase nucleus by visualization of spécifie target DNAs with radioactive and nonradioactive
Ibrahimi O.A., Eliseenkova A.V., Plotnikov A.N., Yu K., Ornitz D.M.,Mohammadi M Structural basis
for fibroblast growth factor receptor 2 activation in Apert syndrome. Proc. Natl. Acad. Sci. USA. 98: 7182-7187., 2001.
in situ hybridization techniques. Hum genêt 1986; 741: 346-5
Kuno Y., Abe A., Emi N., Lida M., Yokozawa T., Towatari M., Tanimoto M., and Saito H Constitutive kinase activation of the TEL-Syk fusion gene in myelodysplastic syndrome with t(9;12)(q22;p12). Blood. 97:1050-1055., 2001.
Kurzrock R., Shtalrid M., Talpaz M., Kloetzer W.S., Gutterman J.U Expression of c-abl in Philadelphia-positive acute myelogenous leukemia. Blood. 70: 1584-1588., 1987.
Lacronique V., Boureux A., Monni R., Dumon S., Mauchauffé M., Mayeux P., Gouilleux F., Berger R., Gisselbrecht S., Ghysdael J., and Bernard O. Transforming properties of chimeric TEL-JAK proteins in Ba/F3 cells. Blood. 95: 2076-2083 A., 2000. Lengauer C., Kinzler K.W., Vogelstein Genetic instabilities in human cancers. Nature. 396: 643-649 B., 1998.
Lacronique V., Boureux A., Valle V. D., Poirel H., Quang C.T., Mauchauffé M., Berthou C., Lessard M., Berger R., Ghysdael J., and Bernard O. A ;A TEL-JAK2 Fusion Protein with Constitutive Kinase Activity in Human Leukemia. Science. 278: 1309-1312., 1997.
MANDEE! I F . , LO COCO F., PELICCI PG, MARTELLI MF, Cytoplasm nucleophosmin in
Martiat P., Mecucci C., Nizet Y., Stul M., Philippe M., Cassiman J.J., Michaux J.L., Van den Berghe
H., Sokal G P190 BCR/ABL transcript in a case of Philadelphia-positive multiple myeloma. Leukemia. 4: 751-754., 1990.
Martin D.B., Nelson P.S From genomics to proteomics: techniques and applications in cancer research. Trends in Cell Biol. 11: S60-S65., 2001.
med., 1998, 339, 591-598.
Mitani K., Ogawa S., Tanaka T., Miyoshi H., Kurokawa MK., Mano H., Yazaki Y., Ohki M., Hirai H.,
Generation of the AML1-EVi-1 fusion gene in the t(3;21)(q26;q22) causes blastic crisis in chronic myelocytic leukemia. EMBO J. 13: 504-510;1994.
Mitani K., Sato Y., Tojo A., Ishikawa F., Kobayashi Y., Miura Y., Miyazono K., Urabe A. Takaku F., Philadelphia chromosome positive B-cell type malignant lymphoma expressing an aberrant 190 kDa BCR-ABL protein. British J. Haematol. 76: 221-2251990.
Mitelman F., Johansson B., Mertens F., Mitelman database of chromosome aberrations in cancer2001. http://cgap.nci.nih.gov/Chromosomes/Mitelman.
Mohammadi M., Schlessinger J. Hubbard R., Structure of the FGF receptor tyrosine kinase domain reveals a novel autoinhibitory mechanism. Cell 86: 577-587; 1996.
Okuda K., Golub T.R., Gilliland D.G., Griffin J.D p210BCR/ABL, p190BCR/ABL, and TEL/ABL activate similar signal transduction pathways in hematopoietic cell lines. Oncogene. 13: 1147-1152;.,.
organization for research and treatment of cancer--childhood leukemia coopérative group. n. engl. 1996j .
OTTEN J., BAKKUS M., THIELEMANS K., CRAN DC H AMP B., VILMER E., Clinical
Pane F., Frigeri F., Sindona M., Luciano L., Ferrara F., Cimino R., Meloni G., Saglio G., Salvatore F.,
Rotoli B Neutrophilic-chronic myeloid leukemia: a distinct disease with a specific molecular marker (BCR/ABL with C3/A2 junction). Blood. 88: 2410-2414., 1996.
PUCCIARINI A., LISO A., VIGNETTI M., FAZI P., MEANI N., PETTIROSSI V., SAGLIO G.,
Rowley J.D The role of chromosome translocations in leukemogenesis. Sem. in Hematol. 36
(supl. 7): 59-72., séries in haematology array (0x3a828c) publication : Novartis. Édit. : g.flandrin - product.
Shannon KThe Ras signaling pathway and the molecular basis of myeloid leukemogenesis1999.. Curr. Op. In Hematol. 2: 305-308., significance of minimal residual disease in childhood acute
lymphoblastic leukemia. European. STARZA R, DIVERIO D., COLOMBO E., SANTUCCI A., BIGERNA B., PAC INI R. 1995.
Stehelin D., Varmus H.E., Bishop J.M., Vogt P.K DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature. 260: 170-173., 1976.
Strissel PS., Strick R., Rowley JD., Zeleznik-Le NJ., An in vivo topoisomerase II cleavage site and a DNase I hypersensitive site co-localize near exon 9 in the MLL breakpoint cluster region. Blood 92: 3793-3803;1998.
Tanaka K., Tanaka T., Kurokawa M., Imai Y., Ogawa S., Mitani K., Yazaki Y., Hirai HThe AML1/ETO (MTG8) and AML1/EVi-1 leukemia-associated chimeric oncoproteins., 1998
Tanaka T., Mitani K., Kurokawa M., Ogawa S., Tanaka K., Nishida J., Yazaki Y., Shibata Y., Hirai H., Dual functions of the AML1/Evi-1 chimeric protein in the mechanism of leukemogenesis in t(3;21) leukemias. Mol. Cell. Biol. 15: 2383-2392;1995.
Tlsty T.D., White A., and Sanchez J Suppression of gene Amplification in human cell Hybrids. Science. 255: 1425-1427.,. to high spécifie activity. Anal biochem 1984; 1371: 266-7 1992. Wells R.A., Catzavelos C., Kamel-Reid S Fusion of retinoic acid receptor alpha to NuMA, the
nuclear mitotic apparatus protein, by a variant translocation in acute promyelocytic leukaemia. Nat. Genet. 17: 109-113., 1997.
Willis TG., Dyer MJS The role of immunoglobulin translocations in the pathogenesis of B-cell malignancies. Blood. 96: 808-822., 2000.
Wilson G., Frost L., Goodeve A., Vandenberghe E., Peake I., Reilly J BCR-ABL transcript with an e19a2 (c3a2) junction in classical chronic myeloid leukemia. Blood. 89: 3064., 1997.






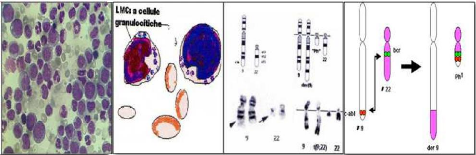
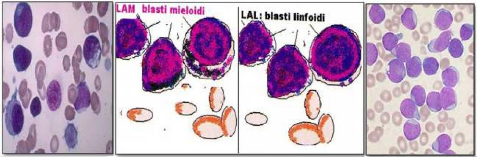
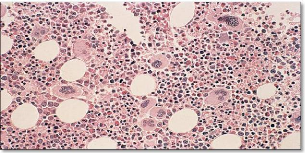

{kind=link}
{kind=link}
{kind=link}
{kind=link}