Page 0 sur 107
UNIVERSITE DE LUBUMBASHI
Faculté de Médecine
Département de Pédiatrie

ETUDE DES MALFORMATIONS CONGENITALES CLINIQUEMENT
VISIBLES A LA NAISSANCE A LUBUMBASHI
Par Monsieur LUBALA Kasole Toni
Docteur en Médecine (Université de
Lubumbashi)
Certifié d'études spécialisées
partielles en Pédiatrie (Université Catholique de
Louvain)
Mémoire présenté et défendu en vue de
l'obtention du grade de Spécialiste en Médecine, Option
Pédiatrie.

Page 1 sur 107
Université de Lubumbashi
Faculté de Médecine
Département de Pédiatrie

Titre :
ETUDE DES MALFORMATIONS CONGENITALES CLINIQUEMENT
VISIBLES A LA NAISSANCE A LUBUMBASHI
Présenté et défendu publiquement par
Monsieur LUBALA KASOLE Toni
(Né à Lubumbashi, le 13 Mai 1981)
Docteur en Médecine (Université de
Lubumbashi),
Certifié d'études spécialisées
partielles en Pédiatrie (Université Catholique de Louvain)
Directeur de Mémoire : Monsieur le Professeur
Docteur LUKUSA TSHILOBO Prosper (Katholieke Universiteit
Leuven)
Co-promoteurs : Monsieur le Chef de Travaux Docteur
MUTOKE NKASHAMA Georges (Université de Lubumbashi)
Monsieur le Professeur Docteur KALENGA MUENZE K.
Prosper (Université de Lubumbashi)
Monsieur le Professeur Docteur GILLEROT Yves
(Université Catholique de Louvain)
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Année académique 2010-2011
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 2 sur 107
Table des matières
PLAN DE REDACTION 4
DEDICACE 6
REMERCIEMENTS 7
LISTE DES ABREVIATIONS, SIGNES CONVENTIONNELS ET ACRONYMES 10
INTRODUCTION 11
1) Problématique 11
2. Objectifs 13
a. Objectif général 13
b. Objectif spécifiques 13
PREMIERE PARTIE: 14
CONSIDERATIONS THEORIQUES 14
CHAPITRE PREMIER : GENERALITES 15
I. BREFS RAPPELS D'EMBRYOLOGIE HUMAINE 15
II. GENERALITES SUR LES MALFORMATIONS CONGENITALES 22
DEUXIEME PARTIE : 40
ETUDE DES MALFORMATIONS CONGENITALES CLINIQUEMENT VISIBLES A LA
NAISSANCE A LUBUMBASHI 40
CHAPITRE DEUXIEME: PATIENTS ET METHODES 41
II.1. TYPE D'ETUDE ET DESCRIPTION DES SITES DE RECHERCHE 41
II.2. TECHNIQUE D'ECHANTILLONNAGE 43
II.3. ICONOGRAPHIE 44
II.4. CLASSIFICATION DES MALFORMATIONS 44
II.5. METHODES STATISTIQUES 44
II.6. CONSIDERATIONS ETHIQUES 45
II.7. DIFFICULTES RENCONTREES 46
CHAPITRE TROIXIEME: RESULTATS 47
III.1. ETUDE EPIDEMIOLOGIQUE 47
III.2. PROFIL CLINIQUE 50
III.3.Facteurs de risque 54
CHAPITRE QUATRIEME : DISCUSSION 56
CONCLUSIONS ET RECOMMANDATIONS 71
Annexes 74
ANNEXE I : Classification internationale des malformations 10
ème révision 75
Annexe II : Iconographie 76
ANNEXE III : Liste des figures 99
ANNEXE IV : Liste des tableaux 99
Bibliographie 100
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 3 sur 107
RESUME
ETUDE DES MALFORMATIONS CONGENITALES CLINIQUEMENT
VISIBLES A LA NAISSANCE A
LUBUMBASHI.
Introduction : L'accouchement d'un enfant malformé
est vécu dans les sociétés Africaines comme un
véritable drame compte tenu tant des considérations
mystico-religieuses qui l'entourent que du véritable poids qu'elle
constitue pour les familles.
Matériel et méthodes : Du mois d'Avril 2010
à Avril 2011, nous avons mené une étude de type analytique
rétrospective (cas témoin) dans la ville de Lubumbashi (RDC)
à travers 11 maternités pour définir le type ainsi que la
fréquence des malformations congénitales cliniquement visibles
à la naissance et identifier les facteurs de risque en vue d'orienter
d'éventuels projets d'intervention visant à prévenir ce
problème de santé à Lubumbashi.
Résultats : Au cours de cette étude, nous
avons dégagé une prévalence de 72 cas pour 12320
naissances soit 58,4 pour 10000 naissances (0,58). Il n'y a pas de
différence statistiquement significative entre la prévalence que
nous avons trouvée et celle trouvée dans une étude
multicentrique à Kinshasa (0,57 %).
Le sexe masculin est plus représenté que le
sexe féminin avec un sex ratio de 2. La commune de Kampemba est la plus
grande pourvoyeuse de cas de malformations avec 30.6% des cas.
Les malformations du système nerveux central sont
les plus fréquentes (20.29 pour 10000 naissances) suivies des
malformations des membres (10.55 pour 10000 naissances), et des fentes
oro-faciales (8.11 pour 10000 naissances).De façon plus précise,
les spina-bifida, les fentes labiales et palatines ainsi que les pieds bots
sont les plus représentés avec respectivement 5.68 ; 6.49 et 5.68
pour 10000 naissances. La trisomie 13 est la malformation chromosomique la plus
représentée dans notre série.
Près de 96% des cas de malformation ont
été découverts à la naissance plutôt
qu'à l'échographie dans le cadre des consultations
prénatales.
Dans notre série, 11.1% des enfants portaient des
malformations incompatibles avec la vie et 32.9% des enfants avec malformations
et vivants à la naissance sont décédés dans les
sept premiers jours de la vie.
Le fait pour un père d'être ouvrier ou
minier, de même qu'un âge maternel supérieur à 35 ans
étaient associés à un risque plus élevé de
donner naissance à un enfant malformé dans notre milieu. Le fait
de ne pas consommer de l'alcool pendant la grossesse, l'absence
d'automédication et de fièvre au premier trimestre de la
grossesse, de même que le fait de résider hors des communes
à forte activité minière semblent constituer un facteur
protecteur par rapport au risque de donner naissance à un enfant porteur
de malformation congénitale.
Mots clés : Malformation congénitale
cliniquement visibles, prévalence, facteurs de risque,
Lubumbashi
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 4 sur 107
PLAN DE REDACTION
Dédicace
Remerciements
I. Introduction
II. Première partie : généralités
II.1. Brefs rappels d'embryologie humaine
II.2. Généralités sur les malformations
congénitales
III. Deuxième partie : Aspects pratiques
III.1. Matériel et méthodes
III.2. Résultats III.3. Discussion
IV. Conclusion
V. Annexes : Iconographie des malformations
congénitales
Page 5 sur 107
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
« Toutes les attitudes médicales
convergent avec un souci
permanent : Voir se terminer une grossesse par la
naissance
d'un bel enfant, vigoureux, dont les structures nerveuses
ont
été préservées en cours de gestation et qui
conserve toutes ses
chances pour l'avenir... »
Jean Henri BAUDET
Professeur au collège de médecine des
hôpitaux de Paris
Mais hélas, bien souvent, la nature en
décide autrement...
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 6 sur 107
DEDICACE
A mon Père,
Professeur François René LUBALA Toto
Ruananza,
(Géologue, Professeur Ordinaire -
Faculté des Sciences - Université de Lubumbashi)
Modèle de responsabilité, de ténacité
et d'intégrité, qui oeuvre sans relâche à faire de
nous un Homme et un Scientifique à son image :
Intègre et Accompli ;
Et
Aux victimes innocentes de malformations
congénitales...,
Je dédie ce travail
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 7 sur 107
REMERCIEMENTS
Une spécialisation en Médecine est un imposant
édifice à la construction duquel de nombreux artisans ont, au fil
des années, apporté leur savoir-faire.
A ma mère, pour ton affection et tes conseils
avisés,... tu m'as tout donné ;
A Fanny, ma tendre épouse, patiente et
prévenante, pour son soutien sans faille et pour les sacrifices qu'elle
a consentis avec le sourire ;
A Maria Eliana, ma joie de vivre,
A Nina, David et Richard, mes complices de toujours,
Aux Autorités académiques de l'Université
de Lubumbashi, en leurs titres et qualités respectifs ; J'adresse des
remerciements particuliers :
A Monsieur le Professeur Prosper Lukusa
Tshilobo, Pédiatre, spécialiste en
cytogénétique de la « Katholieke Universiteit Leuven
» (Belgique) et expert MIDA, pour nous avoir fait l'honneur
d'accepter de nous initier à la génétique clinique et
d'assurer la direction du présent mémoire, apportant à sa
conception et à son élaboration votre expertise et votre
expérience scientifique reconnue.
A Monsieur le Professeur Prosper Kalenga Muenze K.
de l'Université de Lubumbashi (R.D.Congo), pour votre
contribution précieuse à la conception ainsi qu'à la
réalisation de cette étude effectuée dans le cadre de la
mise sur pieds d'un projet de registre des malformations congénitales
à Lubumbashi. Votre rigueur et votre patience ont fait toute notre
admiration.
A Monsieur le Docteur Georges Mutoke Nkashama,
Pédiatre à l'Université de Lubumbashi
(R.D.Congo) et Chef du Département de Pédiatrie. Vous avez
volontiers accepté d'enrichir ce travail de votre expérience et
vous m'avez suivi pas à pas en manifestant un intérêt
toujours renouvelé. Toutes mes réflexions cliniques portent votre
empreinte indélébile : « la physiopathologie au service
d'une clinique et d'une thérapeutique rationnelles ».
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 8 sur 107
A Monsieur le Professeur Yves Gillerot,
Pédiatre à l'Université Catholique de Louvain
(Belgique), Expert de l'Institut de Pathologie et de
Génétique de Loverval et responsable du registre Wallon Eurocat
des Malformations Congénitales, nous sommes également
reconnaissants pour votre contribution à la mise sur pieds d'un registre
des malformations congénitales à Lubumbashi et pour avoir
accepté d'apporter votre expérience de plus de quinze
années d'études et de surveillance des malformations
congénitales en Belgique. J'ai été frappé par votre
disponibilité et par votre curiosité scientifique exemplaire.
Votre apport a été considérable dans la confirmation des
diagnostics des cas retenus dans notre série.
A Monsieur le Professeur Jean Vanderpass
des Facultés Universitaires Notre Dame de la Paix de Namur
(Belgique), pour vos conseils avisés relatifs au choix de la
méthodologie et pour l'aide cruciale que vous avez apportée
à ce travail.
Aux Responsables de MIDA (Migration pour le
développement de l'Afrique) et de l'Organisation Internationale de
Migration pour le soutien qu'ils apportent à la formation post graduate
en Génétique en République Démocratique de
Congo.
A mes Confrères Arthur Munkana, Mick Shongo et
Eugène Twite pour avoir participé activement à la mise en
oeuvre du projet de registre des malformations congénitales à
Lubumbashi.
Aux parents de nos patients, sans qui cette étude n'aurait
jamais eu lieu
A tous mes maitres, qui m'ont initié à l'art de
guérir, j'adresse aussi mes plus sincères remerciements ;
A Monsieur le Professeur Ngwanza Ignace,
clinicien rigoureux et enseignant méthodique, que la mort a hélas
prématurément arraché à nous. Je lui suis
reconnaissant car, alors que j'étais encore indécis, il avait su
me transmettre sa passion pour l'art éminemment noble qu'est celui de
guérir ces petits êtres fragiles et innocents à qui j'ai
choisi de consacrer ma carrière.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 9 sur 107
A Monsieur le Docteur Deogratias Buhendwa
pour m'avoir aidé à consolider mon choix de
carrière. Aujourd' hui, six années après, je m'inspire
encore de son approche exemplaire de la Pédiatrie.
A Monsieur le Professeur Oscar Luboya Numbi,
Pédiatre à l'Université de Lubumbashi pour votre
encadrement clinique et pour votre disponibilité remarquable. Apprendre
à vos côtés était à la fois un plaisir et un
honneur. Vous avez toujours su encourager, canaliser et accompagner mon
émulation pour la recherche scientifique. Votre sagesse, votre sens
clinique et votre rigueur scientifique forcent le respect.
A Messieurs les Professeurs Stanis Wembonyama
Okitotsho et Vincent Kalombo Mupoya, Pédiatres à
l'Université de Lubumbashi, pour m'avoir fait l'honneur de me confier
une part de votre savoir.
La science va sans cesse se raturant elle-même, et ceux
qui s'y intéressent sont des perpétuels apprenants. Votre
savoir-faire me sera donc toujours précieux.
A Monsieur le Professeur Gaston Verellen,
Chef du Département de Pédiatrie de l'Université
Catholique de Louvain pour m'avoir ouvert les portes du très
renommé Hôpital Universitaire Saint Luc de Bruxelles.
A Monsieur le Professeur Didier Moulin, Chef
du Service de Pédiatrie Générale des Cliniques
Universitaires Saint Luc, pour l'inoubliable et solide formation clinique dont
j'ai bénéficié auprès de lui ainsi que de son
équipe spécialisée à la fois très humaine et
remarquablement compétente.
A mes ainés : Docteurs Mudekereza, Ngwej, Yaba,
Assumani, Lukamba et Swana pour votre encadrement irréprochable.
A mes collègues et amis Augustin, Félix, Lewis,
Paul et Adonis pour la collaboration harmonieuse et cordiale durant nos 7
années de spécialisation.
A tous ceux et à toutes celles qui ont
participé, à divers titres et à diverses étapes de
ma formation et de la réalisation de ce travail, je dis
sincèrement merci.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 10 sur 107
LISTE DES ABREVIATIONS, SIGNES CONVENTIONNELS ET
ACRONYMES
ADN Acide
désoxyribonucléique
AFP Alpha foeto-protéine
CMV Cytomégalovirus
EUROCAT European Registry of Congenital
Anomalies and Twins
ICD-10 International classification of
disease 10th revision, 2cd édition
CPN Consultations
Prénatales
Gy Gray
HCG Hormone Chorionique
Gonadotrophique
HSR Heat Shock Response
MCA-eci Multiples Congenital Malformations
e causa incognata
OMS Organisation Mondiale de la
Santé
OR Odds Ratio
PC Périmètre
Crânien
PN Poids de Naissance
RCIU Retard de croissance intra
utérin
RDC République Démocratique
du Congo
TORSCH
Toxoplasmose-Rubéole-Syphilis-Cytomégalovirus-Herpes
USA United States of America
VIH Virus d'Immunodéficience
Humaine
X2 :
Chi-Carré
% : Pourcentage
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 11 sur 107
INTRODUCTION
1) Problématique
Les malformations et les troubles congénitaux
constituent un véritable problème de santé publique au
niveau mondial, compte tenu de leur importance en tant que cause de
mortinatalité et de mortalité néonatale.
Chaque année, plus de 7,9 millions d'enfants, soit 6 %
du nombre total de naissances dans le monde naissent avec un trouble
congénital grave dû à des causes génétiques
ou environnementales. Environ 7% de l'ensemble des décès
néonataux dans le monde sont dus à des anomalies
congénitales (OMS, 2008). En Europe, en 1996, le taux de
mortalité infantile par malformations congénitales était
estimé à 30 %.
Une étude réalisée en milieu hospitalier
aux Etats Unis a montré que les malformations congénitales ainsi
que les anomalies génétiques expliquaient 34% des
décès néonataux ; 16,7% de ces malformations
congénitales étaient d'origine chromosomique (Stevenson et Carey,
2004).
Ces anomalies sont généralement accidentelles,
dues à des défauts de spermatogenèse ou de
l'ovogenèse par non disjonction au moment des méioses (divisions
réductionnelles), et donc non reproductibles dans la fratrie.
Le taux de survie à 20 ans des enfants
présentant des malformations congénitales d'origine chromosomique
dans les pays industrialisés a été évalué
à 85.5% (Rankin, 2010 ; Stone,2010).
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 12 sur 107
Ces survivants constituent bien souvent un véritable
poids social et nécessitent des soins spécialisés à
long terme. Une étude réalisée aux Etats Unis dans une
unité de soins pédiatriques à long terme a montré
que les patients présentant des anomalies congénitales
constituaient 50% des admissions. (Omalley et Hutcheon, 2007).
Au Canada, une étude a montré qu'un nombre
disproportionné de malades restant à l'hôpital plus de 10
jours figure dans le groupe des malformations congénitales. Quant aux
malades admis plusieurs fois à l'hôpital, 70% d'entre eux (soit
3.2% de toutes les entrées) souffraient de maladie
génétique ou de malformation congénitale (Scriver et al. ,
1973).
Très peu d'études fiables sont disponibles dans
nos pays à faible revenu. L'absence de registre de malformations
congénitales dans la plupart de ces pays explique en partie cet
état des choses.
Les quelques données retrouvées en Côte
d'Ivoire (Coulibaly-Zerbo et al., 1997) et au Congo Brazzaville (Mayanda et
al., 1991) attribuent aux malformations congénitales une
fréquence hospitalière de 5 %.
Une étude prospective aux Cliniques Universitaires de
Kinshasa a abouti à une incidence de 2,5 % de naissances vivantes
(Sengeyi et al., 1990) et dans une étude multicentrique
réalisée à Kinshasa en 2004 a dégagé une
incidence de 0,57% (Tabu, 2004).
La même année, à Lubumbashi, au cours
d'une étude rétrospective portant sur une période de 5
ans, menée dans les principales formations médicales de la ville,
166 cas de malformations congénitales ont été
observés sur un total de 22538 naissances soit une prévalence de
0,74% (Kalwaba et al., 2004).
La présente étude, effectuée dans le
cadre de la mise sur pieds d'un registre des malformations congénitales
à Lubumbashi, a pour but d'établir des nouvelles
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 13 sur 107
statistiques sur les malformations congénitales
cliniquement décelables à la naissance et d'en dégager les
facteurs de risque.
2. Objectifs
a. Objectif général
Contribuer à l'amélioration des connaissances sur
les malformations congénitales en vue d'organiser une stratégie
de prévention.
b. Objectif spécifiques
- Déterminer la prévalence des malformations
congénitales cliniquement visibles à la naissance à
Lubumbashi ;
- Identifier chaque type des malformations congénitales
cliniquement visibles à la naissance;
- Identifier les facteurs de risque associés aux
malformations congénitales observées.
-Proposer des stratégies de prévention face
à la problématique des malformations congénitales à
Lubumbashi.
Page 14 sur 107

CONSIDERATIONS THEORIQUES
PREMIERE PARTIE:
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 15 sur 107
CHAPITRE PREMIER : GENERALITES
I. BREFS RAPPELS D'EMBRYOLOGIE HUMAINE
La discussion de l'embryologie humaine pourrait commencer par
n'importe quel point du cycle reproductif.
Nous avons choisi de commencer notre description par la
différenciation des cellules sexuelles mâles et femelles ou
gamètes car l'étude des mécanismes de la
gamétogenèse humaine fournit la base de la compréhension
des anomalies chromosomiques.
I.1. La gamétogenèse
Les cellules germinales matures sont les descendants directs
des cellules germinales primitives qui dérivent de l'épiblaste et
apparaissent chez l'embryon humain dans la paroi du sac vitellin dès la
fin de la troisième semaine de gestation.
Ces cellules migrent ensuite vers la gonade primitive
où elles parviennent vers la fin de la quatrième semaine.
I.1.1. Ovogenèse
Chez l'embryon de sexe féminin, les précurseurs
gonadiques (qui sont des cellules
diploïdes) parvenus à destination au niveau du
futur ovaire embryonnaire se
différencient en ovogonies.
Après une série de mitoses, vers la fin du
3ième mois, des amas de cellules gonadiques s'entourent de cellules
épithéliales aplaties appelées cellules folliculaires, qui
dérivent de l'épithélium qui couvre l'ovaire. La
majorité des ovogonies continuent de se diviser par mitose, mais
certaines se différencient en ovocytes primaires, qui sont bien plus
grands.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 16 sur 107
Dès leur formation, ces derniers entament la prophase
de la méiose I. Le nombre des ovogonies augmente pour atteindre un
maximum, estimé à 7 millions, au 5ème mois,
puis il se produit une dégénérescence progressive jusqu'au
7ème mois, où il ne reste que les ovogonies proches de la surface
de l'ovaire.
Tous les ovocytes survivants sont bloqués en
méiose I et entourés d'une couche de cellules folliculaires,
formant les follicules primordiaux.
Les ovocytes primaires restent bloqués en prophase de
la méiose I jusqu'à la puberté, et ce blocage pourrait
être lié à la sécrétion, par les cellules
folliculaires, d'un « oocyte maturation inhibitor ».
Ce signal n'est pas clairement connu, mais on sait que le
blocage de la maturation ovocytaire est accompagné d'un taux
élevé d'AMP cyclique cytoplasmique. Il semble que GPR3, un
récepteur à 7 segments transmembranaires couplé à
la protéine Gs, exprimé à la surface des ovocytes mais
dont le ligand est inconnu, joue un rôle important.
Au moment de la naissance, le nombre d'ovocytes primaires est
estimé entre 700.000 et 2 millions. Seuls 400.000 sont encore
présents à la puberté et moins de 500 sont ovulés
pendant la vie sexuelle de la femme.
L'augmentation des anomalies chromosomiques avec l'âge
maternel suggère que les ovocytes primaires bloqués en
méiose I ne sont pas à l'abri des lésions
chromosomiques.
Dans le cas typique de la trisomie 21, des études ont
permis d'établir que 90 à 95 % des cas sont dus à une non
disjonction dans la lignée germinale de la mère.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 17 sur 107
I.1.2. Spermatogenèse
La spermatogenèse est l'ensemble
d'événements par lesquels les spermatogonies se transforment en
spermatozoïdes. Contrairement à l'ovogenèse, qui a lieu in
utero, la spermatogenèse ne commence qu'à la puberté.
A la naissance, les cellules germinales mâles sont
visibles dans les cordons spermatiques sous forme de grandes cellules
pâles qui sont entourées de cellules de soutien, homologues des
cellules folliculaires de l'ovaire, appelées cellules de Sertoli.
Peu avant la puberté, les cordons spermatiques se
transforment en tubes séminifères et les cellules germinales
donnent naissance à deux types de spermatogonies.
Les spermatogonies A se divisent par mitose et forment un pool
de cellules souches, dont certaines se divisent pour former des spermatogonies
de plus en plus différenciées.
La dernière division donne naissance aux spermatogonies
B, qui se divisent pour former les spermatocytes primaires.
Ces derniers subissent une division réductionnelle
(méiose) pour donner des spermatocytes de deuxième ordre,
haploïdes.
Ils subissent à leur tour une division pour donner des
spermatides (Merger, 1995). La transformation des spermatides en
spermatozoïdes est appelée spermiogénèse.
Page 18 sur 107
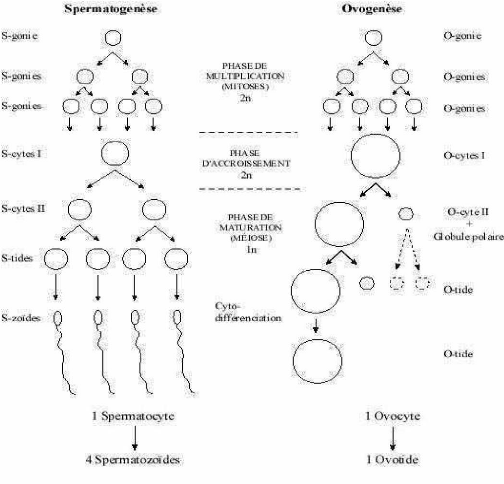
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Figure 1: Gamétogenèse (Gasser,
1975)
Page 19 sur 107
I.2. DE LA FECONDATION A LA NIDATION
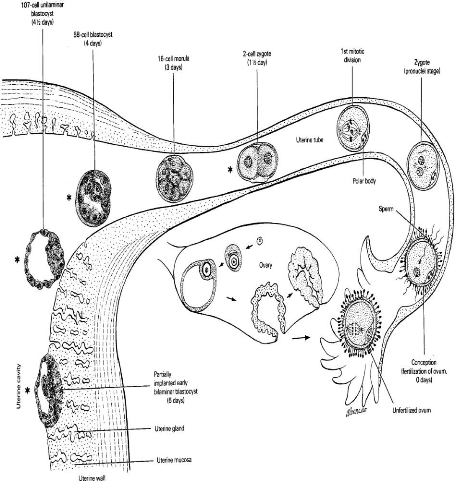
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Figure 2: de la fécondation à la nidation
(Gasser, 1975)
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 20 sur 107
I.3. PERIODE EMBRYONNAIRE
Elle met en place les différents feuillets primordiaux,
puis les ébauches des différents organes
- Stades précoces: de l'oeuf à un disque
embryonnaire, d'abord didermique (DED), puis tri dermique (DET)
1ère semaine : l'oeuf se segmente, se transforme en
morula, puis en une structure creuse, le blastocyste
2ème semaine DED Mise en place de l'ectoblaste et de
l'entoblaste primaire. Le trophoblaste représente l'ébauche du
futur placenta
3ème semaine DET le mésoblaste (3ème
feuillet) apparaît; premières ébauches des principaux
organes.
- Stades ultérieurs : Plicature et
organogénèse précoce
4ème semaine : De l'embryon à 2 dimensions
(Disque) à la fermeture du corps par plicature de 5 à 8/10
semaines : l'organogénèse précoce se poursuit à
partir de la mise en place de l'ensemble des ébauches.
C'est pendant la période embryonnaire que les risques
de malformations congénitales sont les plus grands. Avant cette
période, facteurs tératogènes (anomalies
génétiques ou environnementales) conduisent le plus souvent
à un avortement prématuré, tandis qu'après,
l'incidence des malformations et leur gravité sont plus
réduites.
Les facteurs tératogènes principaux sont les
maladies infectieuses, les substances chimiques et médicamenteuses et
les radiations ionisantes.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 21 sur 107
I.2. PERIODE FOETALE :
Classiquement de 3 mois (en fait de 2mois1/2 à 3 mois 1/2,
selon les organes) jusqu'à la naissance.
L'embryon est constitué. Il a déjà acquis
les caractéristiques de l'espèce humaine (membres, face).
Avec la période foetale, on entre dans une phase de
maturation et de croissance volumique.
Les mécanismes spécifiques de l'embryologie (hyper
prolifération cellulaire, migrations cellulaires, différenciation
cellulaire à partir de cellules souches) tendent à s'estomper
même si certains perdureront toute l'existence.
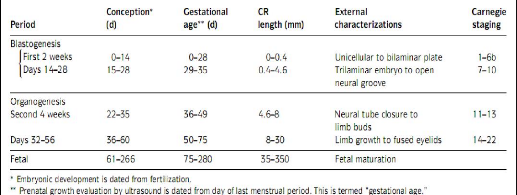
Tableau I : Croissance et développement de
l'embryon humain (Opitz, 2004)
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 22 sur 107
II. GENERALITES SUR LES MALFORMATIONS CONGENITALES
II.1. Historique
L'histoire de l'étude des monstruosités est
extrêmement ancienne. Ainsi, Ctésias, médecin grec à
la cour d'Artaxerxès Mnémon, en Perse, est le premier à
rapporter l'accouchement d'un enfant sans tête par Roxane, épouse
de Cambyse, en 426 av. J.C. (Charon, 2005).
Ambroise Paré (1585) publie le cas d'un monstre femelle
sans tête, né en l'an 1562, premier jour de novembre, à
Villefranche de Beyran, en Gascogne (Charon, 2005).
Jusqu'au XVIIIe siècle, l'opinion a continué
à penser que les malformations congénitales faisaient partie de
la liste des punitions que Dieu infligeait aux couples
désobéissants. Ainsi, les Grecs ont appelé la science qui
s'occupait de ce genre d'enfants « TERATOS » qui signifie «
prodige ». Il s'agissait d'une science d'enfants « prodigieux »
qui sont venus ou revenus dans ces couples pour les discipliner. La
tératologie a commencé à connaître son vrai essor en
1961, avec la tragédie de la thalidomide (Baudet, 1990). La
compréhension de l'étiopathogénie des malformations
congénitales a marqué une avancée significative
grâce aux progrès réalisés par la biologie
moléculaire.
Frédéric et Hausman-Hagemeyer ont
remarqué en 1967 que les anomalies chromosomiques ont été
responsables de la majorité des malformations congénitales. En
1969, « The National Foundation » (USA) a publié une
étude qui a démontré que les malformations
congénitales dues aux aberrations chromosomiques ont été
statistiquement moins nombreuses que celles causées par des troubles
génétiques en général. Avec l'apport de la biologie
moléculaire depuis ces 25 dernières années, les
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 23 sur 107
anomalies génétiques sont
différenciées en deux groupes : 1. mutations géniques ; 2.
aberrations chromosomiques.
II.2. Définitions et Classification
II.2.1. Définitions
Le terme malformation congénitale est essentiellement
générique et vague; il recouvre un nombre très
élevé de situations diverses et variées (Gillerot et Mols,
2009).
En termes généraux, et sans tenir compte de la
cause, une malformation est une déviation morphologique significative
d'un organisme en développement et qui se traduit « in fine »
par une altération importante du phénotype. Une malformation est
une défectuosité d'un champ survenue dès son origine; le
phénotype atteint intrinsèquement en son tout début en est
profondément modifié (Gillerot et Mols, 2009). Ce terme est
à différencier de :
- disruption: c'est l'altération secondaire d'un champ
initialement normal (defect
in field)
- déformation : il s'agit de la modification du
phénotype d'un champ normal mais
ayant subi secondairement l'influence de forces mécaniques
externes.

Figure 3: Différence schématique entre
malformations, disruptions et déformations (Dimmick et Kalousek,
1992)
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 24 sur 107
En ce qui concerne les ensembles malformatifs, l'on distingue
principalement les séquences et les syndromes.
- une séquence est un ensemble d'anomalies survenues en
cascade et étant la
conséquence d'une défectuosité primaire
qui peut être assez insignifiante. L'exemple classique est celui de la
séquence de l'oligohydramnios survenant par exemple à la suite
d'une obstruction urétrale. Ce défect mineur engendre une
séquence malformative dramatique avec la survenue d'une
mégavessie, d'un reflux vésico-urétéral, et la
destruction progressive du parenchyme rénal, une hypoplasie pulmonaire,
des contractures des membres, une compression de la face et une atrophie
progressive de la musculature abdominale donnant plus tard l'aspect en «
prune belly ».
- un syndrome est dû à l'altération
initiale de plusieurs champs de développement
relevant d'un seul agent étiologique. Les anomalies
chromosomiques en sont des exemples classiques. On parle de syndrome disruptif
lorsqu'un processus disruptif survenu assez précocement dans la
grossesse affecte plusieurs champs de développement et peut ainsi «
copier » de façon étonnante un syndrome poly malformatif
classique. Citons ici l'exemple des brides amniotiques.
- Notion de champs embryonnaire : Ce concept avait
déjà été imaginé par Boveri. Il stipule que
le jeune embryon est divisé en « champs » dans lesquels la
formation et la genèse de telle structure anatomique plus ou moins
complexe se fait de façon synchrone, ordonnée et
hiérarchisée.
1. La synchronisation dans le temps fait référence
au réglage précis et simultané
avec lequel se déroulent au sein de ces champs des
événements morphogénétiques contigus de sorte que
l'un ne soit pas déphasé par rapport à l'autre. Exemple :
la synchronisation qui s'opère entre la croissance des doigts et la
disparition des palmures interdigitales.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 25 sur 107
2. L'agencement spatial fait allusion au fait que les
différentes parties de cette
structure complexe assument leur propre
localisation et orientation par l'intermédiaire d'une intrication de
mouvements morphogénétiques actifs et passifs, de croissance et
de relation mutuellement inductrice.
Exemple : la malformation dénommée «
cyclopie » est la conséquence d'une absence de relation
mutuellement inductrice entre le cerveau antérieur et l'étage
moyen de la face.
3. Enfin la hiérarchisation rappelle que le
développement au sein d'un champ se
fait du pluri potentiel
réversible au hautement différencié irréversible,
du non fonctionnel au fonctionnel, du petit au grand, du simple au complexe ...
L'organisation du zygote en un certain nombre de champs de développement
résulte très vraisemblablement de la réduction progressive
au cours de l'évolution des vertébrés du système
métamérique en structures moins nombreuses mais plus
complexes.
II.2.2. Classification
S'agissant de la classification des malformations
congénitales, outre les termes très vagues de malformations
majeures, modérées ou mineures, plusieurs systèmes
existent. Chacun a ses avantages et ses faiblesses.
1. La classification étiologique regroupe les
malformations d'après leur cause; ce qui
est certes séduisant et simple. En effet, il «
suffit » de les classer d'après leur étiologie. L'on
distingue habituellement 5 regroupements étiologiques: chromosomique,
monogénique, multifactoriel, environnemental et inconnu.
Néanmoins ce qui est simple en théorie l'est beaucoup moins dans
la pratique. En effet, la recherche de l'étiologie varie très
fort en fonction des moyens mis en oeuvre et de l'expérience des gens du
terrain. En outre, il n'y a pas encore d'unanimité concernant les
définitions mêmes de certaines étiologies surtout pour ce
qui concerne le groupe le plus important, à savoir
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 26 sur 107
les malformations dites multifactorielles/polygéniques.
Il n'est pas étonnant que, dans ces conditions, les chiffres soient
étonnamment variables d'une série à l'autre.
2. La classification dite anatomique consiste à
recenser les malformations telles
qu'elles se présentent à
l'observateur. Elle est objective et aisée mais n'a pas d'utilité
au regard du conseil génétique et de la prévention; en
outre cette classification ne tient pas compte des malformations
s'intégrant dans des ensembles complexes, par exemple dans le syndrome
de Meckel qui est essentiellement constitué d'un
crânio-rachischisis, d'une polykystose rénale et d'une
hexadactylie post-axiale. Ici le système anatomique recensera
séparément hexadactylie, crânio-rachischisis et reins
polykystiques tandis que la classification étiologique parlera de
syndrome de Meckel.
3. Enfin la classification pathogénique est
intéressante, car elle est à la fois
descriptive mais peut
également être indicatrice de l'étiologie sous-jacente.
Elle se base sur la notion déjà ancienne de champ embryonnaire de
développement (developmental field) redécouverte par le
célèbre généticien américain John Opitz
(Martinez-Frias et al., 1998).
II.3.Epidemiologie
D'après les Statistiques mondiales 2008 (OMS, 2008),
environ 260 000 décès de nouveau-nés dans le monde sont
dus à des anomalies congénitales. Ce chiffre représente
environ 7 % de l'ensemble des décès néonatals, proportion
qui varie cependant de 5 % dans la Région de l'Asie du Sud-Est à
plus de 25 % dans la Région européenne. Les données
disponibles laissent supposer de grandes variations d'un pays à l'autre
: la proportion mentionnée vaut 4 % au Bangladesh, en Guinée
équatoriale, en Ethiopie, au Libéria, au Mali et au Sierra Leone,
8 % selon les estimations pour la Chine et 38 % et plus à Bahreïn,
à Chypre, en Irlande, au Koweït, au Qatar et en République
Arabe Syrienne (OMS, 2008).
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 27 sur 107
Ces anomalies sont aussi une cause majeure de
décès foetal et une cause grandissante de mortalité
néonatale dans les pays subissant la transition
épidémiologique (Chine, par exemple).
Même si elles représentent un plus faible
pourcentage des décès de nouveau-nés et d'enfants d'1
à 59 mois dans les pays à revenus faible ou moyen que dans les
pays riches, plus de 95 % des décès infantiles (OMS, 2008)
résultant d'anomalies congénitales se produisent dans la
première catégorie des pays.
Ceci indique que les anomalies congénitales touchent
toutes les nations et représentent un défi important pour la
santé publique au niveau mondial.
II.4 Aspects étio-pathogéniques des
malformations
II.4.1. Malformations : causes intrinsèques
(constitutionnelles - endozygotiques)
II.4.1.1. Malformations d'origine génique
Mutations mendéliennes :
. Transmission autosomique dominante
Exemple : certaines polydactylies isolées ou
ectrodactylies : absence de 1 ou plusieurs doigts - ectro =avorter (Baudet,
1990).
. Transmission autosomique récessive
Exemple : syndrome de Meckel - Gruber
. Transmission récessive liée à l'X
Exemple : hydrocéphalie par sténose de l'aqueduc de
Sylvius au cours du
Syndrome de BICKERSADAMS.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 28 sur 107
Anomalies dues à une empreinte parentale :
. La disomie uniparentale (isodisomie) est une anomalie
chromosomique dans laquelle une paire de chromosomes est du même parent
(par opposition hétérodisomie = les 2 chromosomes d'une paire
d'une paire proviennent des deux parents).
. L'empreinte parentale est la conséquence de
l'inactivation du gène de l'un des 2 parents (existence de
différences fonctionnelles entre gènes paternel et maternel)
Exemple : Le syndrome de Wiedemann-Beckwith: macrosomie
(viscéromégalie) : macrosplanchnie, omphalocèle,
gigantisme, macroglossie, souvent hémi-hypertrophie, dysmorphie faciale.
Par ailleurs hypoglycémie néonatale, cytomégalie
surrénalienne et pancréatique. Ce syndrome prédispose
à des tumeurs malignes : néphroblastome (pouvant être
bilatéral), hépatoblastome, corticosurrénalome,
gonadoblastome et gliomes. Il est en général sporadique (formes
familiales occasionnelles).
II.4.1.2. Malformations d'origine chromosomique
Elles concernent 1% des naissances. Elles sont dans la grande
majorité des cas accidentels (non-disjonction lors de la méiose)
et donc non reproductibles dans la fratrie sauf dans les remaniements
chromosomiques familiaux du style translocation par exemple.
Exemples de syndromes malformatifs d'origine chromosomique:
? Trisomie 21 (syndrome de Down) ; Trisomie 13 (syndrome de
Patau) ; Trisomie 18 (syndrome de Edwards)
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 29 sur 107
? Le mosaïcisme : présence de deux populations
cellulaires (ou plus) ayant une formule chromosomique différente.
Répartition variable dans l'oeuf : soit généralisée
(à l'embryon et au placenta, soit limitée (confinée) au
placenta, ou à l'embryon (rarissime).
? Une place à part pour les
micro-délétions qui peuvent être à l'origine de poly
malformations.
Il est à noter que les malformations isolées et les
monstruosités ne s'accompagnent généralement pas
d'anomalie chromosomique.
II.4.2. Malformations : causes extrinsèques
Lorsqu'une agression survient au cours de
l'embryogénèse, elle pourra, selon sa gravité,
entraîner un avortement précoce, des «malformations»
gravissimes létales ou enfin donner lieu à des malformations
isolées ou à un syndrome poly malformatif. (Baudet, 1990)
Si une agression survient durant la période foetale, on
aura une foetopathie en général non malformative, mais
s'accompagnant en général d'un retard de croissance
intra-utérin (RCIU). L'agression peut être d'origine maternelle ou
extérieure à la mère.
II.4.2.1. Les causes infectieuses
· Bactériennes : Syphilis congénitale.
· Virales :
- Rubéole : Embryo foetopathie la plus anciennement
connue. Syndrome de Gregg (embryopathie) : atteinte cardiaque, auditive et
oculaire.
Malformations congénitales cliniquement visibles
à Lubumbashi/Mémoire de spécialisation 2010-2011
Page 30 sur 107
- CMV, Herpes Virus, Varicelle, Virus lymphotrope : HIV, ou
encore Parvovirus (induit une érythroblastopénie foetale à
l'origine d'une anasarque foetoplacentaire)
· Parasitaires
Toxoplasmose congénitale : hydrocéphalie
congénitale avec atteinte oculaire (Calcifications
cérébrales en coquille d'oeuf, septicémie).
II.4.2.2. Malformations dues aux agents physiques
. Les radiations ionisantes : «modèle
expérimental» : les enfants nés après l'explosion des
bombes atomiques à Hiroshima et Nagasaki, mais aussi après
l'accident de Tchernobyl. (Baudet, 1990)
Fortes doses : curiethérapies de contact chez une femme
enceinte
Faibles doses : explorations radiographiques chez une femme
qui ne sait pas qu'elle est enceinte.
. Disruptions de l'hyperthermie maternelle :
Expérimentalement l'hyperthermie (fièvre = 38.5°C) peut
avoir un effet tératogène (action comme anti mitotique) =>
troubles du développement du Système nerveux central, anomalies
des membres, hypoplasie médio faciale.
Dix % des anomalies du tube neurale et non neurales pourraient
relever de la fièvre.
Malformations congénitales cliniquement visibles
à Lubumbashi/Mémoire de spécialisation 2010-2011
Page 31 sur 107
II.4.2.3. Malformations chimio-induites Exemples
:
. Thalidomide (hypnotique-sédatif, antalgique,
anti-inflammatoire) : Épidémie de phocomélie survenue en
Allemagne après prise par les femmes enceintes de Thalidomide durant la
période embryonnaire de la gestation.
Il existe encore des indications thérapeutiques
actuellement: Aphtoses (Behcet) et troubles de l'immunité,
lèpre.
Il faut donc garder à l'esprit le risque malformatif
dans la mesure où ce médicament est toujours prescrit et redouter
une reprise des malformations dans les pays où sa délivrance
n'est pas contrôlée, ou prescrite sans précautions
contraceptives.
. Valproate de Sodium (Dépakine) : anti-comitial =>
augmentation du nombre des spina-bifida (traitement préventif
pré-conceptionnel par l'acide folique).
. Isorétinoïne (accutane) : traitement de
l'acné ; si prescrit durant le premier trimestre => avortement,
malformations craniofaciales. Règles de prescription extrêmement
strictes.
II.4.2.4. Facteurs maternels (métaboliques)
. Rôle possible de certaines carences vitaminiques
(acide folique) comme facteur possible des anomalies du tube neural
. Enfant de mère diabétique : si diabète
mal équilibré, ancien et sévère, risque malformatif
important : cardiopathies, de régression caudale, aplasie radiale,
malformations rénales au maximum association VATER.
. Phénylcétonurie maternelle: chez les femmes
porteuses d'une phénylcétonurie traitée durant l'enfance,
risque d'embryopathie malformative qui sera prévenue
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 32 sur 107
par une reprise du régime de manière à
corriger les troubles métaboliques avant la conception (ceux-ci n'ont
jamais disparu mais sont restés sans effet sur la mère
après l'enfance).
II.4.2.5. Pathologie des addictions
. Tabagisme maternel : risque d'avortement, grossesse
compliquée, prématurité, RCIU ; (enfin morts subites du
nourrisson plus fréquentes chez les enfants de mère fumeuse).
. Alcool : La gravité du syndrome d'alcoolisme foetal
est dose-dépendante et dépend de ma période de
l'exposition.
. Toxicomanie : LSD, cocaïne, etc ... II.4.2.6. Les
facteurs mécaniques
. La séquence de rupture amniotique - Adhérence
amniotique.
. La séquence de rupture amniotique (ou maladies des
brides amniotiques) : le primum movens est la rupture de l'amnios.
Il en résulte une mise en contact du mésenchyme
sous-jacent avec le liquide amniotique et la formation de brides dites
amniotiques insérées sur la plaque choriale dénudée
ou flottant dans le liquide amniotique.
Elles provoquent chez le foetus des amputations distales des
membres, des doigts, des syndactylies ou des constrictions.
Si elles sont dégluties, ces brides peuvent aussi
provoquer des fentes faciales. Il n'y a pas de malformation
viscérale.
Malformations congénitales cliniquement visibles
à Lubumbashi/Mémoire de spécialisation 2010-2011
Page 33 sur 107
. Adhérence amniotique : La séquence
foeto-placentaire comporte des lésions cranio-faciales extrêmement
importantes telles une encéphalocèle ou une exencéphalie
recouverte de peau atrophique ; un defect osseux peut être
associé, parfois total (acranie) ainsi que des lésions
difficilement systématisables de l'encéphale, ou des grandes
fentes de la face impliquant la lèvre supérieure, le palais, le
nez et l'oeil.
Il existe aussi des lésions du tronc situées
au-dessus du cordon ombilical : fente sternale, ectocardie.
Cette malformation apparaît comme la mise en
continuité anormale de l'épiderme et de
l'épithélium amniotique, soit juste au-dessus du cordon
ombilical, soit au niveau de la tête ; sa pathogénie est
inconnue.
. Déformation de l'oligo-amnios : Elles sont souvent
la conséquence d'une
pression mécanique sur le foetus : par exemple au cours
de l'oligo-amnios.
II.4.2.7. Disruptions d'origine vasculaire ou
ischémique
Des modifications du flux sanguin peuvent aboutir à
des lésions plastiques d'origine ischémique.
· Ischémiques (clastiques) : certaines formes
d'atrésie intestinales.
· Disruptions au cours de la gémellité.
(Grossesse gémellaire monozygote, monochoriale biamniotique).
Après décès de l'un des 2 foetus, il
survient chez l'autre foetus de lésions ischémiques (infarctus)
=> cerveau (kystes porencéphaliques) et parfois aussi dans d'autres
viscères (rein, poumon,...). Ces lésions sont rapportées
à des
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 34 sur 107
perturbations vasculaires (anastomoses vasculaires entre les 2
secteurs placentaires).
II.4.3. Malformations : causes multifactorielles
En l'absence de cause évidente expliquant la survenue
d'une malformation, on évoque l'intrication de facteurs
génétiques et d'environnement ; exemple : spina-bifida, fente
palatine, voire maladie de Hirschprung.

Figure 4: Périodes à risque d'apparition de
malformations congénitales pendant le développement
humain.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 35 sur 107
II.5. Le diagnostic prénatal
Au cours des dernières décennies, les
progrès de la médecine périnatale ont été
considérables et permettent de détecter des anomalies et,
parfois, d'intervenir in utero.
a. Ultrasons
Bien qu'aucune étude n'ait démontré une
toxicité des ultrasons pour le foetus, certains pensent, sur base
d'arguments incomplets, que la multiplication des examens échographiques
comporterait un risque statistique de troubles du développement
cérébral. Une attitude de bon sens est de considérer que
l'examen échographique reste un examen médical et non une
procédure de confort, et est donc à utiliser sur base
d'indications médicales.
L'échographie bidimensionnelle couplée à
l'échographie Doppler permet de former des images de bonne
résolution et de visualiser le débit sanguin, les mouvements des
valves cardiaques. Elle permet de déterminer l'âge et la
croissance foetale et facilite le suivi des grossesses, réduisant de
manière significative les cas de mort de foetus à risque. Elle
permet de détecter de nombreuses malformations.
b. Dosages dans le sérum maternel
Des recherches sur des marqueurs biochimiques du statut foetal
ont conduit au développement de quelques tests, comme le dosage de
l'alphafoetoprotéine (AFP). L'AFP, l'équivalent de l'albumine
adulte, est produite par le foie foetal et atteint sa concentration
sérique maximale vers 14 semaines. Elle passe le placenta et passe dans
la circulation maternelle. Sa concentration dans le sérum maternel
augmente pendant le second trimestre et décroît après 30
semaines. La concentration d'AFP augmente dans le liquide amniotique et dans le
sérum maternel dans plusieurs malformations, surtout les déficits
de fermeture du tube neural (dysgraphies), mais aussi l'omphalocoele, le
gastroschisis, l'extrophie vésicale et d'autres. A l'inverse, le taux
d'AFP diminue en cas de
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 36 sur 107
trisomie 21 ou 18 et de triploïdie, anomalies qui sont
aussi associées à une diminution des taux de HCG et de oestriol
dans le sérum maternel, ce qui est à la base du "triple test"
(AFP, HCG, oestril) réalisé pour les dépister.
c. Amniocentèse
Pour pratiquer l'amniocentèse, une aiguille est
introduite dans la cavité amniotique sous contrôle
échographique et 20-30 ml de liquide sont aspirés. En raison de
cette quantité de liquide, cette procédure n'est guère
réalisée avant 14 semaines. Le risque de perte foetale est de 1%,
et inférieur lorsque l'amniocentèse est pratiquée dans des
centres spécialisés. Le liquide amniotique ainsi
prélevé est analysé (AFP,
acétylcholinestérase). Les cellules foetales desquamées
sont récupérées et utilisées pour analyses
génétiques (caryotype et autres). Ces cellules se divisent
lentement et il est nécessaire de les mettre en culture en
présence de mitogènes avant de les utiliser, ce qui prend 8
à 14 jours et retarde d'autant le diagnostic.
d. Biopsie villositaire
Pour pratiquer une biopsie villositaire, une aiguille est
introduite, par voie cutanée ou vaginale, dans la masse placentaire pour
aspirer 5 à 30 mg de tissu villositaire. Ces cellules peuvent être
analysées immédiatement, mais la précision des
résultats est limitée par la fréquence relativement
élevée des atypies chromosomiques dans le placenta normal. C'est
pourquoi on sépare les cellules du centre mésodermique des
cellules trophoblastiques par digestion à la trypsine, avant de les
mettre en culture. Le nombre élevé de cellules obtenues fait que
les analyses génétiques peuvent être
réalisées après seulement 2 ou 3 jours, permettant un
diagnostic plus rapide que par amniocentèse. Cependant, le risque pour
le foetus est deux fois plus élevé et des études
suggèrent qu'il existe un risque de malformation des membres.
Page 37 sur 107
En général, toutes ces procédures sont
à réserver aux grossesses à risque, en particulier
lorsque: 1) la mère a plus de 35 ans; 2) il existe une histoire de
défect de fermeture du tube neural dans la famille; 3) une anomalie
chromosomique affecte une grossesse antérieure; 4) un des parents est
affecté d'une anomalie chromosomique; 5) la mère est porteuse
d'une maladie liée à l'X.
II.6. Prévention
La diversité des causes et des déterminants des
troubles congénitaux impose l'application d'une gamme diversifiée
d'approches préventives et thérapeutiques, dont certaines
soulèvent des problèmes éthiques et sociaux. Parmi les
moyens de prévenir les troubles congénitaux figurent :
· l'apport de services de planification familiale et
d'autres services de soins de santé génésique, comme la
prévention et le traitement des infections à transmission
verticale (syphilis, par exemple) ;
· des interventions nutritionnelles, comme la
supplémentation en acide folique ou les programmes d'enrichissement des
aliments pour prévenir les malformations du tube neural (visant toutes
les femmes en âge de procréer) ;
· la vaccination préventive systématique
contre la rubéole et la varicelle ;
· l'éducation dans le cadre communautaire et les
campagnes d'éducation visant les femmes en âge de procréer
à propos des dangers de la consommation d'alcool et de
médicaments pouvant avoir des effets secondaires
tératogènes au début de la grossesse;
· la prestation de services de génétique
communautaire dans le cadre du système de soins de santé
primaires ;
·
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
la surveillance des troubles congénitaux.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 38 sur 107
II.7. La médecine foetale
Lors d'une anémie foetale importante, par exemple en
cas d'incompatibilité Rh, des transfusions foetales peuvent être
réalisées, via un cathéter introduit dans sa veine
ombilicale sous contrôle échographique.
Le traitement des infections, des arythmies cardiaques
foetales et autres problèmes médicaux est
généralement administré à la mère et atteint
le foetus par passage des médicaments à travers le placenta.
Parfois, il est nécessaire d'administrer des médicaments
directement au foetus, soit par injection intramusculaire ou via la veine
ombilicale.
La chirurgie du foetus est possible dans certains cas, dans
des centres spécialisés et lorsque l'indication est formelle.
Lors d'obstruction de l'urètre, on peut être amené à
insérer un cathéter dans la vessie. Le plus important est alors
de faire un diagnostic précoce, avant que des lésions
rénales par reflux ne s'installent. La chirurgie "ex utero" consiste
à ouvrir l'utérus pour opérer directement sur le foetus.
Elle a été utilisée pour réparer des hernies
diaphragmatiques congénitales, pour enlever des kystes du poumon et
réparer des lésions de type spina bifida. Les indications sont
rares et réservées aux cas où le risque foetal est
très important et où l'intervention elle-même
n'entraîne pas de séquelles importantes.
Une curiosité est la cicatrisation de la peau foetale.
Il est impossible de déceler la moindre trace d'une intervention sur un
foetus, dont la peau se cicatrise parfaitement, contrairement à la peau
postnatale. Les raisons de ce phénomène nous échappent,
mais il serait intéressant de les connaître pour en déduire
des procédures aidant à une meilleure cicatrisation des tissus
adultes.
Les cellules souches et la thérapie génique : Le
foetus ne développe pas de compétence immunitaire avant 18
semaines de gestation, et il serait possible avant cette date de lui
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 39 sur 107
transplanter des tissus ou cellules sans engendrer de rejet.
La recherche en cette matière est axée sur les cellules souches
hématopoïétiques pour traiter des déficiences
immunitaires ou hématologiques. La thérapie génique de
maladies métaboliques héréditaires est aussi à
l'étude, mais ces applications ne se traduiront pas en pratique clinique
avant plusieurs années, probablement décennies.
Page 40 sur 107

ETUDE DES MALFORMATIONS CONGENITALES CLINIQUEMENT
VISIBLES A LA NAISSANCE A LUBUMBASHI
DEUXIEME PARTIE :
Malformations congénitales cliniquement visibles
à Lubumbashi/Mémoire de spécialisation 2010-2011
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 41 sur 107
CHAPITRE DEUXIEME: PATIENTS ET METHODES
II.1. TYPE D'ETUDE ET DESCRIPTION DES SITES DE
RECHERCHE
II.1.1. Type d'étude
La présente étude est analytique
rétrospective (cas-témoins). Elle a consisté à
comparer la fréquence d'exposition antérieure à plusieurs
facteurs de risque dans un groupe de « cas » atteints de
malformations congénitales, et dans un groupe de « témoins
» indemnes de celle-ci.
La mesure des facteurs de risque s'est donc effectuée
rétrospectivement.
Elle a été réalisée à
travers la ville de Lubumbashi, dans 11 maternités
sélectionnées parmi les mieux organisées de la ville et
qui comptent environs 1325 naissances mensuelles, soit un nombre de naissance
attendu de plus de 15900 naissances.
II.1.2. Description des Maternités retenues
(Tableau I)
1. Cliniques Universitaires de Lubumbashi : Hôpital
Universitaire situé dans la zone de santé de Lubumbashi,
détenant une maternité d'une capacité de 30 lits, avec un
nombre mensuel moyen d'accouchements de 110.
2. Hôpital Provincial de Référence Jason
Sendwe : Le plus grand Hôpital de la ville de Lubumbashi, appartenant
à l'Etat Congolais. C'est l'hôpital de référence des
cas difficiles de toute la Province. Sa maternité a une capacité
de 60 lits avec un nombre mensuel de naissances de 200.
3. Hôpital SNCC : Hôpital appartenant à la
Société Nationale des Chemins de fer Congolais. Localisé
dans la zone de santé de Tshiamilemba dont il est l'Hôpital
Général de Référence. Sa maternité a une
capacité de 25 lits avec un nombre mensuel de 100 accouchements.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 42 sur 107
4. Hôpital Général de
Référence de la Katuba : Situé au coeur de la zone de
santé de la Katuba, cet hôpital appartient à l'Etat
Congolais. Sa maternité a une capacité de 20 lits et compte en
moyenne 70 accouchements mensuels.
5. Hôpital Général de
Référence de la Kenya : Situé au coeur de de la zone de la
Kenya, commune populaire de la ville de Lubumbashi, cet hôpital
appartient à l'Etat Congolais. Sa maternité a une capacité
de 30 lits et compte actuellement 300 accouchements par mois.
6. Hôpital Général de
Référence de Kampemba: Cet hôpital situé dans la
zone de santé de Kampemba appartient à l'Etat Congolais. Sa
maternité a une capacité maximale de 20 lits. Il compte environ
150 naissances mensuelles.
7. Polyclinique Afia: Hôpital privé situé
dans la zone de santé de Lubumbashi. Sa maternité a une
capacité d'accueil de 10 lits et compte environ 150 accouchements par
mois.
8. Polyclinique Flora : Hôpital privé
situé dans la zone de santé de Lubumbashi. Sa maternité a
une capacité de 10 lits et compte environ 60 naissances mensuelles.
9. Maternité du Centre de santé de
référence Bongonga : Maternité d'un centre de santé
de référence géré par l'ONG Fondation Muntundu. Sa
maternité a une capacité d'accueil de 10 lits et compte en
moyenne 40 accouchements par mois.
10. Maternité Watoto : Centre
mère-enfant situé dans la zone de santé
de Kampemba. Il a une capacité d'accueil de 18 lits et compte environ 45
accouchements mensuels.
11. Hôpital GECAMINES/sud : Cet Hôpital
appartient à la GECAMINES, principale entreprise minière du pays.
Sa maternité a une capacité d'accueil de 32 lits et compte
environ 120 naissances par mois.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 43 sur 107
|
Tableau II: Maternités retenues pour
l'étude
|
|
|
|
STRUCTURE
CLINIQUES UNIVERSITAIRES DE LUBUMBASHI
|
STATUT
État
|
ACCOUCHEMENTS/MOIS
110
|
|
HOPITAL PROVINCIAL DE REFERENCE SENDWE
COMPLEXE HOSPITALO CLINIQUE SNCC
HOPITAL GENERAL DE REFERENCE KATUBA
CENTRE DE SANTE DE REFERENCE BONGONGA
POLYCLINIQUE DON BOSCO
MATERNITE KARIBU WATOTO
HOPITAL GENERAL DE REFERENCE KAMPEMBA
TOTAL
État
Privé
État
État
Privé
Privé
État
État
Privé
Privé
180
100
70
40
120
150
300
45
60
150
1325
La récolte des données a eu lieu du mois d'avril
2010 à avril 2011.
HOPITAL GECAMINES SUD
II.2. TECHNIQUE D'ECHANTILLONNAGE
HOPITAL GENERAL DE REFERENCE KENYA
Notre échantillon a été de convenance.
POLYCLINIQUE FLORA
II.2.1. Critères d'inclusion
ACCOUCH. ESTIMATIF/AN
1320
2160
1200
840
480
1440
1800
3600
540
720
1800
15900
Nous avons considéré comme « cas » dans
notre étude tout nouveau - né ou toute
fausse couche >22 semaines, né ou admis dans l'une des
formations médicales
concernées (cf. Tableau II) durant la période
d'étude, et ayant présenté au moins une
malformation congénitale cliniquement visible.
Nous avons considéré comme
«témoins» des nouveau-nés exempts de malformations
congénitales cliniquement visibles à la
naissance.
Pour chaque « cas » de malformation identifié,
nous avons considéré les 2 naissances
suivantes exemptes de malformation congénitale comme
« témoins ».
Les mères ont été chaque fois
interrogées aussi bien pour les « cas » que pour les
« témoins ».
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 44 sur 107
II.2.2. Taille de l'échantillon
La taille de l'échantillon a été
calculée à l'aide du logiciel épi info version 3.4.1.
Pour un intervalle de confiance à 95% (erreur alpha de
5%) et une erreur béta de 80%. Considérant un cas pour 2
témoins et une fréquence attendue des malformations
congénitales de 20% (odds ratio=4.75), nous obtenons une taille minimale
totale de 186 sujets, soit 62 cas et 124 témoins.
Notre étude a porté sur 72 « cas »
avec malformations congénitales et 144 « témoins »,
soit une population totale de 216 sujets.
II.3 ICONOGRAPHIE
Des photos ont été prises pour les cas des
malformations congénitales observées.
II.4. CLASSIFICATION DES MALFORMATIONS
Nous nous sommes référés à la
Classification Internationale des Maladies (10ème
révision) publiée par l'OMS. (WHO, 2004)
II.5. METHODES STATISTIQUES
II.5.1. Gestion de la base des données
Le logiciel Epi info qui se trouve dans l'environnement
Windows a été utilisé pour la gestion des
données.
II.5.2. Tests utilisés
II.5.2.1. Statistiques descriptives
Celles-ci nous ont permis de calculer les fréquences.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 45 sur 107
II.5.2.2. Statistiques différentielles
La p-value du test du Chi-carré (X2) d'une
part, et l'intervalle de confiance à 95% *IC95%+ de l'ODDS RATIO (qui
donne la précision de l'estimation de l'OR), d'autre part,
effectué sur le tableau de contingence, ont permis de porter un jugement
de signification statistique :
? OR > 1, pX2 < 0.05 et IC
95% supérieur à 1 : risque significativement accru ? OR < 1,
pX2 < 0.05 et IC 95% inférieur à 1
: risque significativement réduit
? pX2 > 0.05 et IC 95% incluant
la valeur 1 : on ne met pas en évidence une relation statistiquement
significative entre le facteur étudié et la maladie.
II.6. CONSIDERATIONS ETHIQUES
- Consentement éclairé : nous avons signifié
aux accouchées qu'elles feraient
partie de l'échantillon de l'étude.
Des photos des nouveau-nés ont été
prises, avec l'accord de leur mère, pour faciliter la description
à posteriori.
- Confidentialité : Les photographies prises seront
réservées strictement à un
usage diagnostic et didactique.
Dans leur usage didactique, en aucun cas l'identité du
patient et de sa famille ne sera révélée.
- Principe de bienfaisance : Les cas ont été
orientés vers des structures de prise en
charge en fonction de la malformation observée.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 46 sur 107
II.7. DIFFICULTES RENCONTREES
a. La principale difficulté rencontrée est
celle relative à la collaboration avec les points focaux des
maternités cibles ainsi que la notification systématique des cas
porteurs de malformations congénitales :
Les malformations multiples spectaculaires ont souvent
été favorisées au détriment des malformations
uniques et mineures, induisant ainsi de façon évidente un sous
rapportage global des malformations.
b. Le biais de mémoire et de sélection induits
par les questions relatives aux antécédents maternels durant la
grossesse (fièvre maternelle, automédication et prise
d'alcool).
Page 47 sur 107
CHAPITRE TROIXIEME: RESULTATS
III.1. ETUDE EPIDEMIOLOGIQUE
III.1.1. Prévalence
Nous avons enregistré 72 cas de malformations pour 12320
naissances, soit une prévalence de 58,4 pour 10000 naissances
(0,58%).
III.1.2. Répartition des cas de malformations en
fonction de la maternité d'origine et de la prévalence par
maternité.
Tableau III : Répartition des cas par maternité
Maternité Nombre de cas Fréquence
(%)/72
Cliniques Universitaires 23 31.9
Hôpital Jason Sendwe 34 47.2
Hôpital SNCC 4 5.6
Hôpital GCM SUD 0 0
Hôpital de référence Katuba 3
4.2
Hôpital de référence Kenya 6
8.3
Hôpital de référence Kampemba 1
1.4
Polyclinique Flora 0 0.0
Polyclinique Afia 0 0.0
Maternité Watoto 1 1.4
TOTAL 72 100
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
47.2 pourcent des cas de malformation ont été
observés à l'hôpital Jason Sendwe
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 48 sur 107
Tableau IV. Prévalence des malformations par
maternité
Maternité
Nombre de cas
Nombre de naissances
Prévalence/ 10000 Nces
Cliniques Universitaires
Hopital Jason Sendwe
Hopital SNCC
Hopital GCM SUD
Hôpital de référence Katuba
Hôpital de référence Kenya
Centre de Santé Bongonga
Hôpital de référence Kampemba
Polyclinique Flora
Polyclinique Afia
Maternité Watoto
TOTAL
23
34
4
0
3
6
0
1
0
0
1
72
1256 2103 931 1030
631 2011 381 1294 530
1710
443
12320
183.1
161.7
43.0
0
47.5
29.8
0.0
7.7
0
0
22.6
La prévalence la plus élevée a
été observée aux Cliniques Universitaires de Lubumbashi et
à l'Hôpital Jason Sendwe.
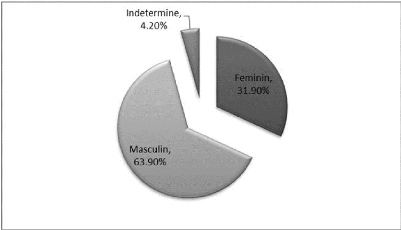
Figure 5: Répartition des cas de malformations
congénitales apparentes en fonction du sexe
Cette figure indique que le sexe masculin est plus
représenté que le sexe féminin avec
un Sex Ratio de 2
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 49 sur 107
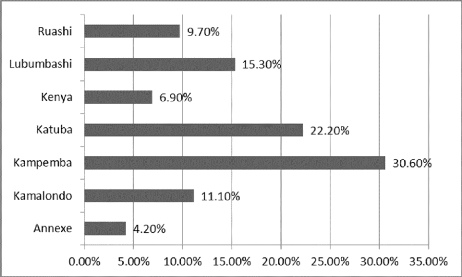
Figure 5: Répartition des cas de malformations
congénitales apparentes en fonction de la commune de provenance
La commune de Kampemba est la plus grande pourvoyeuse de cas
de malformations avec 30.6% des cas.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 50 sur 107
III.2. PROFIL CLINIQUE
III.2.1. Les type des malformations congénitales
Tableau V. Fréquence des malformations
congénitales selon le système (Classification
|
ICD-10)
|
|
|
|
|
|
|
|
|
Système
Peau
Yeux
|
Nombre
0
2
|
Fréquence (%)
/72
0
2.8
|
|
Fréquence/12320
Système respiratoire
Système digestif
Paroi abdominale
Système Urinaire
Oreilles
Fentes oro-faciales
Membres
Système nerveux central
TOTAL
1
4
5
5
7
10
13
25
72
1.4
5.6
6.9
6.9
9.7
13.9
18.12
34.7
100
0
1.6 0.8 3.2 4.1
4.1 5.7 8.1 10.6 20.2
58.4
Les malformations du système nerveux central sont les plus
fréquentes, suivies des
malformations des membres et des fentes oro-faciales.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 51 sur 107
Tableau VI. La fréquence des malformations
congénitales selon le type considéré dans la
classification internationale (WHO, 2004).
|
|
|
|
|
SYSTEM Nervous system
(n=25) Neural Tube Defects: Anencephalus and
similar Encephalocele
Spina Bifida
|
ICD code
Q00
Q01
Q05
|
N
1
1
7
|
|
|
Hydrocephaly
Microcephaly
Arhinencephaly / holoprosencephaly
|
Q03
Q02
Q04.1/Q04.2
|
5
5
6
|
|
|
Anophthalmos
Microphtalmos
|
Q11.0
Q11.1
|
1
|
|
|
|
1
|
|
|
Anotia
Low set ears
Choanal atresia
|
Q16.0
Q30.0
|
1
6
1
|
|
|
Cleft lip with /without cleft palate
|
Q36/Q37
|
8
|
|
|
Cleft palate
Ano-rectal atresia and stenosis
Gastroschisis
|
Q35
Q42
|
2
4
|
|
|
Omphalocele
|
Q79.3
Q79.2
|
2
3
|
|
|
Hypospadias
Indeterminate sex
|
Q54
Q56
|
2
3
|
|
|
Lower limb reduction
|
|
|
|
Frequency/10000
0.8
0.8
5.7
4.1
4.1
4.9
Eye (n=2)
0.8
0.8
Ear (n=7)
0.8
4.9
Respiratory (n=1)
0.8
Orofacial clefts (n=10)
6.5
1.6
Digestive system (n=4)
3.2
Abdominal wall defects (n=5)
1.6
2.4
Genital (n=5)
1.6
2.4
Limb (n=13)
Absence of both lower leg and foot
Club foot - talipes equinovarus
Polydactyly
Q72
Q72.2
Q66.0
Q69
1
7
5
0.8
5.7
4.1
TOTAL
72
58.4
Les spina-bifida, les fentes labiales et palatines ainsi que les
pieds bots sont les plus
représentés.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 52 sur 107
Tableau VII. Fréquence des complexes malformatifs
Syndrome de Down (Trisomie 21)
Syndrome de Patau (Trisomie 13)
Séquence de Prune Belly
Syndrome de Arnold Chiari
MCA-eci
1
4
1
1
1
0.81
3.25
0.81
0.81
0.81
TOTAL
8
6.5
La trisomie 13 a été observée chez 4 cas sur
12320 naissances, soit une prévalence de
3.2 pour 10000 naissances. Un cas de malformations
congénitales multiples d'origine
indéterminée a été décrit.
III.2.2. Diagnostic des malformations
congénitales
Tableau VIII : Période où le diagnostic a
été posé
|
|
|
|
|
|
Naissance
|
Nombre
69
|
95.8
|
|
Antenatal
|
3
|
|
Pourcentage
4.2
100
Dans notre série, 95.8 % des cas de malformation ont
été découverts à la naissance plutôt
qu'à l'échographie dans le cadre des consultations
prénatales.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 53 sur 107
III.2.3. Evolution des foetus et des nouveau nés
malformés
Tableau IX : Type de naissance
Malformés Non
Malformés OR X2 p
N % N %
Mort-né 8 11.1 8 5.6
Type de naissance Vivant à la
naissance 64 88.9 136 94.4 2.13(0.69-6.58) 2.15 0.142
TOTAL 72 100 144 100
Le risque pour un malformé d'être mort-né
est de 2.13; l'intervalle de confiance est compris entre 0.69-6.58. Le
X2 = 2.15 (p = 0,142)
Tableau X : Mortalité néonatale précoce
(7 jours) et malformations congénitales (Mort-nés exclus)
|
|
|
|
Nombre
|
|
|
Décès
Survivants
|
21
43
|
67.1
|
|
TOTAL
|
64
|
|
Pourcentage
32.9
100
32.9% des enfants malformés nés vivants sont
décédés dans les 7 jours qui ont suivi.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 54 sur 107
III.3.Facteurs de risque
III.3.1. Facteurs de risque maternel
Tableau XI. Facteurs maternels et risque de malformations
congénitales.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Malformés
|
|
Non Malformés
|
OR
|
X2
|
|
|
Prise d'alcool
|
Oui
Non
|
N
9
63
|
%
44.4
56.6
|
N
58
85
|
68.8
|
0.21 (0.09-0.48)
|
17.49
|
|
|
Automédication
|
TOTAL
Oui
Non
|
72
11
61
|
100
15.3
84.7
|
144
65
79
|
54.9
|
0.22(0.1-0.47)
|
18.68
|
|
|
Fievre maternelle au premier trimestre
|
TOTAL
Oui
Non
|
72
19
53
|
100
26.4
73.6
|
144
77
67
|
46.5
|
0.31(0.16-0.60)
|
14.19
|
|
|
Fausses couche/avortements spontanés
|
Total
Oui
Non
|
72
32
40
|
100
44.4
55.6
|
144
45
99
|
68.8
|
1.76(0.94-3.29)
|
3.63
|
|
%
31.3
100
45.1
100
53.5
100
TOTAL
72
100
144
31.3
100
Antécédants familiaux de malformation
Oui
Non
TOTAL
4
68
72
5.6
94.4
100
10
134
144
6.9
93.1
100
0.79(0.20-2.87)
0.15
Parité de la mère
1 a 3
> 3
Total
35
37
72
48.6
51.4
100
88
55
144
62.5
38.5
100
0.59(0.32-1.09)
3.25
Age de la mère
> 35 ans
< 35 ans
TOTAL
49
23
72
68,1
31,9
100
53
91
144
36,8
63,2
100
3.66(1.93-6.98)
18.72
Le risque pour une mère âgée de plus de 35
ans de donner naissance à un enfant
porteur d'une malformation congénitale est de 3.66;
l'intervalle de confiance est
compris entre 1.93-6.98. Le X2 = 18.72 (p =
0,00001).
L'absence de fièvre au premier trimestre (OR : 0.31
[0.16-0.62], p=0.001),
d'automédication (OR: 0.22 [0.1-0.47], p=0.00001) et de
prise d'alcool (OR : 0.21 [0.09-
0.48], p=0.00002) semblent constituer des facteurs protecteurs
vis-à-vis du risque de
donner naissance à un enfant porteur d'une malformation
congénitale.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 55 sur 107
III.3.2. Facteurs de risques paternels
Tableau XII. Malformations congénitales et profession du
père
|
Malformés Non
Malformés
|
OR X2 p
|
|
|
|
|
|
|
|
|
|
|
|
Miniers et ouvrier
Autres professions
|
N
44
28
|
%
61.1
38.9
|
N
38
106
|
26.4
|
4.38 (2.30-8.38)
|
24.46
|
|
%
TOTAL
72
100
144
73.6
100
0.0000008
Le risque pour un père minier ou ouvrier d'avoir un enfant
malformé est de 4,38;
l'intervalle de confiance est compris entre 2.30-8.38. Le
X2 = 24.46 (p = 0,0000008).
III.3.3 Facteur de risque environnemental
Tableau XIII : Lieu d'habitation - à proximité
versus éloigné - d'une usine de traitement
de minerais et malformations congénitales
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Malformés
|
|
Non
malformés
|
|
|
Proximité
|
32
40
|
N %
44.4
55.6
|
N
88
56
|
61.1
|
OR
|
X2
5.38
|
|
%
Situation géographique
Eloignées
TOTAL
72
100.0
144
38.9
100.0
0.51(0.28-0.94)
Le fait de résider hors d'une commune à forte
activité minière constitue un facteur
protecteur par rapport au risque de donner naissance à un
enfant malformé : OR : 0.51
(0.28-0.94) ; p<0.05.
III.3.4. Malformations congénitales et
consanguinité
Aucun enfant malformé n'était né d'une union
consanguine.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 56 sur 107
CHAPITRE QUATRIEME : DISCUSSION
Prévalence des malformations congénitales
visibles
Prévalence globale : Des malformations
congénitales ont été observées dans 58,3 pour 10000
naissances vivantes dans les onze maternités de Lubumbashi retenues pour
l'étude. Six années plus tôt, dans leurs études
réalisées à Lubumbashi et Kinshasa, Kalwaba et al. (2004)
et Tabu (2004), avaient évalué la prévalence des
malformations respectivement à 74 et 57 pour 10000 naissances
vivantes.
La prévalence trouvée dans notre étude
est significativement plus faible que celle de Kalwaba et al., en 2004
(X-score=2 >1.96, p<0.05). Cet état des choses s'expliquerait
davantage par un sous-rapportage que par une réelle baisse de la
prévalence des malformations à Lubumbashi.
Ceci est d'autant plus vraisemblable qu'aucune intervention
n'a été réalisée entre
temps.
Cette faible prévalence pourrait en outre être
mise en parallèle avec la baisse drastique de l'activité des
industries extractives minières survenue de 2008 à 2010
consécutivement à la crise financière internationale ayant
entrainé la chute du cours des principaux produits miniers extraits au
Katanga.
Rappelons que notre étude s'est effectuée
d'avril 2010 à avril 2011. Il est cependant important de souligner que
cette dernière hypothèse est à considérer avec
réserve car, à ce jour, aucune étude n'a établi de
façon formelle une relation de cause à effet entre
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 57 sur 107
l'activité minière à Lubumbashi et la
variation des prévalences des malformations congénitales.
En outre, il est intéressant de constater qu'il n'y a
pas de différence significative entre la prévalence
trouvée dans notre étude et celle de Kinshasa rapportée
par Tabu en 2004 (X-score=0.239 <1.96, p>0.05).
Mise à part la possibilité de sous rapportage
dans notre série, ceci pourrait s'expliquer par la présence
à Kinshasa d'autres facteurs de risque tant environnementaux que
parentaux.
Dans un tout autre contexte (pays industrialisé) le
registre des malformations congénitales du Hainaut et Namur, en
Belgique, qui porte sur 15000 naissances vivantes fait état d'une
prévalence de 2.5 % nettement supérieure à celle de notre
étude.
Ceci s'explique à la fois par le recrutement plus
rigoureux des cas, mais aussi par le fait que ce chiffre englobe tant les
malformations visibles que celles nécessitant pour leur diagnostic des
examens complémentaires dont nous n'avons pas pu disposer.
Prévalences spécifiques :
Dans notre étude, les malformations du système
nerveux étaient les plus fréquentes avec 20.29 pour 10000
naissances, suivies des malformations des membres avec 10.55 pour 10000
naissances et des fentes oro-faciales avec 8.11 pour 10000 naissances (Tableau
IV). S'agissant des malformations du système nerveux central, le
registre du Hainaut et Namur rapporte une prévalence quasi
équivalente à la nôtre (24 pour 10000 naissances
vivantes).
Le registre des malformations Eurocat, réalisé
à l'échelle européenne, rapporte quant à lui une
prévalence de 4 pour 10000 naissance vivantes, significativement
inférieure à la nôtre.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 58 sur 107
Dans ce groupe, les malformations par défaut de
fermeture du tube neural (Anencéphalie, spina bifida) dont la
pathogénie est liée aux carences en acide folique occupent une
place importante.
Ceci relance le débat sur la supplémentation en
acide folique chez la femme enceinte dans notre pays. En effet, il est admis
aujourd'hui que l'acide folique est un cofacteur dans la majorité des
réactions enzymatiques qui entrent en jeu dans la formation de nouveaux
tissus.
Un faible taux de folates dans les globules rouges maternels
en période périconceptionnelle a été fortement
associé à la survenue de défects du tube neural. Il a
été démontré que cette survenue était
dose-dépendante (Pass, 2006; Thame et al., 2002).
Eu égard à l'apport de l'acide folique, nos
résultats montrent que les attentes sont déçues.
En effet, malgré une supplémentation
théoriquement inscrite dans les protocoles des consultations
prénatales, les taux de prévalence des anomalies de fermeture du
tube neural et singulièrement du spina bifida sont
élevés.
Il y a donc lieu de s'interroger sur l'efficacité de la
politique nationale de supplémentation en acide folique (Pass, 2006).
Cette question est d'autant plus pertinente qu'en Irlande, la prévalence
des défects du tube neural est passée de 46.9/10.000 naissances
à 11.6/10.000 entre 1980 et 1994 (Thame et al., 2002).
Ces résultats sont attribués à la prise
effective d'acide folique en période périconceptionnelle. En
Suède, la prise d'acide folique en période conceptionnelle est
une stratégie nationale et a permis de baisser de façon
significative la prévalence des malformations du tube neural (Thame et
al., 2002).
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 59 sur 107
The American Academy of Pediatrics, reconnue par l'US Public
Health Service (USPHS) recommande la prise de 400 micros grammes d'acide
folique par jour en période péri-conceptionnelle. Cette mesure
permet de baisser la prévalence des malformations du tube neural de plus
de 50 %.
Une étude multicentrique (11 centres)
d'évaluation de la prise préventive d'acide folique entre 1987 et
1996 rapporte qu'il y a une diminution significative de la prévalence
des défects du tube neural dans des centres comme ceux d'Atlanta,
d'Angleterre, du Pays de Galles, de Hongrie ou du Japon (Wals et al., 2007).
L'application stricte de cette stratégie
nécessite une planification des grossesses. Or, en Amérique du
Nord et en Europe, près de 50 % de toutes les grossesses ne sont pas
planifiées (Gillerot et Mols, 2009). La situation n'est guère
meilleure à Lubumbashi, société pro nataliste.
Ceci pose la question d'une supplémentation
systématique et effective dans l'alimentation, en rappelant que cette
façon de procéder pourrait également avoir un effet
protecteur pour d'autres malformations comme certaines cardiopathies, les
fentes labiopalatines, l'atrésie anale et peut-être même la
trisomie 21 (Gillerot et Mols, 2009).
Cependant, il est important de souligner que le taux de
folates a un déterminisme génétique : il est sous le
contrôle du gène C677T. Dans la forme homozygote d'anomalie de ce
gène, le taux de folates érythrocytaire est de 20 %
inférieur à la normale (Morrison et al.,
1998).
Ceci explique que la prévalence du génotype TT
est élevée de façon significative chez les enfants
porteurs de spina bifida et chez leurs parents (Thamel, 2002). Dans d'autres
études, d'autres gènes (PDGFRA, CBS, MS, MTHFR), seuls ou en
interactions, sont impliqués dans la survenue des défects du tube
neural (Pass, 2006).
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 60 sur 107
S'agissant des fentes oro-faciales, notre étude
rapporte une prévalence de 8.1 pour 10000 naissances (Tableau V). Si on
considère l'ensemble des fentes palatines observées de 1990
à 2004, on relève 132 cas dans le registre Hainaut-Namur,
c'est-à-dire une prévalence de #177; 7 pour 10000 naissances.
Ces taux coïncident avec les chiffres globaux d'EUROCAT
central. En effet, le registre central (Belfast) a enregistré 3.426 cas
de 1980 à 1999, c'est-à-dire une prévalence globale de 5.8
pour 10000 naissances (Gillerot et Mols, 2009).
Ces malformations sont visibles dès la naissance et
aisément identifiables. Corollairement, le risque d'un
sous-enregistrement est faible. La prévalence des fentes oro-faciales
est extrêmement variable dans les pays Africains : de 3 pour 10000
naissances au Nigeria a 16.5 pour 10000 au Kenya (Butali et Mossey, 2009).
Ces prévalences importantes tant à Lubumbashi
que dans les autres pays Africains inquiètent compte tenu du poids
psychosocial et économique considérable des enfants porteurs de
fentes palatines.
Le manque d'infrastructure et de personnel qualifié
allonge la liste des problèmes que posent ces enfants dans les pays en
développement. Il convient en effet ici de souligner que l'équipe
spécialisée multidisciplinaire devrait idéalement
comprendre un chirurgien plastique, un pédiatre, un
généticien, un otorhinolaryngologiste, un dentiste
pédiatrique, une infirmière, un orthophoniste, un audiologiste et
un psychologue (Turgeon et al., 2008).
Il est donc crucial d'identifier les facteurs associés
à la survenue de ces fentes oro faciales afin de les prévenir.
L'hérédité est ici multifactorielle, et
l'acide folique pourrait jouer un rôle « protecteur »,
l'apparition de fentes labio maxillo palatines étant elle aussi
liée à la formation du tube neural au stade embryonnaire.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 61 sur 107
En effet, les parties médianes du visage se forment
à partir de la crête neurale qui, à l'instar du tube
neural, se développe à partir de la plaque neurale (Eichholzer et
al., 2008).
Dans notre série, les anomalies des membres figurent
parmi les plus fréquentes, essentiellement représentées
par les pieds bots avec 5.68 pour 10000 naissances, ainsi que la polydactylie
avec 3.24 pour 10000 naissances (Tableau V).
Les malformations réductionnelles des membres n'ont,
quant à elles, représenté que 0.8 pour 10000 naissances.
Ce groupe d'anomalies est emblématique car il fut en quelque sorte le
coup d'envoi d'EUROCAT par le truchement de l'épidémie de la
Thalidomide.
S'agissant de la prévalence en fonction des
maternités, il ressort des Tableaux II et III que les prévalences
sont élevées dans les hôpitaux Universitaires de la
Ville.
Ces résultats sont certainement influencés par
la présence dans ces hôpitaux d'un personnel plus entrainé
à la reconnaissance des malformations, par leur taux de
fréquentation élevé, mais aussi par leur statut
d'hôpitaux provinciaux de référence.
Dans l'analyse de la prévalence en fonction des
Communes de provenance des patients porteurs de malformations
congénitales, nous avons constaté que la Commune de Kampemba
(30.6%) était la plus représentée suivie de la Commune de
Katuba (22.2%). En outre il ressort de nos résultats que le fait de
vivre hors des communes à forte activité minière de la
ville de Lubumbashi (Ruashi : Usines Ruashi Mining ; Kampemba : Chemaf ; Annexe
: Somika,...) semble être un facteur protecteur vis-à-vis du
risque d'avoir un enfant porteur de malformations congénitales : OR=0.51
(0.28-0.94) p<0.05 (Tableau XIII).
Plusieurs hypothèses pourraient expliquer ces
résultats, notamment une exposition répétée
à des métaux toxiques ou à une radioactivité
anormalement élevée.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 62 sur 107
Parlant de cette dernière hypothèse, une
étude publiée en 1983 rapporte la survenue d'un cluster (6 cas)
de trisomies 21 chez des enfants dont les mères ont vécu leur
adolescence à Dundalk, en Grande-Bretagne, village exposé selon
les auteurs aux retombées radioactives de l'accident de Windscale
survenu le 10 octobre 1957 (Doyle, 2000).
Cependant deux autres études concernant les
malformations congénitales et les mutations germinales dans un rayon de
30 km autour de zones d'émanation radioactive en Hongrie (Doyle, 2000)
et en Roumanie (Bembea, 2001) n'ont retrouvé aucun lien évident
entre les taux de malformations avant et après la mise en service du
site.
A Lubumbashi, le risque d'exposition aux produits miniers
radioactifs, bien que considéré comme faible, n'a pas encore
été évalué et documenté ; d'où la
nécessité de rester prudent quant à son influence sur la
prévalence des malformations congénitales dans la ville (Banza et
al., 2009).
S'agissant de l'hypothèse relative à
l'intoxication par les métaux, les taux élevés de cobalt
et autres métaux toxiques mesurés dans les urines des sujets
issus de la population générale au Katanga confirment que la
population vivant près des usines de traitement de minerais est
exposée à ces toxiques (Banza et al., 2009).
Reconnaissons cependant que la démonstration de la
relation de cause à effet entre cette exposition aux métaux
liée au contexte minier et la majoration de la prévalence des
malformations congénitales est prématurée.
S'agissant de la répartition des cas en fonction du
sexe, nous avons trouvé un sex ratio de 2. Kalwaba a,
quant à lui, rapporté un sex ratio de 1.31 (Kalwaba et al., 2004)
.D'autres études ont montré que le sex ratio est proche de 1 dans
la majorité des malformations.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 63 sur 107
Dans les malformations du système génital
externe, une proportion plus importante de garçons va de soi (Gillerot
et Mols, 2009).
Considérant le moment de la découverte,
l'échographie prénatale est devenue une étape majeure dans
la découverte et la mise au point des anomalies foetales. Elle
s'intègre cependant dans un processus global qui « in fine »
participe à la prévention de ces anomalies et parfois (mais
rarement) à leur traitement.
Cette prévention peut s'entendre de 2 façons :
celle d'une part d'éviter, lors d'une grossesse en cours, la naissance
d'un enfant vivant atteint de malformations létales ou de malformations
telles que la qualité de sa vie en sera gravement altérée,
et celle d'autre part de prévenir, par l'intermédiaire du conseil
génétique, la naissance ultérieure au sein du couple d'un
enfant porteur de malformations identiques à celles du « cas
princeps » (Gillerot et Mols, 2009).
Dans notre étude, seuls 4.2% des cas ont
été diagnostiqués en anté natal (Tableau VIII). Ce
chiffre représente également le nombre des seules gestantes ayant
pu bénéficier d'une échographie durant leurs consultations
prénatales.
Ces résultats posent avec acuité le
problème d'accessibilité tant géographique que
financière des services d'échographie obstétricale dans la
ville de Lubumbashi.
Nous avons analysé la mortinatalité (Type de
naissance) et les décès néonataux survenus avant la fin de
la première semaine de vie des patients porteurs de malformations
congénitales (Décès néonataux).
En effet, les malformations congénitales sont responsables
de 7 % de l'ensemble des
décès néonataux dans le monde. Cette
proportion varie cependant de 5 % dans la Région de l'Asie du Sud-Est
à plus de 25 % en Europe.
Dans notre série, le risque pour un malformé
d'être mort-né est de 2.13 (Intervalle de confiance : 0.69-6.58,
X2 = 2.15, p = 0.142). La différence entre le groupe des
malformés
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 64 sur 107
et des enfants sains n'est cependant pas statistiquement
significative. Ici, il est possible qu'un biais de rapportage ait
influencé nos résultats, les nouveaux nés malformés
et mort nés n'étant probablement pas tous notifiés.
En période néonatale précoce, 32.9% des
enfants nés vivants et porteurs de malformations congénitales
sont décédés endéans les 7 jours. Ce chiffre
reflète l'ampleur du problème mais ne permet cependant pas
à lui seul de dégager la part relative des malformations dans la
mortalité néonatale précoce, étant donné
qu'il n'est pas comparé au taux de mortalité précoce des
nouveau-nés non malformés.
Dans notre série, des complexes malformatifs ont
été observés dans 6.5 cas pour 10000 naissances (Tableau
VI), la principale part revenant aux malformations d'origine chromosomiques
(environs 4 pour 10000 naissances). Ces résultats sont très
différents de ceux obtenus dans une étude Roumaine où la
prévalence était de 13.74 pour 10000 naissances vivantes (Bembea,
2001) et dans une autre étude réalisée dans les provinces
Belges du Hainaut et Namur, avec 33 pour 10000 naissances (Gillerot et Mols,
2009) .
Cette nette différence s'explique probablement par un
sous rapportage des cas, particulièrement des trisomies 21 qui,
semble-t-il, passent encore inaperçus en période néonatale
dans la plupart des maternités de la ville de Lubumbashi.
En outre, il est important de remarquer qu'avant l'apparition
et les progrès des tests biochimiques (triple test) et de
l'échographie anténatale, un certain nombre de trisomies 21,
aujourd'hui diagnostiquées en Europe, passaient inaperçues et
étaient éliminées sous la forme de fausses couches
tardives, voire d'enfants mort-nés macérés. Il n'est donc
pas exclu non plus qu'avant l'ère des tests biochimiques et de
l'échographie précoce, un certain nombre de cas d'anomalies
chromosomiques aient été « manqués » (Gillerot
et Mols, 2009).
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 65 sur 107
Nous pensons que le développement de
l'échographie anténatale à Lubumbashi pourrait permettre
de réduire drastiquement le sous rapportage de nos cas.
Notons avec intérêt qu'un cas de malformation
multiple de cause inconnue (MCA-eci) a été observé dans
notre série. De tels cas sont de la plus grande importance en
épidémiologie car ils traduisent souvent l'introduction d'un
tératogène dans l'environnement.
Selon Yves Gillerot (Gillerot et Mols, 2009), il ne faut pas
attendre qu'il y en ait des dizaines répertoriées pour tirer la
sonnette d'alarme. Un ensemble inédit, non décrit, et
retrouvé ne fût-ce qu'à 2 ou 3 reprises dans une
région donnée doit être considéré comme
très suspect.
Divers facteurs de risque potentiels ont été
analysés dans ce travail.
Comme on le sait, l'âge de la mère constitue un
facteur de risque foetal pour ce qui concerne la mortalité, la
morbidité et les malformations congénitales, avec comme exemple
emblématique la trisomie 21. De nombreuses études ont
montré que ce risque augmente surtout après 35 ans (Gillerot et
Mols, 2009).
Ces résultats concordent avec ceux de notre
étude où 68% des mères porteuses de foetus
malformés avaient plus de 35 ans. (Tableau X). En outre, nous avons
trouvé que, dans notre milieu, le risque pour une mère
âgée de plus de 35 ans de donner naissance à un enfant
porteur d'une malformation congénitale est de 3.66 (Intervalle de
confiance:1.93-6.98, X2 = 18.72, p = 0,00001).
Un consensus existe sur le fait que le risque de survenue de
malformations congénitales augmente avec l'âge maternel.
Néanmoins, des divergences demeurent encore quant à l'âge
limite au-delà duquel ce risque est évident.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 66 sur 107
Une étude saoudienne porte cette limite à 45 ans
(Al Bu Ali, 2011), tandis que la plupart des études européennes,
ainsi que celle réalisée dans nos conditions à Kinshasa
(Tabu, 2004) la situent à 35 ans.
Nous avons également étudié la profession
du père. En effet, la province du Katanga étant essentiellement
minière, nous avons voulu évaluer le risque pour des parents
travaillant dans les mines de cuivre et de cobalt d'engendrer un enfant
malformé.
Nos résultats montrent que le risque pour un
père minier ou ouvrier d'avoir un enfant malformé est très
important : OR= 4.38, IC : 2.30-8.38, X2 = 24.46, p = 0,0000008
(Tableau XI).
Les métaux auxquels les artisans miniers sont
exposés, tels qu'ils ressortent de la revue de la littérature sur
la composition des minerais du Katanga, sont notamment : l'arsenic, le cadmium,
le cobalt, le cuivre, le manganèse, le mercure, le plomb, le zinc,...
(Banza et al., 2009).
Les traces de ces minerais, identifiées et
quantifiées dans les cheveux des mineurs grâce à des
techniques de spectrométrie, ont révélé des teneurs
élevées et permis de matérialiser l'exposition aux
métaux contenus dans la gangue minéralisée (Elenge,
2010).
Toutefois aucune étude n'a à ce jour
formellement documenté un éventuel rapport entre cette exposition
professionnelle à des métaux dans notre milieu, la
toxicité de ces métaux et le risque de donner naissance à
un enfant porteur de malformations congénitales.
En outre, le niveau d'exposition à la
radioactivité des ouvriers des usines minières de Lubumbashi
n'est pas non plus documenté. Il ressort cependant des rapports de
géologues que la quasi-totalité des gisements exploités
ces dernières décennies ont un faible potentiel radioactif. Leur
effet délétère sur la lignée germinale masculine
serait
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 67 sur 107
donc à priori négligeable dans notre milieu. Il
convient néanmoins de rappeler ici que l'effet potentiellement
tératogène de telles substances a été bien
documenté.
En effet, une étude réalisée en 1989
(Rousseau, 1990) à partir de 640 caryotypes de spermatozoïdes de
patients ayant reçu une radiothérapie et/ou une
chimiothérapie montre une fréquence totale d'anomalies
chromosomiques de structure significativement élevée.
Dans une autre étude, on a observé 23% de
spermatozoïdes porteurs d'anomalies de structure chez un patient, 11 et 16
mois après une irradiation (dose testiculaire : 0,25 à 0,45 Gray)
pour séminome testiculaire (Rousseau, 1990).
Des études spermatiques (Rousseau, 1990) chez des
patients porteurs de translocations équilibrées ont montré
que les spermatozoïdes à caryotype
déséquilibré peuvent franchir toutes les étapes de
la fécondation hétérospécifique : attachement
à la membrane plasmique de l'ovocyte, entrée dans le cytoplasme
ovocytaire, décondensation nucléaire, formation de
pronucléus, synthèse d'ADN et condensation du matériel
génétique en métaphase.
Il semble, selon ces études, que la présence
d'anomalies chromosomiques multiples dans un spermatozoïde ne perturbe pas
toujours nécessairement le processus de fécondation dans le
système hétérospécifique.
Si on extrapole cette observation au système de
fécondation homospécifique (qui n'en diffère
essentiellement que par la présence de la zone pellucide), il en
résulterait la constitution d'un oeuf fécondé à
contenu cytogénétique très altéré.
S'agissant de la parité de la mère et des
antécédents de malformation foetale évoqués dans
d'autres études comme facteurs de risque de malformations
congénitales (Tabu, 2004), aucune association statistiquement
significative n'a été observée dans notre
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 68 sur 107
étude, probablement du fait de la petite taille de
notre échantillon en comparaison, par exemple, avec l'étude de
Tabu à Kinshasa (Tabu, 2004).
Quant aux antécédents de fausse
couche/avortement, associés dans l'étude de Tabu (Tabu, 2004)
à un risque plus élevé de malformation (OR=2.5, IC :
1.06-6.02), leur rôle n'a pas pu être mis en évidence dans
notre étude.
En effet, l'analyse du risque pour une mère ayant au
moins un antécédent d'avortement et/ou de fausse couche
spontanée de donner naissance à un enfant porteur d'une
malformation congénitale (Tableau X) n'a pas montré de
différence significative (OR= 1.76 ; IC :0.94-3.29. X2 =
3.63, p=0.05).
S'agissant de la notion d'automédication au premier
trimestre de la grossesse, elle est aujourd'hui admise comme facteur de risque
de survenue de malformations congénitales. Tabu, à Kinshasa
(Tabu, 2004), a montré qu'il existe près de 3 fois plus de risque
pour une femme qui consomme des médicaments sans ordonnance durant sa
grossesse de donner naissance à un enfant malformé.
On sait également que l'utilisation de
médicaments pendant la grossesse est courante. Un sondage de l'OMS a en
effet établi que 86 % des femmes prenaient des médicaments
pendant leur grossesse. Ce chiffre prend bien évidement en compte tant
les médicaments délivrés sur ordonnance que ceux
disponibles en vente libre.
Dans notre série, seuls 15.3% des femmes avec enfants
malformés ont avoué avoir pris des médicaments (hormis
vitamines) durant le premier trimestre de leur grossesse, ce qui pourrait
indiquer une peur de culpabilisation de leur part.
Il apparait néanmoins, au vu de nos résultats
englobant également le groupe contrôle, que le fait pour les
mères de ne pas s' « automédiquer » pendant la
grossesse est un facteur protecteur vis-à-vis du risque de donner
naissance à un enfant porteur de malformations congénitales
(OR=0.22, IC : 0.1-0.47, p=0.00001).
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 69 sur 107
Parmi les médicaments en vente libre couramment
utilisés pendant la grossesse, on trouve l'acétaminophène
(65 %), l'ibuprofène (10 %) et la pseudoéphédrine (15 %).
Mais il est bien inquiétant de reconnaitre qu'à Lubumbashi, tous
les médicaments sont accessibles aux femmes enceintes sans qu'aucune
ordonnance ne soit exigée.
S'agissant de l'hyperthermie, la fièvre maternelle au
premier trimestre de la grossesse constitue le premier agent
tératogène découvert initialement chez l'animal, puis chez
l'homme, avec des conséquences identiques dans les deux groupes (Graham,
1998).
Elle est aujourd'hui reconnue comme étant un agent
physique, à effets dose-dépendants, pouvant être
responsable d'avortements et de malformations (Milunsky et al., 1992). Dans la
littérature, elle est essentiellement incriminée dans la survenue
des anomalies de fermeture du tube neural : RR= 6.2, CI : 2.2-17.2 (Daly et
al., 1995) et des membres (Milunsky et al., 1992).
Ces effets peuvent cependant être atténués
dans certaines circonstances par le « heat shock response » (HSR) qui
confère à l'embryon une protection lorsqu'il est exposé
à une dose initiale de chaleur (Graham, 1998).
Dans notre étude, l'absence de fièvre chez les
mères constitue également un facteur protecteur vis-à-vis
du risque de donner naissance à un enfant malformé (OR=0.31, IC :
0.16-0.62, p=0.001).
Il semble qu'un effort supplémentaire pourrait
être fait dans le sens d'un meilleur suivi des grossesses, surtout dans
les régions de paludisme stable où il sévit sur un mode
hyper endémique, atteignant, au cours de la grossesse, des
prévalences record de 40% (Ehiolé et al., 2008).
La prise d'alcool pendant le premier trimestre de la grossesse
a aussi des effets connus sur le développement foetus (O'Leary et al.,
2010). Mais les conséquences d'une consommation légère
occasionnelle et d'une consommation importante régulière, bien
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 70 sur 107
que dépendant par la dose, ne sont pas toujours les
mêmes dans tous les cas: c'est l'effet « dose et modulation »
par susceptibilités individuelles variables (génome de la
mère et du foetus).
L'alcool est responsable d'une
dégénérescence neuronale par apoptose. Il perturbe
notamment le métabolisme des neurotransmetteurs en bloquant les
récepteurs N-Méthyl-D-Aspartate (NMDA) et en activant les
récepteurs Acide-Gama-Amino-Byturique (GABA).
L'importance de l'apoptose dépend de la quantité
totale d'alcool ingérée et de la durée pendant laquelle
elle est ingérée. Les effets de l'éthanol sur le foetus
peuvent se classer en 4 catégories : retard de croissance, anomalies
neurologiques, dysmorphies faciales et autres malformations (Gillerot et Dan,
2005 ;Gillerot et Mols, 2009).
Dans notre étude, il apparait que le fait pour les
mères de ne pas consommer d'alcool durant leur grossesse constitue un
facteur protecteur par rapport au risque de donner naissance à un enfant
malformé (OR=0.21, IC : 0.09-0.48, p=0.00002).
Il est à noter qu'ici également, seule une
petite proportion (12.5 %) de femmes avec enfant malformé a reconnu,
sans doute aussi par peur de culpabilisation, avoir consommé
régulièrement de l'alcool durant la grossesse.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 71 sur 107
CONCLUSIONS ET RECOMMANDATIONS
Du mois d'Avril 2010 à Avril 2011, nous avons
mené une étude analytique rétrospective
(Cas-témoins) dans la ville de Lubumbashi (RDC) à travers 11
maternités pour définir le type ainsi que la fréquence des
malformations congénitales cliniquement visibles à la naissance
et pour en dégager les facteurs de risque en vue d'orienter
d'éventuels projets d'intervention visant à prévenir leur
survenue à Lubumbashi.
Au cours de cette étude, nous avons obtenu une
prévalence de malformations 58.4 pour 10000 naissances (0.58%),
statistiquement la même que celle trouvée dans une étude
multicentrique à Kinshasa (0.57).
Le sexe masculin est plus représenté que le sexe
féminin avec un sex ratio de 2. La commune de Kampemba est la plus
grande pourvoyeuse de cas de malformations avec 30.6% des cas.
Les malformations du système nerveux central sont les
plus fréquentes (20.29 pour 10000 naissances) suivies des malformations
des membres (10.55 pour 10000 naissances) et des fentes oro-faciales (8.11 pour
10000 naissances).
En analysant plus en détail ces systèmes, les
spina-bifida, les fentes labiales et palatines ainsi que les pieds bots sont
les plus représentés avec respectivement 5.68 ; 6.49 et 5.68 pour
10000 naissances.
La trisomie 13 est la malformation chromosomique la plus
représentée dans notre série.
Près de 96% des cas de malformation ont
été découverts à la naissance plutôt
qu'à l'échographie dans le cadre des consultations
prénatales.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 72 sur 107
Dans cette série, 11.1% des enfants portaient des
malformations incompatibles avec la vie et 32.9% des cas de malformations
vivants à la naissance sont décédés dans les 7
jours qui ont suivi.
Le fait pour le père d'être ouvrier ou minier, de
même que l'âge de la mère supérieur à 35 ans
étaient associés à un risque élevé de donner
naissance à un enfant malformé dans notre milieu.
Par ailleurs, l'absence de fièvre au premier trimestre
de la grossesse et le fait pour les mères de ne consommer ni alcool, ni
médicaments sans ordonnance médicale de même que le fait de
résider en dehors d'une commune où il existe une
activité
minière, ont constitué des facteurs
protecteurs.
S'agissant de ce dernier facteur, la prudence s'impose car
aucune étude à ce jour n'a établi de façon formelle
de relation de cause à effet entre l'activité minière
à Lubumbashi et la variation des prévalences des malformations
congénitales.
Ce travail constitue un véritable plaidoyer en faveur
de la poursuite du projet de registre des malformations congénitales
à Lubumbashi initié en 2009 par les Professeurs Kalenga Muenze de
l'Université de Lubumbashi et Yves Gillerot de l'Université
Catholique de Louvain, responsable du registre des malformations
congénitales Belge du Hainaut et Namur.
Cet enregistrement systématique de tous les cas de
malformations congénitales est très capital dans la mesure
où il joue un rôle de « sentinelle » sur notre
environnement et où il permettra d'envisager une stratégie
nationale et/ou régionale de diagnostic anténatal, de conseil
génétique et de prévention des malformations
congénitales à la naissance.
Nous recommandons une surveillance particulière
d'éventuels nouveaux complexes malformatifs de cause inconnue.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 73 sur 107
Nos résultats nous permettent de recommander
également le renforcement de la politique de supplémentation en
acide folique des femmes en âge de procréer, des programmes de
sensibilisation maternelle au cours des consultations prénatales
vis-à-vis de la consommation d'alcool et de l'automédication
d'une part, et d'encourager la supplémentation
périconceptionnelle en acide folique d'autre part.
Il s'avère également vital de renforcer les
mesures de sécurité dans les exploitations minières au
Katanga pour minimiser le risque pour ces ouvriers d'avoir des enfants porteurs
de malformations congénitales.
La poursuite de ce projet et la mise en oeuvre de ces
recommandations nécessitent des moyens tant humains que financiers. Sa
pérennisation ne peut donc s'inscrire que dans le cadre d'un programme
national avec implication des pouvoirs publics.
Page 74 sur 107

Annexes
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 75 sur 107
ANNEXE I : Classification internationale des
malformations 10 ème révision (WHO, 2004)
SYSTEM ICD code
Nervous system Neural Tube Defects:
Anencephalus and similar Q00
Encephalocele Q01
Spina Bifida Q05
Hydrocephaly Q03
Microcephaly Q02
Arhinencephaly / holoprosencephaly
Q04.1/Q04.2
Eye
Anophthalmos Q11.0
Microphthalmos Q11.1
Cataract Q12.0
Ear
Microtia Q17.2
Anotia Q16.0
Respiratory
Choanal atresia Q30.0
Orofacial clefts
Cleft lip with /without cleft palate Q36/Q37
Cleft palate Q35
Digestive system
Oesophageal atresia and stenosis Q39
Ano-rectal atresia and stenosis Q42
Abdominal wall defects
Gastroschisis Q79.3
Omphalocele Q79.2
Urinary
Bladder extrophy Q641
Genital
Hypospadias Q54
Indeterminate sex Q56
Limb
Upper limb reduction Q71
Complete absence of upper limb Q71.0
Absence of upper arm and forearm with hand present
Q71.1
Absence of both forearm and hand Q71.2
Absence of hand and fingers Q71.3
Longitudinal reduction defect/shortening of arm
Q71.4
Fewer 5 fingers Q71.8
Lower limb reduction Q72
Complete absence of lower limb Q72.0
Absence of thigh and lower leg with foot present
Q72.1
Absence of both lower leg and foot Q72.2
Absence of foot and toe Q72.3
Longitudinal reduction defect/shortening of leg
Q72.4
Split foot Q72.7
Club foot - talipes equinovarus Q66.0
Polydactyly Q69
Syndactyly Q70
Darwfism Q77.4
Other malformations
Arthrogryposis multiplex congenital Q74
Congenital constriction band/ amniotic band
Q79.8
Conjoined twins Q89.4
Disorders of skin Q80-Q82
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 76 sur 107
Annexe II : Iconographie
I.SYSTEME NERVEUX
1. ANENCEPHALIE (ICD10:Q00)
Description: Absence totale ou partielle du tissus
cérébral et de la voute crânienne. La face et les yeux sont
présents.
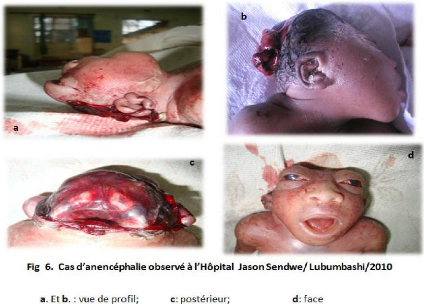
Note: Incompatible avec la vie, la mort survient dans les
premières 48 heures de vie. Malformations associées : Anotie,
Malformations cardiaques, fentes labiales et palatines.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 77 sur 107
4. ENCEPHALOCELE (ICD10:Q01)
Description : Expansion des méninges et du tissu
cérébral à l'extérieur de la boite crânienne
recouverte par une peau normale ou atrophique.
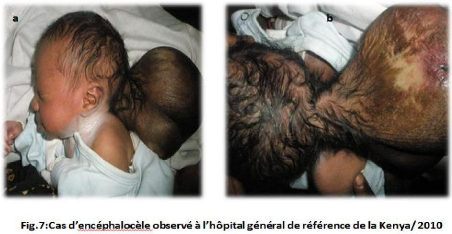
Note: 75% des encéphalocèles se situent dans la
région occipitale. Malformations associées: Fentes labiales,
anophtalmie/microphtalmie, Malformations cardiaques, Malformations
réductionnelles des membres, Polydactylie.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 78 sur 107
5. SPINA BIFIDA (ICD10:Q05)
Description : Exposition de la moelle osseuse et des
méninges résultant d'un défect des arcs postérieurs
des vertèbres.
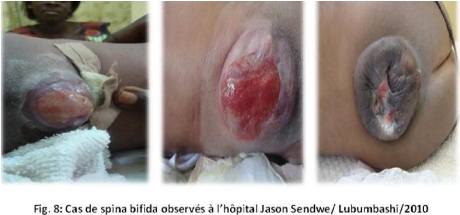
Note: 90% des cas de spina bifida sont de type
myéloméningocèles. 70% des
myéloméningocèles se situent dans la région
lombaire et lombo-sacrée.
Malformations associées: Hydrocéphalie,
Malformations cardiaques, atrésie ano-rectale, défects de la
paroi abdominale, fentes oro-faciales, anophtalmie/ microphtalmie,
malformations réductionnelles des membres.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 79 sur 107
6. HYDROCEPHALIE (ICD10:Q03)
Description: Dilatation des ventricules cérébraux
avec élargissement du crâne. .
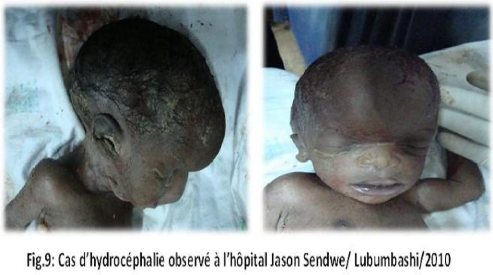
Note: Hydrocéphalie est caractérisée par
une augmentation du périmètre crânien au-delà de la
valeur normale pour l'âge sur les courbes de croissance. La fontanelle
peut être tendue ou bombée avec disjonction des sutures
crâniennes. On peut observer un regard en coucher du soleil (the
«setting sun sign).
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 80 sur 107
7. MICROCEPHALIE (ICD10:Q02)
Description: Le périmètre crânien est plus
petit que la normale pour l'âge.
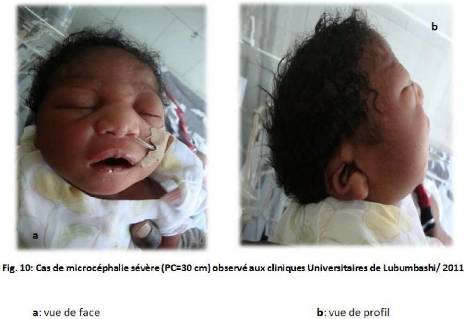
Note: La microcéphalie peut être définie
par un périmètre crânien inférieur à 5
déviations standard par rapport à la valeur moyenne pour
l'âge (Opitz, 1990). Plusieurs mesures successives sont
recommandées, particulièrement lorsque l'anomalie est minime. La
microcéphalie est un signe indirect d'un cerveau de petite taille
(Woods, 2004). Ses causes sont variables et elle entre dans la constitution de
nombreux syndromes génétiques.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 81 sur 107
8. HOLOPROSENCEPHALIE (ICD10:Q04)
Description: Résulte d'une erreur dans le
développement cérébral et dans sa séparation en 2
hémisphères cérébraux et ventricules
latéraux.
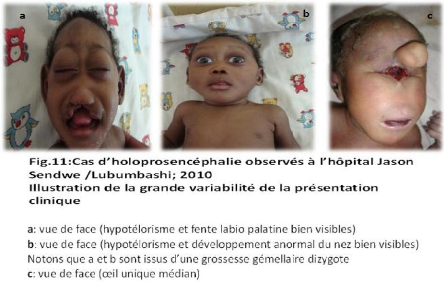
Note: Holoprosencéphalie est accompagnée de
manifestations faciales caractéristiques dans 80% des cas:
hypotélorisme, oeil médian unique, d'anomalies du
développement du nez et plus rarement d'une fente labiopalatine
médiane. L'holoprosencéphalie peut être isolée ou
retrouvée dans le cadre d'un syndrome tel que la trisomie 13.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 82 sur 107
II.YEUX ET OREILLES
1. ANOPHTHALMIE / MICROPHTHALMIE (ICD10:Q11)
Description: Anophtalmie: Absence unilatérale ou
bilatérale du tissu oculaire. Microphtalmie : Yeux avec un
diamètre axial inferieur à la normale.
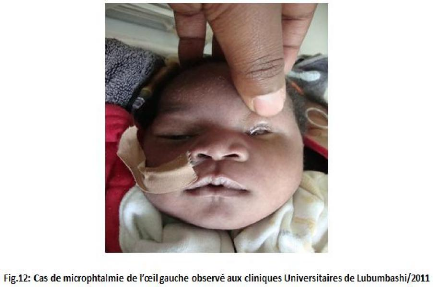
Note: Généralement accompagnée de
malformations cérébrales et de la face. Fréquemment
observée dans les syndromes génétiques.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 83 sur 107
3. MICRO/ANOTIA (ICD10:Q16) Description:
? Anotie: Absence du pavillon de l'Oreille avec ou sans
atrésie du conduit auditif. ? Microtie : Malformation ou hypoplasie de
l'Oreille externe.
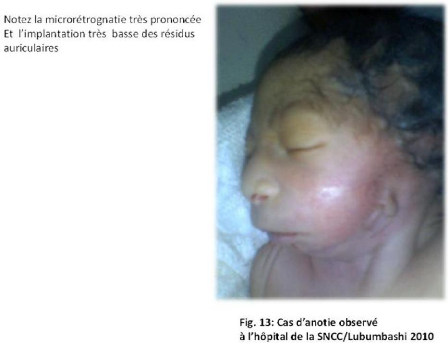
Note: Tout enfant présentant une malformation de
l'Oreille externe devrait bénéficier d'un examen clinique
minutieux a la recherché de dysmorphies crânio-faciales et
d'autres malformations.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 84 sur 107
IV.FENTES ORO-FACIALES
1. FENTES LABIALES AVEC OU SANS FENTES PALATINES
(ICD10:Q36/37)
Description : Fente du maxillaire supérieur et des palais
durs et mous
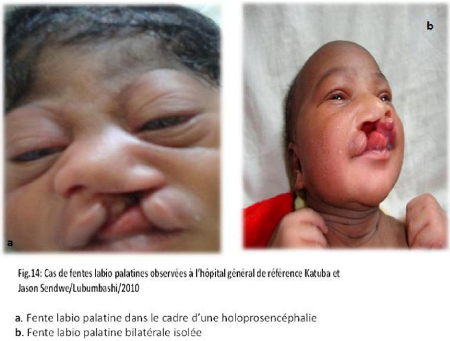
Note: Les fentes labiales peuvent être
unilatérales (80%) ou bilatérales (20%), et quand elles sont
unilatérales, elles sont généralement gauches (70%).
Environs 85% de fentes labiales bilatérales et 70% de
fentes labiales unilatérales sont associés à des fentes
palatines.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 85 sur 107
V.SYSTEME DIGESTIF
1. ATRESIE ET STENOSE ANO-RECTALE (ICD10:Q42)
Description: Imperforation anale ou absence ou sténose
de l'ampoule rectale.
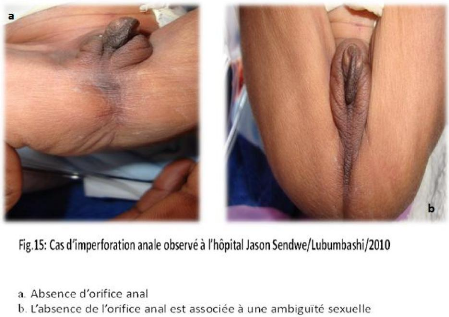
Note: Le diagnostic est posé précocement par
l'absence de l'orifice anal ainsi que par une absence d'élimination du
méconium.
L'atrésie ano-rectale est l'une des malformations
observées dans le syndrome VATER ou l'association
VACTERL (Vertebral, Anal, Cardiac, Trachéo-oesophageal, Renal,
Limb defects) association pouvant inclure des malformations vertébrales
et cardiaques, une fistule tracheo-oesophagienne, ainsi que des malformations
rénales et des membres.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 86 sur 107
VI.DEFECTS DE LA PAROI ABDOMINALE 1. GASTROSCHISIS
(ICD10:Q79.3)
Description: Protrusion du contenu abdominal non recouvert d'une
membrane, à travers un orifice latéral, laissant l'ombilic
intact.
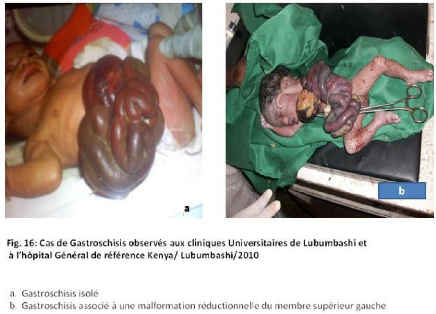
Note: Contrairement à l'omphalocèle, le
gastroschisis est habituellement une malformation isolée. A ce jour, il
ne fait pas parti d'un syndrome rapporté dans la littérature.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 87 sur 107
2. OMPHALOCELE (ICD10:Q79.2)
Description: Herniation du contenu abdominal à travers
l'anneau ombilical. Le contenu est recouvert d'une membrane qui peut se rompre
pendant le travail.
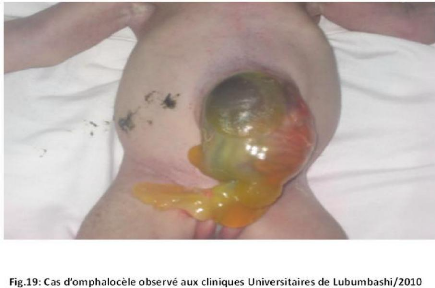
Note: Des malformations congénitales sont
fréquemment associées à l'omphalocele et leur incidence
varie de 40% à 90%: Système nerveux central, Système
cardio vasculaire et Système génito urinaire.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 88 sur 107
VIII.SYSTEME GENITAL
1. HYPOSPADIAS (ICD10:Q54)
Description: Le méat urétral est anormalement
déplacé vers la surface ventrale du pénis.
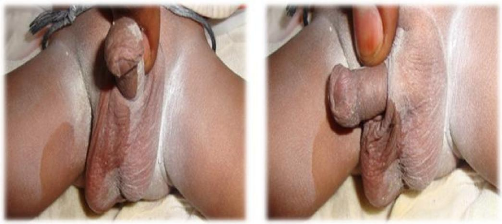
Note: Hypospadias est habituellement une anomalie
isolée. Il peut cependant être observé en association
à d'autres malformations: cryptorchidie, fentes labiales et palatines,
malformations cardiaques, syndactylie, polydactylie.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 89 sur 107
3. AMBIGUITE SEXUELLE (ICD10:Q56)
Ambiguïté sexuelle observée à la
naissance complique la détermination du sexe génotypique
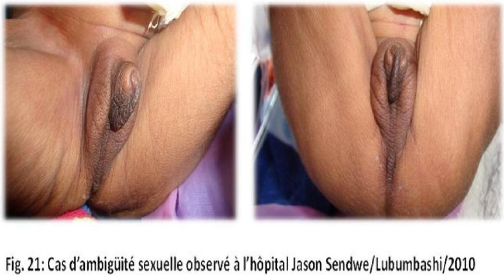
Note: nécessite un bilan malformatif complet et la
réalisation d'un caryotype. En attendant , il est
préférable de différer l'annonce du sexe de l'enfant.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 90 sur 107
IX. MEMBRES
1. MALFORMATION REDUCTIONNELLE DES MEMBRES SUPERIEURS
(ICD10:Q71)
Description: Absence complète ou partielle du bras
(humérus), ou de l'avant-bras (radius et/ou cubitus), ou de la main
(carpes, métacarpes, phalanges).
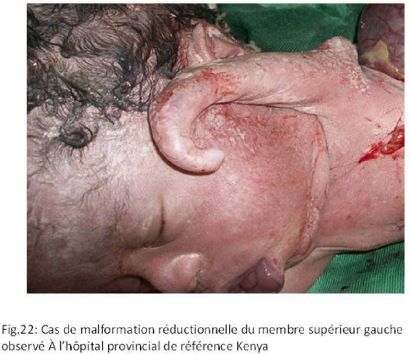
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 91 sur 107
2. MALFORMATION REDUCTIONNELLE DES MEMBRES INFERIEURS
(ICD10:Q72)
Description: Absence complète ou partielle de la cuisse
(fémur), de la jambe (tibia et/ou péroné), des pieds
(tarses, métatarses), ou des orteils (phalanges).
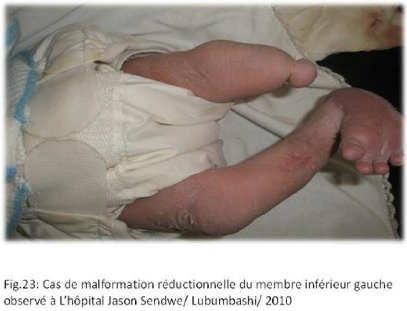
Note: Les malformations des membres supérieurs sont
plus fréquentes que celles des membres inférieurs (60-80% versus
25-40%).
Les malformations généralement associées
sont : cryptorchidie, fentes labio palatines, pieds bots, syndactylie,
agénésie rénale, imperforation anale et
hydrocéphalie.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 92 sur 107
4. PIEDS BOTS VARUS EQUINS (ICD10:Q66)
Description: anomalie du pied avec position équine au
niveau de la cheville, varus du pied et adduction de l'extrémité
du pied.
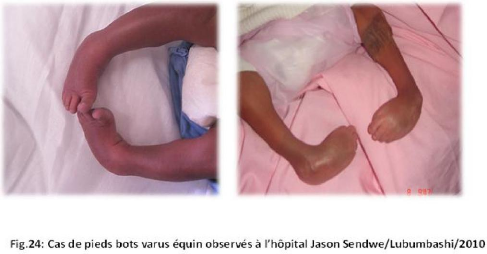
Note: Les pieds bots varus équins peuvent être
associés à d'autres malformations congénitales (telles que
spina bifida cystica) ou syndromes (trisomie 18, Edwards syndrome).
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 93 sur 107
5. POLYDACTYLIE (ICD10:Q69)
Description: Doigts surnuméraires sur la main et/ou les
pieds. Il peut s'agir d'une polydactylie pré-axiale (Côté
radial) ou post-axiales (côté cubital).
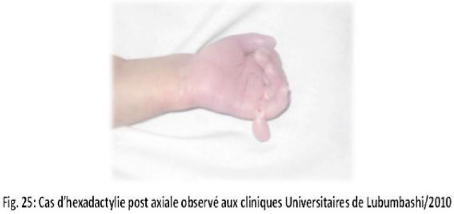
Note: La polydactylie est habituellement isolée (dans
85% à 88% des cas). Plusieurs anomalies majeures ont été
rapportées en association avec la polydactylie : Anomalies des membres,
anomalies des systèmes nerveux central, cardiovasculaire,
gastrointestinal et génito-urinaire, fentes labiales et/ou palatines,
anophtalmie, microtie...
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 94 sur 107
X. MALFORMATIONS D'ORIGINE CHROMOSOMIQUE
1. TRISOMIE 13 : SYNDROME DE PATAU
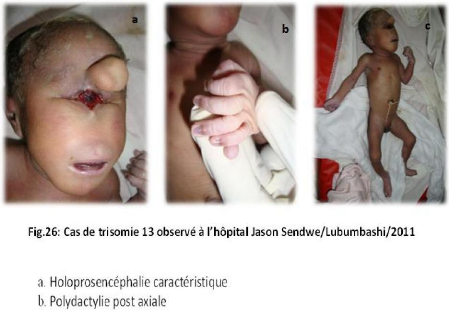
Trisomie 13 : Présence d'un chromosome 13
supplémentaire. Cela s'accompagne de diverses malformations notamment
malformations cérébrales (holoprosencéphalie,
arhinencéphalie), cardiaques (CIA, CIV, CA), crânio-faciales
(Gueule de loup, anophtalmie, proboscis), anomalies des
extrémités (Hexadactylies). Certaines comme la
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 95 sur 107
polydactylie sont peu graves, mais d'autres, qui touchent le
coeur ou le cerveau, sont létales (mortelles) : 97% des enfants atteints
meurent avant l'âge de 6 mois.
2) TRISOMIE 21 : SYNDROME DE DOWN
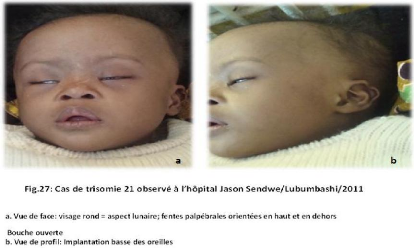
Trisomie 21 : Dysmorphie crânio-faciale :
- Aspect global : cou court, visage rond de face et plat de
profil = aspect
lunaire
- Etage oculaire : fentes palpébrales orientées en
haut et en dehors - nez
court - bouche ouverte, petite, lèvres
épaisses
- Pli palmaire unique.
Formes cliniques :
- Trisomie isolée : forme classique (94% des cas). Origine
: Non disjonction méiotique chez l'un des parents.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 96 sur 107
- Trisomie en mosaïque (2.4%). Origine : Non disjonction
mitotique (post-zygotique)
- Trisomie par translocation (3.6%). Origine :
Ségrégation méiotique non équilibrée
XI. SEQUENCES MALFORMATIVES : Syndrome d'ARNOLD-CHIARI
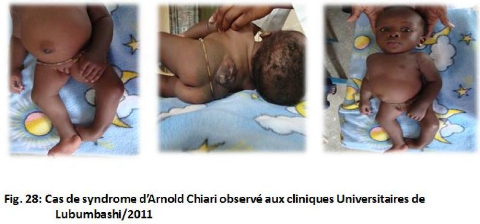
Note : Dans le type II de ce syndrome, le
myéloméningocèle dorso-lombaire est associé
à une hydrocéphalie. Le tronc cérébral et le
cervelet sont situés dans le canal cervical (Turgeon, 2008).
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 97 sur 107
XII. VALVE DE L'URETRE POSTERIEUR/ OU PRUNE BELLY SYNDROME
(ICD10:Q79.4)
Description: Obstruction de l'Urètre associée
à une dilatation de la vessie et une hydronéphrose. Dans les cas
sévères, on observe une distension de l'abdomen.
Prune belly syndrome: Caractérisé par la Triade :
1.Agenesie des muscles de la paroi abdominale; 2. Obstruction empêchant
la vidange vésicale et 3.Cryptorchydie bilatérale.
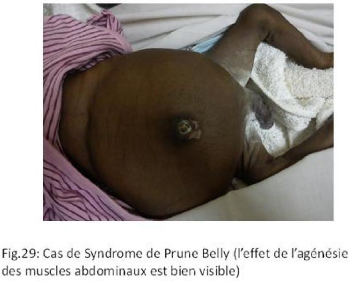
Note: Prune belly syndrome peut être associé aux
trisomies 18 et 21. Les Patients avec prune belly syndrome ont également
une incidence importante de tétralogie de Fallot (TF) et de
malformations ventriculo septales.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 98 sur 107
XIII. MALFORMATION CONGENITALE D'ORIGINE INCONUE (MCA-eci)
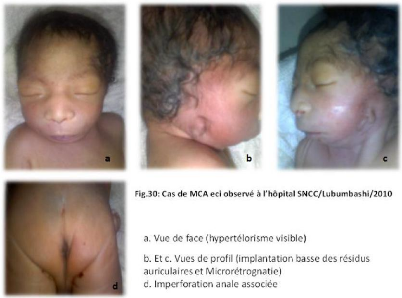
Note : On note une microrétrognatie très
prononcée et une implantation très basse des résidus
auriculaires un peu comme dans l'une des formes légères
d'otocéphalie. L'association de cette malformation complexe à une
imperforation anale fait que nous sommes dans un de ces fameux cas de MCA
eci.
Ici, il ne fait pas de doute que l'atteinte de l'embryon a
été très précoce.
Comme étiologies l'on pourrait penser à une
forme grave et bilatérale de Goldenhar ou syndrome
oculo-auriculo-vertébral (Wilson, 1983). L'association avec
l'imperforation anale doit aussi faire penser à un état
intermédiaire et déjà décrit entre un Goldenhar et
un Townes-Brocks.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 99 sur 107
ANNEXE III : Liste des figures
Numéro figure Titre de la
figure
Fig.1 Gametogenese
Fig.2 de la fécondation a la nidation
Fig.3 Différence schématique entre
Malformations, disruptions et déformations
Fig.4 Périodes à risque d'apparition de
malformations congénitales pendant le développement
humain
Fig.5 Répartition des cas de malformations
congénitales apparentes en fonction du sexe
Fig.6 Répartition des cas de malformations
congénitales apparentes en fonction de la commune de provenance
Fig.9 Cas de spina bifida
Fig.10 Cas d' hydrocephalie
Fig.13 Cas de microphtalmie
Fig.16 Cas d' imperforation anale
Fig.17 Cas de Laparoschisis
Fig.18 Cas d' omphalocele
Fig.19 Cas d' hypospadias
Fig.20 Cas d' ambiguite sexuelle
Fig.7 Cas d'anencéphalie
Fig.21 Cas de malformation reductionnelle du membre
superieur
Fig.8 Cas d'encéphalocèle
Fig.22 Cas de malformation reductionnelle du membre
inferieur
Fig.23 Cas de pied bot
Fig.24 Cas de polydactilie
Fig.11 Cas de microcéphalie
sévère
Fig.25 Cas de trisomie 13
Fig.12 Cas d' holoprosencephalie
Fig.26 Cas de trisomie 21
Fig.27 Cas de syndrome d' arnold Chiari
Fig.14 Cas d' anotie
Fig.29 Cas de syndrome de prune belly
Fig.15 Cas de fentes labio palatines
Fig.30 Cas de MCA eci
ANNEXE IV : Liste des tableaux
Numéro tableau Titre du
tableau
|
II Liste des maternités retenues dans
l'étude
|
I Croissance et développement de l'embryon
humain
III Répartition des cas par maternité
IV Prévalence des malformations congénitales
par maternité
V La fréquence des malformations congénitales
selon le système
VII Fréquence des complexes malformatifs
|
VI La fréquence des malformations congénitales
selon le type
VIII Période ou le diagnostic a été
pose
|
IX Type de naissance
|
X Mortalité néonatale précoce et
malformations congénitales
|
XI Facteurs de risque maternels
|
XII Malformations congénitales et profession du
père
|
XIII Lieu d'habitation et malformations
congénitales
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 100 sur 107
Bibliographie
Al Bu Ali W.H., Balaha M.H., Saleh Al M.(2011) Risk factors
and birth prevalence of birth defects and inborn errors of metabolism in Al
Ahsa, Saudi Arabia . Pan African Medical Journal,
8(14):1-17.
Banza C.L., Nawrot T.S., Haufroid V., Decrée S., De
Putter T., Smolders E., Kabyla B.I., Luboya N.O., Ilunga A.N., Mutombo A.M.,
Nemery B. (2009). High human exposure to cobalt and other metals in Katanga, a
mining area of the Démocratic Républic of Congo.
Environmental research, 109 :745-752.
Baudet J.H., Ronayette D. (1990) Développement de
l'oeuf fécondé In Baudet J.H., Ronayette D.
(eds) Obstétrique pratique. Maloine, Paris: pp 11-21.
Baudet J.H., Ronayette D. (1990)
Généralités sur les malformations congénitales
In Baudet J.H., Ronayette D. (eds) Obstétrique
pratique. Maloine, Paris: pp 457-463.
Bembea M. (2001) Clinical and epidemiological aspects of
congenital anomalies in bihor county in 16 years of activity. Elite
méd, 1: 11-20.
Brent R.L. (2004) Environmental Causes of Human Congenital
Malformations. Pediatrics , 113: 957-968.
Butali A. and Mossey P. (2009) Epidemiology of Orofacial
clefts in Africa: Methodological challenges in ascertainment. Pan African
Medical Journal, 2(5): 1-9.
Caouette-Laberge L., Bernard-Bonin A.C., Hudon C., Arsenault
N., Abelardo L., Quintal M-C. (2008) Fentes labiale et palatine. In
Turgeon J., Bernard-Bonin A.C., Gervais P., Ovetchkine P. et Gauthier
M. (eds) Dictionnaire de thérapeutique pédiatrique Weber 2e
édition. De Boeck, Bruxelles : pp 544-545.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 101 sur 107
Charon P. (2005) Tératologie du tube neural: histoire et
paléopathologie. Antropo, 10 :
83-101.
Coulibaly-Zerbo F., Amorisani-Folquet M., Kacou-Kakou A.,
Sylla M., Noua F., Kramo E. (1997) Etude épidemiologique des
malformations congénitales. Médecine d'Afrique Noire,
44(7): 409-414.
Daly L.E., Kirke N., Molloy A., Scott J. (1995) Folate levels
and neural tube defects implications for prevention. JAMA,
274(21): 1698-1702.
De Wals P., Tairou F., Van Allen M.I., Soo-Hong Uh., Lowry
R.B., Sibald B., Evans J.A., Van den Hof M.C., Zimmer P., Crowley M., Fernandez
B., Lee N.S., Niyonsenga T. (2007) Reduction in Neural-Tube Defects after acid
folic fortification. N Engl J Med, 357: 135142.
Dimmick J.E., Kalousek D.K. (1992) Developmental pathology of
Fetus and embryology. Lippicott Williams and Wilkins, Pennsylvania: pp 132.
Doyle P., Maconochie N., Ronan E., Davies G., Smith P.G.,
Beral V. (2000) Fetal death and congenital malformation in babies born to
nuclear industry employees : report from the nuclear industry fanily study.
Lancet , 356: 1293-1299.
Ehiolé S.P., Bissagnéné E. et Girard P-M.
(2008) Mémento thérapeutique du paludisme en Afrique.
IMEA, Paris : pp 99.
Eichholzer, Camenzind-Frey E., Amberg J., Baerlocher K. (2008)
L'acide folique: indispensable au développement normal de l'embryon.
Office fédéral de la santé publique-OFS :p 29-44.
Elenge M.M. (2010) Santé et sécurité dans
l'exploitation minière au Katanga:Etat des lieux. Livre des
résumés du colloque international : La quête des ressources
en Afrique Centrale-2. Tervuren :p 97-98.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 102 sur 107
Gasser R.F. (1975) Atlas of human embryos. Harper
& Row, publishers Inc. Hagerstown, Maryland.
Gilbert-Barnes E., Debich-Spicer D., Opitz J.M. (2004). The
Human Embryo and Embryonic Growth Disorganisation In
Gilbert-Barnes E. and Debich-Spicer (Eds) Embryo and fetal pathology:
color atlas with ultrason correlation. Cambridge University Press, Cambridge:
pp 1-16.
Gillerot Y. et Dan B. (2005). Le syndrome d'alcoolisme
foetal. Education du patient et enjeux de santé,
23(3):70-72.
Gillerot Y. et Mols M. (2009) Quinze années de
surveillance des malformations congénitales dans le Hainaut et dans la
province de Namur : Enseignements et recommandations. Services publics de
Wallonie : p1-50.
Graham J.M., Edwards M.J., Edwards M.J. (1998) Teratogen
Update: Gestational Effects of Maternal Hyperthermia Due to Febrile Illnesses
and Resultant Patterns of Defects in Humans. Teratology,
58: 209-221.
Kalwaba K., Nyembo M.K., Twite K.E., Djuma R., Kalenga M.K.
(2004) Etude des malformations congénitales cliniquement
décelables à Lubumbasi. Elite Méd,
4: 32-37.
Martinez-Frias M-L., Frias J.L., Opitz J.M. (1998) Errors of
Morphogenesis and Developmental Field Theory. Am. J. Med. Genet,
76: 291-296.
Mayanda HF., Bobossi G., Malonga H. , Djouob S., Senga P.,
Nzingoula S., Loukaka (1991) Malformations congénitales observées
dans le service de Néonatologie du CHU de Brazzaville.
Médecine d'Afrique Noire, 38(7): 505-509.
Merger R. , Lévy J., Melchior J. (2001) Précis
d'obstétrique. 6 ème édition, Masson, Paris : pp 11-25.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 103 sur 107
Milunsky A., Ulcickas M, Rothman K.J., Jick S.S., Jick H.
(1992) Maternal heat exposure and neural tube defects. JAMA,
268(7): 882-885.
Molloy A.M., Kirke P.N., Troendle J.F., Burke H., Sutton M.,
Brody L.C., Scott J.M., Mills J.L. (2009) Maternal vitamin B 12 Status and Risk
of Neural Tube Defects in a Population with High Neural Tube Defect Prevalence
and No Folic Acid Fortification. Pediatrics,
123:917-923.
Molloy A.M., Mills J.L., Kirke P.N., Weir D.G., Scott J.M.
(1999) Folate status and neural tube defects. biofactors,
10(2-3): 291-294.
Morrison K., Papapetrou C., Hol F.A., Mariman E.C.M., Lynch
S.A., Burn J., Edwards Y.H. (1998) Susceptiblity to spina bifida ; an
association study of five candidate genes . Ann Human
Genet,62(5): 379-396.
Mossey P.A., Little J., Munger R.G., Dixon M.J., Shaw W.C.
(2009) Cleft lip and palate. Lancet, 374:
1773-1785.
O'Leary C.M., Nassar N., Kurinczuk J.J., de Klerk N., Geelhoed
E., Elliott E.J., Bower C. (2010). Prenatal Alcohol Exposure and Risk of Birth
Defects. Pediatrics, 126:843-850.
Omalley M. and Hutcheon R.G. (2007) Genetic disorders and
congenital malformations in pediatric long term care. J Am Med Dir Assoc,
8 (5) : 332-334.
OMS (2008). Statistiques mondiales. Organisation Mondiale de la
santé.
Opitz J.M., Holt M.C. (1990) Microcephaly: general
considerations and aids to nosology . J Craniofac Genet Dev Bio!
10(2) : 175-204.
Pass R.F. (2006) Neural Tube Defects: from origine to
treatment. N Eng! J Med 355(5): 532-533.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 104 sur 107
Rankin J. (2010) 20-year survival of children born with
congenital anomalies: a population-based study. The lancet,
375(9715): 649-656.
Rousseau S. (1990) Etude de l'effet des radiations ionisantes
sur les chromosomes de spermatozoides humains. Thèse de doctorat,
Grenoble, Université Joseph Fourier: 20-35.
Sanoussi S., Gamatie Y., Kelani A., Sbai C.I., Abarchi H.,
Bazira L . (2001) Malformations du tube neural au Niger : à propos de
387 cas en 10 ans. Médecine d'Afrique Noire, 48(12)
: 510-515.
Scriver C.R., Neal J.L., Saginur R., Clow A. (1973) The
frequency of genetic disease and congenital malformation among patients in a
pediatric hospital. C.M.A. Journal, 11 : 11-15.
Sengeyi M.A., Tshibangu K., Tozin R., Nguma M., Tandu U.,
Sinamuli K., Mbanzulu P.N., Tshiani K. (1990) Etiopathogénie et type de
malformations congénitales observées à Kinshasa
(Zaïre). Journal de gynécologie obstétrique et biologie
de la reproduction, 19(8): 955-961.
Sheehan P.M.E., Hillary R.B. (1983) An unusual cluster of
babies with Down's syndrome born to former pupils of an Irish boarding school.
BMJ, 287: 1428-1429.
Siffel C., Otos M., Czeizel A.E. (1996) Congenital
abnormalities and indicators of germinal mutations in the vicinity of the Paks
nuclear plant, Hungary. Mutagenesis, 77 : 299-303.
Stevenson D.A. and Carey J.C. (2004) Contribution of
malformation disorders to mortality in a children's hospital. Am J Med
Genet, 126A: 393-397.
Stone D. (2010) Long-term survival of children born with
congenital anomalies. The lancet, 375:614-615.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 105 sur 107
Tabu G. (2004) Profil des malformations congénitales
apparentes à Kinshasa. Mémoire de Spécialisation,
Université de Kinshasa.
Thame G., Guerra-Shinohara E.M., Moron A.F. (2002) Serum
Folate by Two Methods in Pregnant Women Carrying Fetuses with Neural Tube
Defects. Clinical Chemistry, 48 : 1094-1095.
Turgeon J., Mercier C., D'Anjou G. (2008) Hydrocéphalie
In Turgeon J., Bernard-Bonin A.C., Gervais P., Ovetchkine P.,
Gauthier M (eds). Dictionnaire de thérapeutique pédiatrique
Weber, 2e édition. De Boeck, Bruxelles : p 619.
Wilson G.N. (1983) Cranial defects in the Goldenhar Syndrome.
Am. J. Med. Genet, 14:435-443.
Wilson R. D., Allen V.M. , Blight C., Gagnon A., Johnson J-A,
Langlois S., Summers A., Wyatt P. (2007) Principes de tératologie
humaine : Exposition aux médicaments, aux produits chimiques et aux
agents infectieux. J Obstet Gynaecol Can, 29(11) :
918-926.
Wilson R.D., Davies G., Désilets V., Reid G.J., Summers
A., Wyatt P., Young D. (2007) Supplémentation préconceptionnelle
en vitamines / acide folique 2007 : Utilisation d'acide folique, conjointement
avec un supplément multivitaminique, pour la prévention des
anomalies du tube neural et d'autres anomalies congénitales. J
Obstet Gynaecol Can, 29(12): 1014-1026.
WHO (2004). International statistical classification of
diseases and related problems-tenth revision-2cd edition. World heath
organization.
Woods G. (2004). Human microcephaly. Current Opinion in
Neurobiology, 14(1): 112117.
Malformations congénitales cliniquement visibles à
Lubumbashi/Mémoire de spécialisation 2010-2011
Page 106 sur 107


