|
tallographique de l'UMPKeco et de la CKOm



Institut National Agronomique Ecole Nationale
Supérieure des Industries Unité mixte de recherches (UMR)
206
Paris Grignon (INA P-G) Agricoles et alimentaires
(ENSIA) Chimie Biologique INRA / INA P-G
ude cris
Et
Mémoire du Master
Soutenu publiquement le : Jeudi 06 Juillet 2006 à
l'ENSIA par
Ahmed MEKSEM
Master en Sciences Et Technologies Du
Vivant
des Grandes Ecoles du vivant d'Ile de France
Mention : Aliments et
Bio-produits

Spécialité : Nutrition
Santé

Cristallographie de deux enzymes :
- mutant de l'uridine monophosphate kinase d' E. coli
insensible à l'activation allostérique
- cytokinine oxydase de maïs complexée
à différents ligands
Enseignant-Responsable du stage
Pierre BRIOZZO Maître de
conférences en Biochimie, INA P-G
Jury de soutenance
Thierry CHARDOT Directeur de recherches, INRA
(Rapporteur)
Daniel TOMÉ Professeur en Nutrition
Humaine, INA P-G
Dominique FOUQUES Maître de
conférences en Biochimie, INA P-G
Institut National Agronomique Paris Grignon (INA
P-G)
Site de Paris : 16, rue Claude Bernard - 75231
PARIS CEDEX 05
Tél. +33 (0)1 44 08 16 61 - Télécopie +33
(0)1 44 08 17 00
Site de Grignon : BP 1 - 78850 THIVERVAL-GRIGNON
Tél. +33 (0)1 30 81 53 53 - Télécopie +33 (0)1 30 81 53
27
www.inapg.fr
- Membre de ParisTech
Ecole Nationale Supérieure des Industries
Agricoles et
alimentaires (ENSIA)
1, avenue des olympiades 91744 MASSY CEDEX Tél. +33 (0) 1
69 93 50 50
Télécopie +33 (0) 1 69 20 02 30
www.ensia.fr
Remerciements
Au terme de ce travail je tiens à présenter mes
remerciements à :
-Mme Catherine LAPIERRE (Professeur en Biochimie à l'INA
P-G et directrice de l'UCB 206), pour m'avoir accueilli dans son
unité.
-Mr Pierre BRIOZZO pour sa disponibilité au long de la
réalisation de ce travail.
Sans pour autant oublier :
-Mr Amar HADJ KACI (Maître Assistant Chargé de Cours
de Biochimie)
-Mr Abderahmane MATI (Maître de Conférences en
Biochimie)
- Mrs Ramdane HADDOUCHE, Abdelaziz BENDOU et Ramdane
KHATI (Ingénieurs d'état en
Biologie).
A la faculté des Sciences Biologiques et des Sciences
Agronomiques, Département Biochimie- Microbiologie, Université
Mouloud MAMMERI de Tizi-Ouzou (Algérie).
Pour leur soutien moral, leur orientation et leur aide.
Présentation de la structure d'accueil
Ce stage, effectué dans le cadre du Master 2
Nutrition Santé, s'est déroulé à l'UMR
206
Chimie Biologique (UCB), INRA / INA-PG, Centre de Recherches
Versailles-Grignon.
L'UCB comporte quatre équipes :
(1)-Étude des lignines naturelles
et industrielles, de la structure à la valorisation : dirigée par
Pr
Catherine LAPIERRE.
(2)-Étude des lipides de
réserve végétaux organisés en corps
lipidiques, oléosomes et oléosines : dirigée par
Dr Thierry CHARDOT.
(3)- Étude des allergènes
des céréales : dirigée par Dr Michel
LAURIERE (CR, INRA).
(4)- Étude cristallographique des
protéines : dirigée par Dr Pierre
BRIOZZO.
Mon sujet a été réalisé au sein de
l'équipe « cristallographie des protéines », il a
porté sur la biologie structurale d'enzymes bactérienne
(UMPKeco) et végétale (CKOm). Son but est de
déterminer leur structure 3D à résolution atomique
afin mieux comprendre leur fonctionnement et envisager par la suite des
modifications pour des besoins de santé humaine et agronomiques.
CR : Chargé de Recherches
UMR : Unité Mixte de Recherches
Abréviations
aa : acide aminé
ADN : acide désoxyribonucléique AMP :
adénosine monophosphate ATB : antibiotique
ATP : adénosine triphosphate
CKO : cytokinine oxydase
CKOm : cytokinine oxydase de maïs
CMP : cytidine monophosphate
CMPK: cytidine monphosphate kinase
CNRS : centre national de recherche scientifique
CTP : cytosine triphosphate
FAD: flavine adenine dinucléotide
GMP: guanosine monophosphate
GTP: guanosine triphosphate
MM : masse moléculaire
MPD : 2-méthyl-2,4-pentanediol ;
C6H14O2
NAGK : N-acétyl glutamate kinase
NDP : nucléoside diphosphate NMP : nucléoside
monophosphate NTP : nucléoside triphosphate
NDPK : nucléoside diphosphate kinase
NMPK : nucléoside monophosphate kinase pI : point
isoélectrique
PCR: polymerase chain reaction
SDS-PAGE : sodium dodecyl sulphate - polyacrylamide gel
electrophoresis
TMP : thymidine monophosphate
UDP : uridine diphosphate
UMP : uridine monphosphate
UMPK: uridine monphosphate kinase
UMPKeco : uridine monphosphate kinase d'E. coli
UTP : uridine triphosphate
Liste des Encadrés / Figures / Tableaux
Encadrés-----------------------------------------------------------------------------
Encadré 1 : Vérification de la concentration
protéique par absorption dans l'UV...........................
29
Encadré 2 : Concentration de la
protéine............................................................................
29
Figures-------------------------------------------------------------------------------
Figure 1 : Nombre de structures 3D résolues par
cristallographie, déposées et accessibles dans la PDB...
8
Figure 2 : Evolution de la cristallographie des
protéines...........................................................
10
Figure 3 : De la protéine à sa structure
3D...........................................................................
11
Figure 4 : Les NMPK et la phosphorylation des
NMP............................................................. 13
Figure 5: Les trois domaines des
NMPK..................................................................................
15
Figure 6 : Régulation de l'activité de
l'UMPKeco...................................................................
16
Figure 7 : Repliement global de l'UMPKeco : le
monomère......................................................
18
Figure 8 : Repliement global de l'UMPKeco :
l'hexamère fonctionnel..........................................
18
Figure 9 : Formules de quelques cytokinines et d'un
inhibiteur ................................................... 20
Figure 10 : Dégradation des cytokinines par la
CKOm............................................................. 20
Figures 11 a) et b) : Structure 3D et sites de
mutagenèse de la CKOm..........................................
22
Figure 12 : La
cristallisation..................................................................................................
24
Figure 13 : Mosaïcité d'un
cristal.....................................................................................
24
Figure 14 : Diagramme de phase
bidimensionnel...................................................................
25
Figure 15 : Technique de la goutte
assise............................................................................
25
Figure 16 : Photos du robot de Cristallisation et de la
boite Linbro............................................ 30
Figure 17 a): Méthode de cristallisation en goutte
suspendue..................................................... 33
Figure 17 b) : Montage des cristaux sur des boucles en
nylon.................................................... 33
Figure 17 c) : Collecte des données de
diffraction..................................................................
33
Figure 18 : Suivi de la pureté de l'UMPKeco D159N
D93A par SDS-PAGE..................................... 35
Figure 19 : Cristaux de l'UMPKeco D159N
D93A................................................................
36
Figure 20 : Cristaux de la
CKOm.....................................................................................
39
Tableaux-----------------------------------------------------------------------------
Tableau 1 : Structures 3D résolues par les
différentes techniques analytiques déposées dans la
PDB...... 8
Tableau 2 : Résumé de la variabilité
des conditions de cristallisation de l'UMPKeco D159N D93A....... 35
Tableau 3 : Cristaux de l'UMPKeco D159N D93A : Conditions
d'obtention et dimensions................. 35
Tableau 4 : Résumé de la variabilité
des conditions de cristallisation de la CKOm...........................
38
Tableau 5 : Cristaux de la CKOm : Conditions d'obtention et
dimensions..................................... 38
Tableau 6 : Enregistrement des données de
diffraction............................................................
39
Tableau 7 : Résultats du traitement des
données....................................................................
40
Sommaire
A- Introduction
....................................................................................................
7
I)-L'uridine monophosphate kinase d'Escherichia coli
(UMPKeco).................................................
12
A- Les
NMPK...........................................................................................................
12
1- Définition et Rôles
biologiques..............................................................................
12
2-Structures
3D.....................................................................................................
14
B-L'UMPKeco
..........................................................................................................
14
1-Une NMPK atypique
...........................................................................................
13
2-Structures 3D et propriétés
catalytiques.....................................................................
13
3-Rôles
............................................................................................................
17
4- Contexte et objectifs du
stage.................................................................................
17
II)-La Cytokinine oxydase de
maïs........................................................................................
19
A-Les
cytokinines.........................................................................................................
19
1. Nature chimique
..............................................................................................
19
2. Lieu de
synthèse................................................................................................
19
3.
Rôles............................................................................................................
19
B-La
CKOm...............................................................................................................
21
1. Propriétés et Structure
3D...................................................................................
21
2. Objectifs du stage
.............................................................................................
21
III)-La cristallogénèse des
protéines
.........................................................................................23
A-Aspects
généraux......................................................................................................
23
1. Le
cristal............................................................................................................
23
2. Principes de la
cristallogenèse.................................................................................
23
3. Etapes de la
cristallisation.......................................................................................
25
B-Méthodes de
cristallisation..........................................................................................
27
B-Matériel et méthodes
..........................................................................................
28
1. Clonage, surexpression et
purification..........................................................................
28
2.
Cristallisation......................................................................................................
28
..
.
....
...
.
...
..
.
.
.
.....
...
.
....
.
.
..
......
.
....
...
....
3. Etapes
ultérieures................................................................................................
31
C-Résultats et
discussion.......................................................................................
34
A)-Mutant D93A non régulé par le GTP de
l'UMPKeco ..............................................................
34
B)-CKOm native et mutant
D169E.......................................................................................
37
D-
Projets.........................................................................................................
41
Références
bibliographiques................................................................................................
42
Annexes........................................................................................................................
44
Résumé........................................................................................................................
46
A- Introduction
La génomique décrypte des séquences
protéiques en quantité exponentielle (plus de 2 600 000
séquences connues actuellement :
ftp://ftp.ncbi.nih.gov/refseq/release/, 05 Mai 2006). Mais ces
séquences ne renseignent pas plus sur la biologie des systèmes
considérés qu'un annuaire téléphonique
de Paris sur l'organisation et les merveilles de cette
ville. En effet, les fonctions biologiques des protéines
dépendent étroitement de leur structure tridimensionnelle (3D).
Celle-ci ne peut être prédite correctement d'après la
séquence et doit être résolue
expérimentalement, avec trois méthodes possibles : la
microscopie électronique, la résonance magnétique
nucléaire (RMN) et la cristallographie. La microscopie
électronique a une faible résolution : de l'ordre de 20 ? ; la
RMN est surtout efficace pour des molécules d'une masse
moléculaire (MM) inférieure à 30 kDa. La
cristallographie est la seule méthode qui permette la
détermination de la structure 3D à résolution
atomique : de 1 à 2 ?, et l'étude des macromolécules sans
limitation de la MM.
La cristallographie constitue une méthode fondamentale,
en biologie structurale, pour l'étude et l'analyse des structures 3D des
acides nucléiques et complexes : protéine-acides
nucléiques, protéine- protéine et
protéine-ligand, dans leur conformation active (Duée et
Dideberg, 1997 ; Deleu et al,
1998). Cette technique permet d'obtenir une description 3D
détaillée des molécules, et elle apporte des informations
concernant les interactions protéine-ligand. Elle réclame
un équipement lourd et très onéreux : environ 300 000
€ pour le seul ensemble générateur de rayons X /
détecteur. Son principal inconvénient est la
nécessité d'obtenir des cristaux de la protéine
étudiée.
Ses premiers succès datent des années 60 avec la
résolution de la structure 3D d'une protéine monomérique
(myoglobine, J.C. Kendrew, 1960), d'une protéine
oligomérique (hémoglobine, M.F. Perutz, 1962) et
d'une enzyme (lysozyme, D.C. Philips, 1965). Les changements
fondamentaux intervenus dans les techniques propres à la
cristallographie et à la biologie (séquençage des
génomes, biologie moléculaire, purification de
protéines étiquetées, sources intense de rayons X
avec les synchrotrons, détecteurs, informatique...) ont
entraîné une véritable explosion de la
cristallographie
des protéines. Le nombre de structures
résolues ne cesse d'augmenter : 30000 structures
protéiques
(soit 86% du total des structures résolues) sont
disponibles en mai 2006 dans la Protein Data
Bank1 contre 11000 en 2000 (Figure 1 &
Tableau 1). Mais ce chiffre ne correspond en fait qu'à 1,3 %
des 2 millions de séquences protéiques connues
(ftp://ftp.ncbi.nih.gov/refseq/release/).
Cet aspect quantitatif est doublé d'un aspect
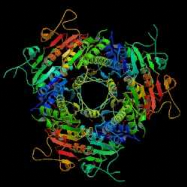
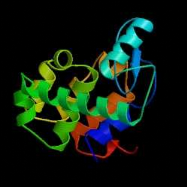
qualitatif : des structures des protéines de plus en plus complexes et
importantes sont résolues : virus, ribosome 70S bactérien (52
protéines, 3 ARN, au total 2500 kDa)...(Figure 2).
Même un nombre non négligeable de protéines
membranaires, malgré
leur insolubilité, ont été
cristallisées et leur structure 3D résolue.
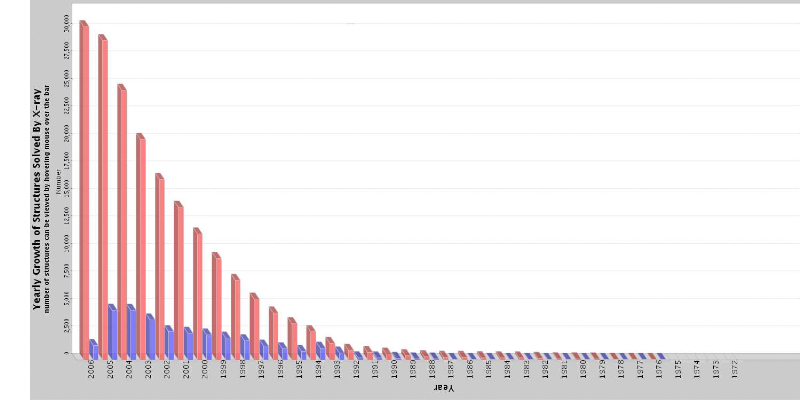
Données actualisées le
28 Mai 2006
Figure 1 : Nombre de structures 3D résolues par
cristallographie, déposées et accessibles dans la
PDB.
En bleu : le nombre de structures déposées dans
l'année, en rose : le nombre total de structure.
Le 28 Mai 2006, le nombre est exactement
de 31 111 Structures
(http://www.rcsb.org/pdb/Welcome.do)
Tableau 1 : Structures 3D résolues par les
différentes techniques analytiques déposées
dans la PDB / Le 28 Mai 2006
|
Type de Molécule
|
|
Protéines
|
Acides
Nucléiques
(AN)
|
Complexes
Protéine/AN
|
Autres
|
Total
|
|
Méthode
Exp.
|
|
Rayons X
|
|
|
28835
|
|
|
899
|
|
|
1349
|
|
|
28
|
|
|
31111
|
|
|
|
|
|
RMN
|
4568
|
699
|
121
|
6
|
5394
|
|
Microscopie
Electronique
|
88
|
9
|
28
|
0
|
125
|
|
Autres
|
73
|
4
|
3
|
0
|
80
|
|
Total
|
33564
|
1611
|
1501
|
34
|
36710
|
Etant pluridisciplinaire, la cristallographie est une
méthode qui nécessite de bonnes
compétences en biologie, en chimie, en physique et en
informatique.
Cette méthode comporte une étape
limitante : la cristallisation, obtention de cristaux stables à
partir des macromolécules pures en solution, de taille suffisante
(environ 0,1 mm) et qui diffractent suffisamment les rayons X. La
connaissance de la physico-chimie des macromolécules n'est pas
suffisante pour établir une méthode rationnelle de
cristallogenèse, qui reste pour une grande part très empirique.
Cette étape délicate et assez longue ne réussit
que pour, environ, un quart des cas. La résolution de la
structure 3D est longue, elle peut prendre plus d'une année. En
revanche, dans les cas faciles et avec les technologies actuelles, les
résultats peuvent être obtenus en quelques mois
(Figure
3) (Stelter, 2003).
Les données déduites de l'étude
structurale, par cristallographie, des macromolécules en
général et des protéines en particulier ont un fort impact
sur l'élaboration des concepts fondamentaux
de la biologie (recherche fondamentale).
Elles permettent aussi de comprendre le mécanisme
d'action des médicaments [antibiotiques, anti-viraux,
anti-cancéreux, antihypertenseurs ...] et des produits phytosanitaires,
et d'en concevoir de nouveaux (« drug design »). Dans ce même
contexte, les principales caractéristiques des enzymes qui
intéressent les industries agro-alimentaires (spécificité
et thermosensibilité) sont comprises et on peut ainsi les modifier
et/ou les optimiser selon les exigences de l'industrie en question
(recherche appliquée).
1 La Protein Data Bank / PDB (Brookhaven, Etats-Unis)
: est la banque de données créée en 1977 pour archiver
les structures 3D des macromolécules, obtenues par diffraction X ou
RMN...
A
B
C
D
Figure 2 : Evolution de la cristallographie des
protéines
Les premières protéines étudiées par
cristallographie étaient classiques : des molécules compactes et
globulaires qui cristallisent facilement (lysozymes, A). Par
le suite, de nouvelles protéines de plus grande taille et plus
délicats à cristalliser (comme celles faisant l'objet de notre
stage) voient leur structure 3D résolue : Cytokinine oxydase du
maïs, B ; Uridile monophosphate kinase d'E.
coli, C. Récemment, les structures de très
grosses molécules ont été résolues : sous
unité 50 S du ribosome bactérien, D :
Structures résolues de plus en plus complexes.
Les étapes de détermination de la
structure 3D d'une protéine par cristallographie
Protéine pure (97 à 99 %) /Stable
ou fraîche / Concentration min =
5 mg/ml / Soluble / Monodisperse
et repliée Cristaux protéiques
très fragiles /
sensibles aux irradiations
I
S O L E
M
E I
N II
Montage sur des cryoboucles
T Surexpression et purification
Environ 1 protéine sur 4 cristallise
I- Cristallisation
Taches de diffraction (de Braggs) des rayons X : direction
hkl/amplitude et phase


















Structure 3D détaillée : vision
atomique détaillée de la protéine
Comprendre : Fonction / Spécificité
/ Interactions dynamiques entre molécules / Régulation
/
Ingénierie des protéines (mutagenèse
dirigée, drug design...) / évolution
moléculaire (relations
Séquence-Evolution-Fonction) /
modélisations moléculaires
II- Enregistrement des données de
diffraction : cliché de diffraction
IV- Construction et affinement
III
Carte de densité électronique
VI
Figure 3 : De la protéine à sa
structure 3D. Toute étude structurale des
protéines par
cristallographie débute par sa purification, se
poursuit par la préparation de cristaux, enregistrement des
données de diffraction des rayons X et l'exploitation des clichés
pour obtenir la structure 3D. Malgré les progrès
considérables réalisés aux différents niveaux
de la cristallographie, l'écueil principal reste la
cristallogenèse.
III- Phasage
I)-L'uridine monophosphate kinase d'Escherichia coli
(UMPKeco)
Les nucléotides sont essentiels à tout
processus vivant. Les enzymes responsables de leur
synthèse constituent depuis plusieurs années
la cible de médicaments antiviraux, antibactériens et
antiparasitaires. Parmi ces enzymes figurent les nucléosides
monophosphate kinases (NMPK).
A-Les NMPK
1-Définition et Rôles biologiques
: Les NMPK sont ubiquitaires, elles représentent une
famille de protéines globulaires relativement homologues du point de
vue structural et catalytique (Yan & Tsai,
1999). Ces enzymes catalysent la phosphorylation
réversible d'un nucléoside monophosphate (NMP)
en un nucléoside diphosphate (NDP) en
présence de Mg2+, le phosphate transféré
provient d'un nucléoside triphosphate (NTP)(Noda, 1973),
[réaction ci-dessous]. Le principal donneur de phosphate
est l'ATP, mais ce n'est pas le seul possible : la
spécificité des NMPK vis-à-vis des donneurs de
phosphate n'est pas très étroite.




Phosphorylation réversible des NMP
NMP + ATPMg2+
NMPK
NDP + ADPMg2+
N est une base azotée : A, G, T, U ou
C
Cette étape métabolique est critique dans la
synthèse des NTP. Ces derniers jouent plusieurs
rôles au sein de la cellule (Vonrhein et al, 1995)
:

ils sont les précurseurs dans la synthèse des
acides nucléiques et des phospholipides ;
 ils
interviennent dans la signalisation cellulaire ; ils
interviennent dans la signalisation cellulaire ;
 ils
jouent un rôle dans le cycle : polymérisation /
dépolymérisation des microtubules ; ils
jouent un rôle dans le cycle : polymérisation /
dépolymérisation des microtubules ;
 ils
constituent une source importante d'énergie cellulaire ; ils
constituent une source importante d'énergie cellulaire ;
 ils
entrent dans la structure du site actif de certaines coenzymes. ils
entrent dans la structure du site actif de certaines coenzymes.
L'échange du phosphate est facilité par
les chaînes très conservées d'arginine ; les NMPK
suivent une cinétique du type bi-bi aléatoire
: ceci implique l'existence de trois formes : libre (sans
substrats), semi fermée (avec le NTP ou le NMP) et
fermée (avec le NTP et le NMP). Le 1er substrat
fixé peut être indifféremment le NTP ou le
NMP.
Les NMPK sont au nombre de 4 chez les eucaryotes (AMP, GMP, TMP
et UMP/CMP kinases) et de 5
chez les procaryotes (AMP, GMP, TMP, UMP et CMP kinases)
(Dreusicke et al, 1988 ; Zhou et al,
1998) (Figure 4). Chez
les eucaryotes l'UMP/CMP kinase phosphoryle avec la même
efficacité l'UMP et la CMP, par contre les chez bactéries
se sont deux enzymes distinctes qui assurent cette fonction. Ces CMP
et UMP kinases bactériennes ont une spécificité et
une structure différentes de celles de leurs homologues
eucaryotes. Cela en fait des cibles attractives pour la recherche et
la
conception de nouveaux antibactériens.
Etude cristallographique de l'UMPKeco et de la
CKOm
Phosphorylation des nucléotides
Chez les procaryotes : une NMPK pour chaque
NMP
NMP
AMP GMP dTMP
UMP
CMP
dCMP
AMP
kinase
GMP
kinase
TMP
Phosphorylation des nucléotides
Chez les procaryotes : une NMPK pour chaque
NMP
AMP GMP dTMP UMP CMP dCMP
AMP GMP TMP UMP kinase UMP/CMP CMP
kinase
kinase kinase kinase bactérienne kinase
bactérienne
eucaryote
ADP GDP UDP CDP
RR RR RR RR
dADP dGDP dTDP dUDP dCDP
Phospho- rylation
oxydative, Nucléoside diphosphate kinase
Glycolyse
...
dATP dGTP dTTP dUTP dCTP
ATP GTP UTP CTP
RR : Ribonucléoside
diphosphate Réductase
NDP dNDP RR




kinase
UMP kinase bactérienne
UMP/CMP
kinase eucaryote
CMP kinase bactérienne
NMPK
ADP GDP
UDP
CDP
RR RR
RR RR
dADP
dGDP
dTDP
dUDP
dCDP
NDP
Phospho-
rylation oxydative, Glycolyse
...
Nucléoside diphosphate kinase
NDPK
dATP
dGTP
dTTP
dUTP
dCTP
ATP GTP
UTP
CTP
NTP
RR : Ribonucléoside
diphosphate Réductase
NDP dNDP RR
Figure 4 : Les NMPK et la phosphorylation des
NMP
La production de NTP dans la cellule à
partir des NMP passe par deux étapes : (1)
phosphorylation
des NMP en NDP par des NMPK spécifiques ;
(2) transformation des NDP en NTP par la NDPK non
spécifique de chaque base azotée.
2-Structure 3D : Les NMPK partagent la
même structure 3D globale comprenant trois domaines : le
domaine CORE, le domaine LID et le domaine NMPbind
(Figure 5). Le domaine CORE, rigide et très
conservé, comporte un feuillet â à cinq brins
parallèles, des hélices á (généralement au
nombre de 8 ou
9) et une P-loop. Le domaine LID (flexible) joue le rôle
de « couvercle » en venant recouvrir le site donneur
de phosphate, permettant l'échange du phosphate ã à l'abri
des molécules d'eau. Le domaine flexible NMPbind lie l'accepteur de
phosphate. (Yan & Tsai, 1999).
B-L'UMPKeco : modèle intéressant de la
régulation enzymatique et cible potentielle de
nouveaux ATB
1-Une NMPK atypique : Avec un point
isoélectrique (pI) de 7,24 et une MM de 156 kDa (28,5kDa / sous
unité), l'UMPKeco représente environ 0,05 % des protéines
totales d'E. coli. Elle est codée par le gène pyrH
découvert en 1992, ce gène très conservé est
propre aux bactéries est sans homologue chez
les eucaryotes (Serina et al, 1995). L'UMPKeco
ne présente aucune similarité significative de
séquence ou de repliement avec les autres NMPK. Elle ressemble
d'avantage aux aspartokinases (30 % d'identité de séquence),
et à un degré moindre (moins de 20% d'identité)
à la N-acétyl glutamate kinase (NAGK) et la carbamate kinase
(Bucurenci et al, 1996). Elle est présente dans le cytoplasme
et près de la membrane bactérienne.
2-Structure 3D et propriétés
catalytiques : A la différence des NMPK classiques,
généralement monomériques [sauf la TK qui est
homodimérique : Haouz et al, 2003], l'UMPKeco est
hexamérique
(Serina et al, 1995). La régulation de son
activité et assez complexe (Figure 6) :
(1) l'UTP est un inhibiteur, cette inhibition est
levée par des concentrations élevées de Mg2+
(2) le GTP agit comme
un activateur allostérique, il se fixe
sur un site allostérique distinct du site actif, indépendamment
de
Mg 2+ (Briozzo et al,
2005).
Une propriété de l'UMPKeco, qui a
empêché pendant plusieurs années la détermination de
ses structures cristallographiques, est sa très basse solubilité
: < 0,1 mg de protéines/ml (Bucurenci et al,
1996).
Nucléoside monophosphate kinases
NMP + ATP-Mg2+ NDP + ADP-Mg2+
Domaines flexibles
LID
NMP-bind
NMP kinases
ADP NMP



-P-
Domaine rigide
- Courtes : AMPK eucaryotes, GMPK, TMPK, UMP/CMPK eucaryotes
Insert
CORE
-Les AMPK des bactéries, plantes et
mitochondries ont un insert supplémentaire dans le
LID
- Longues : AMPK des bactéries, plantes et mitochondries :
grand LID avec insert
CMPK et GMPK ont un NMPbind plus long
-Les CMPK bactériennes et GMPK ont un insert au
niveau du NMP bind
Figure 5 : Les trois domaines des
NMPK.
Toutes les NMPK ont un repliement de base similaire,
organisé en trois domaines. Un domaine
principal et central CORE qui est rigide et qui contient la
P-loop. Un domaine LID et un domaine
NMPbind qui se referme sur le site de
l'accepteur du phosphate.
Excès de NTP
puriques : A et G
Excès de NTP
puriques : A et G
Excès de NTP
Excès de NTP
pyrimidiques : C, T et U
pyrimidiques : C, T et U
GTP
Activation
UTP
?? Inhibition
Allostérique
Allostérique
Site allostérique C
UMPK Bactérienne
ompétitif
Site allostérique
Compétitif
de P




















UMPK Bactérienne
neur de P P Site accepteur
Site don
Site donneur de P
P Site accepteur de P
ADP
ATP UMP
UDP
Figure 6 : L'UMPKeco / impliquée
dans la synthèse de novo des nucléotides
pyrimidiques
Figure 6 : Régulation de
l'activité de l'UMPKeco
3-Rôles : L'UMPKeco est essentielle
à la croissance bactérienne, elle est indispensable à la
synthèse
des nucléotides pyrimidiques en catalysant la
transformation spécifique de l'UMP en UDP. Les CMP
et UMPK bactériennes n'ont pas d'équivalents
chez l'homme, des inhibiteurs pourraient être conçus pour inhiber
ces enzymes et bloquer ainsi la croissance bactérienne, sans
pour autant entraîner des effets secondaires chez le malade.
Cependant, des E. coli sans activité CMPK restent viables
(Fricke et
al, 1995). Ceci s'explique par le fait que ces
bactéries peuvent produire le CTP à partir de l'UTP
grâce
à la CTP synthétase. Au contraire,
l'UMPK est essentielle à la croissance bactérienne et constitue
de
ce fait une cible privilégiée pour la
recherche et la conception (« drug design ») de nouveaux
antibiotiques (ATB) (Kholti et al, 1998 ; Fassy et al,
2004).
En plus de son rôle dans la conversion de l'UMP en UDP,
l'UMPKeco participe à la régulation
de la transcription et joue très probablement un
rôle dans la division des cellules bactériennes (Londais
et al, 1999).
4-Contexte et objectifs du stage :
Avant 2005, il n'existait aucune structure 3D de l'UMPKeco
(faible solubilité / cristallogenèse impossible). Après la
découverte par A.-M. Gilles et al du mutant ponctuel soluble
qui a les mêmes caractéristiques catalytiques que l'enzyme sauvage
: mutant D159N.
P. Briozzo et al ont résolu sa structure 3D
sous différentes formes : avec l'UTP / inhibiteur,
l'UMP / substrat naturel et l'UDP / produit
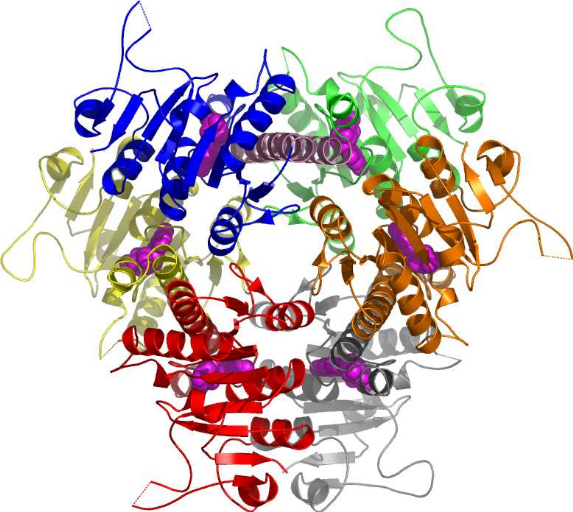
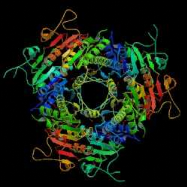
(Figures 7 & 8). Le caractère hexamèrique et
le repliement de son monomère distinguent l'UMP kinase d'E. coli
des autres NMPK. La structure avec l'UTP montre de
manière surprenante que ce nucléotide se
fixe sur le même site accepteur de phosphate que l'UMP et l'UDP (c'est la
première fois qu'un NTP est retrouvé sur ce site). Cela
suggère fortement que l'UTP
est un inhibiteur compétitif, et pas
allostérique comme il a été décrit par les auteurs
travaillant sur les NMPK. En conséquence, seule la structure avec
GTP permet de visualiser et de décrire le site
allostérique. La structure de l'UMPKeco avec le GTP est connue depuis
2005 (article en préparation), mais elle n'explique pas clairement le
phénomène d'allostérie. Nos collaborateurs de l'Institut
Pasteur
ont constaté que le mutant D93A n'est
plus régulé allostériquement par le GTP
(D93 est impliqué dans
les interactions de maintien de l'hexamère).
Résoudre sa structure 3D pourrait nous permettre de mieux
comprendre le phénomène de régulation
allostérique.
7 6
2
C 7 5
9 1
3
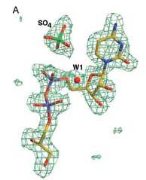
![]()
6 8
1 2 4
7 6
2
7 5
9 1
3
6 8
1 2 4
3
8 4 5
1
3
8 4 5
1
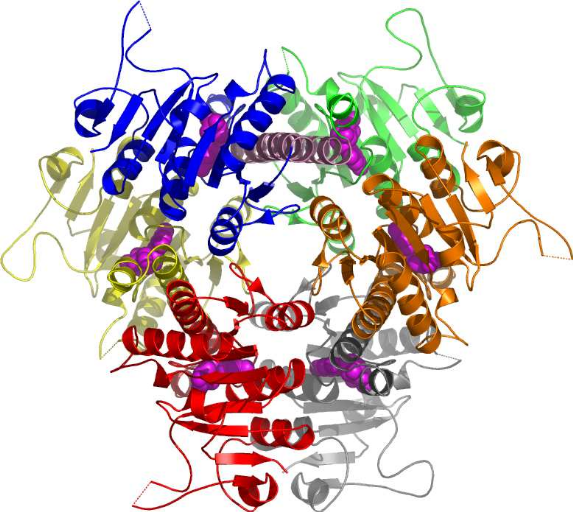
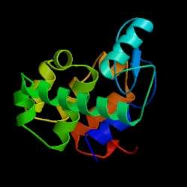
Figure 7 : Repliement de l'UMP kinase d'E.
coli : le monomère avec l'UMP (Briozzo et al, 2005)
Figure 8 : Repliement de l'UMP kinase d'E.
coli : l'hexamère fonctionnel avec l'UMP (Briozzo et al,
2005)
II)-La cytokinine oxydase de maïs (CKOm)
Le développement de plantes est régulé
par des interactions entre l'environnement et les
facteurs endogènes, particulièrement les
hormones. Les phytohormones représentent l'un des éléments
essentiels de la régulation de la croissance et du
développement des plantes [impliquées à tous les
stades depuis la pollinisation jusqu'au contrôle de la floraison, de la
fructification et de la sénescence].
Les effets qu'elles provoquent sont souvent
pléïotropes et résultent d'interactions complexes qui
rendent leur étude difficile. Nous nous intéressons
plus particulièrement aux cytokinines qui constituent une des
six classes d'hormones végétales (cytokinines +
gibbérellines, dormine, éthylène, auxine et
brassinostéroïdes).
A-Les cytokinines
(www.inrp.fr/biotic/morpho/html/cytokinines.htm)
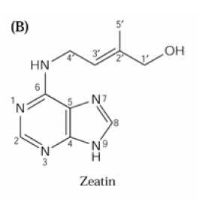
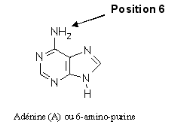
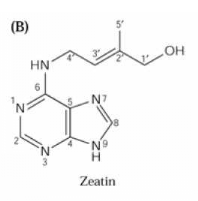
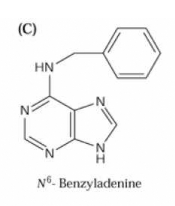
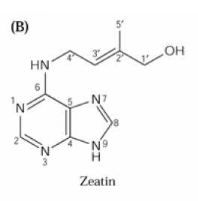
1-Nature chimique (Figure
9): Environ 200 cytokinines ont
été identifiées et isolées (depuis les
années 50). Les plus fréquentes sont la
trans-zéatine et la N6 -isopentényladénine. Leur
formule est à base d'adénine substituée :
au lieu du H du groupement amine en position 6 vient se brancher un autre
groupement. De plus, beaucoup de cytokinines sont sous forme conjuguée :
dans ce cas, il s'agit le plus souvent de glycosideS.
2-Lieu de synthèse : Les
cytokinines sont présentes dans presque tous les
tissus. Elles sont particulièrement abondantes dans les
graines et les fruits. Elles sont, pour l'essentiel, synthétisées
sur place, ce qui fait qu'elles se trouvent généralement
en quantité suffisante sur leur lieu d'utilisation. Cependant,
pour les tissus à prolifération intense, un complément
peut être nécessaire. Il semble que
les tissus proches de l'apex racinaire soient
impliqués dans la production des ces cytokinines
surnuméraires qui vont alors migrer vers les tissus demandeurs via la
sève brute.
3-Rôles : Les cytokinines :
-stimulent la division cellulaire : effet sur la duplication des
chromosomes et sur le recloisonnement cellulaire ;
-peuvent agir sur l'élongation cellulaire (augmentation de
la taille des cellules) au niveau des tiges ou des racines ;
-ont en général un effet inhibiteur sur
l'élongation longitudinale mais favorisent l'extension radiale.
Par ailleurs, la meilleure connaissance des
mécanismes de leur action devrait permettre le
développement d'une utilisation raisonnée des
régulateurs de croissance à activité agoniste ou
antagoniste en agriculture et en arboriculture.
HN
N HN
N
N N
N N
H
HA8
NH N
N6-(2-isopentenyl)adénine: IP
Inhibiteur
Figure 9 : Formules de quelques cytokinines et
d'un inhibiteur
CKOm
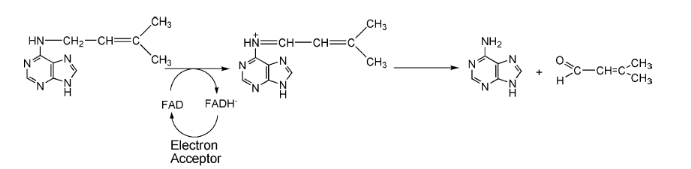
H2O
Cytokinine
Composé intermédiaire
(Quinones)
Adénine
Aldéhyde
Figure 10 : Dégradation des cytokinines
par la CKOm (Malito et al, 2004)
B-La cytokinine oxydase du maïs
(CKOm)
1-Propriétés et Structure 3D
: Dans beaucoup de tissus végétaux l'oxydation
semble être la voie
principale pour l'inactivation des cytokinines. Le
système enzymatique capable de cliver la chaîne N6-
latérale des cytokinines a été découvert en
1971 par Paèes et al. Les cytokinines oxydases de
maïs (CKOm) oxydent les cytokinines en présence
d'oxygène moléculaire [dégradation irréversible
: production de l'adénine et d'un aldéhyde] (Figure
10). La CKOm est une flavoenzyme monomérique,
elle fait partie de la famille des oxydoréductases
à cofacteur FAD (Flavine Adénine Dinucléotide), ce dernier
est lié d'une façon covalente à l'His 87. La MM de la CKOm
est de 69 kDa, sa séquence est constituée de 516 aa.
L'aminoacétonitrile, HA-8 (un inhibiteur irréversible :
molécule suicide) et le cyanure (CN) inhibent son activité
alors que les complexes de cuivre-imidazole l'activent (Kopeèny
et al, 2004).
Le clonage du gène de ckx, fait en 1999, fut
une étape cruciale dans l'élucidation du rôle de la CKOm
dans le développement des plantes (Galuszka et al, 2001 ;
Malito et al, 2004). Des grains de maïs, l'orge et de blé
ont été souvent employés pour la purification de la
CKO.
Les seules structures de la CKO disponibles sur la PDB sont
celles publiées en 2004 par Malito
et al : enzyme seule, complexée avec
Isopentényladénine (IP), avec la Trans-zéatine
(TZ) et avec la
Benzylaminopurine (Figure 11 a)).
2-Objectifs du stage : Plusieurs mutant
ont étés produits par mutagenèse dirigée
(Figure 11 b)). La détermination de la structure 3D
de mutant de résidus proches du FAD et de l'enzyme sauvage
en
présence d'inhibiteurs va permettre de mieux comprendre le
fonctionnement de cette enzyme.
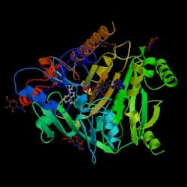
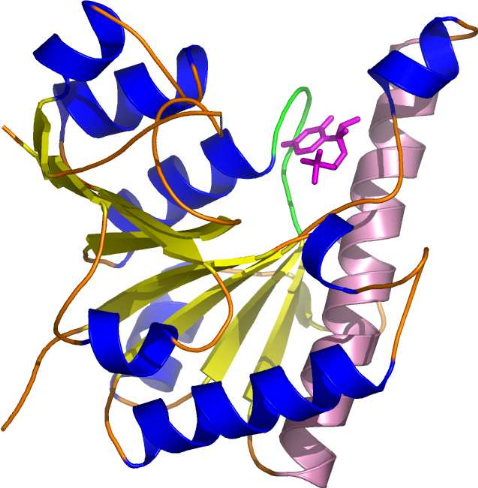
Figure 11 a) : Structure de
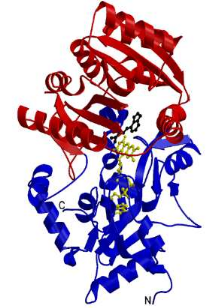
la CKOm
La CKOm est monomérique, elle comporte deux
domaines :
-en bleu le domaine de liaison avec le
FAD
-en rouge le domaine de fixation du
substrat
Le FAD est montré en jaune, le substrat
(IP) est
montré en noir (Malito et
al, 2004).
Figure 11 b) : Mutants de la CKOm
ion du substrat
%o Leu) P427 - hydrop
P427Q (Pro %o
V378 - fixation du substrat
![]()









![]()






V378 L (Val %o Leu)
P427 - hydrophobicité
P427Q (Pro %o Gln)
E288 - interaction avec le
D169
E288Q (Glu %o Gln)
D169 - fixation du substrat
D169 - fixation du substrat
Résidu clef dans la catalyse D169E (Asp %o%o%o%o
Glu)
D169N (Asp %o Asn)
Résidu clef dans la catalyse
D169E (Asp %o
Glu) D169N (Asp %o Asn)
L492 - transfer de protons et reduction du
FAD
L492A (Leu %o Ala)
L492 - transfer de protons et
reduction du FAD
L492A (Leu %o Ala)
Figures 11 a) et b) : Structure 3D et sites de
mutagenèse dirigée de la CKOm
III)-La cristallisation des protéines
Pourquoi cristalliser une protéine
?
Pour résoudre par les méthodes biophysiques des
structures 3D à résolution atomique il faut utiliser
un rayonnement dont la longueur d'onde est proche de la
distance inter-atomique, c'est-à-dire de l'ordre de l'?, donc les
rayons X.
Si on voulait résoudre la structure 3D d'une
protéine isolée en solution en l'exposant aux rayons
X : les ondes diffusées seraient
de faible intensité et l'échantillon se dégraderait
sous l'effet du rayonnement X ; sous l'effet du mouvement Brownien la
protéine n'aurait ni position, ni orientation définies.
D'où l'intérêt d'obtenir un cristal
protéique : manipulable et contenant une quantité énorme
de molécules (1015) ayant la même orientation. Chacune
contribue au signal (interférences constructives entre toutes les
molécules) et la dégradation due aux rayons X est partagée
entre elles.
A-Aspects généraux
1-Le cristal : arrangement périodique
ordonné dans l'espace 3D
Le cristal, dont la taille est de quelques dizaines de microns (u
), est un état solide ordonné : les protéines qui le
composent sont empilées d'une façon périodique et
régulière dans les trois directions
de l'espace. La maille cristalline est
répétée par translation à travers tout le cristal
(Figure 12). La cohésion entre les molécules
protéiques voisines dans le cristal est assurée par des
interactions non covalentes : interactions hydrophobes, liaisons
hydrogènes, de Van der Waals,... La protéine dans le cristal a
la même structure qu'en solution, car étant volumineuses
et globulaires, les protéines, ne peuvent pas s'empiler
très étroitement. Les espaces ménagés par
l'empilement des protéines sont comblés par le solvant
(généralement l'eau) qui représente en moyenne 50 %
du volume du cristal
[cette caractéristique est à l'origine de la
fragilité des cristaux] (Mathews, 1985). Les protéines sont donc
dans un état comparable à celui de la solution.
2-Principe de la cristallogénèse : de la
solution au cristal
La solubilité d'une protéine est fonction de
nombreux paramètres tels que : sa concentration, le pH, la
température, la force ionique, l'effet d'additifs....
Lorsque le cristal protéique est obtenu, se pose
la question de l'évaluation de sa qualité ; l'aspect
visuel peut apporter une première indication mais le véritable
test est l'analyse aux rayons X (Mullin, 1993). parmi les défauts
rencontrés dans les cristaux protéiques figure la
mosaïcité (Figure
13) : structure mosaïque du cristal
qui consiste en une juxtaposition de blocs monocristallins
(Helliwell, 1993 ; Chayen et al, 1996 ; Chernov,
1999).
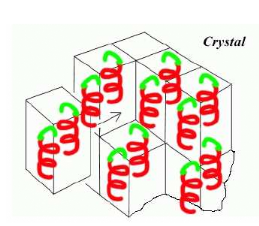
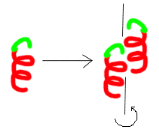
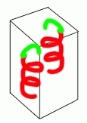
Figure 12 : La cristallisation /
Transition de phases (passage d'un état désordonné
liquide à un état ordonné
solide).
La protéine quitte la solution et passe d'un
état soluble à un état de solide ordonné,
le cristal. Le motif moléculaire
répété par translation le long du réseau cristallin
s'appelle maille cristalline. Elle correspond à une molécule
protéique ou à une sous unité protéique.
L'arrangement périodique des protéines est à
l'origine du phénomène de diffraction observé lorsque le
cristal est placé dans

un faisceau de rayons X. La résolution des
données et la précision finale de la structure
dépendent, directement, de la régularité de
l'empilement cristallin (Sauter et Giegé, 2001).
Figure 13 : Mosaïcité d'un
cristal
Diagramme de phases
Remplacement des interactions Solvant-Protéines
par des interactions Protéines-Protéines
La cristallisation est une transition de phase : la
protéine cristallisée passe d'un état de soluté
[protéine soluble] à une phase
solide [le cristal]. La cristallisation est le
résultat d'un compromis entre les facteurs thermodynamiques
(solubilité) et cinétiques
(nucléation et croissance cristalline).
Le comportement d'une protéine en fonction des variations
de son environnement peut être décrit sous forme d'un
diagramme de phases (Figure 14) (Saridakis et al, 1994).
Sur le diagramme est reportée la
courbe de solubilité qui définit la limite
entre la phase soluble et la phase solide [cristal,
précipité microcristallin ou amorphe]. Pour qu'une
protéine cristallise, elle doit dépasser cette courbe et entrer
dans une zone hors équilibre thermodynamique, elle
se trouve alors dans un état de sursaturation qui lui
permet d'initier la cristallisation : la nucléation. Il existe de ce
fait une courbe de supersaturation, qui sépare
deux zones sursaturées du diagramme : la zone de nucléation
où la sursaturation élevée conduit à la
nucléation du cristal et la zone métastable où
la sursaturation plus faible est juste suffisante pour
que le cristal existant croisse (Sauter et Giegé,
2001).
Le passage de la zone insaturée à la zone
sursaturée est réalisé en faisant varier un ou plusieurs
paramètres physico-chimiques : la concentration protéique et
de l'agent cristallisant [sel, polymères, alcool], le pH et plus
rarement la température (McPherson, 1982 ; McPherson, 1998).
3-Etapes de la cristallisation
3.1-La nucléation : La
nucléation est le point de départ de la cristallisation,
elle correspond à la constitution d'amas moléculaires qui,
lorsqu'ils dépassent une taille critique, donnent naissance au
cristal.
3.2-La croissance cristalline : La
croissance cristalline est le processus physique qui va suivre la
nucléation et permettre l'augmentation de la taille des cristaux.
a)-Cinétique : elle
dépend directement du degré de sursaturation. Pour les
protéines : une couche de protéines s'ajoute toutes les 3
secondes, soit une croissance d'environ 30 ?.s-1. Le
transport des molécules en solution vers le cristal dépend
de la diffusion, la convection et de l'agitation de la solution
(McPherson et al, 1999). Mais aussi des concentrations de la
protéine et de l'agent précipitant,
du pH, de la force ionique, de la température et de la
pureté des agents chimiques et de la protéine
utilisée (Stelter, 2003).
Agents précipitants (cristallisants) usuels
:
Sels : sulfate d'ammonium, phosphate de
potassium et chlorure de sodium
Polymères :
polyéthylène glycérol
Paramètres de la cristallogenèse
:
Concentration protéique
Contaminants non macromoléculaires
Température, pH et Force ionique et nature du
tampon Densité, viscosité, pression, temps d'évaporatoire
se Vibrations, gravité et mouvements de diffusion,
Convection
Nature de l'agent cristallisant
Solvants organiques : non volatil
(méthyl-2,4-pentadiol), volatils (éthanol)
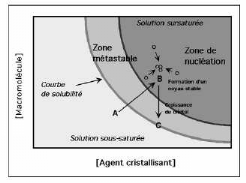
Figure 14 : Diagramme de phase
bidimensionnel d'une macromolécule en fonction de sa propre
concentration et de celle d'un
agent cristallisant. Lors de la
cristallisation, la solubilité de la macromolécule diminue
jusqu'à atteindre l'état de supersaturation favorable
à l'obtention de cristaux, sans toutefois tomber dans la zone de
précipitation.
Au niveau de la courbe de solubilité,
le cristal et la solution sont en équilibre thermodynamique.
Au-delà de cette courbe, la solution est dite
sursaturée. Plus la molécule s'enfonce dans
cette zone A---B plus la sursaturation est élevée. En B, il y'a
formation d'un noyau stable à partir du quel un cristal va
croître. Dans la zone métastable il y'a la croissance
cristalline.
La courbe de supersolubilité
sépare la zone métastable de la
zone de nucléation.
b)-Equilibre et arrêt de la croissance
: l'arrêt de la croissance (mûrissement d'Oswald)
intervient
lorsque le système atteint le point
d'équilibre où la concentration de la molécule est
égale à la solubilité : le nombre de molécules
qui se détachent du cristal est égal au nombre de
molécules qui s'y lient. La présence d'impuretés peut
induire l'arrêt irréversible de la croissance cristalline
avant d'atteindre le point d'équilibre (Sauter et Giegé, 2001 ;
Stelter, 2003).
B-Méthodes de cristallisation :
Plusieurs méthodes sont développées
pour cristalliser les protéines. Elles ont toutes
pour principe « d'amener les protéines sans les
dénaturer en conditions de sursaturation propices à la
nucléation, puis à la croissance du cristal». La
méthode la plus utilisée est la diffusion de vapeur en goutte
suspendue, sa simplicité et son adaptation aux volumes
réduits de solutions expliquent sa popularité (McPherson,
1998). Dans une enceinte close, l'équilibre s'établit entre
la goutte (contenant
la protéine, le tampon et l'agent cristallisant)
et le réservoir (contenant le tampon et
l'agent cristallisant à des concentrations plus
élevées) par diffusion des espèces volatiles de la
goutte (principalement l'eau) jusqu'à ce que la tension de
vapeur soit la même dans la goutte et dans le
réservoir.
Ce processus d'équilibration concentre l'agent
cristallisant et la protéine dans la goutte, ce qui diminue la
solubilité de la protéine et si les conditions sont favorables
l'amène à cristalliser (Sauter et Giegé, 2001). La
cinétique de la diffusion dépend des différences de
concentrations dans la goutte et dans le réservoir mais également
de la géométrie du système, du volume de la goutte et de
sa distance
au réservoir. Dans la Figure 15, est
représentée la méthode de la goutte assise utilisée
dans les tests au
robot.
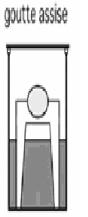

Figure 15 : Technique de la goutte assise
B- Matériel et méthodes
1-Clonage, surexpression et
purification
A-UMPKeco, double mutant D159N D93A [en position
159 : Asp(D) remplacé par Asn (N) / en 93 :
Asp(D) remplacé par Ala(A)] :
Le variant UMPK D159N D93A est construit par la méthode de
double PCR. La souche transformée est cultivée à 37°C
jusqu'à l'obtention d'une absorbance de 1.5 à 600 nm. Puis la
production de protéine est induite par l'ajout de 1 mM
d'isopropyl-thiol--D-galactosidase (IPTG) pendant 3 h à 37°C. Les
bactéries sont ensuite centrifugées. Le culot bactérien
est soit traité immédiatement, soit conservé à
-20°C pour une utilisation ultérieure (Serina et al,
1996).
La purification s'est faite en deux étapes :
-la première met en jeu une chromatographie
d'affinité sur résine de l'acide Ni-Nitrile
Triacétique agarose ;
-la deuxième une chromatographie d'exclusion sur
Séphacryl S-300.
La concentration protéique est
évaluée par la méthode de Bradford. L'enzyme
purifiée est alors conservée dans du Tris-HCl pH 8,5 50
mM et du NaCl 100 mM à 20°C plusieurs semaines. La
pureté du variant UMPKeco D93A est vérifiée par
électrophorèse sur gel de polyacrylamide (12.5 %)
en présence de SDS (SDS-PAGE). A l'issue de cette
étape, l'enzyme doit être très pure, au moins à
97
%, sinon on risque de n'obtenir aucun cristal du fait
des impuretés qui pourraient compromettre la cristallisation. Il
est aussi nécessaire de disposer de quantités suffisantes de
protéines.
B-CKOm sauvage et mutants D169E et D192L :
L'amplification est effectuée par la méthode PCR en
utilisant un plasmide d'E. coli. Le vecteur contenant le gène
d'intérêt : pINA1267,
est ensuite cloné et surexprimé dans la
levure Yarrowia lipolitica. La purification de la
protéine obtenue (CKOm) est effectuée par deux chromatographies
sur colonne échangeuse d'anions. La pureté
de la CKOm est vérifiée par la SDS-PAGE
(Kopeèny et al, 2004).
2-Cristallisation
Avant de procéder à la cristallisation proprement
dite, on a procède à chaque fois à la
vérification
de la concentration protéique en mesurant
l'absorbance à 280 nm (Encadré 1), la
concentration minimale requise pour la cristallogenèse est
d'environ 5 mg/ml. Si la concentration protéique est
inférieure à cette valeur on procède dès lors
à la concentration de la protéine (Encadré 2).
Une fois
que la solution protéique est très pure et
suffisamment concentrée, on peut passer à la phase de
cristallisation qui comprend deux cribles (Figure 16).
A-UMPKeco, double mutant D159N D93A :
Premier crible : tests au robot / essais
préliminaires / goutte assise : La recherche des
conditions prometteuses de cristallisation s'est effectuée en utilisant
les kits Crystal Screen I et II (Annexes 1 &
2) (Hampton Research).
Encadré 1 : Vérification de la
concentration protéique par absorption dans l'UV
La mesure de la concentration protéique s'est
effectuée en utilisant un spectrophotomètre UV/VISIBLE
(BECKMAN DU 640B).
Un rayon lumineux traverse la solution absorbante contenant 2
u l de la solution protéique et 498 u l du
tampon
Tris-HCL pH 8,5 50 mM pour l'UMPK et pH 7,0 100 mM pour la
CKOm, après lecture des absorbances à 280
nm et en appliquant la loi de BEER-LAMBERT on détermine la
concentration en mg/ml de la protéine.
Loi de BEER-LAMBERT : A= log (I / Io) = å.
l . C
A : absorbance, I : intensité de la
lumière incidente, Io : intensité de la
lumière transmise, l est l'épaisseur (en cm)
de la cuve appelé aussi chemin optique, C est la
concentration en mg/ml et å : coefficient d'extinction molaire
caractéristique de chaque protéine à une longueur d'onde
donnée (å est lié à la teneur de la protéine
en Trp, Tyr
et en Phe)
Pour l'UMPKeco : 1/å =2,33 & pour la CKO 1/å =
0,777
Encadré 2 : Concentration de la
protéine
On a utilisé des centricons 10 à contenance
maximale de 2 ml, leur porosité permet à certaines
molécules de passer : sels, petites molécules et retient les
protéines dont la MM est supérieure à 10 kDa. La solution
protéique
est déposée dans la partie supérieure du
tube et repose sur la membrane, le tube est alors centrifugé pour forcer
le liquide et les petites molécules à traverser cette
dernière. Les protéines trop grosses ne peuvent pas
passer et
restent dans la partie supérieure, on peut ensuite les
récupérer pour mesurer la concentration.
Détermination des conditions
prometteuses de la cristallisation
Tests préliminaires
Robot de cristallisation
96 conditions différentes
Robot de cristallisation


![]()
![]()
![]()
![]()




![]()
![]()
![]()
![]()


96 conditions différentes
Tests manuels approfondis dans
les conditions prometteuses
Boite Linbro
Boite Linbro
24 puits
24 puits
Figure 16 : Photos du robot de cristallisation et de la
boite Linbro
Les kits contiennent 96 conditions
différentes : ensemble de précipitants, additifs
et
tampons...le plus fréquemment testés avec
succès dans la PDB. Quoique empiriques, ces kits permettent
d'échantillonner systématiquement un grand nombre de
conditions. Le robot de cristallisation permet d'obtenir des cristaux
rapidement, en utilisant de faibles quantités de la protéine (0,5
u l par test = économie de protéine par rapport aux essais
manuels : 2 u l) ce qui rend possible la réalisation de beaucoup de
tests. Dans les tests au robot, on a testé l'UMPK D159N D93A
seule (enzyme brute), avec l'UMP
(substrat accepteur de phosphate) et avec le GTP
(activateur allostérique). Chaque goutte contient 1 u l de
solution (0,5 u l de solution protéique -enzyme seule ou avec ligand- et
0,5u l du puit).Au total, 288 essais sont
réalisés avec le robot ; la boite est
incubée dans
une enceinte thermostatée à 20 °C.
L'observation des résultats se fait à la
loupe biloculaire le lendemain, après 3 jours puis chaque
semaine. A l'issue de ces essais on détermine et on
retient les conditions prometteuses de cristallisation.
Deuxième crible : tests manuels approfondis
/ méthode de diffusion de vapeur / technique de
la
goutte suspendue (Figures 17) : On
utilise la technique de la goutte suspendue (hanging drop) sur des boites
Linbro. Les essais ont été réalisés de la
façon suivante : un volume A de la solution
SE
(solution de l'enzyme) [UMPK D159N
D93A, ligand GTP et additif ( Glucoside, Glycérol)] est
mélangé avec un volume B de solution
SR (solution du réservoir) [PEG
6000, le NaCl, l'additif et le tampon], A + B = 4ul. Ce
mélange constituant la goutte est déposé sur une
lamelle en verre [ronde, 22 mm de diamètre, siliconé avec
l'aquasil (Pierce) 2,5 % v/v] placée avec précaution au dessus du
puits contenant 1 ml de la SR.
L'étanchéité étant indispensable, on graisse
préalablement le pourtour du puits avec une graisse à vide
à l'aide d'une seringue. Les boîtes ont ensuite été
placées dans l'enceinte thermostatée à une
température de 20°C. L'observation des résultats se
fait se fait comme pour le
robot.
Les travaux menés dans l'UMR de Chimie
Biologique par Briozzo et al, ont montré que l'UMPKeco
cristallise mieux au contact de la graisse, pour chaque paramètre requis
on a effectué des essais en présence et en absence de la graisse.
Le pH est maintenu constant grâce au tampon Tris-HCl
pH 8,5, 50 mM. 266 essais ont étés
effectués manuellement. Au total 554 essais
sont effectués avec
l'UMPK D159E D93A.
B-CKOm sauvage et mutants D169E et
D192L
On n'a pas effectué d'essais au robot pour cette enzyme
car :
(1) nous avons que des petites
quantités de la protéine ;
(2) les mutants de la CKO
cristallisent en général dans les conditions
déterminées en 2004 par Briozzo et al
(précipitant : PEG 1500 à 30% - condition 43 du Crystal
Screen I-, pas de sel, tampon : Tris-HCl pH 7,0 50 mM). Pour les essais
manuels approfondis nous avons employé la méthode de
diffusion de vapeur les boites Linbro. Nous avons varié
dans le puits la concentration du PEG 1500 et
dans la goutte la concentration protéique. Des tests sont
effectués avec la CKOm sauvage brute ( pour
infiltration), avec le mutant D169E-IP et avec le mutant D492L.
Au total 96 essais ont étés effectués avec la CKOm : 42
avec la CKOm native, 30 avec le mutant D169E et 6 avec le mutant L492A. Pour
la CKOm, les concentrations de départ étant
assez importantes (> à 13 mg/ml), on ne l'a pas
concentrée
3- Etapes ultérieures
3.1-Montage des cristaux (Figure : 17 b))
: Les cristaux les plus beaux ont été
montés délicatement dans des boucles en nylon de 0,01 mm de
diamètre pour l'UMPKeco et 0,1 mm pour la CKOm, et serviront à la
diffraction des rayons X. Cette étape est réalisée sous
la loupe biloculaire. Avant de monter les cristaux, une solution correspondant
à la solution de la goutte ou a été
préparée. Cette solution est ensuite déposée
sur la goutte, le cristal a été pêché
soigneusement. La boucle est ensuite montée sur une tête
goniométrique orientable, ce qui permet de centrer et d'orienter
correctement le cristal dans le faisceau de rayons X. Avant l'enregistrement
des données de diffraction,
les cristaux ont été congelés sous flux
d'azote gazeux à 100 K. On augmente ainsi leur durée de vie et
on réduit leur dégradation [modification
chimique et formation de radicaux libres] par les rayons X (faisceaux
très puissants) pendant l'enregistrement. Comme un cristal
contient près de 50 % de solvant, qui est principalement l'eau, la
congélation sans cryoprotectant cristalliserait cette eau sous forme
de microcristaux qui détériorent le cristal et donnent des
anneaux parasites sur les clichés de diffraction. Il est
nécessaire de trouver un cryoprotectant adapté qui
transforme l'eau en solide amorphe (empêchant la formation de cristaux
de glace), sans détériorer le cristal.
On a pêché le cristal avec une boucle de
taille adaptée et on l'a transféré dans la
solution cryoprotectante (pendant environ 1 min). Dans notre cas, le
cryoprotectant utilisé été le glycérol à
20
%. Ces étapes ont lieu au laboratoire d'enzymologie et
biochimie structurales de Gif sur Yvette (91).
3.2-Collecte des données de diffraction
(Figure 17 c)) : Cette étape consiste à soumettre
les cristaux à un faisceau monochromatique de rayons X issu
d'un rayonnement synchrotron intense permettant d'enregistrer à haute
résolution. On choisit la longueur d'onde appropriée et on fait
tourner
le cristal autour d'un axe perpendiculaire au faisceau
incident de rayons X (1° dans notre cas). Les intensités
diffractées son enregistrées par un détecteur placé
lui aussi perpendiculairement au faisceau incident.
3.3-Détermination des paramètres
de maille des cristaux : Les données de diffraction sont
collectées sur la ligne ID14-1 à l'ESRF (European Synchrotron
Radiation Facility, Grenoble) équipée d'un détecteur CCD
(charged coupled device). L'enregistrement étant terminé,
il est nécessaire de déterminer pour chaque
réflexion : son indice hkl, son intensité et l'incertitude
sur sa mesure. Le programme DENZO fait l'indexation sur une image
puis sur toutes les images ; le programme SCALEPACK de HKL est
utilisé pour mettre à l'échelle les intensités de
toutes les réflexions dans un
fichier unique.
![]()
.
Figure 17 a) : Méthode de cristallisation en
goutte suspendue : La méthode de diffusion de vapeur est la
méthode le plus couramment utilisée dans les
cristallisations manuelles. Elle est basée sur la saturation atteinte
par évaporation des solvants volatils dans une enceinte
fermée sans contact entre les solutions liquides.
L'équilibre
![]()
s'établit progressivement par diffusion de la vapeur
d'eau, l'eau passe de la goutte vers le puits et ceci jusqu'à ce qu'on
atteigne les conditions de succès (obtention des cristaux) ou les
conditions de précipitation / agrégation.
![]()
Figure 17 b) : Montage des cristaux sur des
boucles en nylon
Figure 17 c) : Collecte des données de
diffraction
C- Résultats et discussion
A-Mutant D93A non régulé par le GTP de
l'UMPKeco
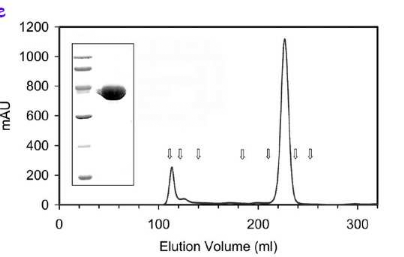
1-Concentration de l'échantillon
purifié : Comme le montre la figure
18, la purification
réalisée par les collaborateurs de l'Institut
Pasteur est très concluante : le matériel protéique
prêt à être utilisé pour la cristallisation est
très pur.
Les pertes en protéines lors de la concentration,
evaluées par la mesure de l'A280 nm du filtrat, sont
négligeables. Le procédé utilisant les centricons 10 est
adapté à l'UMPKeco.
2-Cristallisation :
---Tests au robot : Les essais de Crystal
Screen I & II ont donné les résultats suivants:
? Pour l'enzyme seule (7,41 mg/ml dans la solution
départ et de 3,7 mg/ml dans la goutte): de nombreuses aiguilles
fines sont apparues dans la condition 1 du Crystal Screen II, en
présence de
10 % p/v de PEG 6000 / de NaCl à 2 M et sans tampon (donc
le pH dans la goutte était de 8,5 :
celui de la solution protéique).
? Pour l'enzyme (3,7 mg/ml dans la goutte) avec le GTP (20
mM) : il y a apparition de belles et nombreuses aiguilles dans la
même condition (condition 1 du Crystal Screen II).
? Pour l'enzyme (3,7 mg/ml dans la goutte) avec à
la fois GTP (20 mM) & UMP (10 mM) : aucune forme cristalline n'est
apparue, il y a dans certaines conditions apparition d'agrégats
et de précipités.
Les conditions les plus favorables à ce mutant pour
cristalliser sont donc : PEG 6000 à 10 % et NaCl 2,0 M, avec GTP
à 20 mM, pH 8,5. Par ailleurs, la structure avec GTP est plus
intéressante que celle de l'enzyme brute.
---Tests manuels approfondis : Afin
d'améliorer les formes cristallines, nous avons fait varier :
Dans le réservoir :
-la concentration : du PEG, du NaCl [exercent un effet
salting out (recherché pour la cristallisation] et rendent
les protéines moins solubles en fixant les molécules d'eau) et du
GTP ;
-la concentration et la nature de l'additif (glycérol et
le â-octyle-glycoside).
Dans la goutte : nous avons fait varier
la concentration protéique et le volume de A &
B.
Des aiguilles sont obtenues dans 14 cas
correspondants à des conditions différentes
(Tableau 3
& Figure 19). Le temps de cristallisation de
l'UMPKeco D159N D93A en présence du GTP est à peu près le
même que celui de l'UMPKeco D159N D93A seule (environ une semaine).
L'étude cristallographique (par cocristallisation) de
complexes enzyme/substrat est aléatoire et présente de grandes
difficultés. En effet, il nous est impossible de savoir si le
GTP (ligand) est
réellement présent dans le cristal avant la
résolution de la structure, qui peut prendre plusieurs mois.
![]()
![]()
1 2 3 4 5 6 7
Figure 18 : Suivi de la pureté de
l'UMPKeco D159ND93A par SDS - PAGE
1 - marqueurs
2 - extrait brut
3 - Pass - through colonne de Ni
4 - lavage
5 - Pool après élution colonne de Ni
6 - Pool après colonne Séphacryl S-
300
7 - Pool après concentration
Matériel prêt à être
donné pour la cristallisation
Figure 18 : Suivi de la pureté de
l'UMPKeco D159ND93A par SDS - PAGE
1 - marqueurs
2 - extrait brut
3 - Pass - through colonne de Ni
4 - lavage
5 - Pool après élution colonne de Ni
6 - Pool après colonne Séphacryl S-
300
7 - Pool après concentration
Matériel prêt à être
donné pour la cristallisation
Tableau 3 : Cristaux de l'UMPKeco D159N D93A :
Conditions d'obtention et dimensions.
|
|
PEG 6000
(%)
|
GTP (mM)
|
Graisse
|
NaCl
(M)
|
â-Glucoside
(%)
|
Dimensions (mm)
|
|
|
10
|
0
|
Présence
|
2
|
0
|
0,016 x 0,32
|
|
7
|
0,016 x 0,96
|
|
12
|
20
|
0,016 x 0,8
|
|
10
|
20
|
Absence
|
0,016 x 0,72
|
|
18
|
20
|
Présence
|
0,016 x 0,32
|
|
22
|
20
|
0,25
|
Non Déterminé (ND)
|
|
12
|
20
|
0
|
0,016 x 0,32
|
|
9,5
|
20
|
0,016 x 0,32
|
|
10
|
20
|
ND
|
|
16,5
|
20
|
0,016 x 0,32
|
|
17
|
20
|
0,016 x 0,32
|
|
19,5
|
20
|
0,016 x 0,32
|
|
18,5
|
20
|
0,016 x 0,32
|
|
19
|
20
|
0,016 x 0,32
|
|
|
|
PEG
6000 (%)
|
NaCl
(M)
|
GTP
(mM)
|
MPD
(%)
|
ß glucoside
(%)
|
Glycérol
(%)
|
Concentration protéique
(mg/ml)
|
|
|
3 à 22
|
1, 6 à 2,
6
|
20
|
5
|
0,25 à 0,5
|
0,25
|
3.7 à 7,4
|
|
Tableau 2 : Résumé de la variabilité
des conditions de cristallisation de l'UMPKeco D159N D93A
|
|
Sans Additif
Avec Graisse
Sans GTP
PEG 6000 10 % PEG 6000 7 %
![]()
![]()
![]()
![]()


![]()

![]()

GTP 20mM
PEG 6000 10 % / Sans graisse
PEG 6000 22 % / âGlucoside
à 0,25 %
/ Avec graisse
Figure 19 : Cristaux de l'UMPKeco D159N D93A
0,2 mm
En revanche, le nombre de cristaux obtenus, pour un
nombre d'essais comparables, en
présence du GTP est plus important que celui obtenu avec
l'enzyme brute (12 cas en présence du GTP
contre 2 avec l'enzyme seule) ; le GTP aurait un
effet favorable sur la cristallisation de cette enzyme.
Les meilleurs résultats sont obtenus dans des conditions
variables de PEG (entre 7 et 22 %) et
la même concentration du sel (NaCl à 2M), les
aiguilles les plus belles sont obtenues au voisinage de
10 % de PEG. La variation de la concentration protéique
n'a pas d'effet significatif sur la vitesse et le rendement de la
cristallisation de l'UMPKeco D159N D93A. Sauf pour les concentrations
élevées du précipitant (22 % du PEG), l'ajout d'additifs
(â-octyl-glucoside à 0,25 %) n'influe pas
nettement sur
le processus de cristallisation de cette enzyme.
Le tableau 3 montre que cette enzyme cristallise mieux au contact
de la graisse (1 cas de succès sans graisse contre 13 avec graisse),
comme pour l'enzyme D159N dont la structure a été
publiée.
3-Enregistrement et traitement des données
(Tableau 6) :
Les données de diffraction des rayons X ont
été enregistrées jusqu'à 3 ? de
résolution.120
images sont enregistrées, les taches sont très
rares et peu intenses et la mosaïcité est importante : le jeu
de données semble extrêmement difficile à
traiter.
B-CKOm sauvage et mutants D169E et
D192L
Pour la CKOm, nous avons fait varier dans le
réservoir la concentration du PEG 1500 et le volume du
tampon Tris-HCl pH 7,0. Dans la goutte nous avons fait
varier la concentration protéique
et le volume de A & B (Tableau
4). De gros cristaux colorés en jaune
(à cause du FAD lié covalemment à l'enzyme par
l'His 87) sont obtenues dans 12 cas correspondant
à des conditions différentes: 5 cas avec l'enzyme native, 7
cas avec le mutant D169E, aucun cristal n'a été obtenu avec
le mutant D492L (Tableau 5 & Figure 20). Le
temps de cristallisation de la CKOm est comparable à celui de
l'UMPKeco (environ une semaine). Plusieurs formes cristallines sont
obtenues (maclées, rectangulaires...). Comme on pouvait s'y attendre,
la CKOm cristallise mieux que l'UMPKeco. Enregistrement et
traitement des données de diffraction X : Le
Tableau 6 indique quelques
paramètres de l'enregistrement.
Les données de diffraction de la CKOm sauvage
complexée avec un inhibiteur suicide : HA-
8 et du mutant D169E avec le substrat : IP ont été
traitées pendant ce stage. Plusieurs paramètres ont
été utilisés pour le suivi et la
validation de l'affinement des résultats des diffraction : Rsym
(Rsym maximal toléré est 40 % dans la dernière
tranche de résolution et 10 % pour l'ensemble des
résolutions), complétude (doit être au moins de 80 %)...,
les statistiques obtenues à l'issue de plusieurs
séries d'affinements sont indiqués dans le
Tableau 7.
Tableau 4 : Résumé de la
variabilité des conditions de cristallisation de la CKOm
|
|
CKOm
|
PEG
1500 (%)
|
Tampon
Tris-HCl pH =7,0
|
â-octyl-glucoside
(%)
|
Concentration
protéique
(mg/ml)
|
VB dans la goutte de 4ul le
reste est complété par VA de la
SE
|
|
|
Native
|
16 à 33
|
+
|
0,5
|
7
|
1,92 à 2,92
|
|
Mutant D169 E
|
28 à 33
|
+
|
0,5
|
8
|
1,92 à 2,92
|
|
Mutant D492L
|
28 à 33
|
+
|
0,5
|
5
|
1,92
|
Tableau 5 : Cristaux de la CKOm : Conditions
d'obtention et dimensions.
|
|
Enzyme
|
PEG 1500 (%)
|
â-Glucoside
(%)
|
Dimensions (mm)
|
|
|
Enzyme Native
|
16
|
0,5
|
0,032 x 0,24
|
|
20
|
0,048 x 0,24
|
|
22
|
0,032 x 0,24
|
|
24
|
0,048 x 0,24
|
|
30
|
0,032 x 0,24
|
|
Mutant D169E
|
22
|
0,032 x 0,24
|
|
26
|
0,13 x 0,24
|
|
28
|
0,13 x 0,24
|
|
30
|
0,13 x 0,24
|
|
31
|
0,13 x 0,24
|
|
32
|
0,032 x 0,24
|
|
33
|
0,032 x 0,24
|
|
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

![]()


CKOm native Mutant D169E
Figure 20 : Cristaux de la CKOm (native et mutant
D169E)
Figure 20 : Cristaux de la CKOm (native et mutant
D169E)
Tableau 6 : Enregistrement des données de
diffraction
|
|
Nombre de
cristaux montés sur des boucles
|
Nombre de cristaux qui ont diffracté
|
Conditions retenues
|
Résolution / Ligne
|
Glycérol
(Cryoprotectant)
|
|
|
UMPKeco D93A avec GTP ?
|
3
|
1
|
-18 % de PEG 6000
-2,0 M NaCl
- Protéine à 3,7
mg/ml
|
3 ? / ID 14-1
|
20 % pendant
1 min
|
|
CKOm, mutant D169E avec IP
|
3
|
3
|
-29 % PEG 1500
-0,5 % â-Glucoside
-Protéine à 8 mg/ml
-Tris HCl pH 7,0
100 mM
|
1,9 ? / ID 14-1
|
|
Tableau 7 : Résultats du traitement des
données
|
Jeu de données
|
CKOm sauvage avec HA-8,
Résultats Définitifs
|
CKOm D169E avec IP,
Résultats Préliminaires
du
traitement en cours
|
|
Longueur d'onde des rayons X (?)
|
0,934
|
0,934
|
|
Nombre d'images enregistrées
|
200
|
190
|
|
Angle de rotation lors de
l'enregistrement (°)
|
1
|
1
|
|
Distance Cristal -Détecteur (mm)
|
162,62
|
177,09
|
|
Système cristallin
|
Système monoclinique
|
Système monoclinique
|
|
Groupe d'espace
|
C2
|
C2
|
|
Paramètres de maille
|
a = 254,09 / b = 50,87 / c= 52,26 (?)
á = ã = 90,00 / â = 93,19 (°)
|
a = 251,08 / b =51,27 / c = 51,58 (?)
á = ã = 90,00 / â = 93,27 (°)
|
|
Résolution retenue pour résoudre
la structure 3D (?)
|
Résolution max 25,00
Résolution min 2,4
|
Résolution max 25,00
Résolution min 2,00
|
|
Nombre de réflexions observées
|
287781
|
720276
|
|
Nombre de réflexions uniques
|
22648
|
43460
|
|
Rodandonce
|
12,7
|
16,6
|
|
Mosaïcité
|
1,2
|
2
|
|
Complétude %
|
85,8
|
96,8
|
|
R fact (%) (< 10 %)
|
12,2
|
13,2
|
|
I/ä (> ou = à 2)
|
9,94
|
9,74
|
Les résultats obtenus pour la CKOm-HA-8 sont moins bons
que ceux de la CKOm-D169E-IP.
La structure 3D avec HA-8 ne serait pas assez précise
pour permettre de comprendre et d'expliquer clairement le mécanisme
d'inhibition. En plus, une structure avec un inhibiteur comparable a
été résolue (article en cours de rédaction).
Par contre, celle du mutant D169E-IP, vu les résultats
préliminaires du traitement des données, va certainement
expliquer le rôle de l'Asp 169 dans la catalyse enzymatique. Le
tableau 8 résume les résultats de l'étude
cristallographique des autres mutants de la CKOm cristallisés et
montés par Briozzo et al. La compréhension fine
du rôle et du fonctionnement de la CKOm sera améliorée
si la structure 3D de ces mutants, dont les cristaux sont
déjà obtenus, est résolue.
D- Projets
1-UMPKeco D159N
Notre travail nous a permis d'obtenir des cristaux avec le GTP
d'un mutant du résidu impliqué dans les interactions de maintien
de l'oligomérisation de l'enzyme. La suite du projet consistera d'une
part à poursuivre les études cristallographiques d'autres mutants
ponctuels non régulés par le GTP et d'autre part à
approfondir l'étude des mécanismes de régulation. Cela
pourrait avoir des applications dans le domaine de la santé publique.
2-CKOm
Le traitement des données étant entrepris, il est
nécessaire de résoudre la structure du mutant
D169N avec l'IP et de l'enzyme native avec l'HA-8. Il existe
d'autres structures de mutants ponctuels
en cours d'affinement avec des accepteurs d'électrons. Des
données ont été enregistrées, le même jour
sur la ligne ID14-1, avec les mutants L492A (deux jeux de
données avec des inhibiteurs différents) et E288Q (avec
inhibiteur). L'ensemble de ces structures permettra une meilleure
compréhension des mécanismes de fonctionnement de la
CKOm et cela aura éventuellement des applications
agronomiques.
Références bibliographiques
1. Briozzo P., Golinelli-Pimpaneau B., Gilles
A. M., Gaucher J. F., Burlacu-Miron S., Sakamoto
H., Janin J., and Bârzu O. (1998). Structures of
Escherichia coli CMP kinase alone and in complex with CDP: a
new fold of the nucleoside monophosphate binding domain and insights into
cytosine nucleotide specificity. Structure. 6 :1517 -27.
2. Briozzo P., Evrin C., Meyer P., Assairi L.,
Joly N., Barzu O. and Gilles A.M.(2005). Structure
of Escherichia coli UMP kinase differs from that of
other nucleoside monophosphate kinases and sheds new light on enzyme
regulation. J. Biol. Chem. 280(27): 25533-40.
3. Bucurenci N., Sakamoto H., Briozzo
P., Palibroda N., Serina L., Sarfati R.S., Labesse G., Briand G.,
Danchin A., Barzu O. & Gilles A.M. (1996). CMP kinase from Escherichia
coli is structurally related to other nucleoside monophosphate kinases.
J. Biol. Chem. 271 (5) :2856-
62.
4. Chayen N.E., Boggon T.J., Cassetta A. et
al. (1996). Trends and challenges in experimental macromolecular
crystallography. Quarterly Rev. Biophys. 29, 227-78.
5. Chernov A.A. (1999). Estimates of
internal stress and related mosaicity in solution grown crystals :
proteins. J. Crystal Growth. 196 : 524-34.
6. Deleu M., Wathelet B., Brasseur R. et
Paquot M. (1998). Aperçu des techniques d'analyse
conformationnelle des molécules biologiques. Biotechnol. Agron. Soc.
Environ. 2(4) : 234-247.
7. Dreusicke D., Karplus P.A. &
Schulz G.E. (1988). Refined structure of porcine cytosolic adenylate
kinase at 2.1 Å resolution. J. Mol. Biol. 199 :
359-371.
8. Duée E. et Dideberg O. (1997). La
cristallographie moléculaire en mouvement. La technologie biofutur
173, 3-11.
9. Ducruix A. & Giegé R et
al, (1999). Crystallization of Nucleic acids and Proteins : a
Practical
Approach (second edition). The practical approach series, IRL
Press : Oxford.
10. Fassy F., Krebs O., Lowinski M., Ferrari P.,
Winter J., Collard-Dutilleul V. & Salahbey Hocini
K. (2004). UMP kinase from Streptococcus pneumoniae
: evidence for co-operative ATP
binding and allosteric regulation. Biochem. J. 384(Pt 3)
: 619-27.
11. Fricke J., Neuhard J., Kelln R.A. and
Pedersen S. (1995). The cmk gene encoding cytidine monophosphate kinase
is located in the rpsA operon and is required for normal replication rate
in Escherichia coli. J. Bacteriol. 177( 3) :
517-523

12. Galuszka P., Frébort
I.,  ebela
M., Sauer P., Jacobsen S. and Pe P. (2001). Cytokinin oxidase ebela
M., Sauer P., Jacobsen S. and Pe P. (2001). Cytokinin oxidase
or dehydrogenase? Mechanism of cytokinin degradation in
cereals. Eur. J. Biochem. 268, 450-
461.
13. Haouz A., Vanheusden V., Munier-Lehmann H.,
Froeyen M., Herdewijn P., Van Calenbergh
S., Delarue M. (2003). Enzymatic and structural
analysis of inhibitors designed against Mycobacterium tuberculosis
thymidylate kinase. New insights into the phosphoryl transfer
mechanism. J. Biol. Chem. 278(7) : 4963-71.
14. Helliwell J.R.(1988). Protein Crystal
perfection and the nature of radiation domage. J. Crystal
Growth. 90, 295-72.
15. Kopeèny D., Briozzo P., Joly N.,
Houba-Herin N., Madzak C., Pethe C. & Laloue M.(2004).
Purification, crystallization and preliminary X-ray diffraction
study of a recombinant cytokinin oxidase from Zea mays. Acta.
Crystallogr. D. Biol Crystallogr. D60 1500-1.
16. Kholti A., Charlier D., Gigot D.,
Huysveld N., Roovers M. & Glansdorff N.(1998). pyrH- encoded
UMP-kinase directly participates in pyrimidine-specific modulation of
promoter activity in Escherichia coli. J. Mol. Biol. 280(4) :
571-82.
17. Landais S., Gounon P., Laurent-Winter
C., Mazie J.C., Danchin A., Barzu O., Sakamoto H. (1999).
Immunochemical analysis of UMP kinase from Escherichia coli. J.
Bacteriol. 181(3) :
833-40.
18. Malito E., Coda A., Bilyeu K.D., Fraaije
M.W., Mattevi A. (2004). Structures of Michaelis
and product complexes of plant cytokinin dehydrogenase :
implications for flavoenzyme catalysis. J. Mol. Biol. 341(5)
:1237-49.
19. Matthews B.W. (1985). Determination of
protein molecular weight, hydration, and packing
of crystal density. Methods enzymol. 114 :
176-87.
20. McPherson A. (1982). Preparation and
analysis of protein crystals. Wiley and Sons : New
York.
21. McPherson A. (1998). Crystallization of
Biological Macromolecules. Could Spring Arbor
Laboratory Press : New York.
22. McPherson A., Malkin A.J., Kuznetsov
Y.G., Koszelak S., Wells M., Jenkins G., Howard J. & Lawson G. (1999).
The effects of microgravity on protein crystallization : evidence for
concentration gradients around growing crystals. J. Crystal Growth.
196 : 572-86.
23. Mullin J.W. (1993). Crystallisation.
Beterworth : oxford.
24. Noda L. (1973). In the enzymes.
3rd Edition. (Boyer P.D.). Vol 08, Academic Press. New
york.
279-305.
25. Sauter C. et Giegé R. (2001). La
cristallographie des macromolécules biologiques. Regard sur
la Biochimie. N° 3, 21-31.
26. Serina L, Blondin C, Krin E, Sismeiro O,
Danchin A, Sakamoto H, Gilles A.M. & Barzu O. (1995). Escherichia
coli UMP-kinase, a member of the aspartokinase family, is a
hexamer regulated by guanine nucleotides and UTP. Biochemistry. 1834
(15) : 5066-74.
27. Stelter M. (2003). Détermination
de la structure cristallographique d'une protéine. CESIRE
(IBS), 12 P.
28. Saridakis E.E., Stewart P.D., Lloyd
L.F., Blow D.M. (1994). Phase diagram and dilution experiments in
the crystallization of carboxypeptidase G2. Acta. Crystallogr.
D. Biol. Crystallogr. 50(Pt 3) : 293-7.
29. Vonrhein C., Schlauderer G.J., Schulz G.E.
(1995). Movie of the structural changes during a catalytic cycle of nucleoside
monophosphate kinases. Structure. 3(5) : 483-90.
30. Yan H. & Tsai M.D. (1999).
Nucléoside Monophosphates Kinases : Structure, Mechanism, and
Substrate Speficity. Adv. Enzymol. Related. Areas. Molec.
Biol. 73 : 103-139.
31. Zhou L., Lacroute F. & Thornburg
R. (1998). Cloning, expression in Escherichia coli, and
characterization of Arabidopsis thaliana UMP/CMP kinase. Plant
Physiol. 117: 245-254.
SUR LE WEB:
- NCBI Reference Sequence project :
ftp://ftp.ncbi.nih.gov/refseq/release/
- RCSB Protein Data Bank :
http://www.rcsb.org/pdb/Welcome.do/
- L'essentiel sur les cytokinines :
http://www.inrp.fr/biotic/morpho/html/cytokinines.htm
Annexes
Annexe I : Crystal Screen 1
|
1. 0.02 M Calcium chloride dihydrate, 0.1 M Sodium acetate
trihydrate pH 4.6, 30% v/v (+/-)-2-Methyl-2,4-pentanediol
|
|
2. 0.4 M Potassium sodium tartrate tetrahydrate
|
|
3. 0.4 M Ammonium phosphate monobasic
|
|
4. 0.1 M Tris hydrochloride pH 8.5, 2.0 M Ammonium sulfate
|
|
5. 0.2 M Sodium citrate tribasic dihydrate, 0.1 M HEPES sodium
pH 7.5, 30% v/v (+/-)-2-Methyl-2,4-pentanediol
|
|
6. 0.2 M Magnesium chloride hexahydrate, 0.1 M Tris
hydrochloride pH 8.5, 30% w/v Polyethylene glycol 4000
|
|
7. 0.1 M Sodium cacodylate pH 6.5, 1.4 M Sodium acetate
trihydrate
|
|
8. 0.2 M Sodium citrate tribasic dihydrate, 0.1 M Sodium
cacodylate pH 6.5, 30% v/v 2-Propanol
|
|
9. 0.2 M Ammonium acetate, 0.1 M Sodium citrate tribasic
dihydrate pH 5.6, 30% w/v Polyethylene glycol 4000
|
|
10. 0.2 M Ammonium acetate, 0.1 M Sodium acetate trihydrate pH
4.6, 30% w/v Polyethylene glycol 4000
|
|
11. 0.1 M Sodium citrate tribasic dihydrate pH 5.6, 1.0 M
Ammonium phosphate monobasic
|
|
12. 0.2 M Magnesium chloride hexahydrate, 0.1 M HEPES sodium pH
7.5, 30% v/v 2-Propanol
|
|
13. 0.2 M Sodium citrate tribasic dihydrate, 0.1 M Tris
hydrochloride pH 8.5, 30% v/v Polyethylene glycol 400
|
|
14. 0.2 M Calcium chloride dihydrate, 0.1 M HEPES sodium pH 7.5,
28% v/v Polyethylene glycol 400
|
|
15. 0.2 M Ammonium sulfate, 0.1 M Sodium cacodylate pH 6.5, 30%
w/v Polyethylene glycol 8000
|
|
16. 0.1 M HEPES sodium pH 7.5, 1.5 M Lithium sulfate
monohydrate
|
|
17. 0.2 M Lithium sulfate monohydrate, 0.1 M Tris hydrochloride
pH 8.5, 30% w/v Polyethylene glycol 4000
|
|
18. 0.2 M Magnesium acetate tetrahydrate, 0.1 M Sodium cacodylate
pH 6.5, 20% w/v Polyethylene glycol 8000
|
|
19. 0.2 M Ammonium acetate, 0.1 M Tris hydrochloride pH 8.5, 30%
v/v 2-Propanol
|
|
20. 0.2 M Ammonium sulfate, 0.1 M Sodium acetate trihydrate pH
4.6, 25% w/v Polyethylene glycol 4000
|
|
21. 0.2 M Magnesium acetate tetrahydrate, 0.1 M Sodium cacodylate
pH 6.5, 30% v/v (+/-)-2-Methyl-2,4-pentanediol
|
|
22. 0.2 M Sodium acetate trihydrate, 0.1 M Tris hydrochloride pH
8.5, 30% w/v Polyethylene glycol 4000
|
|
23. 0.2 M Magnesium chloride hexahydrate, 0.1 M HEPES sodium pH
7.5, 30% v/v Polyethylene glycol 400
|
|
24. 0.2 M Calcium chloride dihydrate, 0.1 M Sodium acetate
trihydrate pH 4.6, 20% v/v 2-Propanol
|
|
25. 0.1 M Imidazole pH 6.5, 1.0 M Sodium acetate trihydrate
|
|
26. 0.2 M Ammonium acetate, 0.1 M Sodium citrate tribasic
dihydrate pH 5.6, 30% v/v (+/-)-2-Methyl-2,4-pentanediol
|
|
27. 0.2 M Sodium citrate tribasic dihydrate, 0.1 M HEPES sodium
pH 7.5, 20% v/v 2-Propanol
|
|
28. 0.2 M Sodium acetate trihydrate, 0.1 M Sodium cacodylate pH
6.5, 30% w/v Polyethylene glycol 8000
|
|
29. 0.1 M HEPES sodium pH 7.5, 0.8 M Potassium sodium tartrate
tetrahydrate
|
|
30. 0.2 M Ammonium sulfate, 30% w/v Polyethylene glycol 8000
|
|
31. 0.2 M Ammonium sulfate, 30% w/v Polyethylene glycol 4000
|
|
32. 2.0 M Ammonium sulfate
|
|
33. 4.0 M Sodium formate
|
|
34. 0.1 M Sodium acetate trihydrate pH 4.6, 2.0 M Sodium
formate
|
|
35. 0.1 M HEPES sodium pH 7.5, 0.8 M Sodium phosphate monobasic
monohydrate, 0.8 M Potassium phosphate
monobasic
|
|
36. 0.1 M Tris hydrochloride pH 8.5, 8% w/v Polyethylene glycol
8000
|
|
37. 0.1 M Sodium acetate trihydrate pH 4.6, 8% w/v Polyethylene
glycol 4000
|
|
38. 0.1 M HEPES sodium pH 7.5, 1.4 M Sodium citrate tribasic
dihydrate
|
|
39. 0.1 M HEPES sodium pH 7.5, 2% v/v Polyethylene glycol 400,
2.0 M Ammonium sulfate
|
|
40 0.1 M Sodium citrate tribasic dihydrate pH 5.6, 20% v/v
2-Propanol, 20% w/v Polyethylene glycol 4000
|
|
41. 0.1 M HEPES sodium pH 7.5, 10% v/v 2-Propanol, 20% w/v
Polyethylene glycol 4000
|
|
42. 0.05 M Potassium phosphate monobasic, 20% w/v Polyethylene
glycol 8000
|
|
43. 30% w/v Polyethylene glycol 1500 : Conditions de
cristallisation de la CKOm
|
|
44. 0.2 M Magnesium formate dihydrate
|
|
45. 0.2 M Zinc acetate dihydrate, 0.1 M Sodium cacodylate pH 6.5,
18% w/v Polyethylene glycol 8000
|
|
46. 0.2 M Calcium acetate hydrate, 0.1 M Sodium cacodylate pH 6.5
, 18% w/v Polyethylene glycol 8000
|
|
47. 0.1 M Sodium acetate trihydrate pH 4.6, 2.0 M Ammonium
sulfate
|
|
48. 0.1 M Tris hydrochloride pH 8.5, 2.0 M Ammonium phosphate
monobasic
|
Annexe II : Crystal Screen 2
|
1. 10% PEG 6000, 2.0 M Sodium Chloride : Conditions
de cristallisation de l'UMPKeco D93A
|
|
2. 0.5 M Sodium Chloride, 0.01 M CTAB, 0.01 M Magnesium
Chloride
|
|
3. 25% Ethylene Glycol
|
|
4. 35% Dioxane
|
|
5. 5% iso-Propanol, 2.0 M Ammonium Sulfate
|
|
6. 1.0 M Imidazole pH 7.0
|
|
7. 10% PEG 1000, 10% PEG 8000
|
|
8. 10% Ethanol, 1.5 M Sodium Chloride
|
|
9. 2.0 M Sodium Chloride, 0.1 M Na Acetate pH 4.6
|
|
10. 30% MPD, 0.1 M Na Acetate pH 4.6, 0.2 M Sodium Chloride
|
|
11. 1.0 M 1,6 Hexanediol, 0.1 M Na Acetate pH 4.6, 0.01 M Cobalt
Chloride
|
|
12. 30% PEG 400, 0.1 M Na Acetate pH 4.6, 0.1 M Cadmium
Chloride
|
|
13. 30% PEG MME 2000, 0.1 M Na Acetate pH 4.6, 0.2 M Ammonium
Sulfate
|
|
14. 2.0 M Ammonium Sulfate, 0.1 M Na Citrate pH 5.6, 0.2 M K/Na
Tartrate
|
|
15. 1.0 M Lithium Sulfate, 0.1 M Na Citrate pH 5.6, 0.5 M
Ammonium Sulfate
|
|
16. 2% Polyethyleneimine, 0.1 M Na Citrate pH 5.6, 0.5 M Sodium
Chloride
|
|
17. 35% tert-Butanol, 0.1 M Na Citrate pH 5.6
|
|
18. 10% Jeffamine M-600, 0.1 M Na Citrate pH 5.6, 0.01 M Ferric
Chloride
|
|
19. 2.5 M 1,6 Hexanediol, 0.1 M Na Citrate pH 5.6
|
|
20. 1.6 M Magnesium Sulfate, 0.1 M MES pH 6.5
|
|
21. 2.0 M Sodium Chloride, 0.1 M MES pH 6.5, 0.2 M Na/K
Phosphate
|
|
22. 12% PEG 20,000, 0.1 M MES pH 6.5
|
|
23. 10% Dioxane, 0.1 M MES pH 6.5, 1.6 M Ammonium Sulfate
|
|
24. 30% Jeffamine M-600, 0.1 M MES pH 6.5, 0.05 M Cesium
Chloride
|
|
25. 1.8 M Ammonium Sulfate, 0.1 M MES pH 6.5, 0.01 M Cobalt
Chloride
|
|
26. 30% PEG MME 5000, 0.1 M MES pH 6.5, 0.2 M Ammonium Sulfate
|
|
27. 25% PEG MME 550, 0.1 M MES pH 6.5, 0.01 M Zinc Sulfate
|
|
28. 1.6 M Sodium Citrate pH 6.5
|
|
29. 30% MPD, 0.1 M Hepes pH 7.5, 0.5 M Ammonium Sulfate
|
|
30. 10% PEG 6000, 0.1 M Hepes pH 7.5, 5% MPD
|
|
31. 20% Jeffamine M-600, 0.1 M Hepes pH 7.5
|
|
32. 1.6 M Ammonium Sulfate, 0.1 M Hepes pH 7.5, 0.1 M Sodium
Chloride
|
|
33. 2.0 M Ammonium Formate, 0.1 M Hepes pH 7.5
|
|
34. 1.0 M Sodium Acetate, 0.1 M Hepes pH 7.5, 0.05 M Cadmium
Sulfate
|
|
35. 70% MPD, 0.1 M Hepes pH 7.5
|
|
36. 4.3 M Sodium Chloride, 0.1 M Hepes pH 7.5
|
|
37. 10% PEG 8000, 0.1 M Hepes pH 7.5, 8% Ethylene Glycol
|
|
38. 20% PEG 10,000, 0.1 M Hepes pH 7.5
|
|
39. 3.4 M 1,6 Hexanediol, 0.1 M Tris pH 8.5, 0.2 M Magnesium
Chloride
|
|
40. 25% tert-Butanol, 0.1 M Tris pH 8.5
|
|
41. 1.0 M Lithium Sulfate, 0.1 M Tris pH 8.5, 0.01 M Nickel (II)
Chloride
|
|
42. 12% Glycerol, 0.1 M Tris pH 8.5, 1.5 M Ammonium Sulfate
|
|
43. 50% MPD, 0.1 M Tris pH 8.5, 0.2 M Ammonium Phosphate
|
|
44. 20% Ethanol, 0.1 M Tris pH 8.5
|
|
45. 20% PEG MME 2000, 0.1 M Tris pH 8.5, 0.01 M Nickel (II)
Chloride
|
|
46. 20% PEG MME 550, 0.1 M Bicine pH 9.0, 0.1 M Sodium
Chloride
|
|
47. 2.0 M Magnesium Chloride, 0.1 M Bicine pH 9.0
|
|
48. 10% PEG 20,000, 0.1 M Bicine pH 9.0, 2% Dioxane
|
Résumé
La fonction biologique d'une protéine dépend
étroitement de sa structure 3D. La cristallographie est la seule
méthode qui permette sa détermination à résolution
atomique et sans limitation de la masse moléculaire. Associée
à la mutagenèse dirigée, cette technique constitue un
outil clé pour la compréhension des mécanismes
enzymatiques. Nous l'avons utilisée pour deux enzymes :
Uridine monophosphate kinase d'E. coli
: Cette enzyme est certainement essentielle à la croissance
bactérienne et n'a pas d'homologue chez l'homme, ce qui en fait une
cible potentielle d'antibiotiques. Elle est hexamétrique et
régulée par le GTP, un effecteur allostérique. La
structure de l'enzyme avec GTP n'explique
pas simplement l'allostérie. Des cristaux du mutant D93A
(non régulé par le GTP) ont été obtenus avec du
GTP.
Les données de diffraction des rayons X ont
été enregistrées à une résolution de 3 ?. Le
jeu de données semble très difficile à exploiter.
Cytokinine oxydase du maïs : C'est une
flavoenzyme monomérique à cofacteur FAD, elle clive par
oxydation les cytokinines (phytohormones intervenant dans la croissance et le
développement des plantes) et les rend biologiquement inactives.
Plusieurs mutants de résidus proches du FAD avaient
été produits par mutagenèse dirigée. Des cristaux
de la CKOm sauvage (pour infiltration) et du mutant D169E (avec le substrat,
IP) ont étés obtenus, les données
de diffraction ont été enregistrées à 1,8 ?.
Le traitement des données du complexe CKOm sauvage-inhibiteur
HA-8 (2,4 ?) et du complexe mutant D169E-substrat IP (2,0 ?) a
été réalisé. Le phasage sera entrepris
prochainement.
Mots clés : Protéine,
Cristallographie, Uridine monophosphate kinase d'E. coli,
Cytokinine oxydase de maïs, Mutagenèse dirigée.
Summary
The biological function of a protein depends on its 3D
structure. Crystallography is the only method which allows its
determination at atomic resolution and without limitation of the molecular
weight. Associated
to site-directed mutagenesis, this technique constitutes a
key tool to understand the structural basis of the enzymatic
mechanisms. We applied it for two enzymes:
Uridine monophosphate kinase of E. coli: it is
hexametric and is regulated by GTP, an allosteric effector. This enzyme is
certainly essential to bacterial growth and does not have any counterpart in
human. Crystals of mutant D93A (not controlled by the GTP) have been
obtained with GTP. X-ray diffraction data have been collected to 3 ?
resolution. These data are extremely difficult to process.
Cytokinine oxydase of maize: it is a monomeric flavoenzyme
with FAD cofactor; it cleaves by oxidation
the cytokinines (phytohormones involved in plant growth
and development) and inactivates them. Several mutants of residues
close to FAD have been produced by site-directed mutagenesis. Crystals
of wild-type CKOm (for infiltration) and of D169E variant (with the IP
substrate) have been obtained, the diffraction data have been recorded to 1,8
?. Data processing of CKO-HA-8 (2,4 ?) and D169E variant with IP (2,0 ?) have
been successful. Phasing should be soon achieved.
Key words: Protein, Crystallography, Uridine
monophosphate kinase from E. coli, Cytokinine oxydase
from maize, site-directed mutagenesis.
| 

