|

N° d'ordre : 24/2007-E/CH
République Algérienne Démocratique et
Populaire
Ministère de l'Enseignement Supérieur et de la
Recherche Scientifique
Université des
Sciences et de la Technologie
Houari Boumediene
Faculté de
Chimie.

THESE
Présentée pour l'obtention du diplôme
de
DOCTORAT d'ETAT
En : CHIMIE
Spécialité: Physico-chimie quantique
Par : Mlle TURKI Naïma
sujet
ETUDE DES INTERACTIONS INTERMOLECULAIRES
DANS LES AGREGATS
IONIQUES ET NEUTRES
PAR DIFFERENTES METHODES THEORIQUES
Soutenue le 30 décembre 2007 devant la
Commission d'Examen :
|
Mr A. AIT KACI
|
Professeur
|
U.S.T.H.B
|
Président
|
|
Mlle O. OUAMERALI
|
Professeur
|
U.S.T.H.B
|
Directeur de thèse
|
|
Mr R. MOSZYNSKI
|
Professeur
|
U. Varsovie
|
Examinateur
|
|
Mme S. DJENNANE
|
Maître de Conférence
|
U.S.T.H.B
|
Examinateur
|
|
Mr A. RAHMOUNI
|
Professeur
|
U. Saïda
|
Examinateur
|

Dédicace :
_ A mes parents
- A mes frères et soeurs
- A tous ceux que j'aime
Avant Propos :
- Cette thèse de Doctorat d'Etat a été
préparée au laboratoire de Physico-Chimie Théorique et
Chimie Informatique de la faculté de Chimie de l'Université des
sciences et de la Technologie Houari Boumedienne (U.S.T.H.B), sous la direction
avisée de Mademoiselle Ourida Ouamerali, Professeur à l'U.S.T.H.B
Directrice du Laboratoire, Responsable de l'équipe deux.
Je lui suis reconnaissante d'avoir suivi et orienter mes
travaux de façon efficace et pour ses conseils sans faille. Elle m'a
toujours aidé à trouver des solutions aux multiples obstacles
auxquels je me suis heurtée et ce dans tous les domaines. Qu'elle trouve
ici l'expression de ma profonde et respectueuse gratitude.
- Une partie de ce travail a été
réalisée au laboratoire de Chimie Théorique à
l'Institut de Chimie de Strasbourg, sous la direction de Madame Elise
Kochanski, Directeur de recherches au C.N.R.S.
Pour sa collaboration, son soutien constant et l'aide
précieuse qu'elle m'a apportée continuellement. Je lui exprime ma
profonde reconnaissance.
- Nous remercions très vivement Monsieur Ahmed Aït
Kaci, Professeur à l'U.S.T.H.B, Directeur du Laboratoire de
Thermodynamique et de Modélisation Moléculaire, qui nous a fait
l'honneur d'accepter la présidence de notre Jury de thèse.
- Nous exprimons notre très vive reconnaissance
à Monsieur le Professeur Robert Moszynski, Directeur du Laboratoire de
Chimie Quantique de la Faculté de chimie de l'université de
Varsovie, pour l'honneur qu'il nous a fait en acceptant de venir à
Alger, participer à notre jury et juger notre travail.
Le Professeur R. Moszynski, m'a aidé à
réactualiser et à finaliser le travail de cette thèse en
m'invitant dans son laboratoire, et ce dans les meilleures conditions. Je le
remercie infiniment.
- Madame Sema Djennane, Maître de Conférence
à l'U.S.T.H.B, m'honore de sa présence en étant membre du
jury. Je la remercie très respectueusement.
- J'adresse mes chaleureux remerciements à Monsieur Ali
Rahmouni Professeur à l'université de Saïda, pour l'honneur
qu'il me fait en acceptant de participer au jury de cette thèse et
d'examiner notre travail.
- Je tiens à remercier mes collègues du
laboratoire, de l'équipe deux, Monsieur Maamar REKHIS, Yahia MOUSSAOUI,
Djaffar KHEFFACHE, Amar SAAL, Sofiane MOUSSI et Mahfoud BENALI, pour leurs
précieuses aides.
- Je tiens aussi à remercier tous les membres, des
laboratoires de Strasbourg 1 et de Varsovie pour leurs accueils chaleureux.
- Je voudrais également remercier tout le personnel de la
Faculté de Chimie.
- A mes parents qui n'ont jamais cessé de me soutenir par
leurs encouragements continuels, qu'ils trouvent ici plus que de simples
remerciements.
- Je ne saurais jamais assez remercier mes frères et
soeurs pour leur aide et leur compréhension.
SOMMAIRE
Introduction Générale
8
Chapitre I: Méthodes
Théoriques utilisées pour le
calcul de l'énergie d'interaction
13
I.A/ Le Modèle de la supermolécule au
niveau SCF : Calculs ab intio 13
I.B/ La Méthode Hartree Fock 14
I.B. 1/ Déterminant de Slater 15
I.B.2/ Energie de Hartree-Fock associée à un
déterminant de Slater 16
I.B.3/ L'approximation LCAO 17
I.B.4/ Le Procédé SCF 18
I.B.5/ La limite Hartree-Fock 19
I.C/ Les méthodes Post Hartree-Fock 19
I.C. 1/ L'énergie de corrélation 19
I.C.2/ La Théorie de la perturbation Möller-Plesset
jusqu'à l'ordre n (MPn) 20 I.C.3/ La Méthode coupled-cluster avec
excitations simple, double et triple 21 excitation approchée, CCSD
(T)
I.C.4/ La Méthode multiconfigurationnelle SCF (MCSCF)
23
I.D/ Théorie de la perturbation de la
symétrie adaptée (SAPT) 24
I.E/ Méthode de la fonctionnelle de la
densité (DFT) 28
I.E.1/ Le Formalisme de Hohenberg-Kohn 28
I.E.2/ Le Formalisme de Kohn-Sham 30
I.E.3/ Forme analytique de EXC 32
I.E.4/ Fonctionnelles de la densité 32
I .E.4.a/ Fonctionnelles traditionnelles 34
I.E.4.b/ Fonctionnelles hybrides 34
I.F/ Méthode théorique du calcul de la
polarisabilité du radical OH 35
I.F.1/Polarisabilités multipolaires 35
I.F. 1 .a/Coefficients d'interaction à longue
portée 36
I.F.1.b/Polarisabilités statiques 39
I .F.1 .c/Application à OH, considérations
symétriques 41
I.G/ Erreur de superposition de bases (BSSE)
42
I.H/ Base utilisée 43
Chapitre II: Structures
moléculaires OH- (H2O)n
étudiées 44
II.A/ Géométries des Configurations de
OH- (H2O)n 44
II.A.1/ Structures de OH-(H2O)2 correspondant à
la série R 44
II.A.2/ Structure de OH-(H2O)2 correspondant à
la configuration X2 45
II.A.3/ Structures de OH-(H2O)2 correspondant
respectivement aux séries 45 T et F
Chapitre III: Interprétation
des résultats obtenus
pour les systèmes
OH-(H2O)n 50
III.A/ Résultats obtenus avec les configurations
OH-(H2O)2 de la série
R et de la configuration X2 50
III.A. 1/ Variation de l'énergie à trois corps en
fonction de l'angle kl 50
III.A.2/ Variation des composantes de l'effet à trois
corps en fonction de
l'angle kl 53
III .A.3 / Comparaison des effets à trois corps aux
résultats CCSD(T) 55
III.A.4/ Variation de l'énergie à deux corps 55
III.B/ Résultats obtenus avec les configurations
OH-(H2O)2 du système
OH-(H2O)4 55
III.B. 1/ Variation de l'énergie à trois corps en
fonction de l'angle kl 56
III.B.2/ Variation des composantes de l'effet à trois
corps en fonction de
l'angle kl 57
III .B.3/ Comparaison des effets à trois corps aux
résultats CCSD(T) 60
III.B.4/ Variation de l'énergie à deux corps 60
III.C/ Résultats obtenus avec les configurations
OH-(H2O)2 du système
OH-(H2O)10 61
III.C. 1/ Variation de l'énergie à trois corps en
fonction de l'angle kl 61
III.C.2/ Variation des composantes de l'effet à trois
corps en fonction de l'angle kl 65
III .C.3 / Comparaison des effets à trois corps aux
résultats CCSD(T) 66
III.C.4/ Variation de l'énergie à deux corps 66
III.D/ Résultats obtenus avec la méthode
DFT 66
III .D.1/ Les effets à trois corps DFT 67
III .D.2/ Comparaison des énergies de la première
itération DFT au terme
Heitler-London 69
III.D.3/ Les énergies de délocalisation DFT 71
III.E/ Conclusion 74
III..F/ Méthodes de
génération et d'optimisation des
géométries
des systèmes moléculaires 75
III .F.1/ Méthodes théoriques de
génération des configurations 75
III .F.2/ Méthodes expérimentales de
génération des configurations 75
III. F.3/ Méthodes d'optimisation des configurations 76
Chapitre IV : Interprétation
des résultats pour le
système RbOH 77
IV.A/ Interaction étudiée dans le
Système RbOH 77
IV.B/ Comparaison entre les polarisabilités de OH
calculées avec
les deux Bases 79
IV.C/ Comparaison entre les polarisabilités de OH
calculées et celles
trouvées dans la bibliographie 82
IV.D/ Conclusion 82
Conclusion générale 84
Bibliographie 86
Articles Internationaux 91
Perspectives 91
Introduction générale
L'idée de l'énergie d'interaction est basée
sur la théorie de Born-Oppenheimer (noyaux figés). Nous
définissons l'énergie d'interaction de la configuration R [1]:
E = E -[E +E +E + ]
? R ? R A R ? R ? R
Eest l'énergie électronique du système total
et E , E , ... sont les
? R A R B R
énergies électroniques des sous systèmes
interagissant.
Dans notre étude des interactions
intermoléculaires, nous nous sommes intéressés à
deux types de systèmes moléculaires, les agrégats ioniques
OH-(H2O)n et les molécules RbOH à basse
température ou froides.
Les agrégats moléculaires sont importants, ils
participent à la compréhension de la transition des
environnements de la phase gaz à la phase condensée. L'anion
hydroxyde OH- est trouvé communément dans la nature,
dans la phase gazeuse (en particulier dans l'atmosphère) ou dans les
phases liquides et condensées. L'ion hydroxyde est un excellent
accepteur de liaison hydrogène et est souvent sous forme hydraté.
Les agrégats aqueux de l'anion OH- sont d'une utilité
particulière à cause de leur participation dans les clusters et
les phases condensées de la chimie acide-base. Dans sons ouvrage
'Experimental researches in electricty', M. Faraday, est le premier en
1938[127] à suggérer la présence de particules
conductrices pour expliquer les propriétés électriques
particulières des solutions aqueuses, acides ou basiques. En 1907, H.
Goldschmidt et al[128] parle d'une espèce (H2O, H), qu'il
considère comme le vecteur de la catalyse acide. Des ions
hydratés négatives ont été trouvés dans
l'atmosphère et on a soutenu que les ions négatifs de la chimie
contrôle la conductivité de l'atmosphère. La solvatation
des ions constitue un sujet largement étudié avec les
méthodes expérimentales et théoriques. Les premiers
renseignements structuraux sont obtenus par la spectroscopie IR[110]
. L'attention du monde scientifique pour ces agrégats s'est accrue
quand, des expériences atmosphériques ont montré qu'ils
étaient les ions dominants dans la région D de
l'ionosphère. La présence des agrégats de l'ion hydroxyde
postulée dans le milieu interstellaire démontre que
l'intérêt des études sur ces clusters dépasse les
frontières de notre système solaire Des expériences
atmosphériques ont montré qu'ils étaient les ions
dominants dans la région D de l 'ionosphère[129].
Les agrégats de l'eau autour d'un ion positif ou
négatif est un phénomène important pour une grande
variété de domaines. L'étude des agrégats
OH-(H2O)n est d'intérêt biologique, physique et
chimique.
La compréhension profonde du processus de formation
d'agrégats ioniques s'avère
d'une grande valeur. La
géométrie des agrégats OH-(H2O)n en fonction de
n, est-elle
linéaire, plane, pyramidale, les chercheurs se sont
longtemps posés cette question. Les
géométries des systèmes OH-(H2O)
n trouvées dans la bibliographie varient en fonction du
procédé de génération et la méthode
d'optimisation. En général ces structures ont la forme
chaîne ou cyclique et dans les deux cas elles peuvent appartenir ou pas
à un groupe de symétrie. Voici quelques exemples de structures
OH-(H2O)n trouvées dans la
littérature, dans l'ordre de n croissant:
- n=1, Différentes configurations sont trouvées
dans la littérature[65,75,91].
- n=2, les configurations les plus utilisées dans les
catalogues de chimie ont une structure chaîne, de symétrie
C2V ou Cs [[65-75,76], comme celles que nous avons
utilisées. Pour la structure OH-(H2O)2
notée aussi H5O3 -, cyclique, il y a par exemple, celle appelée
« OH- double proton acceptor»[75],
OH- accepteur double proton.
- n=3, cette structure bien que nous l'avons pas
étudié, nous avons choisi de la citer, car on la trouve dans
plusieurs articles, qui soutiennent tous que cette structure existe dans
l'atmosphère. En effet plusieurs configurations H7O4 - ont
été optimisées, les géométries obtenues pour
les structures chaînes sont de symétrie pyramidale
(C3)[65], la plus probable, où toutes les molécules
réagissent comme donneuses de proton à l'ion hydroxyde et
à l'autre molécule d'eau comme accepteuses de proton, de
symétrie, chaîne branchée (C3V)[75], qui
s'approche de la pyramidal (C3)[93], chaîne plane
(C2h)[75], ou de symétrie C2 . La structure cyclique H7O4 -,
la plus produite par optimisation est de symétrie
(Cs)[65,75], ressemble à une chaise avec OH- comme
dossier de la chaise et accepteur d'un double proton de deux molécules
d'eau.
- n supérieur à 3 molécules d'eau, les
configurations OH-(H2O)4 sont peut présentes dans la
bibliographie. Dans les articles récents, références
[91-93], en particulier la référence [93], l'auteur essaie de
montrer que OH-(H2O)n dans la phase gazeuse, à la
symétrie Cn, pour n = 2 à 6, qu'il justifie par le
fait avec les nombreux travaux qui argumentent, que l'ion OH- se lie
fortement à plus de trois molécules d'eau[65,75,76,78,79,83
et 85]
.
Les molécules RbOH à l'état froid ont un
rôle primordial en physique et en biologie. La connaissance des
énergies d'interaction à grande distance avec le plus de
précision, nous permettrait d'avoir les sections efficaces de collision
ou observables que nous comparons à des résultat
expérimentaux. L'étude des interactions intermoléculaires
à très basses températures pour ces systèmes est
récente.
Pour comprendre les liens intermoléculaires des
agrégats, nous utilisons plusieurs méthodes de calcul des
différentes composantes de l'énergie d'interaction des
systèmes étudiés, dans notre cas : OH-(H2O)2,
OH-(H2O)3, .... OH-(H2O)n, RbOH. L'énergie
intermoléculaire est la somme des contributions des paires, des trois
corps,... jusqu'à n corps :
E inter = E (paire) + E (3 corps) + E(4 corps) + ... + E (n
corps)
Dans le cas des agrégats ioniques OH-(H2O)n,
nous nous sommes intéressés à l'énergie
d'interaction des paires (bi) et à l'énergie d'interaction
à trois corps non additive (na), celle-ci représente un faible
pourcentage par rapport à l'énergie à deux corps mais
augmente de façon importante avec le nombre de molécules d'eau
entourant l'ion hydroxyde dans une même couche.
Les effets non additifs affecte le nombre de coordination,
puisqu'on peut trouver quatre molécules d'eau dans la première
couche de solvatation quand les contributions non additives sont
négligées et seulement trois s'ils sont prises en compte[2], [3],
[4].
Cette estimation du nombre de coordination a été
trouvée aussi dans d'autres systèmes ioniques et est bien connu
maintenant. Cependant la détermination du nombre de coordination dans
les systèmes qui favorisent le transfert de proton reste une question
difficile à résoudre du fait qu'il y a un mélange de
structures variées due à cet effet
[5,6]
.
Différents calculs de l'énergie à trois
corps ont été réalisés dans nos travaux, nous les
avons classés en trois catégories, deux de type ab initio et la
DFT. Dans la première catégorie, utilisant l'approche
supermolécule, il y a la méthode Hartree-Fock, et les
méthodes post Hrtree-Fock, la théorie de la perturbation
Möller-Plesset, calculs jusqu'à l'ordre n (MPn) et coupled-cluster
incluant les excitations simple, double et triple approchée
[CCSD(T)].
La deuxième catégorie est la méthode
symmetry-adapted perturbation theory (SAPT)[7],[8], qui
consiste à perturber chaque monomère du système. Elle a
été développée pour les interactions à trois
corps, elle donne une décomposition de l'énergie non additive en
termes distincts physiquement. Elle a été récemment
généralisée pour l'utilisation des orbitales DFT
[9].
La théorie de la fonctionnelle de la densité
(DFT) [9] a été utilisée pour évaluer
les effets non additifs, les calculs du terme à trois corps ont
été faits avec deux fonctionnelles B3LYP et B3PW91, les
résultats obtenus représentent la troisième
catégorie.
Dans tous nos calculs les molécules d'eau sont
localisées dans la première ou la deuxième couche de
solvatation.
Cette thèse est composée de quatre chapitres.
Dans la première partie, nous décrivons, les différentes
méthodes théoriques de calcul de l'énergie d'interaction
à deux et trois corps: la méthode Hartree-Fock (H F), les
méthodes post Hartree-Fock et la théorie de la fonctionnelle de
densité. La méthode Möller-Plesset [10] ,
améliore le procédé Hartree-Fock en prenant en compte les
effets de la corrélation d'électrons en utilisant la
théorie de la perturbation, généralement au second (MP2),
troisième (M P3) et quatrième ordre (M P4). La méthode
coupled-cluster ou cluster couplé (CC) a apparu comme la plus
précise pour décrire le problème de corrélation
électronique et ainsi l'outil de structure électronique le mieux
développé, elle convient idéalement aux systèmes
étendus. Plusieurs formulations théoriques et des avances
informatiques ont mené à l'état présent de la
théorie CC [1] . Le rapport de la théorie CC avec
d'autres théories électroniques a aussi été
étudié en profondeur. Particulièrement la relation avec la
théorie de la perturbation a été une source importante
d'informations. Cependant beaucoup d'aspects critiques de la théorie
sont toujours sous la formulation. Dans les décennies récentes,
les développements les plus importants ont eu lieu dans les secteurs des
variantes de multi référence de la théorie CC. Ces
versions de la théorie ont favorisé l'application des
méthodes CC aux situations quasi-dégénérées
importantes et exigeantes, à savoir ionisées, excitées,
les états d'électrons attachés ou
les surfaces d'énergie potentielle. Il y a plusieurs
versions de la multi référence de la théorie CC,
fournissant des racines multiples, aussi bien que la racine simple
d'intérêt, qui ont des mérites et des
démérites selon le secteur d'application. Cependant, plusieurs
problèmes théoriques importants restent encore à
résoudre et il y a les portées du progrès et de la
recherche. Les développements informatiques, l'introduction d'effets
relativistes et l'incorporation de fonctions explicitement
corrélées constituent aussi les secteurs stimulants de la
recherche, particulièrement dans le contexte de la version de multi
référence de la théorie. La formulation CC
dépendante du temps a aussi été une direction importante
de la recherche dans ce secteur.
Une formule à n corps [7,12-14] a
été développée par la méthode de la
perturbation de la symétrie adaptée (symmetry-adapted
perturbation theory (SAPT)) pour les interactions intermoléculaires
[15-18]. Dans cette approche, toutes les contributions physiques
importantes du potentiel, telles que électrostatiques, échange,
induction et dispersion sont déterminées et programmées
séparément. En ajoutant un développement de perturbation
dans l'interaction intermoléculaire comme c'est dans la
corrélation électronique intramoléculaire, c'est possible
de faire la somme des contributions de corrélation aux différents
effets physiques.
Puisque les diverses contributions de l'énergie
d'interaction montrent une dépendance différente à la
distance intermoléculaire R, elles peuvent être adaptées
séparément, avec des paramètres ajustables et physiquement
interprétables. Dans plusieurs cas, un excellent accord est atteint en
comparaison avec les potentiels semi-empiriques exacts de certains
systèmes.
Le formalisme de la DFT repose sur le fait que
l'énergie d'un système est une fonction de sa seule
densité électronique, selon le théorème de
Hohenberg et Kohn [19]. Il est difficile de calculer
l'énergie de corrélation. Il y a deux approches, l'une et l'autre
suivent les méthodes de type interaction de configurations (CI, MC, SCF,
CC, etc...) ou vont dans la direction des fonctions corrélées
explicitement. Les premières constituent un obstacle pour prendre en
considération les nombreuses configurations excitées, les
secondes très fastidieuses et le temps de calcul d'intégrales.
Dans les deux cas, on connaît l'Hamiltonien et on bataille pour une
fonction d'onde adéquate. On a une troisième direction, la DFT
qui ne prend pas en considération les configurations, excepté une
et n'a pas le problème d'embouteillage des intégrales difficiles,
par contre nous avons un genre de fonction d'onde dans la forme d'un seul
déterminant de Slater mais nous avons un sérieux problème
à définir l'Hamiltonien propre [1].
L'objectif ultime de la méthode DFT est de calculer
l'énergie totale du système et la distribution de la
densité d'électron de l'état fondamental sans
utilisé la fonction d'onde du système. Ce qui est très
important car en plus du fait que les calculs DFT prennent en compte la
corrélation d'électrons, ils ne sont pas chers, leur coût
est comparable à celui de la méthode HF, un ordinateur de
même puissance, nous permet d'explorer beaucoup plus de molécules
qu'avec les autres méthodes post Hartree-Fock (méthodes de
corrélation).
Dans la méthode Kohn et Sham [10], qui en
permet une exploitation efficace, l'énergie cinétique et
l'énergie d'interaction coulombienne des électrons entre eux et
avec les noyaux sont calculées exactement. Le terme rassemblant les
effets d'échange et de corrélation, dont la forme exacte est
inconnue, est évalué en appliquant des approximations dont va
dépendre la précision des résultats. Diverses
fonctionnelles
pour l'évaluation de l'énergie
d'échange-corrélation ont été
suggérées par différents auteurs [20].
Kohn et Sham [10] ont montré que les
équations monoélectroniques qui permettent de décrire le
système sont des équations de type HF où le potentiel
effectif inclut à la fois l'échange et la corrélation. La
densité électronique utilisée pour calculer
l'énergie totale du système est obtenue à partir des
orbitales monoélectroniques qui sont solutions de ces
équations.
La méthode MCSCF a été utilisée pour
le calcul de la polarisabilité de la molécule OH, celle-ci est
indispensable pour obtenir le potentiel d'interaction.
Nous exposons les différentes configurations de
OH-(H2O)n étudiées dans le deuxième chapitre,
nous citons les diverses méthodes d'optimisation de ces structures.
Les résultats obtenus pour les différentes
configurations de ces structures sont interprétés dans le
troisième chapitre.
Dans la quatrième partie, nous nous intéressons
à l'interaction entre Rb et OH pour des molécules froides. On
décélère OH, c'est à dire on baisse son
énergie cinétique, ensuite on le bombarde avec un alcalin Rb
(Rubidium) pour le décélérer encore plus, OH à
l'approche de Rb perd sa dégénérescence, il y'a
levé de dégénérescence. L'interprétation des
résultats obtenus pour les molécules RbOH sera
présentée dans ce chapitre.
Chapitre I
Méthodes Théoriques utilisées pour
le calcul de l'énergie d'interaction
La nature et l'importance des interactions
intermoléculaires dans les agrégats ioniques
OH-(H2O)n ont été étudiées
par les méthodes de la supermolécule HartreeFock et post
Hartree-Fock, Möller-Plesset, théorie de la perturbation (MPPT),
à l'ordre deux (MP2), trois (MP3) et quatre avec excitations simple,
double et quadruple (M P4SDQ), clusters couplés, simple double
excitations [CCSD(T)] avec une inclusion approchée de la triple
excitation, par la méthode de la perturbation, symmetry-adapted
perturbation theory (SAPT) et la théorie de la fonctionnelle de la
densité (DFT).
Les différentes méthodes utilisées pour
le calcul de l'énergie d'interaction donnent des informations
complémentaires sur les contributions énergétiques non
additives.
I.A/ Le Modèle de la supermolécule au
niveau SCF : Calculs ab intio:
Dans l'approche supermolécule (le système total
étant considéré comme une supermolécule),
l'énergie d'interaction est calculée comme dans l'équation
(1), utilisant n'importe quelle méthode fiable du calcul de
l'énergie électronique. Dans le cadre de la supermolécule,
des calculs ab initio ont été faits aux niveaux SCF, MP2, M P3, M
P4SDQ et CCSD(T).
Dans le modèle de la supermolécule, les
énergies d'interaction, à deux et trois corps non additive, sont
données respectivement, par :
+(1)
M (C
- (3)
st t (
= ---+++(2)
Où l'exposant SM est l'abréviation pour la
méthode de la supermolécule. SM est remplacé par HF pour
la méthode H artree- Fock, MPn pour la théorie de la perturbation
Möller-Plesset à l'ordre n et par CCSD(T) pour les calculs
coupled-cluster simple, double excitations et la triple excitation
approchée.
Nous avons calculé les énergies de la
première itération pour tous les systèmes AB,
AC, BC et
ABC, ce qui nous permet d'obtenir l'énergie d'interaction à trois
corps à la
première itération : . La différence
entre l'énergie à trois corps HF non
s it a
additive, obtenue à la convergence, et est notée
par [22].
F ? n ? st t a HF ? na ?
Cette quantité communément appelée
«l'énergie de déformation Hartree-Fock[23,24]
» ou énergie de délocalisation, nous donne une idée
sur la manière dont se déforme la distribution de charge des
monomères isolés (délocalisée sur toute la
supermolécule) pendant le processus SCF[25].
I.B/ La Méthode Hartree Fock [28]:
L'équation de Schr~dinger non relativiste et
indépendante du temps pour des systèmes polyélectroniques
est résolue de façon approchée via les approximations
suivantes :
L'approximation de Born-Oppenheimer considère les
noyaux figés par rapport aux mouvements des électrons
[9] . Nous pouvons négliger l'énergie cinétique
des noyaux et la répulsion entre noyaux est considéré
comme une constante. Dans ce cas l'opérateur hamiltonien de
l'énergie électronique d'un système à n
électrons et N noyaux, est :

Soit et E la fonction d'onde et l'énergie décrivant
ce système. L'équation de
Schr~dinger s'écrit :
Par la suite, on omettera de mettre l'indice e pour
électronique, dans toutes les équations développées
cette approximation est prise en compte.
La plus importante approximation, est celle de hartree-Fock, qui
consiste à ramener le
problème à n
éléctrons, comliqué, à un problème à
un életrcon où la
répulsion
électron-électron est traîté
comme une moyenne. La fonction d'onde approchée
donnée par
Hartree-Fock en 1928 [32] où la fonction d'onde du
système
est représentée par le produit des fonctions
d'onde monoélectroniques
( )
r
qui ne dépendent chacune que des coordonnées d'un
seul électron est :
r
(4)
Ø ( ) ( r
Dans la théorie de Pauli [33], pour mieux
décrire l'électron, il est nécessaire de
spécifier
son spin. Ceci est possible en introduisant deux fonctions
de spin ,
s e s
correspondant respectivement au spin haut et spin bas. La
fonction d'onde monoélectronqiue, dite fonction spinorbitale, est alors
écrite comme le produit de deux fonctions indépendantes : la
fonction d'espace et la fonction de spin :
)
(5)
? ( r = Ö ( x )
où ri désigne les variables d'espace et de spin, x
la variable d'espace et s la variable
de spin. ne peut prendre que deux
vaeurs . Ces deux
?
J ? ? J 1
e ?
fonctions de spin sont orthonormales :
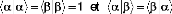
I.B. 1/ Déterminant de Slater:
Compte tenu du principe d'exclusion de Pauli,
l'équation (4) ne reproduit pas l'antisymétrie du système
lorsqu'on permute deux électrons, c'est à dire le fait que la
fonction doit changer de signe si on permute deux électrons, ce qui
revient au principe d'exclusion de Pauli, qui énonce, qu'une spin
orbitale ne peut être occuper que par un seul électron. C'est
ainsi que Slater [34] a proposé une nouvelle fonction qui est
donnée par la somme antisymétrique de tous les produits
d'orbitales possibles :
(6)
avec
|
et où
?
|
, est l'opérateur d' antisymétrie pour les n
électrons, son effet est de
|
construire le déterminant de Slater au delà du
produit d'orbitales sur qui il opère. Les
sont les spinorbitales, produits des orbitales spatiales et de
la fonction spin à un électron ou . P est un opérateur de
permutation qui agit sur la suite 1,2,....,n en échangeant deux
particules à la fois. est un facteur qui vaut +1 ou -1 selon que la
permutation est paire ou impaire et où la sommation sur p
s'étend à toutes les permutations.
La fonction d'onde du système peut s'écrire sous la
forme d'un déterminant de Slater :
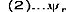
(7)
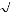
!
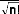
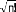
où est le facteur de normalisation et n le nombre
d'électrons.

Pour n'importe quel système moléculaire, il y' a un
nombre infini de fonctions d'onde de la forme (4), mais la fonction d'onde
Hartree-Fock est celle pour qui les orbitales ont été
variées pour produire l'énergie totale la plus basse :
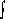
(8)
I.B.2/ Energie de Hartree-Fock associée à un
déterminant de Slater:
L'hamiltonien d'un système à n électrons et
N noyaux s'écrit :
(9)
qui est la somme d'un terme monoélectronique et d'un terme
biélectronique. est
)
l'opérateur associé à l'énergie
cinétique de l'électron , l'opérateur associé
à
l'énergie d'attraction électron-noyau et la
répulsion entre l'électron et
1
l'électron :
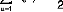
? ? ?
?
? ?
avec 2 opérateur Laplacien
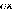
2
2 2 2
? 2
on peut écrire :
(10)
est l'opérateur monoélectronique contenant
l'énergie cinétique de l'électron et
)
la somme des interactions entre cet électron et les
noyaux.
Les résultats des équations Hartree-Fock sont
faciles à obtenir dans le cas où l'expression de l'énergie
dépend des fonctions d'onde d'un seul déterminant. En
remplaçant H et dans (8), on obtient :
(11)
où Ii est l'intégrale
monoélectronique :
(12)
= ? ( d
Jij est l'intégrale biélectronique coulombienne
:
(13)
J Ö )
i
??
et Kij est l'intégrale biélectronique
d'échange :
(14)
? r ? ?
En intrduisant les opérateurs de Coulomb et
d'échange, définis par :
Jà ? ? ? ?
? ? ? ? ( j ?
à K ? ? ? ?
? ? ? ? ?
i
Nous pouvons écrire les équations Hartree-Fock
:
(15)
F Ö ì Ö
( ) ) = (
Où est l'énergie de l'orbitale et [38]
l'opérateur monoélectrique de Fock défini
comme suit :
(16)
Bien que les solutions aux équations Hartree-Fock pour
les systèmes ouverts soient plus difficiles à leurs analogues
à couches fermées car le nombre d'orbitales augmente, les
procédures sont bien établies.
En fait, les méthodes sont maintenant disponibles pour
les solutions des équations Hartree-Fock pour n'importe quel
système dont l'expression de l'énergie implique seulement les
intégrales de Coulomb et d'échange.
I.B.3/ L'approximation LCAO:
Roothaan et Hall considèrent que les orbitales
atomiques situées sur chaque noyau peuvent former une base
approchée. Ces orbitales ne sont pas nécessairement orthogonales
mais sont normées.
Roothaan et Hall [35,36] propose de développer
les fonctions spatiales sur la base des
orbitales atomiques (OA), , c'est à dire les sont des
combinaisons
? ? 1
linéaires d'orbitales atomiques (LCAO en anglais) :
(17)

m est la dimension de la base.
Le problème de la détermination des orbitales
moléculaires se ramène à celui des coefficients
numériques Cij [37] . La méthode de variation qui
consiste à minimiser l'énergie totale, nous permet de les
obtenir.
Si on remplace par (17) dans (15), on a les équations
Hartree-Fock sous forme matricielle, équations de Roothan [35,36]:
(18) d'où :
(19)
m
1 1
Le calcul de l'énergie revient à trouver les
coefficients numériques, ces derniers sont obtenus par le principe
variationnel, de manière à ce que l'énergie totale soit
minimale, c'est-à-dire
I.B.4/ Le Procédé SCF:
Hartree[32] a introduit une méthode pour
construire l'opérateur de Fock et les orbitales grâce à un
procédé itératif, en partant d'un ensemble de fonctions
à l'itération zéro, on détermine un
opérateur initial . La recherche des
{ ) ?
Ö 0
fonctions propres de , notées , permet d'avoir un nouvel
opérateur
)
)
dont on recherche les fonctions . Le processus se
répète ainsi jusqu'à
) )
ce que la différence entre les fonctions et soit
inférieure à , où est
) n )
un paramètre arbitrairement petit, choisi selon la
précision voulu.
L'opérateur de Fock décrit le mouvement d'un
électron dans le champ
)
des noyaux et dans le champ moyen des 2N- 1 autres
électrons. Le procédé itératif consiste à
construire progressivement ce champ moyen. Une fois la convergence atteinte, on
dit que l'on a construit un champ autocohérent, d'où le nom de
méthode Self Consistent Field ou SCF.
I.B.5/ La limite Hartree-Fock:
Différents choix de jeux de bases "basis set" peuvent
donner différentes énergies et fonctions d'onde SCF. Si nous
étendons notre base pour le calcul, en général, nous
pourrons obtenir une énergie plus basse. Si on ajoute des fonctions de
polarisation et qu'on répète le calcul SCF, nous obtiendrons une
énergie encore plus basse. Nous pourrons continuer ainsi mais à
un moment donné l'énergie ne décroît plus ou
décroît très lentement. C'est la valeur de l'énergie
limite qui est la plus basse que nous pouvons atteindre avec le simple
déterminant de fonctions d'onde. Elle est appelée la limite
Hartree-Fock. Les orbitales moléculaires de cette limite sont
appelées les OM Hartree-Fock et le déterminant est appelé
fonction d'onde Hartree-Fock[1].
Nous avons vu que la fonction (7) est un produit de fonctions
monoélectroniques avec une contrainte additionnelle
d'antisymétrie par rapport à l'échange binaire. C'est donc
une fonction approchée dans laquelle le mouvement de l'électron i
est quasi- indépendant de celui des autres électrons. En fait,
cette quasi-indépendance des mouvements est partiellement
limitée. L'antisymétrie de entraîne une corrélation
entre les électrons de même spin, car il y apparaît une
probabilité nulle de rencontrer deux électrons de même spin
proches l'un de l'autre. Par contre, des électrons de spins
opposés restent non corrélés. Ce défaut de
corrélation entraîne donc une surestimation positive de
l'énergie due à ces situations mal décrites par la
fonction HF [28] . Cette constatation a été aussi
faite par Coulson [29] qui a noté en 1960 que "il est
maintenant clair qu'un seul déterminant de Slater doit
inévitablement conduire à une mauvaise énergie". La
différence entre l'énergie calculée par la méthode
de HF et la valeur exacte (non relativiste) est connue sous le nom de
l'énergie de corrélation. Dans
l'approximation non relativiste, on néglige lors du calcul
de l'énergie exacte, la variation des masses en fonction de la vitesse
et le couplage spin-orbite et spin-spin. I.C/ Les méthodes
Post Hartree-Fock :
I.C.1/ L'énergie de corrélation:
L'énergie Hartree-Fock n'est pas aussi basse que
l'énergie réelle du système. La raison mathématique
pour ça est le fait que soit un seul déterminant, ce qui est
restreint. Nous pouvons introduire plus de flexibilité
mathématique en permettant à
de contenir plusieurs déterminants. Une telle
flexibilité nous donne une énergie encore plus basse.
Dans l'approximation Hartree-Fock, nous résolvons les
équations pour le comportement de chaque électron dans le champ
moyen des (n-1) électrons restants. Malheureusement les électrons
réagissent les uns aux autres de façon instantanée via la
loi de Coulomb, c'est à dire les [n(n-1)]/2 mouvements de paires
d'électrons sont corrélés et c'est
précisément cette corrélation d'électrons
qui est définie conventionnellement comme[30] la
différence entre l'énergie exacte et l'énergie limite
Hartree-Fock.:
(21)
E E

Pour cette raison, les calculs de mécanique quantique sont
parfois jugés suivant le pourcentage de l'énergie de
corrélation reproduit.
I.C.2/ La Théorie de la perturbation
Möller-Plesset jusqu'à l'ordre n (MPn):
La méthode de Hartree-Fock permet d'obtenir
l'état du système électronique de la molécule. Un
déterminant de Slater obéit d'une manière convenable
à la statistique de Fermi ce qui signifie que des électrons de
spins différents s'évitent, Cependant, le formalisme HF permet
que des électrons de même spin (état quantique
différent) peuvent être présents simultanément aux
mêmes endroits dans l'espace, ce qui est bien sur inexact. Plusieurs
méthodes dites "post Hartree-Fock" ont essayé de
représenter la corrélation électronique en utilisant des
combinaisons linéaires de déterminants de Slater comprenant des
orbitales occupées et virtuelles. Une des méthodes les plus
simples est la méthode Möller-Plesset MP2. On écrit
l'Hamiltonien comme la somme d'une partie correspondant au déterminant
de Hartree-Fock et d'une perturbation V (corrélation dynamique : les
électrons se repoussent). L'énergie totale sera la somme des deux
énergies associées à ces deux hamiltoniens :
La théorie de la perturbation Möller-Plesset (MP)
est le résultat de l'application des formules de la perturbation
Rayleigh-Schrödinger[39] traditionnels à la
théorie de la structure électronique[10], utilisant la
somme des opérateurs de Fock comme Hamiltonien d'ordre 0.
L'opérateur de Fock habituel pour les systèmes
à couches fermées est :
(22)
où i et l se réfèrent respectivement aux
électrons et aux orbitales occupées. est la
partie monoélectronique de l'Hamiltonien, sont les
opérateurs Coulomb et
à
échange. Les équations Möller-Plesset sont
:
(23)
(24)
Si la perturbation est suffisamment petite, alors
l'énergie et la fonction d'onde peuvent être écrites sous
la forme de séries en puissance :
La substitution de ces séries dans l'équation de
Schr~dinger donne une nouvelle équation:
(25)

La solution à l'ordre zéro de cette
équation donne l'énergie qui est la somme des énergies des
orbitales des électrons. La solution au premier ordre (i=1) corrige
cette énergie et donne l'énergie Hartree-Fock et la fonction
d'onde. Pour aller au-delà du traitement Hartree-Fock, il faut aller
au-delà du premier ordre. Les calculs MollerPlesset, second (MP2),
troisième (M P3) et quatrième ordre (MP4) sont les niveaux
standards utilisés pour les petits systèmes et sont
implémentés dans beaucoup de codes de la chimie quantique. Au
plus haut ordre, les calculs M P existent dans quelques codes et sont rarement
utilisés.
Soit diagonal, l'expression générale pour
l'énergie au second ordre dans la théorie
de la perturbation Rayleigh-Schrödinger est donnée
par :
E(2) = ? Ø 0
? Ø H | Ø
0 0



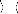

(26)
où n parcourt toutes les fonctions d'état de
configuration (CSF) excepté la référence CSF
[5]. Pour les équations Rayleigh-Schrödinger de la
théorie de la perturbation au plus haut ordre, voir la
référence [10].
I.C.3/ Méthode coupled-cluster avec excitations simple,
double et triple approchée, CCSD(T)[1,126] :
La méthode cluster-couplé (CC), est aujourd'hui
largement reconnue comme l'outil le plus puissant pour une description
précise de la corrélation d'électrons dans les atomes et
les molécules. La fonction d'onde de la méthode CC, donnée
par l'exponentiel Ansatz[40], dépend de paramètres
ajustables (cluster amplitudes). Elle commence par la méthode des
orbitales moléculaires Hartree-Fock et ajoute un terme correcteur pour
prendre en compte la corrélation d'électrons. Certains calculs
très précis pour des molécules de taille petites et
moyennes utilisent cette méthode.
Au début, on introduit un spécial
déterminant de Slater [1], déterminant de
référence
(appelé vacuum state, ça peut
être le déterminant Hartree-Fock) et nous écrivons

que la fonction d'onde exacte de l'état fondamental comme
une exponentiel ansatz [41,
42, 43]:
(27)
p ( ) ?

où est l'opérateur d'onde et lui même est un
opérateur de cluster[44,45,46] ou
opérateur d'excitation, quand il agit sur , il produit une
combinaison linéaire de
déterminants de Slater excités. Dans la
méthode CC, la normalisation intermédiaire de la fonction est
utilisée:
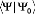
L'opérateur cluster s'écrit comme la somme
d'opérateurs d'excitations [47, 48, 49] :
(28)
= +
T T T
où T1 est l'opérateur de toutes les simples
excitations, T2 est l'opérateur de toutes les doubles excitations, en
général l désigne le rang de l'excitation.
(29)
(30)

Les symboles a,b,...indiquent les spinorbitales occupées
dans et r, s,... celles
inoccupées.
Les t représentent les amplitudes ou les nombres dont la
détermination est le but de la méthode CC.
La fonction d'onde exacte satisfait l'équation de
Schr~dinger:
( T Ö
est l'Hamiltonien du système, et E, la fonction d'onde et
l'énergie de l'état de plus basse énergie.
L'équation de Schr~dinger est :
(31)
( = e
?
On peut écrire :
(32)
( T H p ( ?
Après transformation, nous obtenons :
L'opérateur, le conjugué de est :
? T †
T
e
Qui n'est pas la valeur principale de l'Hamiltonien. La
méthode CC n'est pas
†
variationnelle, si nous multiplions à gauche par , on
obtient :
T
qui a un caractère variationnel.
I.C.3.a/ Les différents types de méthodes
Coupled-cluster (CC):
Les abréviations pour les différents types de
méthodes CC sont en général :
S - pour les simples excitations
D - pour les doubles
excitations
T - pour les triples excitations
Q - pour les quadruples excitations
En CCSD(T), on a :

Le T dans CCSD(T) est entre parenthèses, pour indiquer que
la triple excitation est approchée.
I.C.4/ La Méthode multiconfigurationnelle SCF
(MCSCF) :
Les calculs multi référence interaction de
configuration (MRCI) nécessitant beaucoup de temps de calcul. Nous avons
utilisé la méthode multi configurationnelle (MCSCF) pour
l'étude du système RbOH.
Comme dans la méthode d'interaction de configurations
(IC)[64], dans la méthode MCSCF, le mouvement
électronique est corrélé en décrivant l'état
fondamental du système par une combinaison linéaire de fonctions
, qui correspondent à diverses configurations c'est-à-dire
plusieurs déterminants de Slater:

Les deux méthodes prennent comme point de départ
les solutions HF. Les
déterminants sont construits à partir
des fonctions propres de l'opérateur
Ø
HF. Soit un déterminant de Slater construit à
partir de spinorbitales, on peut
construire d'autres déterminants, monoexcité,
biexcité, tri... :
Ør Ø Ø
rs rst
Une combinaison linéaire de tous ces déterminants
constitue une expression approchée de la fonction d'onde du
système :
Dans la méthode d'interaction de configuration (IC),
lorsque les coefficients sont
déterminés par le principe variationnel, les
coefficients des orbitales moléculaires de l'approximations LCAO, dans
l'Eq. (17) du paragraphe I.B.3, sont gardés fixes. Par contre dans le
calcul multiconfigurationnelle SCF (MCSCF), les deux calculs variationnels sont
faits simultanément, c'est à dire nous optimisons les
coefficients des OM des orbitales qui sont utilisées pour construire les
déterminants en même temps que les coefficients des
configurations, de façon à minimiser l'énergie E du
système selon le procédé SCF varaitionnel. Dans ce cas le
calcul est plus complexe mais le nombre de déterminants utilisés
en MCSCF est généralement plus petit que dans un calcul d'IC.
Les deux méthodes IC et MCSCF sont des techniques de
calcul post-HF très utilisées pour prendre en compte en
considération la corrélation électronique [28]
. Cependant, ces méthodes présentent quelques
inconvénients, coût en temps de calcul élevé et
capacité de stockage énorme. Les calculs MCSCF usuels sont
souvent limités à un certain nombre de configurations
biexcitées, d'énergies les plus basses.
I.D/ Théorie de la perturbation de la
symétrie adaptée (SAPT):
La Théorie de la perturbation de la symétrie
adaptée donne un important aperçu concernant la nature des forces
intermoléculaires, puisqu'elle donne les énergies d'interaction
sous forme d'une somme de contributions bien définies et physiquement
significatives: électrostatique, échange, induction et
dispersion. Ceci nous fourni des informations sur les interactions dues aux
contributions attractives et répulsives. Nous avons utilisé pour
cette méthode, le formalisme développé récemment
pour les interactions à trois corps non additives [7,8].
L'énergie totale d'interaction pour un trimer peut
être diviser comme la somme de contributions de deux corps (additive) et
trois corps (non additive) [59]:
où désigne, en général, la
contribution de K corps dans l'énergie
? K
d'interaction à M corps.
Si on considère le trimer ABC, il est décrit par
l' Hamiltonien H0 = HA+ HB + HC, où HX, X= A, B, ou C, désigne l'
Hamiltonien du monomère X. La fonction d'onde et l'énergie E0,
correspondant à l'état fondamental, sont exprimées en
fonction des fonctions d'onde et des énergies des monomères non
perturbés :
,
A B C B
Ø Ø E E +
Une fois le système perturbé, l' Hamiltonien du
trimer s'écrit :
H(A,B,C) = H0 (A,B,C) + V(A,B,C) (37)
Où
l'opérateur de perturbation est donné par : V
(38)
L'opérateur VXY rassemble toutes les
interactions Coulombienne entre électrons et
noyaux des
monomères X et Y et les paramètres ont en général
des valeurs
ä t
physiques égales à l'unité afin de faciliter
le calcul[7]. L'énergie d'interaction du trimer peut
être écrite comme:
(39)
E ä å ÷
( )


Nous considérons que la fonction d'onde du trimer
satisfait la condition de
normalisation intermédiaire :
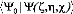
Cette fonction est solution de l'équation de Schr~dinger
paramétrée :
(40)
( ) (
H E Ø ä
|
L'indice RPA dans
|
|
et signifie que dans le calcul de ces
)
PA
|
L'énergie d'interaction paramétrée et la
fonction d'onde et ont
( ä ) ä å )
été développées en séries en
fonction de :
ä et
' A,
,? ?
? ,i 1
) ) ?
, ? ? ?

où
indiquent l'énergie de polarisation et la fonction d'onde
du i ième ordre dans , du j ième dans et k ième ordre dans
. Nous considérons que
Les corrections énergie et fonction d'onde, ,
E 0 Ø
( ijk) k)
p l
peuvent être obtenues des équations de la
théorie de la triple perturbation [62,63].
Après développement nous extrayons les
énergies d'interaction à deux et à trois corps non
additive, nous avons [7]:
(42)
( )
E bi E
n p
, ? ?
(43)
( )
E E
pp L
où est le terme non additif Heitler-London[60]
calculé avec les déterminants
Hartree-Fock des monomères isolés. Ce terme est
identique à celui de l'énergie de la
première
itération HF notée . L'égalité du terme avec
nous
st t a st it a ?
permet d'obtenir ce terme de n'importe quel traitement. , est
l'énergie
)
d'induction au second ordre, considérée
complètement pour la déformation
électrostatique des
orbitales. , , et sont les énergies
) 3 ) )
RPA PA
d'échange-dispersion au second ordre,
d'induction-dispersion au troisième ordre et la
dispersion au
troisième ordre. , est évaluée comme
l'échange-corrélation au
P )
2
premier ordre et les énergies
d'échange-induction-dispersion au troisième ordre
[61].
contributions, les fonctions réponses de l'approximation
phase aléatoire (Randomphase approximation (RPA)), ont
été utilisées [7,61].
La quantité représente une approximation aux effets
échange-induction Hartree-
Fock non additifs. Elle n'a pas été
programmée dans SAPT mais plutôt calculée comme la
différence entre l'énergie d'interaction Hartree-Fock et la somme
des énergies Heitler-London et induction:
(44)
= +
E E ?
Une telle approche, bien qu'approximative est
complètement justifiée, puisqu'on peut montrer que cette
différence est en effet dominée par les effets
d'échange-induction et d'échange-déformation au second
ordre et par les effets échange et induction de plus haut
ordre[61].
La différence entre et est notée [22]:
(45)
E E
HF
Aux bas ordres de la théorie de la perturbation,
l'énergie de délocalisation, , est
donnée par la somme des termes de l'induction et de
l'échange-induction, appelée "énergie de
déformation Hartree-Fock" [23,24] . Cette différence
donne une idée sur comment la distribution de charge des
monomères isolés se déforment (délocalisée
sur toute la supermolécule) durant le processus SCF [25].
De (44) et (45), nous pouvons déduire :
(46)
E E F
=
, désigne les effets échange-induction et
déformation. , représente
La décomposition de l'énergie d'interaction
à trois corps en différents termes de signification physique est
très importante pour l'analyse des phénomènes
d'interaction et la construction de potentiels analytiques. Des relations
similaires peuvent être trouvées pour les énergies
supermolécule et SA PT au niveau corréler.
Comme c'est discuter dans la
référence[61], la composante échange rassemble
la
correction de corrélation intramonomère de l'énergie
Heitler-London, l'énergie
d'échange-dispersion et le terme
échange-induction-dispersion alors que la partie déformation de
MP2 non additive peut être dominée par la contribution induction-
dispersion. Dans notre étude, nous utilisons cette décomposition
pour définir l'énergie d'échange MP2. En accord avec les
références 23 et 24, l'énergie à trois corps MP3
est donnée par la somme de l'énergie de dispersion Hartree-Fock
et des termes échange et déformation.
A noter que les termes d'échange inclus dans les
énergies à trois corps MP2 et M P3 ne peuvent pas être
définis de la même façon que dans la théorie
SAPT[7]. Par conséquence, la convergence du modèle
supermolécule Möller-Plesset et le développement SAPT peut
être quelque peu différente.
I.E/ Méthode de la fonctionnelle de la
densité (DFT) :
En plus des calculs ab initio, des calculs DFT utilisant
diverses fonctionnelles ont également été menés,
c'est une autre méthode de détermination de la
corrélarion. Il y a quelques années, les méthodes
basées sur la fonctionnelle de la densité sont devenues
progressivement connues.
Les meilleures méthodes DFT donnent une plus grande
exactitude que la théorie HF avec une augmentation modeste dans le
coût de calcul. Se rapprochent des résultats M P2 pour les
systèmes moléculaires de taille moyenne et plus grandes, en
incluant certains effets de corrélation d'électrons beaucoup
moins coûteusement que les méthodes traditionnelles de
corrélation.
Thomas [50] et Fermi [51] (TF) sont les
premiers à avoir établi une méthode TF basée
seulement sur la densité électronique dans le
système, Par la suite, d'autres
ñ ( )
approximations ont conduit à d'autres méthodes.
Au-delà du modèle TF,
l'approximation Slater [52], proposée en
1951, consiste à remplacer le potentiel d'échange des
équations HF par un potentiel moyen, fonction de la densité
électronique, d'où le nom de méthode de Hartree-
Fock-Slater (H FS).
I.E.1/ Le Formalisme de Hohenberg-Kohn:
Pour un système à n électrons et N noyaux,
l'opérateur hamiltonien associé à
l'énergie électronique est en u.a :
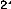
(47)N
1 =1 a
?
· +
=1 a
Le terme qui dépend de la charge nucléaire ou le
deuxième terme, est appelé potentiel
extérieur,
symbolisé par Vext(r), car il résulte de la
présence des champs produits par
des particules non inclus dans le
groupe des électrons.
En 1964, Hohenberg et Kohn (HK) [53] énonce
un théorème où ils démontrent, l'existence d'une
fonctionnelle unique qui permet de déterminer l'énergie de
l'état fondamental, que la densité électronique est
suffisante pour décrire les propriétés de l'état
fondamental, en particulier l'énergie totale.
Le formalisme de Hohenberg et Kohn est basé sur deux
théorèmesqui sont démontrés: 1/ «Le potentiel
externe Vext(r) est à une constatnte près une
fonctionnelle unique de
»
2/ « la valeur exacte de la densité
électronique de l'état fondamental d'un système
polyélectronique est celle qui
minimise l'énergie de cet
état», qui aboutit à l'équation de Euler-Lagrange:
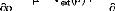
Dans cette théorie l'énergie de l'état
fondamental d'un système d'électrons interagissant dans un
potentiel Vext(r) s'écrit :
(48)
? ? ?
? ? V d
est une fonction universelle de la densité,
indépendante du pontentiel externe,
? K
qui contient tous les effets d'échange et de
corrélation. La forme de FHK n'est pas connue.
Hohenberg et Kohn écrivent :
(49)
G ( = T ( ) +
? ?
où est l'énergie cinétique du système
et l'énergie d'échange-corrélation.
? ) ( ? )
Cette méthode tente de remplir un besoin dans la chimie
théorique, le développement de la nouvelle approche de la chimie
quantique pour l'étude des interactions faibles dans un grand nombre de
systèmes moléculaires, le but est d'atteindre un niveau
d'exactitude des énergies d'interactions, ce qui est prévu avec
les techniques de chimie quantique ab initio corrélées. En 1965,
Kohn-Sham développe cette méthode [54,10].
I.E.2/ Le Formalisme de Kohn-Sham:
Les méthodes DFT programment la corrélation
d'électrons en passant en général par les fonctionnelles
de la densité d'électrons. Les fonctionnelles DFT partagent
l'énergie électronique en plusieurs composantes qui sont
programmées séparément: L'énergie cinétique,
l'interaction électron-noyau, la répulsion Coulomb et le terme
échange- corrélation expliquant le reste de l'interaction
électron-électron (qui est lui même divisé
séparément en échange et composantes de corrélation
dans la plupart des formulations DFT actuelles).
L'énergie DFT d'un système peut s'écrire:
(50)
? r,.
? ?
Où T0 est l'énergie cinétique
sans interaction, Vext représente le champ électrique
créé par tous les noyaux et le troisième terme
l'interaction coulombienne des électrons. Le dernier terme est
appelé fonctionnelle échange-corrélation et comprend la
partie inconnue restante de la fonctionnelle DFT exacte. En principe, la
solution non relativiste (exacte) au problème (incluant les forces de
dispersion London) peut être obtenue si ce terme, était connue
précisément.
Les trois derniers termes de cette équation peuvent
être groupés en un potentiel effectif:
(51)
V V
où
(52)
ñ )
( ?
L'équation (50) peut être écrite sous la
forme:
(53)
? ? ? ?
T
En utilisant le potentiel effectif, les équations du
système à particules indépendantes peuvent être
écrites comme suit:
(54)
qui peuvent être résolues de façon self
consistante pour trouver l'énergie minimum de l'équation (50),
avec:
(55)
où N est le nombre total d'orbitales occupées dans
le système.
Par analogie aux méthodes de fonctions propres, la
fonctionnelle qui relie E à , , peut être partager en une
contribution énergie cinétique, , une
] [ ñ ]
contribution due aux attractions électron-noyau, et les
répulsions électron-
[ ñ ]
électron, . Ce dernier peut être décomposer
en termes de Coulomb et
[ ñ ]
d'échange, et .
ñ ] ]
L'expression finale de l'énergie DFT est :
(56)
E ñ T
[ ] [ +
La fonctionnelle échange corrélation, comprend la
différence entre l'énergie
[ ñ ]
cinétique exacte et , appelée, la correction
énergie cinétique, la partie répulsion
électron-électron (échange), et les
contributions corrélation des deux termes
]
et .
Dans la pratique réelle, les calculs DFT Kohn-Sham
self-consistent (auto- cohérent) sont effectués de
manière itérative, de façon analogue à la
programmation SCF. Cette similarité avec la méthode de la
théorie Hartree-Fock a été montrée par Kohn et
Sham. Les orbitales Kohn-Sham sont fonctions propres de l'hamiltonien effectif
à un électron, qui est presque identique à
l'opérateur de Fock dans les équations SCF. Cependant dans le cas
Kohn-Sham, les opérateurs échange HF sont remplacés par
des fonctionnelles dérivées de l'énergie d'échange
corrélation. Appuyant l'existence de EXC et prévoyant
une densité d'électron initiale, les équations Kohn-Sham
sont résolues pour les orbitales qui peuvent être utilisées
afin de définir une nouvelle densité d'électron et
l'hamiltonien effectif. Ces itérations continuent jusqu'à ce que
la densité converge vers un seuil spécial, qui donne
l'énergie la plus
basse. Cette procédure d'avoir E à partir de reste
incomplète. D'où le
développement, de fonctionnelles approchées, qui
relient l'énergie à la densité d'électron est un
domaine extrêmement actif de la recherche actuelle.

I.E.3/ Forme analytique de EXC :
La forme exacte de EXC n'est pas encore connue,
malgré cela, un grand nombre de fonctionnelles
d'échange-corrélation, approchées, est publié dans
la littérature. Généralement les fonctionnelles
échange-corrélation les plus connues sont divisées en
contributions d'échange pure et corrélation, et .
[ ñ ] [ ñ ]
En effet Hohenberg et Kohn ont démontré que
EXC est déterminé entièrement par (est fonction
de) la densité d'électrons. En pratique, l'expression
approchée la plus souvent utilisé de EXC est sous
forme d'intégrale incluant seulement les densités de spin avec
possibilité ou non de leurs gradients :
est généralement utilisée comme
divisée en deux parties distincts, l'échange et la
corrélation, correspondant actuellement, aux interaction
same-spin (même spin) et mixed-spin (spin mixte),
respectivement:
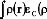
sont les densités d'énergie par particule.
Les trois termes sont fonctionnels de la densité
d'électrons, les deux termes à droite de l'équation sont
respectivement les fonctionnelles échange et corrélation.
I.E.4/ Fonctionnelles de la densité:
La fonctionnelle est définie en mathématique
comme une fonction d'une fonction. Dans la théorie fonctionnelle de la
densité, fonctionnelle est la fonction de la densité
d'électron, elle même une fonction de coordonnées dans
l'espace.
Les deux fonctionnelles échange et corrélation
peuvent être de deux types différents:
local functionals
(fonctionnelles locales) qui dépendent seulement de la
densité
d'électron ou gradient-corrected functionals qui
dépendent de et de son
gradient, . Le mot local ne coincide pas avec le
terme utilisé en mathématiques, les deux fonctionnelles local
et gradient-corrected sont locales dans le sens mathématique).
Locale car la densité électronique prend comme
référence l'électron dans un gaz homogène, elle est
donc trop localisée autour de l'électron de
référence. La contribution d'un système à
l'énergie en chaque point ne dépend que de la seule
densité en ce point ( on néglige
l'inhomogénéité de la densité ou la variation de la
densité.
Si on utilise LSD ou LSDA (Local Spin Density Approximation) pour
tenir
?
compte de la polarisation de spin.
Un exemple de ces fonctionnelles. La fonctionnelle
échange local est définie
pratiquement toujours comme la formule de Dirac:
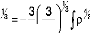
où est bien sûre une fonction de . Cette forme a
été développée pour reproduire
l'énergie d'échange d'un gaz d'électron
uniforme. Cependant, elle ne peut pas décrire les systèmes
moléculaires.
Becke a formulé en 1988 la fonctionnelle échange
gradient-corrigé basée sur la fonctionnelle échange LDA
local, qui est maintenant largement utilisée :
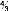
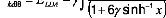
|
où x =
|
|
. est un paramètre choisi de façon
appropriée aux énergies
|
d'échange connues des atomes de gaz inertes. comme le
montre l'équation çi-dessus, la fonctionnelle Becke est
définie comme une correction à la fonctionnelle échange
LDA local et elle réussie à remédier aux nombreuses
déficiences de fonctionnelles LDA local.
Le défaut des fonctionnelles est la surestimation des
énergies d'échange et de corrélation à cause de la
localisation de la densité autour de l'éléctron de
référence.
I.E.4.a/ Fonctionnelles traditionnelles:
- Parmi les fonctionnelles échange et
corrélation locale qui impliquent seulement les valeurs des
densités de spin d'électron, Slater et , sont connues comme les
fonctionnelles d'échange local et le traitement de densité spin
locale de Vosko, Wilk et Nusair (VWN) est largement utilisé comme
fonctionnelle de corrélation locale.
- Pour les fonctionnelles du gradient corrigé qui
impliquent en même temps les valeurs des densités de spin
électron et leurs gradients. De telles fonctionnelles sont aussi parfois
citées comme non Local dans la littérature, elles tiennent compte
de l'inhomogénieté de la densité dans le gaz
d'électrons, . Les
f ? ? ?
fonctionnelles GGA (Generalized Gradient Approximation) sont
utilisées pour corriger les fonctionnelles locales:
La fonctionnelle échange gradient-corrigé la
plus connue est de Perdew et Wang PW86 et celle décrite
çi-dessus, proposée par Becke en 1988, B88. La
fonctionnelle corrélation gradient-corrigé largement
utilisée est la fonctionnelle Perdew P86 et LYP de Lee,
Yang et Parr. La combinaison des deux formes, la B-LYP
méthode est disponible en passant par la BLYP clé en Gaussain. La
différence entre ces fonctionnelles réside dans l'expression de ,
ajustement de paramètres).
NL ( ? ? ? ?
Perdew a aussi proposé certaines fonctionnelles de
corrélation importantes du gradient-corrigé, connues comme
Perdew 86 [56] et Perdew-Wang 91
[57].
I.E.4.b/ Fonctionnelles hybrides:
La théorie Hartree-Fock inclut aussi un terme
d'échange faisant partie de sa formulation. Récemment,
Becke[20] a formulé des fonctionnelles qui incluent un
mélange d'échange Hartree-Fock et DFT qui marche avec la
corrélation DFT, définissant EXC comme:
c E
ybr d HFH
?
a EX F où les coefficients sont constants et
ajustés par Becke pour reproduire les énergies de liaisons d'une
série de
molécules de référence.
Il y a plusieurs fonctionnelles hybrides, qui
définissent la fonctionnelle échange comme une combinaison
linéaire des termes d'échange HF, locale et
gradient-corrigé. Cette fonctionnelle échange est alors
combinée avec la fonctionnelle corrélation locale et/ou gradient
corrigé. La mieux connue de ces fonctionnelles hybrides est la
formulation à trois paramètres de Becke-style [58],
elle est définie par l'expression suivante:
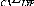
Dans B3LYP, B pour Becke, 3 désigne le nombre de
paramètres et LYP npour Lee Yang et Parr.
Ici, le paramètre c0 permet l'utilisation d'un
mélange d'échange Hartree-Fock et local LDA. De plus, le
gradient-corrigé de Becke pour l'échange local LDA est aussi
inclut, évaluer par le paramètre cX. Les fonctionnelles hybrides
de ce type, sont disponibles en Gaussian en passant par les clés
B3LYP et B3PW91.
De façon similaire, la fonctionnelle de
corrélation local VWN3 est utilisé et peut être
corrigée de manière optionnel par la correction
corrélation LYP à travers le paramètre cC.
Différentes fonctionnelles peuvent être construites de la
même méthode en variant les composantes de la fonctionnelle. Par
exemple, en substituant la fonctionnelle corrélation gradient
corrigé Perdew-Wang 1991 à la LY P et en ajustant les valeurs des
trois paramètres.
Nous avons utilisé les fonctionnels hybrides
B3LYP et B3PW91 pour le calcul des
interactions à trois corps. Bien que les orbitales obtenues par le
calcul DFT sont un peu différentes des orbitales HF, cependant, avec la
méthode DFT, l'énergie de la première itération
n'est pas tout à fait identique au terme Heiltler-London, pour le
démontrer, nous avons calculé pour les interactions non additives
les contributions: ,
t iter )
.
*Les fonctionnelles hybrides Becke, ont prouvé leur
supériorité, sur les fonctionnelles traditionnelles.
I.F/ Méthode théorique du calcul de la
polarisabilté du radical OH :
Pour le calcul des polarisabilités de sous systèmes
dégénérés, dans notre cas, le radical OH, nous
avons utilisé la méthode suivante:
I.F.1/Polarisabilités multipolaires:
Le formalisme du développement du potentiel d'interaction
à deux centres à longue portée[114], a
été étendu aux molécules qui peuvent être
dans un état dégénéré.
La conséquence de la
dégénéréscence est que les coefficients
d'interaction à longue portée communs doivent être
remplacés par les «matrices d'interaction à longue
portée». Le formalisme a été appliqué à
OH-CO et a mené à une description juste d'une partie à
longue portée de ses surfaces de potentiel.
Les coefficients d'interaction à longue portée
sont des oultils précieux pour prédir très exactement, le
comportement asymptotique des surfaces d'énergie potentielle. La partie
à longue portée du potentiel est d'un intérêt
spécial pour la saisie des processus comme les interactions incluant des
radicaux.
Comme c'est bien connu, en particulier dans
l'atmosphère et la chimie de combustion, les radicaux
dégénérés dans les états fondamentaux
2 ð à couche ouverte, jouent un rôle crucial dans
un grand nombre de réactions. Dans ce contexte, les systèmes les
plus importants sont OH et CO. Par conséquent, l'extension
générale du dévéloppement à deux centres du
potentiel d'interaction à longue portée pour les systèmes
qui sont dans un état dégénéré est d'une
grande utilité pour la construction des surfaces de potentiel de tels
cas. Nielson et al[115] ont abordé la question de la
définition des coefficients d'interaction à longue portée
quand une molécule dans un état n
dégénéré est impliquée.
La formulation habituel de la théorie de la
perturbation Raleigh-Schrödinger pour les états
dégénérés semble, ne pas être très
appropriée pour déterminer les coefficients d'interaction
à longue portée à cause du développement en (1/R)
du potentiel d'interaction qui conduit à une multitude
d'opérateurs de perturbation. Il n'est pas possible de définir
les fonctions d'onde d'ordre zéro adiabatiques, puisque ces
opérateurs se fractionnent en groupes avec des propriétés
symétriques différentes.
Dans notre étude une représentation diabatic a
été choisie. Dans cette représentation, chacune des
matrices d'interaction à longue portée est
développée en séries en puissance, fonction de 1/R et une
partie angulaire en termes de matrices de rotation deWigner[116].
Manifestement, la représentation diabatic, est avantageuse dans les
calculs impliquant les fonctions d'onde du rotateur rigide, qui sont aussi des
matrices de rotation de Wigner. Anisi, l'intégration sur les
degrés angulaires de liberté de mouvement peuvent se faire
analytiquement.
I.F.1 .a/ Coefficients d'interaction à longue
portée:
Le potentiel d'interaction de deux systèmes A et B dans
la région à longue portée, c'est à dire l'espace
où le recouvrement des deux fonctions d'onde est négligeable,
peut être déterminer de façon conventionnelle comme
suite:
quand les deux systèmes sont des états non
dégénérés. Dans l'équation (3), les sont les
matrices rotaions de Wigner et , les abréviations des
angles d'Euler qui relient les coordonnées de la
molécule fixe des systèmes à celles dont l'axe z coincide
avec l'axe intermoléculaire joignant les deux centres de masse.
Les coefficients d'interaction à longue portée
peuvent être alors
V n
L L
M M
calculés en utilisant la théorie de la
perturbations habituelle,
Rayleigh-Schrödinger.
la
compilation détaillée de toutes ces formules
pertinentes a été donnée par Spelsberg et
al[117].
Dans ce paragraphe, les changements dans la définition
des coefficients d'interaction à longue portée sont
argumentés comme étant nécessaires quand les
dégénéréscences dans les sous systèmes sont
présentes. Pour la simplicité, le cas considéré est
celui où seulement un système, dit A est in an N-fold (
dans un N- incorporé) état fondamental
dégénéré.
Commençant par:
de même pour B et
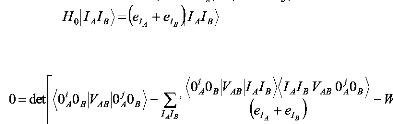
? (6)
Nous obtenons l'équation séculaire N×N:
pour programmer l'énergie d'interaction WAB
au delà du second d'ordre dans VAB. Le
prime indique que
les états IA=0i et IB=0 sont exclus de la sommation.
L'extenstion de
cette théorie, à d'autres situations, comme
l'état excité dégénéré ou la
dégénéréscence
dans les deux systèmes,
est en cours de développement.
Le potentiel VAB est donné en termes
d'harmoniques solides.
(8) 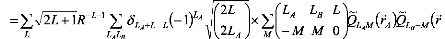
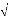
où les sont les opérateurs moment multipolaire
?
Q r ? 4? 2
? ?
l
communs et le signe au-dessus du tilde indique le choix de la
coordonnée espace fixe du système pour .
L'insertion de l'équation (8) dans l'équation (7),
conduit à:
?
? LAMA ,LBe A , ? ? R
(9)
après la modification appropriée de
l'équation (3). Les coefficients
V
n L sont donnés comme suite:
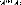
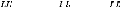
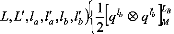
1 (10)
2 (() b
iLM l a
? q
où les coefficients angulaires couplés g1 et g2
sont définis comme:

(11)
et
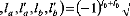
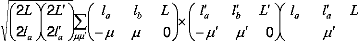
(12)
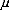
Les éléments de matrice ij qui apparaissent dans
l'équation (10), à cause de la
dégénéréscence sont les abréviations pour
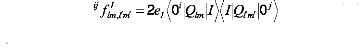
(14)
et
(15)
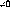
Ici the , et sont respectivements, les moments statiques, les
f I
forces de l'oscillateur et les polarisabilités
statiques.
|
En ajoutant les éléments
|
|
, la matrice interaction à longue portée
(N×N)
|
comme définie dans l'équation (9), peut
être programmée pour une géométrie spécifique
et sa diagonalisation donne le ou les potentiels à longue portée
requis. Les deux premiers termes de l'équation (10) représentent
l'induction et le troisième terme la dispersion.
I.F.1 .b/ Polarisabilités statiques:
En concordance avec l'équation (15), les
polarisabilités dynamiques, peuvent être
généralisées aux états
dégénérés tels que:
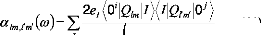
(16)
e
2 ?
(17)
?
i, ? ?
La quantité indispensable, , qui peut être
appelée aussi polarisabilité
l ? ??
dynamique, peut être écrite ainsi:
où les fonctions d'onde perturbées du premier ordre
satisfont[117]:
Suivant ce qui précède, l'équation (19),
peut être résolue pour certaines fréquences
choisies
simultanément avec seulement d'infime modifications du code de
la
méthode moderne d'interaction de configuration
directe[117]. Dans la forme la plus
|
compacte, la référence espace
|
|
consiste à toutes ces substitutions dans les
|
|
|
|
configurations à travers l'espace
référence
|
|
où une des orbitales interne s est
|
remplacée par une seule orbitale perturbée
correspondante. Ces dernières sont déterminées par des
calculs multi configuration champ auto cohérent (MCSSCF).
Dans notre cas, nous nous interessons aux polarisabilités
statiques, dipolaires, diagonales, ce qui implique respectivement :
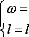

L'équation (16) se réduit à :
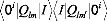
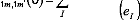
Pour m=m' :
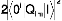
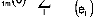
avec eI=Ei-E0
i désigne l'orientation de la fonction d'onde dans
l'espace alors que I indique la symétrie de l'état.
I.F.1 .c/Application à OH, considérations
symétriques:
Pour OH les deux états
dégénérés peuvent être choisis de
façon à ce qu'ils
|
soeint les états propres de Lz,
|
|
|
et
|
|
|
(base sphérique). En
|
|
|
|
|
|
pratique, dans les calculs de chimie quantique, il est plus
commode de travailler avec des combinaisons linéaires réelles de
ces états. Pour le calcul des potentiels d'interaction, c'est aussi la
base appropriée, puisque dans le cas général, la
supermolécule a moins de symétrie que Par conséquent, la
représentation
tesseral[119] est utilisée pour les
états propres et les opérateurs multipolaires quand c'est
convenable:
? ?
1 / 2
? p p (21)
La définition dans l'équation (21) est identique
à celle donnée dans la référence [118].
Dans la
base tesseral, les états propres d'une molécule linéaire
sont aussi états propres
de ózy, la
réflection à travers le plan zy et p indique la valeur propre
correspondante.
|
|
|
|
|
|
|
|
|
Pour simplifier les états propres
dégénérés
|
|
se réfèrent à
|
|
et
|
|
.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
La base tesseral d'harmoniques sphériques notéeest
reliée aux harmoniques
? ? ?
sphériques complexes :
? ? ?
Y ? 1,0
1
0
Y
ei,m ,
?
Y1
Y
m
0 :
(22)
q
?
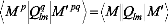
m''i,
,
avec:
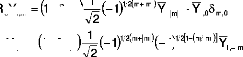
1
R
Y l
I m
Y l
1
Certaines propriétés de la symétrie de OH,
qui sont évidentes dans la base
sphérique ne sont pas évidentes dans la base
tesseral. Les relations symétriques pour
correspondre les
éléments de matrice des opérateurs multipolaires dans la
base tesseral
peuvent être énoncés par l'introduction d'une
matrice d'éléments réduite
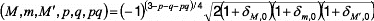
Avec l`equation (21), les coefficients couplés et par
conséquent les relations de symetrie peuvent être établis,
tel que :
A noter que la matrice d'éléments réduite
découlant des états
généralement pas notée. Dans tous les
autres cas, les éléments de matrice
sont marqués tout simplement par les facteurs et 0 pour M,
m et M'
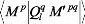
fixés. Les règles de symétrie qui en
dérivent sont données dans la référence [114].
I.G/ Erreur de superposition de bases(BSSE):
Soit la molécule AB :
1) Considérons la base = -'-, comme constituée des
orbitales atomiques
centrées sur les noyaux A et B .
( (
2) Calculons l'énergie du système AB, Eavec ;
l'énérgie del'atome A, E , avec et l'énergie du
monomère B, Eavec , ça apparaît logique mais en
réalité,
nous n'avons pas utilisé la même méthode pour
le calcul des énergies AB, A et B. Il serait plus approprié de
calculer les trois quantités avec la même base [1].
Quand les calculs sont faits pour AB avec la base , nous
calculons implicitement, non seulement l'énergie d'interaction mais
aussi nous permettons aux
sous systèmes de baisser leurs énergies. En faisant
()-()-(), nous
obtenons avec l'énergie d'interaction, un terme non voulu
et non physique (erreur), relier à la baisse de et , quand on calcul,
cette erreur est appelée BSSE [1].
Pour remédier à cette erreur, nous utilisons
aussi pour le calcul des énergies de A et B, respectivement et . Cette
méthode est appelée counter-poise method ou
méthode du contrepoids, introduite au départ par Boys et Bernardi
[21].
Si contamine dans le calcul de l'énergie de A, ou si
contamine dans
le calcul de l'énergie de B, , cette pollution peut
être quantitativement annulée par
la BSSE [31]:
BSSE (AB) = E E A
Toutes les énergies de la supermolécule et DFT
ont été corrigées pour l'erreur de superposition de base
(Basis set superposition error) en utilisant la méthode du
contre poids de Boys et Bernardi.
Pour le modèle de la supermolécule, les calculs ont
été faits avec Gaussian 94[26]. Les calculs SAPT ont
été fait avec le programme SAPT.
Les résultats DFT ont été obtenus à
l'aide du programme Gaussian.
Pour les calculs MCSCF, nous avons utilisé le programme
MOL PRO.
I.H/ Bases utilisées :
Alml~f, Taylor et leurs collègues ont trouvé que
les jeux de base d'orbitales neutres qui dérivent de calculs de
corrélation atomique donne une excellente description des effets de
corrélation moléculaire. Pour décrire les distributions de
charge les plus diffuses des anions, les bases neutres ont été
augmentées en additionnant des fonctions optimisées pour les
atomes d'anions. Les valeurs des énergies baissaient dû à
l'ajout successif de fonctions de corrélation. Ceci a conduit au concept
de corrélation-consistent basis set ou jeu de bases de
corrélation consistante. Une base de fonctions primitives s et p a
été ajoutée aux bases (sp) de chacun des jeux de bases de
corrélation consistante, double zeta (cc-pVDZ), triple zeta (cc-pVTZ) et
quadruple zeta (cc-pVQZ), de valence polarisé de l'atome neutre et les
exposants ont été optimisés pour l'énergie SCF de
l'anion. Ensuite une fonction de polarisation primitive additionnelle a
été ajoutée à chacun des ensembles l-polarisation
présent dans le jeu de base neutre et les exposants de ces fonctions ont
été optimisés pour le calcul de l'énergie
Hartree-Fock de l'anion. Cette procédure a conduit à trois jeux
de base suivant l'ordre de taille et le degré de précision,
The augmented correlationconsistent polarized valence double-zeta,
triple zeta et quadruple zeta.
Les calculs ab intio ont été faits en utilisant
la base aug-cc-pVDZ [71,72], composée d'une primitive (9s4p1d)
contractée en [3s2p1d] pour l'atome d'oxygène et (4s1p)
contractée en [2s1p] pour l'atome d'hydrogène étendue avec
un ensemble de fonctions diffuses, spd sur O et sp sur H. Cela dans le but
d'avoir des valeurs d'énergies très proches de ceux de la
référence (CCSD(T)). Ces jeux de base ont été
utilisés dans la référence [65].
Chapitre II
Structures moléculaires OH-(H2O)n
étudiées
II.A/ Géométrie des Configurations de
OH-(H2O)n:
Nous avons calculé les énergies d'interaction
pour des sous systèmes OH-(H2O)2 appartenant à quatre
séries de configurations OH-(H2O)n, qui sont
notées: R et X, pour n=2, T pour n=4 et F pour n=10. Ces configurations
ont été prises de différentes références.
Dans toutes les configurations OH-(H2O)n
, les molécules d'eau appartiennent à la
première ou à la deuxième couche de
solvatation. Nous considérons qu'une molécule d'eau appartient
à la deuxième couche de solvatation à partir d'une
distance entre l'oxygène de l'ion et l'oxygène de l'eau dik >
3Å.
Nous avons noté :
ctwk : l'angle de la molécule d'eau k
dik=d(Oi-Owk) : distance entre l'oxygène de l'ion et
l'oxygène de la molécule d'eau k. dkl=d(Owk-Owl) : distance entre
les oxygènes des molécules d'eau k et l.
kl= <Owk-Oi-Owl : l'angle formé par l'oxygène de
la première molécule d'eau,
l'oxygène de l'ion et l'oxygène de la
deuxième molécule d'eau
dans OH-(H2O)2, où i désigne l'ion et k
et l sont les indices des
molécules d'eau considérées dans chaque
configuration OH-
(H2O)n voir figures 1, 2 et 3.
II.A.1/ Structures de OH-(H2O)2 correspondant à la
série R (Fig.1):
Les structures OH-(H2O)2 de la série R sont
obtenues par minimisation au niveau MP2, avec les géométries des
sous-systèmes, c'est-à-dire celles des molécules d'eau et
de l'ion hydroxyde figées à leur valeurs expérimentales:
d(OH)= 0.9572 Å ,
ctwa,b =104.5° pour l'eau[73] et
d(OH-)= 0.964Å[74].
La correction de la BSSE n'a pas été prise en
compte dans la procédure
d'optimisation, la meilleure structure donne
une énergie MP2 de -228.2374 a.u,
correspondant à un
angleab= <Owa-Oi-Owb 80.9061°. Ensuite, une des
deux
molécules est tournée d'un angle ab=
<Owa-Oi-Owb 110.5179°, 140.4432°,
170.2524°,
conduisant à la série R110, R140 et R170.
Les autres paramètres géométriques sont :
dia=d(Oi-Owa)= 2.642 Å et dib=d(Oi-Owb)=
2.614Å.
II.A.2/ Structure de OH-(H2O)2 correspondant à la
configuration X2 (Fig.1):
Comme il est important de voir aussi l'effet de relaxation
géométrique des sous- systèmes, la structure X2
trouvée dans la référence [65], a été
obtenue avec une optimisation complète de la géométrie de
la supermolécule au niveau MP2, les paramètres
géométriques des monomères dans le complexe sont
différents de ceux des monomères isolés. En particulier,
les deux distances OH dans la même molécule d'eau ne sont pas
identiques, elles sont égales à 0.965 et 1.033 Å dans l'une
et 0.965 et 1.038 Å dans l'autre. Le stretching (élongation) dans
chacune des molécules conduit à une forte variation des valeurs
des contributions énergétiques, composantes de l'énergie
d'interaction du complexe.
La correction BSSE n'a pas été prise en compte dans
la procédure d'optimisation. L'énergie de cette structure est de
-228.244097 a.u.[65] .
Les paramètres géométriques des
monomères sont : d(OH-)= 0.967Å
d(OaH1a)= 1.033Å ; d(OaH2a)= 0.965Å ;
OEwa= <H1a-Oa-H2a= 101.5°; d(ObH1b)= 1.038Å ;
d(ObH2b)= 0.965Å ; OEwb =<H1b-Ob-H2b= 101 .7°
Les autres paramètres géométriques : dab=
4.375Å
dia=2.591Å ; dib= 2.580Å
?ab= <Owa-Oi-Owb = 115.6°.
* Remarque importante : L'optimisation au niveau MP2 de deux
structures OH-(H2O)2 avec deux procédés
différents, dans un cas les paramètres géométriques
des monomères du système sont fixés à leurs valeurs
expérimentales et dans l'autre une optimisation totale est faite, a
conduit aux configuration R80 dont l'énergie est de - 228.2374 a.u
(-143.2211.10+3 Kcal/mol) et X2 dont l'énergie est de
-228.244097 a.u (- 143.2253.1 0 +3 Kcal/mol), les deux valeurs de
l'énergie des deux systèmes sont proches malgré que les
paramètres géométriques des deux configurations
OH-(H2O)2 soient différents. L'énergie du
système varie avec les paramètres géométriques.

II.A.3/ Structures de OH-(H2O)2 correspondant
respectivement aux séries T et F (Fig. 2 et 3):
Ces séries ont été obtenues par des
calculs Monte Carlo[113] préliminaires [66],
effectués
pour les clusters OH-(H2O)2,
OH-(H2O)4 et OH-(H2O)10. Les géométries des
molécules
d'eau et de l'ion hydroxyde sont fixées à
leurs valeurs expérimentales : d(OH)= 0.9572
Å , OEwa,b =104.5° pour l'eau et d(OH-)=
0.97Å [67-69].
Deux ensembles de potentiels ont été
utilisés, qui ont donné deux séries de
géométries. Une série notée T, a été
obtenue avec les potentiels des paires proposés dans la
référence [66]. La seconde série est notée F, dans
ce cas, l'interaction entre l'ion hydroxyde et la molécule d'eau est
décrite par l'expression des paires de Fornili et al [67] et
le potentiel MCY [70] est utilisé pour l'interaction entre
deux molécules d'eau.
Pour les deux séries, les termes non additifs ne sont
pas inclus. En plus du problème de l'exactitude de ces calculs
préliminaires, nous devons noter que de tels traitements type Monte
Carlo, ne conduisent pas à des géométries
minimisées. En outre, ces calculs sont faits à 300 K, alors que
la minimisation quantique de l'énergie est faite à O K. Les
structures T4 et F10 sont les plus stables générées dans
ces calculs Monte Carlo mais ne correspondent pas au minimum global.
Les autres paramètres sont : Pour la configuration T4 :
dab= 3.300 Å ; dac= 4.659 Å ; dad= 3.451Å ;
dbc= 4.393 Å ; dbd= 4.817 Å ; dcd= 3.582 Å dia= 2.546 Å
; dib= 2.503Å ; dic= 2.505 Å ; did= 2.559Å ;
3ab = 81.6° ; 3ac = 134.5° ; 3ad = 85.1° ; 3bc =
122.6° ; 3bd = 144.2° ; 3cd = 90.0°
Pour la configuration F10 :
Comme il y'a un grand nombre de molécules d'eau dans le
système F, nous détailleront que les paramètres
géométriques des configurations étudiées:
* Les molécules d'eau appartenant à la
première couche sont : a, b, c, d
* Les molécules d'eau appartenant à la
deuxième couche sont : e, f, g, h, i, j
dkl en Å : daj= 3.05, dcj=
3.70, dbg= 5.80, dbj=
5.95, dgf= 3.01, dei= 7.74
dik en Å : dia= 2.77, dib=
2.73, dic= 2.74, did= 2.83
die= 4.05, dif= 3.85,
dig= 3.98, dih= 3.95,
dii= 3.98, dij= 3.96
3aj= 50.1°, 3cj= 64.0°, 3bg=
118.2°, 3bj= 124.3°, 3gf= 45.1°,
3ei= 149.3°
Les paramètres géométriques des
différentes configurations OH-(H2O)2 étudiées,
sont représentés dans un tableau récaputilatif ( Angles en
degré et distances en Angström ).
Tableau récaputilatif des Paramètrs
géométriques des différentes
configurations
OH-(H2O)2 ( Angles en degré et distances en
Angström )
|
Configurations
|
Paramètres des
Monomères
|
3kl
|
Dik
|
|
R(n = 2)
|
d(OH)I=0.964
|
3ab=80.9061
|
dia=2.642
|
|
«On bouge une des
|
d(OH)wa,b=0.9572
|
3ab=1 10.51 79
|
dib=2.614
|
|
deux molécules
|
ctwa,b=104.5
|
3ab=140.4432
|
|
|
d'eau, 3ab varie»
|
|
3ab=170.2524
|
|
|
X(n = 2)
«Relaxtation de
toute
la
géométrie»
|
d(OH)I=0.967 d(OH 1 )wa=0.965 d(OH2)wa=1
.033 d(OH1)wb=0.965 d(OH2)wb=1 .038
|
3ab=1 15.6
|
dia=2.591
dib=2.580
|
|
(X wa=101 .5
|
|
|
|
OEwb=101.7
|
|
|
|
T(n = 4)
|
D(OH)I=0.97
|
3ab=81.6
|
dia=2.546
|
|
d(OH)wk=0.9572
|
3ac=134.5
|
dib=2.503
|
|
OEwk=104.5
|
3ad=85.1
|
dic=2.505
|
|
|
3bc=1 22.6
|
did=2.559
|
|
|
3bd=144.2
|
|
|
|
3cd=90.0
|
|
|
F(n = 10)
|
D(OH)I=0.97
|
3aj= 50.1°
|
dia= 2.77
|
|
«a,b et c
|
d(OH)wk=0.9572
|
3cj= 64.0°
|
dib= 2.73
|
|
appartiennent à la
première couche
|
Ctwk=104.5
|
3bg= 118.2°
|
dic= 2.74
die= 4.05
|
|
de de
solvatation
|
|
3bj= 124.3°
|
dif= 3.85
|
|
l'ion et e, f, g et j à
|
|
3gf= 45.1°
|
dig= 3.98
|
|
la deuxième
couche»
|
|
3ei= 149.3°
|
dij= 3.96
|
i: ion
k et l : indices des molécules d'eau dik: dOi-Owk
ctwk: angle de la molécule d'eau k 3kl : <Owk-Oi-Owl
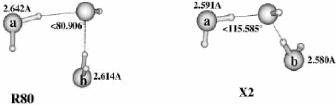
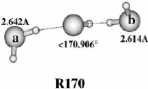
Fig. 1 Structures OH-(H2O)2
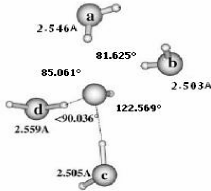
Fig.2 Structure OH-(H2O)4 (T4)
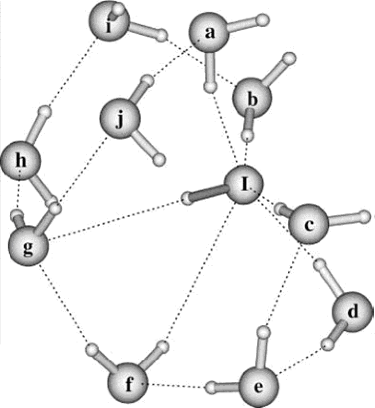
Fig.3 Structure OH-(H2O)10 (F10)
Chapitre III
Interprétation des résultats obtenus pour
les systèmes OH-(H2O)n
Comme les calculs d'interaction de configuration pour des
systèmes d'une telle taille sont difficiles à réaliser,
nous avons utilisé les valeurs CCSD(T) comme
référence[124,125].
Nous avons reporté dans les tableaux I, II et III
toutes les composantes des énergies d'interaction à trois et deux
corps de tous les sous systèmes OH-(H2O)2 des
structures décrites dans le chapitre précédent,
obtenues par les différentes méthodes théoriques
utilisées dans nos calculs, excepté les résultats
DFT[125], parce qu'avec cette méthode nous avons
calculé les énergies de la première itération et
les effets à trois corps seulement pour certaines configurations.
Nous avons gardé les mêmes notations que ceux du
chapitre précédent, pour chacune des configurations
OH-(H2O)2 considérée, nous avons :
- kl= <Owk-Oi-Owl : l'angle formé par l'oxygène
de la première molécule d'eau, l'oxygène de l'ion et
l'oxygène de la deuxième molécule d'eau.
- dik=d(Oi-Owk) : distance entre l'oxygène de l'ion et
l'oxygène de la molécule d'eau k. La molécule d'eau
appartient à la deuxième couche de solvatation pour dik >
3Å. -Les numéros des couches une et deux sont notées
respectivement dans les tableaux par :1 et 2.
III.A/ Résultats obtenus avec les configurations
OH-(H2O)2 de la série R et la configuration X2 (Fig. 1, Tableau I,
Graphe I) :
Les deux molécules d'eau dans ces configurations
appartiennent à la première couche de solvatation de l'ion
hydroxyde.
III.A.1/ Variation de l'énergie à trois corps
en fonction de l'angle kl:
L'angle kl de la série R varie de 80 à
1700. L'énergie de la première itération
, croit de façon attractive régulière
avec l'accroissement de l'angle kl, les calculs que nous avons faits sur la
géométrie X2 issue d'un autre procédé
d'optimisation [65] et où l'angle kl est de 115.6° le
confirment.
L'énergie d'interaction à trois corps HF, est
répulsive et décroît quand on
passe d'un angle kl de 80 à 110°, ensuite elle
croît. Pour la configuration X2 elle est
de 3.42 Kcal/mol, presque le double par rapport aux valeurs de
la série R. Si on compare les configurations de la série R avec
la configuration X2, on trouve que, l'orientation des deux molécules
d'eau dans la série R est différente de celles dans la
configuration X2 et surtout les géométries de la série R
et de la configuration X2 ont été obtenues par optimisation au
niveau MP2 en ne prenant pas en considération la correction BSSE et en
utilisant la base aug-cc-pVDZ mais dans la série R on a figé
les
paramètres géométriques des
molécules d'eau et de l'ion aux valeurs expérimentales alors que
le cluster X2 a été obtenu par minimisation complète du
système. Par conséquent dans la configuration X2 les distances
dik sont inférieures à 2.6Å pour les deux
molécules d'eau alors que pour les configurations de la série R,
elles sont toutes supérieures à 2.6Å, les distances d(OH)
des molécules d'eau de la série R sont différentes de
celles de la configuration X2 et les angles des molécules d'eau de la
série R sont aussi différent des angles des molécules
d'eau de la structure X2 (voir les paramètres
géométriques, paragraphes II.A.1 et II.A.2 ). Ceci montre
l'importance des paramètres géométriques, qui est mis en
évidence si on compare les résultats du cluster R110 à
ceux de la configuration X2 dont les angles kl sont respectivement de
110.5° et 115.6°, donc proches.
Pour vérifier quel paramètre
géométrique a le plus influencé sur la variation de
l'énergie, nous avons fait un autre calcul avec la configuration X2 mais
en prenant comme distances OH pour les molécules d'eau, la valeur
expérimentale 0.9572Å et nous avons laissé les autres
paramètres inchangés, le tableau I montre clairement que la
modification de ce paramètre a changé de façon importante
le comportement général des diverses contributions
énergétiques à trois et deux corps. Toutes les valeurs
énergétiques de la configuration X2a convergent vers celles de la
R110. La configuration X2b est déduite de la X2a en modifiant les angles
des molécules d'eau de la X2a, nous leur avons attribué la valeur
expérimentale de 104.5°, les valeurs des énergies obtenues
sont presque les mêmes que celles de la X2a, le changement des valeurs
des angles des molécules d'eau n'a pas eu le même effet que le
changement des distances d(OH) des molécules d'eau, ce paramètre
a un effet beaucoup plus important.
Nous avons calculé l'énergie de déformation
Hartree-Fock, (paragraphe
I.B, équation (45)), nous avons obtenu la même
variation deen fonction de l'angle kl que pour , les énergies sont un
peu plus répulsives et mêmes constatations concernant les
résultats obtenus pour les configurations X2, X2a et X2b.
Les énergies d'interaction à trois corps non
additives obtenues avec les différentes méthodes
théoriques, M P2, M P3, M P4SDQ, CCSD(T) et SAPT sont toutes
répulsives et ont la même variation en fonction de l'angle ab que
(voir graphe I a)
et mêmes observations concernant les calculs faits avec ces
méthodes sur les configurations X2, X2a et X2b.


Fig.1 Structures OH-(H2O)2
|
a/ Energies à trois corps en Kcal/mol
|
|
|
|
|
|
Conf.
|
R80
|
R110
|
R140
|
R170
|
X2
|
X2a
|
X2b
|
|
N° couches
|
1-1
|
1-1
|
1-1
|
1-1
|
1-1
|
1-1
|
1-1
|
|
kl(°)
|
80.9
|
110.5
|
140.4
|
170.3
|
115.6
|
115.6
|
115.6
|
|
?
=1
|
-0.34
|
-0.46
|
-0.75
|
-0.90
|
-0.62
|
-0.51
|
-0.52
|
|
2.01
|
1.86
|
1.93
|
1.97
|
3.42
|
2.12
|
2.09
|
|
2.35
|
2.33
|
2.68
|
2.87
|
4.04
|
2.63
|
2.61
|
|
1.70
|
1.52
|
1.58
|
1.61
|
2.86
|
1.69
|
1.67
|
|
P2 ?
|
|
|
|
|
|
|
|
|
2.24
|
2.03
|
2.08
|
2.11
|
3.60
|
2.28
|
2.25
|
|
P
|
|
|
|
|
|
|
|
|
1.79
|
1.56
|
1.60
|
1.63
|
2.94
|
1.75
|
1.73
|
|
MP4
|
|
|
|
|
|
|
|
|
1.97
|
1.74
|
1.78
|
1.81
|
3.17
|
1.94
|
1.91
|
|
CC
|
|
|
|
|
|
|
|
|
APT
|
1.79
|
1.59
|
1.70
|
1.78
|
2.95
|
1.77
|
1.75
|
=1+2+3+4+5+6+7
|
APT
|
|
|
|
|
|
=2 2.73 2.87 3.67
|
4.37
|
3.47
|
3.17
|
3.13
|
|
=3 -0.38 -0.54 -0.81
|
-1.50
|
-0.57
|
-0.56
|
-0.52
|
|
=4 0.18 0.05 -0.02
|
-0.06
|
0.10
|
0.05
|
0.05
|
|
=5 0.40 0.76 0.99
|
1.15
|
0.92
|
0.87
|
0.86
|
|
3
|
|
|
|
|
|
=6 -0.93 -1.12 -1.21
|
-1.28
|
-1.56
|
-1.32
|
-1.30
|
|
P2 )
|
|
|
|
|
|
7 0.13 0.04 0.01
|
-0.00
|
0.07
|
0.05
|
0.05
|
|
PA
|
|
|
|
|
|
b/ Energies à deux corps en Kcal/mol
|
|
|
|
|
|
-43.02 -42.56 -40.70
|
-37.11
|
-53.63
|
-44.12
|
-43.79
|
|
-43.88 -43.02 -41.19
CCS
|
-37.83
|
-53.20
|
-44.38
|
-44.14
|
* kl= <Owk-Oi-Owl *Conf. = Configurations

: diminuent.
et
III.A.2/ Variation des composantes de l'effet à
trois corps en fonction de l'angle kl:
, , ,
P2 ) )
PA
La méthode SAPT nous a permis d'avoir les
différentes composantes de l'énergie à trois corps , , ,
,
) 3 )
(paragraphe I.B, équation (43)), la première
constatation est que l'énergie d'induction
au second ordre ,
représente la plus importante contribution énergétique
de
)
l'effet à trois corps non additif, elle est
répulsive, ensuite c'est l'énergie d'échange
M P2 ,
elle est attractive. Nous citons les cinq autres termes de l'énergie
P2 )
d'interaction à trois corps par ordre de contribution
énergétique décroissante:
l'induction-dispersion au
troisième ordre , répulsive ; les effets d'échange-
3 )
RPA
induction et le terme non additif Heitler-London , sont
attractives et du
même ordre de grandeur; enfin l'échange-dispersion
au second ordre et la
)
dispersion au troisième ordre , sont presque
négligeables.
)
PA
Soient la variations de ces composantes en fonction de l'angle kl
qui augmente :
: croit de façon répulsive, des calculs faits avec
les configurations X2a et X2b,
)
donnent des énergies qui convergent vers les
résultats de la R110 (angle kl = 110.5°). : des valeurs attractives
qui augmentent de façon uniforme sauf pour la
P2 )
structure X2 dont l'angle kl est de 115.6°, l'ordre est
perturbé, des calculs faits avec
les configurations X2a et X2b, donnent des énergies qui
convergent nettement vers les résultats de la R1 10.
|
: croit de manière répulsive, les résultats
obtenus avec les clusters X2a et
|
X2b convergent vers l'énergie de la R1 10.
: des énergies négatives qui croissent en valeurs
absolues, les calculs faits avec les configurations X2a et X2b tendent au
résultat obtenu pour la R1 10.
: contributions énergétiques attractives qui
deviennent de plus en plus importantes, les énergies de la X2a et la X2b
convergent vers la valeur de la R110.
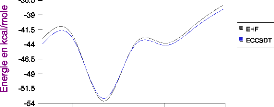
Graphe I
Energies d'interaction : Systèmes OH-(H2O)2
2
a/ Energies à trois corps
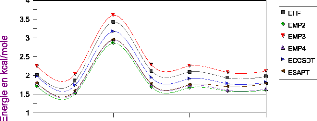

b/ Energies à deux corps
III.A.3 / Comparaison des effets à trois corps aux
résultats CCSD(T) :
Les énergies HF convergent bien vers les
résultats CCSD(T) [124]. La théorie de la perturbation
de la symétrie adaptée (SAPT) et la méthode Moller-Plesset
à l'ordre quatre avec excitations simple double et quadruple (MP4SDQ),
donnent des énergies presque identiques, qui se rapprochent beaucoup des
résultats CCSD(T), les valeurs SAPT sont légèrement
meilleures. Les résultats MP2 et MP3 tendent aussi vers les valeurs
CCSD(T) [124]. Sur le graphe I a), on voit que les courbes M P4 et
Sapt sont presque confondues, les meileurs résultats sont ceux des
méthodes HF et SAPT.
III.A.4/ Variation de l'énergie à deux corps
:
A la fin du Tableau I, nous avons donné les
énergies à deux corps HF et CCSD(T), les énergies des
paires, HF et CCSD(T), convergent aussi voir graphe I b). Les contributions
à trois corps CCSD (T) représentent moins de 4.5% de la somme des
énergies des paires CCSD(T). La somme des interactions à deux
corps est attractive pour chacune des configurations et diminue en fonction de
l'anglekl croissant sauf
pour le système X2. De même que pour les effets
à trois corps, des calculs des énergies des paires faits avec les
clusters X2a et X2b donnent des énergies qui convergent vers la valeur
de la configuration R110.
L'énergie total de ces configurations à trois
corps, qui est la somme des énergies des paires et l'effet à
trois corps non additif, est attractive.
III.B/ Résultats obtenus avec les configurations
OH-(H2O)2 du système OHïH2O)4 (Fig. 2, Tableau II, Graphe II) :
Les quatre molécules d'eau de la structure
OH-(H2O)4 appartiennent à la première couche de
solvatation de l'ion hydroxyde. Nous avons représenté les effets
à trois corps et à deux corps de toutes les configurations
OH-(H2O)2 appartenant à la structure OH-(H2O)4.
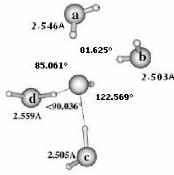
Fig.2 Structure OH-(H2O)4 (T4)
III.B.1/ Variation de l'énergie à trois corps
en fonction de l'angle kl:
Dans la série T les paramètres
géométriques des molécules d'eau ont été
fixés à leurs valeurs expérimentales (paragraphe II.A.3),
l'angle kl varie de 81.6 à 144.2°. Nous avons les énergies
à trois corps E(3corps): HF, MP2, MP3, MP4SQD, CCSD(T) et SAPT, qui sont
répulsives. La variation de ces contributions à l'interaction
intermoléculaire en fonction de l'angle kl croissant est la suivante
:
kl = 81.6 à 85.1°, elles diminuent.
kl = 85.1 à 122.6°, elles augmentent. Pour la
CCSD(T), l'énergie augmente jusqu'à kl = 90.0° et à
partir de cette valeur, elle a de nouveau la même variation en fonction
de l'angle que les résultats obtenus avec les autres méthodes.
kl = 122.6 à 134.5°, elles diminuent.
kl = 134.5 à 144.2°, elles augmentent.
est répulsive comme précédemment et varie de
la même façon que .
On ne peut pas comparer cette variation à celle de la
série R où l'angle d'une configuration à une autre
augmente de 30° alors que dans cette série l'angle varie de 5
à 15°. Cependant, nous pouvons noter, que si nous prenons le
même intervalle de kl,
nous avons la même variation de l'effet à trois
corps dans les deux séries :
Si nous passons de kl = 80.9 à 140.4° dans la
série R et de kl = 81.6 à 144.2° dans la série T, les
valeurs des énergies baissent dans les deux séries.
Pour des valeurs d'angles kl très proches, nous avons
les énergies à trois corps répulsives des configurations
OH-(H2O)2 de la série R, qui sont moins importantes que
celles des sous systèmes OH-(H2O)2 de la série T,
appartenant au cluster OH-(H2O)4 :
Pour kl = 80.9° dans la R80 et kl = 81.6° dans la T4ab,
E(3corps) de R80 est inferieur a E(3corps) de T4ab.
Pour kl = 140.4° dans la R140 et kl = 144.2° dans la
T4bd, E(3corps) de R140 est inferieur a E(3corps) de T4bd.
Les différences entre les configurations
OH-(H2O)2 des séries R et T sont, l'orientation des deux
molécules d'eau dans l'espace, le fait que la configuration de la
série R est un système OH-(H2O)2 et celle de la
série T est un sous-système de la structure OH(H2O)4 et les
distances dik (k désigne l'une des deux molécules
d'eau, dans chaque sous système), elles valent dans la série R :
dia = 2.642 et dib = 2.614. Dans la série T : pour T4ab, dia = 2.546
Å et dib = 2.503 Å. et pour T4bd, dib = 2.503 Å et did =
2.559 Å (voir figures 1et 2). Les distances dik sont
différentes car les procédés d'optimisation sont
différents.
En conséquence, E(3corps) a augmenté, quand
dik a diminuée en passant de la série R à la
série T.
Pour vérifier que ce résultat est bien l'effet de
la distance dik et non le fait que le
nombre de molécules
d'eau augmente dans la première couche quand on passe de la
série R à la série T, nous avons
comparé trois autres configurations dont les
paramètres
géométriques des molécules d'eau sont
les mêmes et l'angle kl est à peu près égal.
Soient les configurations R1 10 (kl = 1 10.5°), X2b (kl =
1 15.6°) et T4bc (kl =
122.6°), les distances dik, sont
respectivement en Å : dia = 2.642, dib = 2.614 ; dia = 2.591, dib = 2.580
et dib = 2.503, dic = 2.505 Å. De la première à la
troisième configuration dik diminue et E(3corps) augmente, et
ce, malgré que la R110 et X2b soient toutes les deux des systèmes
OH-(H2O)2 , c'est-à-dire avec le même nombre de
molécules dans la première couche, la conclusion
précédente est donc à vérifier en faisant d'autres
calculs sur un plus grand nombre de configurations.
Les énergies trouvées attractives dans les
systèmes OH-(H2O)2 le sont dans les sous systèmes
OH-(H2O)2 de la structure OH-(H2O)4 et même chose
pour celles qui sont positives.
III.B.2/ Variation des composantes de l'effet à
trois corps en fonction de l'angle kl:
Nous avons observé la variation des composantes de
l'effet à trois corps obtenues avec la théorie SAPT pour les
configurations OH-(H2O)2 du système OH(H2O)4 en fonction de
l'angle kl, nous avons les résultats suivants :
: Elle est répulsive, diminue de kl = 81.6 à
85.1°, ensuite augmente jusqu'à
)
kl = 122.6°, baisse à kl = 134.5° et augmente
fortement pour kl = 144.2°. La différence entre ces
résultats et les valeurs de la série R, pour des angles kl
à peu près
égaux, est que les énergies d'induction au
second ordre, de ces configurations sont plus importantes malgré que les
géométries des molécules d'eau sont les mêmes. Ceci
est dû aux méthodes d'optimisation différentes dans les
deux séries, qui donnent certains paramètres
géométriques inégaux.
: Elle est attractive, en valeur absolue, elle varie en fonction
de l'angle kl
P2 )
comme la contribution à l'effet à trois corps , par
rapport aux résultats des
)
configurations de la série R, même constatation que
précédemment.
|
: Elle est répulsive, varie comme en fonction de l'angle
kl et mêmes
)
|
remarques vis-à-vis de la série R.
|
: Elle est attractive et varie en valeur absolue comme
|
|
et même conclusion
|
par rapport aux valeurs des configurations de la série
R.
: Elle est attractive, en valeur absolue diminue de kl = 81.6
à 90.0°, augmente à
kl = 122.6°, diminue à kl = 134.5° et
augmente à 144.2°. En valeur absolue les contributions
énergétiques de ces configurations sont aussi plus importantes
que ceux de la série R.
Tableau II: Energies d'interaction des
sous-systèmes OH-(H2O)2
de la structure OH-(H2O)4
a/ Energies à trois corps en Kcal/mol
|
Conf.
|
T4ab
|
T4ad
|
T4cd
|
T4bc
|
T4ac
|
T4bd
|
|
N° couches
|
1-1
|
1-1
|
1-1
|
1-1
|
1-1
|
1-1
|
|
kl(°)
|
81.6
|
85.1
|
90.0
|
122.6
|
134.5
|
144.2
|
|
=1
|
-0.62
|
-0.55
|
-0.34
|
-0.89
|
-0.78
|
-1.05
|
|
2.36
|
1.89
|
2.19
|
2.35
|
2.10
|
2.16
|
|
2.98
|
2.44
|
2.53
|
3.25
|
2.88
|
3.21
|
|
1.94
|
1.50
|
1.76
|
1.81
|
1.63
|
1.71
|
|
P2 ?
|
|
|
|
|
|
|
|
2.66
|
2.15
|
2.48
|
2.53
|
2.23
|
2.32
|
|
P
|
|
|
|
|
|
|
|
2.09
|
1.63
|
1.88
|
1.90
|
1.70
|
1.76
|
|
MP4
|
|
|
|
|
|
|
|
2.33
|
1.86
|
2.13
|
2.11
|
1.88
|
1.97
|
|
CC
|
|
|
|
|
|
|
|
APT
|
2.07
|
1.61
|
1.88
|
1.94
|
1.75
|
1.91
|
|
|
APT
|
=1+2+3+4+5+6+7
|
|
|
|
=2
|
3.39
|
2.75
|
3.41
|
4.45
|
3.95
|
5.20
|
|
=3
|
-0.40
|
-0.31
|
-0.88
|
-1.20
|
-1.07
|
-2.00
|
|
=4
|
0.27
|
0.26
|
0.21
|
0.08
|
0.01
|
-0.01
|
|
= 5
|
0.45
|
0.41
|
0.76
|
1.33
|
1.11
|
1.51
|
|
3
RPA
|
|
|
|
|
|
|
|
=6
|
-1.21
|
-1.10
|
-1.44
|
-1.88
|
-1.51
|
-1.77
|
|
P2 )
|
|
|
|
|
|
|
|
7
)
|
0.20
|
0.15
|
0.16
|
0.06
|
0.04
|
0.02
|

b/ Energies à deux corps en Kcal/mol
-38.89 -30.33 -30.21 -38.82 -39.44 -30.09
|
-39.81 -31.74 -31.81 -39.36 -39.76 -31.06
|
*kl = <Owk-Oi-Owl *Conf. = Configurations
Graphe II
Structure OH-(H2O)4
-2
a/ Energies à trois corps des sous-systèmes
OH-(H2O)2
125 31
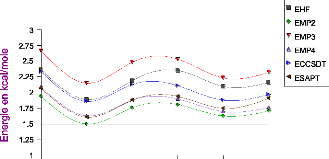
b/ Energies à deux corps
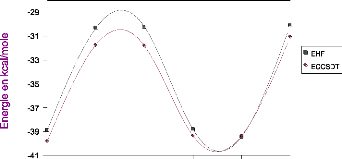
Dans le cas de la série T, les composantes de l'effet
à trois corps et ne
) )
PA
sont pas négligeables, elles sont répulsives et
légèrement supérieures à ceux de la série R
pour des angleskl très proches. Les valeurs de ces énergies
diminuent quand kl augmente.
En général toutes les énergies en valeurs
absolues des configurations OH-(H2O)2 de la structure
OH-(H2O)4 sont plus grandes que celles de la structure
OH-(H2O)2 (série R) pour des angles presque égaux et
des géométries de monomères identiques. Ceci peut
s'expliquer par le fait que les méthodes d'optimisation de ces deux
structures sont différentes et ont donné des distances
dik différentes, nous avons aussi l'ion hydroxyde, dans la
première structure est entouré par deux molécules d'eau
alors que dans la deuxième, il y'a quatre molécules d'eau dans la
première couche de solvatation.
III.B.3 / Comparaison des effets à trois corps aux
résultats CCSD(T) :
Nous obtenons les mêmes conclusions que pour la
série R (voir le paragraphe II I.A.3). Les résultats HF, SAPT et
CCSD(T) sont presque égaux (graphe II a).
III.B.4/ Variation de l'énergie à deux corps
:
Contrairement aux effets à trois corps, les
énergies des paires de la série T, sont en valeurs absolues
infériueres à celles de la série R, c'est à dire
moins attractives. L'énergie totale qui est la somme des énergies
à deux corps et à trois corps reste attractive pour toutes les
configurations OH-(H2O)2. Dans ce cas l'effet à trois corps
atteint 5.85% de l'énergie des paires. Les résultats HF sont en
général très proches de ceux obtenus avec la
méthode CCSD(T) (graphe II b). Toutes les énergies des paires des
configurations OH-(H2O)2 de la structure OH-(H2O)4 sont
attractives. La variation de l'énergie des paires en fonction de l'angle
donne en valeur absolue:
E(2corps) qui diminue de kl = 81.6° à 90.0°,
ensuite E(2corps) augmente dekl = 90.0 à 134.5° et baisse à
kl = 144.2°, nous avons la même variation que les énergies
à trois corps, nous pouvons conclure que plus les énergies
à deux corps sont attractives,
plus l'effet à trois corps est répulsive et
inversement.
Pour des valeurs d'angles kl très proches, nous avons
les énergies à deux corps des configurations OH-(H2O)2
de la série T, qui sont moins importantes que celles des systèmes
OH-(H2O)2 de la série R :
Pour kl = 80.9° dans la R80 et kl = 81.6° dans la
T4ab, E(2corps) de R80 est superieur a E(2corps) de T4ab.
Pour kl = 140.4° dans la R140 et kl = 144.2° dans la
T4bd, E(2corps) de R140 est superieur a E(2corps) de T4bd.
Ce résultat est l'inverse de ce qui a été
trouvé pour l'effet à trois corps.
L'inégalité des énergies à deux
corps de deux configurations ayant le même angle kl et les mêmes
paramètres géométriques des monomères, appartenant
à deux structures différentes, est due aux mêmes raisons
citées pour l'inégalité des effets à trois corps de
ces configurations (paragraphe III. B.1), principalement le paramètre
géométrique dik.
III.C/ Résultats obtenus avec les configurations
OH-(H2O)2 du système OH(H2O)10 (Fig. 3, Tableau III, Graphe III) :
Les molécules d'eau de la structure
OH-(H2O)10 appartiennent à la première et à la
deuxième couches de solvatation de l'ion hydroxyde OH-. Nous
avons noté les molécules d'eau appartenant à la
première couche par : a, b, c et d et les molécules d'eau
appartenant à la deuxième couche par : e, f, g, h, i, j (figure
3). Nous avons quatre molécules d'eau dans la première couche et
six dans la deuxième( Å).
3
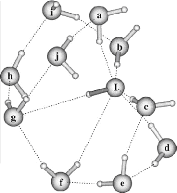
Fig. 3 Structure OH- (H2O)10 (F10)
Nous avons choisi quelques configurations OH-(H2O)2
de la structure OH-(H2O)10 pour calculer les effets à trois
corps et à deux corps, de façon à voir la variation de ces
énergies quand une des deux molécules d'eau appartient à
la première couche de solvatation de l'ion hydroxyde et l'autre à
la deuxième ou quand les deux molécules d'eau appartiennent
à la deuxième couche de solvatation.
III.C.1/ Variation de l'énergie à trois corps
en fonction de l'angle kl:
L'angle kl dans les quatre premières configurations varie
de 50.1 à 124.3° et
les deux molécules d'eau sont situées à la
première et à la deuxième couche de l'hydratation de l'ion
hydroxyde.
Les effets à trois corps obtenus avec les
différentes méthodes théoriques, HF, MP2, MP3, MP4SQD,
CCSD(T) et SAPT sont plus faibles que dans les cas où les deux
molécules appartiennent à la première couche et dans ce
cas, nous avons même des valeurs de l'énergie à trois corps
attractives pour kl = 50.1 et 64.0°.
*Les valeurs de E(3corps) augmentent quand l'angle kl croit.
est attractive ou répulsive et varie de la même
façon que l'effet à trois corps, en
fonction de l'angle kl.
Les structures OH-(H2O)10 et OH-(H2O)4
ont été optimisées avec la même méthode
(Monte Carlo[113]), elles ont toutes les deux quatre
molécules d'eau dans la première couche et les
géométries des monomères dans les deux systèmes
sont identiques. Si nous comparons les résultats de deux configurations
prises des deux structures, avec un angle, presque identique, kl(T4bc) =
122.6° et kl(F10bj) = 124.3°, nous avons:
E(3corps) de F1 0bj est inferieur a E(3corps) de T4bc.
(a)L'effet à trois corps quand les deux
molécules d'eau appartiennent à la première couche est
plus important que quand une molécule d'eau est à la
première couche et l'autre à la deuxième.
Soient les deux configurations OH-(H2O)2 , F10gf et
F10ei où les deux molécules d'eau appartiennent à la
deuxième couche :
E(3corps) est répulsive et baisse quand on passe d'un
angle kl = 45.1 à 149.3°.
Si nous comparons l'effet à trois corps entre deux
configurations, dans l'une les deux molécules d'eau n'appartiennent pas
à la même couche de solvatation et dans l'autre les deux
molécules d'eau appartiennent à la deuxième couche, la
F10aj et la F10gf, qui ont des angles kl très proches, 50.1 et
45.1°, nous avons :
E(3corps) F10aj est inferieure à E(3corps) F10gf.
(b)L'effet à trois corps quand une
molécule appartient à la première couche et l'autre
à la deuxième est moins important que quand les deux
molécules d'eau appartiennent à la deuxième couche de
solvatation.
Maintenant si nous comparons l'effet à trois corps
entre deux configurations, dans l'une les deux molécules d'eau
appartiennent à la première couche de solvatation et dans l'autre
les deux molécules d'eau appartiennent à la deuxième
couche, la T4bd et la F10ei, qui ont des angles kl très proches, 144.2
et 149.3°, nous avons :
E(3corps) F10ei est inférieure a E(3corps) T4bd.
(c) L'effet à trois corps quand les deux
molécules d'eau appartiennent à la deuxième couche est
mois important que quand elles font partie les deux de la première
couche. En conséquence, des résultats (a), (b) et (c) nous
pouvons déduire :
"E(3corps) d'une configuration où les deux
molécules d'eau appartiennent à la première couche est
superieur E(3corps) d'une configuration où les deux molécules
d'eau appartiennent à la deuxième couche est superieur E(3corps)
quand les deux molécules d'eau n'appartiennent pas à la
même couche[124]."
Tableau III: Energies d'interaction des
Sous-systèmes OH-(H2O)2 de la structure
OH-(H2O)10
Les molécules d'eau de la première couche sont : a,
b, c Les molécules d'eau de la deuxième sont : e, f, g, i, j
a/ Energies à trois corps en Kcal/mol
Conf. F10aj F10cj F10bg F10bj F10gf F10ei
N° couches 1-2 1-2 1-2 1-2 2-2 2-2
kl(°) 50.1 64.0 118.2 124.3 45.1 149.3
=1 -0.12 0.04 0.02 -0.06 -0.11 -0.01
-1.09 -0.06 0.14 0.38 0.44 0.11
-0.97 -0.10 0.12 0.44 0.55 0.12








-1.14 -0.01 0.18 0.41 0.47 0.14
-1.09 0.01 0.18 0.45 0.48 0.13
-1.09 0.02 0.18 0.41 0.48 0.14
-1.06 0.07 0.21 0.46 0.51 0.16
-1.15 0.01 0.18 0.43 0.49 0.14

=1+2+3+4+5+6+7
=2 -1.04 -0.21 0.10 0.56 0.56 0.14
=3 0.07 0.11 0.02 -0.12 -0.01 -0.02
=4 0.09 0.03 -0.00 -0.00 0.02 -0.00
=5 -0.22 -0.08 0.04 0.13 0.02 0.02
=6 0.03 0.09 0.00 -0.08 -0.01 0.00
=7 0.04 0.03 0.00 0.00 0.02 0.00
b/ Energies à deux corps en Kcal/mol
-28.19 -29.20 -23.53 -27.44 -13.42 -12.14

-29.39 -29.54 -24.00 -27.58 -14.67 -12.16
*kl= <Owk-Oi-Owl et Conf. =Configurations
Graphe III
Energies d'interaction : Systèmes OH-(H2O)10 a/
Energies à trois corps
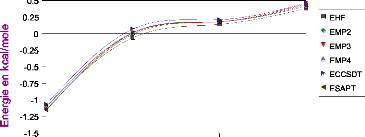
b/Energies à deux corps
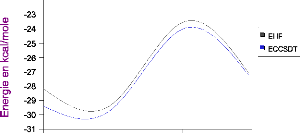
III.C.2/ Variation des composantes de l'effet à
trois corps en fonction de l'angle kl:
La variation des composantes de l'effet à trois pour les
configurations OH(H2O)2 du système OH-(H2O)10 en fonction de
l'angle kl, donne :
: Les énergies des configurations OH-(H2O)2
qui ont une molécule des deux
)
molécules d'eau dans la première couche et l'autre
dans la deuxième, croissent quand kl augmente, contrairement aux
configurations qui ont deux molécules d'eau dans la deuxième
couche.
Pour des comparaisons similaires à ceux faits avec
E(3corps), nous avons la même conclusion :
" d'une configuration où les deux molécules d'eau
appartiennent à la première
)
couche est superieur a d'une configuration où les deux
molécules d'eau
)
appartiennent à la deuxième couche est superieur
quand les deux molécules
)
d'eau n'appartiennent pas à la même couche."
: Elle est attractive ou répulsive, par rapport aux
résultats obtenus avec la série
P2 )
R et T, les valeurs sont presque négligeables. Dans ce
cas, nous avons :
"
P2 ) d'une configuration où les deux
molécules d'eau appartiennent à la première couche est
inferieur a d'une configuration où les deux molécules d'eau
P2 )
appartiennent à la deuxième couche est inferieur a
quand les deux molécules
P2 )
d'eau n'appartiennent pas à la même couche."
|
: attractive ou répulsive, augmente en fonction de
l'angle kl pour les
|
configurations où les deux molécules d'eau
n'appartiennent pas à la même couche de solvatation de l'ion. Pour
les configurations où les deux molécules d'eau sont dans la
deuxième couche, exemple F1 0gf et F1 0ei, cette énergie est
presque nulle. En comparaison aux résultats obtenus avec la série
R et T, Ces valeurs sont presque négligeables et nous avons :
"

d'une configuration où les deux molécules d'eau
appartiennent à la
première couche est superieur a d'une configuration
où les deux molécules
3 )
RP
d'eau appartiennent à la deuxième couche est
superieur a quand les deux
3 )
RPA
molécules d'eau n'appartiennent pas à la même
couche."
: attractive ou répulsive et très faible par
rapport aux autres composantes de l'énergie.
"d'une configuration où les deux molécules d'eau
à la première couche est plus
attractive que d'une configuration où les deux
molécules d'eau appartiennent à la
deuxième couche est plus attractive que quand les deux
molécules d'eau n'appartiennent pas à la même couche."
: Même chose que .
Dans le cas de la série F, les composantes de l'effet
à trois corps et
) )
PA
sont très petites.
"Ces deux composantes de l'effet à trois corps sont
plus importantes quand les deux molécules d'eau appartiennent à
la première couche d'hydratation de l'ion OH- que lorsque les
deux molécules d'eau n'appartiennent pas à la même couche
et dans ce cas, les termes de l'effet à trois corps sont plus grands
lorsque les deux molécules font partie de la deuxième couche."
III.C.3 / Comparaison des effets à trois corps aux
résultats CCSD(T) :
Nous obtenons les mêmes résultats que la
série R et T (voir le paragraphe III.A.3). Graphe III a.
III.C.4/ Variation de l'énergie à deux corps
:
Les énergies des paires de la série F sont
attractives et en valeurs absolues elles sont inférieures à
celles de la série R et T. Les résultats HF sont très
proches de ceux obtenus avec la méthode CCSD(T), graphe III b.
Quand les deux molécules d'eau n'appartiennent pas
à la même couche de solvatation de l'ion, l'effet à trois
corps atteint jusqu'à 3.6% de l'énergie des paires et la
variation de l'énergie des paires en fonction de l'angle kl pour la
série F, est la suivante :
E(2corps) diminue de kl = 50.1 à 64.0°, ensuite
augmente à kl = 118.2° et diminue
de nouveau à 124.3°.
Les énergies des paires des configurations où
les deux molécules d'eau appartiennent à la deuxième
couche de solvatation sont moins attractives que celles des sous
systèmes où les deux molécules d'eau n'appartiennent pas
à la même couche.
III.D/ Résultats obtenus avec la méthode
DFT (Tableau IV, Graphe IV) :
Afin de comparer les calculs DFT[125] obtenus avec
deux fonctionnelles B3LYP et B3PW91 aux énergies des autres
méthodes théoriques, nous avons sélectionné des
configurations OH-(H2O)2 de différentes structures
OH-(H2O)n (n = 2, 4, 10) et nous avons faits les calculs
DFT seulement pour ces configurations.
III.D.1/ Les effets à trois corps DFT:
Les résultats obtenus avec la fonctionnelle B3LYP
convergent aux énergies à trois corps CCSD(T) à 77%, ceux
obtenus avec la B3PW91 se rapprochent à 89% et ce quelque soit la
configuration OH-(H2O)2 (graphe IV).
Les résultats de la DFT B3LYP sont à 84% de ceux
donnés par la B3PW91.
Les résultats HF tendent à 88% à ceux de la
B3PW91 et à 89% aux valeurs CCSD(T). Les valeurs des méthodes
SAPT et M P4SDQ convergent aux effets à trois corps B3PW91 à 79%
et nous avons trouvé qu'ils convergent aux valeurs CCSD(T)
respectivement à 88% et 85%.
Ces données confirment ce qui a été
trouvé précédemment, concernant l'orientation des calculs
obtenus avec les méthodes HF, MP4SDQ et SAPT vers ceux qui
résultent de la théorie CCSD (T). En conclusion, à partir
de nos calculs, nous pouvons classer les méthodes théoriques par
ordre décroissant des pourcentages de convergence des effets à
trois corps aux résultats CCSD(T), comme suit : La DFT en utilisant la
fonctionnelle B3PW91, HF, SAPT et MP4SDQ.
Tableau IV :
Effets à trois corps de sous systèmes
OH-(H2O)2 appartenant à différentes
structures
OH-(H2O)n en Kcal/mol
|
Conf.
|
R80
|
T4cd
|
X2
|
F10bg
|
R170
|
F10gf
|
|
N° couches
|
1-1
|
1-1
|
1-1
|
1-2
|
1-1
|
2-2
|
|
kl(°)
|
80.9
|
90.0
|
115.6
|
118.2
|
170.3
|
45.1
|
|
2.01
|
2.19
|
3.42
|
0.14
|
1.97
|
0.44
|
|
1.70
|
1.76
|
2.86
|
0.18
|
1.61
|
0.47
|
|
2.24
|
2.48
|
3.60
|
0.18
|
2.11
|
0.48
|
|
1.79
|
1.88
|
2.94
|
0.18
|
1.63
|
0.48
|
|
P4nt
|
|
|
|
|
|
|
|
1.97
|
2.13
|
3.17
|
0.21
|
1.81
|
0.51
|
|
CSD
|
|
|
|
|
|
|
|
1.79
|
1.88
|
2.95
|
0.18
|
1.78
|
0.49
|
|
T
|
|
|
|
|
|
|
|
2.26
|
2.40
|
3.35
|
0.29
|
2.05
|
0.73
|
|
LYP
|
|
|
|
|
|
|
|
PW9
|
1.93
|
2.09
|
3.32
|
0.23
|
2.14
|
0.59
|
*kl= <Owk-Oi-Owl
*Conf. = Configurations
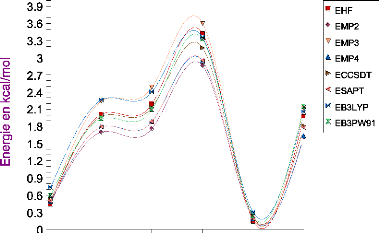
Systèmes OH-(H2O)2 appartenant à
différentes structures OH-(H2O)n
03
Effet à trois corps
III.D.2/ Comparaison des énergies de la
première itération DFT au terme Heitler-London (Tableau V,
Graphe V):
Nous avons mentionné au paragraphe I.B que le terme non
additif HeitlerLondon est identique à celui de l'énergie de la
première itération HF, ,
st it a ?
ceci d'après la référence [60].
Les énergies de la première itération DFT
calculées avec deux fonctionnelles, la B3LYP et la B3PW91 ne sont pas
les mêmes que les valeurs Heitler-London, néanmoins les
résultats obtenus avec la fonctionnelle B3PW91 sont très proches
de ces dernières et ce quelque soit la configuration
OH-(H2O)2, graphe V.
De même que le terme Heitler-London, les énergies de
la première itération DFT sont attractives.
Tableau V :
Energies de la première itération en Kcal/mol
|
Conf.
|
R80
|
T4cd
|
X2
|
F10bg
|
R170
|
F10gf
|
|
N° couches
|
1-1
|
1-1
|
1-1
|
1-2
|
1-1
|
2-2
|
|
kl(°)
|
80.9
|
90.0
|
115.6
|
118.2
|
170.3
|
45.1
|
|
-0.34
|
-0.34
|
-0.62
|
0.02
|
-0.90
|
-0.11
|
|
ter
|
-0.05
|
0.03
|
-0.57
|
-0.00
|
-0.96
|
-0.09
|
|
-0.41
|
-0.40
|
-0.70
|
-0.00
|
-0.97
|
-0.19
|
*kl = <Owk-Oi-Owl *Conf. = Configurations
Energies de la première itération de
différentes
configurations OH-(H2O)2
-038
Effets a trois corps
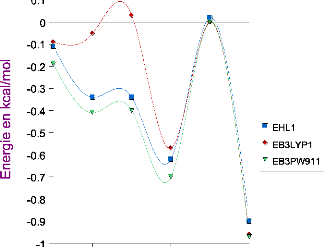
III.D.3/ Les énergies de délocalisation DFT
(Tableau VI, Graphe VI) :
Nous avons vu au paragraphe précédent que
l'énergie de la première itération DFT est
différente du terme Heitler-London. La différence entre
l'énergie d'interaction non additive et l'énergie de la
première itération de chaque méthode théorique est
due à la délocal isation électronique du système,
donc très importante, nous l'avons représenté par dans le
Tableau VI. Il est
E E
M = (
intéressant de comparer la variation de cette
quantité à celle de la méthode
DFT. MT=HF, M P3, M P4SDQ, CCSD(T), SAPT,
B3LYP ou B3PW91.
= dans toutes les méthodes (paragraphe I.B) sauf dans le
cas de la DFT,
er
=ou .
ter
Nous avons trouvé que est répulsive quelque soit la
méthode de calcul.
Le résultat le plus important et étonnant que nous
avons tiré du Tableau VI, est le
|
suivant :
Les valeurs et
MP SDQ
4
|
convergent à 97%, ce qui est normal puisque les effets
à
|
trois corps M P4SD Q et SA PT convergent à 97% et nous
leur avons retranché la même
quantité . Par contre les effets à trois corps DFT,
B3LYP et B3PW91 convergent à
84% et nous ne leur avons pas soustrait la même valeur pour
le calcul et
LYP
, qui convergent à 93%, en plus sans les configurations
de OH-(H2O)2
PW 91
instables, comme la X2, ces énergies auraient
convergé 99% !
Ceci s'explique par le fait que les énergies de la
première itération et les effets à trois corps soient de
signes opposées et que dans le cas de la D FT B3LY P et B3PW91, la B3LYP
donne des énergies de la première itération peu
attractives et des énergies à trois corps plus répulsives
par rapport aux résultats de la B3PW91, ce qui fait, que les
différences ou les énergies de délocalisation des
différentes configurations tendent à avoir les mêmes
valeurs avec les deux fonctionnelles.
Par rapport aux valeurs , les résultats obtenus pour et
,
CSD( )
T LYP PW 91
convergent tous les deux à 86%.
, , et tendent vers les énergies avec les mêmes
T MP SDQ
4 CSD( )
T
pourcentages que dans le cas de la comparaison des effets
à trois corps c'est-à-dire respectivement 89, 88 et 85 %.
Le classement des différentes méthodes
théoriques par ordre décroissant des pourcentages de convergence
des énergies de délocalisation aux résultats CCSD(T) est
le suivant : , , et , graphe V I.
T PW 91 P loc
4
Tableau VI:
Energies de délocalisation à trois corps de sous
systèmes OH-(H2O)2 appartenant
à différentes
structures de OH-(H2O)n en Kcal/mol
|
Conf.
|
R80
|
T4cd
|
X2
|
F10bg
|
R170
|
F10gf
|
|
N° couches
|
1-1
|
1-1
|
1-1
|
1-2
|
1-1
|
2-2
|
|
kl(°)
|
80.9
|
90.0
|
115.6
|
118.2
|
170.3
|
45.1
|
|
-
|
2.35
|
2.53
|
4.04
|
0.12
|
2.87
|
0.55
|
|
-
|
2.04
|
2.10
|
3.48
|
0.16
|
2.51
|
0.58
|
|
-
|
2.58
|
2.82
|
4.22
|
0.16
|
3.01
|
0.59
|
|
-
i4St
-
|
2.13
2.31
|
2.22
2.47
|
3.56
3.79
|
0.16
0.19
|
2.53
2.71
|
0.59
0.62
|
|
CSD
-
|
2.13
|
2.22
|
3.57
|
0.16
|
2.68
|
0.60
|
|
T
-
|
2.31
|
2.37
|
3.92
|
0.29
|
3.01
|
0.82
|
|
LYP tr
-
PW9 ite3
|
2.34
|
2.49
|
4.02
|
0.23
|
3.07
|
0.78
|
*kl= <Owk-Oi-Owl *Conf. = Configurations
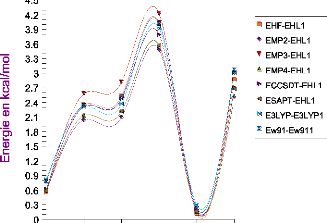
Graphe VI
Energies de délocalisation de
différentes
configurations OH-(H2O)2
09
Effet a trois corps
III.E/ Conclusion :
1/ Les énergies à trois corps sont
répulsives alors que les énergies à deux corps sont
attractives.
2/ L'influence des paramètres
géométriques sur la variation des énergies à trois
et à deux corps est très importante, en particulier les distances
OH dans les monomères des structures OH-(H2O)n.
3/ La stabilité des structures
OH-(H2O)n obtenues par différents
procédés d'optimisation est relative, les résultats
varient d'une méthode à une autre et ce n'est pas facile de
sélectionner les configurations vu la diversité des
références.
4/ Les plus importantes contributions énergétiques
de l'effet à trois non additif sont dans l'ordre, l'énergie
d'induction au second ordre , elle est répulsive et
)
l'énergie d'échange au niveau MP2, qui est
attractive.
5/ Les pourcentages des effets à trois par rapport aux
sommes des énergies des paires dans les configurations,
OH-(H2O)2 et OH-(H2O)4, sont respectivement 4.5 et 5.85%.
Les effets à trois corps dans le cas où quatre molécules
d'eau appartiennent à la première couche de solvatation de l'ion
sont plus répulsives et les énergies des paires plus attractives
que dans le cas où il y'a deux molécules d'eau.
6/ les configurations OH-(H2O)2 où les deux
molécules d'eau appartiennent à la deuxième couche
d'hydratation de l'ion ont des effets à trois corps et à deux
corps moins importants que ceux des configurations où les deux
molécules d'eau n'appartiennent pas à la même couche, qui
sont moins importants que ceux où les deux molécules d'eau
appartiennent à la première couche.
7/ Les signes des différentes contributions
énergétiques sont en général les mêmes dans
toutes les structures.
8/ Dans les méthodes théoriques, HF, MP3,
MP4SDQ, CCSD(T) et SAPT, les énergies de la première
itération sont égaux au terme Heitler London, qui a
été calculé par la méthode SAPT alors que les
énergies de la première itération DFT calculées
avec deux fonctionnelles, B3LYP et B3PW91 sont différentes de cette
énergie. Dans le cas de calculs DFT, les énergies de la
première itération ne sont pas égales à la
composante Heitler London.
9/ Les méthodes théoriques sont classées
par ordre décroissant des pourcentages de convergence des effets
à trois corps et à deux corps aux résultats CCSD(T), comme
suit : La D FT en utilisant la fonctionnelle B3PW91, H F, SA PT et M P4SDQ, ces
deux dernières donnent des résultats presque similaires.
10/ Pour les configurations OH-(H2O)2 où les
deux molécules d'eau appartiennent à la première couche de
solvatation de l'ion hydroxyde, nous avons les énergies des paires de la
série T(n=4), sont en valeurs absolues inférieures à
celles de la série R(n=2), c'est à dire moins attractives. Par
contre les énergies à trois corps de la série T sont plus
répulsives. Les énergies totales de la série T sont moins
attractives que ceux de la série R. Les configurations
OH-(H2O)2 de la série R(n=2) sont plus stables que ceux de la
série T(n=4).
Quand n augmente les sous systèmes OH-(H2O)2
deviennent moins stables.
III..F/ Méthodes de génération et
d'optimisation des géométries des systèmes
moléculaires:
Vu l'importance de l'ion hydroxyde OH- et en
général des agrégats moléculaires[111],
plusieurs études théoriques et expérimentaux ont
été menées [75-105], afin d'essayer de connaître le
processus de réaction ion hydroxyde-molécules d'eau en phase
gazeuse et liquide. Dans ces études, différents
procédés sont utilisés pour générer des
configurations de systèmes OH-(H2O) n et les optimiser par la
suite. Nous citerons, dans ce qui suit, les méthodes les plus
utilisées:
III.F.1/ Méthodes théoriques de
génération des configurations:
- La méthode Monte-Carlo est utilisée pour
générer des configurations OH-(H2O)n, des
calculs avec cette méthode, appuient que le nombre d'hydratation de
l'ion hydroxyde dans les solutions aqueuses, est 5 [81] et 6
[80].
- La méthode ab initio de dynamique moléculaire a
prédit que OH- a un nombre de coordination de 4
molécules d'eau dans les solutions aqueuses[5,6,82,88].
III.F.2/ Méthodes expérimentales de
génération des configurations:
- des informations concernant l'ion hydroxyde OH-
peuvent être extraites d'études IR et Raman[106]
d'hydroxydes en solutions aqueuses. La position de la vibration OH-
dans le spectre IR[107] et la différence des
intensités dans le spectre Raman[110] pour le mode stretching
symétrique maintient la proposition que le groupe OH n'a pas de liaison
hydrogène par son hydrogène mais il existe une forte liaison
hydrogène entre l'oxygène de l'hydroxyde et les hydrogènes
des molécules d'eau.
- Yang et Clasterman[98] ont utilisé un
réacteur à tube à flux pour produire beaucoup de clusters
avec 60 molécules d'eau au moins. Ils ont trouvé que les clusters
de l'ion hydroxyde avec 11, 14, 17, et 20 molécules d'eau augmentent la
stabilité avec l'existence de discontinuités, anomalies (souvent
appeler nombres magiques) dans l'intensité du spectre de masse.
- Arshadi et Kebarle ont utilisé une source d'ions
à haute pression de 2000 eV pour
l'ionisation pour étudier
l'équilibre de formation de clusters et déterminer
les
enthalpies d'hydratation() progressives de : -22.5, -16.4, -15.1, - 14.2
et -
14.1 Kcal/mol pour l'ajout successive, jusqu'à 5
molécules d'eau à OH-, ces valeurs d'enthalpies sont
plus faibles que ceux (non équilibres) de De Paz et al[98]
parce que l'expérience a été faite avec la méthode
d'ionisation continue, par conséquent, ne suit pas la dépendance
du temps des concentrations d'ions.
- Des mesures plus récentes de Meotner(Mautner) et
Speller[79], utilisant un spectromètre de masse à
haute pression, pulseur, ont produit les enthalpies progressives de : -26.5,
-17.6, -16.2, -12.0, -11.5, -11.2 et 10.4 Kcal/mole, pour l'addition
successive, jusqu'à 7 molécules d'eau à OH-.
Ces résultats ont soutenu les estimations de
Kebarle[77,99].
III.F.3/ Méthodes d'optimisation des configurations:
Les configurations générées par moyens
théoriques ou expérimentaux sont optimisées en
général avec des méthodes ab initio. Au cours de
l'optimisation les paramètres géométriques des
monomères peuvent être laissés tels quels ou
remplacés par les valeurs expérimentaux. Les
procédés les plus utilisés pour l'optimisation sont:
- L'optimisation du système tolal au niveau MP2. - La
méthode Monte Carlo[113].
- La méthode d'optimisation Berny[112].

Chapitre IV :
Interprétation des résultats pour le
système RbOH
IV.A/ Interaction étudiée dans le
Système RbOH :
Nous nous intéressons à l'interaction entre Rb
(Rubidium) et OH (radical hydroxyde) pour des molécules froides.
L'objectif de notre travail est d'avoir les observables ou
sections efficaces de collision. Pour cela, nous devons calculer les
polarisabilités de OH et Rb, ensuite déterminer l'énergie
d'interation à grande distance, c'est à dire l'induction et la
dispersion. A partir des énergies, nous pourrons obtenir les
observables. Celles ci sont comparées aux résultats
expérimentaux.
Réaction :
On décélère OH, c'est-à-dire, on
baisse son énergie cinétique, ensuite on le bombarde avec un
alcalin Rb pour le décélérer encore plus «voir
1)», OH à l'approche de Rb perd sa
dégénérescence, il y'a levé de
dégénérescence «voir 2)» :
Rb + OH (2) OH doublement
dégénéré :
ou
1) Cette interaction dépend de R et :
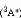
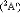
Quand on bombarde OH avec Rb, OH passe de l'état
dégénéré de symétrie 2à
l'état non dégénéré de symétrie et
.
Calcul du potentiel
d'interaction[114] :

La constante , doit être calculer pour avoir le potentiel
:
variable d'intégration [fréquence]
Ce qui revient à calculer la polarisabilité lors du
refroidissement du radical
)
hydroxyde seul, c'est-à-dire quand il passe de
l'état 2à l'état n ou états
excités :
Nous avons calculé pour chaque symétrie de OH :
:
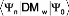
O
= ~ ~ ~
? où
= E
m ou
est le moment de transition.
E0 est l'énergie de l'état fondamental.
En est l'énergie de l'état n.
est la fonction d'onde de l'état n
est la fonction d'onde de l'état fondamentale
la composante w du moment dipolaire, w = x, y ou z
max est le nombre d'états excités
calculés de symétrie n. En général nous essayons
d'avoir le maximum. Pour la symétrie , on ne prend pas m=1 car
l'état fondamental est de symétrie .
Les calculs sont faits avec le programme MOLPRO, méthode
MCSCF. Nous prenons
pour le radical hydroxyl, la symétrie
C2v sous groupe abélien de afin de faciliter
les calculs.
IV. B/ Comparaison entre les polarisabilités de
OH calculées avec les deux Bases :
La durée de vie du radical OH est très courte,
par conséquence les données expérimentales concernant la
polarisabilité sont très rares. Nous avons comparés nos
résultats théoriques avec ce qui a été
publiés(Tableau III). Nous avons choisi, les résultats obtenus
par la méthode CEPA(Coupled electron pair approximation) comme
référence.
La polarisabilité de OH a été
calculée pour différentes distances internucléaires. Nous
avons obtenu avec la base avtz (Tableau I), les données en unité
atomique:
1.79 ROH 1.95 : 10.08 10.16
( ?
et avec la base davtz (Tableau II):
1.87 ROH 1.95 : 7.12 7.36
( ?
Les deux bases, donnent une valeur d'une des composantes
perpendiculaires, de la
polarisabilité, plus faible que l'autre. Avec
la base avtz, c'est la composante qui a
la plus grande valeur et avec la base davtz c'est la composante
.
Les énergies de l'état fondamental de OH (Tableaux
I et II), obtenus avec la base avtz, sont inférieures à celles
obtenues avec la base davtz.
E0 diminue quand ROH augmente et ce quelque
soit la base.
Polarisabilités de OH obtenues avec la bases avtz,
données en ua*
|
ROH
|
( ?
|
( ?
|
yy ( ?
|
E0
|
|
1.79
|
10.08
|
8.36
|
5.92
|
-75.4316
|
|
1.87
|
10.16
|
8.62
|
6.00
|
-75.4355
|
|
1.89
|
10.08
|
8.72
|
6.08
|
-75.4362
|
|
1.95
|
10.08
|
8.58
|
6.16
|
-75.4367
|
Tableau II
Polarisabilités de OH obtenues avec la bases davtz,
données en ua*
|
ROH
|
( ?
|
( ?
|
yy ( ?
|
E0
|
|
1.87
|
7.2
|
8.58
|
4.56
|
-75.4115
|
|
1.89
|
7.12
|
7.26
|
4.56
|
-75.4126
|
|
1.95
|
7.36
|
7.5
|
4.64
|
-75.4138
|
*ua =unité atomique
Polarisabilités trouvées dans la littérature
en ua*
|
Référence
|
ROH
|
Polarisabilité
|
Méthode de calcul
|
[122]
|
1.79
|
z ?
|
=7.541
|
MCSCF
|
|
|
z ?
|
= 8.75
|
|
[114]
|
1.87
|
|
=7.55
|
MCSCF
|
|
|
( ?
|
|
|
|
|
|
=6.37
|
|
|
|
( ?
|
|
|
[123]
|
|
1.89
|
z ?
|
=7.786
|
(U)MP2
|
|
|
|
z
|
=9.42
|
|
[120]
|
1.95
|
|
|
CEPA
|
|
|
|
=7.02
|
|
|
|
( ?
|
|
|
|
|
z ?
|
= 8.77
|
|
[121]
|
|
1.95
|
|
|
TDUHF
|
|
|
|
|
=6.19
|
|

TDUHF: Time-dependent unrestricted Hrtree-Fock CEPA: Coupled
electron pair approximation
*ua =unité atomique
IV.C/ Comparaison entre les polarisabilités de
OH calculées et celles trouvées
dans la bibliographie (Tableau IV):
Toutes les valeurs de la polarisabilité du radical OH
calculées avec la base avtz sont plus proches que les valeurs davtz, de
ce qui été trouvé dans la bibliographie.
Les valeurs les plus proches de ceux de la méthode CEPA,
sont ceux de la méthode TDUHF et celles que nous avons calculé
avec la base avtz et ce pour ROH=1 .95.
Suivants nos résultats et ceux trouvés dans la
bibliographie, nous avons : La polarisabilité de OH qui se situe entre
8.50 et 9.50 ua.
( ?
|
La polarisabilité de OH
|
|
qui se trouve entre 6.50 et 7.50 ua.
|
IV.D/ Conclusion :
1) La base avtz donne des résultats un peu meilleurs que
ceux de la base davtz.
2) Les énergies de l'état fondamental de OH
obtenus avec la base avtz, sont inférieures
à celles obtenues avec la base davtz.
3) E0 diminue quand ROH augmente et ce quelque soit
la base.
Tableau IV
Polarisabilités du radical OH calculées avec
différentes méthodes en ua*
|
ROH
|
( ?
|
yy ( ?
|
Référence
|
|
1.79
|
8.36
|
5.92
|
avtz
|
|
7.54
|
|
[122]
|
1.87
|
8.62
|
6.00
|
avtz
|
|
8.58
|
4.56
|
davtz
|
|
8.75
|
6.37
|
[114]
|
1.89
|
8.72
|
6.08
|
avtz
|
|
7.26
|
4.56
|
davtz
|
|
7.79
|
|
[123]
|
|
|
1.95
|
8.58
|
6.16
|
avtz
|
|
7.5
|
4.64
|
davtz
|
|
9.42
|
7.02
|
[120]
|
|
8.77
|
6.19
|
[121]
|
|
*ua =unité atomique
Conclusion générale
Les travaux menés jusqu'à maintenant sur les
clusters OH-(H2O) n ont beaucoup aidé à la
connaissance de ces systèmes. Ces agrégats sont d'un grand
intérêt, dans les domaines de biologie, de physique, de chimie et
les réactions atmosphériques. Cette étude a
contribué au développement des méthodes théoriques
utilisées.
Les calculs faits sur les clusters de l'ion hydroxyde, ont
montré que l'énergie de corrélation et l'ajout de
fonctions diffuses sont tous les deux nécessaires pour reproduire des
énergies d'interaction précises.
Le pourcentage des effets à trois corps par rapport aux
contributions des paires augmente avec n qui augmente.
La variation du paramètre géométrique,
distance interatomique, de l'un des monomères des configurations
OH-(H2O)2, contribue de façon
importante au changement des valeurs des énergies à trois corps
et à deux corps.
Les contributions dominantes dans l'énergie à
trois coprs sont les composantes et , respectivement dûes à
l'induction répulsive et à l'échange attractive.
) P2 )
Les énergies totales des configurations
OH-(H2O)2 où les deux molécules d'eau appartiennent
à la deuxième couche de solvatation de l'ion sont moins
attractives que celles des configurations où les deux molécules
d'eau n'appartiennent pas à la même couche de solvatation moins
attractives que celles où les deux molécules d'eau appartiennent
à la première couche d'hydratation de l'ion hydroxyde.
Pour des sous systèmes OH-(H2O)2 de
différentes configurations OH-(H2O)n où
toutes les molécules d'eau appartiennent à la première
couche de d'hydratation de l'ion, quand on passe de n=2 à n=4, nous
avons les énergies à trois qui deviennent plus répulsives
et les énergies à deux corps qui sont moins attractives. Les
énergies totales sont moins attractives.
Vu la variété de sources qui nous permettent
d'avoir des structures OH-(H2O)n, nos
résultats restent relatives, d'autres calculs sur un plus grand nombre
de configurations, de préférence, issues de
procédés de génération et d'optimisation meilleurs,
nous permettraient avec une comparaison, à plus de données
expérimentales, d'avoir des résultats plus proche de la nature et
de mieux comprendre les réactions fondamentales où ces
agrégats interviennent.
Pour les systèmes RbOH, la base avtz nous a
donné des valeurs des différentes composantes de la
polarisabilté du radical OH, plus proches que ceux de la base davtz.
Les énergies de l'état fondamental du radical OH
obtenues avec la base avtz, sont inférieures à celles
obtenues
avec la base davtz. L'énergie de l'état fondamental diminue quand
ROH augmente et ce quelque soit
la base.
Nos calculs ont donné une valeur de la composante de la
polarisabilité de OH,
qui se situe entre 8.50 et 9.50 ua et la valeur de la
composante
( ? yy ( ?
entre 6.50 et 7.50 ua. Dans ce cas, d'autres calculs sont
aussi nécéssaires, en utilisant d'autres paramètres. Le
but est d'obtenir une polarisabilité de OH qui se rapproche le plus de
la valeur réelle. Le développement de techniques
expérimentales pour mesurer cette polarisabilité est
nécéssaire afin que nous puissions comparer nos résultats
théoriques aux résultats expérimentaux. Une valeur plus
exacte de la polarisabilté du
radical OH nous conduirait à des valeurs plus
précises des énergies d'interaction à grande distance dans
RbOH, ce qui nous permetterait d'avoir les observables les plus proches de
celles expérimentales.
Bibliographie
[1] Lucjan Piela, Ideas of Quantum Chemistry, Elsevier (2007).
[2] R. Kelterbaum and E. Kochanski, J. Mol. Struct.: THEOCHEM
371, 205 (1996).
[3] E. Kochanski, R. Kelterbaum, S. Klein, M.-M. Rohmer, and A.
Rahmouni, Adv. Quantum Chem. 28, 273 (1997).
[4] E. Kochanski and R. Kelterbaum, J. Phys. Chem.
99, 12493 (1995).
[5] M. Tuckermann, K. Laasonen, M. Sprik, and M. Parrinello, J.
Phys. Chem. 99, 5749 (1995).
[6] M. Tuckermann, K. Laasonen, M. Sprik, and M. Parrinello, J.
Chem. Phys. 103, 150 (1995).
[7] R. Moszynski, P.E.S. Wormer, B. Jeziorski, A van der Avoird,
J. Chem. Phys. 103 8058 (1995); 107 672
(1997).
[8] V. Lotrich and K. Szalewicz, J. Chem. Phys.
106, 9668 (1997).
[9] H.L. Williams, C.F. Chabalowski, J. Phys. Chem. A,
105, No. 3, 646 (2001).
[10] Modern Electronic Structure Theory (Part II), Editor David
R. Yarkony,
(1995), ISBN:9810219601.
[11] Molecular Physics, 89, 81 (1996).
[12] K. Szalewicz, B. Jeziorski, Mol. Phys.,
38, 191, (1979).
[13] Moszynski, R., Jezioski, B. , and Szalewicz, K. , J. Chem.
Phys. , 100, 1312, (1994).
[14] H. L. Williams, K. Szalewicz, R. Mosynski, and B.
Jeziorski, J. Chem. Phys., 103, 4586 (1995).
[15] B. Jeziorski, and W. Kolos, Int. J. Quantum Chem. Suppl. 1,
12, 91 (1977).
[16] B. Jeziorski, and W. Kolos, Molecular Interactions, Vol. 3,
edited by H. Ratajczak and W. J. Orville-Thomas(New York: Wiley), 1 (1982).
[17] T. Cwiok, B. Jeziorski, W. Kolos, R. Mosynski, and K.
Szalewicz, J. Chem. Phys., 97, 7555 (1992).
[18] T. Cwiok, B. Jeziorski, W. Kolos, R. Mosynski, and K.
Szalewicz, J. Molec. Struct. Theochem., 307, 135 (1994).
[19] P. Hohenberg, W. Kohn, W. Phys. Rev. B,
136, 864 (1964).
[20] Exploring Chemistry With Electronic Structure Methods: A
Guide to Using Gaussian (Paperback) by James B. Foresman and AEleen
Frisch.
[21] S.F. Boys and Bernardi, Mol. Phys., 19,
553 (1970).
[22] E. Kochanski, in Intermolecular Forces, The
Jerusalem Symposium on Quantum Chemistry and Biochemistry, edited by B. Pullman
(Reidel, Dordrecht ), Vol. 14, p.15 (1981).
[23] G. Chalasinski, M.M. Szczesniak, and S.M. Cybulski, J.
Chem. Phys., 92, 2481 (1990).
[24] G. Chalasinski , M.M. Szczesniak, and R.A. Kendall, ibid.
101, 8860 (1994).
[25] M. Dreyfus and A. Pullman, Theo. Chim. Acta
19, 20 (1970).
[26] M.J. Frisch, G.W. Trucks, H.B. Schlegel, P.M.W. Gill, B.G.
Johnson, M.A. Robb, J.R. Cheeseman, T. Keith, G.A. Petersson, J.A. Montgomery,
K. Raghavachari, M.A. Al-Laham, V.G. Zakrzewski, J.V. Ortiz, J.B. Foresman,
C.Y. Peng, P.Y. Ayala, W. Chen, M.W. Wong, J.L. Andres, E.S. Replogle, R.
Gomperts, R.L. Martin, D.J. Fox, J.S. Binkley, D.J. Defrees, J. Baker, J.P.
Stewart, M. Head-Gordon, C. Gonzalez, and J.A. Pople, GAUSSIAN 94, Revision
B.2, Gaussian, Inc., Pittsburgh PA, (1995).
[27] Modern Electronic Structure Theory (Part I), Editor David
R. Yarkony,
(1995), ISBN:9810219598.
[28] A. Lembarki, Thèse, U.C.B. Lyon-1, France (1994).
[29] N.H. March, "Electron Density Theory of A toms and
Molecules" , (Academic Press, 1992); S. Lundqvist and N.H. March, "Theory
of the Inhomogenous Electron Gas", (Plenum Press, 1983).
[30] E.P. Wigner, Phys. Rev., 46, 1002
(1934).
[31] E. Clementi, G. Gorongiu, and O.G. Stradella, in MOTTEC
(Modern Techniques in Computational Chemistry), ed., E. Clementi,
(ESCOM Leiden, The Netherlands (1991).
[32] D.R. Hartree, «The Calculation of Atomic
Structures», (Wiley, New York, 1957).
[33] W. Pauli, Z. Physik, 43, 601 (1927).
[34] J.C. Slater, «Quantum Theory of Matter»,
McGraw-Hill, New York, (1951). [35-36] C.C.J. Roothan et Hall, Rev. Mod. Phys.,
23, 69 (1951) et 32, 179 (1960).
[37] R.S. Mulliken, J. Chem. Phys., 46, 497
(1949).
[38] V.A. Fock, Z. Physik, 89, 145 (1935).
[39] Wen-Fa Lu, Chul Koo Kim, Jae Hyung Yee, and Kyun Nahm, The
Americal Physical Society, Physical Review D, 64, 025006
(2001).
[40] Nakatsuji Hiroshi, Journal of Chemical Physics,
115, Issue 6, pp. 2465-2475 (2001).
[41] F. Coester, Nucl. Phys., 7, 421 (1958).
[42] F. Coester and H. Kümmel, Nucl. Phys.,
17, 477 (1960).
[43] A. deShalit and H. Feschbach, «Theoritical Nuclear
Physics», 1, N uclear Structure, Chap. 3, Jhon Wiley
and Sones, New York, (1974).
[44] J.M. Eisenberg and W. Greiner, «Nuclear
Theory», 3, Microscopic Theory of Nucleus, chap.4,
North-Holland, Publishing Co., Amsterdam, (1972).
[45] J. Cizek, J. Chem. Phys., 45, 4256
(1966).
[46] J. Cizek, Adv. Chem. Phys., 14, 35
(1969).
[47] J.A. Pople, R. Krishnan, H.B. Schlegel and J.S. Binkley,
Int. J. Quant. Chem., 14, 545 (1978).
[48] J.A. Pople, M. Gordon, and Raghavachari, J. Chem. Phys.,
87, 5968 (1978).
[49] R. F. Bishop and H.G. Kümmel, Phys. Today,
40, 52 (1987).
[50] L. H. Thomas, Proc. Camb. Phil. Soc., 23,
542 (1927).
[51] E. Fermi, Rend. Accad. Lincei, 6, 602
(1927).
[52] J.C. Slater, Phys. Rev., 81, 385(1951).
[53] P. Hohenberg and W. Kohn, Phys. Rev., B
136, 864 (1964).
[54] W. Kohn, L.J. Sham, Phys. Rev., A 140, 1133 (1965).
[56] J.P. Perdew and W. Yang , Phys. Rev., B
33, 8800 (1986).
[57] J.P. Perdew, in «Electronic Structure of Solids 91,
Eds. P. Ziesche and H. Eschrig, (Academic Veralg, Berlin, 1991).
[58] A.D. Becke, Phys. Rev., A 38, 3098
(1988).
[59] T. Korona, R. Moszynski, and B. Jeziorski, J. Chem. Phys.
105, 8178 (1996).
[60] B. Jeziorski, M. Bulski, and L. Piela, Int. J. Quantum
Chem. 10, 281 (1976).
[61] R. Moszynski, P. E. S. Wormer, T. G. A. Heijmen, and A. van
der Avoird, J. Chem. Phys. 108, 579 (1998).
[62] B. Jeziorski, Ph.D. thesis, University of Warsaw, Warsaw
(1974).
[63] M. Bulski and G. Chalasinski, Theor. Chim. Acta
56, 199 (1980).
[64] I. Shavitt, in «Methods of Electronic Structure
Theory», Vol. 3 of Modern Theoretical Chemistry, eds. H.F. Schaefer
(Plenum Press, New York, 189 (1977).
[65] S.S. Xantheas, J. Am. Chem. Soc. 117,
10373 (1995).
[66] Y. Kazi Aouel, Thèse de Magister, University of
Tlemcen, Algeria (1997).
[67] G. Andaloro, M. A. Palazzo, M. Migliore, and S. L. Fornili,
Chem. Phys. Lett., 149, 201 (1988).
[68] H.L. Friedman and C.V. Krishnan, in Water: A
Comprehensive Treatise, edited by F. Franks, Plenum, New York,
3, 58 (1972).
[69] L.M. Branscomb, Phys. Rev., 148, 11
(1966).
[70] O. Matsuoka, E. Clementi, and M. Yoshimine, J. Chem. Phys.,
64, 2314 (1976).
[71] T. H. Dunning, Jr., J. Chem. Phys., 90,
1007 (1989).
[72] R.A. Kendall, T.H. Dunning, Jr., and R.J. Harrison,
ibid, 96, 6796 (1992).
[73] Interatomic Distances, The Chemical Society of
London, Special Publication No. 11, (1958); Supplement,
Special Publication No. 18 (1965).
[74] J.C. Owrutsky, N.H. Rosenbaum, L.M. Tack, and R.J.
Saykally, J. Chem. Phys. 83, 5338 (1986).
[75] M.D. Newton and S. Ehrenson, J. Am. Chem. Soc. 93,
N° 20, 4971 (1971).
[76] A.M. Sapse, L. OSORIO and G. Snyder, INTERNATIONAL JOURNAL
OF QUANTUM CHEMISTRY, VOL 26, 223 (1984).
[77] M.Arshadi, P.Kebarle, J.Phys.Chem. 74,
1483 (1970).
[78] M.Meot-Ner (Mautner), J.Am.Chem.Soc. 108,
6189 (1986).
[79] M.Meot-Ner (Mautner), C.V.Speller, J.Phys.Chem.,
90, 6616 (1986).
[80] J.D.Madura, W.L.Jorgensen, J.Am.Chem.Soc.
108, 2517(1986).
[81] G.Andaloro, M.A.Palazzo, M.Migliore, S.L.Fornnili,
Chem.Phys.Lett. 201,149 (1988).
[82] M.Tuckerman, K.Lassonen, M. Sprik, M.Parrinello, J.Phys.:
Condens. Matter, 6A93(1994).
[83] I.Tunon, D.Rinaldi, M.F.Ruiz-Lopez, J.L.Rivail,
J.Phys.Chem. 99, 3798 (1995).
[84] S.S.Xantheas, J.Am.Chem.Soc., 117, 10379
(1995).
[85] A.R.Grimm, G.B.Bacskay, A.D.Haymet, Mol.Phys.
86, 369 (1995).
[86] M.Tuckerman, K.Lassonen, M. Sprik, M.Parrinello,
J.Chem.Phys. 103,150 (1995).
[87] M.Tuckerman, K.Lassonen, M. Sprik, M.Parrinello,
J.Phys.Chem. 99, 5749 (1995).
[88] M.Tuckerman, M.Sprik, M.Parrinello, Femtochemistry ,
578(1996).
[89] C.P.del Valle, J.J.Novoa, Chem.Phys.Lett.
269, 40 1(1997).
[90] J.J.Novoa, F.Mota, C.P.del Valle, M.Planas,
J.Phys.Chem. A 101,7842 (1997).
[91] M.Masamura, J.Mol. Struct. (Theochem) 498,
87 (2000).
[92] M.Masamura, J.Comput.Chem. 22,
31(2001).
[93] M. Masamura, ab initio study of OH-(H2O)n and
SH-(H2O)n in the gas phase, accepté dans Chem.Phys.Lett..
Department of Preventive Dentistry, Okayama University Dental School,
Shikata-cho 2-5-1, Okayama, 700-8525 Japan (sous presse).
[94] C.E. Klots and R.N. Compton, J. Chem. Phys.,
69, 1644 (1978).
[95] M. Knapp, O. Echt, D. Kreisle and E. Recknagel, J. Chem.
Phys., 85, 636 (1986).
[96] J.M. van Doren, S.E. Barlow, C.H. Depuy and V.M. Bierbaum,
Int. J.Mass Spectrom. Ion Proc., 81, 85 (1987).
[97] X. Yang and A.W. Castleman, J. Phys. Chem.,
94, 8500 (1990).
[98] M. De Paz, A.G. Giardini and L.J. Friedman, J. Chem. Phys.,
52, 687 (1970).
[99] J.D. Payzant, R. Yamdagni and P. Kebarle, P. Can. J. Chem.,
49, 3308 (1971).
[100] M.M. Szczesniak, S.J. Scheiner, J. Chem. Phys.,
77, 4586 (1982).
[101] C. McMichael Rohlfing, L.C. Allen, C.M. Cook, J. Chem.
Phys., 78, 2498 (1983).
[102] K. Ohta, K. Morokuma, J. Phys. Chem., 89,
5845 (1985).
[103] J.L. Andrés, M. Duran, A. Lledos, J. Bertran, J.
Chem. Phys., Lett., 124, 177 (1986).
[104] E. Carbonell, J.L. Andrés, A. Lledos, M. Duran, J.
Bertran, J.Am.Chem.Soc., 110, 996 (1988).
[105] J.E. De Bene, J. Phys. Chem., 92, 2874
(1988).
[106] P.A. Giguère, Rev. Chim. Miner.,
20, 588 (1983).
[107] V.D. Maiorov, L.V. Bocharova and N.B. Librovich, Russ. J.
Phys. Chem., 56, 67 (1982).
[108] N.B. Librovich and V.D. Maiorov , Russ. J. Phys. Chem.,
56, 380 (1982).
[109] V.D. Maiorov, L.V. Bocharova, N.B. Librovich, G.G.
Bikbaeva and M.L. Vinnik, Russ. J. Phys. Chem., 52, 688
(1978).
[110] W.R. Busing, D.F. Hornig, J. Phys. Chem.,
65, 284 (1961).
[111] Clusters: A new state of matter. LBL Res. Rev.,
16, 2 (1991).
[112] H.B. Schlegel, J. Comput. Chem. 3, 214
(1982).
[113] M. Mascagni, Advanced Monte Carlo Method , Swiss Federal
Institute of Technology Zurich.
[114] D. Spelsberg, J. Chem. Phys., 111, 9625
(1999).
[115] G.C. Nielson, G.A. Parker, and R.T. Pack, J. Chem. Phys.,
64, 2055 (1976).
[116] Dachsel, Holger, J. Chem. Phys., 124,
144115 (2006).
[117] D. Spelsberg, T. Lorenz, and W. Meyer, J. Chem. Phys.,
99, 7845 (1993).
[118] D. Spelsberg, and W. Meyer, J. Chem. Phys.,
108, 1532 (1998).
[119] A.V. Podolskiy, and P. Vogl, Physical Review, B
69, 233101 (2004).
[120] A.D Esposti and H-J Werner, J. Chem. Phys.,
93, 3351 (1990).
[121] S.P. Karna, J. Chem. Phys., 104, 6590
(1996).
[122] L. Adamowicz, J. Chem. Phys., 89, 6305
(1988).
[123] U. Dinur, J. Mol. Struct.: THEOCHEM 303,
227 (1994).
[124] N.Turki, A.Milet, A.Rahmouni, O.Ouamerali, R.Moszynski,
E.Kochanski, and P.E.S.Wormer, J. Chem. Phys. 109, 7
157(1998).
[125] N. Turki , A. Milet, O. Ouamerali, R.
Moszynski,E. Kochanski, J. Mol. Struct. (Theochem) 577, 239
(2002).
[126] K. Jankowski and K. Kowalski, Phys. Rev. Letters,
81, 1195 (1998) J. Chem. Phys. 110, 37-93
(1999), ibid. 111, 2940-2952 (1999).
[127] M. Faraday, Experimental Researches in electricity, Vol.
1, 1844, Vol.2,
1849, Vol. 3, 1855 (1839).
[128] H. Goldschmitt, O. Udby, Z. Physik. Chem.,
60, 728 (1907).
[129] R.Kelterbaum, Thèse de Doctorat, Université
Louis Pasteur, Strasbourg I, (1995).
Articles Internationaux
Le travail de cette thèse a donné lieu à
deux d'articles internationaux publiés:
1) THEORITICAL STUDY OF THE OH-(H2O)2 SYSTEM :
NATURE AND IMPORTANCE OF THREE BODY INTERACTIONS N.Turki,
A.Milet, A.Rahmouni, O.Ouamerali, R.Moszynski, E.Kochanski, and P.E.S.Wormer,
J. Chem. Phys. 109, 7 157(1998).
2) The OH-(H2O)2 SYSTEM : EFFICIENCY OF AB INITIO AND
DFT
CALCULATIONS FOR TWO AND THREE-BODY INTERACTIONS
Turki Naima, Milet Anne, Ouamerali Ourida, Moszynski Robert,
Kochanski Elise, J. Mol. Struct. (Theochem) 577,
239 (2002).
Journal of molecular structure. Theochem ISSN 0166-1280 CODEN
JMOSB4.
· Un autre article, portant sur les interactions
intermoléculaires dans les molécules RbOH sera
concrétisé prochainement.
Perspectives
Le travail réalisé a donné des
résultats qui ont amélioré la connaissance des
systèmes étudiés et qui sont importants dans les domaines
de chimie, biologie et physique. Ceci nous encourage à poursuivre la
modélisation moléculaire pour des systèmes plus difficiles
à étudier, en particulier nous nous interessons aux interactions
intermoléculaires à courte et grande distance dans les complexes
de Van der Waals à couches ouvertes.
| 

