I.C.1/ L'énergie de corrélation:
L'énergie Hartree-Fock n'est pas aussi basse que
l'énergie réelle du système. La raison mathématique
pour ça est le fait que soit un seul déterminant, ce qui est
restreint. Nous pouvons introduire plus de flexibilité
mathématique en permettant à
de contenir plusieurs déterminants. Une telle
flexibilité nous donne une énergie encore plus basse.
Dans l'approximation Hartree-Fock, nous résolvons les
équations pour le comportement de chaque électron dans le champ
moyen des (n-1) électrons restants. Malheureusement les électrons
réagissent les uns aux autres de façon instantanée via la
loi de Coulomb, c'est à dire les [n(n-1)]/2 mouvements de paires
d'électrons sont corrélés et c'est
précisément cette corrélation d'électrons
qui est définie conventionnellement comme[30] la
différence entre l'énergie exacte et l'énergie limite
Hartree-Fock.:
(21)
E E

Pour cette raison, les calculs de mécanique quantique sont
parfois jugés suivant le pourcentage de l'énergie de
corrélation reproduit.
I.C.2/ La Théorie de la perturbation
Möller-Plesset jusqu'à l'ordre n (MPn):
La méthode de Hartree-Fock permet d'obtenir
l'état du système électronique de la molécule. Un
déterminant de Slater obéit d'une manière convenable
à la statistique de Fermi ce qui signifie que des électrons de
spins différents s'évitent, Cependant, le formalisme HF permet
que des électrons de même spin (état quantique
différent) peuvent être présents simultanément aux
mêmes endroits dans l'espace, ce qui est bien sur inexact. Plusieurs
méthodes dites "post Hartree-Fock" ont essayé de
représenter la corrélation électronique en utilisant des
combinaisons linéaires de déterminants de Slater comprenant des
orbitales occupées et virtuelles. Une des méthodes les plus
simples est la méthode Möller-Plesset MP2. On écrit
l'Hamiltonien comme la somme d'une partie correspondant au déterminant
de Hartree-Fock et d'une perturbation V (corrélation dynamique : les
électrons se repoussent). L'énergie totale sera la somme des deux
énergies associées à ces deux hamiltoniens :
La théorie de la perturbation Möller-Plesset (MP)
est le résultat de l'application des formules de la perturbation
Rayleigh-Schrödinger[39] traditionnels à la
théorie de la structure électronique[10], utilisant la
somme des opérateurs de Fock comme Hamiltonien d'ordre 0.
L'opérateur de Fock habituel pour les systèmes
à couches fermées est :
(22)
où i et l se réfèrent respectivement aux
électrons et aux orbitales occupées. est la
partie monoélectronique de l'Hamiltonien, sont les
opérateurs Coulomb et
à
échange. Les équations Möller-Plesset sont
:
(23)
(24)
Si la perturbation est suffisamment petite, alors
l'énergie et la fonction d'onde peuvent être écrites sous
la forme de séries en puissance :
La substitution de ces séries dans l'équation de
Schr~dinger donne une nouvelle équation:
(25)

La solution à l'ordre zéro de cette
équation donne l'énergie qui est la somme des énergies des
orbitales des électrons. La solution au premier ordre (i=1) corrige
cette énergie et donne l'énergie Hartree-Fock et la fonction
d'onde. Pour aller au-delà du traitement Hartree-Fock, il faut aller
au-delà du premier ordre. Les calculs MollerPlesset, second (MP2),
troisième (M P3) et quatrième ordre (MP4) sont les niveaux
standards utilisés pour les petits systèmes et sont
implémentés dans beaucoup de codes de la chimie quantique. Au
plus haut ordre, les calculs M P existent dans quelques codes et sont rarement
utilisés.
Soit diagonal, l'expression générale pour
l'énergie au second ordre dans la théorie
de la perturbation Rayleigh-Schrödinger est donnée
par :
E(2) = ? Ø 0
? Ø H | Ø
0 0



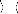

(26)
où n parcourt toutes les fonctions d'état de
configuration (CSF) excepté la référence CSF
[5]. Pour les équations Rayleigh-Schrödinger de la
théorie de la perturbation au plus haut ordre, voir la
référence [10].
I.C.3/ Méthode coupled-cluster avec excitations simple,
double et triple approchée, CCSD(T)[1,126] :
La méthode cluster-couplé (CC), est aujourd'hui
largement reconnue comme l'outil le plus puissant pour une description
précise de la corrélation d'électrons dans les atomes et
les molécules. La fonction d'onde de la méthode CC, donnée
par l'exponentiel Ansatz[40], dépend de paramètres
ajustables (cluster amplitudes). Elle commence par la méthode des
orbitales moléculaires Hartree-Fock et ajoute un terme correcteur pour
prendre en compte la corrélation d'électrons. Certains calculs
très précis pour des molécules de taille petites et
moyennes utilisent cette méthode.
Au début, on introduit un spécial
déterminant de Slater [1], déterminant de
référence
(appelé vacuum state, ça peut
être le déterminant Hartree-Fock) et nous écrivons

que la fonction d'onde exacte de l'état fondamental comme
une exponentiel ansatz [41,
42, 43]:
(27)
p ( ) ?

où est l'opérateur d'onde et lui même est un
opérateur de cluster[44,45,46] ou
opérateur d'excitation, quand il agit sur , il produit une
combinaison linéaire de
déterminants de Slater excités. Dans la
méthode CC, la normalisation intermédiaire de la fonction est
utilisée:
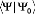
L'opérateur cluster s'écrit comme la somme
d'opérateurs d'excitations [47, 48, 49] :
(28)
= +
T T T
où T1 est l'opérateur de toutes les simples
excitations, T2 est l'opérateur de toutes les doubles excitations, en
général l désigne le rang de l'excitation.
(29)
(30)

Les symboles a,b,...indiquent les spinorbitales occupées
dans et r, s,... celles
inoccupées.
Les t représentent les amplitudes ou les nombres dont la
détermination est le but de la méthode CC.
La fonction d'onde exacte satisfait l'équation de
Schr~dinger:
( T Ö
est l'Hamiltonien du système, et E, la fonction d'onde et
l'énergie de l'état de plus basse énergie.
L'équation de Schr~dinger est :
(31)
( = e
?
On peut écrire :
(32)
( T H p ( ?
Après transformation, nous obtenons :
L'opérateur, le conjugué de est :
? T †
T
e
Qui n'est pas la valeur principale de l'Hamiltonien. La
méthode CC n'est pas
†
variationnelle, si nous multiplions à gauche par , on
obtient :
T
qui a un caractère variationnel.
| 

