I.D/ Théorie de la perturbation de la
symétrie adaptée (SAPT):
La Théorie de la perturbation de la symétrie
adaptée donne un important aperçu concernant la nature des forces
intermoléculaires, puisqu'elle donne les énergies d'interaction
sous forme d'une somme de contributions bien définies et physiquement
significatives: électrostatique, échange, induction et
dispersion. Ceci nous fourni des informations sur les interactions dues aux
contributions attractives et répulsives. Nous avons utilisé pour
cette méthode, le formalisme développé récemment
pour les interactions à trois corps non additives [7,8].
L'énergie totale d'interaction pour un trimer peut
être diviser comme la somme de contributions de deux corps (additive) et
trois corps (non additive) [59]:
où désigne, en général, la
contribution de K corps dans l'énergie
? K
d'interaction à M corps.
Si on considère le trimer ABC, il est décrit par
l' Hamiltonien H0 = HA+ HB + HC, où HX, X= A, B, ou C, désigne l'
Hamiltonien du monomère X. La fonction d'onde et l'énergie E0,
correspondant à l'état fondamental, sont exprimées en
fonction des fonctions d'onde et des énergies des monomères non
perturbés :
,
A B C B
Ø Ø E E +
Une fois le système perturbé, l' Hamiltonien du
trimer s'écrit :
H(A,B,C) = H0 (A,B,C) + V(A,B,C) (37)
Où
l'opérateur de perturbation est donné par : V
(38)
L'opérateur VXY rassemble toutes les
interactions Coulombienne entre électrons et
noyaux des
monomères X et Y et les paramètres ont en général
des valeurs
ä t
physiques égales à l'unité afin de faciliter
le calcul[7]. L'énergie d'interaction du trimer peut
être écrite comme:
(39)
E ä å ÷
( )


Nous considérons que la fonction d'onde du trimer
satisfait la condition de
normalisation intermédiaire :
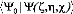
Cette fonction est solution de l'équation de Schr~dinger
paramétrée :
(40)
( ) (
H E Ø ä
|
L'indice RPA dans
|
|
et signifie que dans le calcul de ces
)
PA
|
L'énergie d'interaction paramétrée et la
fonction d'onde et ont
( ä ) ä å )
été développées en séries en
fonction de :
ä et
' A,
,? ?
? ,i 1
) ) ?
, ? ? ?

où
indiquent l'énergie de polarisation et la fonction d'onde
du i ième ordre dans , du j ième dans et k ième ordre dans
. Nous considérons que
Les corrections énergie et fonction d'onde, ,
E 0 Ø
( ijk) k)
p l
peuvent être obtenues des équations de la
théorie de la triple perturbation [62,63].
Après développement nous extrayons les
énergies d'interaction à deux et à trois corps non
additive, nous avons [7]:
(42)
( )
E bi E
n p
, ? ?
(43)
( )
E E
pp L
où est le terme non additif Heitler-London[60]
calculé avec les déterminants
Hartree-Fock des monomères isolés. Ce terme est
identique à celui de l'énergie de la
première
itération HF notée . L'égalité du terme avec
nous
st t a st it a ?
permet d'obtenir ce terme de n'importe quel traitement. , est
l'énergie
)
d'induction au second ordre, considérée
complètement pour la déformation
électrostatique des
orbitales. , , et sont les énergies
) 3 ) )
RPA PA
d'échange-dispersion au second ordre,
d'induction-dispersion au troisième ordre et la
dispersion au
troisième ordre. , est évaluée comme
l'échange-corrélation au
P )
2
premier ordre et les énergies
d'échange-induction-dispersion au troisième ordre
[61].
contributions, les fonctions réponses de l'approximation
phase aléatoire (Randomphase approximation (RPA)), ont
été utilisées [7,61].
La quantité représente une approximation aux effets
échange-induction Hartree-
Fock non additifs. Elle n'a pas été
programmée dans SAPT mais plutôt calculée comme la
différence entre l'énergie d'interaction Hartree-Fock et la somme
des énergies Heitler-London et induction:
(44)
= +
E E ?
Une telle approche, bien qu'approximative est
complètement justifiée, puisqu'on peut montrer que cette
différence est en effet dominée par les effets
d'échange-induction et d'échange-déformation au second
ordre et par les effets échange et induction de plus haut
ordre[61].
La différence entre et est notée [22]:
(45)
E E
HF
Aux bas ordres de la théorie de la perturbation,
l'énergie de délocalisation, , est
donnée par la somme des termes de l'induction et de
l'échange-induction, appelée "énergie de
déformation Hartree-Fock" [23,24] . Cette différence
donne une idée sur comment la distribution de charge des
monomères isolés se déforment (délocalisée
sur toute la supermolécule) durant le processus SCF [25].
De (44) et (45), nous pouvons déduire :
(46)
E E F
=
, désigne les effets échange-induction et
déformation. , représente
La décomposition de l'énergie d'interaction
à trois corps en différents termes de signification physique est
très importante pour l'analyse des phénomènes
d'interaction et la construction de potentiels analytiques. Des relations
similaires peuvent être trouvées pour les énergies
supermolécule et SA PT au niveau corréler.
Comme c'est discuter dans la
référence[61], la composante échange rassemble
la
correction de corrélation intramonomère de l'énergie
Heitler-London, l'énergie
d'échange-dispersion et le terme
échange-induction-dispersion alors que la partie déformation de
MP2 non additive peut être dominée par la contribution induction-
dispersion. Dans notre étude, nous utilisons cette décomposition
pour définir l'énergie d'échange MP2. En accord avec les
références 23 et 24, l'énergie à trois corps MP3
est donnée par la somme de l'énergie de dispersion Hartree-Fock
et des termes échange et déformation.
A noter que les termes d'échange inclus dans les
énergies à trois corps MP2 et M P3 ne peuvent pas être
définis de la même façon que dans la théorie
SAPT[7]. Par conséquence, la convergence du modèle
supermolécule Möller-Plesset et le développement SAPT peut
être quelque peu différente.
| 

