|


Année académique 2012 - 2013
Faculté de Pharmacie
Optimisation de la technique d'échantillonnage
« Headspace» dans le cadre de l'analyse des huiles
essentielles
Mémoire présenté par Laurent SALADE
En vue de l'obtention du grade de Master en Sciences
Pharmaceutiques
Promoteur : Pierre DUEZ
Jury : Jean - Michel KAUFFMANN
Pierre VAN ANTWERPEN

I
Remerciements
Je tiens à remercier les personnes qui m'ont
apporté leur aide pour la réalisation de ce mémoire ainsi
que ceux qui m'ont fait partager leur expérience et leur
réflexion au fil de mon travail. Je pense aussi à ceux qui m'ont
apporté leur énergie, leurs encouragements, en m'accordant un peu
ou beaucoup de leur temps pour me permettre de mener à bien ce
travail.
Merci donc :
- Au professeur Duez, promoteur de ce mémoire, pour son
accueil chaleureux au sein
du laboratoire de Pharmacognosie et pour sa patiente, sa
pédagogie ainsi que pour
toute l'aide qu'il m'a apportée.
- A Marie Faes, pour son aide, son énergie et sa
motivation.
- A Jeremy, pour ses connaissances en statistiques.
- A Olivier, pour l'entretien et la fourniture du
matériel.
- Ainsi qu'à ma famille et mes amis qui m'ont permis de
réaliser ce travail.
II
Résumé
La technique d'échantillonnage « Headspace
» est utilisée depuis une cinquantaine d'années
déjà dans divers domaines relatifs à l'analyse de
composés volatils en association avec la chromatographie gazeuse. Elle
présente l'avantage de pouvoir introduire l'échantillon
directement sous forme gazeuse dans l'injecteur du système
chromatographique. Ainsi, elle se prête bien à l'étude
d'analytes facilement volatilisable présents dans des matrices non
chromatographiables comme par exemple l'analyse d'huiles essentielles
présentes dans certaines plantes.
Ce mémoire porte sur cette dernière application.
Les plantes employées dans cette présentation appartiennent au
genre Ocimum L. et sont analysées à l'aide d'un
appareillage « Headspace » statique. Etant donné les
difficultés rencontrées lors de travaux antérieurs sur des
échantillons de ce type, cette étude tente d'identifier et de
résoudre les problèmes analytiques en question. Deux aspects
distincts sont abordés :
- D'une part, l'influence des différentes
méthodes de préparation de l'échantillon sur les analyses
en « Headspace ».
- Et d'autre part, l'importance et l'effet des
différents paramètres du système « Headspace
».
La technique est donc étudiée de façon
globale mais tout en se limitant au cadre d'une analyse sur matière
végétale.
La validation d'un tel procédé analytique serait
très avantageuse car il est peu coûteux, rapide et permet de
s'affranchir des effets de matrice.
Il est cependant apparu que cette méthode analytique,
relativement simple en théorie, présente une multitude de
paramètres expérimentaux à prendre en considération
et s'avère donc relativement complexe à mettre en pratique. Ce
travail permet déjà d'apporter certaines informations quant au
comportement de l'appareillage « Headspace »
vis-à-vis de l'analyse de plantes aromatiques mais une série
d'essais supplémentaires s'impose afin de pouvoir valider le
processus.
III
Table des matières
|
1.
|
Introduction :
|
1
|
|
1.1
|
Applications :
|
2
|
|
1.2
|
Principe général :
|
3
|
|
1.3
|
Avantages et désavantages :
|
4
|
|
1.4
|
Les différents types de « Headspace » :
|
5
|
|
1.4.1
|
La « Static Headspace Extraction » (SHE):
|
5
|
|
1.4.2
|
La «Dynamic Headspace Extraction» («Purge and
Trap»):
|
6
|
|
1.4.3
|
La « Solid Phase Microextraction » (SPME) :
|
7
|
|
1.5
|
Théorie et application de la « Static Headspace
Extraction » (SHE) :
|
8
|
|
1.5.1
|
Les différents types d'échantillonnage en SHE :
|
8
|
|
1.5.2
|
Le coefficient K :
|
11
|
|
1.5.3
|
Le coefficient Beta :
|
12
|
|
1.5.4
|
La dérivation :
|
13
|
|
1.5.5
|
Préparation de l'échantillon :
|
14
|
|
1.5.6
|
Les différents paramètres :
|
15
|
|
1.5.7
|
La « Multiple Headspace Extraction » (MHE) :
|
18
|
|
1.6
|
Le genre Ocimum :
|
18
|
|
2.
|
But du Travail :
|
20
|
|
3.
|
Matériels et méthodes :
|
21
|
|
3.1
|
L'échantillon :
|
21
|
|
3.1.1
|
Matière végétale :
|
21
|
|
3.1.2
|
Préparation de l'échantillon :
|
21
|
|
3.2
|
Le système « Headspace » :
|
22
|
|
3.2.1
|
Type d'appareillage :
|
22
|
|
3.2.2
|
Protocole :
|
24
|
|
3.3
|
La Chromatographie Gazeuse :
|
25
|
|
3.3.1
|
L'injecteur :
|
26
|
|
3.3.2
|
Colonne et phase stationnaire :
|
26
|
|
3.3.3
|
Détecteur :
|
26
|
|
3.3.4
|
Programme de Température :
|
27
|
|
4.
|
Résultats et discussion :
|
27
|
IV
4.1 Optimisation de la préparation de
l'échantillon : 28
4.1.1 Comparaison des méthodes de broyage : 28
4.1.2 Changement d'échantillon : 31
4.1.3 Ajout d'un solvant : 32
4.1.4 L'effet « Salting-out » : 38
4.2 Optimisation des paramètres d'extraction «
Headspace » : 39
4.2.1 Plan factoriel : 40
4.2.2 Variation de température : 41
4.2.3 Variation du temps de remplissage de la boucle («
Loop Fill Time ») : 43
4.2.4 Optimisation de la pression du vial : 45
4.2.5 Ajouts dosés eau - poudre : 46
4.2.6 Changement d'échantillon : 49
4.3 Evolution générale : Erreur !
Signet non défini.
4.3.1 Modèle linéaire et quadratique : 50
5. Conclusion : 54
6. Références : 56
1
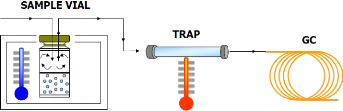
1. Introduction
La découverte du concept chromatographique est
généralement attribuée à un botaniste russe du nom
de Mikhaïl Tswett (Rouessac and Rouessac 2004). Vers 1900, lors de ses
travaux de biochimie végétale, il décide de mettre au
point une technique de séparation mettant en oeuvre une colonne remplie
de carbonate de calcium et un éluant constitué d'un
mélange éther de pétrole - éthanol afin de
séparer la chlorophylle et, les caroténoïdes. Il s'agit
là du premier modèle de chromatographie (Wilkinson 2003).
Vers 1941, Martin et Synge apportent certains
éléments qui vont en quelque sorte révolutionner le
domaine de l'analyse chromatographique et cela, aussi bien au niveau de la
chromatographie liquide que phase gazeuse ou planaire. Ils reçoivent le
prix Nobel en 1952 (Wilkinson 2003).
Mais ce n'est vraiment qu'en 1952 que les premiers travaux sur
la chromatographie gazeuse apparaissent avec James et Martin à la suite
desquels cette méthode connait un réel essor. En effet, les
colonnes capillaires ainsi que des détecteurs à ionisation de
flamme, à ionisation à l'argon et à capture
d'électrons vont alors faire leur apparition. Par après de
nombreuses recherches ont suivi afin de mettre au point de nouvelles colonnes,
de nouveaux injecteurs, etc....
Suite à cela, diverses techniques couplées
à la chromatographie gazeuse, comme la technique « Headspace
», vont faire leur apparition. Sa première utilisation remonte
à 1958 ; par après la méthode va se diversifier et
différentes versions vont être mises au point. Cette
méthode, qui est apparentée à une extraction en phase
gazeuse, est couplée à la chromatographie gazeuse
pour l'analyse de composés volatils présents
dans des matrices complexes. Elle est
largement répandue dans
différents domaines tels que l'agro-alimentaire, la parfumerie, le
milieu pharmaceutique, la justice criminelle, l'environnement (Snow and Slack
2002). Un des pionniers de cette technique, Roman Kaiser, un chimiste suisse, a
longtemps cherché à mettre au point une méthode d'analyse
permettant de travailler sur des senteurs afin de
2
reproduire celles-ci en parfumerie (Sell 2005).

Figure 1 : Roman Kaiser (Hume 2009).
1.1 Applications
L'analyse de composés volatiles dans les
médicaments représente une des applications les plus communes de
la méthode d'extraction « Headspace » en industrie
(Sitaramaraju, van Hul et al. 2008).
Cette méthode a également trouvé son
application dans le domaine de l'analyse biologique, par exemple pour l'analyse
de produits de réactions métabolique ou encore pour l'analyse de
diverses toxines (Snow and Slack 2002).
Dans le milieu médico-légal, elle est notamment
utilisée pour l'analyse de l'alcool dans le sang mais également
pour toute une série d'autres composés de faibles poids
moléculaires présents dans ce dernier (Dills, Kent et al. 1991).
Etant donné sa bonne reproductibilité, sa sensibilité et
sa stabilité, cette technique d'extraction s'avère une
méthode de choix pour les analyses médico-légales (Snow
and Slack 2002).
Pour certaines analyses cliniques elle s'avère
également intéressante, par exemple pour l'évaluation de
la stabilité de l'éther dans le corps dans le cadre d'une
étude portant sur les suicides. Certains chercheurs ont également
étudié la variation des composés volatils responsables des
odeurs corporelles en fonction de l'âge (Snow and Slack 2002).
Elle est aussi utilisée pour analyser divers
arômes naturels et odeurs en parfumerie mais aussi dans le domaine de
l'alimentation. Des travaux ont par exemple été menés pour
analyser la composition en acétaldéhyde de la bière (Tian
2010) ou des substances aromatiques dans le cognac (Snow and Slack 2002).
3
Certains ont même étudié la variation des
substances volatiles présentes dans le fromage blanc en fonction du type
de champignons utilisés pour sa production.
La technique « Headspace » est
également largement appliquée dans le domaine environnemental
notamment pour l'analyse de polluants. C'est le cas du toluène, un
solvant fréquemment utilisé dans l'industrie
pétrochimique, pharmaceutique et textile ; la teneur en toluène
présente dans les urines d'un groupe de travailleurs a ainsi pu
être déterminée (Heidari, Shahtaheri et al. 2008).
En parfumerie, elle est également très
utilisée car elle permet de capturer l'odeur d'une plante sans devoir la
détruire ni même la cueillir car il suffit de placer une cloche en
verre autour de la fleur pour emprisonner directement l'espace de tête au
dessus de la fleur (ce qui est un avantage non négligeable pour les
plantes protégées ou chères)(Anonyme). Elle permet aussi
de capturer et d'étudier des odeurs plus proches, selon les parfumeurs,
de l'odeur réellement dégagée par la plante contrairement
aux odeurs obtenues par hydrodistillation ou via l'extraction par un solvant
organique (Linskens and Jackson 2010). Mises à part les odeurs
dégagées par certaines plantes, la technique peut aussi servir
pour l'étude d'autres senteurs d'intérêt en parfumerie
comme celles présentes sur certains lieux (ex : salon de thé).
Ce type de méthode d'extraction peut donc
s'avérer fort intéressant pour l'analyse de composés
volatils présents dans des matrices complexes (par exemple un parfum
présent dans un savon) et cela sans perte excessive de temps et d'argent
et avec une certaine facilité d'utilisation (Restek 2000).
1.2 Principe général
La méthode d'extraction « Headspace
» est relativement simple et regroupe en fait une famille de
techniques basées sur un principe commun, l'établissement d'un
équilibre entre une phase solide ou liquide une phase gazeuse (Zhu and
Chai 2005). Dans toutes ces techniques, l'échantillon, solide ou
liquide, est introduit dans un flacon en verre qui est ensuite scellé.
Ensuite les analytes volatils sont libérés de la matrice complexe
pour les faire passer en phase gazeuse (c'est cette phase que l'on appelle
« Headspace » ou « Espace de
4
tête). Ensuite un aliquote de la phase gazeuse est
prélevé et introduit dans le chromatographe gazeux où il
est analysé (Grob and Barry 2004).
La taille du flacon doit donc être suffisante pour
permettre à la phase gazeuse de prendre place. L'échantillon peut
être analysé tel quel ou être additionné d'un solvant
de dilution ou d'un agent pouvant modifier la matrice (Restek 2000).
L'étape cruciale se situe au niveau de
l'équilibre qui s'établit avec la phase gazeuse ; lors du
prélèvement de l'échantillon il faut être certain
que l'équilibre soit atteint (Grob and Barry 2004).
Deux grands types de « Headspace » ont
été développés. D'une part la méthode
statique où, une fois l'équilibre atteint, un aliquote de la
phase gazeuse est prélevé et transféré à la
GC. D'autre part la méthode dynamique (aussi appelée «
Purge and Trap ») où l'équilibre n'est jamais
atteint étant donné que la phase gazeuse « Headspace »
est continuellement renouvelée pour être piégée sur
une matière adsorbante (Grob and Barry 2004).
1.3 Avantages et désavantages
La chromatographie gazeuse couplée au « Headspace
» (HSGC) permet de travailler sans utiliser de solvant organique ce qui
n'est généralement pas le cas en GC classique. Ici le liquide
d'extraction est remplacé par un gaz qui est en fait le solvant
idéal pour les composés fortement volatils (Kolb and Ettre
2006).
Elle permet donc d'analyser directement un échantillon
sous forme solide ou liquide tout en évitant l'effet de matrice, ce qui
est avantageux pour la plupart des substrats biologiques ou environnementaux
qui sont relativement complexes (Rouseff, Cadwallader et al. 2001). D'autre
part, ce procédé permet un réel gain de temps au niveau de
la préparation des échantillons, une étape relativement
chronophage (Sitaramaraju, van Hul et al. 2008). En effet, dans la plupart des
laboratoires, cette étape représente en fait les 2/3 du temps
dépensé pour une analyse (la plupart des échantillons
nécessitant un traitement préalable adapté à la
technique analytique). L'HS-GC permet également de diminuer les erreurs
qui peuvent apparaître lors de cette étape de préparation.
Ces erreurs peuvent être causées par des impuretés, des
pertes d'échantillon,...
Sa facilité d'utilisation et son automatisation sont
également deux points positifs (le premier modèle
automatisé est apparu en 1967) (Restek 2000).
Toutefois, pour que ce type d'extraction soit utilisable, il
faut que la substance recherchée soit fortement volatile. Il faut aussi
pouvoir garantir l'établissement de l'équilibre entre la phase
gazeuse et l'échantillon (aussi appelé « phase
condensée ») (Grob and Barry 2004). Le risque
5
de perte de gaz lors du transfert de l'échantillon vers
la chromatographie gazeuse est aussi problématique pour certains types
de « Headspace » (Poole 2009).
1.4 Les différents types de « Headspace
»
1.4.1 La « Static Headspace Extraction
» (SHE)
Il s'agit du type de « Headspace » le plus
ancien et le plus classique ; sa première utilisation remonte à
1958. Dans la littérature, le terme général «
Headspace » est d'ailleurs souvent utilisé pour désigner la
SHE (Hübschmann 2009). C'est la version la plus facilement
automatisée et validée de toutes les différentes «
Headspace » (Snow and Slack 2002). Elle est utilisée pour les
analyses de gammes de concentration plus élevée que les autres
versions reprises ci-dessous ; elle est donc moins sensible.
Dans ce système, l'échantillon solide ou
liquide, accompagné ou non d'un agent modifiant la matrice, est
placé dans un flacon de 10 ou 20 mL qui est ensuite fermé
hermétiquement. Le tout est chauffé pendant un temps donné
pour atteindre l'équilibre entre la phase gazeuse (« Headspace) et
l'échantillon ; la quantité de chaque composé volatil se
trouvant dans le « Headspace » est alors proportionnelle à
celle contenue dans l'échantillon (Papet, Brunet et al. 2010). Au cours
de cette étape, les composés volatils sont donc extraits de la
matrice complexe non volatile. Un aliquote de la phase gazeuse est alors
prélevé, soit via une seringue (mode manuel), soit via un
système de prélèvement automatisé, et est
transféré à la GC pour l'analyse (Grob and Barry 2004).
Dans les installations modernes, la seringue est remplacée par une
boucle de transfert thermostatisée et pressurisée, ce qui permet
un transfert plus inerte et plus « propre » vers la GC (Snow and
Slack 2002).
La version « statique » ou « d'équilibre
» est relativement facile d'utilisation et ne nécessite
généralement aucune préparation supplémentaire au
niveau de l'échantillon pour ce qui est des analyses qualitatives. En ce
qui concerne l'aspect quantitatif, il peut être intéressant de
prendre en compte et de minimiser l'effet de matrice pour augmenter la
linéarité et la précision. Pour ce type d'analyses on
s'orientera donc préférentiellement vers la version «
Dynamique » (cf. ci-dessous) qui s'avère être nettement plus
fiable (Paris 2002-2003). La SHE étant la technique utilisée pour
les diverses analyses reprises pour ce mémoire, elle sera
expliquée plus en détails par après.

6
Figure 2 : Principe de base (Restek, 2000).
1.4.2 La «Dynamic Headspace Extraction»
(«Purge and Trap»)
Cette technique a fait son apparition vers 1970 après
la commercialisation du Tenax® un polymère adsorbant
fréquemment utilisé (Snow and Slack 2002). Le mode dynamique
s'avère être préférable pour l'analyse de traces ou
lorsque l'on recherche une extraction plus approfondie des analytes. Elle est
donc plus sensible que la version statique car, ici, la quasi-totalité
de l'analyte est transférée à la GC contrairement au mode
statique où seulement une certaine quantité est
prélevée.
Comme pour la version « statique », l'extraction
à partir de la matrice est fortement liée à la
volatilité de l'analyte. Pour principale différence, la version
dynamique ne laisse pas l'équilibre s'établir entre
l'échantillon et la phase gazeuse. En effet, un flux continu de gaz
vecteur (par exemple, l'héliumn, le « Purge Gas ») permet de
balayer en permanence soit la phase gazeuse « Headspace » soit
directement l'échantillon empêchant ainsi l'établissement
de l'équilibre. Cette étape de « Purge » est
réalisée pendant une durée bien déterminée.
L'extraction de l'analyte est ainsi facilitée grâce à la
formation d'un gradient de concentration ; les analytes sont
entraînés par le gaz porteur pour être piégés
et concentrés juste avant l'analyse, d'où le nom « Purge
And trap »(Grob and Barry 2004). Le système piégeant
l'analyte doit remplir certains critères, notamment le fait de retenir
essentiellement l'analyte d'intérêt, de permettre une injection
rapide au niveau de la colonne et de ne pas introduire d'impuretés dans
le système.
Ensuite une désorption thermique au niveau du
piège permet à l'analyte de passer dans le GC.
7
Figure 3 : Dynamic Headspace (Bergna, M.
(2011)
1.4.3 La « Solid Phase Microextraction
» (SPME)
Le couplage « Headspace - SPME », apparu en 1993, a
été developpé par la société « Supelco
». Il s'agit d'une nouvelle alternative de « Headspace
» qui peut être également couplée à la
HPLC. Cette technique a fait l'objet de nombreuses recherches durant les
dernières décennies. Elle peut être utilisée pour
une large gamme d'applications pour lesquelles elle a été
validée (Papet, Brunet et al. 2010). Au début de son utilisation,
les scientifiques ont rencontré divers problèmes technique pour
ce qui est de sa reproductibilité, de son automatisation et de son
chauffage mais, aujourd'hui, de nombreuses solutions ont été
apportées et elle est maintenant utilisée pour mener des analyses
complexes et ce, dans des gammes de concentrations relativement faibles. (Snow
and Slack 2002)
La technique consiste à introduire dans l'espace de
tête de l'échantillon une seringue rétractable remplie
d'une fibre en silice fondue imprégnée d'une phase stationnaire
polymérique (par exemple du polyacrylate ou du divinyl benzène).
Celle-ci va piéger et concentrer les analytes se trouvant dans cette
phase confinée. La seringue est ensuite introduite dans l'injecteur
où il y a désorption thermique, ce qui permet à l'analyte
d'être libéré dans le GC. Une même fibre peut
être réutilisée jusqu'à cinquante fois. La SPME peut
être couplée à un mode statique ou dynamique de «
Headspace », leur association permettant d'augmenter la sensibilité
et de diminuer les limites de détection et de quantification. (Papet,
Brunet et al. 2010)
8
Figure 4 : Fonctionnement SPME (Kolb and Ettre
2006)
1.5 Théorie et application de la « Static
Headspace Extraction » (SHE)
La totalité des analyses reprises dans ce
mémoire ayant été menées sur SHE, la théorie
et l'application de celle-ci seront vues plus en détails.
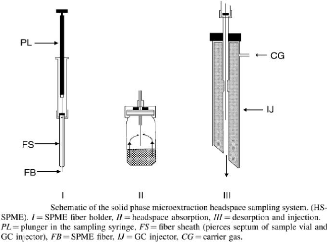
1.5.1 Les différents types
d'échantillonnage en SHE
En SHE divers types d'instrumentation, automatisés ou
non, permettent d'extraire les composés d'intérêts. Parmi
ceux-ci on rencontre 3 grandes classes d'appareillage :
- « Injection à l'aide d'une seringue étanche
aux gaz »
- « Système à équilibre de pression
» - « Système avec boucle - pression »
-
? Injection à l'aide d'une seringue
étanche aux gaz
Dans ce genre de configuration, une seringue étanche
permet de transférer l'échantillon de l'espace de tête
à la GC. Il existe des systèmes à seringues manuels ou
automatisés.
Les systèmes manuels présentent deux
inconvénients majeurs.
D'une part, ceux-ci ne sont pas thermostatisés ; il y a
donc un risque de recondensation dans la seringue lors du transfert de
l'aliquote. Il est d'ailleurs conseillé de préchauffer la
seringue dans un four à 90°C avant d'effectuer le transfert, ce qui
semble peu pratique.
D'autre part, lors du transfert à la GC, la pression au
sein de la seringue n'est pas contrôlée et peut donc varier. Pour
pallier ce problème, des valves ont été
développées, permettant de bloquer le système pour
empêcher les fuites (cf. Valves Luer-Lock) (Kolb and Ettre 2006).
Actuellement, les systèmes automatisés sont
nettement plus répandus. L'échantillon est tout d'abord
thermostatisé à une certaine température et pendant un
certain temps. Une fois l'équilibre atteint, le
prélèvement est effectué via la seringue et injecté
dans la GC. Le point positif est qu'ici la seringue est thermostatisée
(Kolb and Ettre 2006).

9
Figure 5 : Système « Injection par
seringue étanche aux gaz » ( Restek (2000)
? Système à équilibre de pression
Comme pour les autres techniques, l'échantillon est
thermostatisé à une certaine température pendant un temps
donné. Une fois l'équilibre atteint, l'échantillon est
pressurisé à une pression supérieure à celle de la
colonne par apport de gaz vecteur via une aiguille introduite dans le
récipient. Après ces deux étapes d'équilibrage et
de pressurisation, la valve est fermée et l'afflux de gaz vecteur
interrompu. L'échantillon est alors connecté à la GC via
une ligne de transfert chauffée qui reste ouverte durant un certain laps
de temps. Grâce à la différence de pression qui a
été établie, l'échantillon est
transféré à la GC (Kolb and Ettre 2006). Différents
paramètres peuvent être contrôlés comme par exemple
la pression établie dans le vial ou encore le temps de transfert.
Ce genre d'appareillage permet en principe d'obtenir des
résultats présentant une bonne reproductibilité. Cela est
dû notamment au fait que le nombre de pièces utilisées pour
le transfert est relativement réduit ce qui évite en grande
partie les éventuelles pertes par
10
adsorption ou pas fuites.
Ce type d'échantillonnage ne permet cependant pas de
connaître avec exactitude le volume prélevé ; le temps
d'ouverture de la boucle ne définit en effet le volume
prélevé qui dépend aussi de tous les autres
paramètres (pression, temps de pressurisation, température,...)
(Restek 2000).
Une des premières versions automatisées de ce
type d'instrument est apparue en 1967 (Kolb and Ettre 2006).
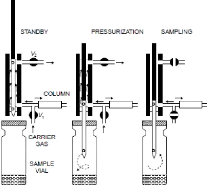
Figure 6 : Système à
équilibre de pression (Grob, R. L. and E. F. Barry (2004)
? Système avec boucle - pression
Une fois l'équilibre atteint, l'échantillon est
pressurisé grâce au gaz vecteur, de manière similaire
à la configuration précédente. Comme principale
différence, l'injection ne se fait pas directement dans la colonne. En
effet, comme son nom l'indique, cette technique fait intervenir une boucle
(« Loop ») qui sert d'intermédiaire entre le vial du «
Headspace » et la GC (Pawliszyn 2002).
Tout d'abord, l'aliquote du « Headspace » va remplir
la boucle grâce à la différence de pression
précédemment établie. Ensuite le contenu de la boucle sera
lui-même introduit dans la GC de manière habituelle. Tout cela
grâce à une valve à 6 entrées (Kolb and Ettre 2006).
Cette version présente certains avantages et désavantages. Sur le
plan positif, le fait de pouvoir thermostatiser la boucle permet de
réduire les pertes des composés de hauts poids
moléculaires par adsorption,... De plus avec ce système, le
volume de la boucle est connu et fixe (contrairement au système
précédent), ce qui permet d'augmenter la reproductibilité
(Restek 2000).
11
Comme principal inconvénient, une analyse
précédente peut contaminer la boucle du système («
Carryover »), pour provoquer l'apparition de pics parasites (« Ghost
Peaks »). Ce dernier modèle est celui utilisé pour les
diverses analyses reprises dans ce mémoire.
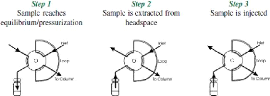
Figure 7: Système boucle-pression (Restek
(2000)
1.5.2 Le coefficient K
La préparation de l'échantillon doit être
effectuée de telle sorte que la concentration de l'analyte au niveau de
l'espace de tête soit maximale et que les contaminations provenant de la
matrice soient minimales (Restek 2000). Ceci est basé sur le coefficient
de partage K qui définit la distribution de l'analyte entre la phase
condensée de l'échantillon et la phase gazeuse. Les
composés ayant un faible coefficient auront plus facilement tendance
à passer dans la phase gazeuse ; ces composés seront plus
facilement détectables (par exemple l'hexane dans l'eau). Et
inversement, pour les composés ayant un coefficient élevé,
ceux-ci présenteront une limite de détection élevée
et conduiront à l'obtention d'un signal faible (par exemple
l'éthanol dans l'eau) (Flanagan, Taylor et al. 2007).
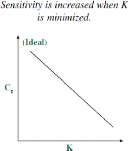
Figure 8 : Influence du coefficient de partage
(Restek (2000)
Plusieurs paramètres permettent de diminuer la valeur du
coefficient K : ? La température du flacon
12
Pour l'éthanol par exemple, le fait de passer de
40°C à 80°C au niveau du vial permet de diminuer d'environ 4
fois le coefficient de partage (Restek 2000).
? L'effet « salting out
»
Le coefficient peut être réduit par l'ajout d'un
sel inorganique. Ceci est valable pour un échantillon de type aqueux et
pour faciliter le passage des substances organiques polaires. L'ajout de sel
permet alors de diminuer leur solubilité et d'augmenter leur
concentration au niveau du « Headspace ». Cet effet n'est
quasiment pas visible sur les composés ayant un coefficient moyennement
élevé (Restek 2000).
Il faut aussi travailler avec des concentrations en sel
relativement élevées pour pouvoir réellement augmenter la
sensibilité.
Certains problèmes peuvent survenir suite à
l'ajout de sel comme l'augmentation de la viscosité des
échantillons aqueux, augmentant ainsi le temps nécessaire pour
atteindre l'équilibre. Certains sels contiennent aussi des
impuretés pouvant contaminer l'analyse. Il a été
suggéré que cette amélioration de la sensibilité
due au « salting out » dans le cadre de l'HS-GC ne serait
pas exclusivement due à l'ajout du sel ; le fait d'ajouter une certaine
quantité de sel permettrait également d'augmenter le volume de
l'échantillon et, de ce fait, d'augmenter la sensibilité (par
diminution du coefficient voir 1.5.3) (Kolb and Ettre 2006).
1.5.3 Le coefficient Beta
Le coefficient f3 est défini comme le volume
occupé par le phase gazeuse « Headspace » par rapport à
celui de la phase condensée de l'échantillon. Une valeur faible
de f3 permet généralement une réponse plus
élevée mais cette relation ne se vérifie pas toujours. Il
convient tout d'abord d'optimiser l'échantillon pour diminuer la valeur
du coefficient K avant même de penser à modifier le coefficient f3
(par exemple en augmentant la taille de l'échantillon) (Kolb and Ettre
2006).
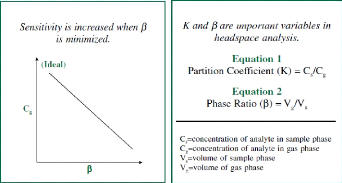
13
Figure 9 : Influence du coefficient Beta (Restek
(2000)
1.5.4 La dérivation
La dérivation est une autre technique permettant
d'augmenter la sensibilité et les performances de l'analyse «
Headspace ». Il s'agit d'une « réaction chimique quantitative
entre un réactif et une molécule peu ou non volatile pour
augmenter sa volatilité, par diminution des tensions internes (liaisons
inter- et intramoléculaires) comme, par exemple, l'estérification
des fonctions carboxyliques » (Sternberg 2003 - 2004).
Elle concerne principalement les alcools, les acides et les
amines qui sont des composés pouvant présenter certaines
difficultés lors des analyses de par la présence de liens
hydrogènes. Ces espèces peuvent se condenser ou s'adsorber au
niveau de l'orifice lors de l'injection ou au niveau de la colonne, ce qui
provoque une diminution de la réponse de l'appareil. Elles
présentent également une grande solubilité au sein des
échantillons aqueux, ce qui provoque une diminution du signal (Restek
2000).
La dérivation peut permettre de diminuer les effets
d'adsorption lors de l'injection dans le GC et également d'augmenter la
volatilité de l'analyte. Pour ce faire, toute une série de
réactions sont possibles comme l'estérification,
l'acétylation, la silylation ou l'alkylation. Ces réactions se
déroulent souvent à hautes températures au sein même
du vial et peuvent créer une pression supplémentaire à
celle tolérée par le septum et le récipient ; il existe
donc des septums spéciaux pouvant tolérer un certain excès
de pression. Il faut toutefois être attentif au fait que ces
réactions de dérivation peuvent dans certains cas faire
apparaître des produits de réactions volatils et perturbent
l'analyse du « Headspace » (surtout dans le cas où
leur temps de rétention est proche de celui de l'analyte) (Restek
2000).
14
1.5.5 Préparation de l'échantillon
La préparation de l'échantillon doit se faire de
telle sorte que la concentration de l'analyte dans la phase gazeuse soit
maximale tout en minimisant les contaminations pouvant provenir de la matrice.
Par exemple l'eau peut poser problème en se recondensant dans la ligne
de transfert.
Les échantillons fortement concentrés peuvent
également être responsables de « carry over1
» ; c'est pourquoi on conseille de procéder par ordre croissant de
concentration lors d'une analyse. Dans le cas où on est certain que
l'échantillon précédent a contaminé la colonne,
celle-ci peut être chauffée à sa température
maximale pour en quelque sorte la « décrasser ».
De même, le fait de travailler avec des
températures plus élevées au niveau de la ligne de
transfert permet de diminuer le phénomène de « carry over
» (Restek 2000).
? Echantillon solide
Les échantillons solides fréquemment
analysés en « Headspace » sont les médicaments, les
matériaux pour emballages imprimés (films plastiques,
aluminium,...), les sols et les polymères (Kolb and Ettre 2006).
Il s'avère parfois difficile pour ce type
d'échantillons d'atteindre l'équilibre (étape
indispensable pour les analyses quantitatives). En effet, les substances
volatiles à analyser peuvent parfois être emprisonnées dans
l'échantillon ce qui nécessite un temps considérable pour
atteindre l'équilibre. Il faut donc pouvoir garantir
l'établissement de celui-ci et ce, dans un laps de temps raisonnable.
Dans un tel cas, il est alors nécessaire de modifier l'état
physique de l'échantillon.
Pour les formes solides, il existe donc trois types
d'approches:
- soit l'analyse du solide tel quel (méthode valable
pour les solides permettant d'atteindre l'équilibre facilement)
- Soit l'ajout d'un solvant (dans le cas où le solide ne
permet pas d'atteindre l'équilibre) - Soit le broyage du solide (dans le
cas où le solide ne permet pas d'atteindre l'équilibre)
L'ajout d'un solvant permet de transférer
l'échantillon dans le liquide, ce qui est intéressant car le
travail sur matière liquide s'avère être nettement plus
simple, l'équilibre étant en
1 Il s'agit d'une contamination provenant d'une
analyse précédente et qui provoque l'apparition d'un pic parasite
sur le chromatogramme.
15
principe atteint nettement plus rapidement. Cependant, cette
approche provoque bien souvent une diminution du signal en comparaison avec
l'analyse sur poudre sèche (à cause de la viscosité du
solvant,...). Il y a donc une certaine perte de sensibilité. Cette
méthode présente un second inconvénient ; en effet, le
solvant organique utilisé entraîne bien souvent une solubilisation
de l'analyte. Ce phénomène va donc provoquer une augmentation du
coefficient de partage (généralement faible dans les
échantillons solides) diminuant ainsi la concentration d'analyte en
phase gazeuse (Grob and Barry 2004).
Le broyage, quant à lui, permet d'augmenter la surface
disponible pour le passage dans le « Headspace ». Pour moudre le
solide sans générer de chaleur excessive, on utilise en
général la technique du cryobroyage qui consiste à broyer
l'échantillon en présence de dioxyde de carbone ou d'azote
liquide. Cette technique permet de réduire nettement la taille de
l'échantillon tout en évitant les pertes et dégradations
pouvant provenir d'une augmentation de température (Kolb and Ettre
2006).
Cette diminution de taille permet de réduire nettement
le temps de diffusion et d'atteindre plus facilement l'équilibre. Il
faut noter que les échantillons préparés par ce
procédé doivent être mélangés avant
d'être analysés s'ils ont été stockés un
certain temps car un gradient de concentration a pu s'établir durant le
stockage (Grob and Barry 2004).
? Echantillon Liquide
La préparation des échantillons liquides en SHE
se fait habituellement de manière assez simple. En
général, l'échantillon est tout simplement introduit dans
le vial qui est ensuite rapidement scellé pour limiter les pertes (Grob
and Barry 2004).
1.5.6 Les différents paramètres
Le choix des divers paramètres doit se faire de
manière judicieuse. En effet, il faut pouvoir maximiser le signal et la
sensibilité tout en minimisant le temps et le matériel
utilisé.
? La thermostatisation
La température à laquelle le vial sera
chauffé va permettre d'influencer la pression de vapeur de l'analyte.
Cette pression de vapeur est une variable très importante pour cette
technique analytique car elle joue sur le passage de l'analyte en phase
gazeuse. La pression varie de manière exponentielle, dès lors,une
faible différence de température peut avoir une très forte
influence sur la réponse obtenue. Le fait d'augmenter la
température permet de diminuer le coefficient K (cf. section 1.5.2) et
donc d'augmenter la pression de vapeur. Ce phénomène est
16
surtout visible pour les composés avec une valeur
élevée du coefficient K (une molécule polaire dans une
matrice aqueuse par exemple) (Muffet 2011). Une augmentation de
température permet donc d'accroître le flux d'évaporation
et de diminuer le temps nécessaire pour atteindre l'équilibre, ce
qui va également influencer la concentration à saturation (Papet,
Brunet et al. 2010).
Ce paramètre a donc une influence directe sur la
sensibilité et sur le temps nécessaire pour atteindre
l'équilibre. La température influence aussi la solubilité
dans le milieu à analyser (Kolb and Ettre 2006).
Toutefois cette règle n'est pas universelle. En effet,
certains composés peuvent être décomposés par la
chaleur ou peuvent être impliqués dans des
phénomènes d'oxydation (Kolb and Ettre 2006). Certains
composés parasites présents dans la matrice peuvent aussi voir
leur volatilité augmenter (Hübschmann 2009).
Il faut aussi se méfier de l'augmentation de pression
dans l'espace de tête pouvant provenir du solvant présent dans le
vial. Cette augmentation peut être responsable de divers problème
analytiques notamment d'un déséquilibre de pression lors du
prélèvement d'un aliquote1. Cette
réalité concerne surtout les solvants à basse
température d'ébullition, ce qui pousse l'analyste à
préférer des solvants présentant des hauts points
d'ébullition. On conseille en général de ne pas travailler
à une température supérieure à 10°C au dessus
de la température d'ébullition d'un solvant (Anonyme 2000) .
Le temps durant lequel l'échantillon est chauffé
joue aussi un rôle important pour le transfert de l'analyte dans l'espace
de tête. Ce paramètre est déterminé en fonction de
la quantité d'échantillon, du type d'échantillon, du
coefficient de partage de l'analyte,... Il existe pour chaque analyte un temps
d'équilibration optimal. En effet il ne faut pas que celui-ci (i) soit
inférieur au temps nécessaire pour atteindre l'équilibre
(ce qui entrainerait des problèmes de reproductibilité) ; et (ii)
soit supérieur au temps optimal car, même sans modification du
signal, il s'agit là d'une perte de temps inutile. Pour
déterminer le temps de chauffe idéal, il est habituellement
conseillé de procéder par balayage en faisant varier la
température pour évaluer l'évolution de l'aire du pic
d'intérêt en fonction du temps de chauffe (Anonyme 2000).
? Le type de vial
Le type de vial choisi doit correspondre à la taille de
l'échantillon. A ce niveau-ci, de nombreuses contaminations peuvent
apparaître suite à un nettoyage incomplet ou encore via
l'adsorption de contaminant.
1 La plupart des systèmes de
prélèvement étant basés sur une différence
de pression.
17
Le type de septum doit aussi permettre d'éviter
d'éventuelles contaminations. Le Teflon® permet en
général de minimiser les risques (Restek 2000).
s Le temps de mélange
Le fait de mélanger l'échantillon lors du
chauffage permet de faciliter le passage à l'état
d'équilibre (Restek 2000). Pour certaines analyses, le temps
d'équilibration a pu être réduit de 45 à 10 min
grâce à cette étape de mélange (Hübschmann
2009).
s Le remplissage de la boucle
Après l'étape de pressurisation, une partie du
« Headspace » est transférée dans la boucle grâce
à la différence de pression préétablie. Il est
possible de régler le temps de remplissage de la boucle (le temps durant
lequel le mélange gazeux « Headspace - Gaz de
pressurisation » traverse la boucle) ainsi que le temps
d'équilibration de la boucle (le temps nécessaire pour permettre
au mélange gazeux d'atteindre la température de la boucle et
permettre de stabiliser la pression) (Anonyme 2000).
s La pressurisation
Pour cette étape, il est possible de définir non
seulement la pression établie dans le flacon mais aussi le temps durant
lequel le tout est pressurisé.
Pour la majorité des échantillons liquides, la
pression développée dans le milieu est suffisante pour permettre
le passage dans la boucle. En revanche pour les matières solides cela
peut poser problème. Il est alors nécessaire de pressuriser et
d'ajouter une pression supplémentaire. Les valeurs usuelles de pressions
dans le flacon sont de l'ordre de 1.5 à 2 Atm. Pour pouvoir
définir la pression optimale pour une analyse donnée il est
conseillé ici également de procéder par balayage (pour des
mêmes valeurs de temps et de température) en portant sur graphique
l'aire du pic en fonction de la pression.
Pour le temps de pressurisation, les valeurs habituelles vont
de 10 à 30 secondes (Anonyme 2000).
s Le temps d'injection
Il s'agit du temps nécessaire pour transférer la
totalité du contenu de la boucle vers la GC. Ce temps doit être
suffisant pour que la totalité de l'aliquote puisse passer. En cas de
temps trop court, une partie de la quantité prélevée
serait perdue, ce qui impliquerait une perte de sensibilité. Il est
dès lors vivement conseillé de fixer cette valeur à un
niveau suffisamment
18
élevé afin de garantir la totalité du
transfert. Il faut aussi noter qu'un temps excessif ne représente pas un
problème en soi (Anonyme 2000).
? La taille de l'échantillon
Le fait d'augmenter la quantité d'échantillon
permet en général d'obtenir une meilleure sensibilité.
Dans le cas où la sensibilité n'est pas primordiale, cette
approche s'avère cependant être peu intéressante car le
temps nécessaire pour atteindre l'équilibre se trouve alors
augmenté.
1.5.7 La « Multiple Headspace Extraction »
(MHE)
La MHE est une méthode quantitative qui consiste
à prélever successivement plusieurs aliquotes d'un même
échantillon. L'aire du pic obtenu va alors diminuer proportionnellement
avec le nombre de prélèvements. Une fois l'analyse
terminée (c'est-à-dire lorsque l'aire du pic vaut zéro),
la somme des aires des pics obtenus est calculée, ce qui fournit la
quantité totale d'analyte présent dans l'échantillon (Kolb
and Ettre 2006). De nos jours, les prélèvements ne se font plus
jusqu'à l'obtention d'une aire de pic nulle vu la perte de temps trop
importante qui en découlerait. Après un nombre limité de
prélèvements (2 à 4) la quantité totale du
composé est alors déterminée au moyen d'une analyse par
régression linéaire (Grob and Barry 2004).
Lors de cette analyse, la valeur du coefficient K ne varie
pas, ce qui ne change donc pas l'affinité de l'analyte pour le milieu.
Cette technique présente l'avantage de s'affranchir des effets de
matrice et s'avère utile également lorsque l'utilisation de
standard est compromise (Jordi 2004).
1.6 Le genre Ocimum
Le genre Ocimum appartient à la famille des
Lamiaceae et à la sous-famille des Nepetoidae. Les
plantes appartenant à ce genre sont utilisée depuis plus de 3000
ans pour un usage médicinal et alimentaire (Johnson and Franz 2002). Au
niveau taxonomique, la place du genre n'est pas très claire, en effet,
à cause de nombreux phénomènes d'hybridation et de
polyploïdie, la classification en est devenue plus complexe (Bhowon,
Jhaumeer-Laulloo et al. 2012). Il comprend au total pas moins de 65
espèces (Paton, Harley et al. 1999) dont le représentant le plus
connu est le basilic (Ocimum basilicum L.) Celui-ci est originaire
d'Inde et peut être utilisé comme condiment frais ou
séché (Klimánková, Holadová et al. 2008).
Mis
19
à part l'assaisonnement de divers plats, il est
également utilisé en médecine traditionnelle pour traiter
les maux de tête, la diarrhée, les troubles
rénaux,...(Grayer, Vieira et al. 2004). Le type de basilic
cultivé mais aussi les conditions environnementales ont une influence
sur les teneurs des différents composés responsables de
l'arôme développé (Jirovetz, Buschbauer et al. 2003). Ces
composés sont, notamment, l'estragole, le linalool, le 1,8
cinéole et le méthyl eugénol (Lee, Umano et al. 2005).
Pour pouvoir étudier la composition de l'huile essentielle de ces
plantes il faut au préalable les extraire. Pour ce faire, il existe
diverses méthodes comme la distillation à la vapeur d'eau,
l'extraction par un solvant ou encore l'extraction en espace de tête. Il
faut noter que les monoterpènes présents peuvent être
facilement perdus ou encore altérés lors de l'extraction. Avec
l'hydrodistillation par exemple, les monoterpènes peuvent subir des
modifications chimiques lors du chauffage en présence d'eau. Avec
l'extraction par un solvant, des pertes peuvent aussi apparaître lors de
l'élimination de ce dernier. Le choix d'une méthode d'extraction
douce et adéquate s'avère donc nécessaire
(Klimánková, Holadová et al. 2008).
20
2. But du Travail
Le couplage HS-GC parait, en théorie, la solution
idéale pour l'analyse de composés volatils présents dans
une matrice non chromatographiable et donc pour l'analyse des huiles
essentielles dans des plantes médicinales. Cependant le nombre de
paramètres à optimiser est conséquent (aussi bien pour la
préparation de l'échantillon (afin d'optimiser l'extraction
« Headspace ») que pour les différents paramètres de
l'appareillage en lui-même). Il faut donc pouvoir s'assurer de travailler
avec les meilleures conditions pour un type d'analyse donné. Cette
optimisation des paramètres permet de garantir l'établissement de
l'équilibre (indispensable pour la reproductibilité d'une analyse
à l'autre) ainsi qu'une extraction et un transfert efficaces des
composés volatils.
Des résultats précédents ont
montré cependant que la technique présentait quelques
disparités au cours des analyses d'Ocimum. En effet, lors de
travaux effectués sur échantillons solides (fragments de plantes
ou poudre végétale), une certaine discrimination entre les
différents composés présents dans l'échantillon est
apparue. Quelques composés présentant les temps de
rétention les plus faibles évoluent de manière
proportionnelle et plus ou moins linéaire avec la quantité de
plante introduite dans le vial alors que les composés plus fortement
retenus, eux, ne suivent pas du tout cette évolution (Michel 2012).
Ce travail vise donc à optimiser la technique «
Headspace ».
Etant donné que l'analyse sur échantillon solide
s'avère être nettement plus complexe (en comparaison avec les
matières liquides), il va falloir aussi bien s'axer sur la
préparation de l'échantillon que sur l'appareillage «
Headspace » en lui-même. L'objectif est donc de diminuer
les effets de matrices (libération des composés volatils,
adsorption éventuelle,...), de permettre un passage homogène des
composés en phase gazeuse (ce passage étant défini par le
« chemin » parcouru par l'analyte pour gagner la phase gazeuse) et,
bien sûr, de définir les valeurs optimales pour les
paramètres de l'extraction en espace de tête (température,
temps d'équilibration,...).
Le but étant aussi de pouvoir observer et comprendre
l'impact qu'a chacun des différents facteurs sur l'analyse des huiles
essentielles en espace de tête.
21
3. Matériels et méthodes 3.1
L'échantillon
3.1.1 Matière végétale
Une première partie des analyses a été
effectuée sur des feuilles d'Ocimum centraliafricanum R.E.
Fries (Paton, Harley et al. 1999) provenant de République
Démocratique du Congo et plus précisément des mines de
Kasonta et Luiswishi. Il s'agit d'une plante herbacée vivace
que l'on trouve essentiellement en Afrique du Sud et qui supporte des sols
exceptionnellement riches en cuivre. Elle mesure entre 15 et 75 cm de hauteur,
les feuilles sont opposées, ascendantes et sessiles. Cet
échantillon a été prélevé et
séché à l'air libre puis introduit dans un sachet
hermétiquement fermé en présence de silicagel (afin de
garantir l'absence d'eau) (Michel 2012).
Cependant, vu la faible rémanence de composés
volatils dans ces échantillons de 2011, il a été
décidé d'opter pour Ocimum basilicum L. que nous avons
acheté en grande surface. Il s'agit du basilic de la marque «
Carrefour® : Selection » (lot : L2282BA, 15 g) et de la marque «
Ducros®» (12 g).

Figure 10 : Feuilles d'Ocimum
centraliafricanum R.E Fries
3.1.2 Préparation de l'échantillon
La quantité de feuilles nécessaire est
prélevée puis coupée à l'aide de ciseaux. Le tout
est introduit dans un récipient en INOX® pour procéder
à un cryobroyage ; de l'azote liquide est versé sur la
matière végétale qui est alors congelée et devient
cassante, le tout est alors broyé
22
à l'aide d'un pilon ce qui donne une poudre
relativement fine et homogène (voir figure 12). Celle-ci est finalement
introduite dans un récipient hermétiquement fermé de 20
mL. Cette technique permet d'obtenir une poudre de fine granulométrie
exempte de solvant et cela sans altérer la matière
végétale. En effet, les température atteintes lors du
broyage restent très faibles, ce qui permet de ne pas altérer les
composés fragiles et instables (Mathieu and Fonteneau 2008).

Figure 11 : Feuilles d'O. centraliafricanum
R.E. Fries.
3.2 Le système « Headspace »
3.2.1 Type d'appareillage
Le modèle de « Headspace »
utilisé dans cette étude est de type statique
équipé d'un système de prélèvement «
Boucle - Pression » (voir point 1.5.1). Il s'agit du modèle «
Agilent® 7694 E Headspace Sampler » et plus
précisément de la version « G1883 ». Cet
appareil analytique est muni d'un plateau tournant dans lequel peuvent
être introduits jusqu'à 12 vials (de 10 ou 20 mL).

Figure 12 : Carrousel du « Headspace
»
Ce modèle dispose de trois zones chauffées
distinctes qui sont le four pour le vial, la boucle et la ligne de transfert.
Au niveau du vial, le système de chauffe est constitué d'un bloc
métallique fonctionnant au moyen d'une résistance. La ligne de
transfert, composée de nickel, sert de jonction entre le «
Headspace » et le GC.
Les pressions de la voie pour le gaz porteur du GC et la voie
servant à la pressurisation du vial peuvent être
réglées manuellement au niveau du « Headspace
».
Cet instrument est également muni d'un bras
robotisé permettant de surélever le vial à analyser et
ensuite de percer le septum du récipient pour le
prélèvement.
Pour rappel, les principales étapes de l'extraction se
succèdent comme tel :
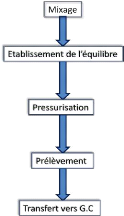
Figure 13 : Succession des étapes lors de
l'extraction « Headspace »
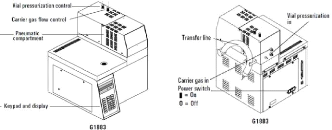
23
Figure 14 : Vue avant et arrière du
« Headspace » (Anonyme 2000)
24
3.2.2 Protocole ? Etalons
Avant de procéder aux diverses analyses, une
série d'étalons externes a été
sélectionnée et analysée selon (Masada 1976). La liste de
ces différents standards est reprise ci-dessous :
|
Composés :
|
T° ébullition (°C)
|
P de vapeur (mm
Hg à 25°C)
|
|
1,8 Cinéole
|
176 - 177
|
1,65
|
|
Citronellol
|
225
|
0,02
|
|
Estragol
|
215 - 216
|
0,21
|
|
Eugenol
|
254
|
0,03
|
|
Limonène
|
176
|
1,98
|
|
Linalol
|
198 - 200
|
0,16
|
|
â-Pinène
|
166
|
2,93
|
|
á-Terpinéol
|
219
|
0,0423
|
Tableau 1 : Liste des étalons externes
(Sigma Aldrich®)
Les étalons ont été prélevés
au moyen de capillaires en verre et une quantité de 5 uL a
été introduite dans chaque vial.
Entre chaque série d'analyses, un essai à blanc a
été effectué afin de détecter d'éventuelles
contaminations (« Carry-Over »).
? Analyse
Pour chaque analyse, une certaine quantité
d'échantillon est introduite dans un vial de type «
Agilent® Technologies, Flat Bottom Headspace Vials » de 10
mL (n° lot : 914-09-12/001) qui est scellé par un septum «
20mm Tan PTFE/White Silicone Septa (Agilent® Technologies)
(n° lot : 122393-1-1) grâce au cerclage en aluminium «
Agilent 20 mm, Crimp Cap Headspace, Safety Release».

25
Figure 15 : Pince, septum et vial.
Le vial est alors introduit dans le carrousel, agité
pendant une minute puis chauffé à 90°C pendant 35 minutes.
Il est ensuite surélevé et percé par une aiguille afin de
prélever un aliquote de l'espace de tête. La suite se
déroule comme expliqué au paragraphe « 1.5.1 » section
« Système Boucle - Pression ». L'ensemble des
paramètres du « Headspace » est repris dans le
tableau ci-dessous.
|
Temps (min)
|
|
Equilibrage du vial
|
34,00
|
|
Pressurisation
|
1,00
|
|
Remplissage de la boucle
|
0,20
|
|
Equilibration de la boucle
|
0,05
|
|
Injection
|
1,00
|
|
Cycle GC
|
33,75
|
|
Mélange
|
1,00
|
|
Température (°C)
|
|
Vial
|
100
|
|
Boucle
|
110
|
|
Ligne de transfert
|
120
|
|
Pression (PSI)1
|
|
Vial
|
22.04
|
|
Gaz porteur
|
21,30
|
Tableau 2 : Paramètres du «
Headspace »
3.3 La Chromatographie Gazeuse
Le système GC appartient à la marque «
Hewlett Packard® », il s'agit du modèle «
6890 Series GC System ».
1 Note : 1 PSI = 0.068045964 Atm / 1 Atm = 14.6959488
PSI
26
3.3.1 L'injecteur
Il s'agit d'un injecteur de type « EPC - Split -
Splitless- Inlet ». Dans la configuration « Split
», le gaz vecteur se mélange à l'échantillon
gazeux et seule une faible quantité est injectée dans la colonne
(la majorité restante étant éliminée). Ce type
d'injecteur permet d'éviter la saturation des sites de rétention
des colonnes à faible capacité (Kauffmann and Van
Antwerpen 2010) .
Le gaz vecteur est l'hélium, à un flux de 2,0 mL
/ min. Le rapport de division (« split ratio ») est
réglé à 1,0 (on travaille donc en mode « Splitless
») et la température de l'injecteur est de
250°C.
3.3.2 Colonne et phase stationnaire
Il s'agit d'une colonne capillaire du type Agilent
19091L-333 HP, 50+50% Phenyl Methyl Siloxane, 30,0 m x 250 um (i.d),
épaisseur de film 0,10 um. Elle résiste à une
température maximale de 310°C. La phase stationnaire appartient
à la famille des polysiloxanes sur lesquels sont greffées des
chaînes alkyles (méthyl et phényl). Ce type de colonne
permet de travailler sur des gammes de température relativement larges
(Rouessac and Rouessac 2004).
3.3.3 Détecteur
Il s'agit d'un détecteur à ionisation de flamme
(« FID ») qui se prête relativement bien à l'analyse des
substances organiques volatiles. Les analytes sont entraînés dans
la flamme alimentée par de l'hydrogène et de l'air, ce qui le
décompose en ions qui créent un courant supérieur à
celui mesuré en présence du gaz vecteur.
Les détecteurs FID sont universels et présentent
une bonne sensibilité ainsi qu'un large champ de
linéarité. Cependant ils impliquent inévitablement la
destruction de l'échantillon et ils sont non spécifiques
(Vaubourdolle 2007).
La température du détecteur est de 250°C et
les débits d'air et d'hydrogène sont, respectivement, de : 400 et
30 mL/min.
3.3.4 Programme de Température
27
Le four commence par chauffer la colonne à 60°C
pendant 1 minute. Ensuite il augmente de 4°C/min jusqu'à
125°C. Puis à raison de 10°C/min jusque 230°C. Et enfin
il redescend à 40°C, température maintenue pendant 6 minutes
(période de rééquilibrage) La durée totale d'un
cycle est de 33,75 minutes. Il est important d'établir un léger
décalage entre le « Headspace » (34,00 min) et le GC
(33,75 min) pour permettre l'enchaînement des analyses.
3.3.5 Analyse Statistique :
Les analyses statistiques ont été
réalisées à l'aide du programme informatique « MODDE
9.1 ». Ce dernier a permis d'établir le plan factoriel et
d'évaluer les effets de chaque paramètre
représenté.
4. Résultats et discussion
Les résultats sont séparés en deux grands
groupes :
28
- La première partie porte essentiellement sur la
préparation de l'échantillon. Elle aborde certains points,
notamment l'influence du type de broyage, de l'ajout d'un solvant ou de l'effet
« Salting-out ».
- La seconde partie s'axe essentiellement sur l'optimisation
des paramètres de l'appareillage «Headspace ». Dans
cette partie, les différents paramètres du « Headspace
» sont étudiés à l'aide d'un plan factoriel afin
de voir l'impact qu'a chacun d'eux sur les analyses. Les valeurs optimales sont
également déterminées pour les paramètres
influençant les résultats chromatographiques.
Pour l'entièreté des tests, le programme du GC
est resté le même comme décrit dans la section «
Matériel et méthodes » ; c'est pourquoi seuls les
paramètres du « Headspace » seront
précisés pour chacune des analyses.
4.1 Optimisation de la préparation de
l'échantillon
Les valeurs initiales des paramètres du «
Headspace » sont reprises ci-dessous (voir tableau 3).
|
Temps (min)
|
|
Equilibrage du vial
|
34
|
|
Pressurisation
|
1
|
|
Remplissage de la boucle
|
0,1
|
|
Equilibration de la boucle
|
0,05
|
|
Injection
|
1
|
|
Cycle GC
|
33,75
|
|
Mélange
|
1
|
|
Température (°C)
|
|
Vial
|
90
|
|
Boucle
|
100
|
|
Ligne de transfert
|
110
|
|
Pression (PSI)
|
|
Vial
|
15,50
|
|
Gaz porteur
|
21,3
|
Tableau 3 : Valeurs initiales des
paramètres du « Headspace ».
4.1.1 Comparaison des méthodes de broyage
L'échantillon a été broyé avant
chaque analyse afin d'obtenir une libération optimale des
composés volatils à partir de la poudre, rendue homogène.
Dans cette section, deux méthodes de broyages sont comparées ; le
broyage à l'aide d'un pilon et le cryobroyage. Une seule des
deux techniques sera sélectionnée pour la suite
des analyses.
Pour rappel, le cryobroyage permet d'obtenir une poudre d'une
fine granulométrie et d'une homogénéité
satisfaisante tout en préservant les composés instables et
fragiles présents dans la matière végétale (Anonyme
2012).
Afin d'évaluer l'éventuelle amélioration
que cette technique peut apporter à une analyse, une étude de
linéarité a été réalisée avec, d'une
part, la méthode de « cryobroyage » et, d'autre part, la
méthode classique avec broyage par pilon en verre. Pour chacune d'elles,
une série de 9 essais a été réalisée avec
trois groupes de masses différentes (3 essais par masse) à savoir
: 100, 200 et 300 mg. Les graphiques de l'aire de pics (en pA.s) en fonction de
la masse ont ensuite été tracés.
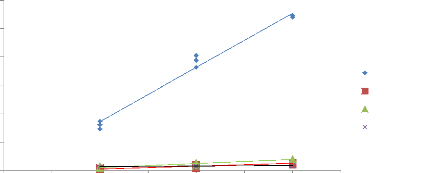
Sur ces graphiques, il apparaît clairement que l'aire
des pics évolue de manière nettement plus uniforme avec la
méthode du « cryobroyage » (figure 16 et 17).
60
50
Temps de rétention :
Surface du pic (pA.s)
40
30
20

R2 = 0.3287
R2 = 0.8786
10
R2 = 0.1971
0
R2 = 0.3133
1,191 1,379 4,263 13,624
29
0 50 100 150 200 250 300 350
Masse (mg)
Figure 16 : Linéarité de 4 pics
différents en fonction de la masse d'un échantillon d'Ocimum
centraliafricanum, broyage à l'aide d'une baguette en verre.
Températures « Headspace » : 90°C (vial) / 100
°C (boucle) / 110°C (ligne de transfert), 3 essais par masse.
Pressurisation du vial : 15,5 PSI, temps de remplissage de la boucle : 0,10
min, temps de pressurisation : 1,0 min
R2 = 0.9841
Temps de rétention :
1,191 1,379 4,263 13,624
R2 = 0.9033
R2 = 0.8621
R2 = 0.6467
|
Surface du pic (pA.s)
|
60 50 40 30 20 10 0
|
0 50 100 150 200 250 300 350
Masse (mg)
Figure 17 : Linéarité de 4 pics
différents en fonction de la masse d'un échantillon d'Ocimum
centraliafricanum à l'aide du cryobroyage. T° «
Headspace » : 90°C (vial) /100°C (boucle) / 110°C
(ligne de transfert), 3 essais par masse. Pressurisation du vial : 15,5 PSI,
temps de remplissage de la boucle : 0,10 min, temps de pressurisation : 1,0
min
Les aires augmentent de manière nettement plus
linéaire, ce qui se traduit par des valeurs de « R2
» plus satisfaisantes (figure 18).
|
Valeur "R2"
|
1.2
1 0.8 0.6 0.4 0.2
0
|
|
pic 1,191 pic 1,379 pic 4,263 pic 13,624 pic 1,191 pic 1,379 pic
4,263 pic 13,624
|
30
pic
|
1,191
|
pic
1,379
|
pic
4,263
|
pic
13,624
|
pic
1,191
|
pic
1,379
|
pic
4,263
|
pic
13,624
|
Figure 18 : Valeurs des « R2
» obtenues pour chacune des deux méthodes de broyage et pour les 4
composés différents (3masses analysées en triplicate).
31
Le fait d'obtenir une poudre plus fine et plus homogène
permet donc une diffusion plus facile et moins aléatoire dans l'espace
de tête. Toutefois, il semble que l'aire du pic le plus retenu (avec le
T.R le plus élevé soit : 4,263 min) présente une valeur du
« R2 » encore faible soit : 0,6467.
Selon (Kolb and Ettre 2006), les composés
présentant un faible coefficient de partage ( qui implique une faible
affinité pour l'échantillon), sont les composés pour
lesquels l'augmentation du volume de l'échantillon aura un effet
significatif au niveau de l'aire du pic. A l'opposé des composés
caractérisés par une valeur élevée du coefficient
ne sont, eux, que peu ou pas influencé par la quantité
d'échantillon. Ceci confirme nos observations ; les composés les
plus volatils (avec les temps de rétention les plus faibles) varient
fortement avec le volume de l'échantillon à l'opposé des
composés présentant une faible volatilité.
4.1.2 Changement d'échantillon
Les prélèvements d'O. centraliafricanum
ayant été effectués il y a plusieurs mois, il est
fort probable que la teneur en composés volatils aie nettement
diminué au fil du temps. Les conditions de conservation n'étant
pas optimales, ceci explique peut-être la faible fréquence des
pics obtenus sur les chromatogrammes ainsi que leur faible intensité. La
solution serait peut-être de congeler les plantes directement
après leur prélèvement et de les conserver ainsi
jusqu'à l'étape de cryobroyage.
L'échantillon a donc été abandonné
et changé pour un basilic (O. basilicum) de la marque
Carrefour® qui a été séché et conservé
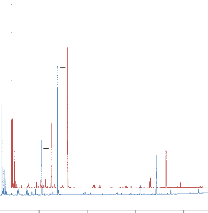
dans des conditions standardisées. Sur les chromatogrammes obtenus
(figure 19), l'intensité et le nombre de pics sont nettement plus
appréciables avec ce nouvel échantillon.
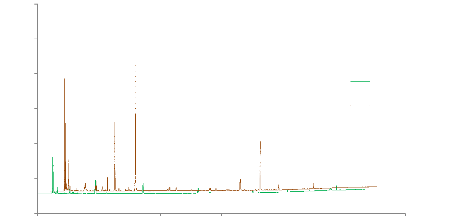
Signal (pA)
120
100
40
80
60
20
0
O. centraliafricanum
O. basilicum
32
0 5 10 15 20 25 30
Temps de rétention (min)
Figure 19 : Comparaison des chromatogrammes de
l'analyse d'Ocimum centraliafricanum et Ocimum basilicum
(Carrefour®). T° « Headspace » : 90°C
(vial) /100°C (boucle) / 110°C (ligne de transfert). Masse : 100 mg,
Cryobroyage. Pressurisation du vial : 15,5 PSI, temps de remplissage de la
boucle : 0,10 min, temps de pressurisation : 1,0 min
4.1.3 Ajout d'un solvant
Par la suite, l'étude s'est portée sur l'effet
provoqué par l'ajout d'un solvant sur la poudre sèche
d'échantillon. Comme expliqué plus haut à la section
« 1.5.5 Préparation de l'échantillon », l'ajout d'un
liquide peut permettre d'atteindre l'équilibre nettement plus
rapidement.
Cependant, cet ajout peut parfois engendrer une perte de
signal provenant de la solubilisation de certains composés, un
ralentissement du passage à l'équilibre de par la
viscosité du solvant ou encore l'apparition d'un pic parasite sur le
chromatogramme.
Dès lors, un solvant adéquat pour les analyses
en espace de tête doit présenter une grande stabilité ainsi
qu'un point d'ébullition élevé.
? Mygliol 812®
Le premier solvant testé est le mygliol 812® qui
est une huile neutre relativement stable par rapport aux
phénomènes d'oxydations et qui présente un point
d'ébullition relativement élevé : entre 240 et 270°C.
La poudre de chaque vial a été additionnée de 500 uL
d'huile neutre et le tout a été scellé
hermétiquement. Les droites de régressions ont été
étudiées de la
même manière que pour le cryobroyage (3 masses
différentes en triplicate).
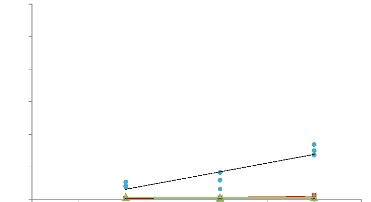
Sur le graphique de linéarité (figure 20) on
peut voir que celle-ci n'est pas améliorée par l'ajout de
liquide.
Quant aux chromatogrammes, deux phénomènes
apparaissent : d'une part, le signal obtenu est fortement diminué par
rapport à l'analyse sur poudre sèche (comme expliqué
précédemment) et, d'autre part, le solvant interfère au
niveau de l'analyse. En effet, un pic important provenant du passage en phase
gazeuse du solvant apparaît sur les chromatogrammes (figure 21).
60
50
Surface du pic (pA.s)
40
30
20
R2 = 0.7684
10
R2 = 0.7973 R2 = 0.0198
R2 = 0.0147
0
Pic 1,379 Pic 2.205 Pic 4.263 pic 1.191
0 50 100 150 200 250 300 350
Masse (mg)
33
Figure 20 : Linéarité obtenue avec
Ocimum basilicum en présence de 500 uL de mygliol 812 ®
pour les 4 composés différents. T° « Headspace
» : 90°C (vial) /100°C (boucle) / 110°C (ligne de
transfert), Cryobroyage, 3 essais par masse. Pressurisation du vial : 15,5 PSI,
temps de remplissage de la boucle : 0,10 min, temps de pressurisation : 1,0
min
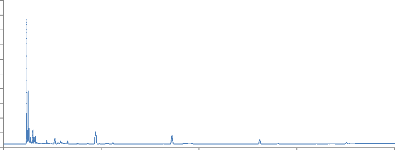
50
45
40
35
Signal (pA)
30
25
20
15
10
5
0
34
0 5 10 15 20
Temps de rétention (Min)
Figure 21 : Chromatogramme obtenu par l'analyse
de 200 mg d'Ocimum basilicum (Carrefour®) en présence de
500 uL de mygliol 812®. T° « Headspace » :
90°C (vial) /100°C (boucle) / 110°C (ligne de transfert),
cryobroyage. Pressurisation du vial : 15,5 PSI, temps de remplissage de la
boucle : 0,10 min, temps de pressurisation : 1,0 min
Ceci est confirmé par le « Blanc » de mygliol
(figure 22).
50
45
40
Signal (pA)
35 30 25 20 15
10
5
0
0 5 10 15 20
Temps de rétention (min)
Figure 22 : Chromatogramme obtenu avec 500 uL
de mygliol 812®. T° « Headspace » : 90°C
(vial) /100°C (boucle) / 110°C (ligne de transfert). Pressurisation
du vial : 15,5 PSI, temps de remplissage de la boucle : 0,10 min, temps de
pressurisation : 1,0 min
Contrairement à ce qui avait été
prévu, une partie des composés présents dans l'huile a pu
passer dans l'espace de tête après le chauffage de 33,75 minutes
à 90°C dans le « Headspace » (il s'agit sans
doute d'acides gras à chaîne courte provenant de la
dégradation
35
du solvant).
Il faut également rappeler que la linéarité
n'a pas été réellement améliorée.
En conclusion, le mygliol 812® ne présente pas de
résultats satisfaisants et n'apporte pas de réelle
amélioration pour l'analyse.
? Eau MilliQ®
Le second solvant testé est l'eau MilliQ
®, qui présente l'avantage de diminuer l'affinité
des composés organiques volatils vis-à-vis de la poudre de
l'échantillon. Etant caractérisée par une faible pression
de vapeur, le risque qu'elle interfère au niveau de la détection
des substances organiques volatiles est donc assez faible (Quiroga, Dong et al.
2009). Cependant, elle risque d'atteindre le détecteur FID et de
provoquer, si la quantité est importante, une extinction de la
flamme.
Pour travailler en présence d'eau, il est
conseillé de porter les températures de la boucle et de la ligne
de transfert à des valeurs supérieures à la
température d'ébullition de l'eau (au-delà de 100°C)
afin d'éviter les phénomènes de recondensation (Anonyme
2000).
Les températures du vial, de la boucle et de la ligne
de transfert ont donc été fixées à 100, 110 et
120°C respectivement. La quantité d'eau introduite dans les vials
est de 500 uL. Sur les chromatogrammes, le signal paraît
légèrement augmenté en présence d'eau (en
comparaison avec la poudre sèche), ceci est surtout remarquable pour le
thymol (voir figure 23).
P-Cymène
B-Caryophyllène
Methyleugénol
150
130
110
Thymol
Linalool
90
? Poudre sèche ? Poudre + H2O
36
5 10 15 20 25 30
Temps de rétention (min)
Figure 23 : Comparaison des chromatogrammes
obtenus avec Ocimum basilicum en présence et en absence d'eau
MilliQ®. T° « Headspace » : 100°C (vial)
/110°C (boucle) / 120°C (ligne de transfert), cryobroyage. Masse :
100 mg. Pressurisation du vial : 15,5 PSI, temps de remplissage de la boucle :
0,10 min, temps de pressurisation : 1,0 min
Par la suite, la répétabilité des analyses
en présence d'eau a été étudiée. Comme
celle-ci présente un point d'ébullition relativement bas (en
comparaison avec le mygliol 812® par exemple), elle peut être
responsable d'un apport de pression supplémentaire au niveau de l'espace
de tête ce qui peut se traduire par des modifications importantes sur le
plan expérimental (Kolb and Ettre 2006).
Pour ce faire, deux séries d'échantillon ont
été préparées : l'une avec la poudre sèche
et l'autre avec la poudre additionnée de 500 uL d'eau MilliQ®.
Chaque série étant constituée de 9 essais avec, pour
chacune, une masse de poudre de 100 mg d'O. basilicum
(Carrefour®). Tous les essais ont été
préparés et analysés le même jour.
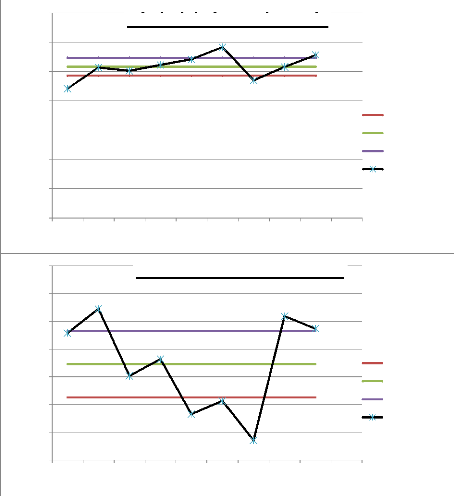
Sur les graphiques (figure 24 et 25), l'ajout d'eau se traduit
très clairement par une diminution de la
répétabilité.
155
150
145
140
135
130
125
120
Surface du pic (pA.s)
1 2 3 4 5 6 7 8 9 10
Essai n°
Répétabilité : Poudre et eau
I.C 95 % Moyenne I.C 95 % Aire P-Cymène
155
150
145
Surface du pic (pA.s)
140
135
130
125
120
1 2 3 4 5 6 7 8 9 10
Essai n°
|
Répétabilité : Poudre
seule
|
|
|
|
|
|
|
|
|
|
I.C 95 % Moyenne I.C 95 % Aire P-Cymène
37
Figures 24 et 25 :
Répétabilité des analyses obtenue par l'analyse
d'Ocimum basilicum en absence et en présence d'eau pour l'aire
du pic de P-Cymène (T.R : 5,288 min). T°
« Headspace » : 100°C (vial) /110°C
(boucle) / 120°C (ligne de transfert), cryobroyage, n = 9 essais.
Pressurisation du vial : 15,5 PSI, temps de remplissage de la boucle : 0,10
min, temps de pressurisation : 1,0 min
En effet, avec la poudre seule, les dispersions autour de la
moyenne sont nettement moins larges qu'en présence d'eau (voir tableau
4).
38
|
T.R (min)
|
C.V % (n=9)
|
Min (pA):
|
Max (pA):
|
|
Poudre sèche
|
5,25
|
3,14
|
142,089
|
147,818
|
|
Poudre + Eau
|
5,25
|
6,13
|
123,537
|
147,252
|
Tableau 4 : Comparaison des coefficients de
variation du P-Cymène pour les des deux séries d'essais (n=9).
Cela s'est également vérifié pour les autres
composés.
En conclusion, l'ajout de ce solvant à
l'échantillon introduit donc une variabilité accrue. Celle-ci
peut, comme expliqué précédemment, provenir d'une
augmentation de pression au sein du vial qui induit des pertes lors du
transfert.
L'eau MilliQ® ne semble donc pas un solvant approprié
pour cette étude.
4.1.4 L'effet « Salting-out »
Lorsque l'on se trouve en milieu aqueux et en présence
de composés organiques relativement polaires, l'ajout
d'électrolyte peut s'avérer fort intéressant pour diminuer
le coefficient de partage et ainsi augmenter la concentration de ces
composés dans l'espace de tête (Zuba, Parczewski et al. 2001).
Pour avoir un effet significatif en analyse « Headspace »,
il est conseillé de travailler avec des concentrations en sel de l'ordre
de 20 % car, en-dessous, les modifications apportées sont minimes, voire
inexistantes.
Par contre, le sel ajouté à l'échantillon
peut parfois contenir certaines impuretés qui vont parasiter le
chromatogramme final. Il peut aussi entraîner une augmentation de la
viscosité de l'eau et modifier la pression de vapeur entraînant
ainsi une augmentation du temps de chauffe nécessaire pour atteindre
l'équilibre (Kott 2010).
Afin d'observer l'impact qu'a l'ajout de chlorure de sodium
sur l'échantillon, 100 mg d'O. basilicum (Carrefour®) ont
été additionnés, d'une part, de 500 uL de solution NaCl
à 20 % et, d'autre part, de 500 uL de solution saturée en
NaCl1. Chaque vial est ensuite analysé. Les
températures du vial, de la boucle et de la ligne de transfert sont
conservées à 100, 110 et 120°C afin d'éviter une
éventuelle condensation lors du transfert.
Il apparaît alors, sur les chromatogrammes obtenus
(figure 26), que l'ajout de sel n'augmente nullement la volatilité des
analytes ; en présence de solution saturée en NaCl une diminution
de celle-ci est même observée.
1 Solubilité NaCl : 357 g / L (Eau, 25
°C)
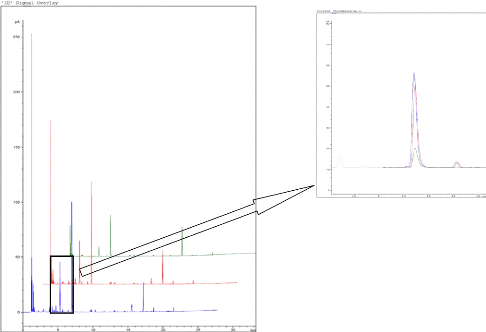
· Poudre sèche
· 20 % NaCl
· NaCl saturée
· Poudre sèche
· 20 % NaCl
· NaCl saturée
39
Figure 26 : Comparaison des chromatogrammes
obtenus par l'analyse d'Ocimum basilicum avec la poudre sèche,
la solution NaCl à 20 % et la solution saturée en NaCl.
T°
« Headspace » : 100°C (vial)
/110°C (boucle) / 120°C (ligne de transfert), cryobroyage,
Résultat d'une expérience. Pressurisation du vial : 15,5 PSI,
temps de remplissage de la boucle : 0,10 min, temps de pressurisation : 1,0
min
Ce phénomène est peut-être causé
par l'augmentation de la viscosité de l'eau qui réduit sans doute
le passage des composés en phase gazeuse.
Dès lors, l'effet « Salting-out »
n'apporte pas d'amélioration significative en termes de
sensibilité pour l'analyse de la plante ; comme expliqué par
(Kolb and Ettre 2006), l'ajout de sel est empirique en analyse «
Headspace », il est difficile de prévoir l'effet produit pour
un type d'analyse donné.
4.2 Optimisation des paramètres d'extraction
« Headspace »
N'ayant obtenu aucun résultat satisfaisant avec l'ajout
d'un solvant, seul la poudre sèche sera analysée pour la suite
des expériences.
40
4.2.1 Plan factoriel
Etant donné le nombre important de paramètres,
une analyse en plan factoriel a été sélectionnée.
Elle permet de prendre en compte plusieurs paramètres tout en effectuant
un nombre réduit d'essais (Groupy 2006).
Un plan d'expérience linéaire constitué
de 5 facteurs et 2 niveaux a été choisi. Les valeurs des niveaux
-1 et+1 (minimum et maximum) ont été fixées pour chacun
des paramètres comme indiqué sur la figure 27. Parmi ces
facteurs, la masse a été choisie car son impact positif sur
l'aire du pic est certain. Elle sert en quelque sorte à « valider
» le plan factoriel. Les 4 autres facteurs testés sont la
température du vial, la pressurisation du vial, le temps de remplissage
de la boucle et le temps de pressurisation du vial. Le plan factoriel se
compose donc d'une série de 8 essais à effectuer selon un ordre
aléatoire. La réponse choisie est l'aire du pic de
P-Cymène en pA.s.
L'entièreté des essais a été
réalisée le même jour. Ensuite les données ont
été représentées sur un graphique (figure 27)
où sont repris les différents paramètres ainsi que leurs
effets sur l'évolution de l'aire du pic. Pour qu'un effet ne soit pas
négligeable, son intervalle de confiance ne doit pas recouvrir
zéro. Il apparaît que la masse a une forte influence sur
l'augmentation de l'aire (comme attendu) ; la température du vial et le
temps de remplissage de la boucle (« Loop Fill time »)
jouent également un rôle significatif. En revanche, la
pressurisation du vial et le temps de pressurisation du vial n'ont qu'un effet
limité, voire inexistant dans la gamme considérée.
Le plan factoriel permet d'avoir rapidement une idée de
l'importance qui caractérise l'effet de chaque paramètre
significatif.
|
Paramètre :
|
Niveau -1 :
|
Niveau +1 :
|
|
Température Vial / Boucle/
Ligne de transfert
|
90
|
/ 100 / 110 °C
|
100
|
/ 110 /120 °C
|
|
Pressurisation du vial
|
|
22,00 PSI
|
|
29,00 PSI
|
|
Temps de remplissage de la boucle
|
|
0,10 Min
|
|
0,20 Min
|
|
Temps de pressurisation du vial
|
|
0,20 Min
|
|
0,50 Min
|
41
Résultats
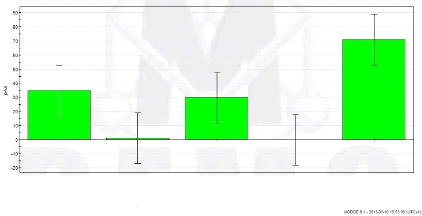
T°
Vial P.
Loop
F. Time
Vial
P. Time
Masse
Figure 27 : Plan factoriel à 5
facteurs et deux niveaux de type linéaire obtenu via le logiciel MODDE
9.1® avec Ocimum basilicum (Carrefour®). Tableau avec les
différents niveaux et graphique des effets en « Plots ».
4.2.2 Variation de température
Sur base du plan factoriel, la valeur optimale pour la
température de chauffe du vial a été définie. Pour
ce faire, un balayage entre 90 et 140°C a été
effectué. Les températures de la boucle et de la ligne de
transfert ont été fixées à des valeurs
supérieures à celle du four du vial afin d'éviter une
éventuelle condensation soit, respectivement, 150 et 160°C.
Généralement, il est conseillé de garder la
température de la boucle et de la ligne de transfert à,
respectivement, 15 et 25°C au-dessus de celle du vial. Et en
présence de solvant, il est vivement conseillé de ne pas
dépasser 10°C au dessus de la température
d'ébullition car cela pourrait engendrer une altération du septum
et des fuites de gaz (Anonyme 2000).
Lorsque la température de chauffe du vial est
augmentée, le passage vers l'espace de tête est facilité et
la concentration de l'analyte en phase gazeuse est accrue. En effet, chaque
composé présent dans la plante est caractérisé par
son propre coefficient de partage ; lorsque l'on
42
augmente la température, la grande majorité de
ceux-ci voit son coefficient diminué et son transfert vers l'espace de
tête augmenté (Hinshaw 2012).
Une augmentation de température permet également
de diminuer le temps d'équilibe (Muffet 2011).
Il faut également noter que la température est
intimement liée à la pression de vapeur des composés et
qu'elle suit une évolution exponentielle en fonction de la
température. Ceci implique qu'une faible variation de température
engendre une importante variation de pression de vapeur et donc une
modification non-négligeable du passage de l'analyte vers la phase
gazeuse (voir figure 28).
Figure 28 : Evolution de la pression de vapeur
de l'eau en fonction de la température (Kolb and Ettre 2006).
En analysant le graphique de l'aire du pic de P-Cymène
en fonction de la température de chauffe du vial (figure 29), il semble
que, jusqu'à 100°C, l'aire évolue de manière
croissante. C'est-à-dire que, jusqu'à ce point, la
quantité d'analyte passant dans l'espace de tête est
43
augmentée.
|
Surface du pic (pA.s)
|
45 40 35 30 25 20 15 10 5 0
|
|
P-Cymène (5,288 min)
|
90 100 110 120 130 140 150 T° du vial
(°C)
Figure 29 : Evolution de l'aire du pic de
P-Cymène en fonction de la température du Vial. Echantillon :
O. basilicum (Carrefour®). T° boucle : 150 °C / T°
ligne de transfert : 160°C, cryobroyage, n = 1, masse : 100 mg.
Pressurisation du vial : 15,5 PSI, temps de remplissage de la boucle : 0,10
min, temps de pressurisation : 1,0 min.
Toutefois, une fois passé ce point, l'aire diminue
fortement sans doute à cause d'une dégradation du composé.
Cela montre bien qu'il y a un juste milieu entre, d'une part,
l'amélioration du transfert vers l'espace de tête et, d'autre
part, la dégradation des composés instables.
Cependant, la structure du P-cymène indique qu'il s'agit
d'un composé relativement stable ; l'hypothèse selon laquelle il
serait dégradé aux températures élevées
paraît, dès lors, peu probable. Il faut également noter que
ce phénomène se produit pour les autres pics, le problème
semble donc provenir de l'appareillage et non de l'échantillon.
Pour la suite des analyses, la température de chauffe du
vial sera donc fixée à 100°C.
4.2.3 Variation du temps de remplissage de la boucle
(« Loop Fill Time »)
Toujours sur base du plan factoriel, un balayage du temps de
remplissage de la boucle a été effectué. Pour rappel, ce
temps correspond à la période durant laquelle la boucle reste
ouverte afin que l'aliquote de l'espace de tête lui soit
transféré. Pour le type de système concerné ici, le
temps conseillé est de 10 secondes (soit 0,16 minutes). Si ce temps de
remplissage est trop long, il y a un risque de retour du gaz vers l'espace de
tête du vial. Si ce temps est court, la
44
pression facilite le passage au niveau de la boucle et la
quantité d'analyte qui passera vers la GC sera plus importante (Anonyme
2000).
Le balayage s'étend de 0,10 min à 0,30 min avec
des espacements de 0,05 min pour chaque essai et avec des masses de 100 mg
(voir figure 30) .
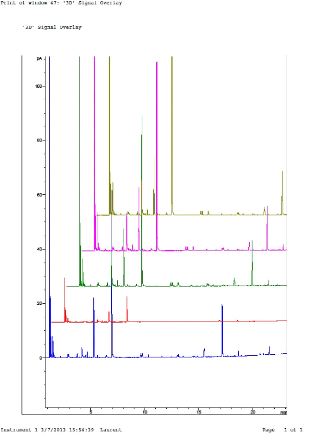
? LFT 0,10 min ? LFT 0,15 min ? LFT 0,20 min ? LFT 0,25 min ? LFT
0,30 min
Figure 30 : Balayage du temps de remplissage de
la boucle avec O. basilicum (Carrefour®). T°vial :
100°C / T° Boucle : 110 °C / T° ligne de transfert :
120°C ; cryobroyage, résultat d'une expérience, masse : 100
mg. Pressurisation du vial : 15,5 PSI, temps de pressurisation : 1,0 min.
La valeur offrant la meilleure sensibilité est 0,20 min ce
qui correspond à un temps de 12 secondes, valeur proche de celle
conseillée à la base (qui est de 10 secondes). Cette valeur sera
conservée pour la suite des analyses.
45
4.2.4 Optimisation de la pression du vial
La pression établie dans le vial doit normalement se
situer aux alentours de 1,5 à 2 Atm pour ce type d'appareillage (Anonyme
2000). Or, la valeur fixée initialement pour les analyses est 15,5 PSI
ce qui correspond à seulement 1,02 Atm. Il se peut donc que la pression
établie au sein du vial soit trop faible que pour permettre un transfert
efficace par différence de pression vers la boucle
d'échantillonnage.
L'objectif de cette expérience est de voir si une
éventuelle augmentation de la sensibilité serait obtenue en
faisant varier la pression entre 18 et 28 PSI. Les autres paramètres du
« Headspace » restent fixes.
La pressurisation du vial est d'autant plus importante pour
les échantillons sous forme solide car, contrairement aux formes
liquides, la pression développée est bien souvent insuffisante
pour permettre un transfert efficace vers le chromatographe gazeux.
En observant le graphique de l'aire du pic de P-Cymène
en fonction de la pression dans le vial (figure 31) et l'évolution du
pic de P-Cymène (figure 32), il semble qu'il y ait une augmentation de
la sensibilité pour une pression de 22 PSI (ce qui équivaut
à 1,497 Atm) mais cela devrait être vérifié par une
série d'essais supplémentaire.
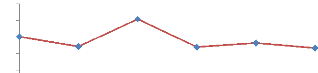
90
80
70
60
50
40
30
20
10
0
18 20 22 24 26 28 30
Pression (PSI)
Surface du pic (pA.s)

Figure 31 : Evolution de l'aire du pic de
P-Cymène en fonction de la pressurisation du vial. Echantillon :
Ocimum basilicum (Carrefour®). T° « Headspace
» : 100°C (vial) / 110°C (boucle) / 120°C (ligne de
transfert). Masse : 100 mg, cryobroyage, résultat d'une
expérience. Temps de remplissage de la boucle : 0,20 min, Temps de
pressurisation : 1,0 min.
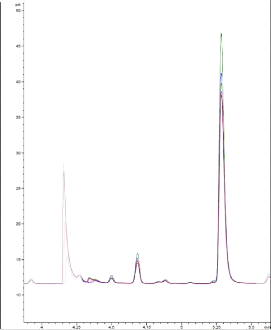
· 18 PSI
· 20 PSI
· 22 PSI
· 24 PSI
· 26 PSI
· 28 PSI
46
Figure 32 : Evolution du pic de P-Cymène
en fonction de la pression dans le vial.
Echantillon : O. basilicum (Carrefour®). T°
« Headspace » : 100°C (vial) / 110°C (boucle) /
120°C (ligne de transfert) ; Cryobroyage, masse : 100 mg, n = 1. Temps de
remplissage de la boucle : 0,20 min, Temps de pressurisation : 1,0 min.
Ceci confirme bien l'hypothèse selon laquelle la
pression établie initialement était trop insuffisante que pour
permettre une extraction efficace. Cette valeur est donc conservée pour
le système « Headspace ».
4.2.5 Ajouts dosés
Cette méthode a permis d'étudier l'influence
réelle qu'exerce notre poudre sur les différents composés
volatils présents dans l'échantillon. Deux séries de 5
essais ont été établies : d'une part, une série
avec 100 mg de poudre sèche et, d'autre part, une série avec une
matrice constituée de 500 uL d'eau MilliQ. Dans chaque série, des
quantités croissantes de standard de 1,8 cinéole (Sigma
Aldrich®) ont été ajoutées (de 1 à 5 uL) (voir
tableau 5).
47
|
Série 1 :
|
Série 2 :
|
|
Matrice :
|
100 mg Poudre
O.basilicum
|
500 uL H2O
|
|
Ajout de
1,8 cinéole (uL)
|
1
|
1
|
|
2
|
2
|
|
3
|
3
|
|
4
|
4
|
|
5
|
5
|
Tableau 5 : Ajouts dosés
Le but de cette expérience est de pouvoir observer les
interactions présentes au sein de la poudre. En effet, si les
composés ne sont que peu ou pas retenus dans l'échantillon les
deux droites obtenues devraient augmenter parallèlement. Toutefois, si
les composés sont sujets à d'importantes interactions avec la
poudre, certaines disparités entre les deux droites pourraient
être rencontrées (Feinberg 2001).
Les deux droites de régression obtenues (figure 33)
présentent, toute deux, un R2 acceptable mais, cependant,
l'évolution des deux séries ne se fait pas de la même
manière.
0 1 2 3 4 5 6
Volume de cinéole (uL)
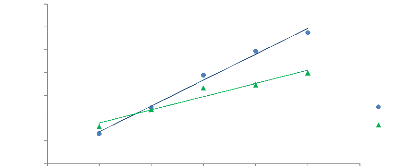
R2 = 0.9918
R2 = 0.9446
Eau Poudre
35000
30000
Surface du pic (pA.s)
25000
20000
15000
10000
5000
0
Figure 33 : Droite de régression du
cinéole obtenue par la méthode des ajouts dosés avec,
d'une part, la poudre (m = 100 mg) telle quelle (O. basilicum
Carrefour ®) et, d'autre part, de l'eau MilliQ® (V = 500 uL).
T° « Headspace » : 100°C (vial) / 110°C
(boucle) / 120°C (ligne de transfert), cryobroyage. Temps de remplissage
de la boucle : 0,20 min, Temps de pressurisation : 1,0 min, pressurisation du
vial : 22,00 PSI.
48
? Test de comparaison des pentes :
Un test d'égalité des pentes de Student a
été réalisé afin de confirmer de manière
statistique cette différence. Ce test compare la valeur « t »
obtenue selon l'équation ci-dessous à celle de la valeur t (0.05;
N1+N2 - 4) trouvée dans les tables pour un seuil de 0,05 et un
degré de liberté de n1+n2-4.

Si la valeur du t calculé est inférieure
à celle des tables, l'hypothèse d'égalité des
pentes des droites est alors acceptée.
|
Poudre et eau
|
|
|
b1
|
5668,4
|
|
Somme des carrés des écarts sur X
|
10
|
|
Somme des carrés des écarts sur Y
|
323960742
|
|
Somme des carrés des écarts (résidus)
|
2276543,4
|
|
Variance des résidus
|
1138271,7
|
|
Poudre sèche
|
|
|
b2
|
2886,5
|
|
Somme des carrés des écarts sur X
|
10
|
|
Somme des carrés des écarts sur Y
|
88206461,1
|
|
Somme des carrés des écarts (résidus)
|
3139427,43
|
|
Variance des résidus
|
1569713,71
|
Tableau 6 : Calculs préliminaires pour le
test sur les pentes.
- La variance résiduelle commune vaut alors :
S2C = 451330,903 - La valeur t calculée vaut : ttest =
9,25931761
- En comparant avec la valeur t des table : t (0.05; 6) =
1,943
- Il y a donc rejet de l'hypothèse de
l'égalité des pentes car :
9,25931761 > 1,943
Il existe donc des interactions entre le matériel
végétal et les composés volatils. Il se peut que la poudre
exerce des effets d'adsorption vis-à-vis des analytes ;
phénomène facilité par la grande porosité de
l'échantillon. Ceci influence directement le passage des composés
dans l'espace de tête étant donné que le coefficient de
partition est lié aux phénomènes d'adsorption
également (Kolb and Ettre 2006). Les systèmes avec adsorption
présentent de nombreuses difficultés pour les analyses
quantitatives.
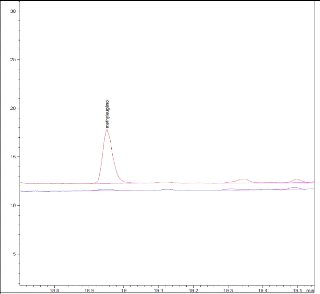
? O. basilicum Carrefour® ? O. basilicum
Ducros®
49
4.2.6 Changement d'échantillon
Pour la suite des analyses, un nouvel échantillon a
été choisi : il s'agit d'O. basilicum de la marque
Ducros®. Les chromatogrammes pour chacune des deux marques
(Carrefour® et Ducros®) ont été comparés et
certaines variations sont apparues. Notamment pour le méthyl
eugénol et l'estragole (figure 34 et 35) qui présentent de
grosses différences.
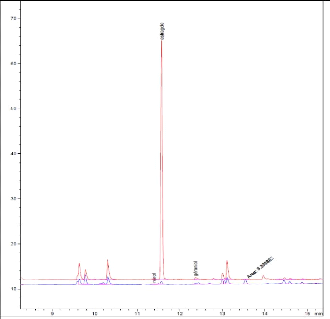
? O. basilicum Carrefour® ? O. basilicum
Ducros®
50
Figure 34 et 35 : Comparaison des pics de
méthyleugénole et d'estragole pour les deux marques d'O.
basilicum (Carrefour® et Ducros®). T° « Headspace
» : 100°C (vial) / 110°C (boucle) / 120°C (ligne de
transfert), cryobroyage, n = 1, masse : 100 mg.
En effet, les teneurs pour le basilic de la marque
Ducros® sont nettement plus élevées. Ce qui prouve bien
qu'en fonction de la variété ou de la qualité, des
conditions de culture et du séchage, les composés volatils
peuvent varier et donc modifier sensiblement l'arôme de la plante.
4.3 Evaluation de l'apport des optimisations à
la qualité des analyses
4.3.1 Modèle linéaire et quadratique
Suite à toutes ces analyses, il a été
décidé d'analyser 3 groupes de masses différentes (100,
200 et 300 mg) en triplicate, tout en fixant l'entièreté des
paramètres du « Headspace » aux valeurs optimales
(voir tableau 7). Ceci permet la comparaison aux données de
départ du (voir tableau 3).
51
|
Paramètres du "Headspace"
|
Valeurs
|
|
Température (°C)
|
Vial
|
100
|
|
|
Boucle
|
110
|
|
Ligne de transfert
|
120
|
|
Temps (min)
|
Remplissage de
la boucle
|
0,20
|
|
|
Equilibration
|
34,00
|
|
Pressurisation
|
1,00
|
|
Equilibration de
la boucle
|
0,05
|
|
Injection
|
1
|
|
GC Cycle
|
33,75
|
|
Pression (PSI)
|
Vial
|
22,04
|
|
|
Gaz vecteur
|
21,30
|
Tableau 7 : Valeurs optimisées des
différents paramètres du « Headspace ». Les
valeurs en rouge ont été modifiées par rapport au tableau
3.
Les droites de régression ont été
tracées sur le graphique pour montrer l'évolution de l'aire des
pics (en pA.s) en fonction de la masse (figure 38). L'évolution suit
plus ou moins le modèle linéaire mais il apparaît, avec les
valeurs des R2, que la linéarité diminue
proportionnellement avec le temps de rétention (comme lors des premiers
tests effectués, figure 16 et 17).
600
R2 = 0.7364
500
400
300
200
100
0
Surface du Pic (pA.s)
0 50 100 150 200 250 300 350
Masse (mg)
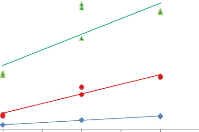
R2 = 0.9481
R2 = 0.9793
Thymol (1.25 min) P-Cymene (5.25 min) Pic 6.95
52
Figure 38 : Droites de régression
obtenues par l'analyse d'O. basilicum (Ducros®) pour 3
composés différents. Paramètres « Headspace
» fixés aux valeurs optimales en fonction des analyses
précédentes, voir ci-dessous « tableau 4 », n = 3 pour
chaque masse.
Une série supplémentaire de 3 essais a
été analysée pour une masse de 200 mg afin de voir si les
valeurs aberrantes obtenues pour cette masse se répétaient.
Ensuite une analyse statistique a été
menée pour voir si ces données suggérent une
évolution différente du modèle linéaire. Pour ce
faire, le graphique des résidus a été tracé ;
lorsque l'on se trouve dans un modèle linéaire, les
résidus doivent se disposer de manière sphérique. Ce qui
n'est clairement pas le cas ici (voir figure 39).
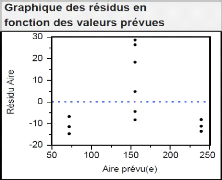
Figure 39 : Graphique des résidus pour le
P-Cymène obtenu par l'analyse de linéarité.
53
En effet, les résidus sont disposés de part et
d'autre de l'axe de façon régulière ce qui tend à
réfuter l'hypothèse selon laquelle les données suivraient
le modèle linéaire.
D'autres modèles ont donc été
testés afin de voir lequel correspond au mieux aux résultats. En
se basant sur l'évolution des résidus qui ne sont pas
répartis de manière aléatoire, le modèle
quadratique a été testé. Avec ce modèle, un
recouvrement des données plus satisfaisant qu'avec le modèle
linéaire a été obtenu (voir figure 40).
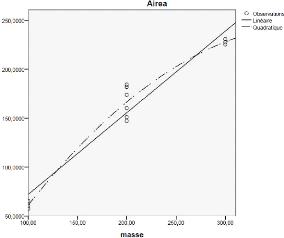
Figure 40 : Comparaison du modèle
linéaire et quadratique pour le P-Cymène
Ceci s'explique en partie par le fait qu'une majorité
des paramètres du « Headspace » varie selon ce
modèle quadratique (comme la température,...). Ce modèle
tend vers une asymptote, aux valeurs de masses importantes, ce qui s'explique
sans doute par la saturation de l'espace de tête lorsque la
quantité de poudre à analyser devient trop grande.
L'hypothèse de départ selon laquelle l'aire des
pics varierait de manière linéaire avec l'augmentation de masse
d'échantillon est en fait peu probable.
54
5. Conclusion
La méthode d'échantillonnage «
Headspace » paraît simple en théorie mais s'avère
nettement plus complexe à mettre en pratique. Le fait de pouvoir
injecter directement l'échantillon sous forme gazeuse dans le GC est
assez avantageux par rapport à l'injection classique mais le nombre
conséquent de paramètres du « Headspace »
ainsi que le mode de préparation de l'échantillon sont deux
aspects à prendre en compte et qui ne sont pas toujours simples à
définir en pratique. C'est pourquoi nous avons réfléchi
à une approche de validation. En réalité, il existe peu de
littérature traitant des analyses en « Headspace »
statique et encore moins dans le cadre de plantes contenant des huiles
essentielles. De plus, aucun protocole de validation n'a été
décrit pour ce type d'analyses.
La version statique paraît, dès lors, plus
adaptée à l'analyse d'échantillons liquides (où
l'équilibre s'établit plus rapidement et uniformément
qu'avec un échantillon solide) et ce, pour des analyses plutôt
qualitatives que quantitatives. L'application de cette technique est fortement
limitée pour les composés sous forme de traces et pour ceux
présentant des tensions de vapeur très faibles (Dijkstra, Massart
et al. 1978).
Pour effectuer des analyses quantitatives, la version SPME
serait peut-être plus appropriée car décrite comme
présentant une meilleure sensibilité ainsi qu'une meilleure
reproductibilité ; faire appel à des techniques comme la
« Multiple Headspace Extraction » (MITE) ou la
méthode des standards internes (qui n'ont pas été
abordées ici par manque de temps) avec lesquelles il serait possible
d'obtenir des résultats plus satisfaisants serait une seconde option
(Moldoveanu and David 2002). Ainsi, l'une de ces deux méthodes
paraît être nécessaire voir même indispensable pour
pouvoir présenter une étude basée sur des données
quantitatives provenant d'une analyse en « Headspace »
statique.
Il reste, toutefois, un certain nombre de paramètres
qui n'ont pas pu être évalués et qui jouent un rôle
difficile à évaluer dans ce type d'analyse comme, par exemple, le
temps écoulé entre la fermeture du vial et l'analyse de
l'échantillon, l'étanchéité du septum au cours
d'une analyse, le nettoyage des vials, la taille du vial employé, les
phénomènes d'adsorption entre l'échantillon et la paroi du
vial,...
Pour conclure, l'hypothèse de départ selon
laquelle l'évolution des aires de pics en fonction de l'augmentation de
la masse se faisait de manière linéaire ne semble pas vraiment se
vérifier dans la pratique. Les données tendent plutôt
à indiquer une évolution selon le modèle quadratique. Pour
confirmer ce modèle, il faudrait procéder à une
série d'analyses supplémentaires et il serait aussi
intéressant d'établir un nouveau plan factoriel basé sur
le
55
modèle quadratique (et non sur le modèle
linéaire) afin d'estimer l'importance de chaque paramètre.
Cependant, ce type d'analyse nécessiterait un nombre d'essais
conséquent (au-delà de la quarantaine pour le plan factoriel).
56
6. Références
Anonyme (2000). Agilent 7694 Headspace Sampler :
Operating Manual. Wilmington, Agilent Technologies, Inc. .
Anonyme, q. (2012). Précis de
Phytothérapie. Paris, Alpen Editions.
Anonyme, t. "Musée International de la
Parfumerie - Les principales techniques."
http://www.museesdegrasse.com/approfondissez
vos connaissances/la documentation/histoire delaparfumerie. Retrieved
28/04, 2013.
Bhowon, M. G., S. Jhaumeer-Laulloo, et al. (2012).
Chemistry for Sustainable Development. London,
Springer.
Dijkstra, A., D. L. Massart, et al. (1978).
Evaluation and Optimization of Laboratory Methods and Analytical
Procedures. Amsterdam, Elsevier Science.
Dills, R. L., S. D. Kent, et al. (1991).
"Quantification of volatile solvents in blood by static headspace analysis."
Talanta 38(4): 365-374.
Feinberg, M. (2001). Validation interne des
méthodes d'analyse. Paris, Ed. Techniques
Ingénieur.
Flanagan, R. J., A. A. Taylor, et al. (2007).
Fundamentals of Analytical Toxicology. Chichester,
Wiley.
Grayer, R.-J., R.-F. Vieira, et al. (2004).
"Characterization of cultivars within species of Ocimum by exudate flavonoid
profiles." Biochemical Systematics and Ecology 32(10):
901-913.
Grob, R. L. and E. F. Barry (2004). Modern
Practice Of Gas Chromatography. Hoboken, John Wiley & Sons,
Inc.
Groupy, J. (2006). "Les Plans d'Expériences."
Revue MODULAD 34.
Heidari, H.-R., S.-J. Shahtaheri, et al. (2008).
"Optimization of Headspace Solid Phase Microextraction Procedure for Trace
Analysis of Toluene." International Journal of Occupational Safety and
Ergonomics 14(4): 365 - 366.
Hinshaw, J.-V. (2012). "Headspace Sampling :
Instrumentation." LCGC Asia Pacific 15(1): 27 - 33.
Hübschmann, H. J. (2009). Handbook of GC/MS :
Fundamentals and Applications. Weinheim, Wiley-VCH.
57
Hume, M. (2009). "Making Scent's of
it."
http://www.time.com/time/specials/packages/article/0,28804,188205918822241882012,00.ht
ml. Retrieved 28/04, 2013.
Jirovetz, L., G. Buschbauer, et al. (2003).
"Chemotaxonomical analysis of the essential aroma compounds of four different
Ocimum species from southern India." European Food Research Technology
217(2): 120 - 124.
Johnson, C. B. and C. Franz (2002). Breeding
research on aromatic and medicinal plants. Binghamton, Haworth Press,
Incorporated.
Jordi, M. (2004). "Multiple Headspace Extraction Gas
Chromatography Mass Spectroscopy." Jordi Labs LLC: 8.
Kauffmann, J.-M. and P. Van Antwerpen (2010).
Analyse Pharmaceutique : méthodes instrumentales et contôle de
qualité, syllabus. Bruxelles, P.U.B.
Klimánková, E., K. Holadová, et
al. (2008). "Aroma profiles of five basil (Ocimum basilicum L.) cultivars grown
under conventional and organic conditions." Food Chemistry 107(1):
464-472.
Kolb, B. and L. S. Ettre (2006). Static
Headspace-Gas Chromatography: Theory and Practice. Hoboken, Wiley -
VCH.
Kott, L. M. C., H. (2010). "Experimental
Considerations in Headspace Gas Chromatography." Pharmaceutical
Technology 34(5): 6.
Lee, S.-J., K. Umano, et al. (2005). "Identification
of volatile components in basil (Ocimum basilicum L.) and thyme leaves (Thymus
vulgaris L.) and their antioxidant properties." Food Chemistry 91(1):
131 - 137.
Masada, Y. (1976). Analysis of essential oils by
gas chromatography and mass spectrometry. New York, John Wiley & Sons
Inc.
Mathieu, M. J. and J. M. Fonteneau (2008). Le
manuel porphyre du préparateur en pharmacie: préparation du BP,
formation continue. Rueil - Malmaison, Ed. Porphyre.
Michel, N. (2012). Le genre Ocymum L. au Katanga
(République Démocratique du Congo) : approche exploratoire de
l'utilisation des huiles essentielles en taxonomie. Laboratoire
d'écologie et biogeochimie. Bruxelles, Université libre de
Bruxelles. Master: 80.
Moldoveanu, S. C. and V. David (2002). Sample
Preparation in Chromatography. Amsterdam, Elsevier
Science.
58
Muffet, R. (2011). "Theory Of Headspace
Sampling."
http://support.owlstonenanotech.com/attachments/token/unvtcilkbxjpxji/?name=Theory+of+Hea
dspace+Sampling.pdf. Retrieved 28/04, 2013.
Papet, Y., B. Brunet, et al. (2010). "Headspace(HS)
et micro-extraction en phase solide (SPME). Théorie et applications."
Annales de Toxicologie Analytique 22(2): 75-79.
Paris, M.-H. (2002-2003). L'analyse des
composés organiques volatils (COV) et leur réglementation dans
les produits cosmétiques. Ingénierie Documentaire. Lyon,
Ecole Nationale Supérieur des Sciences de l'Information et des
Bibliothèques: 72.
Paton, A., M. M. Harley, et al. (1999). Ocimum :
an overview of classification and relationships. Amsterdam, CRC
Press.
Pawliszyn, J. (2002). Sampling and Sample
Preparation for Field and Laboratory: Fundamentals and New Directions in Sample
Preparation, Elsevier Science & Technology Books.
Poole, C. (2009). Handbook of Methods and
Instrumentation in Separation Science. London, Elsevier
Science.
Restek (2000). A Technical Guide for Static Headspace
Analysis Using GC. Bellefonte, Restek Corp.
Rouessac, F. and A. Rouessac (2004). Analyse
Chimique : Méthodes et techniques intrumentales modernes. Paris,
Dunod.
Rouseff, R. L., K. R. Cadwallader, et al. (2001).
Headspace analysis of foods and flavors: theory and practice. New -
York, Kluwer Academic/Plenum Publishers.
Sell, C. J., K. (2005). The Search for Fragrance
Ingredients. The Chemistry of Fragrances. Cambridge, Royal Society of
Chemistry Publishing: 254-293.
Sitaramaraju, Y., A. van Hul, et al. (2008). "Static
headspace gas chromatography of (semi-)volatile drugs in pharmaceuticals for
topical use." Journal of Pharmaceutical and Biomedical Analysis 47(4-
5): 834-840.
Snow, N. H. and G. C. Slack (2002). Head-space
analysis in modern gas chromatography. Trends in Analytical Chemistry
Elsevier Science. 21: 608-616.
Sternberg, R. (2003 - 2004). Petit lexique de
Chromatographie en Phase Gazeuse. Paris.
Tian, J. (2010). "Application of static headspace gas
chromatography for determination of acetaldehyde in beer." Journal of Food
Composition and Analysis 23(5): 475-479.
59
Vaubourdolle, M. (2007). Toxicologie, Sciences
mathématiques, Physiques et Chimiques. Rueil - Malmaison, W.
Kluwer.
Wilkinson, K. (2003). Aspects
généraux de la chromatographie, syllabus, Université
de Genève.
Zhu, J.-Y. and X.-S. Chai (2005). "Some Recent
Depelopments in Headspace Gas Chromatography." Current Analytical
Chemistry 1: 79 - 83.
Zuba, D., A. Parczewski, et al. (2001). Effect of
salt addition on sensitivity of HS-SPME-GC method of volatiles determination.
Faculty of Chemistry. Krakow, Jagiellonian University.
| 

