|
Année Universitaire : 2014-2015

|
UNIVERSITÉCADDI AYYAD FACULTE DES
SCIENCES SEMLALIA MARRAKECH
|
|
Mémoire de Master
Chimie des Matériaux
et ses
Applications Industrielles
Sous le thème :
'Etude de la spéciation chimique de
l'arsenic présent dans les sols pollués
de la ville de Marrakech
Présentépar : Rachid CHIKRI
Sous l'encadrement de : Pr : M. EL MERAY
Pr : A. ABOUELFIDA
Soutenu le : 13 juillet 2015 devant le jury :
Mr M.A.BENNOUNA Professeur à la facultédes
Sciences Semlalia-Marrakech
Mr M.LAARAJ Professeur à la facultédes Sciences
Semlalia-Marrakech
Mr M.EL MERAY Professeur à la
facultédes Sciences Semlalia-Marrakech
Mr A.ABOUELFIDA Professeur à la
facultédes Sciences Semlalia-Marrakech
Remerciements
Je souhaite avant tout remercier ALLAH, le tout-puissant,
qui m'a accordésanté, force et courage pour la rédaction
de ce mémoire.
Ce travail a étéréaliséau sein
du Laboratoire Physico-Chimie des Matériaux et Environnement (LPCME)
sous la direction des Professeurs M. EL MERAY et
A.ABOUELFIDA.
Je leur exprime ici toute ma reconnaissance de m'avoir
donnél'opportunitéde réaliser cette
étude et pour les conseils prodigués et encouragements tout au
long de cette recherche.
Je tiens à remercier les membres de jury :
Mr M.A.BENNOUNA et Mr M.LAARAJ d'avoir
acceptéd'examiner et de commenter ce travail.
Je tiens à remercier Mr A.AITAYOUB pour son aide,
sa présence et sa participation effective durant la réalisation
ce travail.
Je remercie également Mr E.AZIABLE pour son aide
est ses remarques précieuses qu'il m'a apportés pendant la
réalisation de la partie pratique.
Je tiens à remercier particulièrement
Mr A.BOUSSETTA le directeur du laboratoire d'analyses de
REMINEX, et également tous les membres du laboratoire
pour leur contribution à la réalisation de ce travail en
effectuant les analyses par ICP-AES.
Enfin, j'adresse mes remerciements les plus
sincères à tous ceux qui ont contribuéde près ou de
loin à la concrétisation de ce travail.
TABLE DES MATIÈRES
Table des matières
Liste des tableaux 2
Liste des figures 2
Liste des abréviations 3
Introduction générale 4
I ÉTUDE BIBLIOGRAPHIQUE 5
|
1
|
Généralités sur le sol
1.1 Définition
1.2 Constitution du sol
|
6
6
6
|
|
|
1.2.1 La phase solide du sol
|
6
|
|
|
1.2.2 La phase liquide du sol
|
6
|
|
|
1.2.3 La phase gazeuse du sol
|
6
|
|
2
|
Les métaux lourds
|
7
|
|
2.1
|
Généralités
|
7
|
|
2.2
|
Mécanismes de fixation et de relargage des
éléments traces dans les sols .
|
. 7
|
|
3
|
L'arsenic
|
10
|
|
3.1
|
Propriétés
|
10
|
|
3.2
|
Les formes d'arsenic
|
10
|
|
|
3.2.1 Les formes inorganiques
|
11
|
|
|
3.2.2 Les formes organiques
|
11
|
|
3.3
|
Les sources de l'arsenic
|
11
|
|
|
3.3.1 Les sources naturelles
|
11
|
|
|
3.3.2 Les sources anthropiques
|
11
|
|
3.4
|
Spéciation chimique de l'arsenic dans les sols
|
12
|
|
II
1
|
MATÉRIELS ET MÉTHODES
Site d'étude
|
14
15
|
|
1.1
|
Introduction
|
15
|
|
1.2
|
Choix du site d'étude
|
15
|
|
2
|
Paramètres physico-chimiques
|
17
|
|
2.1
|
Préparation du sol
|
17
|
|
2.2
|
pH
|
17
|
1
TABLE DES MATIÈRES
2.3 Conductivitéélectrique 17
2.4 Humidité 17
2.5 Matière organique 18
2.6 Carbone organique total COT 18
2.7 Azote total 19
2.8 Détermination de la teneur totale d'arsenic
«Extraction totale» 20
3 La spéciation chimique 21
3.1 Introduction 21
3.2 Schémas d'extraction séquentielle 21
4 Techniques d'analyse 22
4.1 Diffraction des rayons X 22
4.2 Spectrométrie d'absorption atomique (SAA) 23
4.3 spectrométrie d'émission atomique par plasma
à couplage inductif (ICP-AES) 23
III RÉSULTATS ET DISCUSSIONS 26
1 Paramètres physico-chimiques 27
1.1 pH 27
1.2 Conductivitéélectrique (CE) 27
1.3 Humidité 28
1.4 La matière organique (MO) 29
1.5 Carbone organique total (COT) 29
1.6 Azote total de Kjeldahl (NTK) 30
1.7 Caractérisation par diffraction des rayons X (DRX)
31
1.8 La teneur totale d'arsenic 32
2 Spéciation chimique 33
Conclusion générale et perspective
35
Références 36
Annexes 38
LISTE DES FIGURES
Liste des tableaux
1 Propriétés physico-chimiques de
l'élément Arsenic 10
2 Concentration moyenne en arsenic dans des sols
contaminés par des activi-
tés industrielles [1] 13
3 Les différentes positions de nos échantillons
16
4 Protocole d'extractions séquentielles adaptéde
Tessier et al.,[2] pour 1g du
sol 22
5 tableau récapitulatif des paramètres
physico-chimique étudiés pour les dif-
férents échantillons 31
6 les phases minérales trouvées pour chaque
échantillon 31
7 les teneur en arsenic (en ppm) pour différents
échantillons 34
Liste des figures
1 Principales interactions entre un atome ou une molécule
et un solide àl'interface solide/liquide [3] 9
2 Carte de localisation du site d'étude 16
3 photo du site d'étude par satellite 16
4 Principe du plasma à courant constant 24
5 les valeurs de pH pour les différents
échantillons 27
6 la variation de la conductivitéélectrique en
fonction des échantillons . . . 28
7 les teneurs d'humiditépour les différents
échantillons 28
8 les pourcentages en matière organique pour
différents échantillons 29
9 la variation des pourcentages en carbone organique total pour
différents
échantillons 30
10 les pourcentages en Azote total de Kjeldahl pour les
différents échantillons 30
11 Diffractogrammes de RX des échantillons
étudiés 32
12 les teneurs en arsenic pour différents
échantillons 32
13 les pourcentage d'arsenic dans chaque fraction et pour chaque
échantillon 33
2
3
LISTE DES FIGURES
Liste des abréviations
AAS : Atomic absorption spectroscopy
(Spectrométrie d'absorption atomique)
|
AFNOR
|
: Association fran
|
çaise de normalisation
|
BCR : Bureau Communautaire de Références
CCA : Arséniate de cuivre chromé(ou
chromaté)
CE : Conductivitéélectrique
COT : Carbone Organique Total
DRX : Diffraction des Rayons X
EPA : Environnemental Protection Agency
|
ETM :
|
Élément en traces métallique
|
ICP-AES : Spectrométrie d'émission atomique par
plasma à couplage inductif
IUPAC : Union internationale de chimie pure et
appliquée
MO : Matière Organique
NTK : Azote total de Kjeldahl
4
INTRODUCTION GÉNÉRALE
Introduction générale
La pollution par les métaux lourds est devenue parmi
les préoccupations mondiales, car ces éléments peuvent
s'accumuler à de fortes concentrations dans les sols, sédiments,
eaux et plantes. Certains sont des oligoéléments à faible
concentration et d'autres peuvent être toxiques même à
l'état de traces.
L'arsenic est un élément naturellement
présent dans tous les compartiments de l'envi-
ronnement, il présente des effets nocifs à de
faibles concentrations, en effet; il est employécomme poison
comme en témoignent de nombreuses références historiques
(la théorie de
l'empoisonnement de Napoléon Bonaparte...). De ce fait,
il est très intéressant d'éva-luer sa concentration dans
cet environnement par différentes méthodes d'analyse
physico-chimiques. Tandis que les études de quelques accidents mortels
dûs à la pollution métal-
lique tels que celui de Minamata en 1956 liéau mercure,
et d'Itai-Itai en 1969 attribuéau cadmium ont montréque la
connaissance de la concentration totale d'un métal dans
un milieu donnéest insuffisante pour évaluer sa
toxicitéou sa mobilitévers les chaînes alimentaires.
Dès lors, les recherches scientifiques sont orientées vers la
spéciation des métaux.
La décharge de la ville de Marrakech reçoit
chaque jour des tonnes d'ordures ménagères, des déchets
hospitaliers et industriels. Parmi les métaux lourds que l'on retrouve
l'arsenic comme constituant de quelques pesticides, insecticides, herbicides et
de quelques peintures du bois(CCA)...etc. Sa spéciation dans les sols
contaminés de ce site est devenue intéressante voir indispensable
pour prévoir ses impacts sur le milieu environnant.
Cette étude s'inscrit dans le cadre des travaux
déjàréalisés sur la spéciation des
métaux
lourds comme l'arsenic dans les sols pollués de la ville
de Marrakech pour évaluer le degréde contamination du sol de la
décharge. Pour ce faire, le travail est structuréen trois
parties.
La première partie est consacrée à une
étude bibliographique sur les métaux lourds et leurs
mécanismes de fixation et de relargage, et on termine par les
propriétés de l'arsenic ( ses formes et ses sources...)
La deuxième partie présente les protocoles
opératoires de la caractérisation physico-chimique, la
minéralisation totale et l'extraction séquentielle.
La troisième partie traite les résultats obtenus
et leurs discussion, et on termine ce mémoire par une conclusion
générale et perspectives.
5
Première partie
ÉTUDE BIBLIOGRAPHIQUE
6
Première partie: ÉTUDE BIBLIOGRAPHIQUE
1 Généralités sur le sol 1.1
Définition
Le sol représente la couche externe de l'écorce
terrestre, c' est un milieu dont la phase solide est constituée par des
minéraux et des composés organiques formant des assemblages plus
ou moins volumineux et qui donne au sol sa structure. Cette phase solide n'est
pas continue et délimite un espace poral de géométrie
complexe et de dimensions variées[4].
Depuis longtemps, la seule fonction du sol reconnue
était de permettre la production d'aliments[4], mais
aujourd'hui le sol a beaucoup de fonctions, parmi lesquelles on cite : source
de matériaux, réceptacle de déchets industriels et
urbains...cette dernière est actuellement et malheureusement une
réalité, de nombreux sites industriels et urbains
reçoivent des déchets de toute sorte dans des conditions
incontrôlées ce qui pose le problème de contamination du
sol.
1.2 Constitution du sol
1.2.1 La phase solide du sol
Elle est constituée par des minéraux et des
matières organiques en proportions variables, dont la nature des
minéraux du sol est déterminée à la fois par les
roches sur lesquelles ils se sont formés et par les processus de
pédogenèse, alors que les matières organiques proviennent
principalement des résidus végétaux qui subissent diverses
transformations physiques et chimiques.[4]
1.2.2 La phase liquide du sol
La phase liquide du sol n'est pas de l'eau pure mais est
solution dont la composition est complexe et très variable. On la
désigne souvent par l'expression « solution du sol». En
général, cette dernière est difficile à
décrire et à étudier en raison de sa très grande
variabilitéspatiale et temporelle de sorte qu'il n'existe pas de
composition type. Cependant on peut donner quelques indications
générales en distinguant deux grande catégories de
solutés :
*- les micro-éléments dont la concentration est
inférieure à 1mmol.
m-3(10-6mol.L-1);
beaucoup d'éléments traces métalliques entrent dans cette
catégorie.
*- les macro-éléments dont la concentration est
supérieure à cette limite [4].
1.2.3 La phase gazeuse du sol
La phase gazeuse du sol est encore appelée
l'atmosphère du sol. Sa composition est souvent voisine de celle de
l'air mais elle peut être très variable dans l'espace et dans le
temps. Elle dépend de la proximitéde l'atmosphère, c'est
à dire la profondeur dans le sol et l'activitébiologique,
différentes substances peuvent être présentes dans l'air
du
7
Première partie: ÉTUDE BIBLIOGRAPHIQUE
sol telles que NO, N2O, NH3, CH4, H2S et parfois
quelques composés organiques volatils dont les concentrations
dépendent beaucoup de plusieurs facteurs environnementaux et
anthropiques.[4]
2 Les métaux lourds 2.1
Généralités
On appelle en général métaux lourds les
éléments métalliques naturels, métaux ou dans
certains cas métalloïdes caractérisés par une masse
volumique élevée. Les métaux lourds sont présents
dans tous les compartiments de l'environnement, mais généralement
en très faibles quantités . On dit que les métaux sont
présents « en traces ». Ils sont aussi « la trace »
du passégéologique et de l'activitéde l'homme. La plupart
des scientifiques préfèrent à l'appellation métaux
lourds, l'appellation « éléments en traces
métalliques » -ETM- ou par extension « éléments
traces » [5]; [6]
Ces éléments peuvent s'accumuler à de
fortes concentrations dans les sols, les poussières, les eaux et les
plantes, du fait des rejets liés à des activités
agricoles, industrielles ou à l'exploitation minière. Ainsi,
l'activitéminière peut à elle seule générer
des concentrations en ETM supérieures au fond géochimique dans
les différents compartiments.[7] en arrivant a quelques teneurs
considérés maximales, ces éléments deviennent des
poisons insidieux car ils agissent à plus ou moins long terme; de plus
ils ne sont pas dégradables[2].
La toxicitéde ces éléments a conduit les
pouvoirs publics à réglementer leurs émissions en fixant
des teneurs limites. Cette réglementation n'est cependant d'aucun
secours pour déterminer sans ambiguïtéune liste de
métaux à surveiller car la liste varie selon les milieux
considérés : émissions atmosphériques, rejets dans
l'eau, règles sur l'épandage des boues, des eaux usées ou
la mise en décharge.[8]
Selon l' EPA (Environnemental Protection Agency), les
métaux considérés comme étant à la fois
toxiques et indésirables sont : As, Cd, Cr, Pb, Hg et Ni
2.2 Mécanismes de fixation et de relargage des
éléments traces dans les sols
plusieurs mécanismes physico-chimiques interviennent
à l'interface solide/liquide, permettant le piégeage de ces
éléments dans la phase solide et leurs relargage dans la phase
liquide. dans ce cadre on cite :
V L'échange ionique (ou adsorption non
spécifique) : phénomène de surface par lequel les
cations en solution chargés positivement sont attirés par des
forces électrostatiques de types Van Der Waals ou coulombiennes, vers
les charges négatives de la surface du sol. La présence de
charges à la surface du solide provient
8
Première partie : ÉTUDE BIBLIOGRAPHIQUE
soit de substitutions isomorphiques dans le réseau
cristallin (remplacement d'un cation trivalent par un divalent), soit de
réactions chimiques de surface telles que la protonation :
SOH+ 2 ?? SOH + H+ (1)
SOH ?? SO + H+ (2)
Affin de maintenir l'électroneutralité, la
charge négative de la surface du sol est compensée par celle
d'une quantitééquivalente de cations [9],
[3].
V L'adsorption spécifique ( ou la
chimisorption) : la molécule adhère à la surface
par des liaisons ioniques ou covalentes. Elle est souvent difficilement
réversible et engendre une couche mono-moléculaire. Elle
dépend fortement du pH et reliée à l'hydrolyse des ions
métalliques. Les métaux particulièrement capables de
former des hydroxy-complexes sont plus spécifiquement adsorbés
sur les surfaces déproto-nées chargées négativement
d'oxydes ou d'hydroxydes de fer, d'aluminium ou de manganèse
[9].
M2+ + H2O ? MOH+ +
H+ (3)
= SO + MOH+ ?= S - O
- M - OH (4)
V Complexation
Les ions métalliques peuvent être
complexés avec la matière organique du sol par association entre
les cations métalliques et les groupements fonctionnels des substances
humiques. Une réaction de complexation se produit quand un cation
métallique réagit avec un anion qui fonctionne comme ligand, ces
ligands sont généralement des éléments non
métalliques donneurs d'électrons comme O, N et S. Ces
éléments sont présents dans des groupements de surfaces
basiques : -NH2(amine), = O(carbonyle),
-OH(alcool) et -S - (thioether) et
dans les groupements acides: -COOH(carboxyle),
-OH(nolique) et -SH(thiol).
On peut représenter la complexation d'un cation
métallique Mm+ en solution avec un ligand
anionique = R - Ll de la façon
suivante[9] :
= R - Ll + Mm+
?= R - L - Mm l (5)
V Précipitation : C'est le
passage d'une espèce de l'état dissout à l'état
solide. Les métaux peuvent précipiter dans l'eau des pores ou
à la surface des particules solides. Dans un milieu naturel, les
métaux précipitent principalement sous forme d'hydroxydes, de
carbonates, de phosphates ou de sulfures. Les équilibres de
précipitation sont gouvernés par les produits de
solubilité(Ks) :
Ks =
(A)x.(B)y
où(A) et (B) représentent les activités des
espèces dans le liquide et x et y leur
9
Première partie: ÉTUDE BIBLIOGRAPHIQUE
stoechiométries respectives. Le solide précipite
quand le produit des activités(A) et (B) est supérieur au produit
de solubilité.
V Substitution dans le réseau cristallin
: Un atome peut se substituer à un autre de charge et taille
similaires dans le réseau cristallin. C'est le cas d'un ion
métallique incorporédans le réseau cristallin lors de sa
précipitation, ou bien qui diffuse dans le solide pour remplir un vide
ou remplacer un atome du solide.
V Inclusion (piégeage mécanique)
: Il s'agit d'impuretés piégées
mécaniquement dans des pores non débouchant lors de la croissance
des minéraux. Cela peut être sous forme dissoute ou solide.
Un élément métallique retenu à la
surface d'un matériau (physisorption, complexa-tion,
précipitation, chimisorption) sera plus rapidement mis en solution et
donc présentera davantage de risques de toxicitéque s'il est
insérédans le réseau cristallin du matériau.
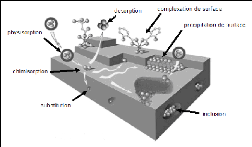
FIGURE 1 - Principales interactions entre un
atome ou une molécule et un solide à l'in-terface solide/liquide
[3]
10
Première partie: ÉTUDE BIBLIOGRAPHIQUE
Parmi les éléments en traces métalliques
que l'on retrouve l'arsenic qui est un polluant ayant une
toxicitéélevée pour la santéet des impacts
dangereux sur l'environnement, pour cela on va s'intéresser par une
synthèse bibliographique sur cet élément.
3 L'arsenic
3.1 Propriétés
L'arsenic est un élément naturellement
présent dans l'environnement puisqu'il occupe le 20
ème rang dans l'ordre d'abondance des
éléments de la croûte terrestre. Le nom arsenic
dérive du terme grec arsenikon, qui signifie « qui dompte
le mâle » en lien avec sa forte toxicité. L'arsenic natif
existe sous trois formes allotropiques : jaune (á); noire (â) et
grise (ã). Cette dernière est la forme la plus stable et la plus
commune, cristallise dans le système rhomboédrique. L'arsenic est
considérécomme un métalloïde en raison de ses
propriétés à la fois de métal et de non
métal. Il appartient au groupe VA de la classification périodique
moderne.
Quelques propriétés physico-chimiques de
l'arsenic sont présentées dans le tableau suivant:
Tableau 1 - Propriétés physico-chimiques de
l'élément Arsenic
Symbole As
Numéro atomique 33
Configuration électronique
[Ar]3d104s24p3
Masse atomique(g.mol-1) 74.9216
États d'oxydation -III, 0, +III, +V
Température de fusion
817C
Densité5.73g/cm3(grise),
1.97g/cm3(jaune)
3.2 Les formes d'arsenic
Plus de trente formes d'arsenic ont
déjàétéidentifiées dans l'environnement,
leur présence et leur répartition dépendant des conditions
du milieu (conditions d'oxydoréduction, pH, cinétiques de
réactions)[10]. Parmi lesquels on cite :
Arsénite As(III)
formes inorganiques
Arséniate As(V)
Acide diméthylarsinique DMAA
Acide monométhylarsonique MMAA formes organiques
Arsénobétaïne AsB
11
Première partie: ÉTUDE BIBLIOGRAPHIQUE
3.2.1 Les formes inorganiques
Les espèces d'arsenic inorganique retrouvées
dans l'environnement présentent des degrés d'oxydation de +III ou
+V, sous forme arsénite As(III)(AsO3- 3 )et
arséniate As(V) (AsO3-
4 ). ces deux formes inorganiques sont facilement
inter-convertibles par réaction d'oxydoréduction, et sont donc
souvent retrouvées ensemble, l' As(V) étant thermody-namiquement
favorisédans les conditions normales oxydantes[10]. Il
existe plus de 200 minéraux contenant de l'arsenic qui correspondent
à des arséniates, des sulfures et sulfo-sels, des
arsénites, des arséniures ou des oxydes(voir annexes).
3.2.2 Les formes organiques
Plusieurs espèces organiques de l'arsenic sont
présentes dans le sol comme des composés arséniés
méthylés (MMAA, DMAA,...). Leur origine peut être soit une
méthylation directe de l'arsenic par les micro-organismes, soit par un
apport anthropique principalement agricole(pesticides, fertilisants...)
3.3 Les sources de l'arsenic
3.3.1 Les sources naturelles
L'arsenic est un composant naturel de l'écorce
terrestre avec une concentration dans la partie superficielle
évaluée à 2 mg/kg, et pouvant atteindre localement 100
mg/kg voire 200 mg/kg dans les dépôts calcaires ou
phosphatés et dans les schistes. On le trouve en proportions plus
considérables dans les roches, oùplus de 99% de l'arsenic
présent se trouve sous forme de minerais de cuivre, de zinc et de plomb,
les plus importants étant l'arsénopyrite (FeAsS), le
réalgar (As4S4), et l'orpiment
(As2S3)
L'érosion, les précipitations et le lessivage
des sols entrainent une redistribution de l'arsenic vers les compartiments
aquatiques sous forme dissoute ou particulaire. Certaines eaux issues de sols
volcaniques peuvent contenir des teneurs atteignant 300 ug/L, mais
plus généralement, l'arsenic dissous dans les eaux superficielles
se trouve à des concentrations comprises entre 0,1 et 10 ug/L.
Dans l'atmosphère, le flux d'arsenic est principalement causépar
l'érosion éolienne et
l'activitévolcanique.[10]
3.3.2 Les sources anthropiques
La production mondiale d'arsenic en 2008 était
estimée à 53500 tonnes, dont 97% sous forme de trioxyde d'arsenic
(As2O3 = As(III)). L'élément
arsenic est produit par pyrolyse de l'arsénopyrite ou par grillage de
sulfure puis réduction de l'oxyde forméavec du carbone. Le
trioxyde d'arsenic est formélors de la fusion du plomb ou du cuivre, par
évaporation et oxydation dans l'air de l'arsenic combinéavec ces
métaux [10]. L'arsenic et ses composés sont
utilisés dans de nombreux domaines :
- En agriculture, il entre dans la composition de nombreux
pesticides, herbicides...etc.
12
Première partie: ÉTUDE BIBLIOGRAPHIQUE
- Dans l'industrie du bois pour la protection contre les
insectes sous forme de complexe cuivre/chrome/arsenic (CCA);
- Dans l'industrie du verre pour favoriser la sortie des
bulles d'air et comme agent décolorant;
- Dans l'industrie des semi-conducteurs, pour la fabrication
de piles photovoltaïques, de diodes électroluminescentes, de
circuits intégrés;
- Dans l'industrie pharmaceutique et
vétérinaire, dans la fabrication d'antibiotiques pour
l'élevage des volailles dans certains pays.
- Dans les industries métallurgiques, pour durcir le
cuivre et le plomb.
3.4 Spéciation chimique de l'arsenic dans les
sols
Dans les sols, l'arsenic est présent principalement
sous forme inorganique, telle que la forme As(V) est majoritaire dans les
conditions d'aération normale ce qui assure une mo-bilitéassez
limitée, car l'As(III) est reconnu pour être plus mobile que
l'As(V). Cependant l'activitémicro-biologique peut conduire à des
réactions de méthylation ou déméthylation, et
également à des réactions d'oxydoréduction, ce qui
engendre un changement des formes d'arsenic.[11]
Au cours des dernières décennies, beaucoup de
recherches scientifiques ont étéeffec-tuées sur l'arsenic,
comme réponse à la croissance de ses effets nocifs sur la
santéet l'environnement [12]. En même temps la
connaissance de la concentration totale en arsenic ne suffit pas pour
évaluer le risque de cet élément. En effet, sa
toxicitéaiguëet chronique dépend de sa spéciation,
c'est-à-dire de la détermination de la forme chimique sous
laquelle il se trouve, et aussi de la voie d'absorption. La toxicitéde
l'arsenic varierait comme suit [1] :
AsH3 gaz, As(-III) >
As(III) >As(V )> composés
méthylés
parmi les études de spéciation
réalisée sur l'arsenic on cite :
V- Une étude de spéciation a
étéeffectuésur plusieurs sols agricoles contaminés
de la zone Aznalcollar (Espagnol) montre que la concentration totale varie
entre 12,6 et 766 mg/kg et que l'arséniate est la principale
espèce dans tous les sols analysés, cependant dans certains
échantillons l'arsénite et les espèces
méthylés pourraient également être
détectés [13].
V- Au Ghana, proche d'une mine d'or (Ashanti), la
spéciation d'arsenic montre que la teneur totale varie entre 189 et 1025
ppm, et que dans les sols a~erobies l'arséniate est l'espèce la
prédominante, alors que l'arsénite se trouve en grande
quantitédans les sols ana~erobies [14].
Dans le tableau suivant on cite quelques exemples de sols
contaminés par l'arsenic :
13
Première partie: ÉTUDE BIBLIOGRAPHIQUE
Tableau 2 - Concentration moyenne en arsenic dans des sols
contaminés par des activités industrielles
[1]
|
Sols contaminés
|
Arsenic
(
mg.kg-1)
|
Auteurs
|
|
Sol de jardin situéà
proximitéd'activités
industrielles,mines fonderies (SO de
l'Angleterre)
|
144 à 892
|
Thornton, 1996
|
|
Sol situéprès d'activités industrielles,
mines et
fonderies de Pb, As...
|
1400
|
Davis et al.,
1992
|
|
Sol situéà proximitéd'activités
métallurgiques
(Freiberg District, Erzgebirge, Allemagne)
|
100 à 1200
|
Bombach et al.,
1994
|
|
Sol de verger à proximitéd'une usine
de
pesticides (Auzon, Auvergne, France)
|
180
|
Eisenlohr et
Laperche, 2001
|
|
Sol agricole situéprès d'activités
industrielles,
fonderies de Zn et As (Reppel, Belgique)
|
113
|
Boisson-
Gruppen, 1999
|
14
Deuxième partie
MATÉRIELS ET MÉTHODES
15
Deuxième partie: MATÉRIELS ET MÉTHODES
1 Site d'étude 1.1 Introduction
Au cours des dernières années, le Maroc a connu
une croissance démographique, un développement économique
et une amélioration du niveau de vie, ce qui a engendréune
augmentation avec complexitéde la quantitéde déchets
solides.
Les déchets sont définis par la loi 28-00
relative à leur gestion et à leur élimination comme :
« tous résidus d'un processus de production, de transformation ou
d'utilisation, toute substance matériaux, produit ou plus
généralement tout bien meuble abandonnéou que son
détenteur destine à l'abandon ou à l'obligation de s'en
défaire dans le but de ne pas nuire à la collectivitéet de
protéger l'environnement ».
Au niveau de la ville de Marrakech la génération
totale de déchets solides est plus de 150 000 tonnes par an, leurs
répartition selon leurs origines comme la suivante :
*- Ordures ménagères : environ 125 000 t/an soit
82%
*- Déchets industriels : environ 27 000 t/an soit
18%
*- Déchets hospitaliers : environ 750 t/an soit 1%
[15]
La collecte des déchets de la ville est assurée
par les services d'assainissement de chaque commune urbaine, 60 % à 70%
des déchets sont collectés et mis à la décharge
publique sans aucun traitement, ni séparation préalable, les
ordures non collectées s'accumulent sur la voie publique ou dans des
décharges sauvages.
A la décharge publique des tonnes des déchets
solides s'accumulent chaque jours, formant des couches très complexes en
terme de composition (matières organiques, métaux lourds ...etc).
Dans des conditions de lumière, de température, de pluie et
d'aération, ces déchets se dégradent en contaminant les
sols de ce site.
1.2 Choix du site d'étude
Le site d'étude choisi est la décharge publique
de la ville de Marrakech. Cette dé-charge(dite de Harbil) est
exploitée depuis 1987, elle se trouve au nord de la ville, en rive
droite de l'oued Tensift, environ 500 m après la traversée du
pont de l'oued Tensift, sur la route de Safi . Il s'étend sur une
superficie de 14 hectares (Commune urbaine de Marrakech, Juin 2006). La
décharge publique de Marrakech est une décharge non
contrôlée, elle reçoit différents types de
déchets(ménagers, industrielles, hospitaliers... ), ce qui
constitue
une source de nuisances pour les populations avoisinantes
à cause des émissions de gaz àeffet de serre et
aussi les odeurs nauséabondes. De même les déchets
présentent des risques
pour la santéet peuvent contribuer à la
dégradation de la qualitédes eaux de la nappe phréatique
et de l'oued Tensift [15].
Notre étude est réalisée sur un
échantillon témoin (témoin1) pris de la région de
Ta-meslouht et un autre de la région de Taouloukoult près de la
ville d'Imintanout (témoin 2) et quatre échantillons E1, E2, E3
et E4 prélevés de différentes positions de l'ancienne
16
Deuxième partie: MATÉRIELS ET MÉTHODES
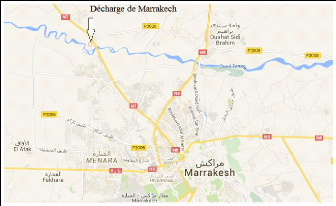
décharge en s'éloignant de la route de Safi, voir
figure 2 et 3
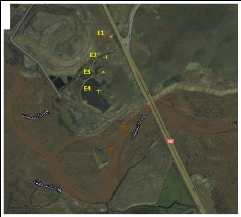
FIGURE 2 - Carte de localisation du site d'étude
FIGURE 3 - photo du site d'étude par satellite
Tableau 3 - Les différentes positions de nos
échantillons
E1 20 m de la route
E2 100 m de la route
E3 200 m de la route
E4 au sommet de la décharge
17
Deuxième partie: MATÉRIELS ET MÉTHODES
2 Paramètres physico-chimiques 2.1
Préparation du sol
Une préparation adéquate du sol joue un
rôle très important pour l'homogénéitéde
l'échantillon et la représentativitédu site
d'étude, pour cela les échantillons prélevés de
différentes positions du site, sont laissés sécher
à l'air libre durant 48 h, tamisés à 2mm et broyés
pour qu'ils soient très fins.
2.2 pH
Le pH est un facteur déterminant pour la
mobilitédes ions métalliques, car un pH acide entraîne la
mise en solution des sels métalliques, la désorption des cations
et l'adsorption des anions, aussi lorsque le pH augmente les cations sont moins
solubles et les anions sont plus solubles. De plus l'augmentation de pH induit
souvent la formation des précipités qui peuvent limiter la
solubilitéde toutes les espèces ioniques
[16].
Mode opératoire
La mesure du pH a étéréalisée
suivant la norme AFNOR NF X31-103(1992) , en préparant
une suspension aqueuse du sol d'un rapport 1/2,5 (masse du sol/ volume d'eau
distillée), ensuite on agite le mélange pendant 30 mn et on le
laisse reposer 2h, le pH est mesurépar un pH-mètre de type
HANNA instruments HI 2211.
2.3 Conductivitéélectrique
La conductivitéélectrique (CE) est une mesure
qui donne une approximation de la concentration des sels solubles
présents dans l'échantillon[16].
Mode opératoire
La mesure est basée sur l'extraction des sels d'un
échantillon, en solubilisant ce dernier dans l'eau distillée dans
un rapport 1/5 (masse du sol/ volume d'eau distillée). Après
30
minutes d'agitation, le mélange est laisséau repos
pendant 2 h, ensuite la conductivitéest mesurée par un
conductimètre de type EUTECH instruments CON 510.
2.4 Humidité
L'humiditéest définie comme la masse
perdue(masse d'eau) après séchage à 105°C d'un
échantillon . Sa mesure permet de déterminer la masse
sèche d'un échantillon du sol. La procédure est de porter
un échantillon pesépréalablement dans l'étuve
pendant 24 h, ensuite il est retiréet laissérefroidir dans un
dessiccateur, la teneur d'humiditéH est exprimée en pourcentage
massique et elle est calculée par la relation suivante :
18
Deuxième partie: MATÉRIELS ET MÉTHODES
Avec :
m0 : la masse initiale de l'échantillon,
m1 : la masse après séchage.
2.5 Matière organique
La méthode la plus répandue pour la
détermination de la teneur en matière organique est la perte au
feu par calcination du sol au four[16], la procédure
est de prendre une masse de l'échantillon, la sécher à
105°C pendant 24 h , puis la calciner à 550°C pendant 4 h. La
teneur en matière organique % MO est calculée par la relation
:
|
Avec :
|
ins - inc
%MO = ins
|
x 100
|
% MO : pourcentage de matière organique
dans l'échantillon sec,
ms : masse de l'échantillon après
passage à l'étuve à 105°C, mc :
masse de l'échantillon après calcination.
2.6 Carbone organique total COT
Le dosage du carbone organique total est effectuépar la
méthode de Walkley et Black, cette méthode est
basée sur l'oxydation du carbone organique par le bichromate de sodium
Na2Cr2O7 en milieu acide sulfurique (source de chaleur).
8H2SO4 + 3C + 2Na2Cr2O7 -+
2Na2SO4 + 2Cr2(SO4)3 + 3CO2 +
8H2O (6)
4 x (Cr6+ + 3e- -+
Cr3+) : rduction
3 x (C -+ C4+ +
4e-) : oxydation
4Cr6+ + 3C -+ 4Cr3+
+ 3C4+
La quantitéde Na2Cr2O7
utilisée excède la quantiténécessaire
pour l'oxydation du carbone organique total. L'excès de
Na2Cr2O7 qui n'a pas réagi est ensuite doséen
retour par une solution titrée d'un réducteur qui est le sulfate
ferreux (sel de Mohr) en présence de diphénylamine. Ce dosage en
retour permet donc de calculer la quantitéde bichromate qui a
éténeutralisée par le carbone
organique[17].
Na2Cr2O7 + 6FeSO4 + 7H2SO4 -+
Na2SO4 + Cr2(SO4)3 +
Fe2(SO4)3 + 7H2O (7)
Réactifs
- Bichromate de sodium (Na2Cr2O7)
- Acide sulfurique H2SO4 (95-97%, d=1,83)
19
Deuxième partie: MATÉRIELS ET MÉTHODES
- Diphénylamine (0,1g du poudre + 4 mL H2O +20 mL
H28O4 concentré)
- Sulfate d'ammonium-fer(II) hexahydraté(sel de
Mohr)((NH4)2Fe(8O4)2, 6H2O).
Mode opératoire
La masse prise pour le dosage est inversement proportionnelle
à la teneur en matière organique de l'échantillon. Plus le
sol est riche matière organique, plus la prise d'essai sera
faible. Dans une fiole conique, on met une masse d'un
échantillon préalablement tamiséà2 mm,
broyéet séché, et on ajoute 10 mL de la solution de
bichromate de sodium (0,1667
M) à l'aide d'une pipette et on agite la fiole d'un
mouvement de va-et-vient, ensuite, on verse d'un coup 20 mL d'acide sulfurique
concentrédans le mélange et on agite pendant une minute. La
solution est laissée au repos pendant 30 minutes pour que l'oxydation
ait lieu. Pour arrêter la réaction on ajoute 150 mL d'eau
distillée et on laisse décanter pendant 2 h. pour le dosage, on
prélève 25 mL du mélange, on la met dans un erlenmeyer de
100 mL et on ajoute 5 mL d'acide orthophosphorique et 3 gouttes de
diphénylamine, et on dose avec le sel de Mohr à 0,1M, la solution
est noire au début, vire au vert clair.
100 100
%C.O.T = [X - C.V ].0,
003.77 .m
Avec :
% C.O.T : pourcentage de carbone organique
total,
X : la quantitéde matière de
bichromate de potassium(oxydante+ excès), C :
concentration de la solution de sel de Mohr
(mol.L-1),
V : volume verséde la solution de sel de
Mohr (mL), m : masse de l'échantillon de sol (g).
2.7 Azote total
La méthode de détermination de l'azote total la
plus utilisée est la méthode de Kjeldahl, le
principe est baséd'abord sur la minéralisation du sol pour passer
de l'azote sous forme organique à l'azote minéral. On
détruit donc la molécule organique par l'at-taque avec l'acide
sulfurique (H28O4) concentré, en présence de catalyseur,
l'azote est transforméen sulfate d'ammonium. Ensuite dans un
distillateur d'azote, l'ion ammonium NH+4 est
déplacépar la soude 40% sous forme d'ammoniac NH3 et
fixépar l'acide borique 2% et enfin titrépar l'acide sulfurique
0,02 N en présence de l'indicateur coloré(Tashiro).
Réactifs
- catalyseur de minéralisation( 2g de sulfate de
potassium K28O4 + 0,2g de sulfate de cuivre Cu8O4
+ 1g de sélénium)
- phénophtaléine :(1g / 100mL d'éthanol
60%)
- soude NaOH 40% (400g /1 litre d'eau distillée)
- acide borique : 10g/1 litre d'eau distillée
- indicateur de Tashiro (0,1g de vert de Bromocrésol +
0,02g de rouge de méthyle +100mL d'éthanol)
20
Deuxième partie: MATÉRIELS ET MÉTHODES
Mode opératoire
Dans un matras Kjeldahl de 250 mL, on introduit environ 5g
d'échantillon avec 0,5g de mélange catalyseur, on ajoute 10 mL
d'acide sulfurique concentrépuis on place le matras sur une rampe
d'attaque avec un entonnoir dans le col du ballon pour condenser les vapeurs
sulfureuses. La digestion se fait à chaud pendant 1h à 180°C
puis pendant 360°C jusqu'àce les échantillons deviennent
blancs. le minéralisât est laissérefroidir puis
récupérédans une fiole jaugée de 250 mL et
complétéavec l'eau distilléau trait pour la distillation.
Dans les mêmes conditions, on réalise un blanc.
Dans un matras de distillation on introduit 20 mL de l'extrait
et 10 mL de lessive de soude 40%, en même temps on introduit 25 mL de
l'acide borique (H3BO3) 1N et 6 gouttes d'indicateur
coloréTashiro dans un erlenmeyer de 200 mL et on le place en dessous du
réfrigérant, la pointe plongeant dans le liquide. Enfin on titre
avec l'acide sulfurique 0,02N.
Le pourcentage d'azote est calculéen utilisant la
formule suivante [16] :
V 100
N(%) = (V2 - V1) x C x
0, 14 x A x g
Avec :
N : Azote total Kjeldahl,
V2 : volume de
(H2SO4) versépendant la titration de
l'échantillon en mL,
V1 : volume de
(H2SO4) versépendant la titration du blanc en mL,
A : aliquote à distiller (20 mL),
V : volume initial (250 mL),
g : la masse de l'échantillon en g,
C : concentration de (H2SO4)
.
2.8 Détermination de la teneur totale d'arsenic
«Extraction totale»
L'extraction totale ou la minéralisation consiste à
mettre en solution en une seule
étape la totalitéde l'élément
métallique concernépar le dosage. La procédure suivie
est
celle décrite par la norme AFNOR NF X31-151
(1993).
mode opératoire
- peser 1g de sol à 0,01 près et le mettre dans un
erlenmeyer.
- ajouter 20 mL d'eau régale (1 3HNO3 + 2
3HCl).
- laisser le contact pendant 2 h.
- chauffer dans un bain de sable jusqu'au sec.
- ajouter un peu de l'eau distillée.
- filtrer sous vide.
- le filtrat recueilli est conservéen ajoutant quelque
gouttes d'acide chlorhydrique
21
Deuxième partie: MATÉRIELS ET MÉTHODES
pour les analyses avec spectrométrie d'émission
atomique par plasma à couplage inductif (ICP-AES).
3 La spéciation chimique
3.1 Introduction
L'accumulation des métaux traces d'origine
anthropogénique dans les sols pose des problèmes de
toxicité, mobilitéet biodisponibilité. La connaissance de
leurs teneurs totales était nécessaire afin de prévoir
leurs impacts environnementaux, mais n'était pas suffisante pour
évaluer leur toxicitéet leur mobilité, car ces
dernières dépendent la forme chimique du métal
[18]. Ensuite la spéciation chimique a pris une place
dans la recherche scientifique. Suivant l'IUPAC (Union internationale de chimie
pure et appliquée), la spéciation est « l'ensemble
des processus qui permettent d'obtenir l'information sur la forme chimique
(ionique ou moléculaire) d'un analyte
».
3.2 Schémas d'extraction séquentielle
La spéciation est basée sur l'extraction
séquentielle d'un élément en fonction de la phase
oùil est relié. Le premier schéma de spéciation des
métaux lourds qui fut mis en évidence est celui de
TESSIER et col [2], il consiste à extraire des phases
successives :échangeable, liée aux carbonates,
réductible, oxydable et résiduelle. Dès lors, de
nombreux schémas ont étéproposé, ils
différent entre eux soit par les réactifs d'extraction, soit par
le nombre de phases à extraire. Le schéma de BCR (Bureau
Communautaire de Références) par exemple consiste à
extraire trois phases :Acido -soluble et soluble dans l'eau,
échangeable, Oxyde de Fer et de manganèse et Organique + sulfure
au lieu de cinq proposés par TESSIER.
Pour notre étude, on a travailléavec le
schéma de TESSIER vue sa simplicitéet son large domaine
d'utilisation. En opérant sur 1g d'échantillon pour extraire les
phases mentionnées dans le tableau suivant :
22
Deuxième partie: MATÉRIELS ET MÉTHODES
Tableau 4 - Protocole d'extractions séquentielles
adaptéde Tessier et al.,[2] pour 1g du sol
|
Phase
|
Réactifs d'extraction
|
T(°C)
|
Agitation
|
t (h)
|
|
Échangeable
|
-8 mL MgCl2 , pH=7
|
ambiante
|
continue
|
2
|
|
Liée au carbonates
(CO2-
3 )
|
-8 mL CH3COONa 1M ajustéambiante
à pH=5 avec CH3COOH
|
|
continue
|
5
|
|
Réductible (liée aux
oxydes de Fe et Mn)
|
-20 mL NH2OH, HCl 0,04M
dans
CH3COOH 25% (V/V)
|
96 #177; 3
|
occasionnelle
|
6
|
|
Oxydable(organique)
|
-8 mL HNO3 0,02M ajustétemps à pH=2 avec
HNO3
+ 5 mL H2O2 30%
|
85 #177; 2
|
en temps
|
2
|
|
+ 3 mLH2O2 30%
|
même T
|
occasionnelle
|
3
|
|
-Après refroidissement on
ajoute 5 mL
CH3COONH4
3,2M dans HNO3 20% (V/V)
|
ambiante
|
continue
|
0,5
|
|
Résiduelle
|
10 mL HF + 2 mL HClO4
|
|
|
à sec
|
Remarque : la séparation est faite par
centrifugation suivie par filtration sur papier-filtre
0,45um
4 Techniques d'analyse 4.1 Diffraction des rayons
X
La diffraction des rayons X sur un matériau permet de
déterminer la nature des phases cristallisées. En effet, les
phases cristallines peuvent être considérées comme des
assemblages de plans réticulaires plus ou moins denses
désignés par leurs coordonnées (h, k, l) et
séparés par des distances caractéristiques de la structure
de la phase considérée. Les pics de diffraction correspondent aux
interférences entre les différents plans réticulaires
peuvent être déterminés selon la loi de
Bragg : 2dsinè = nA
Avec :
d : la distance réticulaire entre deux
plans adjacents d'une même famille,
è : l'angle de
réflexion,
n : l'ordre de réflexion,
A : la longueur d'onde du rayonnement
utilisé.
Les analyses ont étéréalisésur des
échantillons en poudre en utilisant une source de radiation Cu
Ká1,2 et un monochromateur en graphite sur
un diffractomètre XPERT-MPD (FSSM) selon les conditions analytiques
suivantes : pas de 0, 05° 2è, temps de comptage
de 0,4 secondes par pas, sur un domaine angulaire compris entre 5° et
70°
Deuxième partie: MATÉRIELS ET MÉTHODES
2è. L'analyse des diffractogrammes obtenus a
étéréalisée à l'aide du logiciel
X'Pert HighScore Plus.
4.2 Spectrométrie d'absorption atomique
(SAA)
La spectrométrie d'absorption atomique (SAA) est une
technique décrite pour la première fois par Walsh (1955). En
effet, C'est une des principales techniques mettant en jeu la spectroscopie
atomique dans le domaine UV-visible utilisée en analyse chimique. Elle
étudie les absorptions de lumière par l'atome libre. Elle est
très sensible et permet de doser les éléments chimiques
(métaux et non-métaux) même à l'état de
traces . Ses applications sont nombreuses étant donnéqu'on
atteint couramment des concentrations inférieures au mg/L (ppm).
Le principe de SAA est basésur la mesure
d'intensitéde lumière absorbépar un atome. En effet,
lorsque ce dernier est soumis à une radiation, il va l'absorber et passe
d'un niveau d'énergie Eàun niveau
E*. Cette modification d'énergie correspond à
une raie d'absorption.
I0
D'après la loi de BEER-LAMBERT on a : log I =
A = å.C.l
Avec :
I0 : Intensitédu faisceau incident,
I : Intensitédu faisceau transmis
après passage dans la flamme,
A : Absorbance,
å : Coefficient d'extinction molaire de
l'espèce à une fréquence bien déterminé,
C : Concentration du milieu en atome
absorbants,
l : longueur du brûleur.
Ainsi on déduit la concentration de
l'élément étudié.
4.3 spectrométrie d'émission atomique par
plasma à couplage inductif (ICP-AES)
la spectrométrie d'émission atomique par plasma
à couplage inductif(ICP-AES) est une technique largement répandue
pour l'analyse des métaux. Il s'agit d'une spectroscopie
d'émission. L'excitation des niveaux électroniques et la
génération des atomes et des ions est réalisée par
un plasma.
Les plasma sont des gaz partiellement ionisés et
électriquement neutres. Ils sont générés à
haute température entre 7000 K et 15000 K. A partir de ces
températures, on a un
déplacement relativement libre des cations et des
électrons. Ces gaz ont une conductivitéthermique et
électrique plus élevée. Le gaz le plus utiliséest
l'argon.
Ar -? Ar+ + e-
23
Le potentiel d'ionisation très élevéde
l'argon ( 15,76 eV) permet d'ioniser tous les élé-
24
Deuxième partie: MATÉRIELS ET MÉTHODES
ments possédant un potentiel d'ionisation plus faible
dans le plasma. Il y a trois manières de réaliser un plasma:
- Plasma par courant direct (DCP : direct current plasma) ou
plasma-arc : on génère un arc électrique entre deux
électrodes sous atmosphère d'argon, le gaz est ioniséet
génère un plasma.
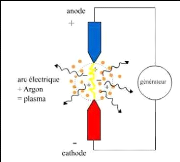
FIGURE 4 - Principe du plasma à courant
constant
- Plasma micro-onde induit (MIP : microwave induced plasma)
:On génère le plasma par micro-ondes qui vont engendrer des
oscillations des atomes de gaz et donc un chauffage et une ionisation des
atomes (collisions) .
- Plasma par couplage inductif (ICP : inductively coupled
plasma) : C'est la méthode la plus utilisée en chimie
analytique.
Le plasma est générédans une torche qui
comporte trois tubes en quartz coaxiaux. Dans le haut de la torche, une bobine
d'induction est parcourue par un courant alternatif à haute
fréquence (entre 5 et 100 MHz). Un flux d'argon tangentiel (tube
externe) va permettre d'isoler, de refroidir la région proche des
bobines et de donner la forme au plasma. Le gaz plasmagène est introduit
dans le tube intermédiaire ce qui permet d'éviter tout contact
proche de la bobine. L'ionisation de ce gaz est amorcée par une
décharge électrique dans le bas de la torche. Puis les ions
crées interagissent avec le champ magnétique (trajectoire
circulaire) au niveau de la bobine d'induction ce qui crée des chocs et
augmente la température (effet Joule) et maintient l'état de
plasma (ionisation).
Après le passage dans un nébuliseur,
l'échantillon est introduit au centre du plasma. La température
extrêmement élevée permet la désolvatation puis la
volatilisation et l'ato-misation des espèces. La température
atteinte permet l'ionisation de la plupart des atomes par chocs avec l'argon
(Ar ou Ar+) ou des électrons :
Ar + M -+ Ar +
M+* + e_
Ar+ + M -+ Ar +
M+*
e_ + M -+ M+* +
2e_
25
Deuxième partie: MATÉRIELS ET MÉTHODES
On observe ensuite la désexcitation des ions ou atomes
par émission de fluorescence.
26
Troisième partie
RÉSULTATS ET DISCUSSIONS
27
Troisième partie: RÉSULTATS ET DISCUSSIONS
Introduction
Dans cette partie on va s'intéresser à la
discussion des résultats de notre travail qu'ils soient obtenus par
caractérisations physico-chimiques ou par diffraction des rayons X (DRX)
ou par spectrométrie d'émission atomique par plasma à
couplage inductif (ICP-AES) .
1 Paramètres physico-chimiques
1.1 pH
Les mesures de pH effectués sur les différents
échantillons montrent que le pH est légèrement alcalin
pour E1, E2, E3 et E4 , et alcalin pour le témoin 1 et le témoin
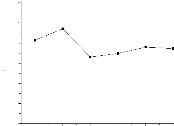
2, la figure 5 montre les valeurs de pH pour différents
échantillons.
p H
Té mo i n 1 Témoin 2 E 1 E2 E3 E4
p H
9 ,0
8 ,5
8 ,0
6 ,5
6 ,0
7 ,5
7 ,0
échantillon
FIGURE 5 - les valeurs de pH pour les
différents échantillons
V- le pH des échantillons est
généralement alcalin, ce qui favorise la précipitation et
la complexation des ETM.
V- selon [19], à pH alcalin
les ETM précipitent ce qui induit un enrichissement intense du sol par
ces polluants.
V- selon [1], à pH<8 l'As(V) est
piégéen forte quantitésur les phases de type oxyhy-droxyde
de fer, cependant à pH>8 certains éléments chimiques
comme les phosphates seront des ions compétiteurs importants
vis-à-vis de l'adsorption de cet espèce.
1.2 Conductivitéélectrique (CE)
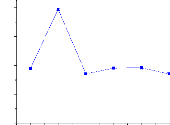
D'après les résultats trouvés on constate
que les échantillons E1, E2, E3 et E4 ont en général des
valeurs élevées de la conductivitéélectrique par
rapport aux échantillons témoin 1 et témoin 2, on constate
aussi que E1 et E3 présentent des maximums pour la
conductivitéélectrique. La figue 6 montre la variation de
conductivitéélectrique en
28
Troisième partie: RÉSULTATS ET DISCUSSIONS
fonction des échantillons.
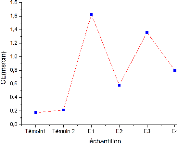
FIGURE 6 - la variation de la
conductivitéélectrique en fonction des échantillons
V- Les résultats trouvés montrent qu'il y a une
quantitéimportante de sels solubles (comme les carbonates...).
V- Cette salinitéexcessive peut nuire aux cultures
envisagées dans le cas de la réhabilitation du site
[20].
1.3 Humidité
Les teneurs d'humiditécalculées
montrent que le sol le plus humide est celui représentépar
témoin 1 (3,89%) suivi par témoin 2 et puis E1, cependant E2, E3
et E4 sont moins humides. La figure 7 montre les teneurs d'humiditépour
les différents échantillons.
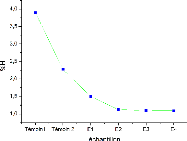
FIGURE 7 - les teneurs d'humiditépour les
différents échantillons
29
Troisième partie: RÉSULTATS ET DISCUSSIONS
V- La teneur en humiditéest élevée
pour l'échantillon témoin 1 suivi par témoin 2 qui sont
des sols agricoles, et cela peut être dûà la
quantitéd'argile contenue.
1.4 La matière organique (MO)
D'après les pourcentages en matière organique
calculés, l'échantillon le plus riche en MO est témoin 2
(7,90%), tandis que les autres échantillons ont des teneurs enter 3 et
4%. La figure 8 présente les pourcentages en matière organique
pour différents échantillons.
% M O
Témo i n 1 Témoin 2 E 1 E2 E3 E4
% M O
8
6
4
2
0
échantillon
FIGURE 8 - les pourcentages en matière
organique pour différents échantillons
V- La grande quantitéen matière organique
est observée pour témoin 2, cela est
dûàl'utilisation des fertilisants naturels.
V- Les teneurs en matière organique pour les
échantillons de la décharge sont faibles par rapport à
celles trouvées dans la décharge de la ville d'Agadir
[20].
1.5 Carbone organique total (COT)
Les résultats des dosages effectués pour
différents échantillons montrent que l'échan-tillon
témoin 2 a la grande teneur en carbone organique total(3,96%), suivi par
E4 (2,27%) puis témoin 1, E1, E2 et E3. La figure 9 montre la variation
des pourcentages en COT pour différents échantillons.
30
Troisième partie: RÉSULTATS ET DISCUSSIONS
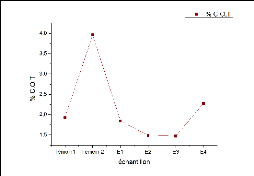
FIGURE 9 - la variation des pourcentages en carbone organique
total pour différents échantillons
V- La teneur en carbone organique est relativement faible,
proche d'un sol normal (2 à 3%) [20].
V- Les teneurs en carbone organique pour les différents
échantillons suivent pratiquement la même évolution que la
matière organique avec un rapport (MO/%COT) de 1,5 à 2,6. La
moyenne des rapports est 2,08 proche à celle trouvée par [21]
(2,01), malgréque le facteur 1,724 est souvent utilisé.
1.6 Azote total de Kjeldahl (NTK)
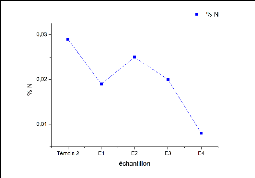
D'après les résultats du dosage d'azote total on
constate que l'échantillon témoin 2 a le pourcentage le plus
élevé(0,029) suivi par E2, E3, E1 et enfin E4, la figure 10
montre les pourcentage en NTK trouvés.
FIGURE 10 - les pourcentages en Azote total de Kjeldahl pour les
différents échantillons
31
Troisième partie: RÉSULTATS ET DISCUSSIONS
V-Les pourcentages en NTK sont très faible par rapport
à ceux trouvés dans les sols de la décharge d'Agadir
[20].
V- Les faibles teneurs en NTK sont dûs à la
dégradation des déchets en entraînant la perte d'azote sous
forme volatile [16].
Tableau 5 - tableau récapitulatif des paramètres
physico-chimique étudiés pour les différents
échantillons
|
Échantillon
|
pH
|
CE(ms/cm)
|
% H
|
% MO
|
% C.O.T
|
% NTK
|
|
Témoin 1
|
8,07
|
0,175
|
3,89906
|
3,79912
|
1,92639
|
-
|
|
Témoin 2
|
8,35333
|
0,215
|
2,2736
|
7,9001
|
3,96247
|
0,029
|
|
E1
|
7,65
|
1,624
|
1,49476
|
3,42851
|
1,84824
|
0,019
|
|
E2
|
7,74333
|
0,583
|
1,12486
|
3,82719
|
1,49022
|
0,025
|
|
E3
|
7,9
|
1,36
|
1,0944
|
3,84785
|
1,47388
|
0,02
|
|
E4
|
7,86333
|
0,8
|
1,08774
|
3,41595
|
2,27347
|
0,008
|
1.7 Caractérisation par diffraction des rayons X
(DRX)
L'analyse de reconnaissance des diffractogrammes des rayons X
par le logiciel X'Pert HighScore Plus montre qu'il y a des phases communes pour
tous les échantillons(Quartz, silice et calcite) et il y a d'autres
présentes dans des sols bien précis (bornite, oxyde
d'uranium...). Le tableau suivant présente les phases de chaque
échantillon.
Tableau 6 - les phases minérales trouvées pour
chaque échantillon
|
Échantillon
|
les phases minérales présentes
|
|
Témoin 1
|
Silice (SiO2), calcite (CaCO3)
|
|
E1
|
Quartz (SiO2), silice (SiO2) , calcite
(CaCO3), oxyde d'uranium UO2, Bornite Cu5FeS4
|
|
E2
|
Silice (SiO2) , calcite (CaCO3)
|
|
E3
|
Quartz (SiO2), silice (SiO2) , calcite
(CaCO3), silicium Si
|
|
E4
|
Quartz (SiO2) silice (SiO2) , calcite
(CaCO3), oxyde d'uranium UO2, Bornite Cu5FeS4,
Fluorite CaF2 , silicium Si, Sodium
erbium fluoride NaErF4, oxyde de praseodymium
PrO1,83
|
On constate que l'arsenic n'apparaît pas dans ces phases
à cause de sa faible teneur et l'absence d'une phase
cristallisées à base de cet élément.
Après une analyse avancée par le logiciel, on
observe l'apparition de quelques phases contenant l'arsenic comme Cu3AsSe3
; CuAsSe2 ; As2O3 ; Pb5Cl(AsO3)3
; Pb3As2S6 ; CoAs3, mais de faible score.
32
Troisième partie: RÉSULTATS ET DISCUSSIONS
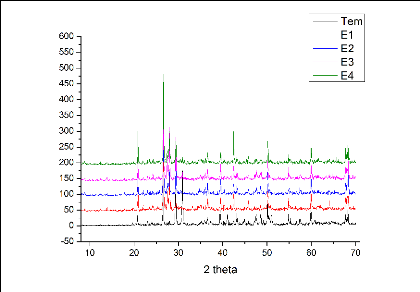
FIGURE 11 - Diffractogrammes de RX des échantillons
étudiés
1.8 La teneur totale d'arsenic
L'analyse des filtrats obtenus après extraction totale
de nos échantillons par ICP-AES montre que Les teneurs sont très
faibles par rapport à la moyenne dans les sols (5mg/kg) [22], en effet
on a 0,496 pour témoin 2 et 0,439 pour E2 et 0,216 pour E3, cependant
les teneurs pour E1 et E4 sont inférieur à la limite de
détection. La figure 12 présente les teneurs en arsenic pour les
différents échantillons.
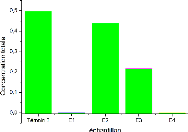
FIGURE 12 - les teneurs en arsenic pour différents
échantillons
33
Troisième partie: RÉSULTATS ET DISCUSSIONS
la discussion de ces résultats va être en
comparaison avec les résultats de spéciation chimique.
2 Spéciation chimique
Les résultats d'analyse des fractions obtenus par la
spéciation chimique de l'arsenic dans les différents
échantillons par ICP-AES montrent que:
-pour témoin 2 : la forte teneur en
arsenic est enregistrée dans la phase réductible (64,68%), suivie
par la phase résiduelle (19,90%) et ensuite la phase oxydable(15,42%),
cependant il n'est pas présent dans la phase échangeable et celle
liée aux carbonates. -pour E1 : on constate que
l'arsenic est présent dans la phase liée aux carbonates avec un
pourcentage de 9,86%, alors que la forte proportion en cet
élément est enregistrée dans la phase
réductible(46,09%) suivie par la phase résiduelle(22,93%) et puis
la phase organique(21,11%), par contre aucune trace dans la phase
échangeable.
-pour E2 : l' arsenic est en proportion
presque égale dans les deux phases : réductible et oxydable
(42,05% et 42,44%) et la même chose pour les deux phases : liée
aux carbonates et résiduelle (7,46% et 8,06%), cependant il est
indétectable dans la phase échangeable. -pour E3
: la teneur en arsenic est maximale dans la phase
réductible(49,69%) suivie de la phase résiduelle(41,23%) et
ensuite la phase oxydable(9,08%). Par contre, ce métalloïde n'est
pas détectédans la phase échangeable et celle liée
aux carbonates.
-pour E4 : la quantitéd'arsenic est
maximale dans la phase réductible (49,79%) suivie par la phase
résiduelle (42,05%) et ensuite la phase organique(8,16%), cependant elle
est inférieure à la limite de détection dans les deux
phases échangeable et liée aux carbonate. La figure 13 montre les
différentes proportions d'arsenic pour chaque échantillon.
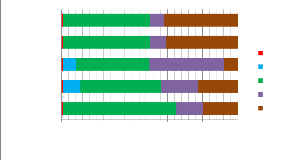
E4
Les échantillons
Témoin 2
E3
E2
E1
phase 1 phase 2 phase 3 phase 4 phase 5
0% 20% 40%
in 2 60% 80% 100%
pourcentage d'As
FIGURE 13 - les pourcentage d'arsenic dans
chaque fraction et pour chaque échantillon
34
Troisième partie: RÉSULTATS ET DISCUSSIONS
V- la teneur d'arsenic est maximale dans la phase
réductible (liée aux oxydes de fer et de manganèse) pour
tous les échantillons, cela peut être expliquépar le
rôle important jouépar les oxydes de fer pour
régulier la rétention de l'arsenic dans les
sols[23], en signalant
que cet élément est retrouvéliéaux
oxydes de fer amorphes en teneur assez élevée qu'aux oxydes
cristallisés.
V-pour la phase organique, il est remarquéque
l'arsenic est liéà cette phase en grande teneur dans
l'échantillon E2, cela peut être dûà la complexation
de cet élément par les groupes fonctionnels des substances
humiques.
V- pour la phase résiduelle, on constate que
l'arsenic est présent en teneur importante et surtout pour les
échantillons E3 et E4, ce qui va limiter sa mobilité.
V- enfin, cet élément est
associéde façon aussi modeste à la phase
carbonatée, alors qu'il n'apparaît pas dans la phase
échangeable ce qui affirme que l'arsenic a une mobilitéassez
limitée dans ces sols.
Tableau 7 - les teneur en arsenic (en ppm) pour
différents échantillons
|
échantillon
|
phase 1
|
phase 2
|
phase 3
|
phase 4
|
phase 5
|
|
Témoin 2
|
0,00
|
0,00
|
2,07
|
0,49
|
0,64
|
|
E1
|
0,00
|
0,48
|
2,23
|
1,02
|
1,11
|
|
E2
|
0,00
|
0,36
|
2,00
|
2,02
|
0,38
|
|
E3
|
0,00
|
0,00
|
4,77
|
0,87
|
3,96
|
|
E4
|
0,00
|
0,00
|
2,71
|
0,44
|
2,29
|
Remarque :
V- lorsqu'on compare la teneur totale en arsenic avec
la somme des proportions dans chaque phase, il apparaît qu'il y a une
grande différence entre eux, cela est dûsoit aux erreurs de
manipulations soit au système ouvert adopté, car il peut y avoir
des composés d'arsenic très volatils comme AsH3.
35
Conclusion générale et perspective
Conclusion générale et perspective
L'objectif de ce travail est d'évaluer le
degréde contamination des sols de la décharge de Marrakech par
l'arsenic d'origine anthropogénique, et d'avoir une idée
générale sur la mobilitéet la toxicitéde ce
métalloïde.
Les résultats de la caractérisation
physico-chimique ont montréque les sols de ce site sont
légèrement alcalins, un peu humides, contiennent des
quantités relativement importantes de sels solubles (les carbonates en
particulier comme le montre l'étude par DRX). Les teneurs en
matière organique sont un peu faibles en comparaison avec celles de la
littérature, sa distribution horizontale est normale sur les
différentes positions d'échantillonnage. Les pourcentages en
carbone organique total sont proportionnels aux teneurs en matière
organique. Les teneurs en azote total de Kjeldahl sont très faibles
à cause de la dégradation des déchets.
L'analyse par DRX montre la présence des phases
cristallines comme le quartz SiO2, calcite CaCO3 et d'autres
contenants l'arsenic comme As2O3 ;
Pb5Cl(AsO3)3... etc., mais de faible score.
Les résultats de la spéciation chimique de
l'arsenic montrent une légère contamination de ces sols par
rapport au sol témoin, et une mobilitéassez limitée de cet
élément puisqu'il n'est pas détectédans la phase
échangeable et en quelques traces dans la phase carbonatée.
Cependant, on ne peut rien dire sur sa toxicité.
Ce travail nous a permis d'avoir une connaissance
préliminaire des caractéristiques phy-sicochimiques de ce site,
et nous a également averti du danger qui peut affecter le milieu
environnant.
Il serait donc très intéressant de poursuivre
cette étude en procédant par une optimisation horizontale et
verticale des positions d'échantillonnage, et par l'utilisation d'autres
techniques de spéciation comme le couplage de HPLC(Chromatographie
liquide haute performance) avec AAS ou avec ICP...etc. qui permettent
d'identifier les ions d'arsenic présents dans le milieu et leur
concentration, et ensuite on peut estimer la mobilitéet la
toxicitéde ce polluant.
36
Références
Références
[1] V Laperche, F Bodénan, MC Dictor, and Ph
Baranger. Guide méthodologique de l'arsenic,
appliquéà la gestion des sites et sols pollués.
Rapport BRGM RP-52066-FR, 2003.
[2] Mohammed Oubaaqua. Etude de
spéciation de métaux lourds ( Cu,Zn,Cd et Cr) dans les eaux
usées et sédiments de la zone d'epandage de marrakech.
thèse de doctorat, Facultédes sciences Semlalia Marrakech
UCA, 1997.
[3] Alain Manceau, Matthew A Marcus, and Nobumichi
Tamura. Quantitative speciation of heavy metals in soils and sediments
by synchrotron x-ray techniques. Reviews in Mineralogy and Geochemistry,
49(1) :341-428, 2002.
[4] Raoul Calvet. Le sol
propriétés et fonctions, volume 1. France Agricole Editions,
2003.
[5] JC Amiard, C Métayer, JP
Baud, and F Ribeyre. Influence de divers facteurs
écologiques sur la bioaccumulation d'éléments
métalliques (cd, cu, pb, zn) chez de jeunes palourdes (ruditapes
philippinarum) au cours du prégrossissement en nourri-cerie. Revue
des sciences de l'eau/Journal of Water Science, 4(4) :441-452, 1991.
[6] Mireille Harmelin-Vivien and Sandrine Ruitton.
Influence des apports anthropiques sur les flux de carbone et de
contaminants dans les réseaux trophiques de poissons de
l'écosystème à posidonia oceanica. OHM Littoral
méditerranéen, 2014.
[7] Sana EL Fadeli. Étude de
l'impact des éléments traces métalliques (Plomb et Fer)
sur l'environnement et la santéd'une population infantile vivant dans la
région de Marrakech. thèse de doctorat, Facultédes
sciences Semlalia Marrakech, 2013.
[8] Nadra Lekouch. Evaluation de
l'exposition de la population humaine aux éléments traces
métalliques dans la région de Marrakech Etude du risque saturnin.
thèse de doctorat, Facultédes sciences Semlalia Marrakech,
2004.
[9] Carole Delmas-Gadras. Influence des
conditions physico-chimiques sur la mobilitédu plomb et du zinc dans un
sol et un sédiment en domaine routier. thèse de doctorat,
Universitéde pau et de pays de l'adour U.F.R sciences, 2000.
[10] Axelle Leufroy. Spéciation
de l'arsenic dans les produits de la pêche par couplage HPLC/ICP-MS.
Estimation de sa bioaccessibilitéen ligne et applications à
d'autres éléments traces métalliques
d'intérêt. thèse de doctorat, L'Institut des Sciences
et Industries du Vivant et de l'environnement, 2012.
[11] Angélique Bossy. Origines de
l'arsenic dans les eaux, sols et sédiments du district aurifère
de St-Yrieix-la-Perche (Limousin, France) contribution du lessivage des phases
porteuses d'arsenic. thèse de doctorat, Universitéde
Limoges, 2010.
[12] PL Smedley and DG Kinniburgh. A review
of the source, behaviour and distribution of arsenic in natural waters.
Applied geochemistry, 17(5) :517-568, 2002.
37
Références
[13] S Garcia-Manyes, G Jiménez, A Padro,
Roser Rubio, and G Rauret. Arsenic speciation in contaminated soils.
Talanta, 58(1) :97-109, 2002.
[14] RJ Bowell, NH Morley, and VK Din.
Arsenic speciation in soil porewaters from the ashanti mine, ghana.
Applied Geochemistry, 9(1) :15-22, 1994.
[15] Karima Belloute. les déchets
solides et leurs impacts sur l'environnement de la ville marrakech(maroc).
mémoire de master, UCA - Facultédes Sciences et
Techniques-Marrakech, 2011.
[16] Bassai Magnoudéwa Bodjona.
Contamination du sol de la décharge finale de la ville de
Lome par les métaux lourds: cas du plomb, du cadmium, du nickel, du
cuivre et du zinc. thèse de doctorat, Universitéde Lome,
2014.
[17] Abderrazzak Baba Ahmed. Etude de
contamination et d'accumulation de quelques métaux lourds dans des
céréales, des légumes et des sols agricoles
irrigués
par des eaux usées de la ville de Hammam boughrara.
thèse de doctorat, UniversitéAbou bekr belkaid -
Tlemcen, 2012.
[18] Ana Fuentes, Mercedes Lloréns,
JoséSáez, Ma Isabel Aguilar, Juan F Ortuño, and Victor F
Meseguer. Comparative study of six different sludges by sequential
speciation of heavy metals. Bioresource technology, 99(3) :517-525,
2008.
[19] Abdellah Elazhari. Etude de la
contamination par les éléments traces métalliques des
sédiments de l'oued moulouya et de la retenue du barrage hassan ii en
aval de la mine abandonnée ze·ýda, haute moulouya.
mémoire de master, UniversitéCadi Ayyad Facultédes
Sciences et Techniques Marrakech, 2013.
[20] N Hafid and M Elhadek. Etude des
déchets urbains déposés dans une ancienne décharge
non contrôlée (study of municipal waste filed in uncontrolled
landfill). Mater environnement sciences, 5(S1) :2145-2150, 2014.
[21] M Giroux and P Audesse. Comparaison de
deux méthodes de détermination des teneurs en carbone organique,
en azote total et du rapport c/n de divers amendements organiques et engrais de
ferme. Agrosol, 15(2) :107-110, 2004.
[22] M Bisson, N Houeix, C Hulot, G Lacroix, JP
Lefevre, S Leveque, H Ma-gaud, and A Morin. Arsenic
et ses dérivés inorganiques. INERIS, fiches de données
toxicologiques et environnementales des substances chimiques, 2006.
[23] Eun Jung Kim, Jong-Chan Yoo, and Kitae Baek.
Arsenic speciation and bio-accessibility in arsenic-contaminated soils
: Sequential extraction and mineralogical investigation. Environmental
Pollution, 186 :29-35, 2014.
38
Annexes
39
Quelques minéraux riches en arsenic
[1]
|
Type
|
Présence
|
Exemple(nom,formule chimique)
|
|
Arséniate
|
Représente 60% minéraux riches en As
|
scorodite
|
FeAsO4.2H2O
|
|
pharmacosidérite
|
Fe4(AsO4)3(OH)3.6H2O
|
|
pharmacolite
|
CaHAsO4
|
|
Sulfure,
suldosel
|
Stabilitéen conditions réductrices. 20%
minéraux arséniés
|
arsénopyrite
|
FeAsS
|
|
orpiment
|
As2S3
|
|
réalgar
|
AsS
|
|
Arsénite
|
Stabilitéen conditions thermodynamique restreints
|
armangite
|
Mn3(AsO3)2
|
|
finnemanite
|
Pb5(AsO3)3Cl
|
|
Arséniure
|
Surtout métallurgie extractive
|
skutterudite
|
CoAs3
|
|
l·ollingite
|
FeAs2
|
|
Oxyde
|
Forte solubilité, forme principale de
commercialisation
|
claudetite arsenolite
|
As2O3 As2O3
|
Le cycle de l'arsenic
L'arsenic est un élément ubiquiste se trouve
dans tous les compartiments de l'environne-ment :
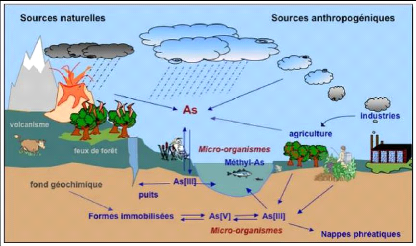
Cycle biogéochimique de l'arsenic[11]
| 

