|
|
UNIVERSITE DE DSCHANG
**********
ECOLE DOCTORALE
**********
UNITE DE FORMATION DOCTORALE
SCIENCES FONDAMENTALES
ET
TECHNOLOGIE
**********
|
|
UNIVERSITY OF DSCHANG
*********
POSTGRADUATE SCHOOL
**********
DOCTORAL TRAINING UNIT
FUNDAMENTAL SCIENCES
AND
TECHNOLOGY
**********
|
DEPARTEMENT DE CHIMIE
DEPARTMENT OF CHEMISTRY

LABORATOIRE DE CHIMIE DES NUISANCES ET DU GENIE DE
L'ENVIRONNEMENT
(LACHINGE)
ADSORPTION DU PARACETAMOL EN
SOLUTION AQUEUSE PAR LES CHARBONS
ACTIFS OBTENUS DES BALLES DE RIZ
MEMOIRE
Rédigé et
présenté en accomplissement partiel en vue de l'obtention
du
diplôme de « Master of Science (M.Sc) » en
Chimie
Option : Chimie Inorganique
Spécialité :
Chimie Analytique
Par :
BOPDA Aurélien
Matricule :
CM04-10SCI1094
Licencié en Chimie
Sous la direction de :

Année 2016
NCHE George NDIFOR-ANGWAFOR (PhD) (Chargé de
Cours)
DEDICACE

A Mes chers parents:
TEKOUTCHOP Joseph
&
MENKEU Anne épouse TEKOUTCHOP

REMERCIEMENTS
Ce travail a été initié et
réalisé au sein du Laboratoire de Chimie des Nuisances et du
Génie de l'Environnement (LACHINGE) de l'Université de
Dschang-Cameroun. Je voudrais remercier très chaleureusement:
Le Dr KAMGAING Théophile, responsable
dudit Laboratoire pour la confiance qu'il m'a accordée en m'accueillant
dans son laboratoire.
Le Pr KETCHA Joseph Mbadcam, responsable du
Laboratoire de Chimie Physique et Théorique (LCPT) de
l'Université de Yaoundé I, laboratoire dans lequel les
matériaux ont été carbonisés.
Le Dr NCHE George NDIFOR-ANGWAFOR, pour avoir
suivi de près et de bout en bout ce travail, pour tous ses conseils et
son apport dans la réalisation de ce travail. Je voudrai aussi le
remercier pour avoir accepté de m'initier à la recherche.
Le Pr NGOUELA Silvère Augustin, Chef
de Département de Chimie, pour sa générosité, son
dynamisme et ses conseils. Je le remercie vivement pour la bonne organisation
du département, qui a permis que ce travail soit réalisé
et soutenu dans les délais.
Les membres du jury pour avoir accepté malgré
leurs multiples occupations, de sacrifier un peu de leur temps pour se
réunir et examiner minutieusement ce travail;
Les enseignants de l'Université de Dschang (Cameroun)
en particulier ceux du département de Chimie pour leurs connaissances
transmises;
M. TCHUIFON Donald R, qui a fait preuve d'une
grande disponibilité et qui m'a aidé lorsque j'en avais besoin.
Merci pour son implication dans mon travail et pour toutes ses remarques
toujours pertinentes qui m'ont permis d'avancer;
Les aînés du laboratoire pour leurs conseils, et
leurs assistances dans la réalisation de ce travail;
Mes camarades de promotion pour leur esprit d'équipe,
de fraternité et pour les discussions scientifiques enrichissantes;
Ma famille, et plus particulièrement à mes
grands parents, mes frères et mes soeurs, ma profonde reconnaissance
pour leur patience et leur réconfort dans les moments de doutes et de
découragements, Je ne saurai passer sous silence l'apport inestimable
des autres membres de ma famille.
Le Dr NGUENA Landry, M. NKEUKOH
Francis, et M. NOUBOUDEM Ervis qui ne cessent de
m'encourager jour après jour et à qui j'exprime toute ma
gratitude et toute ma reconnaissance;
Maman TOUKEM Marceline à qui je dois
une profonde reconnaissance pour son affection et ses efforts consentis pour
mon épanouissement et ma réussite;
Mes chers amis que j'ai rencontrés lors de cette
aventure et avec qui j'ai passé de bons moments, autour d'un café
ou d'un repas. Je leur dis merci pour leur sincère camaraderie et pour
leur soutien durant la réalisation de ce travail;
A tous ceux dont les noms ne figurent pas dans ce document et
qui ont contribué d'une façon ou d'une autre à la
réalisation de cette thèse, qu'ils acceptent l'expression de ma
profonde gratitude.
iv
TABLE DES MATIERES
DEDICACE i
REMERCIEMENTS ii
TABLE DES MATIERES iiiv
LISTE DES TABLEAUX vii
LISTE DES FIGURES viii
LISTE DES ABREVIATIONS x
RESUME xii
ABSTRACT. xiii
INTRODUCTION GENERALE 1
CHAPITRE 1: ETUDE BIBLIOGRAPHIQUE 3
1.1. POLLUTION PAR LES PRODUITS PHARMACEUTIQUES 3
1.1.1. Définitions 3
1.1.2. Sources de contamination par les produits pharmaceutiques
3
1.1.3. Impacts environnementaux des produits pharmaceutiques 4
1.1.4. Cas du paracétamol 4
1.2. LES PROCEDES D'ELIMINATION DES PRODUITS PHARMACEUTIQUES
DANS L'ENVIRONNEMENT 6
1.2.1. Procédés chimiques 6
1.2.2. Procédés Physiques 7
1.3. PHENOMENE D'ADSORPTION 7
1.3.1. Adsorption physique 8
1.3.2. Adsorption chimique 8
1.3.3. Facteurs influençant sur le phénomène
d'adsorption 8
1.3.4. Mécanisme d'adsorption 8
1.3.6. Les isothermes d'adsorption 9
1.3.7. Modélisation de la cinétique d'adsorption
13
1.4. LES CHARBONS ACTIFS (CA) 14
1.4.1. Définitions et description 14
1.4.2. Préparation des charbons actifs 15

1.4.3. Types de charbons actifs 16
1.4.4. Propriétés d'un charbon actif 16
1.4.5. Utilisation des charbons actifs 18
CHAPITRE 2 : MATERIELS, REACTIFS ET METHODES 19
2.1. MATERIAUX 19
2.1.1. Collecte des balles de riz 19
2.1.2. Préparation du charbon actif 19
2.1.3. Méthodes de caractérisations des charbons
actifs 19
2.1.3.1. Spectroscopie IR-TF 19
2.1.3.2. Détermination du point de charge nulle (pHpzc)
20
2.1.3.3. pH du charbon 20
2.1.3.4. Densité de l'échantillon 21
2.1.3.5. Taux d'humidité 21
2.1.3.6. Surface spécifique 21
2.1.3.7. Indice d'iode 22
2.2. Réactifs chimiques et équipements 22
2.2.1. Réactifs chimiques 22
2.2.2. Equipements 23
2.2.3. Adsorption du paracétamol en solution aqueuse 23
2.2.3.1. Principe 23
2.2.3.2. Courbe d'étalonnage 24
2.2.3.3. Optimisation de l'adsorption en mode batch du
paracétamol 25
CHAPITRE 3 : RESULTATS ET DISCUSSIONS 27
3.1. CARACTERISATIONS DES MATERIAUX 27
3.1.1. Spectres IR 27
3.1.2. Densité apparente et taux d'humidité 29
3.1.3. Le pH au Point de charge nulle (pHpzc) et pH des charbons
actifs 29
3.1.4. Surface spécifique à l'acide acétique
30
3.1.5. Indice d'iode 31
3.2. ADSORPTION DU PARACETAMOL EN MODE BATCH 32
3.2.1. Influence du pH de la solution 32
3.2.2. Influence du temps de contact 33
3.2.3. Influence de la masse de l'adsorbant 34
3.2.4. Influence de la force ionique 35
3.2.5. Modèles cinétiques 36
vi
3.2.6. Isothermes d'adsorption 39
CONCLUSION ET PERSPECTIVES 44
REFERENCES BIBLIOGRAPHIQUES 46

LISTE DES TABLEAUX
Tableau 1.1: Propriétés physico-chimiques du
paracétamol . 6
Tableau 1.2: Classification de la forme des charbons
actifs. 16
Tableau 1.3 : Répartition des pores d'un adsorbant.
18
Tableau 2.1: Formules chimiques, noms, pureté et
origines des produits chimiques. 23
Tableau 3.1: Densité apparente et taux
d'humidité des différents charbons 29
Tableau 3.2: pH et pHpzc des différents CA. 30
Tableau 3.3: Paramètre de Langmuir et surface
spécifique à l'acide acétique 31
Tableau 3.4 : Valeurs de l'indice d'iode des deux charbons. 32
Tableau 3.5: Constantes de Vitesse et Coefficients de
Corrélation des Modèles
Cinétiques 38
Tableau 3.6: Constantes d'isothermes 42
Tableau 3.7: Comparaisons de la capacité d'adsorption du
paracétamol avec d'autres
adsorbants 42
LISTE DES FIGURES
Figure 1.1: Structure du Paracétamol 6
Figure 1.2: Structure du N-acétyl-para-benzoquinone imine
6
Figure 1.3: Schéma du mécanisme de transport
d'un adsorbat au sein d'un grain 9
Figure 1.4: Types d'isothermes d'équilibre
d'adsorption pour les systèmes gazeux
(Reungoat, 2007). 10
Figure 1.5 : Structure cristalline : (a) graphite ; (b)
charbon actif 17
Figure 2.1 : Mode opératoire de l'adsorption en mode
batch. 24
Figure 2.2: Droite de calibration du paracétamol en
solution aqueuse. 25
Figure 3.1: Spectre IR des balles de riz brutes. 27
Figure 3.2: Spectres superposés des charbons actifs
CANa1 et CANa2. 28
Figure 3.3: Evolution du pHfinal en fonction du
pHinitial. 30
Figure 3.4: Transformé linéaire de l'isotherme
de Langmuir. 31
Figure 3.5: Influence du pH sur l'adsorption du
paracétamol en solution aqueuse (concentration initial 100 mg/L,
température ambiante, t = 90 min v=150
tr/min, m=0,1g). 33
Figure 3.6 : Influence du temps de contact sur l'adsorption
du paracétamol (concentration initial 100 mg/L, température
ambiante, pH=2, v=150
tr/min, m=0,1g). 34
Figure 3.7: Influence de la masse de l'adsorbant sur
l'adsorption du paracétamol (concentration initial 100 ppm,
température ambiante, pH=2, v=150
tr/min). 35
Figure 3.8: Influence de la force ionique (température
ambiante, pH=2, temps = 100
minutes, v=150 tr/min, m=0,1g). 36
Figure 3.9: Formes linéaires du modèle du
pseudo-premier ordre pour CANa1 et
CANa2. 37
Figure 3.10 : Formes linéaires du modèle du
pseudo-second ordre pour CANa1 et CANa2...37
ix
Figure 3.11: Formes linéaires du modèle de
diffusion intra-particulaire pour CANa1 et
CANa2. 38
Figure 3.12 : Influence de la concentration initiale sur
l'adsorption du paracétamol en solution aqueuse (température
ambiante, pH=2, temps= 100 minutes,
v=150 tr/min, m=0,1g). 39
Figure 3.13 : Modèle linéaire de l'isotherme
Langmuir pour CANa1 et CANa2. 40
Figure 3.14: Modèle linéaire de l'isotherme
Freundlich pour CANa1 et CANa2. 40
Figure 3.15 : Modèle linéaire de l'isotherme
Temkin pour CANa1 et CANa2. 41
Figure 3.16: Modèle linéaire de l'isotherme
de Dubinin-Radushkevich pour CANa1et
CANa2. 41

LISTE DES ABREVIATIONS
AATE: Association Américaine pour le Travail et de
l'Eau
BET: Brunauer, Emmet et Teller
CA: Charbon actif
CAG: Charbon actif en grain
CANa1: Charbon Activé par le NaOH à
450°C
CANa2: Charbon Activé par le NaOH à
500°C
CAP: Charbon actif en poudre
CAT: Charbon actif en tissu
dapp: Densité apparente
IR-TF: Infra Rouge à Transformé de Fourier
OMS : Organisation Mondiale de la Santé
pHpzc: pH au point de charge nulle
POA: Procédés d'oxydation avancée
POC: Procédés d'oxydation classique
U.I.C.P.A: Union Internationale de Chimie Pure et
Appliquée
RTA : Réflexion Totale Atténuée
xi
RESUME
La pollution des eaux par des produits pharmaceutiques
nécessite le recours à des procédés de
dépollutions très performants. L'utilisation des charbons actifs
préparés à partir
des balles de riz, ont été utilisée pour
évaluer sa capacité à éliminer le
paracétamol en solution
aqueuse. Le présent travail a pour but
la préparation et caractérisation de deux charbons actifs, CANa1
et CANa2 à partir des balles de riz obtenus par pyrolyse
précédée d'une imprégnation au sodium hydroxyde.
Les charbons préparés présentent les
caractéristiques suivantes: les pH de charge nul sont de 6,80 pour CANa1
et 6,54 pour CANa2 et les pH des charbons CANa1 et CANa2 sont respectivement de
7,10 et 7,13. Les surfaces spécifiques 178,13m2/g pour CANa1
et 104,82 m2/g pour CANa2 sont obtenues à partir de
l'adsorption de l'acide acétique. La détermination de l'indice
d'iode nous a donné les valeurs respectives de 528,39mg/g et de 494,67
mg/g respectivement pour CANa1 et CANa2. L'élimination du
paracétamol en solution aqueuse par CANa1 et CANa2 a été
étudiée en mode batch. L'influence de plusieurs
paramètres, tels que le temps de contact, la masse du matériau,
le pH de la solution, la concentration initiale et la force ionique, a
été mise en évidence à température ambiante.
L'adsorption maximale a lieu à pH = 2 et un temps de contact de 100
minutes pour les deux adsorbants. La cinétique a été bien
décrite par le modèle du pseudo-second ordre et de diffusion
intraparticulaire. Le modèle de Langmuir décrit mieux
l'adsorption sur CANa1 tandis que le modèle de Freundlich décrit
mieux l'adsorption sur CANa2. Les quantités adsorbées obtenues
sont de 20,964 mg/g et 14,881 mg/g respectivement pour CANa1 et CANa2 par le
modèle de Langmuir. Selon les résultats obtenus, CANa1 et CANa2
peuvent constituer un support efficace pour l'élimination du
paracétamol des eaux usées industrielles.
Mots clés: paracétamol,
adsorption, charbon actif, solution aqueuse.
xii
ABSTRACT
Pollution of water by pharmaceutical products requires a
remedy. The use of the activated carbon prepared from rice husks was used to
evaluate the elimination capacity of paracetamol in aqueous solution. The
purpose of this work is the prepare and characterizate two activated carbons
CANa1 and CANa2 starting with rice husks obtained by a simple process of
pyrolysis preceded by impregnation with sodium hydroxide. The characterization
of the activated carbons showed that pH at zero charge was 6.80 for CANa1 and
6.54 for CANa2 and the pH of activated carbons CANa1 and CANa2 was 7.10 and
7.13 respectively The adsorption of the acetic acid enabled the specific
surface area of 178.13m2 /g for CANa1 and 104.82 m2/g for
CANa2 to be obtained. The determination of the iodine index gave us the
respective values of 528.39 mg/g and 494.67 mg/g for CANa1 and
CANa2respectively.The elimination of paracetamol in aqueous solution by CANa1
and CANa2 was studied in batch mode. The influence of several parameters, such
as contact time, mass of the adsorbent, pH of the solution, initial
concentration and ionic force, was highlighted at ambient temperature. Maximum
adsorption takes place at pH = 2 and a contact time of 100 minutes for the two
adsorbents. The kinetic was described by the model of the pseudo-second order
and diffusion intraparticulate. The model of Langmuir describes better
adsorption on CANa1 while the model of Freundlich describes better adsorption
on CANa2.The adsorbed quantities obtained are 20,964 mg/g and 14,881 mg/g
respectively for CANa1 and CANa2 through Langmuir model. According to results
obtained, CANa1 and CANa2 constitute an effective support for the elimination
of paracetamol in industrial waste water.
Key words: Paracetamol, adsorption, activated
carbon, aqueous solution.
1
INTRODUCTION GENERALE
Les activités anthropiques impactent les
écosystèmes depuis plusieurs décennies, posant de
réels problèmes environnementaux en ce qui concerne la
biodiversité et les ressources, notamment dans le milieu marin,
réceptacle final des polluants chimiques. Certaines substances, dites
émergentes parce que leur détection à l'état de
trace dans les écosystèmes est devenue possible par le
développement de nouvelles techniques analytiques, inquiètent
actuellement la communauté scientifique. Parmi ces contaminants
émergents, les composés pharmaceutiques humains et
vétérinaires deviennent une source de préoccupation
environnementale (Soufan, 2011).
Les produits pharmaceutiques sont principalement
retrouvés dans les eaux usées suite à leur
excrétion métabolique par l'homme et les animaux, notamment via
les effluents hospitaliers ou les effluents agricoles (médicaments
vétérinaires). De plus, les composés pharmaceutiques
assimilés par l'homme et les animaux peuvent se dégrader au sein
des organismes vivants, et les produits de dégradation issus de la
métabolisation, également excrétés dans les urines
ou les fèces, peuvent aussi être considérés comme
micropolluants. Certains d'entre eux sont considérés comme des
micropolluants cancérigènes, mutagène et dangereux
même lorsqu'ils existent sous forme de traces (Soufan, 2011). Parmi ces
produits, nous avons le paracétamol, molécule pharmaceutique la
plus consommée au monde, est aussi celle que l'on retrouve le plus dans
les milieux aquatiques et dans les effluents de stations d'épuration
urbaines (Velichkova, 2014), il se transforme en hydroquinone et benzoquinone
par l'intermédiaire du para-aminophénol qui selon les normes de
l'OMS est cancérigène, ce qui a motivé notre choix pour ce
polluant.
Il est donc nécessaire de procéder au traitement
des effluents chargés en paracétamol afin de minimiser leurs
impacts sur l'environnement et sur la santé. De ce fait,
différentes techniques ont été développées
ces dernières années pour l'élimination des produits
pharmaceutique dans l'eau. Les méthodes usuelles sont:
procédés d'oxydations classiques; procédés
d'oxydations avancées (Alvares et al., 2001); filtration sur
membrane (Robinson et al., 2001); l'adsorption (Fazal &
Rafique, 2012). L'adsorption par les charbons actifs s'est
avérée comme l'une des meilleures techniques (Fazal &
Rafique, 2012).
Le charbon actif est l'adsorbant le plus utilisé pour
l'adsorption avec grand succès grâce à sa grande surface
spécifique (500-2000 m2/g), sa structure microporeuse
(0,2-0,6 cm3/g) et sa capacité d'adsorption
élevée (Kafack, 2012). La biomasse utilisée dans le
cadre
2
de ce travail est la balle de riz, déchet solide
présent en abondance dans les usines de production de riz du Cameroun et
qui est le plus souvent brulée.
Dans l'optique de valoriser la biomasse camerounaise, nous
avons utilisé les charbons actifs préparés à partir
des balles de riz pour réduire la concentration du paracétamol
dans les eaux usées par le processus d'adsorption.
Il est question pour nous, d'évaluer à
température ambiante, la capacité des charbons actifs
préparés à partir des balles de riz à adsorber en
mode batch le paracétamol en solution aqueuse.
De manière spécifique, nous nous proposons de :
y' Préparer les charbons actifs à partir des balles
de riz et les caractériser ; y' Déterminer les paramètres
optimums d'adsorption comme le temps d'équilibre, la
concentration, la masse d'adsorbant, le pH et la force ionique;
y' Étudier l'influence de la concentration initiale de l'adsorbat ;
y' Étudier la cinétique d'adsorption du
paracétamol sur le charbon en utilisant les
modèles basés sur la composition du milieu;
y' Déterminer à température ambiante
l'isotherme appropriée à cette adsorption;
Pour mener à bien ce travail, nous procéderons
comme suit :
Le premier chapitre présentera une revue de la
littérature sur les produits pharmaceutiques et plus
particulièrement le paracétamol, ensuite les différents
procédés de traitement des eaux chargées en produits
pharmaceutiques, le phénomène d'adsorption et enfin les
généralités sur les charbons actifs. Le second chapitre
présente les matériaux, les réactifs et donne une
brève description des équipements et du mode opératoire
utilisés dans le cadre de ce travail. Le troisième chapitre est
consacré à la présentation et à la discussion des
résultats de caractérisation des adsorbants et des
résultats obtenus lors de l'adsorption du paracétamol en solution
aqueuse.
3
CHAPITRE 1: ETUDE BIBLIOGRAPHIQUE
1.1. POLLUTION PAR LES PRODUITS PHARMACEUTIQUES 1.1.1.
Définitions
Les micropolluants dans l'environnement aquatique sont un
problème majeur tant pour la population humaine, utilisatrice des
ressources en eau, que pour les écosystèmes aquatiques. Parmi les
micropolluants émergents, les produits pharmaceutiques ont attiré
l'attention depuis plusieurs années. Ce sont des substances actives qui
peuvent rester longtemps dans l'eau. De plus, de nombreuses recherches montrent
que différentes classes pharmaceutiques, telles que les antibiotiques,
les analgésiques, les anti-inflammatoires, les agents de contraste ou
les antiépileptiques, sont contenues dans les eaux (Soufan, 2011).
Les résidus de médicaments sont rejetés
en continu dans les eaux usées par l'utilisation domestique, les rejets
de l'industrie pharmaceutique, l'agriculture et les établissements de
santé. De plus, les composés pharmaceutiques assimilés par
l'homme et les animaux peuvent se dégrader au sein des organismes
vivants, et les produits de dégradation issus de la
métabolisation, également excrétés dans les urines
ou les excréments, peuvent aussi être considérés
comme micropolluants (Soufan, 2011). Les stations d'épuration, bien
qu'elles se soient significativement améliorées sur le plan
technique ces dernières années, n'ont pas été
conçues pour éliminer la totalité des molécules
pharmaceutiques. Par exemple, même si le paracétamol y est
dégradé à plus de 90 %, on en trouve des traces dans les
eaux usées remises en circulation, puis dans les eaux de surface (Kolpin
et al., 2002). Ces résidus de produits pharmaceutiques,
rejetés dans les eaux de surface, peuvent donc présenter un
risque environnemental non négligeable, de même que leurs
métabolites ou sous-produits de dégradation, parfois plus
dangereux que le produit d'origine (Soufan, 2011).
1.1.2. Sources de contamination par les produits
pharmaceutiques
Après usage, les produits pharmaceutiques sont
excrétés sous leur forme native ou sous forme de
métabolites et peuvent accéder aux systèmes aquatiques par
différentes voies. Ce sont les eaux usées municipales qui
constituent la source principale de contamination de l'environnement par les
médicaments à usage humain. Les effluents des hôpitaux et
de l'industrie pharmaceutique, ainsi que les lixiviats de décharge des
centres d'enfouissement représentent aussi une source non
négligeable. Ces produits pharmaceutiques sont plus ou
4
moins éliminés par les stations de traitement de
l'eau et se retrouvent dans les rivières, les lacs, les estuaires, voire
plus rarement dans les eaux souterraines et l'eau potable. L'épandage de
boues d'épuration contaminées peut également
entraîner la pollution des sols et des eaux de surface par ruissellement.
En outre, les produits pharmaceutiques à usage vétérinaire
peuvent entrer dans les systèmes aquatiques à travers
l'utilisation de fumier dans les champs, mais aussi directement en aquaculture
(Velichkova, 2014).
1.1.3. Impacts environnementaux des produits
pharmaceutiques
Au cours des dernières années, les
composés pharmaceutiques sont considérés comme des
contaminants de l'environnement. Les risques potentiels liés à la
présence de faibles concentrations de composés pharmaceutiques
dans l'environnement sont très mal connus. De même, les risques
possibles dus à la présence de telles substances en
mélange sont actuellement sous le débat (Soufan, 2011).
Toutefois, plusieurs études ont
révélé des cas de féminisation de certains poissons
dans les eaux douces et les milieux marins. Ces phénomènes de
perturbation du système endocrinien pourraient provenir de l'exposition
de certains poissons à des hormones de synthèse féminines
ou d'autres composés d'origine industrielle ou agricole présents
dans les milieux (Soufan, 2011).
1.1.4. Cas du paracétamol
Le paracétamol, dérivé de l'aniline, est
l'analgésique le plus couramment utilisé dans différentes
spécialités médicamenteuses pour le soulagement de la
fièvre, des maux de tête et de certaines douleurs mineures. Il est
transformé dans le foie en métabolites sulfates et
glucuroconjugués, et est excrété dans l'urine. C'est l'un
des médicaments les plus vendus au monde à la fois pour son effet
antalgique mais aussi pour son effet antipyrétique (Driad, 2009). Les
intoxications médicamenteuses restent un problème de santé
publique. Le paracétamol représenterait 4 à 12 % des
intoxications médicamenteuses volontaires. Sa fréquence est
d'autant plus élevée qu'il est accessible rapidement et
disponible sans ordonnance. Compte tenu de la banalisation de son usage, il
passe pour une molécule presque anodine, mais son surdosage demeure un
problème de santé publique important. Le problème
réside dans le fait qu'en cas de surdosage ou de prise volontaire
abusive, le paracétamol peut entraîner des lésions
hépatiques sévères voire mortelles (Driad, 2009). Si ses
effets toxiques à forte dose sont bien connus pour l'homme (dommages
hépatiques), les conséquences sur l'environnement de sa
présence sont moins bien renseignées (Driad, 2009).
5
Lorsqu'il se trouve en solution aqueuse, il est susceptible de
subir une hydrolyse pour former un produit de dégradation primaire:
« para-aminophénol », lui-même
susceptible de donner des produits de dégradation secondaires: «
quinone-imine ». La vitesse de dégradation du paracétamol
croît avec l'augmentation de la température et à la
lumière. Indépendamment de l'hydrolyse, la molécule de
paracétamol subit un autre type de décomposition par formation
d'une quinone-imine susceptible de se polymériser en donnant naissance
à des polymères azotés. Ces polymères et notamment
ceux de N-acétyl para-benzoquinone-imine ont été
décrits en outre comme étant les métabolites toxiques du
paracétamol, notamment cytotoxique et hémolytique. La
décomposition de ce métabolite en milieu aqueux est encore plus
complexe et donne naissance à de la para-benzoquinone et à de
l'hydroquinone (Driad, 2009).
Ils sont très toxiques et peuvent provoquer des
irritations locales. Leur absorption en grande quantité peut provoquer
des modifications neurologiques et musculaires. En expositions
répétées ou prolongées par voie orale, il agit sur
le système nerveux central des hommes et des animaux et provoque une
irritation du pré-estomac, entraine des lésions rénales
sévères et hépatiques (Suresh et al., 2012).
L'utilisation abusive de ce produit en moyenne pendant quatre ans, à des
concentrations élevées sur la peau provoque une pigmentation
inégale de la peau avec des parties complètements
décolorées ou au contraire d'aspect plus sombre, des dermatites
irritantes, des sensations de brulures ou de piqûres (Suresh et
al., 2012).
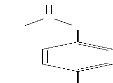
? Structures et propriétés
physico-chimiques
Le paracétamol se présente sous la forme d'une
poudre cristalline blanche, inodore et de goût amer. C'est un acide
faible, ce caractère a pour conséquence qu'il se trouve
essentiellement sous forme ionisée dans l'estomac et l'intestin
grêle, condition favorable à son absorption à ce niveau. Sa
structure, la structure du N-acétyl-para-benzoquinone imine et les
propriétés physico-chimiques du paracétamol sont
données respectivement par la figure 1.1, figure 1.2 et le tableau 1.1
ci-après.
H3C
O
C
NH
OH
Figure 1.1: Structure du
Paracétamol
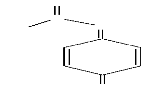
N
H3C
O
C
O
6
Figure 1.2 : structure du
N-acétyl-para-benzoquinone imine
Tableau 1.1: Propriétés
physico-chimiques du paracétamol (Driad, 2009).
|
Paracétamol
|
|
Formule brute
|
C8H9NO2
|
|
Nom scientifique
|
N-acétyl-para-animophénol
|
|
Masse molaire
|
151,2 g/mol
|
|
Point de fusion
|
168-172°C
|
|
PKa
|
9,5
|
|
Solubilité (eau)
|
14 g/L à 20 °C
|
|
Solubilité (alcool)
|
facilement soluble
|
1.2. LES PROCEDES D'ELIMINATION DES PRODUITS
PHARMACEUTIQUES DANS L'ENVIRONNEMENT
1.2.1. Procédés chimiques
? Les procédés d'oxydation classique
(POC)
Les techniques d'oxydation classique utilisent des oxydants
puissants tels que le sodium hypochlorite (NaOCl), l'ozone (O3), l'eau
oxygénée (H2O2) pour dégrader les polluants en solution.
Ces méthodes de traitement sont couramment utilisées pour
l'épuration d'effluents contenant des polluants organiques et
inorganiques en raison de leur mise en oeuvre relativement facile. Cependant,
certains de ces oxydants (le sodium hypochlorite en particulier) sont de moins
en moins utilisés dans ces processus en raison des effets
négatifs qu'ils induisent. Ils réagissent avec les polluants en
solution et donnent lieu à la formation d'amines aromatiques et
d'organochlorés qui sont des composés cancérigènes
(Alvares et al., 2001).
7
? Les procédés d'oxydation avancée
(POA)
Les POA regroupent des méthodes chimiques,
photochimiques ou électrochimiques. Leur développement est en
plein essor depuis environ trois décennies. Elles consistent à
dégrader les molécules de colorants en CO2 et H2O au moyen de
l'UV en présence de peroxyde d'hydrogène. Ces POA regroupent les
technologies qui cherchent à dégrader les polluants organiques
par oxydation via des procédés photocatalytiques susceptibles de
développer des radicaux hydroxyles (
·OH) dont le
pouvoir oxydant est nettement supérieur à celui des oxydants
traditionnels. En plus des systèmes UV-peroxyde, UV-Ozone et du
processus Photo-Fenton, qui a largement démontré leur
efficacité dans l'oxydation des composés organiques, la
photocatalyse hétérogène a aussi émergé
depuis quelques années (Alvares et al., 2001). 1.2.2.
Procédés Physiques
? La filtration sur membrane
Les procédés membranaires sont des techniques de
séparation qui se font à travers une membrane, sous l'action d'un
gradient de pression. La séparation se fait en fonction des tailles
moléculaires des composés, mais aussi en fonction de leur forme
et leur structure (Robinson et al., 2001). Dans ce
procédé, les polluants sont retenus par une membrane
semi-perméable dont le diamètre des pores est inférieur
à celui des molécules à éliminer. Ce
procédé nécessite un post-traitement, ce qui augmente
considérablement le coût de dépollution (Robinson et
al., 2001).
? L'adsorption
L'adsorption est un procédé de traitement, pour
éliminer une très grande diversité de composés
toxiques dans notre environnement. Elle est essentiellement utilisée
pour le traitement de l'eau et de l'air. Au cours de ce processus les
molécules d'un fluide (gaz ou liquide), appelé adsorbat, viennent
se fixer sur la surface d'un solide, appelé adsorbant. Ce
procédé définit la propriété de certains
matériaux de fixer à leur surface des molécules (gaz, ions
métalliques, molécules organiques, etc.) d'une manière
plus ou moins réversible. Au cours de ce processus, il y aura donc un
transfert de matière de la phase aqueuse ou gazeuse vers la surface
solide (Mechrafi, 2002).
1.3. PHENOMENE D'ADSORPTION
L'adsorption est un processus au cours du quelle les
molécules d'un fluide (gaz ou liquide), appelé adsorbat, viennent
se fixer sur la surface d'un solide, appelé adsorbant. Selon
8
la nature des liaisons formées ainsi que la
quantité d'énergie dégagée lors de la
rétention d'une molécule à la surface d'un solide
permettent de distinguer deux types d'adsorption: adsorption physique et
adsorption chimique (Mechrafi, 2002).
1.3.1. Adsorption physique
L'adsorption physique est un phénomène
réversible qui résulte de l'attraction entre les molécules
d'adsorbant composant la surface du solide et les molécules du
soluté de la phase fluide, ces forces attractives sont de nature
physique, comprenant les forces dites de Van der Waals et correspondent
à des énergies faibles qui sont de l'ordre de 5 et 40 kJ/mol. Ce
phénomène consiste essentiellement dans la condensation de
molécules sur la surface du solide et il est favorisé en
conséquence par un abaissement de la température (Mechrafi,
2002).
1.3.2. Adsorption chimique
Les énergies de liaison mises en jeu sont de l'ordre de
40 kJ.mol-1 et plus. C'est un phénomène qui, par sa
spécificité, son énergie d'activation et sa chaleur
dégagée, s'apparente à une réaction chimique entre
molécule en solution et la surface du support. Il y a formation de
fortes liaisons entre adsorbat et adsorbant (covalent par exemple). La couche
adsorbée est au mieux monomoléculaire. Ce phénomène
est plus lent que la physisorption et nécessite une énergie
d'activation (Mechrafi, 2002).
1.3.3. Facteurs influençant sur le
phénomène d'adsorption
L'équilibre d'adsorption, entre un adsorbant et un
adsorbât dépend de nombreux facteurs dont les principaux sont
(Creangã, 2007):
? les caractéristiques de d'adsorbant: polarité,
volume poreux, surface spécifique, fonctions superficielles ;
? les caractéristiques de l'absorbât:
polarité, solubilité, poids et saturation moléculaire ; ?
les paramètres physiques: température, pH.
1.3.4. Mécanisme d'adsorption
Au cours de l'adsorption d'une espèce sur un solide, le
transfert de masse des molécules se fait de la phase fluide vers le
centre de l'adsorbant. Ce processus s'opère au sein d'un grain
d'adsorbant en plusieurs étapes. La figure 1.2 ci-après
représente le mécanisme de transport d'un adsorbat.
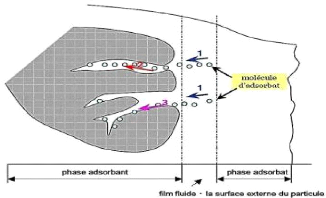
9
Figure 1.3: Schéma du
mécanisme de transport d'un adsorbat au sein d'un grain
( Creangã, 2007).
1. Diffusion externe: Elle correspond au transfert du
soluté (molécules de la phase liquide) du sein de la solution
à la surface externe des particules. Le transfert de la matière
externe dépend des conditions hydrodynamiques de l'écoulement
d'un fluide dans un lit d'adsorbant.
2. Diffusion interne dans la structure poreuse du solide: Les
particules de fluides pénètrent à l'intérieur des
pores. Elle dépend du gradient de concentration du soluté.
3. Diffusion de surface. Elle correspond à la fixation
des molécules sur la surface des pores.
1.3.6. Les isothermes d'adsorption
Une isotherme d'équilibre d'adsorption est la courbe
caractéristique, à une température donnée, de la
quantité de molécules adsorbées par unité de masse
d'adsorbant en fonction de la pression ou de la concentration en phase fluide.
On distingue cinq catégories d'isothermes d'adsorption qui sont : le
type I, II, III, IV, V.
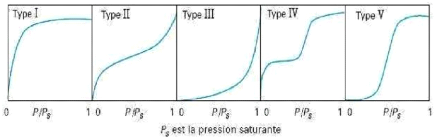
10
Figure 1.4: Types d'isothermes
d'équilibre d'adsorption pour les systèmes gazeux (Reungoat,
2007).
y' Isotherme de type I: l'adsorption est favorable sur des
solides microporeux. La quantité adsorbée est importante
même pour de faibles concentrations de polluant.
y' Isothermes de type II et III: elles sont
généralement observées pour des adsorbants ayant une large
gamme de tailles de pores. Pour chaque système, il y a une progression
continue avec la création de multicouches d'adsorption.
y' Isothermes de type IV et V: dans ces cas, l'adsorption est
dite défavorable puisqu'il faut une forte concentration de
composé en phase liquide pour avoir des quantités
adsorbées importantes.
Plusieurs auteurs ont élaboré des formulations
mathématiques modélisant les isothermes d'adsorption:
+ Isotherme de Langmuir
L'isotherme de Langmuir, fut la première à
être développée en 1918. Elle est définie par une
capacité maximale d'adsorption qui est liée à la
couverture des sites de surface par une monocouche. Les hypothèses de ce
modèle sont les suivantes:
> Les sites d'adsorption sur la surface solide sont
homogènes d'un point de vue énergétique: on parle de
« surface d'adsorption homogène » ;
> Chacun de ces sites peut adsorber une seule molécule,
et l'adsorption est monocouche; > Chacun des sites à la même
affinité pour les molécules en solution;
> Il n'y a pas d'interactions entre les molécules
adsorbées.
L''isotherme est donnée par :
(1.1)
Cette équation est généralement
utilisée sous la forme linéaire:
11
(1.2)
Où Ce est la concentration
de l'adsorbat à l'équilibre dans la solution (mg/g),
Qm est la capacité d'adsorption
maximale de l'adsorbant (mg/g) et K est la constante de
Langmuir. Les constantes Qm et KL
sont calculées de la pente et de l'interception du tracé
linéaire de Ce/Qe en fonction de
Ce. On observera qu'une faible valeur de KL
correspond à une forte affinité du soluté pour la
surface de l'adsorbant (Ajifack et al,. 2014).
? L'isotherme de Freundlich
Le modèle de Freundlich est semi-empirique. Il est
basé sur l'hypothèse d'une surface
hétérogène de l'adsorbant, avec une distribution
exponentielle des sites actifs en fonction des énergies d'adsorption.
L'expression non linéaire de cette isotherme est:
(1.3)
Où Kf est le paramètre relatif à la
capacité d'adsorption (mg1 -1/n L1/n g-1). Sa forme linéaire
obtenue par la transformation logarithmique est :
(1.4)
Cette équation permet de déterminer les constantes
empiriques (Kf et 1/n) de ce modèle.
Partant du tracé linéaire lnQe = f(lnCe), on
pourra déterminer les constantes de Freundlich (Kf et 1/n). Quand la
valeur de 1 /n est inférieure à l'unité, l'adsorption est
favorable ; au contraire quand la valeur 1/n est supérieure à
l'unité, cela indique une adsorption défavorable. La constante Kf
représente l'affinité du solide pour les composés (Ajifack
et al., 2014).
? L'isotherme de Dubinin-Radushkevich
Le modèle de Dubinin-Radushkevich fut
développé pour l'adsorption des substances à l'état
de traces en phase aqueuse sur les solides poreux. Ce modèle est plus
général que celui de Langmuir car il est applicable même
pour les sites d'adsorption non homogènes (Sousa et al., 2012).
Cette isotherme est donnée par l'équation (1.5)
(1.5)
Où Qe est la quantité de
molécules adsorbées par unité de masse d'adsorbant en mg/g
; Qmla capacité maximale d'adsorption de l'adsorbant par
unité de masse mg/g; å le potentiel de Polanyi (kJ/mol), son
expression est donnée par l'équation (1.6)
å= RT ln (1+1/Ce) (1.6)
12
R constante des gaz parfaits (R = 8,314 x 10-3
kJ/mol/K); T la température absolue (T= 298 K) et K': constante
liée à l'énergie d'adsorption
(mol2K/J2).
La forme linéaire de l'isotherme de D-R est donnée
comme suit :
(1.7)
K' est obtenue à partir de la pente du tracé de
lnQe en fonction de å2, et l'énergie moyenne
d'adsorption E (KJ/mol) peut être obtenue à partir de la valeur de
K' en employant l'équation suivante (Sousa et al., 2012):
E = (-2K')-1/2 (1.8)
Les valeurs de l'énergie d'adsorption (E) trouvées
à partir de cette isotherme permettront d'élucider le processus
ou le mécanisme mis en jeu au cours du processus d'adsorption.
E< 8 kJ/mol, la physisorption domine le processus
d'adsorption;
E située entre 8 et 16 kJ/mol, l'échange ionique
est le processus dominant;
E > 16 kJ/mol, l'adsorption est dominée par la
diffusion intra particulaire.
? L'isotherme de Temkin
La dérivation de l'isotherme de Temkin suppose que
l'abaissement de la chaleur d'adsorption est linéaire plutôt que
logarithmique, comme appliqué dans l'équation de Freundlich
(Sousa et al., 2012). L'isotherme de Temkin est donnée par
l'équation
(1.9)
La forme linéaire de ce modèle est donnée
par
(1.10)
Où B= est la Constante de Temkin relative à
l'énergie d'adsorption (KJ/mol); A est la
Constante d'isotherme de Temkin (L/mg) ; R
est la Constante des gaz parfaits (8,314J/mol.K);
Ce la concentration à l'équilibre des
adsorbants (mg/L); Qe est la quantité
adsorbée à l'équilibre en (mg/g); T la
température absolue (K) et ?Q (en kJ/mol) est la
variation de la chaleur d'adsorption.
Le tracé linéaire Qe =
f(lnCe) permet de déterminer la pente B = , puis en
introduisant une valeur de Qm (par exemple issue du
modèle de Langmuir), on peut aisément déterminer la
variation de la chaleur d'adsorption ?Q.
13
1.3.7. Modélisation de la cinétique
d'adsorption
La cinétique d'adsorption donne la vitesse du
processus, le temps minimum nécessaire pour que l'adsorption soit
complète et les informations sur l'étape limitante du processus.
Nous ne décrivons ici ceux qui seront utilisé dans notre
étude.
? Modèle de pseudo-premier ordre
Dans ce modèle la vitesse d'adsorption à
l'instant t est proportionnelle à la différence entre la
quantité adsorbée à l'équilibre Qe et la
quantité Qt adsorbée à cet instant et que l'adsorption est
réversible. La constante de vitesse d'adsorption du premier ordre est
déduite à partir du modèle établi par Lagergren. La
loi de vitesse s'écrit (Ho, 2004):
(Qe-Qt) (1.11)
Où Qe et
Qt sont respectivement les capacités d'adsorption à
l'instant t (min) et à l'équilibre (mg/g) et k1 la Constantes de
vitesse d'adsorption pour le premier ordre (min-1).
L'intégration de l'équation (1.10) donne :
(1.12)
.
La valeur de k1 sera déterminée à partir du
tracé linéaire ? Modèle de la cinétique du
pseudo- second ordre
L'équation du pseudo-second ordre est souvent
utilisée avec succès pour décrire la cinétique de
la réaction de fixation des polluants sur l'adsorbant. Le modèle
du pseudo-second ordre permet de caractériser la cinétique
d'adsorption en prenant en compte à la fois le cas d'une fixation rapide
des solutés sur les sites les plus réactifs et celui d'une
fixation lente sur les sites d'énergie faible:
(1.13)
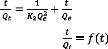
Où k2 (g mg-1 min-1)
représente la constante de vitesse pour une cinétique du
deuxième ordre. L'intégration de l'équation ci-dessus
donne:
. (1.14)
Le tracé de la courbe permet de déterminer la
vitesse initiale (ho)
d'adsorption qui est donnée par (Ho & Mckay, 1999): h0
= K2Qe2
14
? Modèle de diffusion
intraparticulaire
L'équation du modèle de diffusion intraparticulaire
est donné par :
P= kidta . (1.16)
La transformée linéaire de cette équation
donne :
(1.17)
Où P est le pourcentage
d'élimination; kid est la constante de
vitesse de diffusion intraparticulaire ou le facteur de vitesse c'est-à-
dire le pourcentage d'adsorbat par unité de temps (min-1);
a est le gradient de la droite et dépend du
mécanisme d'adsorption
Le tracé de lnP = f (lnt) donne une droite si ce
modèle est applicable et permet de déduire a et kid
qui sont respectivement la pente et l'ordonnée à l'origine.
L'étape limitante est la diffusion des molécules à
l'intérieur des pores de l'adsorbant. Lorsque a tend vers 0
l'agglomération à la surface est importante et lorsque a tend
vers 1, l'agglomération y est faible (Zora et al., 2006).
1.4. LES CHARBONS ACTIFS (CA)
Le Charbon actif est l'un des premiers matériaux
adsorbants utilisés à grande échelle. L'utilisation du
charbon remonte à l'antiquité. Dans le cadre de la
médecine et du traitement des odeurs, les propriétés
d'adsorption des charbons actifs ont été utilisées par les
Égyptiens dans les années 1550 avant Jésus Christ. Un
siècle plus tard, les Phéniciens furent les précurseurs de
son utilisation pour rendre l'eau potable (Tatianne, 2011).
1.4.1. Définitions et description
Les charbons actifs sont les matériaux carbonés
amorphes préparés de manière à exhiber un
degré de porosité élevé et une surface intra
particulaire étendue (Pope, 2001). Ceux-ci ont une
caractéristique essentielle qu'est l'existence d'un réseau
très développé de micropore, lesquels sont à
l'origine de leur pouvoir adsorbant très important. Par
conséquent, ces derniers constituent les adsorbants les plus
fabriqués et les plus utilisés industriellement. Les pores bien
répartis de façon non uniforme à la surface de ces
derniers d'où l'existence d'un important majoritaire des micropores et
mésopores.
Les CA ont des structures moléculaires et cristallines
semblables à celles du graphite. La structure graphitique est
hexagonale, les atomes de carbone s'y trouvant sont distants d'environ 1,45
Å, formant des plans parallèles les uns aux autres. Le charbon
actif est un
15
carbone microporeux inerte qui a subi un traitement permettant
l'augmentation de sa surface interne. Il possède ainsi une très
grande surface spécifique pouvant aller de 200 à 2000
m2/g de charbon actif. Ce qui lui permet ainsi d'avoir une grande
capacité d'adsorption (Zue, 2012).
1.4.2. Préparation des charbons actifs
Le charbon actif est un produit adsorbant obtenu à
partir des matières premières riches en carbone (le bois, la
tourbe, le lignite, l'écorce de coco...). Une fois ces matières
premières sélectionnées, elles sont activées
physiquement ou chimiquement dans des fours d'activation. Par cette activation
on obtient, une structure de carbone hautement poreuse et très active.
La préparation des charbons activés s'effectue en deux grandes
étapes : la carbonisation et l'activation (Slasli, 2002).
? La carbonisation
La carbonisation ou pyrolyse est la décomposition
thermique d'un matériau organique sous vide ou sous atmosphère
inerte à des températures comprises entre 400 et 1000°C. Les
hétéroatomes (oxygène et hydrogène) sont
éliminés sous l'effet de la chaleur et le matériau devient
plus riche en carbone. Les atomes de carbone restants se regroupent en
feuillets aromatiques possédant une certaine structure planaire. Ces
feuillets s'arrangent ensuite d'une manière irrégulière
laissant ainsi des interstices entre eux. Ces interstices donnent naissance
à une porosité primaire du produit carbonisé. Le produit
obtenu par la pyrolyse ne possède qu'une porosité rudimentaire et
ne peut pas être employé comme adsorbant sans une activation
supplémentaire (Slasli, 2002).
? Activation
L'activation consiste à développer la structure
poreuse en éliminant les goudrons qui obstruent les pores, et à
créer des fonctions de surface (généralement
oxydées) qui sont à l'origine des interactions entre le solide et
les molécules adsorbées. Elle peut être physique ou
chimique.
? L'activation physique
Elle comporte deux étapes successives sous
atmosphère contrôlée. Après séchage des
matières premières à 170°C environ, une carbonisation
est effectuée afin d'éliminer les matières volatiles dans
le squelette carboné du matériau. Cette étape se fait
à 600-700°C
16
durant 6 à 8 heures. La seconde étape est
l'activation qui permet de développer les pores existants et d'en
créer d'autres Cette opération se fait à 800-1000°C,
en présence d'un gaz faiblement oxydant (air), du dioxyde de carbone, de
l'oxygène et/ou de la vapeur d'eau, durant 24 à 72 heures
(Cherraye, 2012).
? L'activation chimique
Elle consiste à imprégner le matériau de
départ avec une solution concentrée d'agent très oxydant
et/ou déshydratant (acide phosphorique, chlorure de zinc...). Le
matériau subit ensuite une pyrolyse entre 400°C et 600°C
à l'abri de l'air, puis est lavé et séché. Le
charbon actif est ainsi obtenu en une seule étape. C'est le degré
d'imprégnation du matériau en matière oxydante qui
définit la structure poreuse finale (Slasli, 2002).
1.4.3. Types de charbons actifs
Le CA peut se présenter sous trois formes: en poudre
(CAP), en grain (CAG) et en tissus (CAT), dont chacun présente ses
avantages suivant l'application visée. Le tableau 1.2 ci-apres classifie
la forme des charbons actifs.
Tableau 1.2: Classification de la forme des
charbons actifs (Kafack, 2012).
|
Type de CA
|
Granulométrie
|
Avantages
|
Inconvénients
|
Applications
|
|
CAP
|
Inférieure à
0,2mm
|
Bonne capacité
d'adsorption
Recyclable
|
Quantité Importante
|
Traitement en
phases gazeuse et
aqueuse
|
|
CAG
|
Inférieure à 0,4mm
|
Bon filtre
|
|
Traitement en phase
aqueuse
|
|
CAT s
|
|
meilleure
cinétique
|
risques de déchirement
pour des débits
de
fluides élevés
|
Traitement en phase
Gazeuse
|
1.4.4. Propriétés d'un charbon actif ?
Structure d'un charbon actif
La structure cristalline du CA consiste en un assemblage de
couches planes d'atomes de carbone, ordonnés en hexagones
réguliers, comparables aux cycles aromatiques (figure 1.3 ci-dessous).
L'analyse de diffraction aux rayons X, révèle que sa structure
est graphitique, mais
17
avec quelques différences, notamment par la disposition
de ces couches planes d'atomes de carbone en un agencement
désordonné et par la formation de groupements fonctionnels, dus
à la présence d'hétéroatomes (oxygène,
azote, soufre) dans le réseau cristallin. Ces modifications de structure
pourront engendrer des interactions spécifiques (groupements
fonctionnels) et des interactions non spécifiques
(hétérogénéité du réseau) pour cet
adsorbant (Zue, 2012).
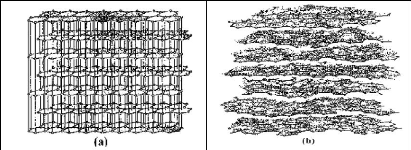
Figure 1.5 : Structure cristalline : (a)
graphite ; (b) charbon actif ? Surface spécifique d'un
charbon actif
Par définition, la surface spécifique d'un
adsorbant est sa surface par unité de masse. Elle est
généralement exprimée en m2/g. Son estimation
est conventionnellement fondée sur des mesures de la capacité
d'adsorption (Qm) de l'adsorbant en question, correspondant à
un adsorbat donné. La molécule adsorbée doit avoir une
surface connue et acceptable. Il suffit à cet effet, de
déterminer la valeur de la capacité de la monocouche à
partir de l'isotherme d'adsorption (Zue, 2012).
? Les tailles des pores et le volume poreux d'un charbon
actif
La classification des pores adoptée actuellement par
l'Union Internationale de Chimie Pure et Appliquée (U.I.C.P.A.) est
fondée sur leurs tailles. Trois catégories de pores ont
été définies dans le Tableau (1.3).
18
Tableau 1.3 : Répartition des pores
d'un adsorbant.
|
Désignation
|
Rayon moyen de
pores (nm)
|
Volume poreux
(Cm3/g)
|
Surface
spécifique
(m2/g)
|
|
Micropores
|
< 2
|
0,2 - 0,6
|
400 - 900
|
|
Mésopores
|
2 - 50
|
0,02 - 0,1
|
20 - 70
|
|
Macropores
|
> 50
|
0,2 - 0,8
|
0,5 - 2
|
1.4.5. Utilisation des charbons actifs
Les CA sont des matériaux poreux couramment
utilisés depuis des siècles dans de nombreuses applications
domestiques et industrielles (purification de l'air, dépollution des
effluents domestiques et industriels, purification ou décoloration de
produits agroalimentaires). Ces applications ont considérablement
évolué, et aujourd'hui, les CA peuvent même être
utilisés comme support de catalyse (Tatianne, 2011).
19
CHAPITRE 2 : MATERIELS, REACTIFS ET METHODES
Ce chapitre présente brièvement les
différents matériaux, les réactifs chimiques, les
équipements ainsi que la description du mode opératoire du
processus d'adsorption du paracétamol en solution aqueuse.
2.1. MATERIAUX
2.1.1. Collecte des balles de riz
Les balles de riz utilisées dans le cadre de ce travail
ont été récoltées dans l'Arrondissent de Ndop
(région du Nord-Ouest Cameroun). Les balles de riz ont été
lavées à l'eau du robinet, ensuite rincées à l'eau
distillée pour éliminer les impuretés et enfin
séchées à température ambiante pendant 72
heures.
2.1.2. Préparation du charbon actif
Apres la récolte les balles de riz lavées ont
été imprégnées. Pour la réaliser, une masse
de 20 g de biomasse a été mise en contact avec 35 mL d'une
solution de NaOH 1M. L'activation a duré 30 minutes et a
précédé le séchage à 105°C pendant 24
heures.
Les échantillons de balles de riz
imprégnés ont été carbonisés pendant 1heure
à 450°C et 500°C à la vitesse de chauffage de
5°C/min en utilisant un four de marque ISUNU. Les produits de la
calcination obtenus ont été lavés à l'eau
distillée dans l'optique d'éliminer l'excès de NaOH ainsi
qu'augmenter la pureté des CA. Les différents charbons actifs
ainsi obtenus, dénommés: CANa1(500°C)et CANa2(450°C),
ont été séchés pendant 24 heures à la
température de 105°C, puis broyés et tamisés pour
obtenir les particules de taille inférieure à 100 um.
2.1.3. Méthodes de caractérisations des
charbons actifs 2.1.3.1. Spectroscopie IR-TF
Les différents charbons actifs ont été
caractérisés par spectroscopie Infra-Rouge à Transformer
de Fourrier (IR-TF). Cette technique permet d'identifier les fonctions
chimiques de surface (Kifuani et al., 2012). C'est une technique
analytique présentant trois principaux avantages à savoir sa
rapidité, sa non destructivité et l'utilisation de faible
quantité d'échantillon.
La méthode de Réflexion Totale
Atténuée (RTA) utilisée dans le cadre de ce travail est
une technique de réflexion dans laquelle on utilise un dispositif
optique de réflexion interne
20
possédant un indice de réfraction
élevé (diamant, germanium, silicium, etc) pour les mesures
(Kenne, 2011). Dans ces travaux, l'enregistrement a été
effectué par le biais d'un spectrophotomètre JR de marque Alpha-P
de la firme Brüker. Dans la pratique, quelques milligrammes de
matériau ont été déposés sur la surface de
mesure constituée d'un cristal RTA diamant. Après avoir recouvert
l'échantillon grâce à un dispositif intégré
à l'appareil, l'enregistrement du spectre a été
directement réalisé.
2.1.3.2. Détermination du point de charge nulle
(pHpzc)
Le pHpzc correspond à la valeur de pH pour lequel la
charge globale de la surface du charbon actif est nulle. Ce paramètre
est très important dans les phénomènes d'adsorption,
notamment lorsque les forces électrostatiques sont impliquées
dans les mécanismes. Le pHpzc permet de déterminer le
caractère acide ou basique d'un charbon et de connaître, selon le
pH de la solution, la charge de surface nette du matériau. Ainsi,
lorsque le pHpzc > pH, la surface du CA est chargée positivement, et
lorsque pHpzc < pH la surface du CA est chargée négativement
(Tatianne, 2011). Il a été déterminé pour
l'ensemble des charbons actifs en utilisant la méthode décrite
par Lopez-Ramon et al., (1999).
Afin de connaitre la charge globale du matériau en
fonction du pH de la solution, des solutions de NaCl 0,1M dont les pH ont
été ajustés entre 2et 10 (par ajout d'une solution aqueuse
de NaOH ou de HCl de concentrations10-3M) ont été
préparées à l'avance. Le pH-mètre (pHS-25) a
été utilisé pour la mesure du pH. Le charbon actif sec
(0,1 g) est mis en contact avec 20 mL de chacune des solutions
précédentes contenues dans des flacons. Les suspensions ont
été ensuite agitées sur une table d'agitation pendant 24
heures à température ambiante. Chaque mélange a
été ensuite filtré à l'aide d'un papier filtre
(Whatman) et une nouvelle mesure du pH est effectuée. Le tracé de
la courbe pHfinal = f (pHinitial) permet de déterminer le
pHpzc (intersection entre cette courbe et la première bissectrice).
2.1.3.3. pH du charbon
Nous avons eu à déterminer le pH de la surface
du charbon en mettant en contact 50 mL d'eau distillée, et 0,5 g de
charbon actif dans un flacon suivi de l'agitation du mélange pendant
24heures à température ambiante. Après ce temps, les
suspensions ont été filtrées et le pH du filtrat a
été déterminé (Tchuifon et al,. 2014a).
2.1.3.4. Densité de
l'échantillon
La densité apparente des charbons a ont
été déterminée en remplissant un flacon de 15mL par
charbon actif jusqu'à la marque, le flacon étant
préalablement pesé. La nouvelle masse obtenue nous a permis de
calculer la différence de masse (Tchuifon et al,. 2014a). La
densité apparente se trouve à l'aide de l'équation
suivante :
(2.1)
2.1.3.5. Taux d'humidité
C'est le rapport, exprimé en pourcentage de la
quantité d'eau contenue dans l'adsorbant, au poids du même
matériau sec. Dans un bécher préalablement pesé,
nous avons introduit 1g de CA. Après avoir séché le
bécher contenant le CA à l'étuve à 110°C
pendant une heure, il a été refroidi dans un dessiccateur.
Après refroidissement, le bécher a été pesé
de nouveau. Le pourcentage de masse obtenu à partir de la
différence de masse nous a donné le taux d'humidité dans
l'échantillon (Tchuifon et al,. 2014a).
(2.2)
Où mi la masse initiale, mf la masse finale et T le taux
d'humidité. 2.1.3.6. Surface spécifique
La surface spécifique d'un adsorbant est sa surface par
unité de masse. Lorsque l'isotherme est exploitée avec
l'équation de BET (Brunauer, Emmet et Teller) on parle de surface
spécifique de BET(SBET) et quand elle est exploitée
avec l'équation de Langmuir, on parle de surface spécifique de
Langmuir (SL). Une autre méthode de détermination de la surface
spécifique est basée sur l'adsorption de la molécule
d'acide acétique (Avom et al., 2001) qui a une surface
moléculaire de 21 Å2 proche de celle de N2 (16,2
Å2).
Pour déterminer la surface spécifique, 20 mL de
solution d'acide acétique de concentration comprise entre
4x10-3et 8x10-3M ont été mis en contact
avec 0,1g de charbon actif. Ce mélange a été filtré
après 60 minutes d'agitation. A l'issue de la filtration, la
concentration résiduelle a été déterminée
par la technique de dosage volumétrique (Avom et al., 2001).
Les surfaces spécifiques SL (en m2/g) sont
déterminées en utilisant l'équation (2.3):
21
SL = Qo.N.SA .(2.3)
22
Où N = 6,02 x 1023mol-1 (nombre
d'Avogadro), SA = 21Å2 (surface moléculaire d'acide
acétique) et Q0 la quantité maximale d'acide adsorbée
obtenue à partir du tracé de l'isotherme de Langmuir.
2.1.3.7. Indice d'iode
L'indice d'iode a été mesuré en utilisant
la procédure établie par la société
américaine pour les tests des matériaux. Il est
défini comme le nombre de milligramme d'iode adsorbé par gramme
de charbons actifs en poudre. Il mesure les micropores de dimensions
inférieures à 2 nm contenus dans le CA.
Dans une fiole de 250 mL a été introduit I2 et
KI de concentration respective 0,02N et 0,04N ensuite dans une seconde fiole de
250 mL a été introduit Na2S2O3.5H2O de concentration
0,005N. Par la suite, 0,1g de chaque CA a été pesé et
introduit dans un flacon, ensuite nous avons ajouté 30 mL d'une solution
de I2 de concentration 0,02N. Le mélange a été
agité, pendant 3 heures puis filtré. Après filtration 10
mL du filtrat ont été titrés par une solution de sodium
thiosulphate de concentration 0,005N (l'indicateur coloré étant
l'amidon) (Tchuifon et al,. 2014b). L'équation de dosage
suivant a été utilisée pour déterminer la
concentration finale d'iode:
2 Na2S2O3 (aq) + I2 ? Na2S4O6 (aq) + 2NaI (2.4)
La quantité adsorbée (indice d'iode) a
été déterminé comme suit:
.(2.5)
Où m : la masse de CA,
Co : est la concentration initiale d'iode, Ct : la
concentration à l'instant t d'iode, V : le volume de la solution
d'iode.
2.2. Réactifs chimiques et équipements
2.2.1. Réactifs chimiques
Les produits chimiques utilisés pour ces travaux sont
regroupés dans le tableau ci-dessous.
23
Tableau 2.1: Formules chimiques, noms,
pureté et origines des produits chimiques.
|
Noms
|
Formules
chimiques
|
Pureté
|
Origines
|
|
Paracétamol
|
C8H9NO2
|
98%
|
BDH
|
|
Acide nitrique
|
HNO3
|
63 %
|
Riedel-de-Häen
|
|
Sodium hydroxyde
|
NaOH
|
98%
|
Fischer
|
|
Sodium nitrate
|
NaNO3
|
98%
|
Fischer
|
|
Diode
|
I2
|
99,90%
|
Fischer
|
|
Sodium thiosulfate eau- (1/5)
|
Na2S2O3.5H2O
|
99,66%
|
Fischer
|
|
Acide acétique
|
CH3COOH
|
99,5%
|
ANALAR
|
|
Acide chlorhydrique
|
HCl
|
36%
|
Phillip Harris
|
2.2.2. Equipements
Dans le cadre de ce travail nous avons utilisé les
équipements et appareils ci-dessous:
V' Quelques matériels du laboratoire couramment
utilisés au laboratoire ;
V' Des papiers filtres de type whatman ;
V' Une balance de précision de marque Sartorius ;
V' Une table d'agitation de marque EDMUND BOHLER GmbH ;
V' Une étuve de marque Binder ;
V' Un four de marque ISUNU ;
V' Un spectrophotomètre UV-Vis de marque JENWAY de
modèle 6715 ;
V' pH-mètre de marque pHS-25 ;
V' Un spectrophotomètre IR-FT de marque Brüker;
2.2.3. Adsorption du paracétamol en solution
aqueuse 2.2.3.1. Principe
Dans un flacon, une solution de paracétamol de volume V
(en mL) de concentration initiale Ci (en mg/L) est mise en contact avec une
masse m (en g) du matériau adsorbant. Le mélange obtenu est
porté sous agitation sur une table d'agitation à une vitesse
v (en tr/min) pendant un temps de contact t précis. A cause de
l'affinité qui pourrait exister entre l'adsorbant et l'adsorbat en
solution, ce dernier est attiré par le matériau. Les liaisons
formées
entre ces deux entités dépendent des
mécanismes mis en jeu. Ce processus prend place jusqu'à ce qu'un
équilibre soit établi entre la quantité de
paracétamol adsorbée et la quantité résiduelle en
solution. A l'équilibre, la solution est filtrée et le filtrat
obtenu est dosé par spectrophotométrie UV-Vis à une
longueur d'onde de 242 nm.
La quantité de paracétamol adsorbée par
unité de masse d'adsorbant (en mg/g) est donnée par la relation
suivante :
(2.6)
C0 (en mg/L) est la concentration initiale du
paracétamol; Ct (en mg/L) est sa concentration résiduelle
à l'équilibre, V (en L) est le volume de la solution et m (en g)
est la masse du l'adsorbant.
Les pourcentages du paracétamol adsorbés %R ont
été calculé à partir de la relation suivante :
(2.7)
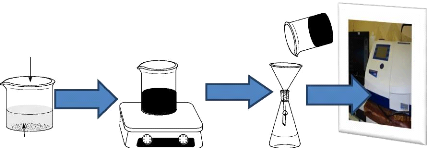
L'adsorption en mode batch du paracétamol se
résume par le schéma présenté à la figure
2.1 ci-après.
Charbon Actif
Agitation
Filtration
Analyse
24
Table d'agitation Filtration simple avec
Spectrophotomètre
un papier filtre UV-Vis
paracetamol
Figure 2.1 : Mode opératoire de
l'adsorption en mode batch.
soluton de
2.2.3.2. Courbe d'étalonnage
Avant adsorption en mode batch du paracétamol en
solution aqueuse, une droite d'étalonnage a été
établie dans le but de convertir à chaque fois les absorbances
obtenues en concentrations résiduelles. Cette droite de calibration a
été établie à partir d'une gamme de concentrations
allant de 10 à 120 mg/Let elle donne une relation directe entre
l'absorbance mesurée et la concentration de l'espèce chimique en
solution.
Les résultats obtenus sont donnés par le tableau
2.2 et la figure 2.2 ci-après :
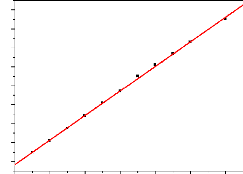
2,0
0,8
0,6
0,4
1,8
1,6
1,4
1,2
1,0
R2= 0,999
Y = 0,013X + 0,3673
0 20 40 60 80 100 120
25
Concentration (mg / L)
Figure 2.2: Droite de calibration du
paracétamol en solution aqueuse.
Cette droite, donnée par la figure 2.2 a pour
équation y = 0,013x +0,367 et son coefficient de corrélation est
égale à R2 = 0,9993. Dans cette équation, y
représente l'absorbance du paracétamol en solution aqueuse et x
représente sa concentration finale.
2.2.3.3. Optimisation de l'adsorption en mode batch du
paracétamol
La procédure expérimentale du processus
d'adsorption a été faite en étudiant l'influence de
quelques paramètres physico-chimiques sur le dit processus. L'objectif
de cette investigation est de réunir les meilleures conditions
expérimentales permettant une rétention optimale du polluant par
les adsorbants. Les paramètres qui ont été mis en
évidence sont : le pH, le temps de contact, la masse de l'adsorbant, la
concentration initiale du paracétamol et la force ionique. Ces
paramètres seront étudiés suivant la procédure
décrite dans les travaux de Tchuifon et al,. (2014b).
? Etude de l'influence du pH
Dans des flacons étiquetés, 20 mL de solutions
de paracétamol de concentration 100 mg/L ont été mises en
contact avec 100 mg de chaque CA. L'effet du pH a été
étudié en ajustant le pH des solutions de paracétamol (2 ;
2,5 ; 3 ; 3,5) à l'aide des solutions de HNO3.
26
Les mélanges ont été portés sous
agitation pendant 90 minutes puis filtrés. Les filtrats obtenus ont
été analysés par spectrophotométrie UV-vis à
242 nm.
? Etude de l'influence du temps de contact :
Dans des flacons étiquetés, 20 mL de solutions
de paracétamol de concentration 100 mg/L ont été mises en
contact avec 100 mg de chaque CA. Les mélanges ont été
portés sous agitation constante, pendant des durées progressives
allant de 5 jusqu'à 150 minutes, puis filtrés. Les filtrats ont
été analysés par spectrophotométrie UV-vis.
? Etude de l'influence de la masse de l'adsorbant
:
Des flacons étiquetés, 20 mL de solutions de
paracétamol de concentration 100 mg/L ont été mises en
contact avec des masses variant de 25 à 450 mg de chaque CA. Les
mélanges ont été portés sous agitation constante,
pendant le temps d'équilibre puis filtrés. Les filtrats ont
été analysés par spectrophotométrie UV-vis.
? Etude de l'influence de la concentration initiale du
paracétamol
Dans les flacons étiquetés, 20 mL d'une solution
de paracétamol dont les concentrations varient de 70 à 120 mg/L
ont été mises en contact avec 100 mg de chaque CA. Les
mélanges ont été portés sous agitation constante,
pendant le temps d'équilibre puis filtrés. Les filtrats ont
été analysés par spectrophotométrie UV-vis.
? Etude de l'influence de la force ionique
Dans les flacons étiquetés, 20 mL d'une solution
de paracétamol de concentration 100 mg/L contenant le NaNO3 dont les
concentrations varient de 0,01 à 0,06 mol/L ont été mises
en contact avec 100 mg de chaque CA afin de faire varier la force ionique du
milieu. Les mélanges ont été portés sous agitation
constante, pendant le temps d'équilibre puis filtrés. Les
filtrats ont été analysés par spectrophotométrie
UV-vis.
27
CHAPITRE 3 : RESULTATS ET DISCUSSIONS
L'objectif principal de ce chapitre est de présenter
les résultats obtenus au cours de la caractérisation des
différents matériaux, ensuite ceux obtenus lors de la variation
de quelques paramètres physico-chimiques influençant le processus
d'adsorption. Il nous permet également d'établir
d'éventuelles corrélations entre ces matériaux et leurs
capacités de rétention envers les molécules du
paracétamol en solution aqueuse et enfin de traiter l'étude des
mécanismes mis en jeu au cours du processus.
3.1. CARACTERISATIONS DES MATERIAUX 3.1.1. Spectres
IR
Les matériaux (balles de riz, CANa1 (500°C) et
CANa2 (450°C)) ont été caractérisés par
spectroscopie IR d'une part dans le but d'identifier les groupements
fonctionnels présents à leurs surfaces et d'autres part dans le
but de savoir quelles sont les modifications fonctionnelles apportées
sur le matériau lors de la préparation des CA. Les spectres
obtenus pour le matériau à l'état brut (balles de riz) et
les charbons actifs (CANa1 et CANa2) sont présentés sur les
figures 3.1 et figures 3.2 ci-dessous.
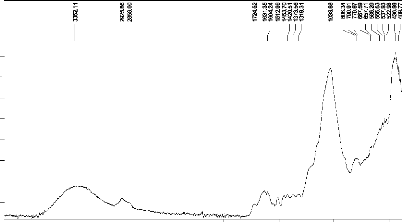
|
3500
|
3000
|
2500
|
2000
|
1500
|
1000
|
500
|
Wavenumber cm-1
Figure 3.1: Spectre IR des balles de riz
brutes.
F:\MEA\RICE HUSK0 RICE HUS
Instrumen te nd / or acce
Le spectre de balles de riz présente une large bande
d'absorption qui apparait dans la région 3650-3200
cm-1attribuable à la vibration d'élongation O-H. Cette
large bande présente un maximum qui est centré autour à
3352 cm-1. Dans la région 2925-2858cm-1, apparait
une
28
bande de vibration de faible intensité
caractéristique des vibrations d'élongation C-H aliphatiques. On
observe également sur ces spectres une bande apparaissant à 1724
cm-1assignée au groupement carbonyles. A 1631
cm-1, apparaît une bande de vibration d'élongation C=O
des carboxylates. Le spectre des balles de riz montre également une
bande d'absorption autour de 1604 cm-1caractéristiques des
vibrations d'élongation C=C du squelette aromatique. Dans la
région spectrale 1300-1000 cm-1, il apparaît une bande
de grande intensité centrée à 1033 cm-1 et
attribuable à la vibration d'élongation C-O. La région
vers 1400 cm-1est attribuable à la vibration
d'élongation C-C des cycles aromatiques (Cherraye, 2012). On note de
plus dans la région 900-600 cm-1des bandes d'absorption
caractéristiques des vibrations de déformation C-H des
systèmes polynucléaires aromatiques. Après l'activation et
la carbonisation des balles de riz, nous avons superposé les
différents spectres des produits obtenus dans le but d'apprécier
les effets de la calcination.
La figure 3.2 ci-dessous présente ces différents
spectres.
3500 3000 2500 2000
1500 1000 500
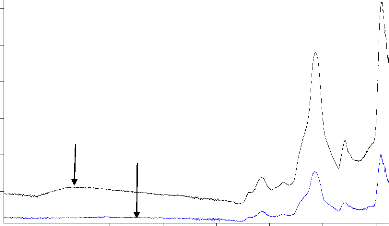
CANa2
CANa1
Wavenumber cm-1
Figure 3.2: Spectres superposés des
charbons actifs CANa1 et CANa2.
Au regard de ces spectres, la bande d'absorption
caractéristique des groupements OH qui apparait autour de 3500 à
3300 cm-1 présente sur le spectre des balles de riz est
présente mais de faible intensité sur le spectre de CANa2 et
absente sur le spectre de CANa1. Ce résultat pourrait s'expliquer par la
carbonisation qui est à l'origine du départ des atomes de C, H, O
sous forme de CO2, H2O, aldéhyde (Ndi & Ketcha, 2013). Aussi la
disparition de la liaison hydrogène montre que le sodium hydroxyde
agirait comme agent déshydratant
29
(Suarez-Garcia et al., 2002). La bande à 2925
cm-1 assigné aux vibrations d'élongations C-H
aliphatiques a disparu dans les charbons actifs et ceci indique que
l'activation enlève une partie significative de la liaison C-H (Hesas
et al., 2013). La perte de la bande à 2925 cm-1 dans
les charbons actifs préparés peut également indiqué
que l'activation chimique coupe plusieurs liaisons dans les espèces
aliphatiques et aromatiques et élimine plusieurs substrats volatiles.
3.1.2. Densité apparente et taux
d'humidité
Les valeurs des densités apparentes et des taux
d'humidités obtenus pour les différents charbons sont
données dans le tableau 3.1 ci-dessous :
Tableau 3.1: Densité apparente et
taux d'humidité des différents charbons
|
Charbons
|
CANa1
|
CANa2
|
|
Densité apparente
|
655,53
|
645,33
|
|
Taux d'humidité
|
7
|
9,09
|
Le tableau 3.1 montre que les valeurs des densités
apparentes obtenues de CANa1 et CANa2 sont largement au-dessus de la limite
minimale de 250 kg/m3 fixée par AATE. Aussi CANa1 est plus
dense que CANa2; ce qui nous permet de conclure que l'augmentation de la
température de carbonisation des charbons favorise la diminution de la
densité apparente (Devi et al., 2012).
Le tableau 3.1 montre également que les taux
d'humidité de CANa1 et CANa2 sont situés au-delà de 5 % ce
qui n'est pas en accord avec les normes de l'AATE. Mais la littérature
nous montre des charbons présentant de bonnes capacités
d'adsorption à des valeurs supérieures à cette norme.
(Tchuifon et al., 2014b).
3.1.3. Le pH au Point de charge nulle (pHpzc) et pH des
charbons actifs
La représentation du pH final en fonction du pH initial
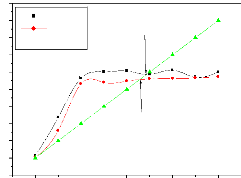
nous a permis de déterminer le pH de charge nulle (pHpzc). La figure
(3.3) ci-dessous nous montre que les pH au point de charge nulle des
différents matériaux sont respectivement de 6,80 et 6,54 pour
CANa1 et CANa2.
11
10
4
2
9
8
7
6
5
3
1
CANa1
CANa2
6,80
6,54
2 4 6 8 10
30
pH intiale
Figure 3.3: Evolution du
pHfinal en fonction du pHinitial.
Les valeurs des pHpzc obtenues nous permettent de
connaître, selon le pH de la solution, la charge nette de surface. Ceci a
une grande importance dans les interactions molécules/matériau
adsorbant en phase liquide.
Les valeurs du pH et du pHpzc sont données dans le
Tableau 3.3. Les valeurs du pH nous permettent de déterminer le
caractère acide ou basique de nos CA et le pHpzc nous permet ainsi de
prédire les types d'interactions pouvant intervenir au cours de
l'adsorption.
Tableau 3.2: pH et pHpzc des
différents CA.
|
Charbons
|
CANa1
|
CANa2
|
|
Ph
|
7,10
|
7,13
|
|
pHpzc
|
6,80
|
6,54
|
Le tableau 3.3 nous montre que le pHpzc< pH,
d'où la surface des CA sont chargée négativement
(Tatianne, 2011).
3.1.4. Surface spécifique à l'acide
acétique
Les valeurs obtenues sont calculées à partir des
données obtenues de l'étude de l'isotherme de Langmuir
présentée par la figure 3.4 ci-dessous. Le modèle de
Langmuir comme celui de D-R donne la quantité maximale de polluant
fixée. En supposant que l'adsorption soit monocouche, nous avons obtenu
la valeur de Qo, ce qui nous a permis de
31
calculer la surface en utilisant la formule de
l'équation (2.3) et les valeurs obtenues sont consignées dans le
tableau 3.4 ci-dessous :
0,0010 0,0015 0,0020 0,0025 0,0030 0,0035 0,0040
0,0045
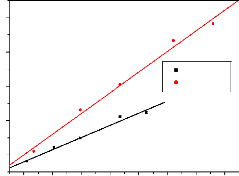
4
2
6
5
3
Y = 1206,2X + 0,794 R2 =
0,990
Y = 709,6X + 1,087 R2 =
0,984
CANa1
CANa2
Ce
Figure 3.4: Transformé
linéaire de l'isotherme de Langmuir.
Tableau 3.3: Paramètre de Langmuir et
surface spécifique à l'acide acétique
|
Paramètres
|
CANa1
|
CANa2
|
|
Q0 (mol/g)
|
1,409 x 10-3
|
8,291 x 10-3
|
|
K
|
652,92
|
1518,89
|
|
R2
|
0,9843
|
0,9904
|
|
SL (m2/g)
|
178,13
|
104,82
|
e
Il ressort de ce tableau que la surface spécifique du
charbon CANa1 est supérieure à celle de CANa2. Ceci permet de
dire que l'augmentation de la température de carbonisation favorise le
développement des pores (Avon et al., 2001)
3.1.5. Indice d'iode
L'indice est habituellement employé pour mesurer la
porosité des matériaux. Ainsi, l'indice d'iode mesuré
permet d'évaluer la capacité d'adsorption des petites
molécules par le charbon actif. Le tableau 3.5 ci-dessous donne les
valeurs de l'indice d'iode des deux charbons (CANa1 et CANa2).
32
Tableau 3.4 : Valeurs de l'indice d'iode des
deux charbons.
|
Charbons
|
CANa1
|
CANa2
|
|
Indice d'iode (mg/g)
|
528,39
|
494,67
|
L'indice d'iode des deux charbons compris est entre 490-530.
Généralement, plus l'indice d'iode est élevé, plus
la capacité adsorption est grande. L'indice d'iode recommandé par
AATE pour un charbon utilisé pour l'adsorption des composés de
faible poids moléculaire est de 500 mg/g. Ainsi, les charbons CANa1 et
CANa2 sont conformes à cette recommandation (Devi et al.,
2012). De plus, au-delà de 500°C, les indices d'iode
décroissent sensiblement. En effet, l'augmentation de la
température engendre sensiblement le développement des micropores
(Baçaoui et al., 2001) entraînant une augmentation de la
capacité d'adsorption des charbons actifs et donc l'indice d'iode. Mais
lorsque la température s'élève au-delà de
500°C, une partie des micropores formés est détruite. Cette
diminution du nombre de micropores s'accompagne d'une réduction sensible
de la capacité d'adsorption du charbon actif (Mohammad et al.,
2007). Ainsi à partir des valeurs obtenues du tableau 3.4, nous pouvons
dire que CANa1 a plus de micropores que CANa2.
3.2. ADSORPTION DU PARACETAMOL EN MODE BATCH 3.2.1.
Influence du pH de la solution
Le pH du milieu est un paramètre qui influence
fortement le processus d'adsorption. L'attraction ou la répulsion
électrostatique entre l'adsorbant et l'adsorbat dépend de la
charge surfacique des deux entités. La figure 3.5 ci-dessous
présente les résultats obtenus au cours de cette étude.
CANa1
CANa2
10 8 6 4
2
33
34
35
1,8 2,0 2,2 2,4 2,6 2,8 3,0 3,2 3,4 3,6
pH
Figure 3.5: Influence du pH sur
l'adsorption du paracétamol en solution aqueuse (concentration initial
100 mg/L, température ambiante, t = 90 min v=150 tr/min et m = 0,1
g).
L'analyse de ces courbes montre que les capacités
d'adsorption des deux matériaux diminuent lorsque la valeur du pH
augmente. A pH 2, on a une adsorption maximale du polluant par les deux
matériaux (10,06 mg/g et 7,25 mg/g). Ce résultat s'explique par
le fait qu'à pH 2, les groupements fonctionnels présents à
la surface des matériaux subissent une forte protonation (Fumba et
al., 2014). Il en résulte une forte attraction
électrostatique entre les groupements fonctionnels chargés
positivement à la surface des adsorbants et les molécules du
paracétamol. Ainsi à ce pH, l'adsorption mise en jeu semble
être de nature électrostatique. L'adsorption diminue
considérablement lorsque le pH augmente. Ceci s'explique par le fait que
les forces d'attraction deviennent plus faibles probablement à cause de
la diminution progressive de la charge positive de la surface du
matériau. En effet, lorsque le pH de la solution augmente, le nombre de
sites chargés négativement augmente contrairement à celui
des sites chargés positivement qui diminue (diminution de la
quantité d'ions H+). La même tendance a
été dévoilée par Fumba et al., (2014).
3.2.2. Influence du temps de contact
Pour déterminer le temps nécessaire pour
atteindre l'équilibre, nous avons tracé la quantité
adsorbée du paracétamol (Q) en fonction du temps. La figure 3.6
représente la variation de la capacité d'adsorption en fonction
du temps de contact.
CANa1
CANa2
12 10 8 6
4 2 0
0 20 40 60 80 100 120 140 160
t ( min )
Figure 3.6 : Influence du temps de
contact sur l'adsorption du paracétamol (concentration initial 100 mg/L,
température ambiante, pH=2, v=150 tr/min et m = 0,1 g).
Les courbes obtenues sur cette figure nous montrent que
l'adsorption se déroule deux phases :
Une phase rapide pendant les 30 premières minutes pour
les deux adsorbants donnant des quantités adsorbées par maximum
de 6,780 mg/g et 5,615 mg/g respectivement pour CANa1 et CANa2. Une telle prise
est due à la présence à la surface des matériaux
des sites d'adsorption libres et disponibles pour la rétention des
molécules du paracétamol.
Dans la deuxième phase, le taux d'adsorption a ralenti
de manière significative jusqu'à ce que l'équilibre soit
atteint à 100 minutes. Ce s'explique par le fait qu'une fois que les
molécules du paracétamol en solution s'y sont fixés, ils
obstruent les pores dont la taille est petite et empêchent ainsi aux ions
restants en solution d'y pénétrer à la même vitesse
que les précédents. C'est la raison pour laquelle à partir
de 100 minutes la quantité de molécule de paracétamol
adsorbé en solution reste constante. La présence du palier
s'explique donc par la saturation de la surface du solide due à une
occupation totale des sites actifs d'adsorption présents à la
surface de l'adsorbant. La même tendance a été
dévoilée par Mukoko et al., (2015).
3.2.3. Influence de la masse de l'adsorbant
Le but de cette étude est de mettre en évidence
l'influence de la masse de l'adsorbant sur le processus d'adsorption. Nous
avons pour cela effectué l'adsorption du paracétamol par les
charbons actifs pour des masses comprises entre 25 - 450 mg. Les
résultats expérimentaux obtenus au cours de cette analyse sont
donnés par la figure 3.7 ci-dessous.
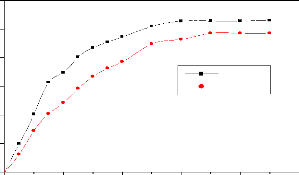
CANa1
CANa2
60 50 40 30
20 10
0
0 100 200 300 400 500
m ( mg )
Figure 3.7: Influence de la masse de
l'adsorbant sur l'adsorption du paracétamol (concentration initial 100
ppm, température ambiante, pH=2, v=150 tr/min).
La figure 3.7 montre que le pourcentage de rétention
des molécules du paracétamol augmente avec la masse des
adsorbants. Ce phénomène est dû au fait qu'une augmentation
de la masse entraine une agglomération des cellules et par
conséquent une réduction des distances intercellulaires, ceci
produit un «effet écran» conduisant à la protection des
sites de liaison de l'adsorbat. D'où l'augmentation de la masse de
l'adsorbant en solution favorise l'augmentation des sites d'adsorption.
À des masses plus élevées d'adsorbant, le taux
d'augmentation de l'adsorption de l'adsorbant ralentit, ceci peut être
dû à l'agrégation ou au recouvrement partiel du charbon
actif qui a pour conséquence une diminution des sites d'adsorptions
(Mukoko et al., 2015).
De plus CANa1 adsorbe mieux que CANa2 ce qui est en accord
avec les résultats de la surface spécifique et de l'indice d'iode
qui ont montré que CANa1 présente plus de pores et une
microporosité supérieure à celle de CANa2.
3.2.4. Influence de la force ionique
La présence d'autres ions en solution influence
fortement la capacité d'adsorption de La molécule cible. La
capacité d'adsorption d'un adsorbant dépend de la
solubilité des espèces chimiques présentes dans la phase
liquide (Kaur et al., 2012). L'étude de l'influence de la force
ionique a été effectuée afin d'apprécier l'effet du
NaNO3 sur la capacité d'adsorption du paracétamol par les
différents charbons actifs. Les résultats expérimentaux
obtenus au cours de cette analyse sont donnés par la figure 3.8
ci-dessous.
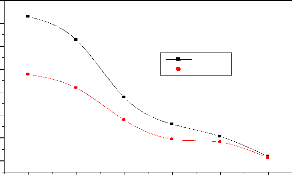
CANa1
CANa2
7
6
5
4
3
2
1
0
0,01 0,02 0,03 0,04 0,05 0,06
C ( mol / L )
36
Figure 3.8: Influence de la force ionique
(température ambiante, pH=2, temps = 100 minutes, v=150 tr/min et
m=0,1g).
La figure 3.8 montre que l'augmentation de la concentration du
NaNO3 diminue la capacité d'adsorption des molécules du
paracétamol. Ce phénomène s'explique par l'existence
effective d'une compétition entre les ions sodium et les
molécules du paracétamol pour l'occupation des sites d'adsorption
sur CANa1 et CANa2. Nous pouvons également expliquer ces
résultats par le fait que l'addition du sel entraîne une
augmentation de la solubilité des molécules du
paracétamol. Ainsi, plus la substance sera soluble et moins elle sera
adsorbée (Kaur et al., 2012) ce qui par conséquent
diminue la capacité d'adsorption du paracétamol en solution. Ce
qui est en accord avec les résultats obtenus par Anguile et
al., (2013).
3.2.5. Modèles cinétiques
Les modèles cinétiques ont été
exploités afin de déterminer les différents
paramètres cinétiques liés aux processus d'adsorption mis
en jeu. A cet effet, trois modèles cinétiques (pseudo-premier
ordre, pseudo-second ordre, diffusion et intraparticulaire) ont
été appliqués aux données expérimentales
obtenues au cours de l'étude de l'influence du temps de contact sur
l'adsorption. Les transformées linéaires relatives à
chacun de ces modèles sont représentées par les figures
3.9, 3.10, 3.11 et 3.12 ci-après.
-0,2
2,0
0,8
0,6
0,4
0,2
0,0
1,8
1,6
1,4
1,2
1,0
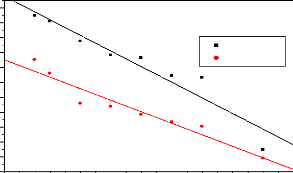
Y = -0,015X + 1,226 R2 =
O,926
Y = -0,021X + 2,051 R2 =
0,917
CANa1
CANa2
0 10 20 30 40 50 60 70 80 90
t ( min )
Figure 3.9: Formes linéaires du
modèle du pseudo-premier ordre pour CANa1 et CANa2.
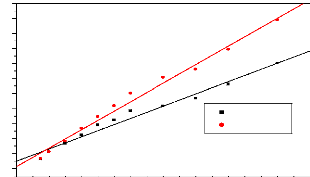
22
20
18
16
14
12
10
4
8
6
2
0
Y = 0,123X + 1,491 R2 =
0,987
Y = 0,081X + 1,792 R2 =
0,976
CANa1
CANa2
0 20 40 60 80 100 120 140 160
t ( min )
37
Figure 3.10 : Formes linéaires du
modèle du pseudo-second ordre pour CANa1 et CANa2.
3,4
3,2
3,0
2,8
2,6
2,4
2,2
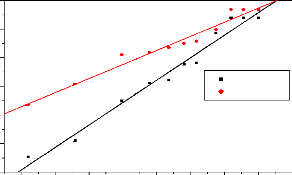
Y = 0,196X + 2,361 R2 =
0,965
Y = 0,313X + 1,744 R2 =
0,976
CANa1
CANa2
38
1,5 2,0 2,5 3,0 3,5 4,0 4,5 5,0 5,5
lnt
Figure 3.11: Formes linéaires du
modèle de diffusion intra-particulaire pour CANa1 et CANa2.
Les différentes constantes obtenues à partir de
ces modèles sont regroupées dans le tableau 3.5 ci-dessous
Tableau 3.5: Constantes de Vitesse et
Coefficients de Corrélation des Modèles Cinétiques
|
Modèles
|
Paramètres
|
CANa1
|
CANa2
|
|
Pseudo premier
ordre
|
R2
|
0,917
|
0,926
|
|
Qe (mg/g)
|
7,775
|
3,407
|
|
K1 (min-1)
|
0,021
|
0,015
|
|
pseudo-second
ordre
|
R2
|
0,976
|
0,987
|
|
Qe (mg/g)
|
12,345
|
8,130
|
|
K2 (g/
min.mg)
|
0,0036
|
0,010
|
|
Diffusion intra
particulaire
|
R2
|
0,976
|
0,965
|
|
Kid (min-1)
|
5,720
|
10,601
|
|
a (mg/g)
|
0,314
|
0,196
|
Les résultats présentés dans le tableau
ci-dessus montrent que l'adsorption du paracétamol par les CANa1 et
CANa2 est mieux décrite par les modèles cinétiques de
pseudo-second ordre et de diffusion intra particulaire également pour
CANa2 donnant de très bons coefficient de corrélation, ce qui
traduit respectivement un mécanisme de chimisorption
39
(Ho & Mc Kay, 1999) et une étape limitante de la
diffusion des molécules du paracétamol à
l'intérieur des pores de l'adsorbant (Zora et al., 2006).
En considérant les valeurs de Qe
déterminées à partir du modèle de pseudo-second
ordre (12,345 mg/g pour CANa1 et 8,130 mg/g pour CANa2), on peut une fois de
plus affirmer que les CANa1 présentent les meilleures performances
d'élimination comparées à celles des CANa2.
3.2.6. Isothermes d'adsorption
Les isothermes d'adsorption obtenues pour des solutions de pH
initial 2 sont représentées par la figure 3.13 ci-après
:
12,0
11,5
11,0
10,5
10,0
9,5
9,0
8,5
8,0
7,5
7,0
6,5
6,0
5,5

CANa1
CANa2
50 60 70 80 90 100 110 120
Ce ( mg / L )
Figure 3.12 : Influence de la concentration
initiale sur l'adsorption du paracétamol en solution aqueuse
(température ambiante, pH=2, temps= 100 minutes, v=150 tr/min et
m=0,1g).
L'isotherme d'adsorption sur les CANa1 et CANa2 est de type I,
caractéristique d'une adsorption chimique, que l'on obtient aussi dans
le cas des adsorptions physiques limitées à la formation d'une
monocouche. Le fait que l'adsorption se limite à la formation d'une
monocouche, laisse envisager que les interactions adsorbant/adsorbat sont de
natures chimiques ou de van der Waals (Anagho et al., 2013).
Les transformées linéaires de quelques
modèles d'isothermes sont représentées par les
figures3.13, 3.14, 3.15 et 3.16 ci-après:
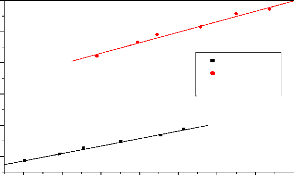
Y = 0,047X + 3,331 R2 =
0,995
Y = 0,067X + 7,912 R2 =
0,982
CANa1
CANa2
16 14 12 10
8 6
50 60 70 80 90 100 110 120
1 / Ce
Figure 3.13 : Modèle linéaire
de l'isotherme Langmuir pour CANa1 et CANa2.
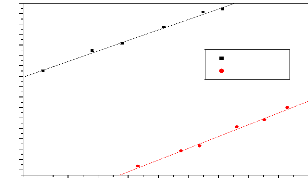
2,50
2,45
2,40
2,35
2,30
2,25
2,20
2,15
2,10
2,05
2,00
1,95
1,90
1,85
1,80
1,75
1,70
Y = 0,503X + 0,207 R2 =
0,994
Y = 0,565X - 0,684 R2 =
0,992
CANa1
CANa2
3,9 4,0 4,1 4,2 4,3 4,4 4,5 4,6 4,7 4,8
lnCe
40
Figure 3.14: Modèle linéaire
de l'isotherme Freundlich pour CANa1 et CANa2.
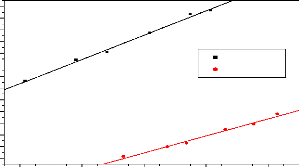
Y = 3,636X - 9,904 R2 =
0,987
Y = 5,188X - 11,54 R2 =
0,994
CANa1
CANa2
12,0 11,5 11,0 10,5
10,0 9,5 9,0 8,5 8,0
7,5 7,0 6,5 6,0 5,5
41
3,9 4,0 4,1 4,2 4,3 4,4 4,5 4,6 4,7 4,8
lnCe
Figure 3.15 : Modèle linéaire
de l'isotherme Temkin pour CANa1 et CANa2.
4 5 6 7 8 9 10 11 12
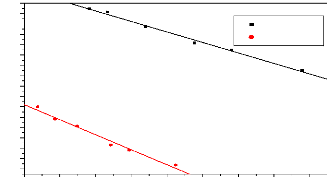
2,50
2,45
2,40
2,35
2,30
2,25
2,20
2,15
2,10
2,05
2,00
1,95
1,90
1,85
1,80
1,75
1,70
Y = -0,050X + 2,766 R2 =
0,993
Y = -0,072X + 2,299 R2 =
0,982
CANa1
CANa2
Figure 3.16: Modèle linéaire
de l'isotherme de Dubinin-Radushkevich pour CANa1et CANa2.
£2
Les constantes relatives à ces modèles sont
regroupées dans le tableau 3.7 ci-dessous :
42
Tableau 3.6: Constantes d'isothermes
|
Modèles
|
Paramètres
|
CANa1
|
CANa2
|
|
Langmuir
|
Qmax (mg/g)
|
20,964
|
14,881
|
|
KL (L/mg)
|
0,0143
|
0,0085
|
|
R2
|
0,9951
|
0,9826
|
|
Freundlich
|
1 /n
|
0,5035
|
0,5656
|
|
KF (L/g)
|
1,2299
|
0,5042
|
|
R2
|
0,9947
|
0,9928
|
|
Temkin
|
(kJ/mol)
|
10,0049
|
10,1399
|
|
KT (L/mg)
|
0,1081
|
0,2979
|
|
R2
|
0,9949
|
0,9875
|
|
Dubinin-
Radushkevich
|
Qmax (mg/g)
|
15,9028
|
9,9721
|
|
E (kJ/mol)
|
3,1403
|
3,7164
|
|
R2
|
0,9932
|
0,9822
|
Au regard du tableau 3.7 ci-dessus, on se rend compte que les
coefficients de corrélations obtenus sont supérieur à 0,9
pour les modèles étudiés, donc le phénomène
d'adsorption du paracétamol est en adéquation avec les
modèles de Langmuir, de Freundlich, Temkin et Dubinin-Radushkevich.
L'adsorption du paracétamol par CANa1 et CANa2 est
décrite respectivement par le modèle de Langmuir (R2 =
0,9951) et le modèle de Freundlich (R2 = 0,9928). Le
modèle de Langmuir prévoit une répartition des
molécules de paracétamol sous forme d'une monocouche (Mukoko
et al., 2015). Les valeurs de 1/n du modèle de Freundlich sont
inférieures à 1 et les valeurs des constantes du modèle de
Temkin sont inférieures à 5 ce qui permet de dire que le
processus de fixation du paracétamol sur l'adsorbant est une
chimisorption (Sousa et al., 2012). Les valeurs de l'énergie
moyenne d'adsorption pour les deux adsorbants sont positives, ce qui signifie
que le processus d'adsorption est exothermique (Tchuifon et al.,
2014b).
Les capacités maximales d'adsorption des CA obtenues
à partir du modèle de Langmuir ont permis de les classer suivant
leurs performances : Les CANa1 (Qmax = 20,964 mg/g) sont plus efficaces que les
CANa2 (Qmax= 14,881 mg/g).
43
Tableau 3.7: Comparaisons de la
capacité adsorption du paracétamol avec d'autres
adsorbants
|
Adsorbants
|
Quantité adsorbé en mg/g
|
Références
|
|
Charbon actif à partir des
coques de
noisette
|
|
13,88
|
|
Demirba°, 2009
|
|
Charbon actif à partir des
déchets de
fruits d'huile de
palmier
|
|
0,84
|
|
Rafeah et al., 2009
|
|
Charbon actif à partir des
peaux d'orange,
sciure et
bagasses
|
3,19
|
; 3,12;
|
2,89
|
Ahsan et al., 2007
|
|
CANa1
|
|
20,964
|
|
Présent travail
|
|
CANa2
|
|
14 ,881
|
|
Présent travail
|
De ce tableau nous constatons que les quantités
adsorbées des molécules du paracétamol en mg/g par CANa1
et CANa2 sont supérieures à celles obtenues par trois
différents matériaux. Ceci nous permet de conclure que les CA
préparés dans ce travail sont efficaces pour l'adsorption du
paracétamol.
44
CONCLUSION ET PERSPECTIVES
L'objectif de ce travail était de préparer et de
caractériser deux charbons actifs à partir des balles de riz et
d'évaluer leurs capacités à éliminer les
molécules du paracétamol en solution aqueuse. Pour atteindre cet
objectif, les CA étant préparés, plusieurs
caractérisations à l'instar de la spectroscopie IR, la surface
spécifique par la méthode d'adsorption de l'acide
acétique, le point de charge nulle, la densité apparente, le taux
d'humidité et l'indice d'iode ont été effectuées
pour élucider ses propriétés. Il en ressort des
résultats de la spectroscopie IR que la transformation du
matériau brute en charbon actif à entrainer la disparition du
groupement fonctionnelle OH présent sur ces derniers. La surface
spécifique augmente avec l'augmentation de la température, de
même que la microporosité.
La méthode de dépollution proprement dite
utilisée dans le cadre de ce travail était l'adsorption en mode
batch. Pour ce fait, plusieurs paramètres à savoir le pH
(L'adsorption s'est avérée favorable lorsque le pH du milieu
était de 2), le temps de contact (100 minutes), la masse de l'adsorbant
(Les pourcentages d'adsorption étant de 52,87 et 48,56 pour la
même masse de 350 mg pour CANa1 et CANa2 respectivement), la
concentration initiale (100 mg/L) et la force ionique ont été
mises en en évidence.
Quelques modèles isothermes et cinétiques
d'adsorption ont par suite été appliqués aux
données expérimentales. Ceci a respectivement permis de
définir les mécanismes de fixation du paracétamol et les
différents paramètres cinétiques liés aux processus
d'adsorption mis en jeu. L'équilibre d'adsorption a montré de
bonnes corrélations avec les modèles de Langmuir et de Freundlich
respectivement pour CANa1 et CANa2. Ceci a été clairement
confirmé par les valeurs des coefficients de corrélation
déterminés à partir de ces modèles. Le processus
d'adsorption mis en jeu sur l'ensemble des deux matériaux est la
chimisorption. A l'équilibre, les molécules du paracétamol
forment une monocouche sur des adsorbants présentant une surface
hétérogène et des sites d'adsorption
énergétiquement différents. Les capacités
d'adsorption maximales déterminées à partir du
modèle de Langmuir ont permis de classer les adsorbants suivant leurs
performances : les CANa2 moins efficaces que les CANa1. Les valeurs de
l'énergie moyenne d'adsorption pour les deux adsorbants est positive par
les modèles de Tenkim, ce qui signifie que le processus d'adsorption est
exothermique.
L'étude cinétique a montré d'une part que
le modèle de pseudo-second ordre et diffusion intra particulaire est le
plus indiqué pour décrire l'adsorption du paracétamol par
les CA.
Les travaux futurs viseront à :
? Caractériser les matériaux par d'autres
techniques ;
45
? Etablir des méthodes de traitement permettant la
récupération du paracétamol et par conséquent la
régénération des adsorbants ;
? construire une colonne capable de traiter efficacement les
effluents des industries qui exploitent le paracétamol.
46
REFERENCES BIBLIOGRAPHIQUES
Abiko, H.; Furuse, M. & Takano, T. (2010). Quantitative
evaluation of the effect of moisture contents of coconut shell activated carbon
used for respirators on adsorption capacity for organic vapors. Industrial
Health, 48, 52-60.
Ahsan, H.; Nazrul, I.; Anarul, I. & Shafiqul, A.M. (2007).
Removal of copper from aqueous solution using orange peel, sawdust and bagasse.
Pakistan Journal of Analytical Environmental Chemistry,
8(1), 21-25.
Ajifack, G.L.; Ghogomu, J.N.; Noufame, T.D.; Ndi, J.N. &
Ketcha, J.M. (2014). Adsorption of Cu(II) ions from aqueous solution onto
chemically prepared activated aarbon from theobromacacoa. British Journal
of Applied Science &Technology, 4, 5021-5044.
Alvares, A. B. C.; Dlaper, C. & Parsons, S. A. (2001).
Partial oxidation by ozone to remove recalcitrance from wastewaters - a review.
Environmental Technology, 22, 409-427
Anagho, S.G.; Ketcha, J.M.; Tchuifon, T.D.R. & Ndi, J.N.
(2013). Kinetic and equilibrium studies of the adsorption of mercury (II) ions
from aqueous solution using kaolinite and metakaolinite clays from Southern
Cameroon. International Journal of Research in Chemistry and
Environment, 3(2), 1-11.
Anguile, J.J.; Ketcha, M.J.; Dingka'a, N.D. & Dongmo, S.
(2013). Effect of solution parameters on the adsorption of cobalt (II) ions on
smectite from Cameroon: Equilibrium studies. Journal of Academia and
Industrial Research, 2, 2278- 5213.
Avom, J.;Ketcha, J, M.; Matip, M, R, L.; & Germain, P.
(2001). Adsorption isotherme de l'acide acétique par des charbons
d'origine végétale. African Journal of Science and
Technology, 2(2), 1-7.
Baçaoui, A.; Yaacoubi, A.; Dahbi, A.; Bennouna, C.;
Luu, R.; Maldonado-Hodar, F.; RiveraUtrilla, J. & Moreno-Castilla, C.
(2001). Optimization of conditions for the preparation of activated carbons
from olive-waste cakes. Carbon, 39, 425-432.
Bouras, O. (2003). Propriétés adsorbantes
d'argiles pontées organophiles: Synthèse et
caractérisation. Thèse de Doctorat, Université de
Limoges, Limoges, France, 160 P.
Cherraye, R. (2012). Préparation par voie chimique d'un
charbon actif à partir des déchets de café (effet de taux
adjuvant). Mémoire de Master, Université Kasdi Merbah
Ouargla, Algérie, 35P.
Creangã, C. (2007). Procédé AD-OX
d'élimination de polluants organiques non biodégradables (par
adsorption puis oxydation catalytique). Thèse de Doctorat,
Institue de Toulouse, Toulouse, France, 294P.
47
Demirba°, E. (2009). Adsorption of cobalt(II) Ions from
Aqueous Solution onto Activated Carbon Prepared from Hazelnut Shells.
Adsorption science & technology, 21, 951-963.
Devi, B.V.; Jahagirdar, A.A. & Ahmed, Z. M.N. (2012).
Adsorption of Chromium on Activated Carbon Prepared from Coconut Shell.
International Journal of Engineering, 2, 364- 370.
Driad, Y. (2009). Stabilité du paracétamol :
Application à un sachet produit en industrie pharmaceutique.
Thèse de Doctorat, Universite Henri Poincare - Nancy 1, Nancy,
France, 102P.
Fazal, A. & Rafique, U. (2012). Mechanistic Understanding
of Cadmium Sorption by Sulfonated and Esterified Spent Black Tea.
International Journal of Chemistry and Environmental Engineering,
3, 230-237.
Fumba, G.; Essomba, J. S.; Tagne, G. M.; Ndi, N. J.;
Bélibi, B. P. D. & Ketcha, M. J. (2014). Equilibrium and Kinetic
Adsorption Studies of Methyl Orange from Aqueous Solutions Using Kaolinite,
Metakaolinite and Activated Geopolymer as Low Cost Adsorbents. Journal of
Academia and Industrial Research, 3(4), 2278-5213.
Hesas, R.H.; Arami-Niya, A.; Daud, W.M.A.W. & Sahu, J.N.
(2013). Preparation and caracterization of activated carbon from apple waste by
microware-assisted phosphoric acid activation: Application in methylene blue
adsorption. Bioresources, 8, 2950-2966.
Ho, Y. S. (2004). Citation review of Lagergren kinetic rate
equation on adsorption reactions. Scientometrics,
59(1), 171-177
Ho, Y.S. & McKay, G. (1999). Pseudo-second order model for
sorption processes. Process Biochemistry, 34, 451 -
465.
Kafack, T.F. (2012). Etude des performances des charbons
actifs préparés à partir de biomasses tropicales pour
l'élimination du chrome et diuron en milieu aqueux. Thèse de
Master, Institut International d'Ingénierie de L'Eau et
l'Environnement, Ouagadougou, Burkina Faso, 79P.
Kaur, S.; Rani, S. & Mahajan, R. K. (2012). Adsorption
kinetics for the removal of hazardous dyes Congo red by biowaste materials as
adsorbents. Journal of Chemistry, 2013, 540552.
Kenne, D.G. (2011). Biosorption du paraquat par des
matériaux lignocellulosiques et élaboration de capteurs
électrochimiques à film de sciure de bois. Thèse de
Doctorat, Université de Yaoundé 1, Yaoundé, Cameroun,
218P.
48
Kifuani, K.M.A.; Noki, V.P.; Ndelo, D.P.J.; Mukana, W.M.D.;
Ekoko, B.G.; Ilinga, L.B. & Mukinayi, J. (2012). Adsorption de la quinine
bichlorhydrate sur un charbon actif peu coûteux à base de la
Bagasse de canne à sucre imprégnée de l'acide
phosphorique. International Journal of Biological Chemical Society,
6, 1337-1359.
Kolpin, D.W.; Furlong, E.T.; Meyer, M.; Thurman, E.M.; Zaugg,
S.D.; Barber, L.B. & Buxton H.A.T. (2002). Pharmaceuticals, hormones and
other organic wastewater contaminants in US streams, 1999-2000: A national
reconnaissance. Environmental Science & Technology,
36, 1202-1211.
Lopes-Ramon, M.V.; Stoeckli, F.; Moreno-Castilla, C. &
Carrasco-Marin, F. (1999). On the characterization of acidic and basic surface
sites on carbons by various techniques. Carbon, 37,
1215-1221.
Mechrafi, E. (2002). Adsorption, désorption et
mobilité des herbicides au contact des adsorbants organiques et
minéraux. Thèse de doctorat, Université Mohammed
V, Faculté des Sciences, Rabat, Maroc, 183P.
Mohammad, A.; Mohammad, A.R.; Mohammad, A.M. & Mohammad,
B.S. (2007). Adsorption Studies on Activated Carbon Derived from Steam
Char. Indian Society for Surface Science and Technology,
23, 73-80.
Mukoko T, Mupa M, Guyo U, Dziike F., (2015). Preparation of
rice hull activated carbon for the removal of selected pharmaceutical waste
compounds in hospital effluent. Journal of Environmental and Analycal
Toxicology S7: 008. doi:10.4172/2161-0525.S7-008
Ndi, J.N. & Ketcha, M. J. (2013). The Adsorption
Efficiency of Chemically Prepared Activated Carbon from Cola Nut Shells by
ZnCl2 on Methylene Blue. Journal of Chemistry, 2013,
1-7.
Pope, J.P. (2001). Activated carbon and some Applications for
the remediation of Soil and Ground Water Pollution. Journal of Serbian Chemical
society, 67, 464- 476.
Rafeah, W.; Zainab, N. & Veronica, U.J. (2009). Removal of
mercury, lead and copper from aqueous solution by activated carbon of palm oil
empty fruit bunch World. Applied Sciences Journal, 5,
84-91.
Reungoat, J. (2007). Etude d'un procédé hydride
couplant adsorption sur zéolithes et oxydation par l'ozone. Application
au traitement d'effluents aqueux industriels. Thèse de
Doctorat, Université de Toulouse, Toulouse, France, 186P.
Robinson, T.; Mcmullan, G.; Marchant, R. & Nigam, P.
(2001). Remediation of dyes in textile effluent: a critical review on current
treatment technologies with a proposed alternative. Bioresource
Technology, 77, 247-255.
49
Slasli, M. (2002). Modélisation de l'adsorption par les
charbons microporeux : Approches
théorique et expérimentale, Thèse de
Doctorat, Université de Neuchâtel, Suisse, 134P. Soufan, M.
(2011). Oxydation par le chlore de composés pharmaceutiques.
Thèse de
Doctorat, Université de Poitiers, France.
173P.
Sousa, N.V.O.; Tecia, V.C.; Honorato, S.B.; Gomes, C.L.;
Baros, F.C.F.; Araujo, S.M.A.; Freire, P.T.C. &Nascimento, R.F. (2012).
Coconut bagasse treated by thiourea ammonia solution for cadmium removal:
kinetics and adsorption equilibrium. Biochemistry Resource,
7, 1504-1524.
Suárez-García, F.;Martínez-Alonso, A.;
Tascón, J.M.D. (2002). Pyrolysis of apple pulp: effect of operation
conditions and chemical additives. Journal of Analytical and Applied
Pyrolysis, 62, 93-109.
Suresh, S.; Srivastava, V. C.&
Mishra,I.M. (2012). Adsorption of catechol, resorcinol, hydroquinone, and their
derivatives. International Journal of Energy and Environmental
Engineering, 3(32), 2251-6832.
Tchuifon, T.D.R.; Anagho, S.; Ketcha, J.M.; Ndifor-Angwafor, G.N.
& Ndi, J.N. (2014b). Kinetics and equilibrium studies of adsorption of
phenol in aqueous solution onto activated carbon prepared from rice and coffee
husks. International Journal of Engineering and Technical Research,
2, 166-173.
Tchuifon, T.D.R.; Anagho, S.G.; Njanja E.; Ghogomu, J.N.;
Ndifor-Angwafor, N.G. & Kamgaing, T. (2014a). Equilibrium and kinetic
modelling of methyl orange adsorption from aqueous solution using rice husk and
egussi peeling. International Journal of Chemical Science,
12(3), 741 -761,
Velichkova, A. F. (2014). Vers un procédé fenton
hétérogène pour le traitement en continu d'eau
polluée par des polluants pharmaceutiques. Thèse de
Doctorat, Université de Toulouse, Toulouse, France. 220P
Zhang, H.; Tang, Y.; Cai, D.; Liu, X.; Wang,X.; Huang, Q.
& Yu, Z. (2010). Hexavalent chromium removal from aqueous solution by algal
bloom residue derived activated carbon: Equilibrium and kinetic studies.
Journal of hazardous materials, 181, 801-808.
Zora, M. & Snezana, B. (2006). Kinetic Studies and
Spectrophotometric Determination of Co(II) Ions by the Oxydation of Ponceau 4R
by Hydrogen Peroxyde. Journal of the Serbian Chemical Society,
71(2),181-196.
Zue, M.M. (2012). Élimination des ions Mn(II) des
solutions aqueuses par adsorption sur des charbons actifs
préparés à partir des coques de noix de noisette.
Mémoire de Master, Université des Sciences et
Techniques de Masuku, Franceville, Gabon, 74P.
| 

