Abstract
We re-examine here the phase separation between phospholipids and
adsorbed polymer chains on a fluid membrane with a change in some suitable
parameter (temperature). Our purpose is to quantify the significant effects of
the solvent quality and of the polydispersity of adsorbed loops formed by
grafted polymer chains on the segregation phenomenon. To this end, we elaborate
on a theoretical model that allows us to derive the expression for the mixing
free energy. From this, we extract the phase diagram shape in the
composition-temperature plane. Our main conclusion is that the polymer chain
condensation is very sensitive to the solvent quality and to the polydispersity
of loops of adsorbed chains.
PACS numbers: 87.16.Dg, 47.57.Ng, 64.60.F
1. Introduction
Biological membranes are of great importance to life, because
they separate the cell from the outside environment and separate the
compartments inside the cell in order to protect important processes and
specific events.
Nowadays, it is largely recognized that biological membranes
are present as a lipid bilayer composed of two adjacent leaflets [1, 2], which
are formed by amphiphile molecules possessing hydrophilic polar-heads pointing
outward and hydrophobic fatty acyl chains that form the core. The majority of
lipid molecules are phospholipids. These have a polar-head group and two
non-polar hydrocarbon tails, whose length is of the order of 5 nm.
0031-8949/11/065801+06$33.00 Printed in the UK & the USA 1
(c) 2011 The Royal Swedish Academy of Sciences
Also, the cell membranes incorporate another type of lipid,
cholesterol [1, 2]. The cholesterol molecules have several functions in the
membrane. For example, they give rigidity or stability to the cell membrane and
prevent crystallization of hydrocarbons. The biomembranes also contain
glycolipids (sugars), which are lipid molecules that microaggregate in the
membrane, and may be protective and act as insulators. Certain kinds of
molecules are bounded by sphingolipids such as cholera and tetanus toxins.
Sphingolipids and cholesterol favor the aggregation of proteins in microdomains
called rafts. In fact, these play the role of a platform for the
attachment of proteins while the membranes are moved around the cell and also
during signal transduction.
Proteins (long macromolecules) are another principal component
of cell membranes. Transmembrane proteins are amphipathic and are formed by
hydrophobic and hydrophilic regions having the same orientation as other lipid
molecules. These proteins are also called integral proteins. Their function is
to transport substances, such as ions and macromolecules, across the membrane.
There exist other types of proteins that may be attached to the cytoplasm
surface by fatty acyl chains or to the external cell surface by
oligosaccharides. These are termed peripheral membrane proteins. They have many
functions; in particular, they protect the membrane surface, regulate cell
signaling and participate in many other important cellular events. In addition,
some peripheral membrane proteins (those having basic residues) tend to bind
electrostatically to negatively charged membranes, such as the inner leaflet of
the plasma membrane.
Phys. Scr. 83 (2011) 065801 M Benhamou
etal
We note that the majority of macromolecules forming the
bilayer are simply anchored on the membrane and form a soft branched
polymer brush [3, 4].
The study of grafted polymers on soft interfaces was motivated
by the fact that they have potential applications in biological materials, such
as liposomes [5-8]. These soft materials were discovered by A Bangham.
Currently, they are major tools in biology, biochemistry and medicine (as drugs
transport agents). Liposomes are artificial vesicles of spherical shape that
can be produced from natural nontoxic phospholipids and cholesterol. But the
lipid bilayers have a short lifetime, because of the weak stability of the
vesicles and their extermination by white blood cells. To have stable vesicles
in time, one useful method consists of protecting them with a coat of flexible
polymer chains (coat size of the order of 50 nm [1]), which prevents the
adhesion of marker proteins [6, 7]. In fact, these polymer chains stabilize the
liposomes, due to the excluded volume forces between monomers [8]. Liposomes
can also be synthesized from A-B diblock-copolymers immersed
in a selective solvent that prefers to be contacted by the polymer A.
The hydrophobic B-polymer chains then aggregate and form a thin
bilayer, while the hydrophilic polymer chains A float in the solvent.
These copolymer-based liposomes have properties that are slightly different
from those of the lipid ones [9] (high resistance, high rigidity and weak
permeability to water). Depending on the choice of copolymers, these liposomes
are resistant to detergents [10].
The grafting of polymers onto lipid membranes was considered
in a very recent paper [11]. More precisely, the purpose was to investigate of
the phase separation between phospholipids and anchored polymers. As
assumptions, the aqueous medium was assumed to be a good solvent, and in
addition, the polymer chains were anchored to the interface only by one
extremity, a big amphiphile molecule. The latter is chemically different from
the phospholipid molecules. In this paper, however, we assume that the polymer
chains may be adsorbed on the membrane by many monomers that are directly
attached to some polar-heads of the host lipid molecules (mobile anchors), and
then organize in polydisperse loops. The surrounding liquid may be a good
solvent or a theta solvent. As we shall see below, the loops' polydispersity
and solvent quality drastically affect the phase behavior of the system. Under
a change in a suitable parameter, such as temperature, the phospholipids and
adsorbed polymer chains phase separate into macroscopic domains alternately
rich in the two components. For the determination of the phase diagram shape,
we elaborate on a theoretical model that takes into account both the loops'
polydispersity and the solvent quality.
This paper is organized as follows. In section 2, we derive
the expression for the mixing free energy of the phospholipid-anchor mixture.
To investigate the phase diagram shape is the aim of section 3. Finally, some
concluding remarks are given in section 4.
2. Mixing free energy
Polymers can adhere to biomembranes in several ways: (i) by
lipid anchors; that is, the polymer is covalently
2
bound to the polar-head of a lipid molecule (for many
proteins, the lipid anchors are glycosylphosphatidylinositol units) [12, 13];
(ii) by hydrophobic side-groups of the polymers which are integrated into the
bilayer [14, 15]; (iii) by membrane spanning hydrophobic domains of the polymer
(membrane-bound proteins, for example); and (iv) by a strong adsorption that
drives the polymer from a desorbed state to an adsorbed one [16]. For a single
polymer chain, the adsorption (on solid substrates) can be theoretically
studied using a scaling argument [17], for instance. In addition to the
polymerization degree of the polymer chain, N, and the excluded volume
parameter, v, the study was based on an additional parameter,
8, which is the energy (per kBT unit) required to
adsorb one monomer on the surface. For a strong adsorption, 8-1
defines the adsorbed-layer thickness. An adsorption transition takes
place at some typical value 8* of 8 scaling as [17]:
8* ~ R-1 F ~ aN-VF
(VF = 3/5), where RF is the Flory
radius of the polymer chain. For dilute and semi-dilute polymer solutions, the
adsorption phenomenon depends, in addition to the parameter 8, on the
polymer concentration.
In this paper, we consider situation (iv), where each monomer
has the same probability of being adsorbed on the membrane surface
[18]1. More precisely, a given monomer is susceptible to becoming
linked to a polar-head of a phospholipid molecule (anchor). As a result, the
polymer chains form polydisperse loops (with eventually one or two tails
floating in the aqueous medium). In fact, this assumption conforms to what was
considered in [19]. The case where no loops are present (adsorption only by one
extremity) was considered in [11]. Contrary to the adsorption on the solid
surface, the anchored polymer chains are mobile on the host membrane and may
undergo the aggregation transition that we are interested in. To be more
general, the fluid membrane is assumed to be in contact with a good solvent or
a theta solvent.
The purpose is to write a general expression for the mixing
free energy. The latter will allow the determination of the phase diagram
related to the aggregation of anchored polymer chains.
First, we start with the free energy (per unit area) of the
polymer layer (for solid substrates), which is given by [20]
|
F0 ' kBT
|
Z N (
1 [b2S (n)]â
+ [-b2S0 (n)] ln
b2 1
|
~ l\
- S0 (n) dn.
S1
(1)
|
For the configuration study, Guiselin [21] considered that
each loop can be viewed as two linear strands (two half-loop). Here, S(n)
is the number of strands having more than n monomers per unit
area, and b represents the monomer size. The number N denotes
the length of the longest strand. Hereafter, we shall use the notation
S1 = /a, which denotes the total number of grafted chains per
unit area, with 8 being the volume fraction of anchors and a
their area. If the mixture is assumed to be incompressible, then 1 - 8
is the volume fraction of the phospholipid molecules.
Let us come back to the free energy expression (1); note that
the first term of the right-hand side represents the
1 The adsorption of an adequate polymer on a fluid membrane
made of two phospholipids of different chemical nature may be a mechanism of
phase separation between these two unlike components as was pointed out.
Phys. Scr. 83 (2011) 065801 M Benhamou et
al
contribution of the (two- or three-body) repulsive
interactions between monomers belonging to the polymer layer. The second term
is simply the entropy contribution describing all possible rearrangements of
the grafted chains in the polymer layer. There, the exponent â depends on
the solvent quality [20]. When the grafting is accomplished in a dilute
solution with a good solvent or a theta solvent, the values of â are
â = 11/6 (blob model) or â = 2, respectively.
For polydisperse polymer layers, the distribution
S(n) is known in the literature [20]. This can be obtained by
minimizing the above free energy with respect to this distribution.
Without presenting details, we simply sketch the general
result that [20]
8
S(n) ~ an1/(â-1) . (2)
For usual solvents, we have
8
8
S(n) ~
(good solvents), (3)
(theta solvents). (4)
an
We shall rewrite the distribution S(n) as
8
S(n) = f (n), (5)
a
with f (n) ~
1/n1/(â-1), for polydisperse
polymer layers, and f (n) = 1 (for all n), for
polymer brushes. Therefore, F0 can be approximated by
F0
= a
kBT
-1u8â
+ç(N)8, (6)
where the linear term in 8 describes the contribution of
entropy to the free energy, where the coefficient ç(N) is as
follows (see the appendix):
lç(N) = a-1 [â
+ln(â - 1)] (1 - N1/(1-â)~
Explicitly, we have
æ(N) = 1 (polymer brushes), (11)
æ(N) ~ Nâ/(1-â) (polydisperse
systems). (12)
Note that æ(N) < 1, since, in all cases,
â > 1. Therefore, the polydispersity of loops decreases the repulsive
interaction energy.
Now, we have all the ingredients for the determination of the
expression for the mixing free energy. To this end, we imagine that the
interface is present as a two-dimensional (2D) Flory-Huggins lattice [17, 22],
where each site is occupied by an anchor or by a phospholipid molecule. Hence,
we can regard the volume fraction of anchors 8 as the probability that a given
site is occupied by an anchor. Therefore, 1 - 8 is the occupation probability
of phospholipids.
Before the determination of the desired mixing free energy
(per site), we note that the latter is the sum of three contributions, which
are the mixing entropy (per site), the volume free energy (per site) and the
interaction energy (per site) coming from the membrane undulations. Actually,
the induced attractive forces due to the membrane undulations are responsible
for the condensation of anchors. These forces balance the repulsive ones
between monomers along the connected polymer chains. We then write
F
kBT = 8ln8+(1-8)ln(1-8)+÷8(1-8)
+u8â +ç(N) 8. (13)
We note that, for polymer chains anchored by one extremity, a
big amphiphile molecule (the polymer brush case), the first contribution of
entropy, 8 ln 8, should be divided by some factor q, which represents
the ratio of the anchor area to the area of polar-heads of the host
phospholipids. In the present case, we have q = 1. In equality (11),
÷ accounts for the Flory interaction parameter
|
+ N1/(1-â) ln N }. (7)
â -1 JJJ
|
÷ = ÷0
|
(14)
1 D
A2 C -kBT
,
|
In the asymptotic limit N ? 8, this coefficient goes
to
ç(N) ? a-1 [â
+ln(â - 1)], (8)
provided that â > 1 (note that â =
11/6, for good solvents, and â = 2, for theta ones). This asymptotic
limit is always positive definite and inversely proportional to the polar-head
area a. In fact, the positive sign of the coefficient
ç(N) agrees with the entropy loss due to the polymer chains
grafting. As we shall see below, this linear contribution does not change the
phase diagram in the composition-critical parameter plane. Here, the coupling
constant u is as follows:
u
(9)
with
æ(N) =
=(b2 1f-1 a) Næ(N),
1N
N J1[ f
(n)]â dn. (10)
3
where the positive coefficients C and D are
such that (in dimension 2) [23]
D = -ð f°°
rU(r) dr, C = ð2
ó2 (covolume). (15)
Here, U(r) is the pair potential induced by
the membrane undulations ([11]; [24] and references therein)
cr
i
U(r) =
8, r < ó,
-AH(
ól4 (16)
/ , r >
ó, r
where ó is the hard disc diameter, which is
proportional to the square root of the anchor area a. There, the
potential amplitude AH plays the role of the Hamaker constant. It was
found that the latter decays with the bending rigidity constant according to
([11]; [24] and reference therein)
AH ~ ê-2. (17)
Phys. Scr. 83 (2011) 065801 M Benhamou et
al
But this amplitude is also sensitive to temperature. We remark
that the above mixing free energy is not symmetric under the change 8 ? 1-8.
Some straightforward algebra gives the following expression for
the attraction parameter D:
ð
2ó2 ~
D = AH
ê-2
ó2 . (18)
Therefore, those membranes with a small bending modulus induce
significant attraction between anchors.
In formula (12), ÷0 > 0 is the Flory interaction
parameter describing the chemical segregation between amphiphile molecules that
are phospholipids and anchors. This segregation parameter is usually written
as
ã0
÷0 = á0 + T , (19)
where the coefficients á0 and ã0 depend on the
chemical nature of the various species. Also, the total interaction parameter
÷ can be written as
ã
÷ = á + T , (20)
with the new coefficients
C D
á = á0 A2, ã =ã0+
kB A2 . (21)
These coefficients then depend on the chemical nature of the
unlike components and on the membrane's characteristics through its bending
modulus ê.
If we admit that the coupling constant u has a slight
dependence on temperature, we will draw the phase diagram in the plane of
variables (8, ÷). Indeed, all of the temperature dependence is contained
in the Flory interaction parameter ÷. interactions widen the compatibility
domain, and then, the separation transition appears at low temperature.
Now, to see the influence of the solvent quality, we rewrite
the interaction parameter u as u =
u0æ(N) ~ u0Nâ/(1-â) <
u0, where u0 is the interaction parameter relative to a
monodisperse system. Thus, the polydispersity of loops has a tendency to reduce
the compatibility domain in comparison with the monodisperse case.
The critical volume fraction, 8c, can be obtained
by minimizing the critical parameter ÷(8) with respect to the 8-variable.
We then obtain
1 1
(1 - 8c)2 82 c
+â(â - 1) (â - 2)
u8â-3
c = 0. (23)
For good solvents (â = 11/6), we have
1
(1 - 8c)2
1
216u8-7/6
55 c = 0. (24)
82 c
Therefore, the critical volume fraction is the abscissa of the
intersection point of the curve of the equation (2x - 1)/
x5/6(1 - x)2 and the
horizontal straight line of the equation y = (55/216)u. Note
that this critical volume fraction is unique, and in addition, it must be
greater than the value 1/2 (for mathematical compatibility). The coordinates of
the critical point are (8c, ÷c), where 8c
solves the above equation and ÷c = ÷(8c).
The latter can be determined by combining equations (19) and (21). For theta
solvents (â = 2), the coordinates of the critical point are exact,
8c = 12, ÷c =
2+u, (25)
where the interaction parameter u scales as
u =
b2 N-1. (26)
a
3. Phase diagram
With the help of the above mixing free energy, we can
determine the shape of the phase diagram in the (8, ÷)-plane that is
associated with the aggregation process that drives the anchors from a
dispersed phase (gas) to a dense one (liquid). We focus only on the spinodal
curve along which the thermal compressibility diverges. The spinodal curve
equation can be obtained by equating to zero the second derivative of the
mixing free energy with respect to the anchor volume fraction 8; that is,
?2F/?82 = 0. Then, we obtain the following
expression for the critical Flory interaction parameter:
÷(8) = 2 (8(11 8)
+â(â - 1) u8â-2 I. (22)
Above this critical interaction parameter appear two phases:
one is homogeneous and the other is separated. Of course, the linear term in 8
appearing in equality (11) does not contribute to the critical parameter
expression.
We remark that, in usual solvents, this critical interaction
parameter is increased due to the presence of (two-or three-body) repulsive
interactions between monomers belonging to the polymer layer. This means that
these
The above relation clearly shows that the polymer chains'
condensation rapidly takes place only when the characteristic mass N
is high enough. The same tendency is also seen in the case of good
solvents.
4
It is straightforward to show that the critical fraction and
the critical parameter are shifted to lower values in the case of polydisperse
systems, whatever be the quality of the surrounding solvent.
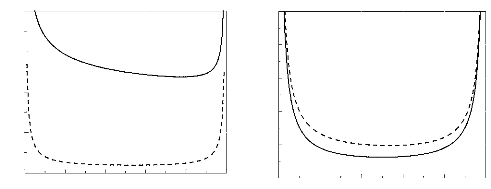
In figure 1, we present the spinodal curve for monodisperse
(with no loops) and polydisperse (with loops) systems, with a fixed parameter
N. We have chosen the good solvents situation. For theta solvents, the
same tendency is seen.
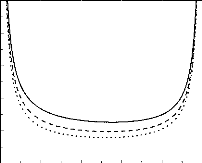
We present in figure 2 the spinodal curve for a polydisperse
system (with loops), at various values of the parameter N. As expected
the critical parameter is shifted to higher values when we augmented the
typical polymerization degree.
Finally, we compare, in figure 3, the spinodal curves for a
polydisperse system (with loops) for the case of theta solvents and those for
the case of good solvents, at fixed parameters b and N. All
the curves in the figure reflect our discussions above.
Phys. Scr. 83 (2011) 065801 M Benhamou
etal
÷
80
60
40
20
0
0,0 0,2 0,4 0,6 0,8 1,0
÷
20
16
12
8
4
0
0,0 0,2 0,4 0,6 0,8 1,0
Ö
Figure 1. Spinodal curves (in a good solvent)
for monodisperse (dashed line) and polydisperse (solid line) systems, when
N = 100, with the parameter b2 = 0.5a.
÷
20
16
12
8
4
0
0,0 0,2 0,4 0,6 0,8 1,0
Ö
Figure 2. Spinodal curves for a polydisperse
system, when N = 50 (solid line), 100 (dashed line) and 150 (dotted
line), with the parameter b2 = 0.5a . We assumed
that the surrounding liquid is a good solvent. For theta solvents, the tendency
is the same.
4. Discussion and conclusions
This paper is devoted to the thermodynamic study of the
aggregation of the polymer chains adsorbed on a soft surface. Such an
aggregation is caused by competition between the chemical segregation between
phospholipids and grafted polymer chains, their volumic interactions and the
membrane undulations. More precisely, we addressed the question of how these
polymer chains can be driven from a dispersed phase (gas) to a dense one
(liquid), under a change in a suitable parameter, e.g. the absolute
temperature.
To be more general, we achieved the study in a unified way;
that is, we have considered more realistic physical situations: the solvent
quality (good or theta solvents) and the polydispersity of the adsorbed loops
formed by the grafted
Ö
5
Figure 3. Superposition of spinodal curves for a
polydisperse system for the case of theta solvents (solid line) on those for
the case of good solvents (dashed line). For these curves, we chose N
= 100 and b2 = 0.5a.
polymer chains. The latter were taken into account through the
well-known form of the chains' length distribution.
In doing so, we first computed the expression for the mixing
free energy by adopting the Flory-Huggins lattice image usually encountered in
polymer physics [17, 22]. Such an expression shows that there is competition
between four contributions: entropy, chemical mismatch between unlike species,
interaction energy induced by the membrane undulations and the interaction
energy between monomers belonging to the grafted layer. Such a competition
governs the phases succession.
We emphasize that the present work and a previous work [11]
differ from another previous work that was concerned with the same problem, but
in which the substrate was assumed to be a rigid surface [3]. Therefore, the
membrane undulations were neglected. As we have seen, these undulations
increase the segregation parameter ÷ by an additive term,
÷m, scaling as ê-2. This means that
the phase separation is accentuated due to the presence of thermal
fluctuations. Compared with the previous work [11], which was concerned with
soft brushes with monodisperse end-grafted polymers, the present work is more
general, since it takes into account both the polydispersity of loops forming
the adsorbed polymer chains and the solvent quality. Thus, the present study
was achieved in a unified way. As we have seen, these details drastically
affect the phase diagram architecture.
Now, let us discuss further the influence of solvent quality
on the critical phase behavior. We recall that the solvent quality appears in
the free energy (11) through the repulsion parameter u, defined in
equation (7). We find, in the N ? 8 limit, that ug
~ N1/5uè, where the
subscripts g and è stand for good and theta solvents,
respectively. This implies that the good solvent plays the role of a stabilizer
regarding the phase separation.
Finally, this work must be considered as a natural extension
of a study reported previously [11], which was concerned with monodisperse
end-grafted polymer chains
Phys. Scr. 83 (2011) 065801 M Benhamou
etal
trapped in a good solvent. Therefore, the present work
presents a wide perspective on the phenomenon of segregation between the host
phospholipids and grafted polymer chains on bilayer membranes.
Acknowledgments
We are indebted to Professors T Bickel, J-F Joanny and C
Marques for helpful discussions during the First International Workshop on
Soft-Condensed Matter Physics and Biological Systems (14-17 November 2006,
Marrakech, Morocco). MB thanks Professor C Misbah for fruitful correspondence
and the Laboratoire de Spectroscopie Physique (Joseph Fourier University of
Grenoble) for their kind hospitality during his visit. We are grateful to the
referee for critical remarks and useful suggestions that helped us to improve
the scientific content of this paper.
Appendix
The aim is to determine the coefficient ç(N)
appearing in formula (6). We start with the entropy contribution to the free
energy
|
Fentropic
kBT
|
Z N
1
(~-b2S0(n)~ ln
'
b2 1
|
~ ~~
- S0(n)
S1
|
dn, (A.1)
|
where the loop-size distribution is defined in equation (2).
Then, the above expression can be rewritten as
= ç(N) 8, (A.2)
Fentropic kBT
with
Z N ( 1 ])
ç(N) = a-1 1
nâ/(1-â) ln â - 1nâ/(1-â)
dn.
â - 1 1
(A.3)
Straightforward algebra gives
{ç(N) = a-1 [â
+ln(â - 1)] (1 - N1/(1-â)~
~
â
+ â - 1 N1/(1-â) ln N. (A.4)
This concludes the determination of the coefficient
ç(N).
References
[1]
6
Lipowsky R and Sasckmann S (ed) 1995 Structure and
Dynamics of Membranes: From Cells to Vesicles vol 1A (New York:
Elsevier)
[2] Benhamou M 2008 Lipids Insights 1
2
[3] Nicolas A and Fourcade B 2003 Eur. Phys. J.
E 10 355
[4] Aubouy M et al 2000 Phys. Rev.
Lett. 84 4858 Aubouy M et al 1996 Macromolecules 29
7261
[5] Ringsdorf H and Schmidt B 1986 How to bridge the gap
between membrane, biology and polymers science Synthetic Membranes:
Science, Engineering and Applications ed P M Bungay et al
(Dordrecht: D Reidel) p 701
[6] Lasic D D 1992 Am. Sci. 80 250
[7] Torchilin V P 1996 Effect of polymers attached to
the lipid head groups on properties of liposomes Handbook of Nonmedical
Applications of Liposomes vol 1, ed D D Lasic and Y Barenholz (Boca Raton,
FL: RCC Press) p 263
[8] Joannic R, Auvray L and Lasic D D 1997 Phys.
Rev. Lett. 78 3402
[9] Discher B M et al 1999 Science 284
1143
[10] Elferink M G L et al 1992 Biochim.
Biophys. Acta 1106 23
[11] Benhamou M, Joudar J and Kaidi H 2007 Eur.
Phys. J. E 24 343
[12] Blume G and Cev G 1990 Biochim. Biophys. Acta
1029 91
[13] Lasic D D, Martin F J, Gabizon A, Huang S K and
Papahadjopoulos D 1991 Biochim. Biophys. Acta 1070 187
[14] Decher G et al 1989 Angew. Makromol.
Chem. 166 71
[15] Simon J, Kuhner M, Ringsdorf H and Sackmann E
1995 Chem. Phys. Lipids 76 241
[16] Garel1 T, Kardar M and Orland H 1995 Europhys.
Lett. 29 303
Chatellier X and Andelman D 1995 Europhys.
Lett. 32 567 Lipowsky R 1995 Europhys. Lett. 30 197
Xie A F and Granick S 2002 Nature Mater. 1
129
[17] de Gennes P-G 1979 Scaling Concept in Polymer
Physics (Ithaca, NY: Cornell University Press)
[18] Binder W H, Barragan V and Menger F M 2003
Angew. Chem. Int. Edn Engl. 42 5802
[19] Manghi M and Aubouy M 2001 Adv. Coll. Interf.
Sci. 94 21
[20] Manghi M and Aubouy M 2003 Phys. Rev. E 68
041802
[21] Guiselin O 1992 Euro. Phys. Lett. 17 225
Guiselin O 1992 PhD Thesis Paris VI University
[22] Flory P J 1953 Principles of Polymer Chemistry
(Ithaca, NY: Cornell University Press)
[23] Balian R 2006 From Microphysics to
Macrophysics--Methods and Applications of Statistical Physics (Berlin:
Springer)
[24] Marchenko V I and Misbah C 2002 Eur. Phys.
J. E 8 477
elhasnaouikhalid@gmail.com
elhasnaouikhalid@hotmail.fr
| 

