PLAN DE TRAVAIL
INTRODUCTION
Première Partie : BIBLIOGRAPHIE
CHAPITRE I : GENERALITES SUR LE PALUDISME ET LES
MEDICAMENTS ANTIPALUDIQUES
I. LA MALARIA
A. Historique et définition
1. Brève historique
2. Définition de la malaria
B. Les Plasmodiums
1. Définitions
2. Classification des Plasmodiums
3. Les Plasmodiums et leur paludisme
C. Cycle du développement des
Plasmodiums
1. Cycle évolutif chez l'homme (schizogonie)
2. Cycle évolutif chez le moustique (sporogonie)
D. Réactions immunitaires de l'hôte
humain et mécanismes de survie du parasite
1. Réactions immunitaires
Immunité non adoptive (naturel)
Immunité acquise (adoptive)
2. Mécanismes de survie parasitaire
E. Les formes cliniques de la
malaria
F. La prise en charge de la
malaria
1. Diagnostic de l'infection palustre
2. Traitement
II. LES MEDICAMENTS ANTIPALUDIQUES
A. Considérations
générales
1. Définition générale des
médicaments
2. Catégories des médicaments
3. Médicaments génériques
4. Médicaments essentiels
5. Médicaments contrefaits ou de qualité
inférieure
a) Définition
b) La Contrefaçon et le Marché des
Médicaments
6. Médicaments Pré- qualifiés
7. La Péremption et la Dégradation des
Médicament
a) La Péremption des Médicaments
b) La Dégradation des Médicaments
8. Stabilité et durée de vie des
Médicaments (ou Produits finis)
B. Définitions des
antipaludiques
1. Définition des antipaludiques
2. Historique des antipaludiques
3. Classification des antipaludiques
a) Schizonticides tissulaires
b) Schizonticides sanguins
c) Gamétocides
d) Les sporontocides
C. Les antipaludiques
1. La Quinine et ses dérivés
2. Les dérivés 4-aminoquinoleines
3. Les dérivés 8-aminoquinoléines
4. L'artémisinine et ses dérivés
5. Autres antipaludiques et associations antipaludiques :
a) Les amyl aminoalcools
b) Les antifoliques
c) Les antifoliniques
d) Quelques Associations antipaludiques
CHAPITRE II. GENERALITES SUR LES ACT ET
L'ARTESUNATE
I. LES ACT OU CTA (Combinaisons
Thérapeutiques à base d'artémisinine)
A. Introduction des ACT dans la thérapie
antipaludique
B. Les ACT en RDC
C. Le pourquoi et l'avantage des ACT comme
associations médicamenteuses
D. Le coût des ACT
II. L'ARTESUNATE
A. Origine de l'artesunate
B. Quelques aspects
pharmacologiques de l'artesunate
C. Importance de l'artesunate dans le traitement du
paludisme
CHAPITRE III. LA RESISTANCE AUX
ANTIPALUDIQUES
A. Définition de
Pharmacorésistance
B. Origine et historique de la résistance
aux antipaludiques
1. Origine de la pharmacorésistance aux
antipaludiques
2. Historique de la pharmacorésistance aux
antipaludiques
C. Quelques antipaludiques présentant la
résistance
Deuxième Partie :
EXPERIMENTATION
CONCLUSION
REFERENCES BIBLIOGRAPHIQUES
« Contribution à l'évaluation de
la stabilité des comprimés à base d'Artésunate en
co-blister avec l'Amodiaquine de la spécialité ARSUCAM®
provenant des sources pré- qualifiées une année
après la péremption: cas de la province du Bas-Congo
(Kinsantu) »
CHAPITRE I : GENERALITES SUR LE PALUDISME ET LES
MEDICAMENTS ANTIPALUDIQUES
I. LA MALARIA
A. Historique et
définition
1. Brève historique
Les sociétés de l'époque primitive
considéraient la malaria comme une manifestation du châtiment
divin car elle causait beaucoup des victimes. Les Egyptiens savaient
déjà que cette fièvre survenait après des
inondations et des pluies, et déconseillaient l'exposition en plein air,
des individus durant ces périodes. (1)
La médecine grecque connait bien les fièvres
palustres, en particulier celles qui se répètent à des
intervalles réguliers. Hippocrate décrivit les formes cliniques
palustres (frissons et températures). En 1630 un aristocrate espagnol,
Don Francisco Lopez, apprit des indiens du Pérou les
vertus thérapeutiques des écorces de quinine contre la malaria.
(1)
Depuis l'époque où les Indiens utilisaient
l'écorce de quinquina (antipaludéen naturel) contre les
fièvres, de nombreux progrès ont été
réalisés dans la compréhension du paludisme. En 1920
Pelletier et Caventou isolent des écorces du quinquina une trentaine
d'alcaloïdes dont les célèbres quinines et quinidines.
Laveran découvrit en 1880 que cette maladie
était due à un parasite, le plasmodium. Les premiers
médicaments antipaludéens synthétiques furent quant
à eux mis au point en 1940. Mais dès 1960, une résistance
à la chloroquine est apparue en Asie du Sud-est. (2)
2. Définition de la malaria
La malaria est une maladie parasitaire due à un
hématozoaire du genre Plasmodium, transmis par un moustique
anophèle femelle. Etymologiquement, paludisme vient de palus
qui veut dire marrais et mal aria qui signifie mauvais air. (3)
Cette maladie est présente dans plus de cent pays du
monde : en Afrique, en Asie, en Océanie, en Amérique
centrale et du sud et dans certaines ils caraïbes. Comme
approximativement 60% de la population mondiale vivent dans ces pays, on estime
à plus de deux milliards le nombre de sujets exposés, avec deux
cent millions de malades et deux millions de morts chaque année. (4)
Pour l'Afrique noire seule, on évalue sa
mortalité à un million par an et fait partie de trois grandes
causes de morbidité et de mortalité en R.D.CONGO. (3)
III. Les
Plasmodiums
1. Définitions
Découverts par Alphonse LAVERAN en
1880, les Plasmodiums sont des parasites intracellulaires de forme
amoeboïde qui présentent au cours de leur cycle une alternance de
reproduction asexuée (schizogonie) chez l'hôte
vertébré et de reproduction sexuée (sporogonie) chez
l'hôte invertébré. Il existe 146 espèces de
Plasmodium dont quatre seulement (P.falciparum, P. vivax, P. malaria et P.
ovale) parasitent l'homme comme l'hôte vertébré et le
moustique hôte invertébré. Ils sont les agents causaux de
la malaria et le moustique est leur vecteur. (2)
2. Classification des Plasmodiums
Selon qu'ils disposent ou non des endosymbiontes, selon leurs
modes de locomotion ou de reproduction, les protozoaires sont subdivisés
en sept taxons. Les plasmodiums forment le troisième taxon qui regroupe
les protozoaires caractérisés par la présence d'une
mitochondrie, d'un complexe apical d'induction d'endocytose, d'une reproduction
sexuée (sporogonie) et asexué (schizogonie). (2)
De manière brève la classification est la
suivante:
- Ordre : Haemasporida
- Sous-ordre : Haemasporidae
- Famille : Plasmodiae
- Genre : Plasmodium
Le P. falciparum appartient au sous genre laverania
parce que les gamétocytes sont falciformes et le reste de forme
arrondie. (2)
3. Les
Plasmodiums et leur paludisme
Paludisme à Plasmodium vivax : étant
largement répandu, il détermine habituellement des accès
fébriles sans gravité. Les infestations massives et
répétés engendre parfois des tableaux
sévères tels que : fièvres rémittentes et
paludisme viscéral évolutif mais jamais d'accès
pernicieux. (3)
Paludisme à Plasmodium malariae : est
surtout remarquable par la périodicité de ses accès
intermittents et sa longévité. (3)
Paludisme à Plasmodium ovale : proche du
paludisme à P. vivax par sa bénignité, sa durée
d'incubation varie de quinze jours à plusieurs mois. L'accès de
reviviscence schizogonique évolue sur rythme de tierce ; si le
malade n'est pas traité, les accès bénins se
répéteront à court-terme mais les rechutes tardives sont
rares (5 ans au minimum). (3)
Paludisme à Plasmodium falciparum :
sévissant intensément sous les tropiques, il peut tuer, par
accès pernicieux, les sujets non prémunis ne se soumettant
à aucune chimioprophylaxie (enfants autochtones de 4 mois à 4 ans
surtout mais aussi les européens récemment transplantés en
zone endémique). (3)
En revanche la longévité du P. falciparum ne
dépassant guère trois mois, il n'existe pas de véritables
rechutes. Mais en région impaludée, les réinfestations
sont fréquentes. Trois tableaux peuvent être
individualisés: accès simple, accès pernicieux et celui du
paludisme viscéral évolutif. Quant à la fièvre
bilieuse hémoglobinurique, ce n'est pas à proprement parler une
manifestation palustre.
Il faut noter que seul le paludisme à P. falciparum
peut tuer car il est le seul à conduire aux accès pernicieux.
(3)
IV. Cycle du développement des
Plasmodiums
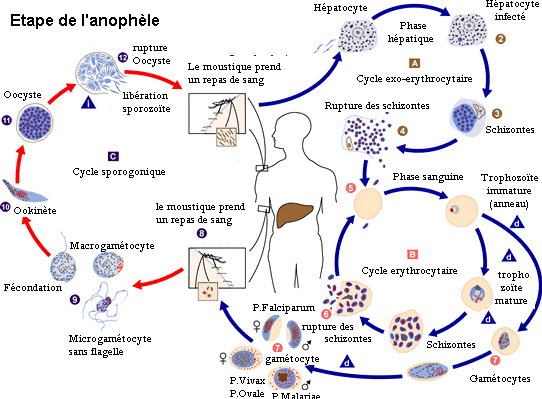
En fonction de ses deux hôtes, le vertébré
(homme) et l'invertébré (moustique) on divisera le cycle
évolutif du Plasmodium en deux grandes parties à savoir la
schizogonie et la sporogonie.
1. Cycle évolutif chez l'homme (schizogonie)
Suivant les tissus qu'ils occupent majoritairement chez
l'homme ce cycle se divise en cycle pré-érythrocytaire
(hépatique ou symptomatique) et cycle érythrocytaire, ce dernier
conduit à la fièvre suite à l'éclatement des
globules rouges. (3)
Cycle
pré-érythrocytaire (hépatique ou
asymptomatique)
Les plasmodiums sous forme de sporozoïtes sont
inoculés lors de la piqûre d'un anophèle femelle
infesté et gagnent les hépatocytes. En se multipliant, ils se
transforment en schizonte extraérythrocytaire ou intra-hépatique
(corps bleu). L'éclatement des corps bleu libère des
mérozoïtes. (3)
Cycle érythrocytaire (symptomatique)
Les mérozoïtes gagnent le sang
périphérique et parasitent les globules rouges en devenant, au
fur et à mesure de leur croissance, trophozoïtes, schizontes
sanguins puis corps en rosace.
Les corps en rosace éclatent libèrent des
mérozoïtes. L'apparition des éléments à
potentiel sexué ou gamétocytes est plus tardive. (3)
2. Cycle évolutif chez le moustique (sporogonie)
Les gamétocytes aspirés avec le sang par le
moustique lors de son repas, gagnent son estomac pour se transformer en
gamète. Après fécondation les gamètes femelles
deviennent des ookinètes libres, puis oocystes fixes.
L'éclatement des oocystes libère des sporozoïtes qui gagnent
les glandes salivaires de l'anophèle à partir desquelles ils
peuvent être retransmis à un autre hôte
vertébré (homme).
Il est à noter que P. ovale et P. vivax avant de passer
à la forme schizonte hépatique, leur mérozoite peut
passer par la forme hypnozoïte qui est susceptible de se réveiller
après plusieurs mois ou plusieurs années. (3)
D. Réactions immunitaires de l'hôte humain et
mécanismes de survie du parasite
1. Réactions immunitaires
Immunité non adoptive (naturel) :
La compatibilité hôte-parasite ne dépend
nullement des facteurs sériques, mais de la présence des
récepteurs spécifiques présents sur les membranes des
parasites et des érythrocytes. Il peut être observé dans
certains cas une résistance au paludisme. (2)
Tous les plasmodiums en général ne
résistent pas chez les individus ayant un déficit en
6-phosphoglucose réductase (6-GPDH).Le P. vivax ne subsiste pas chez les
individus non porteurs du gène Duffy qui assure la synthèse des
récepteurs nécessaires à la pénétration de
sa forme merozoïte dans le globule rouge. (2)
Les facteurs intra-érytrocytaires seraient responsables
du ralentissement de l'acquisition de l'immunité des blancs par rapport
aux noirs dans la malaria à P. falciparum. L'hémoglobine S inhibe
le développement de la schizogonie du P. falciparum. C'est pourquoi on
n'observe pas d'accès pernicieux chez les sujets drépanocytaires
hétérozygotes. (2)
Les anomalies du cytosquelette telles que l'ovalocytose ou
ellipocytose, s'opposent à l'induction de l'endocytose du plasmodium
dans l'érythrocyte. (2)
Immunité acquise (adoptive)
La réponse immunitaire exprimée par la
production de différentes classes d'immunoglobulines semble
principalement être dirigée contre les formes
érythrocytaires asexuées du plasmodium. Les anticorps produits
sont de faible affinité et la mémoire immunologique induite est
de très courte durée (1 à 2 ans pour le P. falciparum).
(2)
Dans les six premiers mois de la vie, l'enfant né d'une
mère immune est relativement protégé contre les
accès palustres grâce à l'immunité passive, avec les
immunoglobulines, transmise par la mère via le placenta. Ayant par la
suite perdu cette immunité passive, il devient, avant d'avoir
réussi à constituer ses progrès en défenses
immunitaires, avant l'âge de cinq ans, un sujet particulièrement
vulnérable. C'est cette strate de la population en zone endémique
qui paie le plus lourd tribut au paludisme. (2)
L'état de grossesse, surtout le premier trimestre et le
second dans une certaine mesure, induit une immuno suppression dite
physiologique qui amplifie malencontreusement la pathogénicité de
P. falciparum pour cette strate de la population, et compromet ainsi la
prémunition passive au nouveau-né. (2)
2. Mécanismes de survie parasitaire
Les plasmodiums présentent au cours de leur cycle
évolutif différents motifs antigéniques superficiels qui
sensibilisent de façon variée le système immunitaire de
l'hôte : sporozoïtes, schizontes sanguins, schizontes
hépatiques et gamétocytes. (2)
Chacun de ces antigènes induit une réponse
immunologique spécifique de faible amplitude qui ne suffit nullement
à conférer au sujet infecté une
immunité vraiment efficace et de longue durée, d'où
il y aura une ré-infection habituelle des sujets déjà
immuns. Et ceci s'observe dans les zones endémiques où circulent
plusieurs variants antigéniques différents d'un isolat à
l'autre et contre les quels l'hôte n'est pas nécessairement
immunisé. (2)
La recombinaison des gènes par la reproduction
sexuée dans l'estomac du moustique, participe au brassage et au
renouvellement de cette capacité de variation antigénique qui
paralyse la machinerie immunitaire de l'hôte. Toutes les 48 heures le P.
falciparum peut modifier environ 20% de la structure primaire de ses
protéines de surface, on dit qu'il joue au dé avec notre
système immunitaire. (2)
Certaines souches de P. falciparum acquièrent la
capacité d'expurger de leur cytoplasme certaines substances
antipaludiques, notamment la chloroquine, 40 à 50 fois plus vite que ne
ferait une souche normale, ce qui explique leur résistance
vis-à-vis de ces produits. (2)
E. Les formes cliniques de la malaria (4)
Les signes de l'infection palustre dépendent des
espèces du parasite et l'état immunologique du patient. Parmi les
multiples formes que présente la malaria on en citera que quelques
unes ;
- Accès simple : simple fièvre
- Accès pernicieux (accès aigu de
paludisme) : fièvre pernicieuse
- Hyperthermie : au dessus de 38,5°C pouvant
atteindre 42°C
- Anémie et thrombocytémie : la
destruction des globules rouges selon la durée et l'intensité
peut entraîner une anémie qui s'accompagne souvent de
l'hémostase due à la thrombocytopénie.
- Fièvre bilieuse hémoglobinurique :
manifestation grave du paludisme à P. falciparum observée chez
les sujets non prémunis ou à chimioprophylaxie
irrégulière par la quinine. Elle se manifeste par une
hémolyse brutale avec frissons, fièvre, vomissements,
ictère, anémie et hémoglobinurie.
- Neuropaludisme (malaria cérébrale) : est
un accès pernicieux cérébral qui est polymorphe dans ses
manifestations, il a comme signes les troubles de conscience ou de
comportement, les convulsions ou coma.
- Néphrites et insuffisance rénale
- Hypoglycémie
- Troubles électrolytiques et acido-basiques :
dus à l'hypovolémie, déshydration et d'acidose
respiratoire ou métabolique avec élévation du taux de
lactate dans le sang et dans le liquide céphalo-rachidien.
- Oedème pulmonaire aigu
- Collapsus cardio-vasculaire
F. La prise en charge de la malaria
1. Diagnostic de l'infection palustre
Le diagnostic du paludisme est loin d'être toujours
facile à dresser. Hormis la forme classique, il peut en effet prendre de
nombreux aspects trompeurs. On peut ainsi aisément le confondre avec une
simple turista, une grippe, une hépatite
virale et, dans les cas d'atteintes cérébrales, on peut
hésiter sur un diagnostic de méningite ou
d'abcès cérébral.
Le retard du diagnostic et d'un traitement adéquat
ayant parfois de graves conséquences, il importe de penser à
pratiquer un examen complémentaire, c'est-à-dire un frottis
sanguin, face à un patient fébrile revenant d'un pays
infesté. Le traitement, à condition qu'il soit rapidement
entrepris, permet d'éviter le passage à la forme grave,
appelé « accès pernicieux palustre ». (5)
Donc, Il n'existe malheureusement pas d'examen de laboratoire
qui permet de poser le diagnostic de paludisme-maladie car même si le
frottis sanguin se révèle positive ce n'est pas une
preuve de l'accès palustre. Puisqu'il faudra recourir aux examens
cliniques comme l'épreuve thérapeutique. (2)
2. Traitement
Il est essentiellement basé sur la chimie, avec la
chimioprophylaxie et chimiothérapie qui utilisent les médicaments
antipaludiques seuls ou en association. Mais leur choix est fonction de la
tolérance et de la pharmacorésistance. (3)
II. LES MEDICAMENTS ANTIPALUDIQUES
A. Considérations
générales
1. Définition générale des
médicaments
Le médicament est toute substance qui possède
des propriétés curatives ou préventives à
l'égard des maladies. Par extension on le considère comme tout
produit pouvant être administré à l'homme en vue
d'établir un diagnostic médical, ou de restaurer, corriger ou
modifier une fonction organique. (6)
2. Catégories des médicaments
Suivant l'origine de leurs formules de préparation on
a :
- Médicament magistral
C'est toute préparation réalisée par le
pharmacien dans son officine sur base d'une formule détaillée
d'une prescription médicale. (6)
- Médicament officinal
Il s'agit d'une préparation dont la composition et le
mode de préparation sont inscrits dans la pharmacopée ou dans un
formulaire national. (6)
- Spécialité pharmaceutique
C'est un médicament préparé à
l'avance, présenté sous un conditionnement particulier, mis au
marché sous une dénomination spéciale et destiné
à être dispensé dans plusieurs officines. (6)
3. Médicaments génériques
C'est une copie d'un médicament original,
utilisé dans la plupart des pays pendant une durée suffisante
pour que les brevets de propriété industrielle de sa
molécule soient tombés dans le domaine public. Bref c'est une
copie légale. Ils sont souvent fabriqués par des petites
structures, avec des frais généraux réduits et une absence
de dépenses de recherche et de promotion, c'est pourquoi ils ont un prix
réduit dans lequel le coût des matières premières
devient le facteur prédominant. (7)
«Les médicaments essentiels sont ceux qui
satisfont aux besoins de la majorité de la population en matière
de soins de santé ; ils doivent donc être disponibles à
tout moment, en quantité suffisante et sous la forme
thérapeutique appropriée". (21) p.52
4. Médicaments essentiels
Ce concept fut lancé par l'O.M.S. dans les
années 1975, a permis de déterminer une liste évolutive
des produits de base surtout des médicaments génériques,
qui laisse à chaque pays une possibilité d'adaptation selon ses
spécificités, besoins et potentiels.
5. Médicaments contrefaits ou de qualité
inférieure
a) Définition
Les médicaments de qualité inférieure
sont des produits dont la composition et les principes ne répondent pas
aux normes scientifiques et qui sont par conséquent inefficaces et
souvent dangereux pour le patient. La qualité inférieure peut
être le résultat d'une négligence, d'une erreur humaine, de
ressources humaines et financières insuffisantes ou d'une
contrefaçon. (8)
Selon l'OMS, un médicament contrefait est :
« un produit étiqueté frauduleusement de manière
délibérée pour en dissimuler la nature et/ou son origine.
La contrefaçon peut concerner aussi bien des produits de marque que des
produits génériques. Ils peuvent contenir des principes actifs
authentiques mais avec un emballage imité, d'autres principes actifs,
aucun principe actif ou des principes actifs en quantité
insuffisante ». (9)
Le problème des médicaments contrefaits
s'inscrit dans le cadre plus large des produits pharmaceutiques de
qualité inférieure. La différence tient à ce qu'ils
soient étiquetés frauduleusement de manière
délibérée pour en dissimuler la nature et/ou la source. La
contrefaçon peut concerner aussi bien des produits de marque que des
produits génériques, et les médicaments contrefaits
peuvent comprendre des produits qui contiennent les principes actifs
authentiques mais un emballage imité, ou d'autres principes actifs,
aucun principe actif ou des principes actifs en quantité insuffisante.
(8)
Dans les pays plus riches, la contrefaçon concerne le
plus souvent des médicaments coûteux tels que les hormones, les
corticoïdes et les antihistaminiques. Dans les pays en
développement, les médicaments qui font le plus souvent l'objet
de contrefaçons sont ceux qu'on utilise contre des affections
potentiellement mortelles comme le paludisme, la tuberculose et le VIH/SIDA.
(8)
b) La contrefaçon et le marché des
médicaments
Les contrefaçons représentent 10% du
marché mondial des médicaments et les recettes mondiales de la
vente des médicaments contrefaits et de qualité inférieure
atteignent plus de US $32 milliards par an. La Food and Drug Administration
(FDA), autorité sanitaire américaine, estime que 25% des
médicaments consommés dans les pays les plus pauvres sont des
faux et ont une réduction de prix de 45%. Ces pays pauvres sont ceux de
l'Afrique, Asie et Amérique du sud. Ainsi 10% des médicaments au
Brésil sont contrefaits. Des testes d'échantillons d'artesunate
collectés au Cambodge, Laos, Birmanie (Myanmor) Thaïlande et
Vietnam de août 1999 et août 2000 ont montre que 38% des
échantillons ne contenaient aucune trace d'artesunate. (8)
Il ressort aussi d'une récente étude parue dans
The Lancet que jusqu'à 40 % des produits supposés contenir de
l'artésunate (le meilleur médicament disponible aujourd'hui
contre le paludisme chimiorésistant) ne contenaient pas en fait de
principe actif et n'avaient aucun effet thérapeutique. (8)
6. Les Médicaments Pré
-qualifiés
Définition
Le programme de pré- qualification des Nations Unies
est un plan d'action visant à élargir l'accès aux
médicaments des personnes atteintes par le VIH/SIDA, la Tuberculose ou
le Paludisme et d'assurer la qualité, l'efficacité et la
sécurité de ces médicaments dans toute la chaîne de
fabrication et de distribution. (22)
C'est l'Organisation Mondiale de a Santé (OMS) qui
gère et organise ce projet pour le compte des Nations Unies. Elle lui
sert donc de support technique et scientifique en garantissant les normes et
standards internationaux qu'on utilise pour l'évaluation, l'inspection
et le contrôle ainsi que la conduite du programme. Le FNUAP, l'UNICEF,
L'ONUSIDA et la Banque Mondiale sont des partenaires mais les acteurs sont
majoritairement les évaluateurs, inspecteurs et laboratoires de
contrôle des autorités compétentes des pays membres de
l'ICH ou CIH (Conférence internationale sur l'harmonisation des
exigences techniques relatives à l'homologation des produits
pharmaceutiques). (22)
Ainsi un Médicament Pré- qualifié est
celui dont le fabricant et lui-même font partie de façon
permanente du programme de pré- qualification de l'OMS.
Lancé en 2001, jusqu'en 2005, seulement 2 anti-
paludéens, 96 anti-rétroviraux et 8 anti-tuberculeux
étaient pré- qualifiés, car la majorité des
produits fabriqués, distribués et administrés ne
répondent pas aux standards internationaux de qualité,
efficacité et sécurité. Il existe des différences
notables selon les pays. (22) En 2007 seuls trois anti-malariques furent
ajoutés à cette liste. (23)
Les objectifs de la pré-qualification
(22) :
- Proposer une liste de produits et de fabricants,
pré-qualifiés dont la qualité et l'efficacité ont
été évaluées, inspectées et
contrôlées selon des standards internationaux.
- Apporter l'assurance que des normes de qualité
internationales ont été appliquées à toutes les
étapes de la pré-qualification.
- Accélérer l'accès à des
médicaments de qualité.
- Assurer le suivi permanent des produits et des fabricants et
leur re-qualification périodique ainsi que la mise à jour des
informations et la prise en compte des variations.
- Développer les possibilités locales de
production ainsi que les capacités des autorités nationales de
réglementation, à assurer l'évaluation, l'inspection et le
contrôle selon des normes de qualité reconnues
internationalement.
7. La Péremption et la Dégradation des
Médicaments
a) Péremption des Médicaments
Définition : La Péremption est un
état de destruction partielle ou totale de l'activité d'un
médicament un certain temps après sa préparation. (17)
- Délai ou Date de Péremption :
C'est un temps ou une période au cours de la quelle un médicament
est supposé perdre une quantité raisonnable et permise de son
activité. (17) Cette date n'indique pas nécessairement que le
médicament n'est plus stable après cette période, mais que
le médicament est encore utilisable à la date indiquée.
Elle doit toujours et obligatoirement figurée à l'emballage
extérieur des spécialités pharmaceutiques, et
généralement on la formule en termes de mois et d'années.
(18)
- Détermination de la Date de
Péremption : Elle s'effectue à partir des études
de dégradation accélérée et des études de
stabilité en temps réel. (18) La dégradation ou
vieillissement accéléré consisté à remplacer
le facteur temps par la température en se basant sur la règle
empirique qui stipule que, la vitesse de réaction double à chaque
élévation de température de 10°C. (17)
- Médicament périmé : est
celui qui est supposé avoir perdu plus que la quantité
raisonnable et permise de son activité. (17) En règle
générale, c'est un médicament dont le titre initial en
principe actif a diminué de 10%. Ce chiffre étant défini
par consensus international, peut être réduit à 5%, et
parfois moins, lorsque les produits de dégradation sont très
toxiques (cas des tétracyclines) ou lorsque la marge
thérapeutique est étroite (cas des anticancéreux,
théophylline, digoxine,...). (18)
- Médicament Périmé : est
celui qui a déjà perdu plus que la quantité raisonnable et
permise de son activité. (17)
b) Dégradation des Médicaments
La dégradation d'un médicament au cours du
temps correspond à une perte de stabilité du principe actif et/ou
des excipients ; elle est fonction des caractéristiques
physicochimiques de ces constituants et des conditions de conservation. (18)
Les principaux processus de dégradation sont
l'hydrolyse, l'oxydation et la photo dégradation. Par conséquent,
les facteurs responsables de la dégradation des médicaments sont
l'oxygène, l'eau, la lumière et la température (une
augmentation de la température entraîne en particulier une
élévation de la vitesse de l'hydrolyse et une
accélération des phénomènes d'oxydation). (18)
La dégradation d'un médicament peut conduire
à une réduction de l'efficacité thérapeutique, et
parfois à une formation des produits à l'origine d'effets
indésirables ou toxiques. (18)
8. Stabilité et durée de vie des
Médicaments (ou Produits finis)
La stabilité d'un médicament peut être
définie comme son aptitude à conserver ses
propriétés chimiques, physiques, microbiologiques et
biopharmaceutiques dans des limites spécifiées pendant toute sa
durée de validité (20).
La stabilité des préparations pharmaceutiques
dépend de paramètres extrinsèques (température,
humidité et exposition à la lumière) et
intrinsèques. Parmi ces derniers, il faut différencier les
facteurs liés aux matières premières, à la forme
pharmaceutique et au conditionnement. (21) p.54
Il existe deux types d'études de stabilité
(20):
· Les études de dégradation
accélérées, destinées à augmenter la
vitesse de dégradation chimique ou physique d'un médicament en le
soumettant à des conditions de stockage extrêmes dans le cas du
programme réglementaire des études de stabilité,
· Les études de stabilité en temps
réel : études expérimentales des
caractéristiques physiques, chimiques, biologiques et microbiologiques
d'un médicament pendant sa durée de validité et
d'utilisation prévue et au-delà, dans des conditions de stockage
prévues pour le marché auquel il est destiné.
Pour les pays du tiers-monde les études de
stabilité dans des conditions de forte humidité sont
particulièrement importantes du fait du risque de dégradation
dû à la semi-perméabilité de certains
conditionnements (21) p.54 :
· Les conditions climatiques dans de nombreux pays en
développement sont très différentes de celles des pays
tempérés où sont fabriqués les
médicaments,
· Une mauvaise stabilité peut entraîner
l'apparition de produits de dégradation toxiques ou une diminution
progressive de l'activité,
· La stabilité ne peut pas être
évaluée par un simple contrôle de qualité sur le
produit fini.
Il est donc nécessaire d'effectuer des études de
stabilité en fonction des conditions climatiques du pays de destination.
A cet effet, le monde a été divisé selon les
recommandations de l'OMS (20) en quatre zones climatiques, avec pour chaque
zone des caractéristiques de température et d'humidité
destinées à standardiser ces études de
stabilité.
Les fabricants européens n'étant pas
obligés de répondre aux spécifications de la zone IV
(climat chaud et humide) pour les études de stabilité en vue
d'une AMM européenne, s'ils exportent vers un pays aux conditions
climatiques extrêmes, il peut se produire des phénomènes de
dégradation imprévus du principe actif. Les faits sur le terrain
confirment ce phénomène puisque de nombreux problèmes de
qualité rencontrés in situ sont dus à des problèmes
de stabilité et non à la qualité intrinsèque du
produit constatée par un contrôle sur produit fini à
réception par l'importateur. (21) p.55
Un médicament n'est pas un simple mélange
d'ingrédients chimiques, c'est un équilibre très complexe
avec de très nombreuses possibilités d'interactions, il
nécessite l'utilisation d'un système d'assurance qualité
d'un bout à l'autre de la chaîne pharmaceutique depuis son
développement, sa fabrication, son contrôle, sa distribution,
jusqu'à son usage rationnel. (21) p.55
B. Définitions des
antipaludiques
1. Définition des antipaludiques
Les antipaludiques sont des médicaments actifs
vis-à-vis de l'infection de l'organisme par quatre espèces
d'hématozoaires du genre Plasmodium. La quinine et l'artémisinine
sont les seuls antipaludiques naturels alors que les autres sont d'origine
synthétique. (10)
2. Historique des antipaludiques
Déjà depuis l'antiquité les chinois
utilisaient l'Artémisia annua (armoise ou Quinghausu) comme
antipyrétique mais l'artémisinine ne fut isolé qu'en 1971.
La chloroquine fut synthétisée après la
2ème guerre mondiale. C'est en 1630 que Don Francisco Lopez
apprend des indiens du Pérou les vertus de l'écorce du quinquina
(Cinquona rubra), mais c'est en 1820 que Pelletier et Caventou en
isolèrent l'alcaloïde actif (la quinine). (3)
3. Classification des antipaludiques
a) Schizonticides
tissulaires
Ce sont des médicaments qui tuent les plasmodiums au
stade de la schizogonie exo-érythrocytaire dans les hépatocytes
chez l'homme. Le proguanil, la pyriméthamine, la primaquine, la
tétracycline en sont des exemples. (4)
b) Schizonticides sanguins
Ils combattent le paludisme clinique en tuant les plasmodiums
au stade de schizonte érythrocytaire. C'est le cas de la quinine, la
chloroquine, la méfloquine, l'artésunate, la
sulfadoxine-pyrimethamine, l'halofantrine. (4)
c) Gamétocides
Ils tuent les gamètes mâles et femelles
(gamétocytes) des plasmodiums. Exemples : tous les
8-aminoquinoleines y compris la primaquine. La Primaquine est aussi active
contre les formes latentes de P. vivax et P. ovale (Hypnozoïtes), elle est
donc hypnozoïtocide. (4)
d) Les
sporontocides
Ils tuent les spores les plasmodiums au stade de la
sporogonie. (4)
C. Les antipaludiques
1. La Quinine et ses dérivés
La Quinine est un alcaloïde extrait du quinquina et est
considérée comme un antipaludique majeur. Dans l'accès
pernicieux la Quinine reste irremplaçable. Seule, elle a une
activité suffisamment rapide et constante pour lutter contre le
paludisme suraigu. (3)
En ce qui concerne sa structure chimique, la quinine a un
noyau isoquinoléique, c'est un stéréo-isomère de la
quinidine qui est active sur le coeur. (3)
Le mécanisme d'action de la quinine s'explique par
son accumulation dans les lysosomes (vacuoles digestives acides) des schizontes
sanguins où l'hémoglobine est digérée, inhibant par
la suite l'enzyme qui assure la polymérisation d'hémozoïne
(produit de dégradation de l'hémoglobine) qui est toxique pour
le parasite. (4)
Les principaux effets indésirables de la quinine
sont l'hypoglycémie, l'anémie et les bourdonnements d'oreilles.
(4)
2. Les dérivés
4-aminoquinoleines
Les 4-aminoquinoléines sont des dérivés
de synthèse parmi lesquels on trouve la chloroquine et l'amodiaquine.
Ils ont l'avantage d'avoir une action rapide.
Leur mécanisme d'action est presque similaire à
celui de la quinine.
Leurs effets indésirables sont
modérés avec peu de risque même chez la femme enceinte.
L'amodiaquine cause l'agranulocytose parfois mortelle. (4)
3. Les dérivés
8-aminoquinoléines
Ce groupe est essentiellement composé de la primaquine
et la pamaquine. Il est de nos jours abandonné à cause de la
toxicité aiguë à laquelle il conduit.
La primaquine est particulièrement active sur les
formes sous croissance des plasmodiums (gamétocytes et
Hypnozoïtes). Ses effets indésirables sont sanguins, notamment
l'hémolyse intravasculaire aiguë chez les personnes atteintes de
déficience en 6-GPDH. (4)
4. L'artémisinine et ses
dérivés :
L'artémisinine est un sesquiterpène lactone
peroxyde isolé de Artémisia annua. Ses dérivés les
plus courants sont l'Artéether, l'Artéflene, l'Artémether,
l'Artésunate, et la Dihydroartémisinine.
L'Artémisinine et ses dérivés ont une
activité rapide mais brève, ils agissent sur les souches de P.
falciparum multirésistantes et chloroquino-resistantes, même en
cas d'accès pernicieux. Leur activité antipaludique repose sur
leur structure peroxyde (trioxane) et les effets indésirables sont peu
nombreux et relativement sans danger. (4)
L'artémisinine n'est pas une nouvelle découverte
car les Chinois l'ont déjà utilisée depuis au moins deux
millénaires. Issue d'une plante chinoise, cette substance a largement
prouvé son efficacité en Asie. Des études menées
sur 2 millions de cas traités dans de nombreux pays impaludés,
démontrent à la fois l'efficacité de l'artémisinine
et sa facilité d'administration : le médicament est disponible
sous forme injectable ou par voie orale en une seule dose quotidienne. Il
élimine plus rapidement les parasites présents dans le sang. Ce
qui représente un atout majeur en phase épidémique car la
substance "casse" la transmission épidémique. Pour augmenter son
effet mais aussi retarder l'apparition des résistances, elle est
administrée en association avec une autre molécule à
savoir la SP ou sulfadoxine/pyriméthamine, amodiaquine ou
méfloquine. C'est cette association que l'on appelle ACT, les
combinaisons thérapeutiques. La faible parasitémie restante
grâce à l'effet de l'artémisinine est
éliminée par le deuxième antipaludéen d'action plus
prolongée. La guérison rapide est assurée à plus de
97% des cas. La notion de rapidité présente toute son importance
dans les accès de paludisme chez l'enfant.
5. Autres antipaludiques et associations
antipaludiques :
a) Les aminoalcools
La méfloquine appartient à ce groupe, elle est
active contre les souches multirésistantes de plasmodium et notamment
les chloroquino-résistantes. (3)
Leur mécanisme d'action est similaire à celui de
la quinine et à la chloroquine. Les effets indésirables sont la
psychose aiguë, l'encéphalopathie transitoire avec des convulsions.
(4)
b) Les antifoliques
Les sulfamides (sulfadoxine et sulfamethoxazole) et les
sulfones (Dapsone) ne sont pas indiqués en monothérapie à
cause de leur action lente et à la chimiorésistance face à
certaines couches de P. falciparum. En association avec la
pyriméthamine, les sulfamides ont une activité schizonticide et
offrent l'avantage d'un traitement à dose unique. (3)
En ce qui concerne leur mode d'action, les sulfones et les
sulfamidés ont un mécanisme identique qui consiste à
inhiber la transformation de l'acide para-amino-benzoïque (PABA) dont
l'hématozoaire a besoin pour sa croissance en bloquant l'activité
de la dihydrofolate synthétase. (3)
Le syndrome de Stevens-Johnson est un de leurs Effets
indésirables redoutables. (4)
c) Les antifoliniques
Les diguanides (proguanil) et les diamino pyrimidines
(pyriméthamine et triméthroprime) ont une activité
schizonticide. Ils empêchent le passage du dihydrofolate en
tetrahydrofolate en inhibant la dihydrofolate réductase. (3)
d) Quelques Associations
antipaludiques
q Sulfadoxine-Pyriméthamine (Fansidar*)
q Sulfaméthoxazole-Triméthoprime (Bactrim*)
q Sulfadoxine-Pyriméthamine-Méfloquine
(Fansimef*)
q Dapsone-Pyriméthamine (Maloprime*)
q Quinine-Tétracycline
q Quinine-Doxycycline
q QuinineClindamycine
q Artésunate-Amodiaquine
q Artéméther-Lumefantrine (Coartem*)
q Proguanyl-Atovaquone (Malarone*)
CHAPITRE II. GENERALITES SUR LES ACT ET
L'ARTESUNATE
I. LES ACT OU CTA
(Combinaisons Thérapeutiques à base
d'artémisinine)
A. Introduction des ACT dans la
thérapie antipaludique
Lors d'une consultation technique de l'OMS sur les
combinaisons thérapeutiques antipaludiques à Genève
(Suisse) les 4 et 5 avril 2001. Il a été reconnu que le
traitement du paludisme au moyen d'associations médicamenteuses est une
stratégie dont l'intérêt potentiel permet d'en faire une
option viable pour améliorer l'efficacité thérapeutique et
retarder l'apparition et la sélection des parasites résistants.
(11)
Ces combinaisons doivent être basées sur des
molécules novatrices dont le mode d'action est différent
de ceux qui présentent déjà la chimiorésistance.
Ce sont des combinaisons à base d'artémisinine ont de nets
avantages en ce sens qu'elles permettent une guérison clinique et
parasitologique rapide, qu'on ne leur connaît pour l'instant aucune
résistance parasitaire, qu'elles réduisent la charge
gamétocytaire et qu'elles sont généralement bien
tolérées. (11)
Sans tenir compte des considérations de coût, ces
combinaisons à base d'artémisinine sont :
i. artéméther-luméfantrine
(CoartemTM);
ii. artésunate (3 jours) plus amodiaquine;
iii. artésunate (3 jours) plus SP dans les régions
où la SP conserve une bonne efficacité;
iv. SP plus amodiaquine dans les régions où la SP
et l'amodiaquine conservent une bonne efficacité.
N.B : SP signifie sulfadoxine et pyrimethamine.
Les combinaisons à éviter sont :
i. associations à base de chloroquine (CQ + SP et CQ +
artésunate);
ii. traitement d'un jour par l'artésunate + SP;
iii. associations à base de méfloquine (par exemple
méfloquine plus artésunate) dans les zones où la
transmission est intense; et
iv. traitement d'un jour par l'artésunate plus la
méfloquine lors de la phase aiguë d'une situation d'urgence
complexe ou d'une épidémie de paludisme.
B. Les ACT en RDC
En juillet 2004, Médecins Sans Frontières (MSF)
décidait de changer le protocole de traitement contre le paludisme dans
tous ses projets en République démocratique du Congo (RDC), pour
mettre en place un traitement plus efficace: les "CTA". C'est dans cet
objectif qu'entre novembre 2004 et janvier 2005, que MSF travailla en
collaboration avec le Programme national de lutte contre le paludisme (PNLP).
Et c'est en février 2005, le gouvernement congolais décidait de
changer le protocole national de traitement et de passer aux CTA. Le CTA
utilisé dans le nouveau protocole est une combinaison
d'artésunate et d'amodiaquine. (11)
C. Le pourquoi et l'avantage des ACT comme
associations médicamenteuses
Les dérivés de l'artémisinine figurent
parmi les schizonticides les plus puissants. Leur brève demi-vie (14
à 18 h), qui n'expose pas les parasites à des concentrations
infra-thérapeutiques, les protège- en principe- du risque de
chimiorésistance. En Chine toutefois, où la monothérapie
par l'artémisinine a été pratiquée depuis le
début des années 80, des études effectuées au
Yunnan semblent indiquer un amenuisement de la sensibilité à
l'artémisinine et à ses dérivés. Un autre avantage
du produit est son action sur les gamétocytes, réduisant la
transmission et limitant le risque de voir émerger des
résistances. Cependant la très brève demi-vie ne permet
pas la destruction totale de la masse parasitaire et les rechutes à 28
jours sont fréquentes, dépendant de l'intensité de la
charge parasitaire, de la dose reçue et de la durée du
traitement. Une dose totale de 600 mg d'artésunate ou
d'artéméther n'entraîne que 88% de guérisons; il
faudrait sept jours de traitement pour en obtenir 100%, ce qui est susceptible
de réduire l'observance du traitement. Pour la faciliter et majorer
l'efficacité des traitements, il a été envisagé
d'associer de nombreuses molécules à une artémisinine : la
S-P, la chloroquine, l'amodiaquine, la doxycycline ou la tétracycline,
la méfloquine, la pyronaridine. (12)
D. Le coût des
ACT
Les traitements ACT ont été recommandés
par l'OMS depuis 2002, année où l'organisation a publié
une recommandation claire sur la nécessité d'utiliser les ACT
dans les pays touchés par les résistances aux traitements
classiques du paludisme. Mais ils présentent encore un obstacle de
taille à cause de leur coût. Produit en faibles quantités,
le médicament est plus cher que la chloroquine. Un traitement classique
coûte entre 0,2 et 0,5 dollar alors qu'un traitement ACT oscille entre
1,2 et 2,4 dollars, soit cinq à six fois plus cher que le "classique".
(11)
D'ailleurs les participants de la Consultation Technique de
l'OMS des 4 et 5 avril 2001 à Genève avait admis que, dans le cas
des associations à base d'artémisinine, le coût du
traitement sera sensiblement supérieur à celui des
monothérapies traditionnelles comme celles qui reposent sur la
chloroquine ou la sulfadoxine-pyriméthamine (dans une proportion pouvant
atteindre un facteur 10). Pour Médecins Sans Frontières, la
référence du coût d'un traitement ACT est de 1 dollar plus
0,5 dollar pour le test rapide. (11)
Le 8 avril 2005 à Paris (France), la
réjouissance de MSF sur l'annonce faite par la DNDi (Drugs for Neglected
Diseases initiative) et Sanofi-Aventis sur l'arrivée prochaine sur le
marché d'un nouveau produit (co-formulation
artésunate-amodiaquine) pour la prise en charge des malades atteints du
paludisme en Afrique et qui devait arrivé au marché en 2006, fut
une lueur d'espoir car cette co-formulation n'étant pas breveté
(non exclusif), simple d'utilisation (sous forme de comprimé avec un
traitement de 6 comprimés en 3 jours) et surtout par ce qu'elle est
moins cher puisque l'objectif de la DNDi et Sanofi-Aventis est d'atteindre
moins d'un dollar par adulte et par traitement (0,5 dollar pour les enfants).
(11)
II. L'ARTESUNATE
A.
Introduction
1. Origine
Après l'émergence et la multiplication des
souches chloroquino-résistances de Plasmodium falciparum, la
découverte de nouveaux antipaludiques est devenue une priorité
sanitaire mondiale. Au cours de deux décennies qui ont suivi
l'apparition de cette résistance, des moyens considérables ont
été consacrés à la découverte des nouveaux
antipaludiques tant sur le plan de synthèse chimique que sur
l'exploitation des produits de phytothérapie. (13)
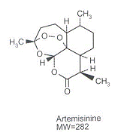
L'artémisinine est extraite de l'Artémisia
annua ou Quinghaosu, une armoise chinoise actuellement
cultivée. Le Quinghaosu est une plante de la famille des
Asteraceae qui est utilisée depuis longtemps en médecine
traditionnelle chinoise, et faisant partie de la pharmacopée chinoise
depuis plus de 2000 ans. Il a fallu les travaux des chercheurs chinois pour
connaître la molécule mère après son isolement en
1971. (13)
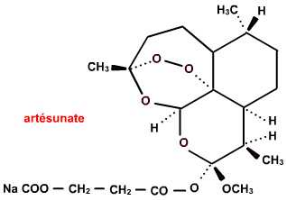
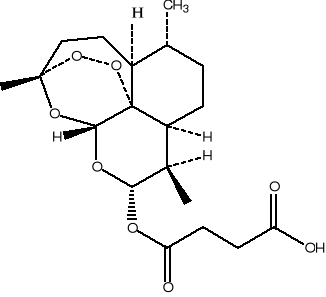
2. Présentation
L'artésunate est un dérivé
synthétique de l'artémisinine, c'est un 10-á-
hemisuccinate de dihydroartémisinine. Sa formule brute est
C19H28O8 et son poids moléculaire est
387. Alors que l'artémisinine est une lactone sesquiterpénique
naturelle contenant un pont peroxyde qui n'a aucune parenté structurale
avec quelques antipaludéens connus. (13)
L'artésunate est une poudre blanche ne se
présentant pas sous forme d'un sel quelconque. Mais il faut signaler que
le suffixe « ate » qui fait référence à la
fonction ester n'a rien à voir avec une fonction de sel sodique comme
utilisé par certains auteurs. Car l'artésunate sodique n'existe
pas sous forme cristalline, mais peut se former en solution si
l'artésunate est dissout dans un milieu contenant par exemple la soude
caustique. L'artésunate est soluble dans le méthanol,
l'éthanol, l'eau et est injectable par voie intraveineuse.
L'artésunate est soluble dans l'eau et injectable par voie veineuse.
Sauf qu'en solution aqueuse il s'hydrolyse rapidement avec perte du radical
succinate. (13)
B. Quelques aspects pharmacologiques de
l'artésunate
1. Propriétés
L'Artésunate a un spectre d'action plus large que
beaucoup d'autres antipaludiques. Il tue aussi bien les schizontes que les
gamétocytes. (13)
L'Artésunate possède également une action
antimicrobienne contre les différentes souches de Schistosomes, de
Babesia, le Pneumocystis carinii et le Toxoplasma gondii. Une importante
activité a été décrite contre le virus cytomegale.
(13)
L'Artésunate n'affecte apparemment aucun organe humain
à la dose thérapeutique notamment le système
cardiovasculaire et même le système nerveux. (13)
Il faut signaler qu'aucune activité à l'encontre
des étapes hépatiques du cycle parasitaire n'a été
démontrée et n'empêche non plus l'inoculation dans le foie
après l'injection par l'anophèle de ses parasites dans le sang.
L'artésunate est éliminé par voies biliaire et urinaire.
(13)
2. Mode d'action
C'est au niveau érythrocytaire que l'artésunate
agit en pénétrant dans le parasite grâce à sa
lipophilie suivant un mécanisme d'absorption non précis. (13)
Une fois à l'intérieur du parasite et sous l'effet
catalyseur de l'ion ferrique provenant de l'hème, le pont peroxyde est
ouvert et il y a libération de l'oxygène en état de
naissance. (13)
Faute de ne pas trouver un partenaire identique, l'oxygène
naissant provoquera des phénomènes d'oxydation car c'est un
puissant oxydant. (13)
Les lipides des membranes sont époxydés, puis
hydrolysés, le tout suivi par une rupture de la chaîne des acides
gras et par la désintégration des membranes. La première
atteinte est la vacuole centrale, suivie de la membrane mitochondriale et de la
membrane nucléaire. De ce faite la survie du parasite devient alors
impossible. (13)
Au-delà de cette action oxydante, une fois le pont
peroxyde ouvert, la molécule se comporterait comme un radical libre qui
pourrait établir une liaison covalente avec les protéines des
parasites et, par conséquent, exercer une action cytotoxique
supplémentaire. L'effet combiné des deux mécanismes est
probablement responsable de la mort rapide du parasite. (13)
3. Posologie
En monothérapie et en cas de paludisme non
compliqué, il était préconisé la dose de 4mg/kg le
premier jour, suivie de 2mg/kg une fois par jour pendant 6 jours et cela par la
voie orale. De nos jours, l'OMS a interdit l'usage de l'Artésunate en
monothérapie, afin de retarder l'apparition de la résistance
à ces médicaments d'espoir.
4. Effets secondaires
L'Artésunate est responsable de peu d'effets secondaires,
dont les plus documentés sont les suivants :
v une augmentation passagère des transaminases,
v une baisse passagère du nombre de
réticulocytes,
v une légère baisse de la fréquence
sinusale.
Il faut signaler que, plusieurs effets secondaires
constatés lors du traitement par l'artésunate sont probablement
d'avantage imputable aux symptômes de la malaria qu'au
médicament.
C. Importance de l'artésunate dans le
traitement du paludisme
Associée à un autre antipaludique (chloroquine,
amodiaquine, sulfadoxine-pyriméthamine), l'artésunate constitue
un médicament de première intention dans le traitement des
accès simples du paludisme à Plasmodium falciparum. (11)
Pour augmenter son effet, mais aussi retarder l'apparition
des résistances, l'artémisinine est donc administrée en
association avec une autre molécule antipaludique. C'est la combinaison
thérapeutique à base de dérivé
d'artémisinine (CTA) ou Artemisinin-based Combination therapy (ACT).
(11)
En 2002, l'OMS a publiée une recommandation claire sur
la nécessité d'utiliser les ACT dans les pays touchés par
les résistances aux antipaludéens classiques. (11)
L'OMS sur l'avis d'experts internationaux, recommande
l'introduction des poly thérapies pour remplacer les mono
thérapies dans le traitement du paludisme et préconise en
particulier le recours à des associations médicamenteuses
contenant des dérivés d'artémisinine. (11)
CHAPITRE III. LA RESISTANCE AUX ANTIPALUDIQUES
A. Définition de
Pharmacorésistance
La pharmacorésistance est définie par l'O.M.S.
comme l'aptitude d'une souche de plasmodium à survivre ou à se
reproduire malgré l'administration d'un médicament à des
doses égales ou supérieures aux doses ordinairement
recommandées mais comprises dans les limites de tolérance du
sujet ; la métabolisation normale du médicament (absorption
à vitesse normale du médicament lié aux protéines)
assurant sa biodisponibilité ; le contact du parasite avec une dose
adéquate du médicament durant un laps de temps suffisant. (2)
La pharmacorésistance n'est pas synonyme d'échec
thérapeutique, mais constitue une de ses causes. En effet l'échec
thérapeutique a pour causes l'erreur de diagnostic, le non respect
de la posologie, le médicament non conforme sur le plan pharmacologique,
la biodisponibilité insuffisante (métabolisme accru,
vomissement...) et la pharmacorésistance. (2)
La définition, par l'OMS, de la
«chimiorésistance» comme «l'aptitude d'une souche de
parasites du paludisme à survivre ou à se reproduire
malgré l'administration et l'absorption d'un médicament
employé à des doses égales ou supérieures aux doses
ordinairement recommandées mais comprises dans les limites de
tolérance du sujet», remonte en 1973, à l'époque
où ni la technique de mise en culture in vitro de
Plasmodium falciparum ni la chromatographie liquide de haute
performance n'était au point et que le génotypage et la
détection des mutations étaient encore un domaine naissant. Il
n'est donc pas étonnant que la définition de la
chimiorésistance citée ci-dessus soit fondée sur une
observation clinique. (14)
Afin de mieux comprendre les notions de
«chimiorésistance» et d'adapter la stratégie de riposte
pour retarder et limiter la dissémination des parasites
résistants, le test in vivo, le test in vitro, le test
dit moléculaire et le dosage de médicaments apportent toujours
des informations complémentaires, sachant que chacune de ces
méthodes aborde le phénomène de la chimiorésistance
sous un angle différent et complémentaire. (14)
La chimiorésistance des parasites constitue un des
obstacles majeurs qui entravent les programmes nationaux de lutte contre le
paludisme depuis des décennies. La situation mondiale du paludisme est
marquée par la résistance des parasites à des
antipaludiques majeurs, notamment à la chloroquine, le moins cher des
médicaments antipaludiques. (14)
B. Origine et historique de la
résistance aux antipaludiques
1. Origine de la pharmacorésistance
L'acquisition de la résistance par une souche de
plasmodium vis-à-vis d'un antipaludique donné est un processus
spontané lié aux aléas de recombinaison génique
(mutation).
Par contre, l'apparition à grande échelle de la
pharmacorésistance dans la population plasmodiale dépend, elle,
de la pression sélective exercée par le médicament qui
favorise la promotion des mutants capables de survivre en présence de ce
médicament par ce qu'utilisant des voies métaboliques qui ne sont
pas bloquées par ce dernier. (2)
Pour autant que ces mutants échappent à l'action
destructrice de l'immunité, ils vont se propager via les
anophèles à d'autres hôtes. La propagation de la
pharmacorésistance dépend de la conjonction de plusieurs
facteurs, dont le plus important est sans aucun doute la large utilisation des
médicaments antipaludiques auxquels le plasmodium est devenu peu
sensible.
Secondairement, il faut tenir compte aussi de la
réduction de la probabilité d'hybridation entre les souches
sensibles et les souches insensibles dans l'estomac de l'anophèle, qui
ne pourrait malheureusement qu'amplifier le phénomène. (2)
Dans le cas de la pharmacorésistance à la
chloroquine, certaines souches de P. falciparum résistantes ont acquis
la capacité d'expurger le médicament de leur cytoplasme plus vite
que ne le ferait un plasmodium non-résistant ou normal. Ceci est
prouvé avec l'utilisation de la vérapamil* (inhibiteur des pompes
calciques) qui en neutralisant cette action restitue à la chloroquine
toute son efficacité face aux plasmodiums chloroquino-résistants.
(2)
Il existe également des souches de P. vivax
chloroquinoresistantes en Nouvelle Guinée. (4)
2. Historique
Les premières souches de P. falciparum
résistantes à la chloroquine ont été
signalée en Thaïlande en 1957, puis en Colombie 1960, au Kenya 1978
et en RDC en 1983. Dès lors cette pharmacorésistance s'est
répandue dans toutes les régions impaludées du globe.
(2)
C. Quelques antipaludiques présentant la
résistance
La perte de sensibilité des plasmodiums concerne
à des degrés divers les autres antipaludiques (amodiaquine,
quinine, sulfadoxine-pyrimethamine, mefloquine, etc.) en dehors de la
chloroquine. Une souche de P. falciparum qui est résistante à
plusieurs antipaludiques est dite multirésistante. (2)
CHAP. IV : GENERALITES SUR LE CONTROLE DE QUALITE
DES MEDICAMENTS
IV.1. Contrôle de qualité et Assurance
qualité
Le contrôle de qualité a constamment pris de
l'importance ces dernières années, surtout dans le domaine du
médicament. Jusqu'au début des années 60, la
qualité des médicaments était orientée
conformément aux pharmacopées nationales. (15)
L'objectif principal du contrôle de qualité est
d'étudier les normes pour les propriétés du produit,
d'évaluer les résultats et de rejeter les produits qui
n'atteignent pas les normes. C'est ainsi qu'il a été
établi (et cela a été confirmé dans la
réglementation de l'OMS) que, pour garantir l'objectivité, le
personnel doit travailler de façon indépendante. Ainsi pour des
raisons d'organisation les fabricants ont séparés le
contrôle des autres départements. (15)
La gamme des activités de contrôle de
qualité s'est étendue bien au-delà des contrôles
ponctuels lors du déroulement des fabrications. Ces activités
incluent le « contrôle en cours de fabrication » en
vue d'atteindre une qualité de produit la plus haute possible.
L'automatisation complète des procédés, depuis l'exclusion
des erreurs dans la fabrication jusqu'à la vérification que le
produit fini répond bien aux normes requises, tout cela est recouvert
par le terme « Assurance qualité ». (15)
IV.2. CARACTERISTIQUES DES MEDICAMENTS
En se référant aux pharmacopées, il se
dégage que les caractéristiques les plus importantes pour
établir la qualité d'un médicament sont :
l'identité, la pureté, l'activité, l'uniformité, la
biodisponibilité et la péremption. (15)
En ce qui concerne l'identité des constituants du
médicament, le principe actif, l'excipient et l'adjuvant
déclarés doivent être présents dans le produit. De
même la forme pharmaceutique doit correspondre à ce qui est
annoncé sur l'emballage. (15)
Quant à la pureté, en dehors des principes
actifs, les excipients et les adjuvants, les médicaments ne doivent pas
contenir de substances potentiellement toxiques. Ces dernières peuvent
provenir du processus d'obtention du principe actif ou excipients, de mauvaises
conditions de conservation pouvant donner naissance aux produits de
dégradation inactifs ou nocifs. (15)
L'activité du médicament est due au principe
actif qu'il contient. Le principe actif du médicament doit avoir une
action thérapeutique confirmée à celle
déclarée sur l'étiquette du produit. Lorsque le
médicament contient plusieurs principes actifs, cette information doit
être mentionnée sur l'emballage. (15)
La qualité d'un médicament peut être aussi
évaluée par l'uniformité de sa forme pharmaceutique. En
effet la couleur, la taille, le poids et la forme du médicament ne
doivent pas varier dans un même lot ou d'un lot à l'autre.
La biodisponibilité est représentée par
la mesure de la fraction d'une dose administrée d'un médicament
qui atteint effectivement la circulation générale et la vitesse
avec laquelle le médicament parvient dans la circulation
générale. C'est paramètre extrêmement important qui
permet de comparer deux médicaments contenant le même principe
actif en prenant un d'entre eux comme référence. (15)
La date de péremption est une caractéristique
importante et légale qui doit figurer de façon explicite sur tout
médicament. C'est une date au-delà de laquelle le fabricant ne
garantit plus l'efficacité et l'innocuité du médicament et
décline toute responsabilité en cas d'effets non attendus,
indésirables ou dangereux survenus lors de l'utilisation du produit.
(15)
IV.3. CONTROLE DE QUALITE DES MEDICAMENTS
IV.3.1. GENERALITES
Lors de contrôle de la qualité des
médicaments, il est question de déceler les défauts
éventuels de fabrication et d'autres dommages tels que,
l'altération, la falsification, la souillure, la contrefaçon dont
les produits ont fait l'objet tout au long de son processus
d'élaboration. (13)
Il s'agit de rechercher tout ce qui rend le médicament
non conforme à sa monographie consignée dans la
pharmacopée de référence et qui le rendrait impropre
à la consommation. La conformité aux normes
préétablies par une pharmacopée ou une législation
officielle ou agréée est obligatoire. (13)
Les opérations de contrôle de qualité de
médicaments peuvent être reparties à deux niveaux :
Le premier correspondant à l'examen organoleptique
comprend toutes les activités relatives à la vérification
des paramètres palpables à l'aide des organes de sens sans
recourir aux appareils de mesure plus ou moins complexes. (13)
Le second concernant le contrôle des paramètres
non palpables fait recours aux réactifs et/ou aux appareils de mesure.
Ces opérations font suite et complètent l'examen des
caractères organoleptiques de l'étiquette et de l'état
physique du produit. (13)
Le rejet d'un médicament de part les paramètres
palpables fait l'objet d'une décision prise avec beaucoup de bon
sens.
IV.3.2. CONTROLE DES PARAMETRES PALPABLES
Dans le contrôle des paramètres palpables, on
utilise son sens d'observation qui permettra de déceler à
première vue un certain nombre d'anomalies au niveau de
l'étiquette et des caractères organoleptiques. L'examen visuel
joue ici un rôle très important. (13)
L'étiquette fournit plusieurs informations utiles
telles que les caractères d'imprimerie utilisés pour leur
codification, la nature du contenu et même les conditions de
conservation. Elle renseigne sur la dénomination du produit, date de
fabrication et de péremption, nom et adresse du fabricant ou de
l'exploitation, etc. Permettant ainsi de se faire une idée sur
l'authenticité, l'origine et l'identité du produit. (13)
Le recours aux organes de sens (la vue, le goût,
l'odorat, le toucher, la taille, la forme et les gravures distinctives pour les
comprimés marqués) permet de déterminer les
caractères organoleptiques et l'état physique des
médicaments. Il suppose une meilleure connaissance du médicament
à analyser et de certaines de ses propriétés physiques
particulières. (13)
L'état physique concerne particulièrement
l'étude de l'intégrité physique du médicament, la
recherche des marques extérieures de manipulation, d'abrasions ou tout
autre signe assimilable à un défaut de qualité. (13)
IV.3.3. CONTROLE DES PARAMETRES NON PALPABLES
Le contrôle par rapport au précédent,
consiste à déceler les modifications plus ou moins profondes des
médicaments qui les rendent impropres à la consommation au
regard des normes établies. (13)
Il consiste ainsi à la vérification de certaines
normes de conformité qui échappent à l'examen des sens.
Puisque ces normes peuvent légèrement varier d'une
pharmacopée de référence à une autre, il y a une
impérieuse nécessité d'indiquer la pharmacopée
auquel le fabricant a fait recours. (13)
La monographie de cette pharmacopée décrit les
essais qualitatifs et quantitatifs, les essais d'uniformité de masse, de
dureté de fiabilité et d'usure, de délitement, de
dissolution, de dosage du principe actif, de stérilité de pH, de
recherche d'impuretés et les conditions de conservation. (13)
L'exigence de ces tests en dehors des épreuves
qualitatives et quantitatives est fonction de la forme galénique. Le
test d'uniformité de masse par exemple sera recommandé pour
certains comprimés alors qu'il ne l'est pas pour d'autres. Elle
dépend aussi d'une pharmacopée à l'autre et de
l'évolution des connaissances scientifiques sur le médicament,
c'est le cas par exemple du test de vitesse de dissolution du principe actif
qui est exigé actuellement pour la plupart de formes solides
administrées par voie orale. (13)
Les tests d'uniformité de poids et l'évaluation
de la qualité chimique du principe actif par chromatographie sur couche
mince d'adsorption figurent parmi les méthodes de contrôle des
paramètres non palpables les plus utilisées. (13)
CHAP. V : METHODES GENERALES D'ANALYSE DES
MEDICAMENTS
V.1. CONTROLE DE L'ETIQUETTE
L'étiquette est un élément important
quant à l'assurance et au contrôle de qualité des
médicaments. Elle constitue la carte d'identité pour chaque
médicament, permettant ainsi :
v l'identification du médicament par son nom, sa forme
galénique, son numéro de lot, le nom et l'adresse du fabricant,
le pays d'origine, les dates de fabrication et d'expiration, le numéro
d'enregistrement dans le pays d'origine (Autorisation de Mise au
Marché).
v la connaissance, pour le médicament des conditions de
sa conservation, et de sa manutention, des instructions ainsi que d'autres
informations pour son meilleur usage. (15)
L'étiquette comporte les éléments
fondamentaux ci-après :
1. le nom du médicament,
2. la composition, la forme galénique, la
quantité du produit dans le récipient,
3. le numéro du lot de fabrication,
4. les dates de fabrication et d'expiration (explicitement ou
sous forme de code)
5. les conditions de conservation et de manutention du
médicament si cela s'avère nécessaire,
6. les instructions et précautions pour le meilleur
usage du médicament,
7. l'adresse complète du fabricant en indiquant surtout
le lieu et le pays d'origine,
8. l'AMM dans le pays d'origine. (15)
D'une matière générale, les
éléments de l'étiquette peuvent être imprimés
sur un papier pour être collés sur le récipient ou
placés à l'intérieur de l'emballage sous forme de notice
ou encore directement sur le récipient. (15)
Par ailleurs, les informations de l'étiquette sont
aussi reprises sur l'emballage et même sur les emballages unitaires. Si
ces derniers sont trop petits pour porter une étiquette complète,
on doit y signaler au moins le nom du produit, la teneur en principe actif, le
numéro du lot de fabrication, les dates de fabrication et d'expiration
ainsi que le nom du fabricant. (15)
Normalement, ces différentes spécifications pour
un médicament donné ne changent pas sans une raison valable et
sans avis du fabricant. Par conséquent, tout changement observé
lors de l'examen de ces éléments peut aider à
découvrir certaines anomalies qui peuvent jouer un rôle important
quant à la décision à prendre sur la qualité du
médicament. Toutefois, il y a lieu de s'informer directement par
l'intermédiaire de l'organisme importateur. (15)
V.2. CONTROLE DES COMPRIMES
Plusieurs tests sont à réaliser pour le
contrôle de qualité des comprimés. Il s'agit des
caractères organoleptiques, des essais d'uniformité de masse, de
pureté, de friabilité, de délitement ou de
désagrégation, de dissolution, et de dosage. (15)
V.2.1. Caractères organoleptiques
Le contrôle des caractères organoleptiques des
comprimés permet de réunir à titre indicatif des
données concernant leur identification et leur différenciation.
(15)
Il est aussi important de connaître les changements que
peuvent subir les caractères organoleptiques, lesquels changements
peuvent constituer :
v de falsification, de contrefaçon ou de
malfaçon (une mauvaise préparation). La forme, la taille, les
marques distinctes d'un lot de comprimés provenant d'un fabricant
habituel ne peuvent être modifiées sans avis préalable de
ce dernier. La présence de comprimés qui s'effritent au toucher
ou cassés et réduits en poudre, une surface rugueuse au lieu
d'être brillante et lisse, le manque d'homogénéité
de la couleur en surface et dans la masse du comprimé cassé...
peuvent susciter des doutes sur la qualité des comprimés
concernés.
v d'altération : les comprimés sont le
souvent, exposés aux influences d'un certain nombre de facteurs
nuisibles aux principes actifs (l'air, la lumière, l'humidité,
etc....) et dont les effets peuvent s'observer par un examen à l'aide
des organes de sens. On peut également observer la prolifération
des moisissures. (15)
V.2.2. Test d'uniformité de poids
Le test d'uniformité de poids concerne les formes
pharmaceutiques solides particulièrement les comprimés, les
capsules, les suppositoires et les ovules. Il permet de déterminer les
variations de poids entre les unités d'une préparation
pharmaceutique d'un seul et même lot. (15)
Certains comprimés peuvent quelques fois
présenter un poids moyen ou individuel de loin inférieur à
celui des principes actifs annoncés par le fabricant indiquant ainsi le
manque d'homogénéité de la population des comprimés
concernés. L'inverse est également vrai, bien que rare. En effet
des anomalies au niveau de l'uniformité de poids peuvent être
tellement évidentes qu'on est obligé d'arrêter la
poursuite des opérations de contrôle de qualité. (15)
Principe et normes du test d'uniformité de poids des
comprimés
Le test d'uniformité de poids de comprimés
s'effectue en prélevant 20 comprimés d'un même lot et les
pesant individuellement un à un à l'aide d'une balance de
précision convenable. (15)
Le calcul du poids moyen des comprimés permet
déterminer en pourcentage la variation de poids positive et
négative du comprimé le plus lourd et le moins lourd par rapport
au poids moyen. (15)
Les normes préconisent que le poids individuel de deux
ou plus de 20 unités peut s'écarter du poids moyen d'un
pourcentage plus élevé que celui indiqué, mais le poids
d'aucune unité ne peut s'écarter de plus de double de ce
pourcentage.
Par rapport au poids moyen, les variations de poids suivantes
sont généralement acceptées :
- comprimés de poids moyen inférieur à
140mg : #177;10%
- comprimés de poids moyen compris entre 140 et
300mg : #177;7,5%
- comprimés de poids moyen supérieur à
300mg : #177;5%. (15)
V.2.3. Test de délitement
Cet essai est destiné à la détermination
du temps de désintégration des comprimés dans un milieu
liquide sous agitation. La désintégration est atteinte lorsqu'il
n y a plus de résidu solide, c'est-à-dire lorsque le
résidu n'est constitué que d'une masse molle, ne comportant pas
d'agrégats palpables et non imprégnée par des fragments
d'enrobage. (15)
V.2.4. Analyses qualitative et qualitative
L'analyse qualitative est réalisée
généralement après extraction du ou des principes actifs
dans le comprimé à l'aide des solvants appropriés. Les
réactifs d'identification sont indiqués pour chaque type de
principe actif. Pour arriver à réaliser cette analyse, on recourt
souvent aux méthodes chimiques et aux techniques de chromatographie.
(15)
Toutefois, les fabricants, les formulaires et
pharmacopées s'arrangent pour mettre au point des méthodes et
techniques très simples, applicables à tout moment tant pour
l'identification des principes actifs que pour la recherche des
impuretés. (15)
Pour ce qui est de l'analyse quantitative, les
procédés de dosage sont indiqués pour chaque monographie
en fonction de la nature des principes actifs. Diverses méthodes ont
été mises au point pour arriver à réaliser cela,
dont on peut citer certaines : la volumétrie, la
complexométrie, la spectrophotométrie UV/visible, les techniques
chromatographiques et bien d'autres.
Pour ce qui est de l'artésunate, nous avions retenu les
méthodes ci-après :
· la chromatographie sur couche mince pour l'analyse
qualitative,
· la spectrophotométrie UV/visible pour l'analyse
quantitative. (15)
V.3. CHROMATOGRAPHIE SUR COUCHE MINCE (CCM)
V.3.1. Introduction
Le botaniste russe MIKHAEL TSWETT est
considéré comme le père de la chromatographie. En effet,
c'est lui qui a utilisé pour la première fois la chromatographie
pour séparer et isoler les pigments végétaux entre autre
les chlorophylles et xanthophylles en se servant d'une colonne à base
de craie pulvérisée ou carbonate de calcium (CaCO3).
C'est une chromatographie d'adsorption. (15)
La chromatographie selon Ergon STAHL, est une
technique physico-chimique pour la séparation des mélanges des
substances basées sur les différents temps de rétention
des substances individuelles dans les phases stationnaire et mobile
primitivement limitées aux substances colorées. (16) p.29
La chromatographie est la méthode de séparation
la plus souple et la plus efficace qui puisse se présenter aux chimistes
(analystes). (15)
V.3.2. Définition et Principe
Définition
La Chromatographie sur Couche Mince (CCM) selon Ergon
STAHL, est une méthode de séparation physico-chimique. La couche
mince (phase stationnaire), constituée d'une substance finement
pulvérisée, est appliquée ou fixée sur une plaque
de verre, de métal ou sur une feuille appropriée. La solution du
mélange inconnue est déposée à la ligne de
départ sous forme d'un point. La plaque ou la feuille est introduite
dans une cuve étanche contenant l'éluant approprié (phase
mobile). (16) p.13 et 14
La phase mobile ou éluant est un moyen de transport,
qui est constituée d'un ou plusieurs solvants. Elle monte par
capillarité dans la phase stationnaire, c'est-à-dire la couche
poreuse. (16) p.17
La CCM est particulièrement indiquée pour la
recherche analytique des en pharmacie. (16) p.13
Principe
La séparation des constituants du mélange
s'effectue grâce à l'ascension par la phase stationnaire
(développement). Ensuite, les substances incolores seront rendues
visibles (détection). (16) p.13 et 14
Selon la nature des phases stationnaires la séparation
des substances en CCM se fait en fonction les phénomènes
suivants :
v Phénomène Adsorption sur phase stationnaire
imprégnée sur un support (Chromatographie d'adsorption)
v Phénomène de Partage entre une phase
stationnaire imprégnée sur un support (Chromatographie de
partage)
v Phénomène d'Echange d'ions (Chromatographie
d'échange d'ions)
v Phénomène d'exclusion stérique
(Chromatographie d'exclusion stérique) (15)
V.3.3. Choix du système
En chromatographie, au moins trois éléments
interviennent dans le choix du système chromatographique. Il s'agit de
la phase stationnaire, la phase mobile et le mélange de substances
à séparer. (15)
Le choix de la méthode chromatographique sur couche
mince (adsorption, partage et échange d'ions) est
déterminé par la nature de la phase stationnaire utilisée.
La phase mobile est choisie en fonction de l'activité de la phase
stationnaire et de l'affinité de celle-ci vis-à-vis des
substances à séparer. (15)
Cette affinité résulte des
caractéristiques structurales les plus importantes, en particulier des
différences de structure des substances à étudier.
L'influence des dimensions de la molécule est plus faible dans la
méthode par adsorption que dans celle de partage où les
différences de solubilité, dépendant de la grandeur de la
molécule se manifestent très nettement. (15)
En CCM, plusieurs séries d'éluants sont
essayées avant de procéder à un changement de phase
stationnaire. Souvent deux substances non séparées par un
éluant, le sont par le voisin immédiat dans la série
éluotrope. (15)
Cependant, l'utilisation de mélanges trop complexes
peut nuire à la reproductibilité des valeurs de rapport frontal,
à cause des modifications de la composition de l'éluant des dues
à des volatilités différentes des solvants
utilisées. (15)
V.3.4. Chromatographie sur couche mince
d'adsorption
a) Choix de la phase stationnaire
En chromatographie d'adsorption, presque tous les oxydes et
oxydes hydratés employés comme phase stationnaire. Les substances
adsorbées sont des composés polarisables, elles sont maintenues
à la surface de l'adsorbant par des forces électrostatiques et
des liaisons hydrogènes qu'elles forment avec les oxydes
hydratés. La polarisation extrême peut conduire à
l'ionisation de la molécule adsorbée. (15)
L'activité des adsorbants usuels est très
variable, elle peut d'ailleurs être modifiée suite à
certains facteurs en particulier la teneur en eau. Cette dernière est
d'une grande importance dans l'activité d'un adsorbant car les
molécules d'eau sont très polaires, facilement adsorbées
et bloquent la surface des sites de l'adsorbant. (15)
Les adsorbants les plus communs sont : le gel de silice
et l'albumine sous forme oxydé. (15)
L'alumine est surtout utilisée pour la
séparation des mélanges basiques (alcaloïdes par exemple),
les stéroïdes, les hydrocarbures à haut poids
moléculaire, les terpènes et les glycérides de bas poids
moléculaire. (15)
Le gel de silice est la phase stationnaire la plus importante
et la plus utilisée. Il en existe de différentes sortes suivant
qu'il contient ou non un agent liant ou un indicateur de fluorescence. (15)
Chimiquement, le gel de silice est constitué
d'anhydride polysilicique sous forme de grains durs et poreux. Il est
particulièrement adapté à la chromatographie des
substances polaires par suite de la possibilité pour ces
dernières de former des liaisons hydrogènes avec les hydroxyles
attachés au squelette silicié. (15)
Borke et Kirsh ont
été les premiers à utiliser n gel formé de silice,
d'oxyde de magnésium et de sulfate calcium dans les proportions
(10/10/4) et imprégné d'une solution tampon de
déhydrogénophosphate de potassium de pH en 1953. (15)
b) Choix de la phase mobile
Le choix de la phase mobile (qui est un solvant ou un
mélange de solvants) dépend avant tout de la polarité des
constituants de l'échantillon et de phase stationnaire. Ces deux phases
doivent avoir des polarités opposées. (15)
V.3.5. Rapport frontal et avantages de la CCM
Le rapport frontal (Rf) exprime le rapport entre la distance
parcourue la substance et la distance parcourue par le front de la phase
mobile. Ces distances sont mesurées à partir de la ligne de
départ correspondant au centre de dépôt initial du
mélange à séparer jusqu'au centre du ou des spot(s) et au
front du solvant. Il faut noter que chaque substance possède un Rf dans
un système chromatographique donné. (15)
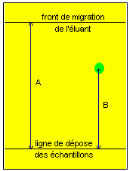
Distance
parcourue par la substance (B)
Rf=-----------------------------------------------------------
Distance parcourue par le front du solvant
(A)
La CCM présent les avantages ci-après :
- la rapidité d'exécution (1 à 2 heures),
- la simplicité d'exécution,
- un coût modeste,
- la sensibilité de l'ordre des microgrammes (ug).
V.3.6. Application de la CCM en Pharmacie
Dans le domaine pharmaceutique, le principal
intérêt de la chromatographie en couche mince est de permettre la
détermination, de faibles concentrations d'impuretés dans les
substances médicinales. Lorsqu'on l'utilise à des fins
d'identification, cette technique permet de comparer le comportement
chromatographique de la substance à identifier avec celui d'une
substance étalon qui est généralement un spécimen
authentique du produit à examiner. (15)
Grâce à la grande variété des
couches que l'on utilise en association avec divers solvants, on peut faire
varier le pouvoir séparateur d'une manière presque infinie et
c'est ce qui rend la CCM aussi utile en Pharmacie. (15)
Pour ce qui est des déterminations quantitatives, elles
peuvent être effectuées directement sur la plaque soit
après récupération du produit sur la plaque en grattant la
tâche et en utilisant un solvant approprié pour l'extraire de
l'adsorbant. Après récupération, le dosage peut se faire
par une méthode suffisamment sensible telle que la
spectrophotométrie UV/Visible, soit directement, soit après
dérivatisation ou réaction chimique (méthode de
Green). (15)
V.4. SPECTROPHOTOMETRIE UV/VISIBLE
La spectroscopie d'absorption moléculaire dans
ultraviolet, le visible et l'infrarouge est largement utilisée pour
l'identification et le dosage d'innombrables espèces inorganiques et
organiques. La spectroscopie d'absorption ultraviolet-visible est surtout
employée en analyse quantitative et est probablement plus
utilisée que toutes les autres méthodes dans les laboratoires
d'analyses chimiques ou médicinales du monde entier. (15)
V.4.1. Absorption par les composés
organiques
L'absorption de rayonnement par les molécules
organiques dans le domaine de longueurs d'onde compris entre 180 et 780 nm
(nanomètre) résulte des interactions des photons qui participent
directement à la formation de la liaison `et qui sont donc
associés à plus d'un atome) avec ceux qui sont localisés
sur des atomes tels que l'oxygène, le soufre, l'azote et les
halogénures. (15)
Les longueurs d'onde d'absorption d'une molécule
organique dépendent de l'énergie de liaison de ses
différents électrons. Les électrons qui forment des
liaisons simples carbone-carbone ou carbone-hydrogène absorbent à
des longueurs d'onde au dessous de 180 nm (l'ultraviolet lointain) et ceux
compliqués dans les doubles ou triples liaisons absorbent à des
longueurs d'onde au dessus de 181 nm jusqu'à 780 nm (l'ultraviolet
proche et le visible). (15)
Les groupements fonctionnels organiques insaturés qui
absorbent dans l'ultraviolet et le visible sont appelés des
chromophores. (15)
V.4.2. Avantages
La spectroscopie d'absorption ultraviolet-visible
présente les avantages ci-après :
- un vaste champ d'application,
- une grande sensibilité,
- une grande sélectivité,
- une bonne exactitude,
- une facilité de mise en oeuvre. (15)
V.4.3. Applications qualitatives et quantitatives
Pour les applications qualitatives, la
spectrophotométrie dans l'ultraviolet détecte les groupements
chromophores ou des atomes tels que le soufre ou les halogénés
par l'apparition d'un ou plusieurs pics dans le domaine de 200 à 400 nm.
Les spectres dans l'ultraviolet ne présentent cependant pas de
structures suffisamment fines pour permettre l'identification certaine d'un
analyste, mis ils doivent être complétés par d'autres
données physiques ou chimiques fournies par d'autres méthodes
spectroscopiques. (15)
Pour les applications qualitatives, la spectroscopie
d'absorption dans l'ultraviolet et le visible est un des outils les plus
utilisés par les chimistes et les biologistes en analyse quantitative.
(15)
Deuxième Partie :
PARTIE EXPERIMENTALE
INTRODUCTION
Le présent travail vise l'évaluation de la
stabilité chimique de l'Artésunate contenu dans les
comprimés en co- blister avec ceux de l'Amodiaquine dans la
spécialité ARSUCAM® provenant des sources
pré qualifiées.
Cette évaluation a été effectué
grâce aux tests d'identification de l'Artésunate tels que la
Chromatographie par Couche Mince, la recherche des produits de
dégradation et l'évaluation pharmaco techniques des
comprimés.
Les résultats obtenus sont rendus et
discutés.
CHAPITRE I. MATERIEL ET METHODES
I.1. Echantillons
Trois échantillons de trois lots différents
d'une même spécialité ARSUCAM®
collectés après la péremption des sources
préqualifiées (centrales de distribution régionale de
Kisantu).
Le tableau I donne les détails relatifs à ces
échantillons.
Tableau I : Caractéristiques des
échantillons d'Artésunate-Amodiaquine
collectés
|
N°
|
Dénomination
|
N° de Lot
|
Date de fabrication et de péremption
|
Codification
|
|
1
|
ARSUCAM® (7 à 13
ans)
|
081
|
03/2005 et 03/2007
|
KIS/AS-AQ/07/01
|
|
2
|
ARSUCAM® (7 à 13
ans)
|
086
|
03/2005 et 03/2007
|
KIS/AS-AQ/07/02
|
|
3
|
ARSUCAM® (7 à 13
ans)
|
075
|
03/2005 et 03/2007
|
KIS/AS-AQ/07/03
|
I.2. Matériel et réactifs
Le matériel et les réactifs suivants ont
été utilisés pour notre étude :
- Capsule en porcelaine
- Balance analytique Mettler H20
- Erlenmeyers 25 ml, 50 ml, 100 ml, 250 ml, 500 ml, 1000
ml,
- Pipettes 5 ml, 10 ml, 25 ml
- Tubes à essai
- Pieds gradués 50 ml, 100 ml
- Pilon et mortier
- Plaque chauffante
- Entonnoir
- Béchers 25 ml, 50 ml et 100 ml
- Chambre (cuve) chromatographique (Boite de Mayonnaise)
- Microcapillaires 2ul et 1ul
- Papier filtre « VWROME »130, 413, 90
nm
- Capsules en verre
- Pince
- Spatule
- Pissette 100 ml
- Phase stationnaire : Plaque CCM Sil G-25 UV254 (Gel de
silice 60 F254)
- Phase mobile : Toluène/Acétate
d'éthyle/Ethanol (6/3/1)
- Méthanol
- Ethanol
- Toluène
- Acétate d'éthyle
- Vanilline/Ethanol/H2SO4
- Eau distillée
- Poudre d'Artésunate (matière première)
I.3. Détermination du poids moyen et des
variations de poids des comprimés
Principe
La détermination du poids moyen des comprimés se
fait en prélevant 20 comprimés au hasard dans un même lot
et les peser un à un au moyen d'une balance à précision. A
l'aide de la formule ci-dessous, on calcule le poids moyen et les variations de
poids.
?Pi
Poids Moyen (PM)= ------
20
Poids minimal-Poids moyen
Ecart (-) = ---------------------------------------x100
Poids moyen
Variation de Poids
(V.P)
Poids maximal-Poids moyen
Ecart (+) = --------------------------------------x 100
Poids moyen
Normes : Comprimés : PM<140 mg=>
VP = + 10 %
PM (140-300 mg) => VP = + 7 %
PM> 300 mg => + 5%
I.4 Mode opératoire de l'analyse par CCM
d'adsorption
I.4.1. Préparation de la phase
stationnaire
A partir d'une plaque à base de Silicagel G254 20x20cm,
des morceaux de 5x10cm ont été découpés. Nous avons
apprêté ces plaques en traçant deux lignes
parallèles et horizontales, l'une à 1,5 cm du bord
inférieur et l'autre à 1 cm du bord supérieur de la
plaque.
Les points de dépôt d'échantillons sur la
plaque ont été séparés de 1 cm en prenant soin de
laisser 1 cm aux extrémités de chaque plaque.
Les détails relatifs à la préparation des
plaques chromatographiques sont montrés par la figure 1.
Les plaques de chromatographie sur couche mince ainsi
apprêtées sont soumises à l'activation de l'adsorbant en
les chauffant légèrement sur une plaque chauffante à 100
°C. Elles deviennent ainsi prêtes à recevoir les
dépôts d'échantillons à analyser.
1cm 1 cm
10 cm
1,5 1,5cm
5cm
Figure 1 :
Représentation d'une plaque de CCM
I.4.2 Préparation des
échantillons
A partir de la poudre fine des comprimés
d'artésunate (pulvérisée au mortier) de chaque lot
à évaluer, une prise d'essai d'environ 1mg d'artésunate
est dissoute dans 3ml d'alcool, après agitation 7ml d'alcool sont
ajoutés. (à dissoudre plutôt dans 2ml de
méthanol) ??
Après agitation et filtration, le filtrat
collecté est évaporé et le résidu repris par 10ml
de toluène. La solution ainsi obtenue est utilisée pour le
dépôt d'échantillon sur la plaque à chromatographie
sur couche mince.
La matière première d'artésunate a servi
de témoin. En effet, 1mg de poudre dissoute dans 10ml de toluène
a donné une solution qui a été utilisée comme
étalon dans le développement chromatographique.
I.4.3. Préparation de la phase
mobile et saturation de la chambre chromatographique
La phase mobile était constituée du
mélange Toluène/Acétate d'éthyle/Ethanol dans les
proportions 6 :3 :1.
Les différentes cuves chromatographiques utilisées
dans notre étude ont été soigneusement lavées et
séchées. Elles ont ensuite été bien rincées
au préalable et saturées avec la phase mobile pendant au moins
deux heures en les fermant hermétiquement.
I.4.4. Préparation du
révélateur à base de vanilline
La solution utilisée comme révélateur est
préparée comme suit : peser 1g de vanilline en poudre et le
placer dans un ballon jaugé de 100ml. Ajouter quelques millilitres
(environ 20 ml) d'éthanol pour la dissolution et porter au volume avec
de l'éthanol. Enfin, ajouter goutte à goutte 2 ml d'acide
sulfurique concentré.
La solution ainsi obtenue ne peut être utilisée
que dans les 48 heures qui suivent sa préparation.
I.4.5. Développement
chromatographique
Sur les plaques de Silicagel préparées, nous
avons déposé 4 ul des solutions des échantillons aux
points indiqués et à l'aide des microcapillaires de 1ul et 2ul
(c'est-à-dire on spot quatre fois de suite ou deux fois de suite) les
différents échantillons.
Après un temps de séchage, les plaques ont
été placées dans les chambres chromatographiques
préalablement saturées pour leur développement.
A la fin du développement (fin de la migration), les
plaques ont été retirées de la chambre chromatographique,
puis séchées à l'air libre pendant 15 minutes.
I.4.6. Révélation des spots
après développement chromatographique
La révélation des substances analysées se
fait en pulvérisant sur les plaques développées
préalablement séchées, la solution du
révélateur (vanilline dans l'éthanol et acide sulfurique).
Puis, on expose les plaques à la chaleur (à l'étuve ou
à une plaque chauffante) pendant 3 à 5 minutes.
Il se développe une coloration rouge en présence
de l'artésunate ou d'un autre dérivé de
l'artémisinine.
CHAPITRE II. RESULTATS ET DISCUSSIONS
II.1 RESULTATS
Les échantillons des comprimés
d'artésunate collectés ont fait l'objet de développement
chromatographique en vue de l'évaluation de la stabilité chimique
du principe actif par la recherche des produits de dégradation dans nos
conditions opératoires.
Avant de procéder au développement proprement
dit, quelques essais ont permis d'apprécier la qualité
pharmacotechnique des comprimés, notamment la détermination du
poids moyen et des variations des poids. Les résultats de ces
déterminations sont repris dans le tableau II
Tableau II : Poids moyen et variation de poids
des comprimés des échantillons collectés
|
N°
|
Dénomination
|
N° de Lot
|
Poids moyen
|
Variation de poids
|
|
1
|
ARSUCAM® (7 à 13
ans)
|
081
|
275,5 mg
|
-1,633% à +1,758%
|
|
2
|
ARSUCAM® (7 à 13
ans)
|
082
|
273,33 mg
|
-1,763% à +1,876%
|
|
3
|
ARSUCAM® (7 à 13
ans)
|
075
|
272,42 mg
|
-1,361% à +2,129%
|
Quant à la CCM, dans nos conditions opératoires,
l'identité de l'artésunate contenue dans les comprimés a
été confirmée sur les chromatogrammes
développés pendant plus d'une année d'étude. En
effet, ainsi que le montre les résultats consignés dans le
tableau III, les échantillons testés ont montré les
mêmes Rf que les solutions d'artésunate utilisées comme
témoin.
Tableau III : Rf des échantillons
collectés par rapport aux témoins en fonction du
temps
|
N°
|
Dénomination
|
N° de lot
|
Rf Témoin
|
Rf Echantillon
|
|
1
|
ARSUCAM® (7 à 13
ans)
|
081
|
|
|
|
2
|
ARSUCAM® (7 à 13
ans)
|
082
|
|
|
|
3
|
ARSUCAM® (7 à 13
ans)
|
075
|
|
|
II.2 DISCUSSION
Tous les échantillons des comprimés
d'artésunate ayant fait l'objet de notre étude ont
été présentés sous blister et leur
étiquetage a été conforme à l'examen visuel. Les
mentions obligatoires que sont la dénomination commune internationale
(DCI), le dosage, les dates de fabrication et de péremption, le
numéro de lot etc. Étaient marqués sur l'emballage.
La détermination du poids moyen et des variations des
poids effectuée sur les comprimés a montré que des
échantillons collectés présentaient des variations de
poids conformes aux normes généralement admises par les
pharmacopées, c'est-à-dire #177; 7,5% pour les comprimés
de poids moyen compris entre 140 et 300 mg .
L'identification du principe actif dans les comprimés
d'artésunate a été réalisée au moyen de la
chromatographie sur couche mince. Cet essai a permis à la fois de
confirmer la présence de l'artésunate dans tous les
échantillons analysés, mais aussi de détecter quelques
produits de dégradation dans certains échantillons dans nos
conditions opératoires. En effet, la présence des spots
supplémentaires sur certains chromatogrammes suggère la
présence des substances autres que l'artésunate mais qui peuvent
lui être apparentées par le rapprochement des Rf.
Toutefois, pour certains échantillons testés,
l'absence des spots supplémentaires sur les chromatogrammes pourrait
s'expliquer par la présentation des médicaments sous blister qui
améliore peut-être la conservation. Cependant, au fur et à
mesure que la durée de vie du médicament est largement
dépassée, l'apparition des produits de dégradation
pourrait être proportionnelle.
CONCLUSION
Notre travail consistait en l'évaluation
préliminaire de la qualité chimique de l'artésunate dans
des comprimés après la date de péremption par la technique
de chromatographie sur couche mince d'adsorption.
Cette technique a permis l'identification et la confirmation
de la présence de l'artésunate dans les comprimés
après la date de péremption. Elle a en outre permis de mettre en
évidence la présence de quelques produits de dégradation
dans certains échantillons par la présence des spots
supplémentaires sur certains chromatogrammes dans nos conditions
opératoires.
Les difficultés rencontrées sont des limitations
d'ordre technique avec des échantillons qui contenaient moins de quatre
comprimés par blister, ne permettant pas ainsi d'effectuer certains
essais.
Au regard de ces résultats, il est souhaitable qu'une
étude de grande envergure soit menée sur la recherche des
produits de dégradation dans les comprimés d'artésunate
dotés d'une date de péremption supérieure à deux
ans (24 mois).
Les résultats à obtenir pourront éclairer
l'autorité sanitaire nationale dans le rejet de tous les
médicaments à base d'artésunate soumis à
l'enregistrement dans le pays avec des durées de vie supérieure
à 24 mois.
REFERENCES BIBLIOGRAPHIQUES
1. BASCO K. L., 2004. Molecular epidemiology of malaria
in Cameroon. XIX. Quality of antimalarial drugs used for
self-medication. Am J Trop Med Hyg, 70, 245-250
2. BOURE P., Ng.Van, Ph.Taugoudeau et ANH, LE
PALUDISME
Ed. SmithKline Beecham SB 1993 p.2
3. CURTIS, SUTTER, WALKER et HOFFMAN, Editions De Boeck, 2001,
PHARMACOLOGIE INTEGREE De Boeck 2001
4. GENTILLINI M., MEDECINE TROPICALE
5ème édition
Médecine-Sciences Flammarion 1993
5. GREEN D.M et al., Dec 2001, Authentification of
artemether, artesunate and dihydroartemisinin antimalarial tablets using a
simple colorimetric method.
Trop Med and Int Health, Vol 6, 12 pp980-982
6. MACE Gordon, GUIDE D'ELABORATION D'UN PROJET DE
RECHERCHE
2ème édition Paris Bruxelles De
Boeck université 1997 p.79-89
7. MULUMBA MADISHALA, 2004, UNIKIN, ELEMENTS DE
PROTOZOOLOGIE, Editions BIOMETRIX, Notes de cours
8. PIERRE Bernard et Geneviève, 1999, Editions De Boeck,
DICTIONNAIRE MEDICAL POUR LES REGIONS TROPICALES.
9. TAMBA VEMBA, 2004, UNIKIN, LEGISLATION ET DEONTOLOGIE
PHARMACEUTIQUES, Notes de cours
10. Organisation Mondiale de la Santé, Genève,
2004, PALUDISME EN AFRIQUE 2003
11. Organisation Mondiale de la Santé, Genève 2003,
Place des antipaludiques dans la contrefaçon des
médicaments. WHO/EDM 2003
12. ANONYME : RAPPORT DU MINISTERE DE LA SANTE
2000
13. ANONYME : ELEMENTS DE PARASITOLOGIE
MEDICALE 3ème édition
Flammarion Sciences 1978 p.298-342
14. ANONYME : COURS DE PHARMACOLOGIE Paris
éd. Ellipse 1987 p.517
15. Encyclopédie médical pratique
copyright c 1994, 1995, 1996, 1997 The Learning Company Inc. TLC-Edusoft
16.
www.ulb.be/erasme/edu/travelcliniere/malaria
17.
www.chmp.org (Fiche n°11)
18.
www.glaxosmithkline.fr/gsk/gsk_France/quali_assu
19.
www.ordre.pharmacien.fr/fr/bleu/index4_1_5
20.
www.ordre.pharmacien.fr/gsk_France/quali_assu
21.
http://www.pasteur.mg/sppalub.html
22.
www.mmv.org/IMG/doc/Trafiquants_d.doc
23.
www.who.int/mediacentre/factsheets
(Aide mémoire n°275 novembre 2003)
24.
http://www.paho.org/French/AD/DPC/CD/mal-adct.htm
(Rapport d'une consultation technique de l'OMS sur les combinaisons
thérapeutiques antipaludiques)
25.
http://www.msf.be/fr/terrain/pays/afrique/congo_news_42.shtml
26.
http://www.agoravox.fr/article.php3?id_article=20250
27.
http://www.santetropicale.com/actualites/ac0205_3.htm
28.
http://www.essentialdrugs.org/emed/archive/200504/msg00030.php
29.
http://edisan.timone.univ-mrs.fr/edisan/Lettre/Textes/LED19.html
Pratiques des combinaisons d'antipaludiques et la
prévention du paludisme :
A. Bourgeade, Y. Nosny, J. Delmon
32. LOPANGU KAPANDA et al., Contribution à la
Surveillance de la Qualité des Médicaments Antipaludiques en
République Démocratique du Congo : Evaluation des Conditions
de Fonctionnement des Officines Ouvertes au Public dans la Ville de
KINSHASA, Travail de Fin de Cycle, Faculté des Sciences
Pharmaceutiques, UNIKIN, 2005, inédit.
33. LANY Nesmy Anouk, Evaluation préliminaire
de la qualité des comprimés à base d'artésunate
disponibles sur le marché pharmaceutique congolais : cas de la
ville de Kinshasa, Mémoire, Faculté des Sciences
Pharmaceutiques, UNIKIN, 2006, inédit.
34. Egon STAHL, Analyse chromatographique et
microscopique des drogues (Manuel pratique pour les pharmacopées
européennes), Entreprise moderne d'édition, 1975, p.13,
14, 17 et 29.
35. Rapport du comité OMS d'experts des
spécifications relatives aux préparations
Pharmaceutiques, 1996.
36. AMIPS Info, Association des Médecins des
Industries des Produits de Santé, N ° 6 8, 1er et 2e
trimestres 2004.
37. Olivier Gross, Programme OMS de
Pré-qualification, Organisation Mondiale de Santé,
Paris, 2005.
38.
http://healthtech.who.int/pq/info_general/documents/2007_PQSummary.pdf
| 
