
Republique Algérienne Démocratique et Populaire
Ministère de l'Enseignement Supérieur & de
la Recherche Scientifique Université Mohamed Cherif Massaadia
Souk-Ahras
Année universitaire 2012-2013
Faculté des Sciences et de
Technologie
Département sciences de la matière
MEMOIRE
Présenté en vue de l'obtention du diplôme
de MAGISTER
Option: Chimie analytique et physique
Etude des couches minces à base de terre
rare
destinées à la protection des matériaux contre la
corrosion
Par: Melle Sameh ATHMANI
Directeur de mémoire: Y. HAMLAOUI M.C. Univ.
Mohamed-Cherif Messaidia S/A
Président: Mr. A. GHEID Pr. Univ. Mohamed Cherif
Messaidia-S/A
Rapporteur: Mr. R. REHAMNIA Pr. Univ. Badji Mokhtar-Annaba .
Rapporteur Melle. S. CHOUCHANE M.C. Univ. Badji Mokhtar-Annaba
.
Membre invité: Melle. S. BOURANANE M.C. Univ.
Mohamed Cherif Messaidia-S/A
REMERCIEMENTS
En préambule à ce mémoire de Magister, je
veux vivement et sincèrement exprimer mes remerciements et ma profonde
reconnaissance à toux ceux et à toutes celles qui m'ont
apporté, directement et/ou indirectement, leurs aides et qui ont
contribué activement de quelque manière que ce soit à
l'élaboration et à l'aboutissement de ce travail.
Je tiens, en particulier, à exprimer, ici, toute ma
gratitude à Monsieur HAMLAOUI Youcef Maître de conférence
« A » à l'université de Souk-Ahras
Mohamed-Chérif MESSAIDIA qui a bien voulu accepter de diriger les
travaux de mon mémoire et à lui demander de bien vouloir croire
et pour toujours à mes remerciements sincères et cordiaux pour la
plus précieuse des richesse qu'on puisse donner le
savoir.
Monsieur HAMLAOUI, vous avez été toujours
à mon écoute et vous avez fait preuve à mon égard
de votre disponibilité permanente en mettant à ma disposition
durant tout le temps de préparation de ce mémoire, vos grandes
capacités scientifiques et pédagogiques; vous m'avez
consacrée tout votre temps, vous m'avez abreuvée de
précieux conseils; vous m'avez aussi encouragée dans les moments
d'angoisses qu'il m'est arrivée de traverser et à m'inciter
à affiner la qualité de mon travail de laboratoire pour
consolider, me dites-vous, les connaissances que j'ai acquises durant tout le
parcours de préparation de ce mémoire.
Je ne peux pas non plus omettre de remercier
sincèrement:
- Monsieur le Professeur GHEID Abdelhak de l'université
de Souk-Ahras Mohamed-Chérif MESSAIDIA non seulement pour m'avoir fait
l'honneur d'accepter de présider le jury de mon mémoire mais
aussi pour avoir ouvert et organisé le concours de post-graduation et
donc l'existence de cette promotion de magistère. Je sais combien, sont
pénibles, couteuses en temps et laborieuses les démarches
administratives et la mise en place de l'encadrement scientifique
nécessaires à l'organisation et au démarrage d'une
formation scientifique de ce type. Je lui dois, à ce titre, toute ma
reconnaissance.
- Monsieur le Professeur REHAMNIA Rabah de l'université
d'Annaba Badji-Mokhtar qui n'a pas, lui aussi, hésité à
mettre à ma disposition l'ensemble des moyens de son laboratoire
d'électrochimie où tous les travaux expérimentaux de mon
mémoire y ont été réalisés, mais aussi, il
m'a souvent aidé avec générosité et
compétence avérée à dissiper mes angoisses en
m'aidant à trouver les solutions quant il m'est arrivée de buter
devant mes questionnements dans mes travaux de laboratoire.
- Mme CHOUCHENE Sabiha MC de l'université d'Annaba
Badji-Mokhtar pour l'amabilité qu'elle me fait d'avoir accepter
d'examiner mon mémoire et d'être membre du jury, qu'elle en soit
sincèrement et vivement remerciée.
- A mon invitée de marque, Mme BOURANANE MC de
l'université de Souk-Ahras Mohamed-Chérif MESSAIDIA que je
remercie pour l'honneur qu'elle me fait d'avoir accepter de sacrifier un peu de
son temps précieux et de rehausser de sa présence la
présentation et la soutenance de mon mémoire.
J'exprime, ici, mes remerciements cordiaux et fraternels à
:
- Mon amie, je peux même dire ma soeur Madame OUCHENANE
Sihem (Doctorante en chimie à l'université Badji-Mokhtar
d'Annaba) à qui, je souhaite, non pas, de réussir la soutenance
de sa thèse, je sais qu'elle en est amplement capable, mais que
l'obtention de ce titre lui ouvre les portes de tout le bonheur qu'elle
mérité. Ma chère amie Sihem, je te prie de croire, pour
toujours, à ma reconnaissance sincère pour les conseils et les
précieuses informations que tu m'as souvent donnés.
- Amel, ma camarade de promotion, mon autre soeur qui ne lui
manque que le nom patronymique pour être ma soeur biologique. Je veux que
tu saches les sentiments de fraternité et d'amitié profonde que
j'éprouve à ton égard. Je te souhaite de soutenir, avec
brio, comme il est dans tes habitudes, ton mémoire de magistère
et à bientôt, Incha-Allah, pour le Doctorat.
- A mes amies les plus chères, Fatima, Nabila, Amel G
et Amel S, Roubila, Saïda à qui je leur adresse mes tendres et
affectueuses pensées.
- A mes parents pour leur contribution, leur soutien et leur
patiente, à mes frères et soeurs sonia et meryouma , à mes
neveux et à mon unique nièce Sarah, à mes cousines et
cousins, à mes camarade de promo, enfin, à tous mes proches qui
m'ont toujours soutenue et encouragée au cours de la réalisation
de ce mémoire et en particulier à mon fiancé Yacine.
Introduction générale 01
CHAPITRE I : Partie théorique
I. 1. Corrosion et protection 03
I. 2. Généralités sur
l'électrodéposition d'oxydes métalliques 05
I.3. Electrodéposition cathodique des oxydes
métalliques 06
I.3.1. Déposition cathodique électrolytique
08
I.3.2. Application 09
I. 4. Que sont les terres rares 10
I.4.1. Généralités sur les terres rares
et l'oxyde de cérium 10
I.4.2. L'oxyde de Cérium 11
I.4.3. Structure cristallographique 12
wwRéférences bibliographiques
13
CHAPITREII : Procédure expérimentale et
techniques de caractérisation
II.1. Méthode d'élaboration des
revêtements 16
II.1.1.Principe de la méthode 17
II.1.2. Mécanismes de
l'électrodéposition 19
II.2. Caractérisation des revêtements
à base d'oxyde de cérium 21
II.2.1. Techniques expérimentales
21
II.2.2. Techniques stationnaires 21
II.2.2.1. Suivi du potentiel à circuit ouvert (OCP)
21
II.2.2.2. courbes de polarisation 22
II.2.2.3. Méthode des droites de Tafel
23
II.2.2.4. Méthode de la résistance de
polarisation 24
II.2.2.5. Voltampérométrie cyclique
25
II.2.3. Techniques non stationnaires 26
II.2.3.1. Spectroscopie d'impédance
électrochimique(SIE) 26
II.2.3.1.1. Définition et principe
26
II.2.3.1.2. Impédance des phénomènes
pouvant se produire à l'interface
électrode/électrolyte 28
II.2.3.1.3. Exploitation des résultats
31
II.2.4. Application de la SIE aux revêtements
32
II.2.5. Représentation des données
34
II.2.6. Avantages et Inconvénients de la SIE
36
Références bibliographiques
37
CHAPITRE III : Résultats et discussion
III.1. Introduction 41
III. 2. Elaboration des revêtements
41
III. 3. Comportement électrochimique des
substrats dans NaCl 44
III. 3. 1. Evolution du potentiel à circuit
ouvert du substrat nu dans NaCl 44
III. 3. 2. Evolution du potentiel à circuit ouvert
des substrats revêtus dans NaCl 47
III. 3. 3. Courbes de polarisation des substrats dans
NaCl 49
III.3.4 Diagrammes SIE enregistrés sur des
substrats dans NaCl 55
III.4. Comportement électrochimique des
substrats dans eau industrielle 61
III.4. 1. Milieu d'essai 61
III. 4. 2. Evolution du potentiel à circuit
ouvert du substrat nu 63
III. 4. 3. Evolution du potentiel à circuit
ouvert du substrat revêtu 64
III.4. 4. Courbes de polarisation des substrats dans
l'eau industrielle 65
III.4. 5. Diagrammes SIE enregistrés sur des
substrats dans l'eau industrielle 69
III.5. Comparaison entre les deux milieux
73
Références bibliographiques
75
CONCLUSION GENEARLE
Résumé : L'utilisation des
revêtements pour la protection contre la corrosion des métaux est
une alternative aux traitements aux chrnomates, proscrits du fait de leur forte
toxicité. Les procédés de conversion de chromatation
montrent à l'égard de la corrosion des aciers et les aciers
galvanisés les meilleurs résultats mais l'utilisation du Cr (VI)
pose des problèmes environnementaux, le chrome hexavalent étant
toxique, cancérigène et mobile dans l'environnement.
Un des traitements alternatifs de passivation utilise des
oxydes de terres rares, notamment le CeO2. A l'heure actuelle, peu de travaux
se sont intéressés aux revêtements sur acier bas carbone
à usage générale. Le traitement de passivation est
réalisé par déposition cathodique à partir de
solutions de sels de cérium. Dans ce dernier cas, la formation des
couches minces est généralement associée à la
formation d'oxydes ou d'hydroxydes de cérium sur les sites cathodiques
de la surface métallique. La caractérisation
électrochimique des revêtements a été
évaluée dans des milieux agressifs tels que NaCl 0,5M et l'eau
industrielle. Ainsi, la résistance à la corrosion des
revêtements obtenus sur acier a été étudiée.
Pour l'ensemble des échantillons, la durée de protection a
été évaluée grâce à la mesure de
Ecorr. En parallèle, la caractérisation des revêtements est
suivie par la mesure de la résistance de polarisation, de la
densité de courant de corrosion extrapolée des droites de Tafel
de la spectroscopie d'impédances électrochimique. En conclusion,
les résultats des différentes techniques électrochimique
et analytique ont montré que l'état de fissuration des
revêtements obtenus sur acier a mis en cause leur bonne tenue
vis-à-vis de la corrosion.
Mots clés : Couches minces, terres rares,
électro-déposition cathodique, oxyde de cérium,
Corrosion.
F% âÏ1 *éãH®Üß'
,üÌß Þþ î3/4 ÞÛß'
éþéäß F/éÌäàß
!üÄß' E'·3' F%
'®Èç æÜß H
,úéèß' ÞÀÓ#
æÔàÜäß' 0ûîÔß' H
0ûîÔß' F/éÌã
ðàË *®Ç#
*éãH®Üß' Þþî
*éÿàäË ®Àã
+îàã H Fé ®'àß ê''ã
«ç# »Óé% »ÿOEÿ
ÞÛéã -®Äþ J#172;ß'
«ã'·3' H ò3''ß'
EH®Üß' »ÿä'ß
.»ÿßéÌß'
«ÿä'ß '®Èç '1îÈã
cents'»#«àÌ· éã
.»OEÿ'ßé
Þÿà× ®éß'
-×îß' òÓ .
Eîþ®ÿ'ß' ÿ'Û# éèÿ
æã )1/éèß' »®÷'
ÿ3éÛ# E·' »àþ'ß'
*é·üÌß' £# F% Þ·# æã
H Béÿ'ß' '#172;3/4 òÓ
H.Fî®Üß' 3/4Ô·èã
0ûîÓ centsÄ3 ðàË
*éØ' ÞÿÜ -ä3/4' òß'
*é3'1ß' ò3/4 »ßéß'
G#172;3/4 òÓ Eîþ®ÿ'ß'
-üã# Dü§ æã êßé'ß'
êÄØß' »Øþ®Ä
Eîþ®ÿ'ß' ÿ'Û# æã
»Ô߆ã *éØ' ÞÿÜ
centsÄ3 æã Ê×'îã
ðàË Eîþ®ÿ'ß'
*'ÿ'ÛH1ÿ3/4 H# ÿ3éÛ# ÞÿÜ
Â'®ã »Øÿ×®ß'
»ÿÏ÷' ÞÿÜ FîÜþ
.FÌäß'
centsàã éèÿ æã
»ÿç'HÌß'
Þÿßéäß' æã
æÿËîç òÓ â
»àÜäß' *éØ'Äàß
òéÿäÿÛH®Üß'
âÿÿØß' FàÓ
«ÿàË H ÞÛß'
»ãHéØã »3'1/ -ä
âÿÿØß' Dü§
.»ÿËéè» Géÿã H
D/Dîã 0,5 °ÿÛ® H0
Eîþ/î1/4ß' þ1îàÛ
» éèß' 1éÿß'
»ÓéoeÛ 3éÿ× éÀþ#
H *éèÿÌß' Êÿä ß
FÌäß' centsÄ3 ðàË
Eîþ®ÿ'ß' ÿ'Û# æã
»àÜäß' *éØ'Äàß
.(éÄØ3û' »ãHéØã
3éÿ× »ÿàäË I®§#
»· æã H »· æã (TAFEL )
òÔÿÄß' Þÿàß'
»ÿàäË æã
F#' »àäÌ'äß'
»ÿàÿàß' H
»ÿéÿäÿÛH®Üß'
*éÿèØß' Òà·ã
Dü§ æã éÿàË
Þ1/4äß' úéèß' *®Ç#
'®ÿ§# .»ÿç'HÌß'
Þÿßéäß' òÓ
'Ûàß »ãHéØã H
1'®Ø3û' ,FÌäß' centsÄ3
ðàË éÿàË
Þ1/4äß' »àÜäß'
*éØ'Äß'
Eîþ®ÿ'ß' ÿ'Û# ,
êßé'ß' êÄØßé
êÿÛ®H®Û c )1/éèß'
»ÿ1÷' ®»éèÌß' c
»Øÿ×1 »ÔàÏ#
:-éÔã *éäàÛ
.ÞÛß'
Abstract: The use of coatings for
metals protection against corrosion is an alternative to chromate treatment,
restricted because of their high toxicity. Indeed, the chromate conversion
processes show, the best results, however the use of Cr (VI) poses
environmental problems. Hexavalent chromium is toxic, carcinogenic and mobile
in the environment. A green passivation treatment uses rare earth oxides, such
as CeO2. At present, few studies have focused on coatings on low carbon steel.
The passivation treatment is carried out by cathodic deposition from solutions
of salts of cerium. Indeed, the formation of thin films is generally associated
with the formation of cerium hydro-oxides on cathodic sites of the surface
metal. The electrochemical characterization of the coatings was evaluated in
aggressive media as NaCl 0.5 M and industrial water. For all samples, the
characterization of the coatings is monitored by measurement of the open
circuit potential, polarization resistance as DC measurement and by
electrochemical impedance spectroscopy as AC measurement. The electrochemical
and analytical results have shown that the observed cracks of the coatings
obtained on steel have implicated their good behavior against corrosion.
Keywords : thin films, rare earth, cathodic electrodeposition,
Cerium oxide , Corrosion .

1
Introduction générale
Introduction générale
Les traitements de surfaces répondent à des
besoins d'amélioration des propriétés mécaniques et
anticorrosion des matériaux. Depuis quelques années, les couches
minces font l'objet d'un intérêt croissant visant à
répondre à ces deux exigences, puisque ceux-ci semblent
présenter des comportements physiques et chimiques attirants.
Les systèmes de faibles dimensions comme les couches
minces nanostructurées présentent des caractéristiques
structurales tout à fait particulières du fait de leur taille
réduite. Ils offrent la possibilité d'induire des
phénomènes nouveaux et exotiques.
De plus, ces couches sont d'une importance fondamentale dans
les applications technologiques. Plusieurs études
montrent que les imperfections de surface affectent d'une certaine
manière les propriétés magnétiques des
métaux de transition. Cependant, les imperfections de
surface de matériaux sous forme de couches minces est accru à
mesure que leur épaisseur décroît. En effet, afin de
réduire au maximum ces irrégularités de surface les
couches minces sont élaborées principalement par des
méthodes telles que l'épitaxie par jet moléculaire, la
pulvérisation et électrodéposition cathodique. Il s'agit
de méthodes reconnues et élégantes, qui demeurent
toutefois souvent contraignantes et onéreuses. L'emploi de ces
revêtements à l'échelle industrielle nécessite un
procédé d'élaboration économiquement
intéressant. De ce fait, nous avons retenu une voie
électrochimique pour l'élaboration des couches minces [1].
Un des traitements alternatifs de passivation sans Cr(VI)
utilise des oxydes de terres rares, notamment l'oxyde de cérium CeO2. A
l'heure actuelle, peu de travaux se sont intéressés aux
revêtements sacrificiels d'alliages de Zn. Le traitement de passivation
est réalisé par immersion dans des solutions de sels de
cérium ou par déposition cathodique. Dans ce dernier cas, la
formation des couches minces est généralement associée
à la formation d'oxydes ou d'hydroxydes de cérium sur les sites
cathodiques de la surface métallique [2].
En effet, l'électrodéposition possède
plusieurs avantages (simplicité de mise en oeuvre, possibilité de
géométries complexes pour les substrats, gamme de
températures et de vitesses de synthèses souples&), en
particulier, elle ne nécessite pas une logistique onéreuse et
présente donc des facilités pour le transfert industriel. Les
couches minces électrodéposées présentent des
grains en surface de dimensions mésoscopiques. La caractérisation
de la surface nécessite donc une méthode d'analyse très
précise.
Ainsi, l'objectif premier de ce travail étant
l'élaboration de couches minces de passivation de CeO2, les
différents mécanismes mis en jeu pour élaborer des films
adhérents avec différente
2
Introduction générale
microstructure seront étudiés sur un substrat en
acier. La caractérisation des couches mettra en oeuvre plusieurs
techniques d'analyse microstructurale. La relation microstructure /
résistance à la corrosion sera abordée lors
d'études de corrosion dans différents milieux (NaCl, eau
industrielle)[2].
Ce manuscrit est constitué de trois chapitres :
§ Le premier établit les bases théoriques
des processus électrochimiques dans l'élaboration des couches
minces des oxydes métalliques, ainsi que une présentation des
caractéristiques de l'oxyde de cérium.
§ Le deuxième chapitre, est divisé en 02
parties, dans la première partie nous avons présenté la
procédure expérimentale suivie durant notre travail pour
élaborer des revêtements d'oxydes de cérium sur acier
à bas carbone à partir de solution de nitrate de cérium
(0,01 et 0,1 M). Le contenu de la seconde partie est à été
orienté vers la description des méthodes et les techniques utiles
pour l'évaluation du comportement des revêtements à la
corrosion et ainsi à l'influence des différents paramètres
tels que la densité de courant, la concentration, et le milieu
corrosif.
§ Dans le dernier chapitre nous exposons les
résultats expérimentaux ainsi que leur interprétation.
L'ensemble du travail à été parachevé par une
conclusion générale
Chapitre I

Synthèse bibliographique
CHAPITRE I
Dans ce chapitre, on présente une synthèse
bibliographique sur les revêtements métalliques exposés aux
problèmes de la corrosion et des conséquences qu'elle induit dans
le secteur industriel en général. Le domaine d'application
s'étend aussi bien aux propriétés protectrices
qu'optiques, électriques et mécaniques.
Les revêtements à base de terre rare est une
alternative aux procédés de conversion de chromatation.
L'utilisation du Cr(VI) pose des problèmes environnementaux, le chrome
hexavalent étant toxique, cancérigène et mobile dans
l'environnement. Pour ces raisons les dépôts à base d'oxyde
de terre rare, notamment le CeO2 déposés sur un substrat est d'un
grand intérêt notamment dans les applications concernant les
revêtements de surfaces dans la lutte contre la corrosion.
Le procédé de déposition
électrolytique sur lequel repose notre travail a connu de récents
développements .Cette évolution au cours des dernières
décennies a été possible grâce aux perfectionnements
des méthodes et de l'appareillage électrochimique. La
possibilité de contrôler et de modifier aisément les
différents paramètres.
3
Chapitre I
I. 1. Corrosion et protection:
La corrosion (du latin corrodere, qui signifie
ronger, attaquer) traduit la réactivité de la plupart des
matériaux industriels vis-à-vis des environnements au contact
desquels ils sont placés au cours de leur emploi. On estime qu'elle
détruit un quart de la production annuelle mondiale d'acier, ce qui
représente environ 150 millions de tonnes par an ou encore 5 tonnes par
secondes. Or, la corrosion ne se limite pas à l'acier, mais affecte tous
les métaux ainsi que les polymères et céramiques. Elle
résulte d'interactions chimiques et/ou physiques entre le
matériau et son environnement. Voici quelques exemples de
phénomènes de corrosion :
V' transformation de l'acier en rouille,
V' fissuration d'un laiton en présence d'ammoniaque,
V' corrosion à chaud d'un superalliage dans une turbine
à gaz,
V' dégradation du PVC par le rayonnement ultraviolet,
V' attaques des briques réfractaires par les laitiers,
V' attaque d'un verre minéral par une solution
alcaline.
Selon le point de vue de l'ingénieur, la corrosion est
une dégradation du matériau ou de ses propriétés
par réaction chimique avec l'environnement [1]. Cette définition
admet que la corrosion est un phénomène nuisible elle
détruit le matériau ou réduit ses
propriétés, le rendant inutilisable pour une application
prévue.
Parfois, la corrosion est un phénomène bienvenu,
voire souhaité. Elle détruit et élimine un grand nombre
d'objets abandonnés dans la nature. Certains procédés
industriels font également appel à la corrosion. Par exemple
l'anodisation de l'aluminium est une oxydation de la surface du métal
pour former un film d'oxyde décoratif qui protège de la corrosion
atmosphérique.
La corrosion métallique est le phénomène
suivant lequel les métaux et alliages ont tendance, sous l'action de
réactifs chimiques ou d'agents atmosphériques, à retourner
vers leur état originel d'oxyde, de sulfure, de carbonate ou de tout
autre sel plus stable dans le milieu ambiant [2]. La corrosion d'un
métal ou d'un alliage pouvant être de différentes formes
uniforme, localisée&.etc, peut se développer selon
différents processus, qui caractérisent chacun un type de
corrosion. On peut distinguer trois types de corrosion: chimique, corrosion
électrochimique ou corrosion bactérienne.
Le coût occasionné par la dégradation
annuelle des matériaux, a provoqué la mise en oeuvre de
méthodes de protection. Ces dernières visent une économie
de matière et d'énergie auxquelles s'additionnent de nombreux
avantages environnementaux.
4
Chapitre I
Toute solution efficace et durable ne peut être qu'un
compromis tenant compte de l'ensemble de ces facteurs, notamment des
contraintes économiques et des critères scientifiques.
En matière de protection contre la corrosion, il est
possible d'agir sur le matériau lui même (choix judicieux, formes
adaptées, contraintes en fonction des applications,...), sur la surface
du matériau (revêtement, peinture, tout type de traitement de
surface,...) ou sur l'environnement avec lequel le matériau est en
contact.
Dans le monde industrialisé, l'utilisation de l'acier
comme matériau de construction est grandement répandu .Un large
domaine d'alliages disponible permet d'obtenir des matériaux
adaptés a chacun des usages. De plus, le cout compétitif de
l'acier est aussi un facteur important qui encourage son utilisation, et ce au
détriment d'autres métaux tels l'aluminium ou l'acier inoxydable.
Par contre, l'acier doux est un alliage qui est plus susceptible d'être
dégradé par des phénomènes de corrosion.
L'acier maintien dans un milieu alcalin
bénéficie généralement d'une couche oxyde passif,
cette couche adhère fortement à la surface de l'acier et
élimine tout contact que pourrait avoir l'acier avec l'oxygène et
l'eau. Ce phénomène d'autoprotection de l'acier est appelé
passivation de l'acier. La passivation disparait cependant lorsque le milieu
environnant l'acier subit une réduction du pH. Un des mécanismes
favorisant la corrosion des aciers est la réduction du pH, l'acier n'est
plus protégé par la couche passive à sa surface. La
protection des aciers contre la corrosion est un phénomène bien
contrôlé dans les structures modernes.
En effet le principal mécanisme du processus de
corrosion des aciers dans les milieux salins est associé à la
pénétration des ions chlore. Les aciers sont exposés aux
ions chlore dans les milieux marins. Les chlorures ont deux effets dans les
mécanismes de corrosion, détruire la couche passive
superficielle, ils facilitent le transport des ions en diminuant la
résistivité de l'électrolyte.
Dans le domaine de la corrosion, le chercheur
s'intéresse par l'étude des mécanismes visant une
meilleure connaissance des causes de la corrosion et des moyens à mettre
en oeuvre pour prévenir et/ ou diminuer la fâcheuse et couteuse
conséquence qui en résultent.
L'ingénieur applique les connaissances scientifiques
à la diminution des effets de la corrosion par des moyens pratiques et
économiques. Par exemple l'ingénieur spécialisé
dans le domaine de la corrosion, emploie la protection cathodique sur une
grande échelle pour prévenir la corrosion des conduites
enterrées ou il essaie et recherche de nouvelles et meilleures
peintures, dosages nécessaires d'inhibiteur de corrosion ou recommande
le dépôt métallique
5
Chapitre I
correct. Le chercheur, tour à tour, précise le
meilleur critère de protection cathodique, détermine la structure
moléculaire ou les composés chimiques qui se comportent le mieux
comme inhibiteurs, synthétise les alliages résistant à la
corrosion et recommande le traitement thermique et les variations de
composition des alliages qui améliorent leur qualité. Les points
de vue du chercheur et de l'ingénieur se complètent dans les
diagnostics des méfaits de la corrosion et prescrit les prescriptions de
remèdes appropriés [3]
I. 2. Généralités sur
l'électrodéposition d'oxydes métalliques:
La corrosion des métaux regroupe de multiples
phénomènes liés au milieu dans lequel elle se
déroule. Ces phénomènes dépendent d'un grand nombre
de facteurs qui interviennent non pas individuellement, mais en relation plus
ou moins complexe les uns avec les autres (la nature et la structure du
matériau, l'environnement et ses caractéristiques
chimiques&etc.) [4,5].
Les moyens de lutte contre la corrosion sont multiples et
doivent être appropriés à chaque cas. La prévention
passive demeure la meilleure solution pour éviter la corrosion des
installations métalliques.
La manière la plus commune d'empêcher un
métal de se corroder ou de retarder sa corrosion est d'appliquer un
revêtement imperméable sur sa surface. Si la couche du
revêtement fournit une barrière protectrice parfaite au
métal dans un environnement corrosif, alors ni l'oxygène, ni
l'eau ne pourront atteindre sa surface et la corrosion sera finalement
empêchée. Le rôle du revêtement est donc de limiter le
flux de ces produits (oxygène, eau) en créant une barrière
physique.
L'utilisation des composés à base de chrome a
été largement utilisée, la plupart des revêtements,
en conséquence les revêtements d'alliages de Zn possèdent
une meilleure résistance à la corrosion que les revêtements
de Zn pur. Cependant, ces dépôts nécessitent un traitement
de passivation postérieur afin de réduire la formation de
produits de corrosion blanchâtres.
Les procédés de conversion de chromatation
montrent à cet égard les meilleurs résultats, mais leur
utilisation est devenue strictement contrôlée et parfois interdite
à cause de leur haute toxicité. Des larges études ont
été effectuées pour remplacer ces composés toxiques
par des composés écologiques et amis de l'environnement. Des
composés organiques ont été proposés, les
carboxylates, les phosphates et les sulfonâtes et des composés
inorganiques comme les molybdates, les nitrites et les terres rares. Un des
traitements alternatifs de passivation utilise des oxydes de terres rares,
notamment l'oxyde de cérium CeO2. A l'heure actuelle, peu de travaux se
sont intéressés aux revêtements sacrificiels d'alliages de
Zn [6].
6
Chapitre I
Le traitement de passivation est réalisé par
immersion dans des solutions de sels de cérium ou par déposition
cathodique dans ce dernier cas, la formation des couches minces est
généralement associée à la formation d'oxydes ou
d'hydroxydes de cérium sur les sites cathodiques de la surface
métallique.
Les techniques usuelles de préparation de la poudre et
des revêtements d'oxyde de cérium les plus utilisées sont:
la déposition chimique en phase vapeur et électrochimique [7,8],
la précipitation chimique et hydrodynamique [9,10], Sol-gel [11,12] et
l'électrodéposition (ELD et EPD) [13,17].
Les premiers travaux consistaient en l'immersion du substrat
dans des solutions de nitrate ou de chlorure de cérium durant plusieurs
jours [18,19]. La longue durée d'immersion rend cette méthode
commercialement pas intéressante. A cet effet, plusieurs auteurs ont
tenté de réduire le temps de déposition par
l'élaboration des films d'oxyde de cérium par immersion dans un
bain à des températures plus élevées avec un
prétraitement anodique de la surface dans Na2MoPO4 [20,21].
Malgré les résultats encourageants de cette procédure, sa
mise en ouvre à l'échelle industrielle présente quelques
difficultés liées à l'évaporation de la solution et
le coût du chauffage.
L'électrodéposition des oxydes de cérium
souvent appelée déposition électrolytique (ELD)
est plus intéressante pour son faible coût et la
possibilité de contrôle des caractéristiques des particules
des dépôts [22]. En effet, l'électrodéposition offre
l'avantage et la particularité dans le développement des
matériaux nanostructurés. Durant les dix dernières
années, un intérêt particulier a été
manifesté pour l'électrodéposition des films minces de
céramique. La faisabilité de l'électrosynthèse du
CeO2 [23], AgO [24], ZrO2 [25], TiO2 [26], CuO [27], complexe d'oxydes de
titane [28,29] et autres oxydes monométalliques et composites [30,33] a
été démontrée. De plus, il été
rapporté que la composition et la morphologie des composites peuvent
être façonnées selon les exigences demandées pour
plusieurs applications électrochimiques, biomédicales et
catalytiques [34,35].
I.3. Electrodéposition cathodique des oxydes
métalliques:
L'étude des films minces métalliques (quelques
Å à 1000 Å d'épaisseur) a démarré il
y'a plus d'une vingtaine d'années. Son essor a été
considérable parce que les techniques de fabrication ont permis
d'atteindre des degrés de précision élevés dans le
contrôle des épaisseurs déposées. On distingue deux
grandes catégories de méthodes d'élaboration des couches
minces : les méthodes physiques telles que la pulvérisation ou
l'évaporation, et les méthodes chimiques telles
l'électrodéposition cathodique.
7
Chapitre I
Dans le domaine des films minces, il faut maîtriser des
dépôts ayant des épaisseurs contrôlées et ceci
est beaucoup plus facile à réaliser avec les méthodes se
basant sur l'électrodéposition cathodique. En effet,
l'électrodéposition cathodique offre un contrôle rigide de
l'épaisseur des films, une bonne uniformité des films et une
vitesse de déposition considérable. Et elle est
intéressante pour le faible coût relatif des
équipements.
Cette technique est intéressante pour obtenir des
dépôts sur des surfaces de substrat de forme assez
compliquées, pour l'imprégnation de surfaces poreuses, et pour la
déposition dans des aires bien spécifiques, comme dans le cas de
l'électrolyse au tampon. De plus, elle est considérée
comme une des méthodes les plus importantes dans les
procédés d'élaboration des films en céramique. Pour
la fabrication des films par électrodéposition cathodique, on
utilise soit la déposition électrophorétique durant
laquelle des suspensions de particules sont utilisées, soit la
déposition électrolytique durant laquelle on utilise des
solutions de sels métalliques [6]. Les principales
caractéristiques des deux méthodes
d'électrodéposition cathodique, EPD (electrophoretic
deposition ) et ELD (electrolytic deposition), sont regroupées
dans le Tableau I.1.
Tableau I.1: Conditions expérimentales
et caractéristiques de la déposition EPD et ELD des
matériaux en céramique [34].
|
Déposition électrophorétique
|
Déposition électrolytique
|
|
Milieu
|
Suspension
|
Solution
|
|
Espèces en mouvement
|
Particules
|
Ions ou complexes
|
|
Réactions d'électrode
|
Aucune
|
Génération des OH- et neutralisation des
espèces cationiques
|
|
Solvant préférentiel
|
Organique
|
Aqueux ou mixte
|
|
Conductivité du solvant
|
Faible
|
Elevée
|
|
Vitesse de déposition
|
1-103 um/min
|
10-3 - 1 um/min
|
|
Homogénéité des dépôts
|
Limitée par la taille des particules
|
A l'échelle nm
|
|
Stoechiométrie des dépôts
|
Contrôlée par la stoechiométrie de la poudre
de départ
|
Peut être contrôlée par le
précurseur
|
En ce qui concerne notre travail, et compte tenu des avantages
de coût et d'utilisation qu'elle présente, le recours au moyen
sera de l'électrodéposition cathodique électrolytique
(ELD) sera avantagé eu égard aux spécificités sus
évoquées.
8
Chapitre I
I.3.1. Déposition cathodique
électrolytique
En général, la déposition cathodique
électrolytique fait intervenir les réactions
d'électrogénération de base à la surface de
l'électrode:
2H2O + 2 e- Ä H2 + 2OH- I-1
NO3 - + H2O + 2 e- Ä NO2 - +2OH-
I-2
O2 + 2H2O + 4 e- Ä 4OH- I-3
Les trois réactions consommant de l'eau,
génèrent les ions hydroxydes, et par conséquent elles
induisent une augmentation du pH interfacial. Les ions et les complexes
métalliques s'hydrolysent par la base
électrogénérée (OH-) pour former des
dépôts d'hydroxydes, oxydes ou peroxydes métalliques sur
les sites cathodiques du substrat. Les hydroxydes et les peroxydes peuvent
être convertis en oxydes correspondants par traitement thermique. Les
réactions d'hydrolyse résultent en l'accumulation des particules
colloïdales à l'interface. En se basant aussi sur la théorie
de stabilité colloïdale «DLVO», il peut
être conclu que la formation des dépôts est causée
par la floculation introduite par l'électrolyte. La coagulation des
particules prés de la surface de l'électrode est peut être
augmentée par le champ électrique, l'écoulement
électrohydrodynamique et la pression résultant de la formation de
nouvelles couches. On note que le processus d'électrodéposition
électrolytique est gouverné par la loi de Faraday. Ainsi, la
quantité déposée sera normalement contrôlée
par la variation du temps de déposition et la densité de courant
appliquée [6].
Les dépôts obtenus par ELD sont
caractérisés par des particules de faible taille et une meilleure
agglomération comparée à ceux obtenus par EPD.
Contrairement aux processus basés sur EPD, le solvant
utilisé dans la déposition électrolytique est aqueux ou
mixte (Alcool-Eau). Cependant, la présence d'une quantité d'eau
pour l'électrogénération de base et la prévention
de la formation des oxydes non-stoechiométriques est nécessaire
[26].
Le Tableau I.2 résume les conditions nécessaires
pour la déposition électrolytique de quelques oxydes et complexes
d'oxydes métalliques. D'un autre côté, il a
été montré que les vitesses de déposition des
hydroxydes monométalliques sont égales permettant la formation
d'un dépôt d'hydroxydes de stoechiométrie souhaitée
[6].
9
Chapitre I
Tableau I.2: Conditions expérimentales de
la déposition de quelques oxydes métalliques par ELD [6].
|
Solution
|
Conditions expérimentales
|
|
précurseur
|
additifs
|
solvant
|
Température
(°C)
|
Densité de courant
(mA/cm2)
|
|
SL1
|
5 mM TiCl4
|
0,02 M
112O2
|
Méthanol-
Eau (3:1)V
|
1
|
20
|
|
SL2
|
5 mM
ZrOCl2
|
|
Eau
|
20
|
20
|
|
SL3
|
5 mM
Al(NO3)3
|
|
Ethanol-Eau
(19:1)V
|
20
|
5
|
|
SL4
|
2,5 mM
TiCl4 + 2,5
mM ZrOCl2
|
0,02 M
112O2
|
Méthanol-
Eau (3:1)V
|
1
|
20
|
|
SL5
|
0,02 mM
SnCl4
|
0,02 M
112O2
|
Ethanol-Eau
(19:1)V
|
20
|
10
|
I.3.2. Application
Durant cette dernière décennie,
l'intérêt porté à l'électrodéposition
cathodique des films de céramiques ne cesse de croître. En effet,
l'électrodéposition est utilisée pour la
préparation des films ferroélectriques [29, 36],
piézoélectriques [37], matériaux magnétiques
[38,39] semi et supraconducteurs [40] d'une épaisseur mince par ELD
[41] ou épaisse par EPD [42]. La méthode de
déposition par ELD et EPD dans le domaine
biomédicale est très appliquée, et ceci grâce au
degré de pureté et la stoechiométrie
contrôlée des dépôts obtenus [43], ce qui est
difficilement atteint par d'autres méthodes de déposition.
L'électrodéposition électrolytique et
électrophorétique est considérée comme une
méthode spécialement attractive pour la conception des cellules
solaires [44], les applications microélectroniques [41], la
préparation des composites à fibres renforcés et les
batteries [42]. Des revêtements de protection contre la corrosion ont
été aussi obtenus par ELD [34, 45]. Les oxydes de Ti,
Ru, Sn et Nb et d'autres films composites obtenus par déposition
électrolytique sont considérablement intéressant pour la
fabrication des anodes stables et autres applications électrochimiques
et catalytiques [46]. Une caractéristique importante de cette
méthode est l'habilité à l'imprégnation des
particules dans les substrats poreux et la consolidation des composites. De
plus, la déposition cathodique électrolytique est
considérée comme un outil performant pour la formation des
matériaux nanostructurés [47].
10
Chapitre I
I. 4. Que sont les terres rares:
Dysprosium, terbium, cérium& au nombre de 17, les
lanthanides, plus communément appelés terres rares, sont un
groupe de minerais aux propriétés chimiques et
électromagnétique exceptionnelles, indispensables aux
technologies de pointe et aux énergies vertes. Comme le charbon au XIX
siècle et le pétrole au XX siècle, ces métaux sont
aujourd'hui le moteur d'une nouvelle révolution industrielle. Sans
elles, pas d'iPad, d'écran plat, de voiture hybride ou encore d'ampoule
basse consommation. Les terres rares sont également utilisées
dans le secteur de la défense, notamment pour la construction de
missiles guidés et de radars. La plupart ne sont pas si rares que cela,
puisque présentes dans le sol en quantité bien supérieure
à l'iode ou à l'argent. Mais il faut traiter des tonnes de
minerais pour en extraire quelques précieux kilos. En vingt ans,
grâce à des couts d'exploitation dérisoire et au
mépris des considérations environnementales, la chine s'est
arrogée prés de 95% de la production mondiale. Un marché
hautement stratégique de 130.000 tonnes annuelles, qui risque fort
d'augmenter avec la demande croissante en énergies propres.
I.4.1. Généralités sur les terres
rares et l'oxydes de cérium:
Le premier minerai de terre rare le « Cérite»
a été découvert en 1750 ; mais considéré
d'abord un composé de
tungstène.il a fallut plus de
cinquante ans pour que, simultanément, Klaproth et Berzelius mettent en
évidence un oxyde encore inconnu, le « Cérine » En
1794, Johan Gadolin avait découvert le premier minerai de terre rare,
dénommé d'abord « Yterbia » puis « Yttria »
l'étude de la Céria et de l'Yttria démontrèrent par
la suite qu'en réalité ces substances étaient des
mélanges complexes de plusieurs éléments inconnus mais
chimiquement identifiés.
Les terres rares forment une série de quinze
éléments métalliques de propriétés chimiques
très semblables. Dans la classification périodique des
éléments, les terres rares sont classées dans les groupes
des lanthanides et occupent, avec le lanthane, une seule et même
période du tableau périodique. Cette particularité
résulte de leur structure électronique qui est identique pour les
couches extérieures et ne diffère au suivant que par addition
d'un électron dans la couche profonde 4f.
Les minéraux de lanthanides sont très nombreux.
On en a décrit plus de deux cents de compositions très diverses
avec prédominance, soit du groupe cérique soit, du groupe
yttrique. Parmi les principaux, citons les carbones et les fluocarbones
(lanthanide, bastnaésite, parasite) les phosphates (monazite et
xénolite) Mais deux d'entre eux seulement forment des gisements
importants exploités industriellement: la Monazite à la fois
minerai de terres rares et le Thorium que l'on rencontre en dépôt
de sable au Brésil, en Inde, au Etats-Unis, en Afrique du
11
Chapitre I
sud, en Australie, la Malaisie, la Corée du sud,
l'Indonésie mais aussi et surtout la bastnaésite dont il existe,
en Californie et en Chine, des gisements qui constituent, sans doute, la plus
grande réserve naturelle de terres rares au monde [1].
I.4.2. L'oxyde de Cérium:
Le cérium a été découvert par
Jöns Jakob Berzelius et Wilhelm Hisinger (Suède) en 1803 et
indépendamment par Martin Heinrich Klaproth (Allemagne) Le nom de «
Cérès» a été donné à
l'astéroïde qui a été découvert en 1801 (soit
deux ans avant l'identification du cérium).
Le Cérium est très abondant dans les terres
rares et on le trouve dans plusieurs minerais par exemple dans la sable
monazite [Ce(PO4)] le cérium est un métal gris-fer,
malléable et ductile. Il ternit au contact de l'air et réagit
facilement avec. Il est aussi réactif aux acides. Lorsqu'il est soumis
à l'action de la chaleur il s'enflamme. Il est aussi fortement
réducteur et possède donc de fortes capacités
thermodynamiques qui apparaissent sous forme oxydée.
Ce sont d'ailleurs les oxydes de cérium qui trouvent une
importante application industrielle notamment dans le domaine, entre autres,
des catalyseurs, des piles à combustibles mais aussi comme agent
inhibiteurs dans le revêtement anticorrosion pour divers métaux et
alliages métalliques. Ses principales propriétés physiques
sont résumées dans le tableau suivant: Tableau I.3
: Principales caractéristiques d éi
|
Cérium - Ce
|
|
|
Numéro atomique
|
|
|
Masse atomique
|
140,12 g.mol -1
|
|
Electronégativité de Pauling
|
1,1
|
|
Masse volumique
|
6,76 g.cm-3 à 20°C
|
|
Température de Fusion
|
799 °C
|
|
Température d'ébullition
|
3426 °C
|
|
Rayon atomique (Van der Waals)
|
0,181 nm
|
|
Rayon ionique
|
0,102 nm (+3) ; 0,087 nm (+4)
|
|
Isotopes
|
4
|
|
Configuration électronique
|
[ Xe ] 4f2 6s2
|
|
Energie de première ionisation
|
526,8 kJ.mol -1
|
|
Potentiel standard
|
- 2,48 V ( Ce3+ / Ce )
|
12
Chapitre I
I.4.3. Structure cristallographique:
L'oxyde de cérium possède, dans des conditions
de température et de pression normale, une structure cristalline de
symétrie cubique à face centrée (CFC) appartenant au
groupe Fm3m et se cristallise dans la structure fluorite cubique. La structure
fluorite consiste en une matrice à face centrée de cations avec
des interstices remplies d'ions d'oxygène. Il existe quatre cations et
huit oxygènes par unité de cellule primitive. La structure
fluorite est très stable avec une possibilité de départ ou
de déplacement d'oxygène. A cet effet, il est très facile
d'observer un remplacement de 20% des cations dans le système par des
accepteurs triples-chargés et qui peuvent être compensés
par la formation de lacunes d'oxygène double-chargés. De plus, il
a été rapporté que CeO2 maintient la structure fluorite
même avec 10% d'insuffisance dans la teneur en oxygène [2,3].
13
Chapitre I
REFERENCES BIBLIOGRAPHIQUES
V. S. Agarwala, Proc. Int. Cong. Metallic Corros., 1,
(1984)380
[1] F. R. Longo, J.J. Dellucia, V.S. Agarwala, Proc. 6th
European Symposium on Corrosion Inhibitors, Univ. Ferrara, Italy, (1985)155.
[2] H.H. Uhlig, Chemical and EngineeringNews, (1949) 27
[3] Cefracor. Matériaux
métalliques, phénomènes de corrosion. CEFRACOR (2003)
33-51
[4] N. Lebozec. Réaction de
réduction de l'oxygène sur les aciers inoxydables en eau de mer
naturelle, influence du biofilm sur les processus de corrosion. Thèse de
doctorat Paris, (2000)7-10.
[5] Y. Hamlaoui, Thèse de doctorat, Elaboration et
caractérisation de revêtements d'oxyde de cérium sur acier
et acier électro zingué. Univ Badji mokhtar-ANNABA(2008).
[6] J. F. Jue, J. Jusko, A. V. Virkar, J. Electrochem. Soc.,
76(1993)1577- 1583.
[7] M. J. Capitan, A. Paul, J. . L. Pastol, J. A. Odriozola.
Oxid. Met., 52(1999) 447-450
[8] P. L. Chen, I. W. Chen . J. Am. Ceram. Soc., 76 ( 1993)
1577-1583.
[9] M. Hirano. E. Kato, J. Am. Ceram. Soc., 79 ( 1996) 777-
780.
[10] N. Ozer, Sol. Energy Mater. Sol. Cells., 68 ( 2001) 391.
[11] I. Zhitomirsky, A. Petric, Ceram. Int., 27 ( 2007)
149-155.
[12] I. Zhitomirsky, A. Petric, Mater. Lett.,
40(1999)263-268.
[13] M. Balasubramaniam, C. A. Melendres, A. N. Mansour, Thin
solid Films, 347( 1999)
178-183.
[14] B. R. W. Hinton, D. R. Arnot, N. E. Ryan, Mater., Forum 9 (
1989)162.
[15] F. Mansfeld, S. Lin, S. Kim, H. Shih, Corr. Sci., 27 (1987)
997- 1000.
[16] M. Dabala, L. Armelao, A. Buchberger, I. Calliari, Appl.
Surf. Sci., 172 (2001) 312-322
[17] F. Mansfeld, Y. Wang, Mater. Sci. Eng., A 198 (1995)
51-61.
[18] L. Arurault, P. Monsang, J. Salley, R. S. Bes, Thin Solid
Films., 466(2004) 75-80.
[19] J. A. Switzer, Bull. Am. Ceram. Soc., 66 (1987) 1521.
[20] B. E. Breyfogle. C. J. Hung, M. G. Shmsky, J. A. Switzer,
J. Electrochem. Soc., 143
(1996) 2741-2746.
14
Chapitre I
[22] L. Gal-Or, T. Silberman, R. Chaim, J. Electrochem. Soc.,
138 ( 1991) 1939-1942.
[23] T. Zhitomirsky, L. Gal-Or, A. Kohn, H. W. Hennicke, J.
Mater. Sci., 30( 1995) 5307-5312.
[24] A. E. Rakhshani, J. Varghese, Thin Solid Films., 157 (
1988) 87-96.
[25] T. Zhitomirsky, L. Gal-Or, S. Klein, J. Mater. Sci. Lett.,
14 ( 1995) 60-62.
[26] T. Zhitomirsky, A. Petric, Ceram. Tnt., 27 ( 2007)
149-155.
[27] T. Zhitomirsky, A. Petric, Mater. Lett.,
40(1999)263-268.
[28] M. Balasubramaniam, C. A. Melendres, A. N. Mansour, Thin
solid Films, 347( 1999)
178-183.
[29] B. R. W. Hinton, D. R. Arnot, N. E. Ryan, Mater., Forum 9 (
1989)162.
[30] F. Mansfeld, S. Lin, S. Kim, H. Shih, Corr. Sci., 27 (1987)
997- 1000.
[31] M. Dabala, L. Armelao, A. Buchberger, T. Calliari, Appl.
Surf. Sci., 172 (2001) 312-322
[32] F. Mansfeld, Y. Wang, Mater. Sci. Eng., A 198 (1995)
51-61.
[33] L. Arurault, P. Monsang, J. Salley, R. S. Bes, Thin Solid
Films., 466(2004) 75-80.
[34] J. A. Switzer, Bull. Am. Ceram. Soc., 66 ( 1987) 1521.
[35] B. E. Breyfogle. C. J. Hung, M. G. Shmsky, J. A. Switzer,
J. Electrochem.Soc.,
143(1996) 2741-2746.
[36] L. Gal-Or, T. Silberman, R. Chaim, J. Electrochem. Soc.,
138 ( 1991) 1939-1942.
[37] T. Zhitomirsky, L. Gal-Or, A. Kohn, H. W. Hennicke, J.
Mater. Sci., 30( 1995) 5307-5312.
[38] A. E. Rakhshani, J. Varghese, Thin Solid Films., 157 (
1988) 87-96.
[39] T. Zhitomirsky, L. Gal-Or, S. Klein, J. Mater. Sci. Lett.,
14 ( 1995) 60-62.
[40] T. Zhitomirsky, J. Eur. Ceram. Soc., 19(1999) 2581-2587.
[41] G. H. A. Therese. P. V. Kamath, Chem. Mater.,
12(2000)1195.
[42] T. Zhitomirsky, Bull. Am. Ceram. Soc., 79(2000) 57.
[43] T. Zhitomirsky and L. Gal-Or, Mater. Lett., 38
(1999,10-17.
[44] T. Zhitomirsky,Mater.Lett., 37 ( 1998), 72-78.
[45] J. Mizuguchi, K. Sumi, and T. Muchi, J. Electrochem. Soc.,
130 ( 1983),1819-1825.
15
Chapitre I
[46] B. V. Derjaguin and L. Landau, Acta Physicochim. USSR,14 (
1941),633-652
[47] E. J. W. Verwey, J. Th. G. Overbeek, Theory of Stability of
Lyophobic Colloid, Amsterdam, Netherlands, (1948),35
Chapitre II

CHAPITRE II
PROCEDURE EXPERIMENTALE
ET TECHNIQUES
DE CARACTERIASATION
Ce chapitre a pour but de présenter les méthodes
expérimentales électrochimiques utilisées dans cette
étude. Une description du matériau, de l'électrolyte, et
du montage effectués permet, dans un premier temps, de fixer une
démarche expérimentale assurant une bonne reproductibilité
des résultats. Les techniques électrochimiques sont à leur
tour présentées, de manière à souligner leur
intérêt et leur patience dans l'étude de
l'élaboration et la caractérisation des revêtements d'oxyde
de cérium.
16
Chapitre II
II.1. Méthode d'élaboration des
revêtements:
Comme nous avons cité auparavant, plusieurs techniques
sont mises au point pour déposer. L'électrodéposition par
rapport aux autres techniques, est une technique non couteuse, facile à
mettre en oeuvre avec une possibilité de déposer des larges
surfaces et elle ne nécessite ni vide ni température
élevée, ces dernières caractéristiques, font de
l'électrodéposition l'une des méthodes les plus
sollicitées pour élaborer des différentes couches
minces.
Dans notre travail, nous avons utilisé cette technique
pour élaborer des couches minces d'oxyde de cérium, en se servant
d'un système à trois électrodes à savoir
l'électrode de travail, l'électrode de référence,
et une contre électrode. Le bain électrolytique est une solution
de nitrate de cérium hydraté [Ce(NO3)3.6H2O] de concentration
0,01 M et 0,1 M.
Générateur électrique
Bain électrolytique Ce(NO3)3.6H2O
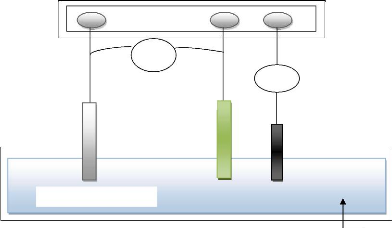
Cathode (acier A366)
U
Electrode de référence
I
Contre électrode
Figure II.1 : Schéma du montage électrochimique
à trois électrodes [1].
17
Chapitre II
Le dispositif expérimental utilisé pour la
réalisation des mesures, est un Voltalab 40 assisté par
ordinateur et relié à une cellule électrochimique.
Le matériau testé dans cette étude est un
acier à faible teneur en carbone conforme aux normes SAE1010, ASTM
A-366. Sa composition chimique élémentaire en
éléments majeurs et en % massique est indiquée dans le
tableau II.1.
Tableau II.1 : Composition chimique en (%
m ) de l'acier A366

Teneur en % m
Carbone(X)
Manganèse (X)
Phosphore(X)
Soufre (X)
0,08-0,13
0,30-0,60
0,04
0,05
La caractérisation électrochimique à
été effectuée dans deux différents milieux:
1- Une solution aqueuse contenant 29.22 g/l de NaCl
correspondant à une concentration de 0,5M équivalent à la
teneur en Cl- contenue dans l'eau de mer.
2- Une eau industrielle récupérée
fraichement de l'entreprise FERTIAL juste avant chaque essai.
II.1.1.Principe de la méthode:
L'électrodéposition est une méthode
permettant de réduire les espèces présentes en solution
afin de réaliser un dépôt sur un substrat. Le schéma
de principe (Figure II.1) présente les différents
éléments d'un montage d'électrodéposition elle
consiste à imposer un courant électrique entre deux
électrodes plongées dans une solution contenant un sel
métallique du métal à déposer. Suivant les
conditions d'élaboration (bain d'électrolyse, pH,
conductivité, température, additifs, densité de courant,
régime continu, régime pulsé, &), il est possible
d'obtenir des tailles de grains nanométriques.
A l'électrode de travail (cathode) se produit la
réduction de l'ion métallique suivant la demi-
réaction : Mz+ + z e- ' M(s)
II-1
Pour que cette réaction de réduction soit active, le
potentiel de l'électrode de travail est abaissé par rapport
à sa valeur d'équilibre. La surtension (h) correspond à la
différence entre le potentiel appliqué « E » à
l'électrode et son potentiel d'équilibre « Eeq ». Elle
dépend de différents facteurs tels que la nature de
l'électrolyte, la densité de courant cathodique et le
métal déposé [2].
La vitesse de formation du dépôt dépend
directement de la densité de courant qui traverse la cellule qui est
fonction de la surtension. Le temps nécessaire à
l'élaboration d'un revêtement
18
Chapitre II
est calculé à l'aide de la Loi de Faraday, en
supposant un rendement faradique de 100%, les
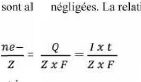
réactions parasites ors n ion utilisée est alors
:
Q = n e- . F = I . t II-2
n métal déposé = =
Q : Quantité d'électricité I : Courant
imposé
F : Constante de Faraday (96485 C.mol-1) t : Temps de
dépôt
ne-: nombre de moles d'électrons échangés z
: Nombre d'électrons échangée
L'électrodéposition est souvent accompagnée
par la réduction des protons, selon la réaction :
2H+ + 2 e- ' H2(g) II-3
Ce phénomène implique qu'une partie du courant
appliqué est consommée par cette réaction.
Les
conséquences de ce dégagement de dihydrogène sont la
formation de piqûres dans le
revêtement, une diminution du rendement cathodique, une
augmentation du pH pouvant engendrer la précipitation d'hydroxydes
métalliques, une fragilisation du revêtement et un changement des
propriétés mécaniques [3,4].
En général, il est préférable de
se placer à des surtensions suffisamment faibles afin de limiter
l'influence de cette réaction. Sur la contre-électrode (anode),
une réaction d'oxydation a lieu,
par exemple l'oxydation de l'eau, une autre réaction
suivant le milieu ou l'oxydation du métal constituant la contre
électrode. Dans le cas d'une contre-électrode en nickel,
l'oxydation de cette dernière conduit à la libération
d'ions métalliques (relation II-4 ) en solution permettant ainsi
d'éviter l'appauvrissement de la solution en cations métalliques
au cours du temps :
Ni(s)' Ni2+ + 2 e- II-4
Différents paramètres peuvent influencer les
caractéristiques de dépôts, ces paramètres sont
généralement classés suivant deux
catégories : les conditions initiales qui sont imposées par
l'expérimentateur et les conditions dites temporelles
qui dépendent de l'évolution du système tableau II.2. Le
rôle de ces paramètres est complexe et dépend du
système considéré. La
densité de courant affecte fortement la structure et la
morphologie des films électrodéposés.
En jouant sur la vitesse de dépôt, la taille et
la distribution des cristallites peuvent être modifiées [5]. La
pureté des sels utilisés pour la fabrication du bain
d'électrolyte, la pureté de
la contre-électrode (montage utilisant une anode
soluble), une mauvaise préparation du substrat, ou une
dégradation du bain d'électrolyse [6] sont susceptibles
d'introduire des impuretés au sein du film
électrodéposé. Ces impuretés (métalliques et
organiques [6] sont à l'origine de la fragilisation du
dépôt, du changement des propriétés
mécaniques, de formation de taches en surface, &)
19
Chapitre II
Tableau II.2 : Conditions
influençant les caractéristiques des revêtements
|
Conditions initiales
|
Conditions temporelles
|
|
- nature du substrat
|
- électrolyte (concentration, pH, &)
|
|
- préparation du substrat
|
- évolution de la surface
|
|
- type d'anode
|
- paramètres électriques
|
|
- électrolyte (concentrations, pH, additifs,
|
- réactions parasites
|
|
pureté des sels, &)
|
- autres conditions
|
|
- paramètres électriques
|
|
|
- distance entre cathode et anode
|
|
|
- agitation
|
|
|
- température du bain
|
|
II.1.2. Mécanismes de
l'électrodéposition
Lors d'une réaction d'électrodéposition
en milieu aqueux, les cations présents dans l'électrolyte sont
transférés sur la surface de la cathode pour former le
dépôt métallique. Il est couramment admis que ce
procédé fait intervenir plusieurs étapes
intermédiaires dont les principales sont décrites ci dessous
[3,7].
V' Transport de l'ion métallique hydraté ou du
complexe hydraté du centre de la solution vers la cathode.
V' Perte d'une partie de l'hydratation de l'ion métallique
hydraté à la surface de la cathode. V' Transfert de charge avec
la formation d'adatome (ou adions) à la surface de la cathode.
V' Formation de germes à la surface de la cathode par
diffusion des adatomes à la surface de
l'électrode.
V' Rassemblement de plusieurs germes afin de minimiser
l'énergie de surface pour former des noyaux stables.
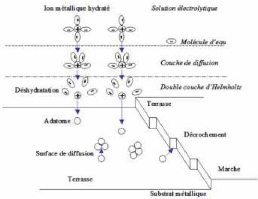
Figure II. 2 : Schéma du mécanisme de
l'électrodéposition [4].
20
Chapitre II
Les différents mécanismes qui interviennent lors
de l'électrodéposition sont schématisés sur la
figure (II- 2) dans le cas d'une surface idéale. Les ions
métalliques présents en solution sont généralement
hydratés ou complexés. Ceux-ci sont transportés vers la
cathode à travers la couche de diffusion et la double couche
d'Helmholtz. En se rapprochant de la cathode, les ions métalliques se
déshydratent ou se décomplexent. Une fois
déshydratés ou décomplexés, ceux-ci se
déchargent en combinant leurs électrons avec ceux de la cathode
pour former des atomes métalliques à la surface de la cathode
(formation des adatomes). Par suite, les adatomes diffusent sur la surface pour
trouver des sites stables (marches, coin, émergence de dislocations,
&)
Dans certaines conditions, la formation de l'édifice
cristallin se fait par un processus de germination/croissance, (figure II-3).
La morphologie et la microstructure du dépôt dépendent
alors principalement de la compétition de ces deux mécanismes
mais aussi des phénomènes de coalescence.
Les adatomes formés à la surface se regroupent
sous forme de clusters. Ces clusters vont alors croître
perpendiculairement par rapport au substrat mais aussi parallèlement,
afin de former des grains. Cependant la croissance de certains grains peut
être bloquée par la croissance plus rapide des grains environnants
ou des phénomènes de coalescence peuvent se produire.
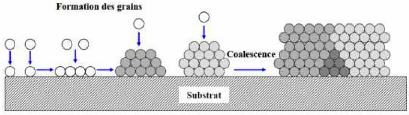
Figure II.3 : Processus de germination / croissance et
coalescence des grains [8].
L'électrodéposition du cérium sur acier est
obtenue en mode potentiodynamique
(galvanostatique) dans un bain de Ce(NO3)3.6H2O
à 0,01M et 0,1M pendant une durée de 20
mn. Les densités de courant appliquées sont: 0,25
-0,5- 1-1,5 et 3 mA/cm2.
Pour cela une cellule électrochimique est placée
qui comprend:
y' Une électrode de référence Ag/AgCl
saturé de KCl.
y' Une contre électrode en platine à fil.
y' l'électrode de travail est sous forme d'un disque de
surface S= 1,4 cm2.
21
Chapitre II
Les échantillons revêtus ainsi obtenus sont
ensuite rincés dans l'éthanol, puis séchés à
l'air pendant 24h. Le choix de l'éthanol comme milieu de rinçage
a été retenu, afin d'absorber la quantité maximale d'eau
piégée et de chasser les nitrates libres dans le
revêtement.
II.2. Caractérisation des revêtements
à base d'oxydes de cérium:
II.2.1. Techniques expérimentales
Les techniques électrochimiques ont permis
d'appréhender l'étude selon deux points de vue. D'un point de vue
phénoménologique d'abord, la caractérisation de
l'adsorption est possible soit par suivi dans le temps du potentiel en circuit
ouvert, caractéristique de la modification de l'interface entre un
métal et son environnement, soit par voltamétrie cyclique
à vitesse de balayage élevée. L'aspect quantitatif
(courbes de polarisation à vitesse de balayage modérée,
spectroscopie d'impédance,...) permet, quant à lui,
d'accéder à des vitesses de réaction et à des
valeurs de paramètres physiques décrivant l'état du
système (capacité de double-couche, résistance de
transfert, capacité du film,...).
Les méthodes électrochimiques peuvent être
classées selon deux groupes distincts à savoir les
méthodes stationnaires et les méthodes non-stationnaires dites
transitoires.
II.2.2. Les techniques stationnaires
Les techniques stationnaires permettent d'étudier un
système situé quasiment dans un état d'équilibre
thermodynamique. Elles prennent en compte tous les couples redox dans la
solution. Ces méthodes sont largement utilisées en laboratoire
dans le cadre de l'étude de la corrosion [9].
II.2.2.1. Suivi du potentiel à circuit
ouvert(OCP)
Un métal plongé dans un milieu
électrolytique quelconque tend à se dissoudre et à se
charger électriquement avec création d'une double couche
électrochimique assimilable à un condensateur électrique.
Au bout d'un temps suffisamment long pour qu'un régime stationnaire soit
établi, l'électrode métallique prend par rapport à
la solution un potentiel, appelé potentiel de corrosion. Ce potentiel ne
peut être connu en valeur absolue. Il est repéré par
rapport à une électrode de référence [10]. Cette
technique simple apporte des informations préliminaires sur la nature
des processus en cours, à l'interface métal/électrolyte :
corrosion, passivation, et renseigne sur l'évolution de la noblesse des
échantillons. Cette mesure permet également de connaître la
durée d'immersion nécessaire à l'établissement d'un
régime stationnaire indispensable aux mesures potentiodynamiques ou
d'impédance électrochimique [11,12]. Dans le cas des
métaux revêtus, la surface du métal exposée à
l'électrolyte est en fonction avec la perméabilité et
à l'intégrité du film à l'eau et aux ions.
22
Chapitre II
Les travaux sur le potentiel de corrosion des métaux
revêtus remontent à 1970 [13,14]. Ces travaux ont montré
l'importance du potentiel de corrosion et comment il se change avec le temps.
Le potentiel positif indique l'absence de la corrosion et le potentiel
négatif indique sa présence.
Ces mesures empiriques ont été suivies par des
études très complètes par Wormwell et Brasher[15]. Ils ont
noté que la forme de la courbe potentiel- temps pendant les
premières heures ou les premiers jours d'immersion, donne des
informations très importantes sur les propriétés
barrières des revêtements.
II.2.2.2. Courbes de polarisation
Le tracé des courbes de polarisation potentiodynamique,
consiste à suivre la réponse en courant de l'échantillon
à une rampe de potentiel permettant de le déplacer lentement de
manière linéaire de son état d'équilibre.
A l'aide d'un générateur extérieur et d'une
contre électrode, on fait passer un courant à travers
l'électrode métallique. Son état stationnaire est
modifié et sa surface prend une nouvelle valeur du potentiel. Les
courbes E =f (I) ou I = f (E) constituent les courbes de polarisation. Ces
méthodes présentent un double avantage, d'une part, elles
permettent de déterminer la résistance de polarisation, et
d'autre part, de déterminer le mécanisme de corrosion.
Pour tout couple redox, l'équation de Butler-Volumer
(Eq.II-6), pour le cas où les échanges à l'interface sont
contrôlés par l'étape de transfert de charge, donne une
relation entre le potentiel interfacial et les densités de courant.
Par exemple, si l'on considère une réaction
électrochimique interfaciale de la forme :
B ox + n e- Bred II-5
L'équation de
Butler-Volmmer permet de relier le courant interfacial en fonction du
potentiel
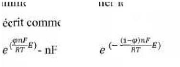
électrochimique et elle s' e suit :
I = Ia + Ic = n F KaCred,s Kc
Cox,s II-6
Avec:
I : courant global de la réaction Ic : courant cathodique
de la réaction
Ia : courant anodique de la réaction n : nombre
d'électrons d'échange
T : température E : potentiel ou surtenstion
Æ: facteur de symétrie. R : constante des gaz
parfaits
23
Chapitre II
Ka, Kc : constantes dépendant des vitesses des
réactions interfaciales élémentaires.
Cred ,s : concentration de l'espèce Bred au niveau de
la surface de l'électrode
Cox ,s : concentration de l'espèce Box au niveau de la
surface de l'électrode.
A l'équilibre, le courant global est nul et Ia = -Ic =
Io. où Io est appelé courant d'échange. On
peut alors introduire la surtension de polarisation (·)
et B- et B+ coefficients anodique et

cathodiq ment.
B- = + = ( · = E - E rev II-7
les conceons des espèces sont les mêmes dans la
solution et à la surface de
Alors, l'équation de Butler-Volmer (Eq.II-6 ) peut
s'exprimer en fonction de ces paramètres dans le
l'électro
I = Io Io II-8
Cette relation (Eq.II-8) est très importante et
très souvent utilisée. Elle donne la relation entre le courant,
c'est-à-dire la cinétique de la réaction, et le potentiel
appliqué E (ou ·).
L'établissement ainsi que l'étude des courbes de
polarisation · = f (i) permettent de déterminer
expérimentalement les grandeurs I0 et (n,Æ) pour les
réactions électrochimiques contrôlées par les
réactions de transfert (ou régime d'activation) car le transfert
électronique est un processus activé [16].
II.2.2.3. Méthode des droites de Tafel:
Il s'agit d'une méthode d'extrapolation basée
sur l'équation E = f (I) de Butler- Volmer (Eq.8). Cette technique
utilise des portions de la courbe de polarisation situées loin de
l'équilibre où le système est fortement polarisé,
c'est-à-dire les portions pour lesquelles les surtensions ·
supérieurs à100 mV (domaine anodique) et · inférieurs
à -100 mV (domaine cathodique).
Si on représente la courbe de polarisation (figure
II-4) obtenue en coordonnées E = f (Log i), l'intersection des droites
anodique et cathodique extrapolées au potentiel de corrosion, donne la
densité de courant de corrosion Icorr. Cette technique permet de
séparer les deux processus anodique et cathodique donnés par la
loi de Butler - Volumer.
24
Chapitre II
Lorsque l'un des deux courants devient négligeable,
l'équation (II.8) s'exprime par :
· = a - + b_ log (i) pour la partie anodique
Avec a-= -2,3 B- log io et b- = 2,3B-
· = a+ - b+ log (i) pour la partie cathodique II-9
Avec a+ = 2,3B+ log io et b+ = 2,3 B+
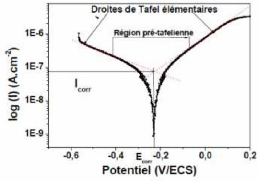
|
Figure II.4 : Représentation graphique des droites de
Tafel [17].
|
Cette représentation permet de déterminer
(Io, B+ et B-), paramètres très importants dans
l'étude des phénomènes de corrosion.
II.2.2.4. Méthode de la résistance de
polarisation
Cette méthode consiste à écarter
légèrement le métal de son potentiel de corrosion. Une
polarisation de quelques millivolts autour du potentiel de corrosion (figure
II-4) suffit pour déterminer la résistance de polarisation d'une
électrode. Il est alors possible d'exprimer le courant de corrosion par
l'équation (II-10). La vitesse de balayage du potentiel à
laquelle est effectuée la perturbation aura aussi une grande influence.
C'est pourquoi, il faut qu'elle soit la plus faible possible pour permettre aux
réactions d'atteindre instantanément un état
d'équilibre.
Concrètement pour calculer une Rp, nous effectuons une
mesure potentiodynamique avec une variation du potentiel de +20 mV ou +10 mV
par rapport au potentiel de corrosion, à une vitesse de 0.166 mV/s et
nous mesurons la densité de courant résultante. [18,19].
Chapitre II

La résistance de Stern et Geary
I = Icor [e II-10
( L
irr = II-11
L
Ba + Bc II-12
0
.Bc
Rp = ( L avec (L
L L
Pente de la courbe I = f (E) au potentiel de corrosion, lorsque
AE est petit et lorsque le balayage est infiniment lent [20].
La mesure de la résistance de polarisation Rp est une
technique peu perturbatrice de l'interface métal/ milieu. Elle permet
donc un suivi de l'évolution de la vitesse de corrosion en fonction du
temps d'immersion de l'échantillon.
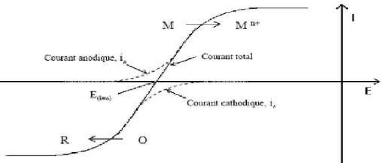
25
Figure II.5 : Courbe de polarisation I = f (E) [21].
II.2.2.5. Voltampérométrie cyclique:
II. 2. 2. 5. 1. La méthode à balayage en
potentiels :
Les mesures de voltamétrie cyclique reviennent à
effectuer des balayages linéaires en potentiels autour d'une position
donnée et d'observer les éventuelles apparitions et/ou
disparition des phénomènes électrochimiques (oxydation
et/ou réduction). Certains paramètres, tels la vitesse de
balayage, permettent de rendre compte de la réversibilité de
certaines réactions.
Le principe de la mesure consiste à perturber un
système électrochimique, qui se trouve initialement à
l'équilibre, par un signal électrique. La voltamétrie
cyclique permet d'imposer un balayage linéaire de potentiel en fonction
du temps. Le potentiel décrit un aller-retour de
26
Chapitre II
part et d'autre du potentiel normal du système redox du
couple étudié. Le voltammogramme représente le courant en
fonction du potentiel appliqué.
Par ailleurs, il est très important de rester dans une
limite raisonnable des potentiels. Si on part dans des potentiels trop
négatifs, on constate un dégagement de H2, qui correspond
à la réduction de l'eau. Au contraire, pour des potentiels trop
positifs, on constate un dégagement d'oxygène correspondant
à l'oxydation de l'eau [23].
II.2.3. Techniques non stationnaires:
Les méthodes électrochimiques classiques
présentent l'inconvénient de négliger certaines
composantes caractéristiques de l'interface métal/solution et de
ne pas pouvoir séparer et analyser les étapes
élémentaires d'un processus complexe. L'utilisation des
techniques transitoires devient alors indispensable. [9,22].
II.2.3.1. Spectroscopie d'impédance
électrochimique (SIE) :
Dans le cadre de l'étude des interfaces
électrode/électrolyte, ce qui est le cas en corrosion aqueuse,
différentes techniques électrochimiques sont couramment
utilisées. Elles mettent toutes en jeu des mesures de potentiel et/ou de
courant, et peuvent être classées en deux groupes. Le premier
regroupe les techniques dites stationnaires, comme la
chronopotentiométrie, la chronoampérométrie, la
voltampérométrie). Ces techniques permettent de recueillir des
informations liées à la thermodynamique du système
étudié et quelquefois à sa cinétique.
Néanmoins, elles sont sujettes à des limitations, notamment dans
le cas de systèmes très résistants ou pour l'étude
des mécanismes réactionnels. De plus, certaines d'entre elles
entraînent la destruction de l'échantillon. Pour contourner ces
limitations, il a été mis au point un certain nombre de
techniques dites transitoires, basées sur l'utilisation des fonctions de
transfert et dont la STE fait partie.
II.2.3.1.1. Définition et principe
La STE repose sur la mesure d'une fonction de transfert suite
à la perturbation volontaire du système électrochimique
étudié. Ce système peut être considéré
comme étant une « boîte
noire » qui réagit en émettant un signal
y(t) quand il est soumis a une perturbation x(t) (figure TT-5) . Les deux
signaux x(t) et y(t) sont alors reliés par une fonction de transfert
H(w) telle que Y (w) = H(w)X(w), X(w) et Y (w) étant respectivement les
transformées de Fourier de x(t) et y(t).
Chapitre II

x (t) y (t)
Système électrochimique
27
Figure II.6 : Schéma d'une fonction de transfert
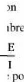
Classiquement, la perturbation imposée est
sinusoïdale. Le signal appliqué est donc de la forme x(w) =
Asin(wt) et la réponse du système est y(t) = B sin(wt + ø)
avec une fréquence f, une pulsatio w= 2Àf et un déphasage
ø. L'impédance électrochimique se définie comme
étant le nome complexe Z(w) résultant du rapport :
?
Z (u) = II- 13
?
où, en mode otentiostatique, AE(w) est la perturbation
imposée à un potentiel choisi E0, et AI(w) la réponse en
courant du système étudié avec une composante continue I0
. Il est aussi possible d'utiliser le mode galvanostatique. Dans ce cas, c'est
une perturbation en courant de faible amplitude qui est appliquée au
système et c'est la réponse en potentiel qui est mesurée.
L'impédance Z) est un nombre complexe qui peut être écrit
sous deux formes équivalentes :
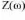
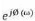
Z(ù) = | Z(ù)| ou Z(ù) = Zr (ù) + j
Zj (ù) avec j = v-1
II-14
| Z | étant le module de l'impédance, ø le
déphasage, Zr la partie réelle et Zj la partie
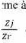
imaginaire. Pour passer d'une for l'autre, il suffit d'utiliser
les relations suivantes :
|Z|2 Z2r + Z2j et Ø = tan
-1 ou Zr = | Z | cos Ø et Zj = | Z | sin Ø II-15
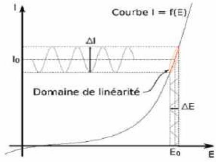
Figure II. 7 : Schéma d'un système
électrochimique non linéaire soumis à une
perturbation
sinusoïdale.
28
Chapitre II
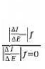
Les systèmes électrochimiques étudiés
n'étant généralement ni linéaires, ni stables dans
le temps, un certain nombre de précautions s'impose. Il faut s'assurer
que le système reste «quasistable» sur toute la durée
de la mesure, et que l'amplitude de la sinusoïde soit suffisamment faible
pour que la fonction I = f(E) soit linéaire dans le domaine
perturbé (Figure II-7).
Le tracé de la fonction : ?
= ' ( A E ) II-16
permet de s'assurer que l'amplitude est adaptée pour
que la perturbation reste dans le domaine linéaire.
II.2.3.1.2. Impédance de différents
phénomènes pouvant se produire à l'interface
électrode/électrolyte
Lors de la mise en contact d'une électrode et d'un
électrolyte, différents phénomènes physiques et
physico-chimiques s'amorcent, chacun suivant sa propre cinétique, et
conduisent le système vers un équilibre thermodynamique.
Ces phénomènes vont dépendre en partie du
potentiel de surface de l'électrode et de celui présent au sein
de la solution. Du côté de l'électrode, le potentiel est
constant en tout point de la surface. Par contre, dans la solution, il est
donné par la résolution de l'équation de Laplace :
? CD2= 0 II-17
Où CD représente le
potentiel. Ceci a pour conséquence de créer une variation de
potentiel et de courant dans l'électrolyte, qui conduit au concept de
chute ohmique. En SIE, l'électrode de référence et la
contre électrode sont placées relativement loin de la surface de
l'électrode de travail. A haute fréquence, la répartition
des courants secondaires peut donc être négligée, à
l'exception de certains cas particuliers discutés par Huang et al.
[24]. La chute ohmique est alors classiquement décrite comme
étant une résistance d'électrolyte Re [25].
L'impédance de
la chute ohmique est : Z Re (w) = Re II-18
Un autre phénomène observé à
l'interface électrode/électrolyte est la formation d'une double
couche d'ions [26] (voir figure II-8). L'application d'une perturbation
sinusoïdale lors de la mesure d'impédance entraîne la charge
et la décharge de cette couche qui se comporte alors comme un
condensateur électrique. La réponse de cette double couche
génère d'un courant Idc qui dépend de la fréquence
de perturbation. Ce type de processus peut être
généralisé à tous les phénomènes qui
entraînent la charge et la décharge de deux zones
séparées par un diélectrique.
29
Chapitre II
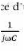
L'impédanc 'un condensateur de apacité C est
donnée par l'équation :
ZC (co) = - avec C = EE0 II-19
où C est la capacité, u la permittivité
relative du diélectrique, u0 la permittivité diélectrique
du vide, A l'aire de réaction et e l'épaisseur du
diélectrique.
Figure II.8 : Représentation schématique de la
double couche par Kauffman
(les points dans l'électrode
représentent des charges négatives) [27].
Il peut aussi se produire des processus faradiques,
c'est-à-dire des réactions d'oxydation ou de réduction
d'espèces à la surface de l'électrode. En
considérant l'équation suivante :
Kf
Ox + ne- Red II-20
kb
deux cas sont à prendre en considération : soit la
cinétique de réaction est strictement contrôlée par
le transfert de charge, soit la cinétique est de type
activation-diffusion avec un
contrôle diffusionnel. Dans le cas où la
cinétique de réaction est limitée uniquement par le
transfert de charge, la contribution faradique If au courant mesuré est
indépendante de la fréquence et ne joue que sur l'amplitude de la
réponse du système. En faisant l'hypothèse que
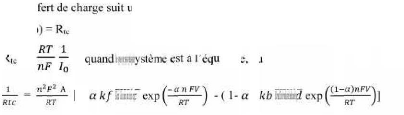
le transf une loi exponentielle (loi de Tafel),
l'impédance mesurée est donc :
Z Rtc (w avec,
R = à l'ilibre ou
L- ) II-21
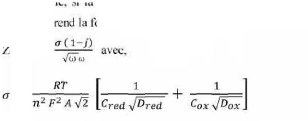
Chapitre II
Dans les équations précédentes, R
représente la constante molaire des gaz, T la température, n le
nombre d'électrons mis en jeu dans la réaction, F la constante de
Faraday, I0 le courant d'échange à l'équilibre, #177; le
coefficient de transfert, kf et kb les constantes de réaction, A l'aire
de la surface sur laquelle se produisent les réactions, cox et cred les
concentrations à l'état stationnaire des espèces
oxydées et réduites et V = (E - E0), E étant le potentiel
auquel est effectuée la mesure d'impédance et E0 le potentiel
standard du système étudié. Si la cinétique est
contrôlée par un processus mixte d'activation-diffusion, il faut
prendre en compte le courant lié au phénomène de
relaxation des éléments actifs dans la couche de diffusion en
fonction de la fréquence de perturbation en plus du courant faradique.
L'impédance de diffusion est connue sous le nom d'impédance de
Warburg. Selon l'hypothèse choisie pour décrire la variation de
concentration des éléments dans la couche de diffusion,
l'impédance de Warburg peut avoir plusieurs expressions
différentes.
Par exemple si la couche de diffusion est
considérée comme semi-infinie, l'impédance de
Warburg pr orme suivante [28] :
ZW (w) =
II-22
=

30
Ci, Cred et Cox sont les
concentrations en solution des espèces appartenant au couple redox et
Dred et Dox sont leurs coefficients de diffusion
respectifs. La couche de diffusion peut aussi avoir une épaisseur finie.
Ceci se produit quand la variation de concentration des espèces actives
suit le modèle de Nernst. L'impédance de Warburg est alors
donnée par l'équation suivante [28] :
Zw (w) = Rd II-23
Dans cette équation, Rd est une résistance de
diffusion, et 'n l'épaisseur de la couche de diffusion selon
le modèle de Nernst. D'un point de vue pratique, l'épaisseur de
cette couche de diffusion peut être contrôlée en ajustant la
vitesse de rotation d'une électrode tournante.
Quand w ' 0, l'impédance prend alors une valeur
particulière appelée résistance de polarisation ou Rp. Ce
paramètre peut aussi être obtenu à partir de la courbe
courant-tension I = f(E) du système. En effet, le calcul de la pente
dE/dI autour de potentiel de corrosion E0
31
Chapitre II
donne Rp. Quand la résistance de polarisation est
obtenue, le diagramme d'impédance est complet en basse
fréquence.
II.2.3.1.3. Exploitation des résultats
Utilisation des schémas électriques
équivalents
Les différents processus se déroulant à
l'interface électrode/électrolyte peuvent être
modélisés par la construction d'un circuit électrique
équivalent. Chacun des composants utilisés, branchés en
série ou en parallèle, représente un
phénomène physique particulier. Ces modèles sont ensuite
utilisés pour ajuster les diagrammes expérimentaux afin
d'extraire les paramètres nécessaires à la
compréhension du système étudié. Bien entendu,
cette façon de faire est une simplification et suppose que les
différents phénomènes sont indépendants les uns des
autres, ce qui n'est pas le cas dans la réalité. Toutefois,
l'erreur introduite par l'utilisation de cette forme de modèle est
suffisamment faible pour que cette simplification puisse être
considérée comme acceptable.
Dans la construction de ces circuits électriques
équivalents, certains composants utilisés sont identiques
à de véritables composants électriques, comme la
résistance R, la capacité C ou même l'inductance L dont
l'impédance est donnée par la relation suivante :
ZL (w) = j L ù II-24
qui peut servir
dans le cadre d'impédance mesurée sur des systèmes
complexes (batteries par exemple [29]. D'autres composants sont
spécifiques aux processus électrochimiques comme
l'impédance de Warburg vue précédemment ou le CPE
(constant phase element).
Il existe plusieurs modèles de circuits
équivalents fréquemment rencontrés. Le plus simple sert
à modéliser le comportement d'électrodes bloquantes,
c'est-à-dire que l'électrode est placée dans des
conditions telles qu'il ne se produit pas de réaction faradique. Ce
circuit est constitué d'une résistance d'électrolyte Re
branchée en série avec une capacité interfaciale, C0 ou un
CPE (Q0,#177;) si le comportement est non idéal (figure II-9 a et b).
Selon le type d'échantillon, cette capacité peut être une
capacité de double couche, de film d'oxyde.
Quand il y a réaction faradique, le modèle
devient plus complexe. Ainsi, s'il n'y a pas de contrôle diffusionnel, le
schéma classiquement utilisé est celui présenté sur
la figure (II-9c). C'est une évolution du modèle de
l'électrode bloquante où une résistance Rtc traduisant le
transfert de charge est branchée en parallèle avec la
capacité de double couche. Par contre, en cas de contrôle
diffusionnel, il faut ajouter, en série avec la résistance de
transfert de charge, une impédance de Warburg (W) comme il est
indiqué sur la figure( II-9 d). Ce circuit est connu sous le nom de
modèle de Randles. Le choix du type d'impédance de Warburg se
fait en fonction des conditions expérimentales.
32
Chapitre II
Figure II.9 : Schémas de circuits électriques
équivalents fréquemment rencontrés :
a)
électrode bloquante idéalement polarisable,- b) électrode
bloquante avec
comportement CPE, - c) électrode avec réaction
faradique sans contrôle diffusionnel, -
d) modèle de Randles et
- e) modèle du film de peinture [30].
Il existe bien d'autres types de circuits équivalents,
chacun d'entre eux décrivant un système particulier. La
manière dont est branché chaque composant ainsi que l'ordre de
leur apparition sont importants, à la fois pour le calcul de
l'impédance et pour la lisibilité du modèle. Il faut
suivre la logique physique du système : les processus successifs sont
branchés en série alors que les processus simultanés sont
branchés en parallèle.
II.2.4. Application de la SIE aux revêtements
Dans le cas idéal, un revêtement protège
le substrat métallique contre la corrosion car il est isolant,
adhérant et étanche (figure II-10). Le revêtement se
comporte donc comme une capacité pure Crev, et l'interface se comporte
comme un circuit électrique RC en série. La résistance Re
correspond à la résistance de l'électrolyte.
Figure II.10 : Modèle physique du revêtement
parfait, et le circuit électrique
équivalent [32].
33
Chapitre II
Cependant, dans la réalité, on arrive
généralement à mesurer le potentiel de corrosion du
métal. Ceci est dû à la diffusion des ions de
l'électrolyte à travers les pores du revêtement [20,31].
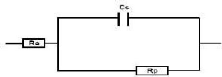
Lorsqu'il y a une dégradation significative du
revêtement, il se crée une résistance Rp reflétant
la porosité et provoquant une fuite de courant, et le modèle
proposé précédemment n'est plus applicable. Ce qui
nécessite l'utilisation d'un nouveau schéma électrique
équivalent figure II-11[33,34].
Figure II.11 : Schéma électrique
équivalent d'un système avec revêtement
poreux sans
interface métal / électrolyte [36].
Dans le cas des revêtements dégradés,
l'électrolyte entre assez rapidement en contact avec le métal, et
une nouvelle interface apparaît. Il se forme une double couche dont le
comportement électrique est équivalent à un circuit RC
parallèle placé en série avec la résistance
d'électrolyte, Re. (La figure II.12) présente le circuit
équivalent et le modèle physique d'un revêtement poreux,
après la formation d'une interface métal/film. On notera Cdc la
capacité de la double couche formée, et RP la résistance
de polarisation ayant lieu au niveau de cette interface.
|
|
b- Relation entre le circuit équivalent et les
propriétés de corrosion et du revêtement
|
|
a- Circuit équivalent
|
|
Figure II -12 : Modèle électrique
équivalent d'un revêtement poreux avec formation
d'une double
couche en surface du métal [37].
Avec:
Crev Capacité du revêtement
Cdc Capacité de la double couche
Re Résistance de
l'électrolyte
Rp Résistance de polarisation Rpore
Résistance de pore
34
Chapitre II
Lorsque les ions atteignent la surface du métal par
diffusion et si l'on suppose que les phénomènes de corrosion
résultent de la réaction de l'électrolyte avec le
métal, alors il est indispensable de tenir compte du transport des
espèces réactives dans ce même électrolyte. Si cette
diffusion est une des étapes lentes (limitantes), elle contribue dans
l'expression de la vitesse de corrosion du métal sous revêtement
[37,38].
La figure II-13, présente le schéma
électrique équivalent d'une électrode, lorsque celle-ci
est le siège simultané d'une réaction de transfert de
charge et d'un transport de matière par diffusion.
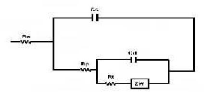
Figure II.13 :Schéma électrique équivalent
du système électrochimique
lors d'un processus de transport de
charge et de matière [39].
II.2.5. Représentation des données:
Habituellement, il existe deux modes de représentation
des diagrammes d'impédance électrochimique. Ils peuvent
être tracés en coordonnées cartésiennes dans le plan
complexe de Nyquist en plaçant les valeurs Zr(É) en abscisse et
--Zj(É) en ordonnée (contrairement aux conventions
utilisées en électrotechnique). La représentation de Bode
est l'autre représentation classique pour visualiser les diagrammes.
Dans ce cas, le module de l'impédance |Z| (représenté en
échelle logarithmique) et le déphasage sont tracés en
fonction de la fréquence, elle aussi représentée en
échelle logarithmique. Ces deux visions différentes d'un
même résultat ne sont pas en compétition, elles sont
complémentaires ; chacune d'entre-elles montre un aspect particulier du
diagramme d'impédance. La représentation de Nyquist permet de
voir les différentes « boucles et droites » du diagramme mais
masque les résultats à haute fréquence alors que la
représentation de Bode offre la vision complète du domaine de
fréquence, tout en étant moins parlante pour identifier certains
phénomènes caractéristiques. Pour illustrer ce qui a
été présenté ci-dessus, des diagrammes
d'impédance électrochimique tirés d'un article
écrit par Orazem et al. [37].
sont donnés sur la figure II-14 dans le plan complexe
de Nyquist et sur la figure II-15 selon la représentation de Bode.
35
Chapitre II
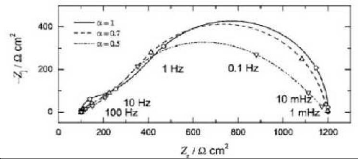
Figure II.14: Représentation des diagrammes
d'impédance dans le plan
complexe de Nyquist [8].
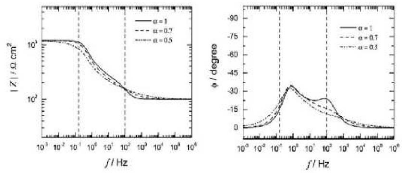
Figure II.15 : Tracés des diagrammes d'impédance
selon la
représentation de Bode[8].
36
Chapitre II
II.2.6. Les avantages et les inconvénients de la
SIE
Les techniques de polarisation linéaire sont valables
pour les métaux nus dans beaucoup de situations, elles demeurent
défectueuses pour l'évaluation des revêtements organiques
déposés sur des métaux. La STE résout ces
problèmes.
La technique d'impédance électrochimique apporte
une analyse plus complète au pouvoir protecteur des revêtements,
comparativement aux méthodes stationnaires, puisqu'elle permet de
séparer les mécanismes de cinétiques différentes.
Les caractéristiques physiques du film protecteur peuvent, en
particulier, être distinguées du mécanisme de transfert de
charge intervenant à l'interface substrat/électrolyte et
quantifiées en fonction des différents paramètres
imposés au système.
L'inconvénient majeur de l'impédance
électrochimique réside dans l'interprétation des
résultats car il est parfois difficile de trouver le circuit
électrique équivalent correspondant le mieux à l'interface
électrode/solution. Un circuit idéal n'est pas toujours
adéquat pour décrire la réponse électrique du
système [22,40].
37
Chapitre II
REFERENCES BIBLIOGRAPHIQUES
[1] A. Godon, Thèse de doctorat, Relations structure
composition propriétés de revêtements
électrodéposés de nickel de taille de grain
nanométrique. Univ. De la Rochelle (2010).
[2] J-C. Puippe, Theory and Practice of Pulse Plating, American
Electroplaters and Surface Finishers Society (1986) 1-243.
[3] J. Amblard, Electrocristallisation Aspects fondamentaux,
Techniques de l'Ingénieur D906 (1976).
[4] T. Watanabe, Nano-plating Microstructure Control theory of
platd film and Data base of Plated film Microstructure, Elsevier (2004) 697
[5] P. Costa, Nanomatériaux Structure et
élaboration, Techniques de l'Ingénieur M4026(2001).
[6] P. Benaben, F. Durut, Nickelage électrolytique
,Mise en oeuvre, Techniques de l'Ingénieur M1611(2003).
[7] N. Kanani, Electroplating, Basic Principles, Processes
and Practrice , Elsevier (2004) 353 .
[8] S. Amokrane et J. P. Badiali , Modern aspects of
electrochemistry, chapitre Analisys of the capacitance of the metal solution
interface, N° 22, Plenum press (1992) 1-91
[9] P. Bommerbah , Evolution des propriétés
d'un film inhibiteur de corrosion sous l'influence de la température et
des conditions hydrodynamiques caractérisation par techniques
électrochimiques, Thèse de doctorat, INSA de Lyon, N°
(05ISAL 0060),( 2005) 13-24.
[10] B. Assouli , Etude par émission acoustique
associée aux méthodes électrochimiques de la corrosion et
de la protection de l'alliage Cuivre-Zinc (60/40) en milieu neutre et alcalin,
Thèse de doctorat, université IBN TOAFIL (KENITRA-MAROC) et INSA
(Lyon-France).N° (02ISAL0103), (2002)45-54.
[11] S. Payan , Comportement à la corrosion galvanique
de matériaux composites à matrice d'alliage d'aluminium
renforcée par des fibres de carbone haut module, Thèse de
doctorat Unive de Bordeaux , 242(2002)34-48.
[12] L.S . Hernandez, G. Garcia, B. Delamo, R. Romagnoli, C.
Lopez, Evaluation of non toxic Alkyd primers by Electrochimical Impedance
Spectroscopy. Corrosion (1998), N° (303) NACE (Houston, Texas)2.
38
Chapitre II
[13] J. Wolstenholme,Electrochemical methods of assessing the
corrosion of painted metals- a review. Corrosion science, Vol. 13,
(1973)521-530.
[14] H. Zahn, Laboratory evaluation of corrosion resistance,
pigments and vehicles. Corrosion-national association of corrosion engineers
Vol 3, (1947)233-240.
[15] F. Wormwell, D.M. Brasher, Electrochemical studies of
protective coatings on metals, part II,Resistance and capacitance measurements
on painted steel immersed in sea water, Journal of the iron and steel
institute,164( 1950)141-148.
[16] M. Mekki Daouadji, Les techniques de mesures de la vitesse
de corrosion(2004).
[17] Mike O'donoghue, Terry J. Aben, Clive hare, EIS
investigations of alkyd and epoxy coatings as they are chemically stripped from
steel panels, N°(03057).corrosion (2003)15.
[18] E. L. Din-stirbu, Comportement à la corrosion des
alliages d'aluminium utilisés dans l'industrie automobile pour la
fabrication de radiateur de chauffage ,Thèse de doctorat. INSA de Lyon ,
(2005)46-48.
[19] Tzu-yu chen. A Ransient, technique to determine solution
resistance for corrosion rate measurement in low conductivity solution.
Corrosion (1996), N° (341), NACE (Houston, Texas)1/341-3/341.
[20] Stephane gastaud, Influence de l'état du
revêtement hydrocarboné sur le risque de corrosion des
canalisations enterrées de transport de gaz. Thèse de
doctorat,INSA de Lyon, N°(02ISAL009) (2002).
[21] N. LEBOZEC, Réaction de réduction de
l'oxygène sur les aciers inoxydables en eau de mer naturelle, influence
du biofilm sur les processus de corrosion. Thèse de doctorat. Paris,
(2000)7-10.
[22] Jaubert lionel, Etude de la corrosion uniforme d'aciers
non alliés et inoxydables: utilisation conjointe de l'émission
acoustique et des techniques électrochimiques. Thèse de doctorat,
INAS de Lyon,( 2005)15-31.
[23] H. Mayet, Etude de la corrosion par piqure des alliages
d'alluminium par l'analyse des fluctuations de courant et de potentiel à
l'abandon. Thèse Science et Genie des Matériaux.Grenoble Institut
National Polytechnique de Grenoble(1992). 172.
[24] V. M.-W. Huang, V. Vivier, M. E. Orazem, N.
Pébère et B. Tribollet , The Apparent Constant-Phase-Element
Behavior of an Ideally Polarized Blocking Electrode. J.Electrochem.
Soc.,(2007)154
39
Chapitre II
[25] J. Newman ,Frequency dispersion in capacity measurements at
a disk electrode. J.Electrochem. Soc., (1970)198-203
[26] A. M. Kauffman ,Understanding Electrochemical Cells,
Rapport technique 17, Solar-tron, (1997).
[27] C. Gabrielli . Identification of electrochemical processes
by frequency response analysis. Rapport technique 004/83, Solartron, (1998).
[28] S. Hong et L. Tai-Chin , Electrochemical Impedance
Spectroscopy for Battery Research and Development. Rapport technique 31,
Solartron, (1996).
[29] L. Beaunier, I. Epelboin, J. C. Lestrade et H. Takenouti ,
Etude électrochimique, et par microscopie electronique a balayage, du
fer recouvert de peinture. Surface Technology, (1976) 237-254
[30] Christophe Mendibide, Caractérisation de
revêtements PVD nanostructurés à base de nitrures de
métaux de transition pour application mécanique , aspect
chimiques, mécaniques et tribologiques, Thèse de doctorat, INSA
de Lyon, N°( 03ISAL0039) (2003) 34-43.
[31] G. J. Brug, A. L. G. Van Den Eeden, M. Sluyters-Rehbach
et J. H. Sluyters .The analysis of electrode impedances complicated by the
presence of a constant phase element.
J. Electrochem., 176(1984) 275-295
[32] F. Zou, D. Thiery, Application of localised
electrochemical impedance spectroscopy to the study of the degradation of
organic coating. ACS Symposium series 689.washington (1998)23-30.
[33] M. kning, J. Scully, Basic aspects of electrochemical
impedance, application for the life production of organic coatings on metals.
Corrosion vol. 46, N° 1(1990)22-29.
[34] D. Loveday, P. Peterson, B. Rodgers, Evaluation of
Organic Coatings with Electrochemical Impedance Spectroscopy Part2 application
of EIS to coating,(2004)88-93
[35] Melha. Nazaf ,Développement de nouveaux primaires
anti-corrosion à base de polymères conducteurs pour la protection
de l'acier contre l'agressivité des milieux aqueux chlorurés.
Mémoire de magister, Sonatrach/Institut Algérien du
Pétrole,Boumerdes, (2005).
[36] G.W.walter, Application of impedance measurements to
study performance of painted metals in aggressive solution. Journal of
electrochem., 118(1981)259-273.
[37] M. E. Orazem, N. Pébère et B. Tribollet ,
Enhanced Graphical Representation of Electrochemical Impedance Data. J.
Electrochem. Soc., (2006)153(4)
40
Chapitre II
[38] Bounoughaz Moussa,Contribution à l'étude
de l'influence de l'argent et du sélénium sur la passivation des
anodes de cuivre durant l'électroraffinage, Ecole des gradues, Unive
LAVAL(1993)23-25.
[39] P. Agarwal, M. E. Orazem et L. H. Garcia Rubio ,
Measurement Models for Electrochemical Impedance Spectroscopy. I. demonstration
of applicability. J. Electrochem. Soc., 139(1992) 1917-1927
[40] T. Pajkossy, T. Wandlowski , D. M. Kolb , Impedance aspects
of anion adsorption on gold single crystal electrodes. J.
Electrochem., 414(1996) 209-220,
Chapitre III

CHAPITRE III
RESULTATS
ET
DISCUSSION
Dans ce chapitre nous exposons les résultats sous forme
de courbes, obtenus grace aux méthodes de caractérisation
électrochimiques, afin d'évaluer l'influence des conditions
opératoires à savoir, la concentration, la densité de
courant et le milieu.
41
Chapitre III
III.1. Introduction:
Ce chapitre est constitué de deux parties, dont la
première concernera la préparation des électrodes de
travail nécessaires à l'élaboration du revêtement
d'oxyde de cérium, quant à la seconde partie, elle portera sur
l'étude et le comportement de la résistance à la corrosion
de l'acier ainsi revêtu au cours de son immersion dans NaCl 0,5M et dans
de l'eau industrielle.
Métal / film
film / solution
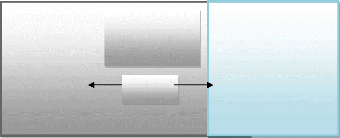
barrière
Couche
'film
Figure III.1 : Modèle schématique d'une
électrode revêtu [1].
L'ensemble des échantillons revêtus est
contrôlé par la mesure de la résistance de polarisation et
de la densité de courant de corrosion obtenue par extrapolation des
droites de Tafel, la technique de la spectroscopie d'impédance
électrochimique (SIE) permet de déterminer la résistance
de polarisation à la corrosion et la capacité du
revêtement, qui renseignent sur la performance du système
(métal/revêtement).
III. 2. Elaboration des revêtements:
Le paramètre mesurable qui détermine la vitesse
d'électrodéposition est la densité de courant cathodique.
La relation qui lie la densité de courant au potentiel de
l'électrode permet d'obtenir des informations sur les mécanismes
réactionnels. Chacune des étapes du processus
d'électrodéposition possède sa propre cinétique et
suivant le système considéré peut devenir limitante. Le
ralentissement de la cinétique d'une réaction
électrochimique se traduit par la présence d'une surtension, qui
est d'autant plus grande que la cinétique est ralentie. La surtension
totale peut être considérée comme la somme des surtensions
associées à chaque étape de la réaction
d'électrodéposition, en d'autre terme la surtension de
réaction (lorsque les réactions chimiques sont impliquées
dans le processus global), la surtension de transfert de charge (transfert des
porteurs de charge à travers la double couche), la surtension de
42
Chapitre III
cristallisation (processus lié à l'incorporation de
l'adatome à réseau cristallin) et la surtension de diffusion
(traduit le transport de l'ion métallique du centre de la solution
à l'électrode). Mais ces différentes contributions ne sont
pas mesurables, et seule la surtension totale est accessible
expérimentalement [2].
L'étude expérimentale consiste à
l'élaboration d'oxyde de cérium sur acier pour ensuite
étudier son comportement électrochimique lorsqu'il est en contact
avec une solution à 0,5 M NaCl d'une part et de l'eau industrielle
d'autre part et dont le résultat attendu sera de déterminer la
qualité de la protection de l'acier. Toutes les couches minces ont
été réalisées sur acier à bas carbone en
mode potentiodynamique (galvanostatique) à partir d'un
électrolyte aqueux. La solution à été obtenue par
la dissolution du nitrate de cérium dans l'eau distillée. Le
dépôt à été effectué à une
concentration de 0,01M et 0,1 M, à température ambiante sous
agitation magnétique. Pendant une durée de 20 minutes
durée qui a été préalablement déjà
sélectionnée lors de travaux antérieurs et à cause
des avantages qu'elle présente pour les besoins de notre étude
[3].
Les densités de courant cathodiques imposées
sont: 0,25 -0,5- 1-1,5 et 3 mA/cm2. Dans le cadre de la mise en
application des étapes sus décrites, une cellule
électrochimique est placée et celle-ci est constituée
d'une : électrode de référence Ag/AgCl saturée de
KCl, une contre électrode en platine et l'électrode de travail
sous forme d'un disque de surface 1,4cm2. Les échantillons
revêtus sont ensuite rincés dans l'éthanol, puis
séchés à l'air pendant 24h.
Les courbes chronopotentiométriques enregistrées
au cours de la formation d'un dépôt à partir d'une solution
Ce(NO3)3.6H2O à différentes densités de courant sont
représentées sur la (figure III-2).
Chapitre III
0 200 400 600 800 1000 1200
E ( V / Ag/AgCl )
-0,4
-0,6
-0,8
-1,0
-1,2
-1,4
-1,6
0.25 mA/cm2 0.5 mA/cm2 1 mA/cm2
1,5 mA/cm2 3 mA/cm2
Temps d'électrodéposition (sec)
Figure III.2 : Courbes chronopotentiométriques
enregistrées au cours de la formation
d'un dépôt sur
acier à partir d'une solution Ce(NO3)3.6H2O 0,01M.
L'allure des courbes temporelles nous ont permis de constater
qu'a différentes densités de courant celles-ci se
caractérisent par une décroissance monotone et rapide du
potentiel vers des valeurs très cathodiques suivies d'un pseudo
stabilité qui se définie par une faible variation du
potentiel.
Avec le temps, la variation monotone du potentiel se traduit
par les réactions de réduction se déroulant à
l'interface où l'anion participe dans les réactions cathodiques
selon les équations suivantes :
- +
NO3+ 10 H+ + 8 e- NH4 + 3 H2O III-1
-
+ H2O + 2 e- NO2
+ 2OH - III-2
-
NO3
43
A cet effet, les ions hydroxydes peuvent être produits
par les trois réactions de réductions à savoir,
l'oxygène dissout, l'eau et les nitrates:
H2O + 2 e- H2+ 2OH- III-3
O2 + 2 H2O + 4 e- 4OH- III-4
Ainsi, la
production des ions (OH-) favorise la formation d'un
précipité d'hydroxyde de cérium sous forme de Ce(OH)3 et
/ou Ce(OH)22+ .
Finalement la formation de l'oxyde de cérium CeO2 se
fait par l'intermédiaire de l'oxydation de Ce(OH)3 et/ou l'hydrolyse de
Ce(OH)22+
Ce(OH)3 CeO2 + H3O+ e- III-5
Ce(OH)22+ + 2OH- CeO2 + 2H2O III-6
44
Chapitre III
III. 3. Comportement électrochimique des
substrats dans NaCl
La caractérisation des revêtements à base
d'oxydes de cérium à été réalisée
dans deux
milieux à savoir NaCl 0,5M et l'eau industrielle.
Le comportement électrochimique des échantillons
nus et revêtus à été établit par plusieurs
techniques électrochimiques en mode stationnaire et
fréquentiel nous nous sommes intéressés
en particuliers :
§ Au suivi du potentiel en fonction du temps d'immersion.
§ Au tracé des courbes de polarisation.
§ la méthode de la voltamètrie cyclique.
§ Au tracé des diagrammes de spectroscopie
d'impédance électrochimique (SIE).
III. 3 .1. Evolution du potentiel à circuit ouvert
du substrat nu dans NaCl Introduction:
Le milieu corrosif dans lequel baigne le métal agit
d'abord par sa nature même (acide, basique, salin) et aussi par la
pression, la concentration, la pureté, la température, la
viscosité et l'état de repos ou d'agitation.
Lorsque l'on mesure OCP d'un métal, on observe qu'il
n'atteint pas en général immédiatement une valeur
stationnaire. En effet, au moment où le métal est plongé
dans la solution, celle-ci ne contient pas d'ions de métal, si bien
qu'un potentiel stationnaire est long à atteindre, d'autre part
l'interface métal-solution peut être modifiée par formation
d'un produit de corrosion insoluble ou d'un gaz comme l'hydrogène. Les
courbes potentiels-temps peuvent présenter différents aspects, la
figure III.3 présente la variation libre à circuit ouvert en
fonction du temps, où chaque courbe caractérise un comportement
cinétique propre.
Figure III.3: Variation du potentiel libre à circuit
ouvert en fonction du temps [1].
45
Chapitre III
Courbe a : le potentiel devient de plus en plus noble, il y a
passivation du métal par formation à la surface d'un produit de
corrosion insoluble protecteur. C'est le cas du fer plongé dans l'acide
nitrique concentré.
Courbe b : le potentiel devient de moins en moins noble, ou
plus négatif, il y a attaque du métal c'est le cas de l'aluminium
plongé dans la soude.
Courbe c : le potentiel devient d'abord plus négatif,
puis tend vers des valeurs plus positives, il y a attaque suivie de
passivation. C'est le cas de l'aluminium plongé dans une solution
d'acide nitrique à 15 ou 20 %.
Courbe d : le potentiel devient plus noble puis se
déplace vers des valeurs plus négatives, c'est le cas lorsqu'au
moment de son immersion, le métal est recouvert d'une couche
protectrice.
Le mécanisme de la corrosion peut être
décomposé en plusieurs étapes et notamment lors de
l'adsorption des ions Cl- à la surface du film passif, suivie
de la migration des ions Cl- à travers le film passif et en
fin terminé par la propagation de la piqûre dans le
métal.
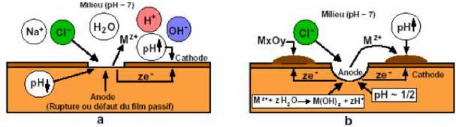
Figure III.4 : Amorçage (a) et propagation (b) d'une
piqûre [8].
En effet leur concentration locale induit une acidification du
milieu et d'autre part le film ainsi formé en surface du métal
devient moins efficace car plus instable et donc plus susceptible de se rompre
localement. Un autre effet de la salinité sur la corrosion
résulte de son influence sur la conductivité du milieu aqueux. La
résistivité du milieu aqueux corrosif limite la distance sur
laquelle le couplage est effectif. Cependant, la forte conductivité des
milieux salins tels que l'eau de mer (NaCl 0,5 M) implique que la
résistivité de l'électrolyte va jouer un rôle mineur
dans la cinétique des réactions de corrosion [4].
L'évolution du potentiel libre (potentiel d'abandon) au
cours d'un test de corrosion, est un premier indice pour estimer
l'évolution de sévérité de la dégradation
pendant l'immersion. L'échantillon est immergé dans une solution
NaCl à 0,5M. Son évolution donne des
Chapitre III
informations sur les phénomènes qui se
déroulent à l'interface métal/solution, ce qui permet
d'évaluer la stationnarité du système.
La figure III-5 Présente le chronopentiogramme de
l'acier nu immergé pendant 05 heures dans une solution NaCl à
0,5M.
|
-0,35
-0,40
|
|
|
|
|
|
1ier
Changement
|
|
|
|
E ( V/Ag/AgCl)
|
-0,45
|
|
|
|
-0,50
-0,55
|
|
|
2éme
Changement
|
|
|
|
|
|
|
-0,60
-0,65
|
|
|
|
|
|
|
|
|
|
46
0 50 100 150 200 250 300
Temps ( min)
Figure III.5 : Suivi du potentiel à circuit ouvert de
l'acier nu
dans une solution NaCl 0,5M
L'allure de la courbe de la figure III-5 nous a permis de
constater que le potentiel de corrosion n'atteint pas immédiatement une
valeur stationnaire, la stabilisation du potentiel à une valeur de -0,65
V/Ag/AgCl est atteinte après 05 heures d'immersion.
Avant la stabilisation du potentiel de corrosion, nous avons
constaté, en premier lieu, une augmentation relativement
remarquée du potentiel pendant les premières minutes d'immersion,
ensuite le potentiel diminue en deux temps différents , le premier
changement qui est après 01 heures d'immersion , la courbe est
caractérisée par une décroissance rapide du potentiel de
corrosion vers des valeurs très cathodiques, le potentiel devient de
moins en moins noble, ou plus négatifs, il ya une attaque provoquant une
dissolution du métal. En revanche, le deuxième changement c'est
un pseudo stabilité caractérisé par une faible variation
de potentiel de corrosion.
Le changement de l'allure de la courbe avec le temps
d'immersion est lié aux réactions de réductions se
déroulant à l'interface, généralement
l'évolution du potentiel vers des valeurs plus
électronégatives caractérise un phénomène de
dissolution spontané avant de se stabiliser
47
Chapitre III
au bout d'un certain temps d'immersion dans la solution
corrosive. C'est une situation de corrosion continue d'un métal.
Lors de la dégradation de l'acier, différents
produits de corrosion (lépidocrocite, goethite, magnétite) sont
susceptibles de se former. Les dépôts de produits de corrosion
formés peuvent jouer un rôle actif de barrière plus ou
moins efficace selon leur nature.
Cependant, ces couches, pas toujours homogènes dans
leur épaisseur, et leur nature change dès lors qu'elles sont
extraites du milieu dans lequel elles se sont formées [5,6].
Tout métal recouvert d'un film passif est sensible
à la corrosion par piqûres. Celle-ci se produit quand le
matériau est mis en contact avec un milieu aqueux contenant les
halogénures: F-, Cl-, Br-,
I- , les solutions contenant des ions Cl- demeurent les
plus
agressives et l'apparition de piqûres s'initient sur les
points faibles de la couche d'oxyde par défauts mécaniques et/ou
en présence de composés intermétalliques cathodiques et
dans un milieu jouant le rôle d'électrolyte [7].
III. 3. 2. Evolution du potentiel à circuit ouvert
des substrats revêtus dans NaCl
L'évolution du potentiel à circuit ouvert pour
les substrats revêtus à différentes densités de
courants élaborés dans un bain du nitrate de cérium
à 0,01 M et 0,1 M est représentée sur la figure III.6a et
III.6b. Le potentiel du circuit ouvert a été suivi pendant trois
heures d'immersion en milieu NaCl à 0,5M.
Pour la figure III.6a, l'allure des courbes nous à
permis de les classer en trois types de courbes. Pour les substrats
revêtus à 0,25 et 1,5 mA/cm2 les potentiels
enregistrés au début d'immersion indique clairement la formation
d'un film protecteur à la surface de l'acier. En effet le potentiel
enregistré était -0,55V/Ag/AgCl alors que le potentiel de l'acier
nu était de l'ordre -0,4 V/Ag/AgCl. De plus, la chute du potentiel au
début de l'immersion est liée à une première
interaction du revêtement avec le milieu agressif induisant une faible
dégradation, indiqué par les faibles valeurs des potentiels
enregistrées. D'un autre coté, un palier
caractérisé par une stabilité des potentiels est
observé juste après quelques minutes d'immersion indiquant une
parfaite protection de la surface.
En revanche, l'allure des courbes relatives aux
revêtements obtenus à 0,5 et 1 mA/cm2 indique une
passivation instantanée c'est-à-dire un comportement
idéale du revêtement.
48
Chapitre III
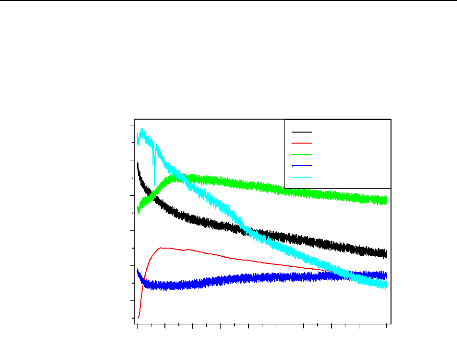
revêtu à 0,25mA/cm2 revêtu à
0,5mA/cm2 revêtu à 1 mA/cm2 revêtu
à 1,5mA/cm2 revêtu à 3 mA/cm2
-0,57
-0,60
-0,63
-0,66
(a)
E( V/Ag/AgCl)
0 20 40 60 80 100 120 140 160 180
-0,51
-0,54
Temps (min)
Figure III.6a : Suivi du potentiel à circuit ouvert de
l'acier revêtu à partir de Ce(NO3)3.6H2O 0,01M dans NaCl 0,5M
E ( V/Ag/AgCl)
-0,56
-0,58
-0,60
-0,62
-0,64
-0,66
-0,68
-0,70
-0,72
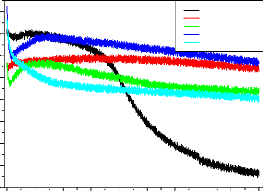
(b)
revêtu à 0,25mA/cm2 revêtu à
0,5mA/cm2 revêtu à 1 mA/cm2 revêtu
à 1,5mA/cm2 revêtu à 3 mA/cm2
0 20 40 60 80 100 120 140 160 180 Temps(min)
Figure III.6b : Suivi du potentiel à circuit ouvert de
l'acier revêtu à partir de Ce(NO3)3.6H2O 0,1M dans NaCl 0,5M
49
Chapitre III
Le suivi du potentiel à circuit ouvert de l'acier
revêtu dans un bain du Ce(NO3)3.6H2O à 0,1M à
différentes densités de courants présenté dans la
figure III.6b, permet de conclure que l'allure des courbes dans la gamme de
densité de courant allant de 0,5 - 1 et 1,5 mA/cm2
présente une évolution de potentiel dans les premières
minutes vers des valeurs négatives il apparait être identifier une
destruction d'une couche externe préexistante à la surface du
substrat, ensuite le potentiel évolue vers des valeurs positives avant
de se stabiliser, cette différence est peut être liée
à la nature, la qualité et les propriétés du
revêtement. A noter que le revêtement est constitué d'une
couche externe riche en oxyde et d'une couche interne riche en hydroxyde de
cérium on pense donc que la couche interne est de nature à
protéger l'acier. La courbe relative au dépôt obtenu
à 0,25 mA/cm2, est caractérisée par une chute
de potentiel après 01heure d'immersion vers des valeurs négatives
Le film formé dans ces conditions serait peut être de nature
à protéger l'acier.
Pour le revêtement obtenu à 3mA/cm2 la
chute monotone et continue du potentiel en fonction du temps nous laisse croire
que l'attaque du revêtement continue de l'être. Ceci est dû
soit à la discontinuité du revêtement ou à la faible
épaisseur.
III. 3. 3. Courbes de polarisation des substrats dans
NaCl
Le comportement électrochimique des électrodes
élaborées lorsqu'elles sont en contact d'une solution de NaCl
0,5M et dans de l'eau industrielle à été observé
par différentes techniques électrochimiques, elles mettent toutes
en jeu des mesures de potentiel et/ou de courant.
Le matériau à été polarisé
dans un bain de sel NaCl 0,5M. Le balayage à été
effectué à une vitesse de 60mV/mn entre #177; 250 mV/E
Ag/AgCl pour les tracés de Tafel et prés de Ecorr
jusqu'à + 100 mV/ E Ag/AgCl en revenant en E initiale pour
les balayages aller- retour. Notons que pour tous les essais une période
d'attente de 10 mn après immersion correspondant au temps de formation
et stabilité de la double couche électrique.
Les figures III-7 et III-9 présentent
l'évolution de la caractéristique courant - tension (tracé
de Tafel) de l'acier nu et celles des électrodes revêtues à
différentes densité de courant lorsqu'elles sont en contact d'une
solution de NaCl 0,5M.
Les paramètres cinétiques déduits sont
regroupés dans les tableaux III-1 et III-2
50
Chapitre III
-900 -800 -700 -600 -500 -400 -300 -200 -100
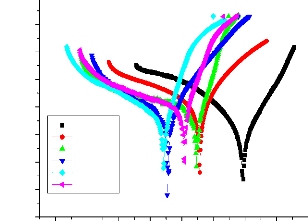
Log I (A/cm2)
- 2,1
- 2,8
- 3,5
- 4,2
- 4,9
- 5,6
- 6,3
- 7,0
substrat nu 0,25mA/cm2 0,5mA/cm2 1
mA/cm2 1,5mA/cm2 3 mA/cm2
E ( mV/Ag/AgCl)
Figure III.7 : Courbes de polarisation enregistrées sur
acier nu et acier revêtu
à partir de Ce(NO3)3.6H2O 0,01M dans
NaCl 0,5 M
-700 -600 -500 -400 -300 -200 -100 0 100 200
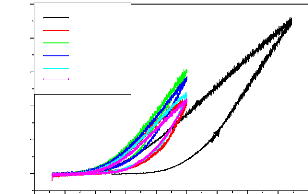
I ( mA/Cm2)
40
50
30
20
10
0
substrat nu 0,25 mA/cm2 0,5 mA/cm2 1
mA/cm2 1,5 mA/cm2 3 mA/cm2
E ( mV/Ag/AgCl)
Figure III.8 : Voltammogrammes enregistrés sur acier nu
et acier revêtu
à partir de Ce(NO3)3.6H2O 0,01 M dans NaCl 0,5
M
51
Chapitre III
-900 -800 -700 -600 -500 -400 -300 -200 -100
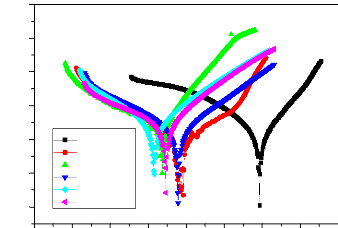
logI(A/cm2)
-1
-2
-3
-4
-5
-6
-7
substrat nu 0,25mA/cm2 0,5mA/cm2 1mA/cm2
1,5mA/cm2 3mA/cm2
E(mV/Ag/AgCl)
Figure III.9 : Courbes de polarisation enregistrées sur
acier nu et acier revêtu
à partir de Ce(NO3)36H2O 0,1M dans
NaCl 0,5 M
I(mA/cm2)
40
50
30
20
10
0
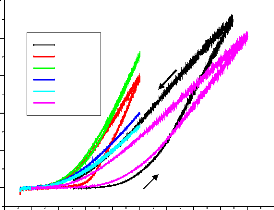
substrat nu 0,25mA/cm2 0,5mA/cm2
1mA/cm2 1,5mA/cm2 3mA/cm2
-700 -600 -500 -400 -300 -200 -100 0 100 200
300
E(mV/Ag/AgCl)
Figure III.10 : Voltammogrammes enregistrés sur acier nu
et acier revêtu
à partir de Ce(NO3)3.6H2O 0,1 M dans NaCl 0,5
M
52
Chapitre III
Tableau III-1: Paramètres
physico-chimiques déduits des courbes de polarisations obtenues sur
acier nu et revêtu à partir de nitrate de cérium 0,01M dans
NaCl 0,5M.
|
Echantillon
|
Paramètres physico-chimique
|
|
Ecorr
mV
|
Icorr
uA/cm2
|
ba
mV
|
bc
mV
|
Rp
&.cm2
|
C
mA.V
|
|
Acier nu
|
-307
|
86
|
76,1
|
93,5
|
212
|
29,38
|
|
Acier revêtu à
0.25mA/cm2
|
-443
|
120
|
93,8
|
216,4
|
237
|
6,82
|
|
Acier revêtu à
0,5mA/cm2
|
-454
|
68
|
39,6
|
88,4
|
174
|
10,37
|
|
Acier revêtu à
1mA/cm2
|
-547
|
97
|
87,8
|
208,5
|
277
|
9,32
|
|
Acier revêtu à
1,5mA/cm2
|
-557
|
124
|
44,4
|
151,5
|
120
|
7,21
|
|
Acier revêtu à
3mA/cm2
|
-492
|
171
|
50,3
|
292,6
|
109
|
7,76
|
Tableau III-2 : Paramètres
physico-chimiques déduits des courbes de polarisations obtenues sur
acier revêtu à partir de nitrate de cérium 0,1M dans NaCl
0,5M.
|
Echantillon
|
Paramètres physico-chimique
|
|
Ecorr
mV
|
Icorr
uA/cm2
|
ba
mV
|
bc
mV
|
Rp
&.cm2
|
C
mA.V
|
|
Acier revêtu à
0.25mA/cm2
|
-513
|
84
|
100,6
|
79
|
227
|
9,42
|
|
Acier revêtu à
0,5mA/cm2
|
-563
|
136
|
66,5
|
186,8
|
157
|
12,63
|
|
Acier revêtu à
1mA/cm2
|
-521
|
133
|
108,6
|
124,5
|
190
|
7,85
|
|
Acier revêtu à
1,5mA/cm2
|
-584
|
225
|
122,1
|
138
|
125
|
6,53
|
|
Acier revêtu à
3mA/cm2
|
-553
|
218
|
93,2
|
166,3
|
119
|
39,19
|
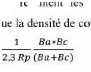
Les tableaux III-1 et III-2 résument les paramètres
cinétiques extraits des courbes de
polarisation, il est à noté qu urant à
été extraite à l'aide de l'équation de Stern
and Geary [10] I corr = III-07
faisant intervenir la résistance de polarisation et les
deux pentes de Tafel. La résistance de polarisation à
été mesurée indépendamment des courbes de
polarisation en mode Tafel.
53
Chapitre III
Divers travaux réalisés pour étudier le
mécanisme de corrosion de l'acier en milieux neutres aérés
[11, 12] ont montré que le processus de corrosion est
contrôlé par la réduction de l'oxygène. Cette
réaction est sous contrôle diffusionnel ou mixte selon le temps
d'immersion. Ainsi, le transport de l'oxygène a lieu dans la phase
liquide et/ou à travers une couche poreuse de produits de corrosion [13,
14].
L'analyse de la branche cathodique de la courbe de
polarisation, montre un domaine relatif à un palier (pseudo palier),
provenant de la réduction de l'oxygène dissous suivant la
réaction:
-
2H2O + O2 + 4é 4OH III-08
La portion anodique, qui se caractérise par une
augmentation monotone de la densité de courant, correspond au passage
des cations ferreux en solution. Dans ces conditions, les produits de corrosion
libérés lors de la dissolution anodique ne forment pas un film
protecteur sur la surface du métal, cette dissolution est due à
la corrosion qui en résulte à cause de l'agressivité du
milieu qui contient une grande proportion en chlorure.
En ce qui concerne les échantillons revêtus,
plusieurs auteurs [3] ont montré que les dépôts à
base d'oxydes de cérium constituent une protection cathodique de la
surface des substrats. Cependant, cette protection dépend
essentiellement de plusieurs facteurs liés à la qualité du
revêtement. En effet un revêtement non recouvrant
l'intégralité de la surface peut inverser la situation en mettant
à nu une faible surface (zone anodique) alors que le reste de la surface
constituant la zone cathodique reste couvert. Il est connu que le rapport de la
densité de courant et surface dans le domaine anodique et cathodique est
toujours vérifié. Ce qui induira une augmentation importante des
densités de courant cathodiques.
Ainsi dans le but d'éviter la formation d'un
revêtement non recouvrant toute la surface; les revêtements ont
été élaborés à des densités de
courant cathodique croissantes, l'influence de ce paramètres a
été vérifié par plusieurs auteurs [22].
En revanche l'augmentation de la densité de courant
appliqué peut non seulement induire une augmentation de
l'épaisseur des revêtements mais aussi rendre l'état de
fissuration des dépôts plus important. Ceci rend certainement les
dépôts plus exposés à l'infiltration du milieu
agressif à travers les fissures et les craquelures. Hamlaoui et al ont
montré que l'état de fissuration est non seulement dû
à l'augmentation de l'épaisseur mais aussi aux tensions internes
évacuées lors du séchage des dépôts est qui
sont une conséquence directe de la réaction de réduction
de l'eau (dégagement H2)[3].
54
Chapitre III
En analysant les résultats figurants dans les tableaux
III-1 et III-2 on s'aperçoit que quelle que soit la concentration, on
observe que par rapport à l'acier nu, les potentiels d'abandon des
substrats revêtus tendent vers des potentiels cathodiques ceci confirme
bien l'existence du film protecteur et son pouvoir de protection cathodique,
les valeurs de la pente des droites de Tafel confirme bien cette conclusion. De
plus, on constate que les potentiels relatifs aux revêtements obtenus
entre 0,5 et 1,5 mA/cm2 sont presque stables (dans les alentours de
500mV/Ag/AgCl). Dépassant cette valeur (à 3mA/cm2), le
potentiel déplace dans le sens inverse (dans le sens anodique). Ceci
nous laisse croire que les revêtements élaborés à
0,5 et 1 mA/cm2 à partir de Ce(NO3)3.6H2O 0,01M et les
revêtements élaborés à 0,25 et 1 mA/cm2
à partir de Ce(NO3)3.6H2O 0,1M sont les plus efficaces.
De plus, on remarque une importante affectation des branches
cathodiques avec une légère modification des branches anodique
figures III.7et III.9 corrélant parfaitement les travaux
précédemment énoncés. Ceci est en parfait accord
avec des travaux précédents [2].
En effet les revêtements à base d'oxydes de
cérium constituent une barrière physique affectant directement le
processus cathodique.
Les figures III.8 et III.10 montrent les voltammogrammes
obtenus sur le substrat nu et les substrats revêtus à
différentes densités de courant, On constate que pour l'ensemble
des substrats revêtus à partir de Ce(NO3)3.6H2O 0,01M y compris le
substrat nu l'existence d'une hystérésis dont sa largeur est
très importante pour le substrat nu. En revanche, les plus faibles
hystérésis, ou inversement pour les couches de compactage sont
relatives aux revêtements obtenus entre 1 et 1,5 mA/cm2. Pour
les revêtements obtenus à partir de Ce(NO3)3.6H2O 0,1M on constate
presque la même chose avec une légère différence. En
effet pour le substrat nu et le substrat revêtu à 3
mA/cm2, l'hystérésis est très importante. De
plus pour le substrat revêtu à 1 mA/cm2 le tracé
aller se superpose au tracé retour traduisant une parfaite
stabilité de la surface.
Les surfaces de l'hystérésis mentionnés
dans les tableaux indique que la couche de compactage formé sur les
substrats revêtus entre 0,5 et 1 mA/cm2 et entre 0,25 et 0,5
mA/cm2 à partir de 0,01 M et 0,1M respectivement sont les
plus représentatives d'un revêtement stable. Cet écart peut
être justifié par le faite de passer de faible à forte
concentration, on dépose plus d'hydroxyde de cérium. De plus sur
les voltammogrammes aucun pic de réduction ou d'oxydation n'a
été observé durant le balayage aller ou de retour
respectivement, ceci nous permis de conclure que le revêtement est
composé d'oxy-hydroxydes stable.
55
Chapitre III

Le com couche de corrosion représente la quantité
du produit adhérant ou de la
surfa u produit de corrosion par unité de courant de
corrosion [15].
C = III-09
L'analyse des deux concentrations précédemment
évoquées conclue que les électrodes revêtues entre
0,5 et 1 mA/cm2 et les électrodes revêtus à 0,25
et 1 mA/cm2 à partir de 0,01 M et 0,1M respectivement
présentent les valeurs de la vitesse de corrosion les plus faibles et
conséquemment elles affichent les revêtements les plus
appropriés.
III.3.4 Diagrammes SIE enregistrés sur des
substrats dans NaCl
Introduction:
Plusieurs études électrochimiques [16-18] ont
montré l'intérêt d'utiliser la spectroscopie
d'impédance électrochimique (SIE). Elle permet de séparer
les phénomènes élémentaires susceptibles de se
développer à l'interface métal/solution en fonction de
leur cinétique respective.
Dans le cas des métaux revêtus, les courants
mesurés sont très faibles, ce qui rend les méthodes
stationnaires classiques peu fiables. La technique de l'impédance
électrochimique permet de surmonter cette difficulté puisqu'elle
est fondée sur la mesure des variations alternatives du potentiel ou du
courant. Cette méthode appliquée au potentiel de corrosion, est
non destructive et permet un contrôle continu de l'état de
dégradation des métaux revêtus. Dans ce travail
l'étude des métaux revêtus immergés dans un milieu
agressif montrent que la détermination de l'impédance
électrochimique dans un large domaine étendu de fréquence
permet de dissocier les composants liés au revêtement
lui-même (pénétration de la solution dans le
revêtement) de celles liées au processus faradique
(phénomène de corrosion du métal, intervenant à
l'interface métal/revêtement).
Le balayage des fréquences a été
effectué des hautes fréquences (HF) (100KHz) vers les basses
fréquences (BF) (10 mHz) avec une perturbation sinusoïdale de 5 mV
d'amplitude autour du potentiel libre. Sur cet intervalle de potentiel le
système est quasi stationnaire [19]. Les diagrammes SIE ont
été obtenus sur des revêtements élaborés
à partir du nitrate de cérium 0,01 et 0,1 M, afin de
préciser et de séparer les différents
phénomènes mettant en jeu la surface métallique, le
revêtement et l'électrolyte. La simulation des diagrammes SIE
à été réalisées à l'aide du logiciel
de simulation le Zview et EC-lab.
Ensuite, l'extraction des différents paramètres
électrochimiques à été réalisée dans
le but de déterminer les paramètres électrochimiques
liés à ces phénomènes en particulier la
résistance de polarisation et la capacité de la double couche
Cdl.
56
Chapitre III
Les figures III.11 et III.12 présentent les diagrammes
SIE en représentation Nyquist du substrat nu et les revêtements
élaborés à une concentration de 0,01M et 0,1 M de nitrate
de cérium à différentes densités de courant.
0 50 100 150 200 250 300 350
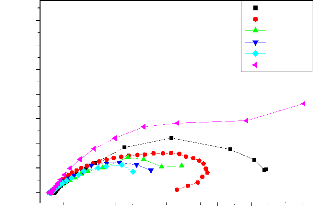
Zi (Ohm.cm2)
350
300
250
200
150
100
50
0
substrat nu 0,25mA/cm2 0,5mA/cm2
1mA/cm2 1,5ma/cm2 3mA/cm2
Zr (Ohm.cm2)
Figure III.11 : Diagrammes d'impédances
enregistrés sur acier nu et acier revêtu à partir
de
Ce(NO3)3.6H2O 0,01M dans NaCl 0,5 M
0 50 100 150 200 250 300
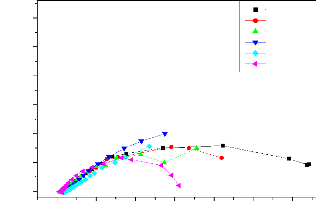
Zi(Ohm.cm2)
250
200
300
150
100
50
0
substrat nu 0,25mA/cm2 0,5mA/cm2
1mA/cm2 1,5mA/cm2 3mA/cm2
Zr(Ohm.cm2)
Figure III.12 : Diagrammes d'impédances
enregistrés sur acier nu et acier
revêtu à partir de
Ce(NO3)3.6H2O 0,1 M dans NaCl 0,5 M
57
Chapitre III
L'aspect des diagrammes montre l'existence d'une boucle
capacitive, une exception à été observée pour
l'électrode revêtue à 0,25mA/cm2 à une
concentration 0,01M du Ce(NO3)3.6H2O, où le diagramme est
constitué d'une boucle capacitive à HF et l'initiation d'une
boucle inductive à BF qui peut être liée à une
variation du taux de recouvrement de la surface induite par des
réactions d'adsorption ou désorption. En revanche, pour les
revêtements obtenus à partir de solutions plus concentrées
0,1M, l'arc inductif disparait.
En ce qui concerne les revêtements
élaborés à partir de 0,01M à une densité de
courant entre 1 et 1,5mA/cm2, les diagrammes STE sont presque
similaires en aspect et en valeur de la résistance de polarisation. Par
ailleurs, le diagramme relatif au revêtement obtenu à
3mA/cm2 est composé d'une boucle capacitive et l'initiation
d'un autre temps de relaxation lié soit à une diffusion ou un
nouveau phénomène capacitif caractérisé soit par
une droite linéaire ou d'une nouvelle boucle capacitive respectivement.
Ceci concorde assez bien avec les résultats des techniques
stationnaires. En effet, nous avons déjà montré que le
meilleur comportement à été observé pour les
revêtements obtenus entre 0,5 et 1 mA/cm2. Ceci est aussi
montré par les résultats des techniques fréquentielles. De
plus la boucle inductive BF observée sur le diagramme relative au
revêtement obtenu à 0,25mA/cm2 est signe d'un
revêtement non recouvrant. Cependant l'initiation de nouveau temps de
relaxation à BF sur le diagramme relatif au dépôt obtenu
à 3mA/cm2 est à priori due à la diffusion de
l'oxygène vers l'interface à travers les fissures du
revêtement.
les travaux effectués antérieurement [3] ont
conclus que les fortes densités de courant forment des
dépôts en deux périodes dont la première est
attribuée à la formation des grains de tailles fines, recouvrant
toute la surface, la deuxième est liée à la superposition
de nouvelles couches engendrant ainsi l'augmentation de l'épaisseur qui
rend les dépôts denses et fissurés, l'augmentation des
fissures est liée au processus de dégagement d'H2, entrainant un
accès plus rapide des ions chlorures à la surface de
l'électrode. Par ailleurs, à de faibles densités de
courant, la formation de gros cristaux retardent le recouvrement total de la
surface, il est à noté qu'a des faibles densités de
courant et notamment à 0,25mA/cm2, la surface de
l'électrode contient de nombreux produits de corrosion,
l'épaisseur du dépôt obtenu à forte densité
de courant les rend invisibles alors qu'ils peuvent exister.
Dans le mécanisme électrochimique les
réactions se déroulant dans un bain de nitrate de cérium
où la production des ions (OH- )favorise la formation d'un
précipité d'hydroxyde de cérium sous forme Ce(OH)3, ainsi
la formation de l'oxyde de cérium se fait par l'intermédiaire de
l'oxydation de Ce(OH)3 de ce fait les revêtements élaborés
à différentes densités de courant sont composés
d'une structure du type bicouches, couche externe d'oxyde
58
Chapitre III
de cérium et une couche interne d'hydroxyde de
cérium , cette apparente différence peut être une
conséquence de la dissolution progressive et
l'inhomogénéité des couches lors de l'immersion dans les
milieux agressifs.
En augmentant la concentration en précurseur 0,1M, les
diagrammes SIE relatifs aux revêtements obtenus à une
densité de courant comprise entre 0,25 et 1 mA/cm2 sont
presque identiques avec un comportement plus meilleur pour ceux obtenus
à 1mA/cm2. Notons que le comportement du revêtement
élaboré à 1,5mA/cm2 est similaire à
celui obtenu à 3mA/cm2 dans 0,01M de Ce(NO3)3.H2O.
Finalement, pour le revêtement obtenu à
3mA/cm2, les très faibles valeurs des résistances de
polarisation indique certainement un revêtement très
fissurés ou peut être dû au détachement au cours de
son immersion dans NaCl 0,5M.
Une cellule électrochimique peut être parfois
représentée par un modèle construit à partir de
dipôles purement électroniques. Dans ce cas, l'interface d'une
électrode fonctionne comme un circuit électrique avec des
résistances, des capacitances des inductances. On peut donc essayer de
trouver un circuit équivalent au système électrochimique
étudié.
Le circuit généralement admis pour les
revêtements électrochimiquement actifs est de type :
Re CPEc CPEdl

Rc
Rtc
Figure III. 13 : Circuit modèle d'un substrat revêtu
par un revêtement actif
- CPEc : capacité de la double couche relative au
revêtement.
Re Fixed(X) 0
- Rc : résistance du revêtement.
CPEcP
- Rél : résistance de l'électrolyte.
Rc
- CPEdl : capacité de la double couche .
CPEdl-P Fi
- Rtc : résistance de transfert de charge.
Rtc Fixe
Sachant d'une part que des paramètres différents
permettent un bon ajustement avec les
Daa File:
diagrammes d'impédance et que d'autre part le coefficient
d'aplatissement « n » est
Cc Me e:
F
difficilement accessible, il faudra interpréter ces
paramètres avec une intuition physique en
Mximum Iteations: 100
accord avec les observations optiques. Il faudra être
d'autant plus prudent que d'autres cas,
ptn on:
T f Fittg: Cl
conduisant à des diagrammesType of
Weighting:présentant un seul arc decercleCalc-Modulus peut aussi
être envisagé.
Dans tous les cas on détermine les paramètres
à l'aide du logiciel, en utilisant les valeurs expérimentales et
en postulant le circuit électrique équivalent. Les calculs en
fait, sont
59
Chapitre III
effectués non pas avec une capacité pure
d'impédance mais avec un élément à phase constante
d'impédance, les éléments à phase constante sont
généralement introduits dans les circuits équivalents
lorsque les diagrammes de Nyquist sont excentrés. D'un point de vu
physique, ces distorsions sont en relation avec diverses
inhomogénéités de la surface.
Dans notre travail et à la base de l'allure des spectres
obtenus on a :
Le circuit équivalent lié au substrat nu et aux
électrodes revêtues est de type :
Re CPEc

Rc CPEdl
Rct

Figure III.15 : Circuit équivalent d'une
électrode nue et revêtue
Concernant le revêtemnt élaboré à
0,25mA/cm2 à partir de solution 0.01M, le circuit
Re Fixed(X) 0 N/A
équivalent donnant la même réponse peut
être représenté comme suit :
CPEcP Fixed(X) 1
Re CPEc
|
Rc CPEdl
Rct R muL1
|
|
Figure III-16 : Circuit équivalent de l~électrode
revêtue à
Type of Fitting Complex
tig
ment
0,25mA/cm2à partir de Ce(NO3)3.6H2O 0,01M.
Freedom Value Eror
|
L'ajustement avec les circuits à donné ls
paramètres des tableaux ci-dessous
CPEc-P Fxed(X) 1 N/A
60
Chapitre III
Tableau III- 3 : Valeurs des
paramètres déduits des diagrammes STE obtenus sur substrat nu et
revêtus à partir de Ce(NO3)3.6H2O 0,01M.
|
Echantillon
|
Paramètres physico-chimiques
|
|
Re
((c))
|
CPEdl
(F.s1/n)
|
n1
|
Rct
((c))
|
CPEc
(F.s1/n)
|
n2
|
Rc
((c))
|
L
(H)
|
|
Acier Nu
|
11,38
|
2,1.10-3
|
0,645
|
100,1
|
2,5.10-4
|
0,604
|
214
|
/
|
|
Revêtu à
0,25mA/cm2
|
7,46
|
1,8.10-2
|
0,73
|
45
|
3,02.10-2
|
0,44
|
200
|
295
|
|
Revêtu à 0,5
|
4,17
|
21,75.10-3
|
0,77
|
10
|
8,65.10-3
|
0,5
|
268
|
/
|
|
Revêtu à 1
mA/cm2
|
5,49
|
6,24.10-3
|
0,647
|
4,15
|
1,09.10-2
|
0,641
|
214,7
|
/
|
|
Revêtu à 1,5
mA/cm2
|
5,51
|
1,6.10-2
|
0,483
|
6,33
|
5,03.10-3
|
0,867
|
264,4
|
/
|
|
Revêtu à 3
mA/cm2
|
5,63
|
2,41.10-3
|
0,668
|
7,95
|
1,06.10-2
|
0,61
|
303,7
|
/
|
Tableau III- 4 : Valeurs des
paramètres déduits des diagrammes STE obtenus sur substrat
revêtus à partir de Ce(NO3)3.6H2O 0,1M.
|
Echantillon
|
Paramètres physico-chimiques
|
|
Re
((c))
|
CPEdl
(F.s1/n)
|
n1
|
Rct
((c))
|
CPEc
(F.s1/n)
|
n2
|
Rc
((c))
|
|
Acier revêtu à
0,25mA/cm2
|
3,70
|
1,1.10-2
|
0,58
|
41,18
|
5,97.10-3
|
0,70
|
167,5
|
|
Acier revêtu à
0,5 mA/cm2
|
3,26
|
5,5.10-3
|
0,63
|
3,69
|
1,5.10-2
|
0,58
|
185,7
|
|
Acier revêtu à
1mA/cm2
|
6,73
|
1,3.10-2
|
0,57
|
5,31
|
2,4.10-2
|
0,65
|
335,5
|
|
Acier revêtu à
1,5mA/cm2
|
8,13
|
5,1.10-3
|
0,61
|
2,55
|
3,83.10-2
|
0,52
|
354,8
|
|
Acier revêtu à
3mA/cm2
|
3,63
|
3,7.10-3
|
0,67
|
2,22
|
3,66.10-3
|
0,75
|
118,5
|
D'où, Re présente la résistance
d'électrolyte, Rct correspond à la résistance de transfert
de charge, CPdl à la capacité de la double couche, n le
coefficient de linéarités, L : l'inductance.
61
Chapitre III
III.4. Comportement électrochimique des
substrats dans l'eau industrielle.
III. 4. 1. Milieu d'essai
L'eau industrielle utilisée dans cette étude a
été fournie par l'entreprise FERTIAL. La dite eau est d'origine
maritime (eau de mer) et parmi les diverses méthodes de traitement elle
a subit un traitement par déminéralisation préalablement a
son utilisation, ainsi avant la déminéralisation de l'eau de mer,
celle-ci doit être d'abord distillée à l'effet de
réduire sa teneur en salinité, ensuite elle est canalisée
dans l'installation de déminéralisation qui fournit une eau
d'appoint aux condensas de retours avant leur passage dans un dégazeur
thermique et le retour aux chaudières après conditionnement.
Le poste de déminéralisation est muni d'un
filtre à charbon actif ayant pour but la filtration et
l'élimination du chlore en excès dans l'eau et les autres
composés à l'effet de ne pas empoisonner les résines
échangeuses d'ions.
Le choix de traitement dépend de la nature des
impuretés et des autres composés présents dans l'eau, dans
le cas de l'entreprise Fertial, le procédé fait appel :
Décarbonatation à la chaux:
La décarbonatation à la chaux est le
procédé de précipitation le plus largement utilisé,
son but est d'éliminer la dureté bicarbonatée liée
au calcium et au magnésium, c'est-à-dire la présence dans
l'eau d'hydrogénocarbonates et carbonates de Ca+2 et de
Mg+2, en ajoutant de la chaux hydratée à l'eau qui
précipite CaCO3, Mg(OH)2.
Les réactions chimiques de base sont les suivantes :
Ca(OH)2 + Ca(HCO3)2 2CaCO3 + 2 H2O III-10
Ca(OH)2 + Mg(HCO3)2 MgCO3 + CaCO3 + 2 H2O III-11
Adoucissement:
Par échange d'ion, on utilisant deux types
d'échangeurs d'ions l'un est cationique capable d'échanger
Ca+2, Mg+2, K+, l'autre est anionique capable
d'échanger Cl-, SO4 -2, NO3 -.
Pour augmenter le pH une double injections de morpholine ensuite
le bisulfite de sodium est effectuée pour réduire la teneur en
O2.
L'eau traitée à la sortie des chaînes de
déminéralisation, présente les caractéristiques
repris dans le tableau ci-dessous :
62
Chapitre III
Tableau III.5 : composition de l'eau
industrielle:
|
prélèvement
|
pH
|
TH °F
|
TA
°F
|
TAC °F
|
Cl _
(mg/l)
|
SiO2
(mg/l)
|
P2O5
(mg/l)
|
NaOH (mg/l)
|
Mg+2
(mg/l)
|
Na+
(mg/l)
|
SO4-2
(mg/l)
|
|
02/01/2012
|
10,2
|
0
|
3,2
|
6,4
|
0.90
|
8.10
|
22.01
|
1.15
|
1,18
|
1,42
|
0,30
|
|
09/01/2012
|
10,4
|
0
|
2,6
|
6
|
0,80
|
7,15
|
22,91
|
1,24
|
1,20
|
1,44
|
0,31
|
|
16/01/2012
|
10,3
|
0
|
2,7
|
5,7
|
0,70
|
8,10
|
27,27
|
1,92
|
1,19
|
1,42
|
0,30
|
|
22/01/2012
|
10,2
|
0
|
2,25
|
5,2
|
0,90
|
8,00
|
26,72
|
1,65
|
1,22
|
1,43
|
0,29
|
|
30/01/2012
|
10,3
|
0
|
2,7
|
5,5
|
0,80
|
6,50
|
23,92
|
1,10
|
1,17
|
1,42
|
0,30
|
Les résultats d'analyse affichés dans le tableau
ci-dessus démontrent une stabilité du pH à
caractère basique, tandis que le titre hydrométrique montre que
la qualité de l'eau est de caractère doux dont la plage de
valeurs du titre hydrotimétrique pour une eau considéré
douce varie entre 0 à 5 °F .
Pour définir la tendance agressive ou entartrante de
l'eau, on utilise la relation de l'indice de RYZNAR dont le calcul indique un
indice variant entre 6.4 < IR < 6.65 cette valeur rend le
caractère de l'eau moyennement corrosif. Ce résultat est
manifestement consécutif à l'imperfection de la procédure
de traitement de l'eau lors de l'élimination des carbonates au cours de
sa déminéralisation.
63
Chapitre III
III. 4. 2. Evolution du potentiel à circuit ouvert
du substrat nu
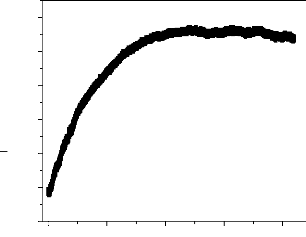
0 50 100 150 200
E ( V/Ag/AgCl)
-0,18
-0,20
-0,22
-0,24
-0,26
-0,28
-0,30
Temps ( min)
Figure III.17 : Suivi du potentiel à circuit ouvert de
l'acier nu dans l'eau
industrielle.
L'évolution du potentiel de corrosion libre en fonction
du temps de l'acier nu dans l'eau industrielle est représentée
sur la figure III-17. On constate que l'évolution du potentiel à
circuit ouvert se fait vers des valeurs positives et par conséquent
l'acier est de moins en moins actif.
Les études réalisées sur la corrosion de
l'acier en milieu neutre montrent des comportements différents selon le
temps d'immersion du métal dans la solution agressive [20,21]. Au
début de l'immersion, toute la surface du métal est active, ce
qui signifie que toute la surface du métal est disponible aux
réactions cathodiques et anodiques intervenant lors de la corrosion des
aciers. En revanche, après quelques heures d'immersion une partie de la
surface est bloquée par les produits de corrosion insolubles. En effet
l'analyse expérimentale démontre que les ions (SO4 -2,
Cl- ,CO3 -2, Ca+2, Mg+2,
K+, Fe+2) présents dans le milieu eau
industrielle, et en particulier la présence des sels carbonatés
constituent la source de la présence du dioxyde de carbone (CO2), ce
dernier intervient selon le mécanisme de réaction suivant :
2 HCO3 - CO2 + CO32- + H2O III-12
CO32- + H2O CO2 + OH - III-13
64
Chapitre III
Ainsi plusieurs auteurs proposentle mécanisme de
réaction du fer en présence de carbonates, dont celui de Castro
et al. [23] proposent la formation du complexe FeHCO3 + selon:
Fe+2 + HCO3 - FeHCO3 + III-14
Alors que Davies
et Burstein [24] proposent la formation du complexe Fe(CO3)2 -2
selon:
Fe+2 + 2 HCO3 - Fe(CO3)2 -2 +
2H+ III-15
Ces derniers justifient leur choix en étudiant
la solubilité de la sidérite avec la concentration en carbonate
à pH fixé. Dans ces conditions, ils montrent que la concentration
en ions ferreux est proportionnelle à la concentration de bicarbonate et
en accord avec l'équilibre suivant :
FeCO3 + HCO3 - Fe(CO3)2 -2 + H+
III-16
III. 4. 3. Evolution du potentiel à circuit ouvert
du substrat revêtu
La figure III-18 présente l'évolution du
potentiel en fonction du temps pour les échantillons revêtus
à différentes densités de courant. Le potentiel à
circuit ouvert à été suivi pendant 03 heures d'immersion.
L'allure des courbes nous a permis de constater l'existence de trois formes
différentes. En effet, pour le revêtement élaboré
à 0,25mA/cm2, on observe une chute continue du potentiel vers
des valeurs cathodiques puis une stabilité du potentiel.
0 20 40 60 80 100 120 140 160 180
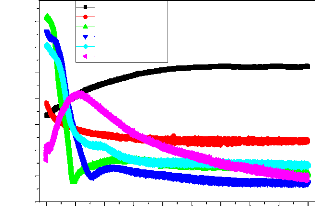
E ( V/Ag/AgCl)
-0,10
-0,15
-0,20
-0,25
-0,30
-0,35
-0,40
-0,45
substrat nu
revêtu à 0,25mA/cm2 revêtu à
0,5mA/cm2 revêtu à 1mA/cm2 revêtu
à 1,5mA/cm2 revêtu à 3mA/cm2
Temps(mn)
Figure III.18 : Suivi du potentiel à circuit ouvert de
l'acier revêtu à partir de
Ce(NO3)3.6H2O 0,1M dans une eau
industrielle
65
Chapitre III
Alors que pour les revêtements élaborés
à 0,5-1-1,5 mA/cm2 les allures des courbes suivent la
même tendance dans le temps, une chute importante du potentiel vers des
valeurs cathodiques puis une léger augmentation du potentiel vers des
valeurs anodiques suivi d'une stabilité du potentiel. L'évolution
du potentiel dans ce cas traduit que les revêtements formés
à ces densités de courant présentent les meilleurs
comportements à la corrosion.
Tandis que la courbe relative au dépôt obtenu
à 3mA/cm2 est caractérisée par une augmentation
du potentiel vers des valeurs anodiques puis d'une diminution jusqu'à
une stabilisation. Ceci confirme que les revêtements obtenus à
3mA/cm2 sont fissurés et qu'après immersion
(dissolution) la formation des produits de corrosion remplit le vide.
L'étude en milieu eau industrielle sera faite suivant
les mêmes conditions expérimentales que celles qui ont
été utilisées dans le milieu NaCl 0,5M, ceci nous
permettra d'étudier l'influence du milieu sur le comportement
électrochimique des revêtements.
III.4. 4. Courbes de polarisation des substrats dans l'eau
industrielle
Les courbes de polarisation pour les deux concentrations 0,01
et 0,1 M de l'acier nu et revêtu, sont reportées sur les figures
III.19 et III.21, de plus, les paramètres électrochimiques tels
que le potentiel de corrosion (Ecorr) et la densité de courant de
corrosion (Icorr), extraits à partir de ces courbes sont
présentés dans les tableaux III-6 et III-7.
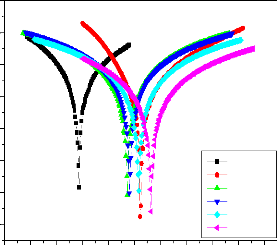
substrat nu 0,25mA/cm2 0,5mA/cm2
1mA/cm2 1,5mA/cm2 3mA/cm2
|
LogI(A/cm2)
|
1,0 0,5 0,0 -0,5 -1,0 -1,5 -2,0 -2,5
|
-400 -350 -300 -250 -200 -150 -100 -50 0 50 100
E ( mV/Ag/AgCl)
Figure III.19 : Courbes de polarisation enregistrées sur
acier nu et acier revêtu à
partir de Ce(NO3)3.6H2O 0,01M dans
l'eau industrielle
66
Chapitre III
-400 -300 -200 -100 0 100 200
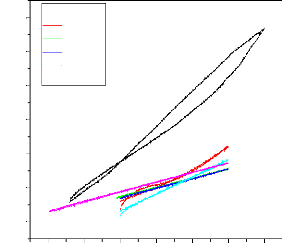
I(uA/cm2)
25
20
15
10
-5
5
0
substrat nu 0,25mA/cm2 0,5mA/cm2
1mA/cm2 1,5mA/cm2 3mA/cm2
E(mV/Ag/AgCl)
Figure III.20 : Voltammogrammes enregistrés sur acier nu
et acier revêtu à partir
de Ce (NO3)3.6H2O 0,1 M dans l'eau
industrielle
Log I (A/cm2)
-0,5
-1,0
-1,5
-2,0
-2,5
-3,0
0,5
0,0
1,0
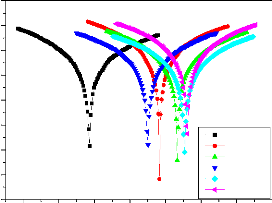
substrat nu 0,25mA/cm2 0,5mA/cm2
1mA/cm2 1,5mA/cm2 3mA/cm2
-350 -300 -250 -200 -150 -100 -50 0
E (mV/Ag/AgCl)
Figure III.21: Courbes de polarisation enregistrées sur
acier nu et acier revêtu à partir de
Ce(NO3)3.6H2O 0,1M dans
l'eau industrielle
67
Chapitre III
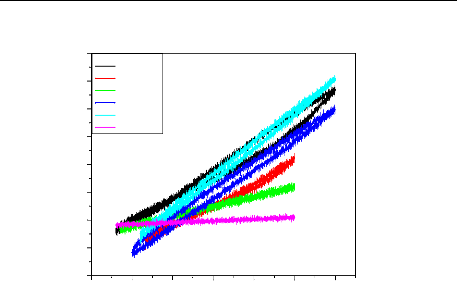
-400 -300 -200 -100 0 100 200
30
25
20
15
10
5
0
-5
-10
I (uA/cm2)
substrat nu 0,25 mA/cm2 0,5 mA/cm2 1
mA/cm2 1,5 mA/cm2 3 mA/cm2
E (mV/Ag/AgCl)
Figure III.22 : Voltammogrammes enregistrés sur acier nu
et acier revêtu à partir de
Ce (NO3)3.6H2O 0,1 M dans l'eau
industrielle
Tableau III -6 : Paramètres
cinétiques déduits des courbes de polarisation
enregistrées dans l'eau industrielle sur acier nu et acier revêtu
d'oxyde de cérium à partir de Ce (NO3)3.6H2O 0,01
M.
|
Echantillon
|
Paramètres physico-chimiques
|
|
Ecorr
mV
|
Icorr
uA/cm2
|
ba
mV
|
bc
mV
|
Rp
&.cm2
|
C
mA.V
|
|
Acier nu
|
-253
|
1,52
|
153,5
|
131,9
|
20195
|
0,014
|
|
Acier revêtu à 0.25mA/cm2
|
-139
|
0,57
|
157,5
|
91,2
|
43652
|
1,33.10-3
|
|
Acier revêtu à 0,5mA/cm2
|
-164
|
0,48
|
185,4
|
184,5
|
83372
|
0,56.10-3
|
|
Acier revêtu à 1mA/cm2
|
160
|
0,38
|
148,7
|
147,9
|
84908
|
0,53.10-3
|
|
Acier revêtu à 1,5mA/cm2
|
-141
|
0,25
|
137,6
|
136,4
|
120754
|
0,27.10-3
|
|
Acier revêtu à 3mA/cm2
|
-118
|
0,22
|
148,4
|
145,4
|
146857
|
0,19.10-3
|
68
Chapitre III
Tableau III -7 : Paramètres
cinétiques déduits des courbes de polarisation
enregistrées dans l'eau industrielle sur acier revêtu d'oxyde de
cérium à partir de Ce (NO3)3.6H2O 0,1 M.
|
Echantillon
|
Paramètres physico-chimiques
|
|
Ecorr
mV
|
Icorr
uA/cm2
|
ba
mV
|
bc
mV
|
Rp
&.cm2
|
C
mA.V
|
|
Acier revêtu à 0.25mA/cm2
|
-157
|
1,87
|
155,9
|
139,8
|
17055
|
3,36.10-3
|
|
Acier revêtu à 0,5mA/cm2
|
-131
|
0,85
|
152,3
|
149,7
|
38470
|
2,83.10-3
|
|
Acier revêtu à 1mA/cm2
|
-173
|
0,82
|
140,2
|
147,9
|
38078
|
0,010
|
|
Acier revêtu à 1,5mA/cm2
|
-121
|
0,58
|
147,1
|
148,4
|
55371
|
0,015
|
|
Acier revêtu à 3mA/cm2
|
-118
|
0,99
|
137,6
|
137,8
|
30140
|
2,45.10-3
|
D'après les résultats expérimentaux
obtenus on note que la composition du milieu influe sur le potentiel et la
densité de courant de corrosion. En effet, on remarque d'une part, le
déplacement des potentiels vers des valeurs anodiques par rapport au
potentiel de l'acier nu, d'autre part les valeurs du courant de corrosion sont
plus faibles comparées à celles obtenues dans le milieu NaCl
0,5M, aussi on observe un changement où on note que pour les
revêtements obtenus à 1,5 - 3mA/cm2 et 1 -
1,5mA/cm2 à partir de Ce(NO3)3.6H2O 0,01M et 0,1M
respectivement sont les plus représentative d'un revêtement
stable. Il apparait que le rôle des dépôts, en
présence de CO2 fait diminuer les vitesses de corrosion, cela est peut
être expliquée par la croissance d'un dépôt
protecteur à la surface et aux caractéristiques variables du
revêtement (morphologie, structure, homogénéité,
épaisseur).
Diverses études [25] rapportent que la consolidation
d'une barrière constituée d'une couche d'oxyde et d'hydroxyde de
cérium, génère l'extension de l'oxyde sur la surface et la
précipitation de sels aux endroits présentant des points de
faiblesse de l'oxyde ; ces sels, étant des produits de corrosion,
renforcent le revêtement engendrant une adhérence
améliorée et conséquemment engager le processus de
ralentissement de la corrosion. Les films ainsi constitués semblent
présenter de bonnes barrières de protection anticorrosion.
Par ailleurs, des essais de polarisation cyclique ont
été réalisés dont les figures III-20 et III-22
montrent les voltammogrammes obtenus sur le substrat nu et les substrats
revêtus à différentes densités de courant. En
générale on constate que l'allure des voltammogrammes
présente des hystérésis relativement étroites par
rapport à celles observées dans le milieu NaCl 0,5M. De plus, on
constate une absence totale des phénomènes
électrochimiques (pic d'oxydation et/ou pic de réduction) ou
d'activité de surface. Ce qui indique peut être que les
revêtements sont
69
Chapitre III
constitués d'oxy-hydroxydes stables ne montrant aucune
activité chimique dans l'eau industrielle.
Quelque soit la concentration du bain, l'intensité de
courant de corrosion des électrodes revêtues par rapport à
l'électrode nue est relativement faible, à l'exception au
revêtement élaboré à 0,25mA/cm2 à
partir de Ce(NO3)3.6H2O 0,1M où on note une élévation de
Icorr.
La mesure du coefficient du compactage permet, entre autre,
d'estimer le compactage du film protecteur. Les valeurs des coefficients de
compactage mesurés sont plus faible comparés aux coefficients
mesurés au milieu NaCl 0,5M.Rappelent que la couche de compactage
indique la quantité des produits de corrosion durant le balayage.
L'examen des résultats expérimentaux
démontrent que, dans l'eau industrielle, l'électrode qu'elle soit
nue ou revêtue est moins exposée aux ions et en particulier aux
sels carbonatés qui constituent la source de la présence de
CO2.et par conséquent une protection
améliorée.
III.4. 5. Diagrammes SIE enregistrés sur des
substrats dans l'eau industrielle
Les figures III.23 et III.24 présentent les diagrammes
d'impédance électrochimique (SIE) en représentation
Nyquist de l'électrode nue et les électrodes revêtues
à une concentration de 0,01M et 0,1 M de nitrate de cérium et
à différentes densités de courant.
Zi ( Kohm.cm2)
140
120
100
40
80
60
20
0
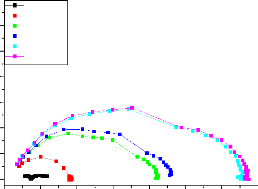
substrat nu 0,25mA/cm2 0,5mA/cm2
1mA/cm2 1,5mA/cm2 3mA/cm2
0 20 40 60 80 100 120 140
Zr ( Kohm.cm2)
Figure III.23 : Diagrammes d'impédances
enregistrés sur acier nu et acier revêtu
d'oxyde de
cérium à partir de Ce(NO3)3.6H2O 0,01 M dans l'eau
industrielle
)
Re CPEc
CPEdl

Rc
Rct
Chapitre III
0 20 40 60 80 100 120
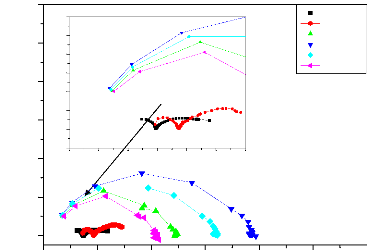
Zi(Kohm.cm2)
120
100
40
80
60
20
0
Zi(Kohm.cm2)
30
25
20
15
10
-5
5
0
0 5
10 15 20 25
Zr(Kohm.cm2)
30
substrat nu 0,25mA/cm2 0,5mA/cm2
1mA/cm2 1,5mA/cm2 3mA/cm2
Zr(Kohm.cm2)
Figure III.24 : Diagrammes d'impédances
enregistrés sur acier nu et acier revêtu d'oxyde
de
cérium à partir de Ce(NO3)3.6H2O 0,1 M dans l'eau
industrielle
Le circuit équivalent adéquat donnant la
même réponse en impédance pour les différents
revêtements est schématisé dans la figure III.25.Notant que
pour l'électrode nue et revêtue d'un dépôt
élaboré à partir de Ce(NO3)3.6H2O 0,1M à une
densité de 0,25mA/cm2 semble que le circuit le plus
approprié est donné dans la figure III.26.
Re CPEc

Rc CPEdl
Rct
70
Figure III.25 : Circuit équivalent d'une
électrode revêtu
|
Figure III.26 : Circuit équivalent d'une
électrode nue et une électrode
CPEdl-P Fixed(X) 1 N/A N/A
revêtue à 0,25mAcm2 à partir de
Ce(NO3)3.6H2O 0,1 M
FixeX) /A
ent Freedom Value Error
|
71
Chapitre III
L'ajustement avec les circuits proposés à
donné les paramètres dans les tableaux ci-dessous:
Tableau III- 8 : Valeurs des paramètres
déduits des diagrammes STE obtenus sur substrat nu et revêtus
à partir de Ce(NO3)3.6H2O 0,01M.
|
Echantillon
|
Paramètres physico-chimiques
|
|
Re
((c))
|
CPEdl
(F.s1/n)
|
n1
|
Rct
((c))
|
CPEc
(F.s1/n)
|
n2
|
Rc
((c))
|
|
Acier Nu
|
4070
|
0,163.10-3
|
0,70
|
6794
|
3,601.10-9
|
0,79
|
6469
|
|
Acier revêtu à 0,25 mA/cm2
|
3106
|
0,169.10-9
|
1
|
8020
|
0,264.10-3
|
1
|
22446
|
|
Acier revêtu à 0,5 mA/cm2
|
3775
|
0,181.10-9
|
1
|
8472
|
1,81.10-6
|
0,32
|
46631
|
|
Acier revêtu à 1 mA/cm2
|
3607
|
0,178.10-9
|
1
|
12000
|
1,145.10-6
|
0,26
|
47971
|
|
Acier revêtu à 1,5 mA/cm2
|
5083
|
0,177.10-9
|
1
|
19398
|
0,377.10-6
|
1
|
82642
|
|
Acier revêtu à 3 mA/cm2
|
5014
|
0,172.10-9
|
1
|
21000
|
0,265.10-6
|
1
|
84958
|
Tableau III-9 : Valeurs des
paramètres déduits des diagrammes STE obtenus sur substrat
revêtus à partir de Ce(NO3)3.6H2O 0,1M.
|
Echantillon
|
Paramètres physico-chimiques
|
|
Re
((c))
|
CPEdl
(F.s1/n)
|
n1
|
Rct
((c))
|
CPEc
(F.s1/n)
|
n2
|
Rc
((c))
|
|
Acier revêtu
à 0,25
mA/cm2
|
9064
|
2,798.10-9
|
0,94
|
4194
|
0,234.10-3
|
0,8
|
9621
|
|
Acier revêtu
à 0,5 mA/cm2
|
3561
|
0,188.10-9
|
0,99
|
4054
|
0,404.10-6
|
1
|
29921
|
|
Acier revêtu
à 1 mA/cm2
|
4192
|
0,184.10-9
|
1
|
2665
|
0,174.10-9
|
0,17
|
45742
|
|
Acier revêtu
à 1,5 mA/cm2
|
3840
|
0,179.10-9
|
1
|
2707
|
0,338.10-9
|
0,26
|
37399
|
|
Acier revêtu
à 3 mA/cm2
|
3042
|
0,169.10-9
|
0,99
|
4583
|
0,718.10-6
|
1
|
25680
|
Dans la figure TTT.23 le spectre d'impédance de
l'électrode nue décrive deux boucles capacitives plus au moins
découplée, ce qui nous laisse croire que la première
boucle est peut être attribuée aux transferts de charges à
l'interface et la deuxième boucle est liée au
phénomène d'adsorption des produits de corrosion accumulés
à la surface de l'électrode. En revanche les spectres
d'impédances relatifs aux électrodes revêtues à
partir de Ce(NO3)3.6H2O 0,01 sont composés essentiellement que d'une
boucle capacitive et les tailles
72
Chapitre III
de ces dernières augmentent avec l'augmentation de la
densité de courant. Cela est peut être attribuée à
une augmentation de l'efficacité du film liée à une
augmentation de son épaisseur. En effet, la résistance du
revêtement (Rc) à la pénétration de
l'électrolyte dans les pores augmente avec l'augmentation de la
densité de courant appliquée et par conséquent à
l'épaisseur du revêtement. Ceci est aussi confirmé par la
diminution de la capacité de la double couche du revêtement (CPEc)
elle-même liée directement à l'augmentation de
l'épaisseur.
La diminution de la valeur de la capacité de la double
couche (CPEdl) avec l'augmentation de la résistance de transfert de
charge ( Rtc) est peut être expliquée d'une part par un blocage
croissant de transfert de charge à la surface de l'électrode et
d'autre part une diminution de la surface de contact liée à
l'adsorption du revêtement et l'accumulation des produits de corrosion
à la surface de l'électrode.
Dans la figure III.24 relative aux revêtements obtenus
à partir de Ce(NO3)3.6H2O 0,1 M, on observe un changement dans l'allure
de la boucle de l'électrode revêtue à
0,25mA/cm2, cette allure est comparable à celle obtenue pour
l'électrode nue. Pour les électrodes revêtues à 1,5
et 3 mA/cm2 la taille de la boucle capacitive se trouve diminuer,
Notons que la taille de la boucle est synonyme de la croissance de la
résistance de polarisation.
Les fluctuations observées sur les courbes obtenues,
pourrait être attribuées à un état métastable
de la surface. En effet pour des densités de courant allant de 0,25
-0,5- 1mA/cm2 on note une augmentation de la Rc conjointement
à une diminution de CPEc. Cette évolution est peut être due
à une pénétration de l'électrolyte dans les pores
du dépôt, en effet l'état de fissuration devient important,
on pense que les produits de corrosion colmatent certains pores de
dépôt. A ces mêmes densités de courant on note une
diminution de Rtc et une diminution de CPEdl cette évolution est peut
être attribuée à un blocage croissant de transfert de
charge à la surface de l'électrode et d'autre part une diminution
de la surface de contact liée à l'adsorption du revêtement
et l'accumulation des produits de corrosion à la surface de
l'électrode. Inversement, à une densité de courant de 1,5
et 3 mA/cm2 on observe une diminution de Rtc et le
dépôt formé qui semble afficher une résistance
à l'agressivité du milieu est donc de médiocre nature
morphologique (structure, homogénéité) apte à
assurer son rôle de protection de l'acier.
73
Chapitre III
III.5. Comparaison entre les deux milieux : NaCl 0,5M
et Eau industrielle.
Quelque que soit le milieu (NaCl 0,5M ou eau industrielle),
l'allure des courbes chronopotentiométriques enregistrées au
cours de la formation du dépôt sur acier à partir de la
solution Ce(NO3)3.6H2O à 0,01M et 0,1M, se caractérisent par une
décroissance monotone du potentiel vers des valeurs cathodiques puis
d'un pseudo stabilité qui est la conséquence d'un blocage de
surface, ceci indique la formation d'un processus
d'électrodéposition de l'oxydes de cérium (CeO2).
La stabilisation du potentiel de corrosion, dans le milieu
NaCl 0,5M, est atteinte après 05 heures d'immersion au terme de
laquelle, la courbe affiche une décroissance du potentiel de corrosion
vers des valeurs cathodiques et le potentiel devient de moins en moins noble.
Alors que dans le milieu eau industrielle, la stabilisation du potentiel est
atteinte après 03 heures d'immersion, l'évolution du potentiel
tend vers des valeurs anodiques et l'acier est de moins en moins actif. Cette
différence est attribuée à la nature des produits de
corrosion formés et à l'agressivité du milieu.
Pour les substrats revêtus à partir d'une
solution Ce(NO3)3.6H2 0,01 M et 0,1M, des différences sont à
noter entre les deux milieux. En effet, dans le milieu NaCl 0,5M les
revêtements élaborés entre 0,25 et 1 mA/cm2 sont
de nature à assurer leur rôle de protection de l'acier. Dans l'eau
industrielle, les revêtements formés entre 1 et 3
mA/cm2 présentent les meilleurs comportements à la
corrosion.
Les résultats obtenus à partir des courbes de
polarisation, nous permettent de conclure que dans le milieu NaCl 0,5M et
quelle que soit la concentration, on observe que, par rapport à l'acier
nu, les potentiels d'abandon des substrats revêtus tendent vers des
potentiels cathodiques alors que dans l'eau industrielle le déplacement
du potentiel se fait vers des valeurs anodiques. D'autre part, les valeurs du
courant de corrosion sont plus faibles comparées à celles
obtenues dans le milieu NaCl 0,5M.
Il apparait que la composition du milieu eau industrielle
influe simultanément sur le potentiel et sur la densité de
courant de corrosion; le rôle des dépôts, en présence
de CO2, fait diminuer les vitesses de corrosion. Concernant
l'étude voltamétrique, l'allure des voltammogrammes
présentent des hystérésis relativement étroites par
rapport à celles observées dans le milieu NaCl 0,5M. Le
tracé aller se superpose presque au tracé retour, les valeurs des
coefficients de compactage mesurés sont plus faibles comparés aux
coefficients mesurés en milieu NaCl 0,5M.
74
Chapitre III
Dans les mesures impédancemétriques, il à
été constaté des changements dans les allures des spectres
d'impédances. Les spectres et les valeurs expérimentaux obtenus
dans l'eau industrielle montrent un comportement amélioré par
rapport au milieu NaCl 0,5M. En effet, dans l'eau industrielle, la composition
et les ions présents dans la solution peuvent influer sur la
qualité du dépôt cathodique. Ces films semblent être
de bonnes barrières à la corrosion. On peut donc conclure que
l'efficacité du revêtement, objet de cette étude, assure
efficacement sa fonction de protection améliorée contre la
corrosion en milieu eau industrielle.
75
Chapitre III
REFERENCES BIBLIOGRAPHIQUES
[1] J. B. Bessone, D. R. Salinas, C. E. Mayer, M. Ebert.
[2] W. J. Lorenz. Electrochem. Acta., 37(1992) 2283.
[3] Y. HAMLAOUI, Thèse de doctorat, Elaboration et
caractérisation de revêtements d'oxyde de cérium sur acier
et acier électro zingué. Univ Badji mokhtar-ANNABA(2008)
[4] C. Vargel, corrosion de l'aluminium,Ed., Dunod,(1999).
[5] M. J. Capitan, A. Paul, J. L. Pastol, J. A. Odriozola, Oxid.
Met., 52(1999) 447-450
[6] M. Hirano,E. Kato, J. Am. Ceram, Soc., 79 ( 1996) 777-
780.
[7] B. Normand, N. Pébère, C. Richard, M. Wery.
Prévention et lute contre la corrosion (2004) Presses
Polytéchniques et université Romandes, CH- 1015 Lausanne.
[8] N. Ozer, Sol. Energy Mater. Sol. Cells., 68 ( 2001) 391
[9] C. Rochaix, Electrochem, Thermodynamique et
cinétique, Ed. Nathan Paris 1996.
[10] M. Mekki daouadji. Les techniques de mesures de la vitesse
de corrosion. Séminaire de l'institut algérien du pétrole,
Boumerdes, 20 au 24 mars (2004).
[11] ] Cefracor. Matériaux
métalliques. phénomènes de corrosion, quatrième
partie les différentes formes de corrosion aqueuse. CEFRACOR 2003,
75-86.
[12] C. agrafiotis. A. Tsetsekou, C. J. Stournaras, A. Julbe, L.
Dalmazio, C. Guizard, J. Eur. Ceram. Soc., 22 ( 2002) 15-25.
[13] P. Stefanov, G. Atanasova. D. Stoychev, T. S. Marinova,
Surf. Coat. Technol., 446(2004) 180-181.
[14] I. Zhitomirsky, A. Petric, Ceram. Int., 27 ( 2007)
149-155.
[15] I. Suzuki, Corr. Sci., 25 ( 1985) 1029.
[16] V. S. Agarwala, Proc. Int. Cong. Metallic Corros., 380
(1984).
[17] S. Payan, Comportement à la corrosion galvanique de
matériaux composites à matrice d'alliage d'aluminium
renforcée par des fibres de carbone haut module, Thèse de
doctorat ,Unive de Bordeaux I, (2002). N° (242) 34-48.
[18] H. Mayet, Etude de la corrosion par piqure des alliages
d'alluminium par l'analyse des fluctuations de courant et de potentiel à
l'abandon. Thèse Science et Genie des Matériaux.Grenoble Institut
National Polytechnique de Grenoble(1992). 172.
[19] Stephane Gastaud, Influence de l'état du
revêtement hydrocarboné sur le risque de corrosion des
canalisations enterrées de transport de gaz. Thèse de doctorat,
INSA de Lyon, (2002), N°( 02ISAL009).
[20] D. You, N. Pébère et F. Dabosi ,An
investigation of the corrosion of pure iron by electrochemical techniques and
in situ observations. Corros. Sci., 34(1), 5-15,( 1993).
76
Chapitre III
[21] G. J. Brug, A. L. G. Van Den Eeden, M. Sluyters-Rehbach
et J. H. Sluyters , The analysis of electrode impedances complicated by the
presence of a constant phase element. J. Electroanal. Chem., 176(1-2), (1984).
275-295
[22] L. Bonou. Thèse, Alliages cuivre-nickel
électro déposés Cinétique
d'électro
cristallisation Comportement à la corrosion. Unive
de Ouagadougou(1993).
[23] E.B. Castro, C.R. Valentini, C.A. moina, J.R. Vilche,
A.J. Arvia, "The influence of ionic composition on the electrodissolution and
passivation of iron electrodes in potassium carbonate-bicarbonate solutions in
the 8,4-10,5 pH range at 25°C", Corrosion Science, (1986)781-793.
[24] D.H. Davies, G.T. Burstein, "The effects of bicarbonte
ions on the corrosion and passivation of iron, Corrosion, (1980) 416-422.
[25] C. A. Palacios, J. R. Shadley, Corrosion 47, 122,
(1991).
Conclusion Générale
Ce travail mené dans le cadre de la
préparation du mémoire de magistèr est consacré
à l'étude des couches minces à base de terres rares
destinées à l'élaboration de revêtements protecteurs
contre les milieux corrosifs.
L'étude expérimentale est divisée en deux
parties dont la première est dédiée à
l'élaboration des revêtements électrolytiques à base
d'oxydes de cérium sur acier à bas carbone « A366 »
à partir de solutions de nitrate de cérium de concentrations
0,01M et 0,1M, les densités de courant
appliquées sont de 0,25 - 0,5 - 1 - 1,5 et 3
mA/cm2.
La seconde partie est consacrée à l'étude
comportementale électrochimique des échantillons nus et
revêtus et dans ce but, plusieurs techniques électrochimiques en
modes stationnaire et transitoire ont été utilisées et
parmi celles-ci, nous nous sommes intéressés en particuliers :
- au suivi du potentiel en fonction du temps d'immersion.
- au tracé des courbes de polarisation linéaire
et cyclique.
- et au tracé des diagrammes de spectroscopie
d'impédance électrochimique (SIE).
La caractérisation des revêtements à
été réalisée dans deux milieux en l'occurrence,
NaCl 0,5M et l'eau industrielle.
Sur la base des résultats obtenus par les
différentes techniques électrochimiques, au cour de
l'élaboration des revêtements, l'allure des courbes temporelles
chronopotentiométriques enregistrées permet de constater qu'a
différentes densités de courant celles-ci se caractérisent
par une décroissance monotone et rapide du potentiel vers des valeurs
très cathodiques suivies d'un pseudo stabilité qui se
définie par une faible variation du potentiel, indiquant la formation
d'un film à la surface.
Le suivi du potentiel de corrosion avec le temps d'immersion
de l'acier nu montre que le changement de l'allure de la courbe avec le temps
d'immersion est lié aux réactions d'oxydo-réductions se
déroulant à l'interface, l'évolution du potentiel vers des
valeurs électronégatives caractérise un
phénomène de dissolution spontané, les dépôts
de produits de corrosion formés peuvent jouer un rôle actif de
barrière plus ou moins efficace. L'évolution du potentiel
à circuit ouvert pour les électrodes revêtus à
différentes densités de courants élaborés dans un
bain du Ce(NO3)3.6H2O à 0,01 M et
0,1 M immergés dans la solution NaCl 0,5M nous permet
de conclure que pour les électrodes revêtus à 0,25 et 1,5
mA/cm2, les potentiels enregistrés au début
d'immersion indiquent clairement la formation d'un film protecteur à la
surface de l'acier, la chute du potentiel est liée à une
première interaction du revêtement avec le milieu agressif
indiqué par les faibles valeurs des potentiels enregistrées .En
effet, un palier caractérisé par une stabilité des
potentiels est observé indiquant une parfaite protection de la surface,
d'un autre côté les courbes relatives aux revêtements
obtenus à 0,5 et 1 mA/cm2 indiquent une passivation
instantanée c'est-à-dire un comportement plus meilleur que celui
observé précédemment.
Il est à noter que pour les dépôts obtenus
à partir de Ce(NO3)3.6H2O 0,1M, des modifications ont été
observées. En effet, l'allure des courbes dans la gamme de
densité de courant de 0,5 à 1,5 mA/cm2, le potentiel
évolue vers des valeurs négatives lié à la
destruction de la couche externe préexistante à la surface du
substrat, ensuite le potentiel évolue vers des valeurs positives avant
de se stabiliser. Cette différence est vraisemblablement liée
à la nature, la qualité et les propriétés du
revêtement. Tandis que la courbe relative au dépôt obtenue
à 0,25 mA/cm2, est caractérisée par une chute
de potentiel après une heure d'immersion vers des valeurs
négatives. Le film formé dans ces conditions serait peut
être de nature à protéger l'acier.
Pour le revêtement obtenu à 3mA/cm2 la
chute monotone et continue du potentiel en fonction du temps laisse croire que
l'attaque du revêtement continue de l'être. Ceci est dû soit
à la discontinuité du revêtement ou à sa faible
épaisseur.
En analysant les mesures de la résistance de
polarisation on s'aperçoit que quelle que soit la concentration, on
observe que, par rapport à l'acier nu, les potentiels d'abandon des
substrats revêtus tendent vers des potentiels cathodiques et ceci
confirme bien l'existence du film protecteur et son pouvoir de protection
cathodique. En outre, on constate de plus en plus, que les potentiels relatifs
aux revêtements obtenus entre 0,5 et 1,5 mA/cm2 sont quasiment
stables et en dépassant cette valeur (à 3mA/cm2) le
potentiel se déplace dans le sens inverse et tend à
démontrer que les revêtements élaborés dans ce
domaine de densité de courant sont les plus efficaces.
En plus les voltammogrammes obtenus confirment bien les
résultats déjà obtenus, ils ne montrent aucun pic de
réduction ou d'oxydation ce qui permet de conclure que les
revêtements sont essentiellement composés d'oxydes stable.
Par ailleurs les mesures d'impédance
électrochimique, ont montré que pour les revêtements
élaborés à 0,25mA/cm2 à partir du Ce
(NO3)3.6H2O 0,01M, la boucle inductive BF obtenu indique un revêtement
non recouvrant.
Le diagramme relatif au revêtement obtenu à
3mA/cm2 est composé d'une boucle capacitive et l'initiation
d'un autre temps de relaxation est le résultat soit d'une diffusion ou
d'un nouveau phénomène capacitif.
En augmentant la concentration à 0,1M, les diagrammes
relatifs aux revêtements obtenus entre 0,25 et 1 mA/cm2 sont
presque identiques, avec un meilleur comportement que ceux obtenus à
1mA/cm2. Pour le revêtement obtenu à
3mA/cm2, les très faibles valeurs des résistances de
polarisation indique certainement un revêtement très
fissurés et est potentiellement dû au détachement au cours
de son immersion dans NaCl 0,5M.
Le suivi du potentiel de corrosion avec le temps d'immersion
de l'acier nu dans le milieu eau industrielle démontre que
l'évolution du potentiel se fait vers des valeurs positives et par
conséquent l'acier est de moins en moins actif.
En effet l'analyse expérimentale démontre que
les ions présents dans le milieu eau industrielle, et en particulier la
présence des sels carbonatés, constituent la source de la
présence du dioxyde de carbone (CO2), qui attribue à la formation
des produits de corrosion.
L'évolution du potentiel à circuit ouvert des
électrodes revêtues à différentes densités de
courants élaborés dans un bain de Ce(NO3)3. 6H2O 0,1 M
immergés dans l'eau industrielle permet d'aboutir à la conclusion
suivante. A des densités de courant allant de 0,25 à 1,5
mA/cm2 l'évolution du potentiel vers des valeurs cathodiques
révèle que les revêtements formés à ces
densités de courant présentent les meilleurs comportements
à la corrosion. Tandis que la courbe relative au dépôt
obtenu à 3mA/cm2 et qu'après immersion la formation
des produits de corrosion remplit le vide.
L'examen des résultats expérimentaux des courbes
de polarisation obtenues par l'électrode nue et par les
électrodes revêtues à différentes densités de
courants, pour les deux concentrations 0,01 et 0,1 M du Ce(NO3)3.6H2O montrent
le déplacement du potentiel vers des valeurs anodiques, une diminution
dans les valeurs du courant de
corrosion comparées à celles obtenues dans le
milieu NaCl 0,5M. I' influence des dépôts, en présence de
CO2 fait diminuer les vitesses de corrosion, et conséquemment, il
engendre la croissance d'un dépôt protecteur à la
surface.
Par ailleurs; les voltammogramme ainsi obtenus
présentent des hystérésis relativement étroites, le
tracé aller se superpose presque au tracé retour, aucune
observation d'apparition des phénomènes électrochimiques
(pic d'oxydation et/ou pic de réduction).
Pour les mesures d'impédance, le spectre de
l'électrode nue décrit deux boucles capacitives laissant penser
que la première boucle est peut être attribuée aux
transferts de charges à l'interface et la deuxième boucle est
liée au phénomène d'adsorption des produits de corrosion
accumulés à la surface de l'électrode .Quand aux
électrodes revêtues à partir d'une concentration de 0,01 M
du Ce(NO3)3.6H2O, le spectre affiche une boucle capacitive, les tailles des
boucles augmentant avec l'augmentation de la densité de courant celle-ci
est manifestement attribuée à l'augmentation de
l'efficacité du film et à son épaississement.
A partir de 0,1 M du Ce(NO3)3.6H2O, on observe un changement
dans l'allure de l'électrode revêtue à
0,25mA/cm2 elle est comparable à celle obtenue pour
l'électrode nue.
Pour des densités de courant variant de 0,25 -0,5-
1mA/cm2 on note une augmentation de la Rc conjointement à une
diminution de CPEc ( capacité de la double couche du revêtement).
Cette évolution est peut être due à la
pénétration de l'électrolyte dans les pores du
dépôt. En effet, l'état de fissuration devenant important,
on peut donc penser que les produits de corrosion colmatent certains pores de
dépôt. Dans les mêmes densités de courant, on note
une diminution de Rtc et une diminution de CPEdl cette évolution est
peut être attribuée à un blocage croissant de transfert de
charge à la surface de l'électrode, et d'autre part une
diminution de la surface de contact causée par l'adsorption du
revêtement et à l'accumulation des produits de corrosion à
la surface de l'électrode. Inversement, à une densité de
courant de 1,5 et 3 mA/cm2 on observe une diminution de Rtc et le
dépôt formé qui semble afficher une résistance
à l'agressivité du milieu est donc de médiocre nature
morphologique (structure, homogénéité) apte à
assurer son rôle de protection de l'acier.
Pour l'ensemble des revêtements
caractérisés dans les deux milieux en l'occurrence, NaCl 0,5M et
l'eau industrielle, la résistance semble être se comporter de
manière identique avec cependant, une légère
différence dans les valeurs des résistances de polarisation et
une meilleure efficacité des revêtements dans l'eau industrielle.
On peut donc conclure que les deux revêtements présentent de bons
facteurs de résistance vis-à-vis de la solution
corrosive.


