|
UNIVERSITE DE KINSHASA
FACULTE DE MEDECINE
DEPARTEMENT DE PEDIATRIE
VARIATIONS PHYSIOPATHOLOGIQUES DE LA LEUCOCYTOSE DANS
LA MORBI-MORTALITE CHEZ L'ENFANT DREPANOCYTAIRE
Par Emmanuel Derbo KIZUNDA PULULU
Mémoire présenté en vue de l'obtention du
grade de Spécialiste en Pédiatrie
Promoteur : Prof. Dr. LONGO-MBENZA
Benjamin
Décembre 2006
PRELUDE
Ps 23 :1. L'Eternel est mon
berger : je ne manquerai de rien.
Ps 116 :12. Comment rendrai-je à
l'Eternel tous ses bienfaits envers moi ?
DEDICACE
A Dieu le père Tout Puissant, Maître de l'univers
visible et invisible ;
A ma chère et tendre épouse Pathy TONDUANGU
KIZUNDA, pour tant d'amour, tu es pour moi une vraie compagne de vie.
A mes chers enfants : Benedicte, Emmanuella et David
KIZUNDA, vous êtes une immense source de bonheur pour moi.
A ma petite soeur, Madame Lili NZANZA, à ma belle soeur
Denise TONDUANGU, aux parents, amis et connaissances, à tous les enfants
drépanocytaires ;
Je dédie ce modeste travail.
REMERCIEMENTS
Au terme de notre formation en pédiatrie, nous
voudrions de tout coeur et vivement remercier tous ceux qui ont
contribué de loin ou de près, d'une façon ou d'une autre
à notre formation et à la réalisation de ce travail.
Nos remerciements s'adressent
particulièrement :
- Au Professeur Benjamin LONGO-MBENZA, promoteur du
présent mémoire, vous avez accepté malgré vos
multiples responsabilités de nous prendre en charge et c'est avec
beaucoup de compétence, de rigueur scientifique et une maîtrise
parfaite des statistiques que vous l'avez fait. Nous vous sommes infiniment
reconnaissant.
- Au Dr Réné NGIYULU, co-promoteur du
présent mémoire, votre souci nous de voir émerger ainsi
que vos sages conseils ont beaucoup contribué pour la réalisation
de ce travail.
- Au Professeur Jean-Marie MBUYI MUAMBA, pour avoir fait du
CMASS un sanctuaire scientifique. Merci pour nous avoir permis de parfaire
notre formation. Soyez rassuré de notre gratitude.
- Au Professeur Lucien MBENSA MASABI, Chef de
Département honoraire de Pédiatrie, pour nous avoir ouvert
paternellement et largement les portes du Département, votre culture
scientifique nous a beaucoup marqué.
- Au Professeur Bruno TADY MUYALA, Chef de Département
de Pédiatrie des Cliniques Universitaires de Kinshasa, vous êtes
pour nous un vrai père, soucieux de nous voir acquérir les
connaissances nécessaires à l'exercice de la pédiatrie.
Vous avez utilisé tout votre savoir pour cela , nous gavant
même avec une dose d'humour.
- Au Professeur Emmanuel NKIDIAKA DUMBU, vous êtes le
porte étendard de la pédiatrie sociale. Vous nous avez appris
à nous intéresser à tous les problèmes de l'enfant.
Nous n'oublierons pas votre souci de la pédagogie universitaire.
- Au Professeur Pierre TSHIBASSU MANYANGA, votre souci de
l'excellence, votre passion pour la science et la perfection nous ont
marqué. Vous êtes pour nous un modèle. Nous vous en
remercions.
- Au Professeur Gilbert NGANDU KABEYA, votre rigueur
scientifique, votre sagesse et votre refus de médiocrité ont
été pour nous des points de repère. Soyez rassuré
de notre profonde gratitude.
- Au Professeur Joseph SHIKU DIAYISU, vous avez forgé
notre admiration par votre esprit d'écoute, de patience et votre
sérénité. Vous êtes un exemple de simplicité
dans la connaissance. Veuillez trouver l'expression de notre gratitude.
- Au Professeur GINI EHUNGU, Directeur scientifique honoraire
de l'Institut de Recherche en Sciences de la Santé, c'est à vos
cotés que nous avons ébauché nos premiers pas sur le
chemin de la recherche et grâce à vous, nous avons publié
le premier article de notre carrière. Nous vous sommes très
reconnaissants.
- Nos remerciements s'adressent également aux Dr.
M.KAPEPELA, Dr.P.BUNGA, Dr.A.NKUANDIOLANDU, Dr.M.F.KIABILUA, Dr.C.NSIBU,
Dr.A.MUPWALA, Dr .L.LUKUNI , Dr.T.BISIELELE, Dr.A.KAZADI, Dr.M.ALONI,
Dr.J.SENGA pour votre encadrement et vos conseils combien précieux et
utiles tout le long de notre formation.
- Aux Dr. J.BODI, Dr. L. MAVINGA, Dr. P. BABAKAZO, Dr. S.
IZZIA, Dr. MAGOGA et Dr AKETI vous nous avez vraiment encouragé et nous
n'oublierons pas la famille que nous formons.
- Aux collègues assistants : Dr MASSAMBA, Dr.
LUMBALA,
Dr. KAMATHE, Dr. MATATA, Dr. SESE, Dr. NZAMBIMPUNGU, Dr.
KUNUANUNUA, Dr. EKULU, Dr. KATABUKA, Dr. KASONGO, Dr. NZOKO, Dr. ENGO, Dr.
KONDANI, Dr. MATUMONA et Dr. NSANGU pour les très bons moments
passés ensemble et le climat de famille. C'est un souvenir
présent en nous.
- A tout le personnel paramédical, administratif et
technique de notre Département, nous vous exprimons également
notre profonde gratitude pour le climat de collaboration.
- Aux Mrs MBUNGU, VANGU , MILONGO et l'assistant NGEOKWE,
pour la saisie et le traitement de texte, veuillez trouver les sentiments de
notre reconnaissance.
- A la grande famille AKUBA, voici plusieurs années que
nous marchons ensemble du Collège de MBANSA MBOMA, passant par le
Collège Moderne Scientifique (C.M.S) et l'Université
jusqu'à la vie professionnelle. Vous êtes pour moi un
réconfort inestimable.
- Aux familles NZITA ANDRE et MAKABI MARC, nous ne savons
combien vous remercier pour tout le soutien spirituel, moral, matériel
et financier que nous avons bénéficié de vous, preuve de
votre amour pour nous. Soyez rassuré de notre profonde reconnaissance.
ABREVIATIONS
2-3 DPG : 2-3 Diphosphoglycerate
AVC : Accident Vasculaire Cérébral.
Ca++ : Ion Calcium.
CD : Classe de différenciation.
Elts : Eléments.
CMASS : Centre de Médecine Mixte et d' Anémie
SS.
Epo : Erythropoiètine.
Fe 3+ : ion Fer ferrique.
Géne S : gène Sickle .
GB : Globule Blanc.
GR : Globule Rouge .
Hb : Hémoglobine .
Hb S : Hémoglobine Sickle ( falciformée)
HLA : Human Leucocyte Antigen.
ICAM : Inter Cellular Adhesion Molecule.
IL : Interleukine.
K+ : ion Potassium.
um : micromètre.
mm3 : millimètre cube
Na+ : ion Sodium.
PH : Puissance en Hydrogène.
TNF : Tumor Necrosis Factor.
VCAM : Vascular Cell Adhesion Molecule.
VLA-4 : Very Late activation Antigen 4
PLAN DU TRAVAIL
Ø INTRODUCTION
Ø CHAPITRE 1. GENERALITES
Ø CHAPITRE 2. MATERIEL ET METHODES
Ø CHAPITRE 3. RESULTATS
Ø CHAPITRE 4. DISCUSSION
Ø CONCLUSION
Ø RECOMMANDATIONS
INTRODUCTION
Les crises douloureuses aigues caractérisant la
drépanocytose ont été décrites en 1872 par
Africanus Horton (1). C'est, bien après, en 1910, que James Herrick
décrit les globules rouges falciformés chez un étudiant
noir en sciences dentaires, venant de l'île de la Grenade dans les
caraïbes (2-4).
Depuis la mise en évidence en 1944 par Pauling de
l'hémoglobine S(5), la drépanocytose est la première
maladie moléculaire chez l'homme (6). Et plusieurs travaux lui ont
été consacrés tant au plan fondamental que clinique
(7).
La drépanocytose est une maladie essentiellement de la
race noire (8), mais on la retrouve également autour du bassin de la
Méditerranée, au Nord de la Grèce, au Sud-est de la
Turquie, à l'Ouest de l'Arabie Saoudite, dans les Antilles, en
Amérique du Nord et du Sud ainsi qu'en Europe (9).
On rapporte 300.000 nouveaux-nés drépanocytaires
homozygotes par an dans le monde (1), 250 nouveaux-nés SS par an en
France et 300 naissances SS par an en Jamaïque (9). En Afrique, on estime
entre 120.000-200.000 nouveaux-nés SS par an (3). La
drépanocytose est un problème de santé publique pour les
pays subsahariens (10). La prévalence du trait drépanocytaire est
estimée entre 5 et 40% (3). Au Togo, 16 % de la population portent le
trait S dont 3% des homozygotes (10).
En République Démocratique du Congo, la
fréquence du gène S varie de 4 à 30 % et la
prévalence de la drépanocytose homozygotes est d'environ 2 %
(11).
Il existe un contraste marqué entre le polymorphisme
clinique et le mécanisme physiopathologique fondamental de la
drépanocytose (la polymérisation de l'hémoglobine)
(7,12).
PROBLEMATIQUE
La drépanocytose ne concerne pas seulement l'anomalie
de l'hémoglobine et du globule rouge, mais concerne aussi celle de
l'endothélium vasculaire, de la fonction des globules blancs, de la
coagulation et de la réponse inflammatoire (13-15). La
drépanocytose est une maladie inflammatoire (16-22) et vasculaire (17,
22) .
L'adhérence des globules rouges à
l'endothélium vasculaire a été corrélée
à la sévérité de la maladie (23).
Déjà incriminée dans les maladies
cardiovasculaires (24), l'hyperleucocytose en absence d'infection est
aujourd'hui considérée comme prédicteur de la survenue
d'accident vasculaire cérébral (AVC), de syndrome thoracique aigu
et de la mortalité aux Etats-Unis et en Grande Bretagne (4, 15,25-28).
En République Démocratique du Congo,
l'équipe de Longo MBENZA a rapporté une relation significative
entre le nombre de leucocytes et certaines pathologies chez les
drépanocytaires SS (29,30).
Ainsi, le taux élevé des globules blancs
était un facteur de risque de cholélithiase chez les enfants
drépanocytaires de Kinshasa (29). Une corrélation significative a
été rapportée entre la pression artérielle,
l'hypertension artérielle et le nombre de leucocytes chez le jeune
adulte drépanocytaire de Kinshasa (30).
L'absence de données locales relatives au rôle
des leucocytes sur la sévérité de la morbidité, la
résistance au paludisme et la mortalité liées à la
drépanocytose homozygote justifie l'initiation de la présente
étude.
BUT
L'intérêt de ce travail réside dans
l'utilisation ultérieure de ses données à mieux comprendre
la physiopathologie des globules blancs dans la prise en charge adéquate
des drépanocytaires SS.
OBJECTIF GENERAL
L'objectif général de la présente
étude vise à analyser les variations physiopathologiques des
leucocytes dans l'histoire naturelle de la drépanocytose homozygote.
OBJECTIFS SPECIFIQUES
Pour atteindre cet objectif général, les
objectifs spécifiques suivants ont été
précisés :
- analyser l'augmentation ou la diminution de taux de
leucocytes au regard des caractéristiques biocliniques de la
drépanocytose homozygote ;
- déterminer le seuil des leucocytes définissant
l'hyperleucocytose et la leucopénie chez le drépanocytaire
homozygote en phase critique et stable ;
- vérifier le rôle aggravant et létal de
l'hyperleucocytose, du sepsis et d'autres infections chez le
drépanocytaire homozygote ;
- préciser le sens de la variation des leucocytes
liés à la protection du drépanocytaire homozygote
vis-à-vis du paludisme compliqué et sans atteinte neurologique et
de l'encéphale.
HYPOTHESES
Plusieurs hypothèses ont été
émises dans ce travail relatif à la morbimortalité
liée aux variations des leucocytes chez le drépanocytaire
homozygote.
Il existe un gradient biologique leucocytaire entre les
témoins AA ; les drépanocytaires SS en phase critique et les
drépanocytaires homozygotes en phase post critique et stable. Ainsi, le
taux bas des leucocytes étant chez les témoins AA, le taux
intermédiaire chez les drépanocytaires SS en phase post critique
et stable et le taux le plus élevé chez les
drépanocytaires SS en phase critique.
Le sepsis, altérant l'adhérence des leucocytes
chez les drépanocytaires SS détermine un renforcement de
l'hyperleucocytose moins importante que chez les témoins AA.
La relation entre le paludisme et le taux des leucocytes chez
les drépanocytaires homozygotes est différent de celle
observée chez les témoins AA.
Le risque de décès en cas de
drépanocytose homozygote et d'hyperleucocytose sans considérant
du temps, s'estompe au sein de la fonction de survie.
CHAPITRE 1. GENERALITES
1.1. La
drépanocytose
1.1.1. Définition
La drépanocytose est une maladie
héréditaire de l'hémoglobine à transmission
autosomique récessive due à une mutation ponctuelle et unique du
gène â globine sur le bras court du chromosome 11. La mutation du
6ème cordon de l'exon I (GAG ? GTG) entraîne la
substitution en position 6 de la â globine de l'acide glutamique,
hydrophile, présent dans l'hémoglobine A (normale) par la valine,
hydrophobe, (hémoglobine S de mauvaise qualité) (6,9,19,23,
30-33).
1.1.2. Physiopathologie
Cette mutation d'un seul acide aminé dans la
chaîne â de la globine va entraîner des nombreuses
manifestations cliniques (35).
Classiquement, au niveau moléculaire ,l'anomalie
structurale de l'hémoglobine est responsable de la
polymérisation, de la gélification, et de la diminution de la
solubilité de l'hémoglobine S en état d'hypoxie sans
altération de déformabilité du globule rouge (36-38). Le
processus est d'abord réversible puis devient irréversible (36).
La polymérisation est influencée par certains facteurs tels que
la température, le PH, la composition ionique, la concentration du
2-3DPG ainsi que la concentration intra corpusculaire de l'hémoglobine S
(36,38-40). Il faudrait considérer aussi la notion de temps de latence
(delay time) nécessaire à la polymérisation qui, dans les
conditions physiologiques, est supérieur au temps de passage dans la
microcirculation (40-42). Toute cause de ralentissement circulatoire provoque
la polymérisation de hémoglobine S (42). Le globule rouge subit
plusieurs cycles de désoxygénation/réoxygénation
avant que la déshydration cellulaire ne soit suffisante pour que
s'amorce la falciformation (41).
Au niveau cellulaire,la polymérisation de
l'hémoglobine S dans le globule rouge induit une série des
modifications (36). La déformation physique de la cellule entraîne
la libération des microvésicules et une déshydration
contemporaine d'anomalies du transport ionique membranaire, avec une
augmentation de la perméabilité de la membrane aux ions (Na+,
Ca++ et K+). Deux canaux sont impliqués dans ce phénomène
de déshydration. Il s'agit du co-transport K-Cl et du canal Gardos,
canal activé par le calcium( 5, 42). Un micro environnement oxydant
apparaît avec libération de Fe3+, création d'un
cycle d'auto oxydation de l'HbS et retentissement sur les autres
protéines du GR (36, 42). Il y a remaniement de phospholipides
membranaires. Ce qui modifie les interactions du globule rouge avec son
environnement plasmatique et cellulaire (36,43). En effet, la
drépanocytose prend une nouvelle dimension vasculaire et
rhéologique (38). Il faut noter le phénomène
d'adhérence des globules rouges, surtout les cellules jeunes, les
réticulocytes de stress, à cause de la stimulation permanente de
l'érythropoïèse par l'anémie chronique. Les globules
rouges expriment deux protéines adhésives la glycoprotéine
CD36, décrite d'abord comme la glycoprotéine plaquettaire IV
(GPIV) et la protéine VLA-4 (ou á4â1)
de la super famille des intégrines. Tandis que la cellule
endothéliale exprime surtout la glycoprotéine CD36 et la
protéine VCAM-1 de la super famille des immunoglobulines.
Des protéines plasmatiques normalement présentes
et libérées par les plaquettes comme la thrombospondine, la
glycoprotéine CD36 et le facteur Von Willebrand sont capables de
promouvoir l'adhérence des GR drépanocytaires à
l'endothélium (41, 42). Le phénomène d'adhérence
concerne aussi les GB (5, 6, 13, 22, 33, 42, 44, 45). Les sélectines, P-
et E- sélectines fixent les neutrophiles (41, 42). Des mécanismes
différents seraient mis en jeu selon les circonstances et le territoire
vasculaire (42). Différents agents inflammatoires comme le TNF, les
interleukines, particulièrement IL-1, des médiateurs de type
prostacyclines, sont souvent augmentés chez les drépanocytaires
et un excès de thrombine crée un état procoagulant (36,
41). On observe également une hyperplasie de l'intima associée ou
non à un événement thrombogène surtout au niveau
des artères cérébrales où il rappelle la maladie de
Moyamoya. Il faut signaler aussi le rôle de l'endotheline ou le monoxyde
d'azote dans la régulation du tonus vasculaire (36). Cette
vaso-occlusion est à la base de la symptomatologie de la
drépanocytose (37, 38).
La physiopathologie de la drépanocytose est un
mécanisme complexe qui implique toutes les cellules sanguines
circulantes, les protéines plasmatiques et l'endothélium (41).
La drépanocytose se caractérise en plus par une
grande susceptibilité aux infections (38, 42, 46, 47). Les infections
peuvent aussi être responsables de déshydratation, d'hypoxie et de
fièvre, et donc favoriser l'apparition de crises vaso-occlusives. Les
mécanismes physiopathologiques qui expliquent cette
susceptibilité aux infections sont encore imparfaitement
expliqués. L'asplénie fonctionnelle, une diminution de la
capacité d'opsonisation par anomalies des lymphocytes et probablement de
la voie alterne du complément (46,47 ) ( l'altération concerne
une déficience en properdine et en protéine C3B
entraînant un défaut de fixation de C3 ). Le
déficit immunitaire des drépanocytaires explique la
spécificité des bactéries telles que les pneumocoques et
les salmonelles. Une corrélation statistique a mis en évidence un
allèle protecteur DRB 1*15, et un allèle de susceptibilité
DQB1*3 (48).
1.1.3. Globules blancs et
drépanocytose
La drépanocytose est une maladie inflammatoire dont
l'un des marqueurs est la leucocytose (19). L'augmentation du nombre et
l'activation des leucocytes sont des médiateurs importants de
l'inflammation dans la drépanocytose (49, 50). Les leucocytes peuvent
adhérer les uns aux autres, aux érythrocytes falciformés
ou non, aux plaquettes et à l'endothélium vasculaire.
L'inflammation récurrente ainsi que les cytokines inflammatoires
augmentent la capacité d'adhésion des leucocytes (51, 52).
Les données de l'expérimentation animale et
humaine indiquent que l'adhésion des leucocytes aux autres
éléments figurés du sang et à l'endothélium
vasculaire est très importante dans la pathogénie des crises
vaso-occlusives de la drépanocytose (23, 53). En adhérant
à l'endothélium vasculaire, les leucocytes réduisent la
lumière des vaisseaux, ainsi ils diminuent le flux sanguin. A cause de
leur grande taille, les leucocytes réduisent davantage la lumière
vasculaire que les érythrocytes. Le monocyte a un diamètre de
14-20 ìm soit 3 fois celui de l'érythrocyte, tandis que le
lymphocyte et le neutrophile mesurant respectivement entre 10-16 ìm et
10-14ìm soit 2 fois le diamètre de l'érythrocyte (7,2
ìm). L'adhésion leucocytaire à l'endothélium
vasculaire est la première étape de la diapédèse
qui se produit au niveau des veinules post capillaires siège de la
vaso-occlusion des GR. Ainsi les leucocytes contribuent à la
pathogénie de la vaso-occlusion dans la drépanocytose.
L'endothélium vasculaire est activé par l'hypoxie, les infections
et les cytokines inflammatoires telles que le TNFá, IL-1â et
IL-6. Ces stimuli activent les leucocytes qui à leur tour activent
l'endothélium vasculaire ainsi apparaît un cercle vicieux. Cette
double activation endothéliale et leucocytaire conduit à une
expression beaucoup plus importante des molécules adhésives
telles que les intégrines, les sélectines et les ICAM-1, 2, 3
qui contribuent davantage à l'obstruction de la lumière des
vaisseaux (19).
L'expression importante de ces molécules
adhésives est associée à la sévérité
de la drépanocytose (28, 54), par exemple le taux élevé de
la L-sélectine (CD62) de lymphocyte est corrélée à
la survenue de l'AVC tandis que celui de â2 intégrine (CD18) de
neutrophile prédispose à la néphropathie
drépanocytaire (28).
CHAPITRE 2. MATERIEL ET
METHODES
2.1. Nature et
période de l'étude
La présente étude cas témoins a
décrit et analysé de manière rétrospective les
dossiers médicaux des patients hospitalisés entre le
1er janvier 1989 et le 30 décembre 2004 soit une
période 15 ans.
Le premier volet concernait la morbidité et le second
la mortalité.
2.2. Cadre
d'étude
Le Centre de Médecine Mixte et d'Anémie SS, dans
la commune de Kalamu a servi de cadre à la présente
étude.
Le choix de ce centre a été justifié par
le caractère d'hôpital de référence pour les enfants
drépanocytaires SS (CMASS).
2.3. Population
d'étude
Etaient éligibles (population cible) tous les enfants
noirs disposant du résultat de l'électrophorèse de
l'hémoglobine, âgés de 1 an à 20 ans et
hospitalisés dans le cadre d'étude.
2.3.1. Critères
d'inclusion
Etaient inclus dans la présente étude, les
patients éligibles répondant aux critères
suivants :
- être un cas défini comme drépanocytaire
SS au cours de l'hospitalisation ;
- être un témoin non drépanocytaire.
Seuls les drépanocytaires SS avec données
relatives à la phase intercritique stable et à la phase critique,
avaient constitué le sous-groupe étudié pour la
morbidité.
Les témoins AA étaient appariés aux
drépanocytaires SS pour le sexe, l'âge, le cadre et la
période d'étude.
2.3.2. Critères
d'exclusion
Etaient exclus de la présente étude, les
patients éligibles avec des paramètres d'intérêt
manquant dans les dossiers médicaux.
2.4. Matériel
Les actifs ayant servi à la présente
étude étaient les suivants :
- les fiches, les protocoles de récolte (en
annexe) ;
- les registres médicaux ;
- les certificats de décès ;
- une règle plate ;
- 10 stylos à bille ;
- Une calculatrice de marque Casio MS -808 V ;
- Un micro-ordinateur personnel portable de marque RM.
2.5. Approche
méthodologique
Le volet relatif à la morbidité a analysé
les dossiers médicaux des témoins AA et des
drépanocytaires SS en phase post critique stable (intercritique) et
ayant développé par la suite des complications de la
drépanocytose (phase critique).
Le volet relatif à la mortalité a porté
sur l'analyse des dossiers des patients de la période d'étude
avec issue vitale précisée à la suite de
l'hospitalisation.
2.5.1. Organisation et
déroulement du travail
La période du 31/01/2002 au 01/03/2002 a
été consacrée à l'obtention des autorisations
administratives auprès des autorités du centre. Cette
période a aussi servi à concevoir le protocole d'étude.
Aussi le travail proprement dit a commencé le
15/05/2002 et s'est poursuivi jusqu'au 30/12/2004.
2.5.2. Collecte des
données
La collecte des données a consisté en
l'enregistrement sur des fiches préalables et précodées
(annexe).
Les données recueillies étaient
regroupées en paramètres d'intérêt de la
manière suivante :
- données épidémiologiques :
âge, sexe ;
- données anamnestiques : nombre de transfusions
sanguines ;
- données cliniques : crises
hyperhémolytiques , crises douloureuses ;
- données hématologiques : Hb, GB,
FL ;
- autres données paracliniques :GE, L.C.R.,...
- Issue vitale : sortie vivant (survivant) ou
décédé (mortalité hospitalière).
2.6. Définitions
opérationnelles
Le paludisme compliqué était de forme grave
(sans manifestations encéphaliques) et de type neuropaludisme.
Les valeurs usuelles (de référence) des
leucocytes ont été définies par le seuil de croisement des
courbes de distribution des leucocytes des SS et des témoins AA :
la leucopénie coïncidant avec le seuil des GB à l'endroit
des croisements des courbes à gauche et l'hyperleucocytose à
l'endroit des croisements des courbes à droite.
La phase intercritique, elle correspondait à l'absence
d'un processus pathologique aigu (pas de crises douloureuses et
hémolytiques, ni d'infection dans les quatre semaines
précédant l'examen et pas de transfusion dans les trois mois
précédant l'examen) (55).
Le delta leucocytaire était la différence entre
le taux des leucocytes des décédés et des vivants.
2.7. Analyses
statistiques
Après validation, les données ont
été saisies et analysées sur microordinateur en utilisant
les logiciels EPI-INFO version 6.04 et SPSS sur Windows version 10.0. Les
logiciels R sur Internet (56) et Excel ont servi à construire les
graphiques.
Les données étaient synthétisées
dans des tableaux et représentées par des graphiques
(figures).
Les données qualitatives ont été
représentées sous forme de proportions (%) et les données
quantitatives (continues ou discontinues) sous forme de moyenne,
médianes et écarts-types (ET) avec leurs extrêmes.
Le test Chi carré de Mantel-Haenszel a servi à
comparer les proportions avec application du test exact de Fischer et du test
corrigé de Yates là où c'était approprié. Le
test -t de Student a servi à comparer les moyennes de deux groupes ayant
des distributions normales et le test U de Massen et Whitney en cas de
distribution asymétriques.
L'analyse des variances (ANOVA avec application du test F en
cas de distributions asymétriques) a servi à comparer les
moyennes entre plusieurs groupes.
Pour l'analyse univariée (unidimensionnelle), l'Odds
ratio (OR) et son intervalle de confiance (IC) à 95% pour
maîtriser la précision ont été calculés par
l'application du test Chi carré de Mantel-Haenszel.
L'analyse logistique appliquée à l'ensemble de
la population étudiée sur l'équation suivante :
P(M+/X1 ; X2 ;
etc.) ou Y=e (â0+â1X1+
â2X2... ânXn )
Dans laquelle :
- P (M+) est la probabilité de
décès ;
- â0 une constante ;
- X1 , X2 , ....Xn
sont des covariables ;
- e : exponentiel ;
- â1,
â2...ân sont des coefficients de
régression partielle des covariables correspondantes ;
- Les exponentiels de â1,
â2...ân sont des OR présentés
avec leur IC à 95%.
L'Odds ratio du risque multivarié permettant
d'identifier les déterminants indépendants et significatifs de
prédiction des variations de mortalité aux soins intensifs.
La fonction de survie, fonction linéaire, a
été évaluée au cours du séjour aux soins
intensifs par les courbes de Kaplan Meier au cours de l'hospitalisation
complète. La régression par le modèle de Cox a permis de
calculer le Hazard ratio (proportionnalité).
La valeur de p<0,05 était considérée
comme le seuil de signification ; p<0,01 comme seuil hautement
significatif ; et p<0,001, p<0,0001 et p<0,00001 comme seuil
très significatif.
CHAPITRE 3. RESULTATS
Les résultats de la population d'étude (n=504)
concernaient la morbidité et la mortalité
hospitalière ; le taux d'exhaustivité (réponse)
étant de 90% (504/560 : 56 dossiers exclus).
3.1. Morbidité
Ce volet a analysé les dossiers de 252 patients (sous
groupe de la population d'étude) dont 126 drépanocytaires
SS (63 de sexe M et 63 de sexe F) et 126 témoins AA (63 sexe M
et 63 de sexe F). L'age moyen était de 10,1#177;5,5 ans (extrêmes
1 an et 20 ans ; médiane :10 ans).
3.1.1. Données
anamnestiques
Le nombre moyen de transfusion sanguine
bénéficié par 236 patients était de 35 #177; 6
transfusions (extrême 1 et 34 transfusions sanguines).
Ces 236 patients sont repartis selon le groupe de transfusion
sanguine dans la figure1.
n= 72 = 63
=45 =56
Groupe de transfusion
Effectif
%

Figure 1. Répartition de 236 patients selon les groupes de
nombre de transfusions sanguines.
3.1.2. Données
hématologiques
Le tableau 1 résume les valeurs moyennes de l'Hb, des
GB, des neutrophiles et des lymphocytes selon l'hémogramme
réalisé à l'admission.
Tableau 1. Données hématologiques
à l'admission dans la population d'étude
|
Variables
|
Moyennes #177; ET
|
Extrêmes
|
|
Hémoglobine (g%)
|
7,9 #177; 2,4
|
2,2 - 15,6
|
|
Globules blancs (éléments/mm3)
|
13123 #177; 8351
|
2800 - 84000
|
|
Neutrophiles (%)
|
58,7 #177; 17,3
|
6 - 90
|
|
Lymphocytes (%)
|
39,7 #177; 17
|
8 - 94
|
3.1.3. Tableau clinique
Le tableau relatif à la phase critique comprenait
différentes entités morbides propres à la
drépanocytose homozygote : crise vaso-occlusive
ostéoarticulaire chez 80 patients, crise hyperhémolytique chez 60
patients, crise abdominale chez 15 patients , syndrome mains pieds chez 9
patients ,syndrome thoracique aigu chez 8 patients, accident vasculaire
cérébral chez 5 patients.
Le tableau clinique était caractérisé par
les infections et affections accompagnant la phase critique des
drépanocytaires SS et l'hospitalisation des témoins AA .
Principalement le paludisme simple et compliqué (n=150),le sepsis(n=42),
la fièvre au long cours (n=39), la bronchopneumonie (n=34), la
pneumonie(n=30), la méningite (n=17), l'infection urinaire (n=17) ,la
tuberculose (n=15), l'ostéomyélite (n=15), la bronchite (n=13),
l'entérite fébrile (n=10), la fièvre thyphoide (n=10),
l'insuffisance cardiaque (n=10) et l'hépatite virale (n=8).
3.1.4. Globules blancs et
morbidité
3.1.4.1. Variations des globules
blancs
3.1.4.1.1. Moyennes des globules
blancs
Les taux sanguins des GB variaient de manière
très significative (ANOVA, test de Kruskall-Wallis ;p< 0,00001)
entre les différents types de morbidité ; le taux le plus
élevé étant observe chez les drépanocytaires
homozygotes SS en phase critique, le taux intermédiaire chez les
drépanocytaires SS en phase intercritique, et le taux le plus bas chez
les témoins AA (Figure2).
GB/mm3
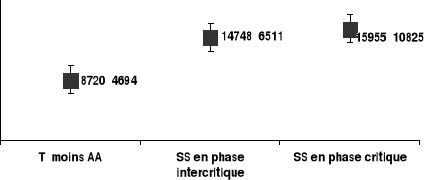
Figure 2. Variations très inégales
des taux sanguins des globules blancs entre les différents groupes des
patients.
3.1.4.1.2. Quartiles des globules
blancs et types de morbidité
Les pourcentages des patients drépanocytaires en phase
critique augmentaient de manière très significative(p<0,00001)
du Quartile I au Quartile IV des GB (présence d'un gradient biologique)
(Figure3).
QI
QII QIII
QIV
SS en phase critique
%
Quartiles des GB/mm3

Figure 3. Relation effet-dose dépendant
entre les proportions des drépanocytaires SS en phase critique et les
taux sanguins des globules blancs.
Il existait une variation inégale mais très
significative (p<0,00001 sans effet dose dépendant) des pourcentages
des drépanocytaires SS en phase post critique entre les
différents Quartiles des globules blancs (Figure 4).
Quartiles des GB/mm3
QI
QII QIII
QIV
SS en phase post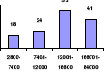 critique critique
%

Figure 4. Variations inégales des
proportions des drépanocytaires SS en phase post-critique entre les
quartiles des globules blancs.
Par contre, les proportions des témoins AA diminuaient
très significativement (p<0,00001 avec relation inverse ou
négative) du Quartile I au Quartile IV des Globules blancs (Figure5).
Témoins AA
Quartiles des GB/mm3
QI
QII QIII
QIV
%

Figure 5. Relation inversement proportionnelle
entre la répartition des témoins AA et les Quartiles des globules
blancs.
3.1.4.1.3. Manifestations
critiques et Quartiles des globules blancs
Il y avait respectivement plus des cas de syndrome mains pieds
et des crises abdominales (Figure 6) dans les groupes Quartiles III et IV que
dans les groupes Quartiles I et II : variations significatives (p<0,05)
des cas des syndromes mains pieds entre les Quartiles des globules blancs et
variations très significatives (p<0,0001) des cas de crises
abdominales entre les Quartiles.
Quartiles des GB
%
Effectif
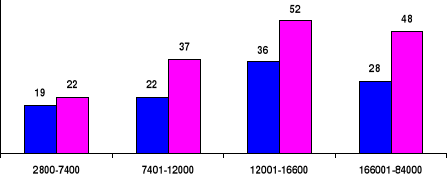
Figure 6. Répartition des cas de syndrome
mains-pieds ( ) et de crises abdominales ( ) entre les Quartiles
des globules blancs.
Les proportions des cas d'AVC, de Syndrome thoracique aigu, de
crise hyperhémolytique ne variaient pas (p>0,05) et de manière
respective entre les Quartiles des globules blancs (résultats non
présentés).
Par contre il était observé une augmentation
directement proportionnelle et de manière respective des cas des crises
vaso-occlusives (p<0,01) et des crises hyperhémolytiques (p<0,01)
(Figure 7).
Quartiles des GB
Effectif
%
%
Effectif
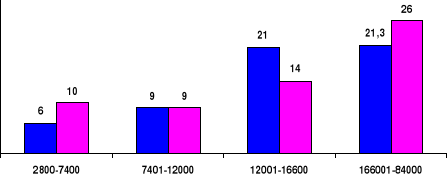
Figure 7. Relation respective avec effet-dose
dépendant entre les crises vaso-occlusives ( ), les crises
hyperhémolytiques ( ) et les Quartiles des globules blancs.
3.1.4.2. Infections
bactériennes et sepsis
Les taux de méningite, de sepsis, de pneumonie,
d'ostéomyélite, d'hépatite, d'infection urinaire et
neuropaludisme ne montraient aucune variation significative (p>0,05) et
respectivement entre les différents Quartiles des GB au sein de la
population totale (résultats non présentés).
3.1.4.3. Paludisme,
sévérité de la drépanocytose et globules blancs.
Chez les drépanocytaires SS toutes phases confondues,
une relation curvilinéaire en forme de U et significative (p<0,05)
était démontrée entre le taux de paludisme grave et les
Quartiles des GB (Figure8).
Quartiles des GB/mm3
QI
QII QIII
QIV
Paludisme grave
%

Figure 8. Relation en U entre le paludisme
grave et les quartiles des globules blancs chez les drépanocytaires SS
toutes phases confondues.
Mais la relation linéaire était négative
et très significative (p<0,001) entre le taux de paludisme grave et
le taux sanguin des globules blancs chez les témoins AA (Figure 9).
Quartiles des GB/mm3
QI
QII QIII
QIV
Paludisme grave
%

Figure 9. Relation inversement
proportionnelle entre les taux de paludisme grave et les taux sanguins des
globules blancs chez les témoins AA.
Il y avait respectivement moins de cas de paludisme grave chez
les drépanocytaires SS en phase critique que chez les
drépanocytaires SS en phase post critique (p <0,001) et chez les
témoins AA (p<0,0001) ; les taux de paludisme grave des
témoins et des drépanocytaires SS en phase critique étant
identiques (p>0,05) (Figure 10).
NS
p<0,00001
p<0,001
Paludisme grave
%

Figure 10. Comparaison des taux de paludisme
grave par types de morbidité.
Les taux de paludisme grave des drépanocytaires en
phase critique et des drépanocytaires SS en phase post critique
étaient respectivement identiques (p>0,05) aux Quartiles I - III des
GB ; mais il y avait moins des cas (p<0,01) de paludisme grave chez
les drépanocytaires SS en phase critique que les drépanocytaires
SS en phase post critique au Quartile IV des GB (Tableau 2).
Tableau 2. Taux de paludisme grave selon la
phase de la drépanocytose SS et les quartiles des globules blancs.
|
Quartiles des globules
blancs/mm3
|
SS en phase critique
%
|
SS en phase post-critique
%
|
p
|
|
QI 2800 - 7400
|
22
|
41
|
NS
|
|
QII 7401 - 12000
|
0
|
14
|
NS
|
|
QIII 12001 - 16600
|
12
|
29
|
NS
|
|
QIV 16601 - 84000
|
17
|
41
|
<0,01
|
Quelque soit les Quartiles des globules blancs
considérés, les drépanocytaires SS en phase critique
souffraient moins (p<0,05 et p<0,01) de paludisme grave que les
témoins AA (Tableau 3).
Tableau 3. Taux de paludisme grave des
drépanocytaires SS en phase critique et des témoins AA selon les
quartiles des globules blancs.
|
Quartiles des globules
blancs/mm3
|
SS en phase critique
%
|
Témoins AA
%
|
p
|
|
QI 2800 - 7400
|
22
|
46
|
<0,05
|
|
QII 7401 - 12000
|
0
|
28
|
<0,01
|
|
QIII 12001 - 16600
|
12
|
41
|
<0,05
|
|
QIV 16601 - 84000
|
17
|
60
|
<0,05
|
Ainsi la protection très significative des
drépanocytaires SS en phase critique vis-à-vis du paludisme
était plus élevée, plus forte en comparaison avec les
témoins AA( p<0,00001) qu'en comparaison avec les
drépanocytaires SS en phase post critique(p<0,0001) (Figure 11).
Risque élevé
Protection ou moindre risque
SS en phase critique versus SS en phase post-critique
SS en phase critique versus Témoins AA
0,41
0,22
0 0,12 0,17 0,33 0,62
OR avec
IC95%
1
Référence
Pas de risque
Figure 11. Protection des
drépanocytaires SS en phase critique vis-à-vis du paludisme.
Cette protection des drépanocytaires SS en phase
critique vis-à-vis du paludisme grave et en comparaison avec les
témoins AA, augmentait très significativement avec l'augmentation
des taux sanguins des GB (Figure 12).
Risque élevé
Protection ou moindre risque
Quartile IV 16601 - 84000
0,14(0,02 - 0,9)
Quartile III 12001 - 16600
0,20 (0,03-0,90)
Quartile II GB 7401 - 12000
0 (0 - 0,38)
Quartile I Gb 2800 - 7400
0,32 (0,10 à 0,97)
0 1 OR
Référence (IC95%)
Figure 12. Influence de l'augmentation des
taux sanguins des globules blancs sur la protection des drépanocytaires
SS en phase critique vis-à-vis du palud isme grave en comparaison avec
les témoins AA.
3.1.4.4. Infections, sepsis et
globules blancs
3.1.4.4.1. Drépanocytaires
en phase critique versus drépanocytaires en phase post critique
Les taux de pneumonie de bronchopneumonie de tuberculose
pulmonaire et de fièvre typhoïde étaient identiques
(p>0,05) entre les drépanocytaires SS en phase critique et les
drépanocytaires SS en phase post critique (résultats non
présentés).
Par contre, il y avait plus des cas de sepsis, de
méningite aigue, d'entérite fébrile et moins d'infection
urinaire chez les drépanocytaires SS en phase critique que chez les
drépanocytaires SS en phase post critique (Tableau 4).
Tableau 4. Sepsis, méningite
aiguë, entérite fébrile et infection urinaire selon la
phase de la drépanocytose SS.
|
Variables
|
SS en phase critique
n (%)
|
SS en phase post-critique
n (%)
|
p
|
|
Sepsis
|
31 (24,6)
|
14 (11,1)
|
<0,01
|
|
Méningite aiguë
|
9 (7,1)
|
0 (0)
|
<0,01
|
|
Entérite fébrile
|
5 (5)
|
0 (0)
|
<0,05
|
|
Infection urinaire
|
1 (1)
|
12 (12)
|
<0,001
|
3.1.4.4.2. Drépanocytaires SS en phase critique versus
témoins AA
Comparés aux témoins AA, les
drépanocytaires SS en phase critique présentaient autant de cas
de méningite aiguë, d'hépatite aigue virale, de
bronchopneumonie, d'entérite fébrile, de tuberculose pulmonaire
(résultats non présentés), mais plus de cas de sepsis,
d'ostéomyélite, de pneumonie et moins de cas d'infection urinaire
(Tableau 5).
Tableau 5. Sepsis,
ostéomyélite et infection urinaire chez les
drépanocytaires SS en phase critique et les témoins AA.
|
Variables
|
SS en phase critique
n (%)
|
Témoins AA
n (%)
|
p
|
|
Sepsis
|
22 (17,5)
|
2 (1,6)
|
<0,0001
|
|
Ostéomyélite
|
7 (5,6)
|
0 (0)
|
<0,01
|
|
Infection urinaire
|
1 (1)
|
9 (8,9)
|
<0,01
|
|
Pneumonie
|
32 (25,4)
|
6 (4,8)
|
<0,00001
|
3.1.4.4.3. Drépanocytaire
SS en phase post critique versus témoins AA
Les drépanocytaire SS en phase post critique
(intercritique) et les témoins AA présentaient des taux
identiques (p>0,05) des méningites aiguës, de pneumonie, de
sepsis, de broncho-pneumonie, d'entérite fébrile, d'infection
urinaire, de tuberculose pulmonaire et de fièvre typhoïde
(résultats non présentés).
3.1.4.4.4. Influence des
globules blancs sur la susceptibilité aux sepsis
ostéomyélite , pneumonie et infection urinaire
En considérant les Quartiles des GB, les variations du
taux de sepsis entre les différents types de morbidité
était seulement significative (p<0,05) au regard du Quartile I des GB
(le taux le plus bas), le taux de sepsis montrant une variation égale
entre les différents types de morbidité au regard des Quartiles
II- Quartile IV (Tableau 6).
Tableau 6. Diminution des globules blancs et
susceptibilité au sepsis chez les drépanocytaires SS en phase
critique
|
Quartiles des globules
blancs/mm3
|
SS en phase critique
%
|
SS en phase
postcritique
%
|
Témoins AA
%
|
p
|
|
QI 2800 - 7400
|
22
|
6
|
4
|
<0,05
|
|
QII 7401 - 12000
|
20
|
14
|
7
|
NS
|
|
QIII 12001 - 16600
|
27
|
10
|
6
|
NS
|
|
QIV 16601 - 84000
|
28
|
14
|
20
|
NS
|
L'augmentation des GB ne montrait pas de différence
significative sur les variations respectives des taux
d'ostéomyélite pneumonie et d'infection urinaire entre les
différents types de morbidité (résultats non
présentés).
Comparés respectivement aux drépanocytaires SS
en phase intercritique et aux témoins AA, les drépanocytaires SS
en phase critique présentaient un risque élevé ou une
protection moindre devant ces différentes affections avant et
après stratification avec les groupes des globules blancs (Tableau 7) et
(tableau 8).
Tableau 7. SS en phase critique versus SS en
phase intercritique : susceptibilité aux infections
|
Risque ou protection devant les affections
|
OR
(IC95%)
sans pondération
|
OR
(IC95%)
pondéré avec groupes des globules
blancs
|
|
Fièvre au long cours
|
13,3****
(4,3 - 56,2)
|
12,5****
(3,6 - 43,4)
|
|
Pneumonie
|
0,7
(0,4 - 1,1)
|
|
|
Paludisme compliqué
|
0,3***
(0,2 - 0,7)
|
0,3***
(0,2 - 0,6)
|
|
Sepsis
|
2,6**
(1,3 - 5,3)
|
2,6**
|
Tableau 8. Susceptibilité des SS en
phase critique versus Témoins AA
|
Risque ou protection de
|
OR
(IC95%)
sans pondération
|
OR
(IC95%)
pondéré avec groupes des globules
blancs
|
|
Pneumonie
|
6,8****
(2,8 - 18,4)
|
6,95****
(2,6 - 18,4)
|
|
Fièvre au long cours
|
13,3****
(4,3 - 56,2)
|
18****
(4,8 - 67,9)
|
|
Paludisme compliqué
|
0,2****
(0,1 - 0,4)
|
0,16****
(0,1 - 0,4)
|
|
Sepsis
|
5,5****
(2,4 - 14,1)
|
4,4****
(1,8 - 11,9)
|
|
Méningite
|
1,1
(0,4 - 3,2)
|
|
|
Neuropaludisme
|
0,12***
(0,02 - 0,5)
|
|
3.1.4.5. Hémoglobine,
anémies et globules blancs
Il existait une relation négative significative entre
les taux sanguins des GB et d'Hb chez les témoins AA (r=-0,264 ;
p<0,05). Mais il n'existait aucune relation significative( p<0,05) entre
les taux sanguins des GB et d'Hb chez les drépanocytaires SS tant en
phase critique qu'en phase post critique stable.
Au regard des Quartiles I et II des GB, le taux
d'anémie était identique (p>0,05) entre les différents
types de morbidité ; mais au Quartile III des GB, le taux
d'anémie était le plus élevé chez les
drépanocytaires SS en phase critique alors qu'il l'était au
Quartile IV chez les témoins AA (Tableau 9).
Tableau 9. Anémie et globules
blancs
|
Quartiles des globules
blancs/mm3
|
SS en phase critique
%
|
SS en phase
postcritique
%
|
Témoins AA
%
|
p
|
|
QI 2800 - 7400
|
17
|
6
|
7
|
NS
|
|
QII 7401 - 12000
|
20
|
5
|
7
|
NS
|
|
QIII 12001 - 16600
|
15
|
0
|
6
|
<0,01
|
|
QIV 16601 - 84000
|
19
|
0
|
20
|
<0,01
|
Chez les drépanocytaires SS en phase critique, les
anémiques et les non anémiques présentaient des taux
sanguins identiques (p>0,05) des GB. Il en était de même chez
les témoins AA (Figures 13 et 14).
NS
Anémie
GB/mm3

Figure 13. Taux sanguins de globules blancs
en présence et en absence d'anémie chez les
drépanocytaires SS en phase critique.
NS
Anémie
GB/mm3

Figure 14. Taux sanguins de globules blancs
en présence et en absence d'anémie chez les témoins AA.
Par contre, chez les drépanocytaires SS en phase
postcritique et stable, le taux sanguin moyen des globules blancs des
anémiques était plus bas (p<0,05) que celui des non
anémiques (Figure 15).
p<0,05
Anémie
GB/mm3

Figure 15. Taux sanguins de globules blancs
en présence et en absence d'anémie chez les
drépanocytaires SS en phase post-critique et stable.
3.2. Globules blancs et
mortalité hospitalière
L'étude sur la mortalité était relative
à 504 patients dont 252 drépanocytaires SS (113 de sexe masculin
et 139 de sexe féminin) et 252 témoins AA (113 de sexe M et 139
de sexe F).
Dans cette population d'étude, 178 patients
étaient décédés dont 126 drépanocytaires SS
et 52 témoins SS.
3.2.1. Globules blancs et
issue vitale
La Figure 16 souligne le sursaut des taux sanguins des GB en
cas décès des drépanocytaires SS et des témoins AA
et la variation très significative (p<0,00001) des taux sanguins des
GB entre les survivants et les décédés : le taux le
plus élevé chez les drépanocytaires SS
décédés, le taux le plus bas chez les témoins AA
survivants, et le taux de GB 150000/ mm étant le seuil discriminant les
décédés des survivants.
15000
GB/mm3

Figure 16. Taux sanguins de globules blancs
selon le gène S et l'issue vitale.
Le delta leucocytaire était plus important chez les
témoins AA(Ä=6107GB/mm3) que chez les
drépanocytaires SS (Ä=1207GB/mm3).
3.2.2. Sepsis, globules blancs
et issue vitale
Les taux sanguins des GB variaient de manière
très significative (p<0,0001) entre les différents types de
morbidité selon le gène S et selon la présence du sepsis
(Figure 17).
Les témoins AA sans sepsis et les
drépanocytaires SS sans sepsis présentaient des taux sanguins
moyens des GB< 15000 éléments/ mm3. Mais les
témoins AA avec sepsis, les drépanocytaires SS sans sepsis
décédés, les drépanocytaires SS avec sepsis
survivants et les drépanocytaires SS avec sepsis
décédés, présentaient respectivement des taux
sanguins moyens des GB > 15000 éléments/mm3. Le
décès et la présence du sepsis entraînaient un
sursaut des sanguins des GB plus marqués chez les témoins AA que
chez les drépanocytaires SS.
15.000
GB/mm3
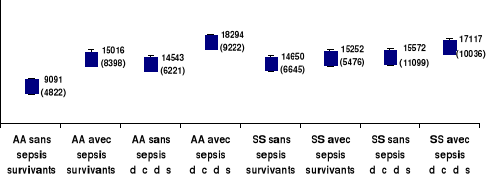 Figure 17. Sepsis, gène S, issue vitale et taux
sanguins des globules blancs. Figure 17. Sepsis, gène S, issue vitale et taux
sanguins des globules blancs.
3.3. Déterminants
indépendants de la mortalité hospitalière après
analyse multivariée au sein de la population totale et sans
considération du temps
A la 14ème étape de la
régression logistique ayant la mortalité hospitalière
comme variable dépendante,le risque multi varié (OR)
ajusté pour plusieurs facteurs de pronostic potentiel, était
significativement et indépendamment déterminé par la
drépanocytose SS, l'AVC, le sepsis l'entérite fébrile, le
taux sanguin des GB>15000elts/mm3 et l'âge >10 ans
(Tableau 10).
Tableau 10. Prédicteurs significatifs
et indépendants de la mortalité hospitalière dans la
population totale
|
Variables indépendantes
|
Coefficient Bêta
|
Es
|
Wald
|
OR
(IC95%)
|
p
|
|
Drépanocytose SS
|
0,855
|
0,236
|
13,091
|
2,4
|
<0,0001
|
|
AVC
|
1,763
|
0,837
|
4,434
|
5,8
|
<0,05
|
|
Sepsis
|
0,959
|
0,456
|
4,417
|
2,6
|
<0,05
|
|
Entérite fébrile
|
2,152
|
1,129
|
3,633
|
8,6
|
<0,05
|
|
Globules blancs>15000/mm3
|
1,144
|
0,234
|
23,985
|
3,4
|
<0,0001
|
|
Age < 10 ans
|
0,471
|
0,228
|
4,259
|
1,6
|
<0,05
|
|
Constante
|
-12,826
|
3,024
|
17,984
|
|
<0,0001
|
Y= -12,826+0,855 gènes+1,763 AVC+0,959 sepsis+2,152
Entérite fébrile+1,144 hyperleucocytose+0,471 Age.
3.4. Fonction de survie aux
soins intensifs
Dans la population totale, 50% étaient encore en vie au
10ème jour d'hospitalisation aux soins intensifs (Figure
18).

Figure 18. Fonction de survie de la
population totale aux soins intensifs selon la courbe de Kaplan Meier.
L'hyperleucocytose de la population totale était
caractérisée par une moindre survie au 10ème
jour d'hospitalisation (22% ; p<0,0001) en comparaison avec un taux des
globules < 15.000 elts/mm3 (Figure 19).

Figure 19. Courbes de Kaplan-Meier selon
l'hyperleucocytose (GBAKI=1,0000) et le taux des globules blancs <
15000/mm3 (GB1KI=2) dans la population totale.
La probabilité de survie au 10ème
jour d'hospitalisation aux soins intensifs était médiocre chez
les drépanocytaires SS (20% ;p<0,01) par rapport aux
témoins AA (60%) (Figure 20).
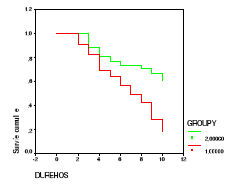
Figure 20. Courbes de Kaplan-Meier chez SS
(Groupy=1) et les AA (Groupy=2).
En introduisant dans la régression par modèle de
Cox tous les prédicteurs indépendants de la mortalité
hospitalière retenus par la régression logistique, seules
l'hyperleucocytose et la drépanocytose SS prédisaient la
mortalité hospitalière de la population totale
indépendamment de l'AVC, du sepsis, de l'entérite fébrile
et de l'âge (Tableau 11).
Tableau 11. Régression par
modèle de Cox pour prédire la mortalité
hospitalière aux soins intensifs à partir des prédicteurs
de la régression logistique
|
Variables indépendantes
|
B
|
ES
|
Wald
|
Hazard ratio
(IC95%)
|
p
|
|
Globules blancs>15000/mm3
|
0,744
|
0,194
|
14,8
|
2,1
(1,4 -3,1)
|
<0,0001
|
|
Drépanocytose SS
|
0,473
|
0,200
|
5,6
|
1,6
(1,1 à 2,4)
|
<0,01
|
Exclus : AVC, sepsis,entérite fébrile,
âge <10 ans.
Mais en considérant la durée d'hospitalisation
aux soins intensifs et en salle d'hospitalisation, seule l'hyperleucocytose
prédisait la mortalité hospitalière dans la population
totale et indépendamment de la drépanocytose SS, du sepsis, de
l'AVC et de l'âge (Tableau 12).
Tableau 12. Régression par
modèle de COX pour prédire la mortalité
hospitalière aux soins intensifs en fonction du temps
|
Variables indépendants
|
B
|
ES
|
Wald
|
Hazard ratio
(IC95%)
|
p
|
|
Globules blancs>15000/mm3
|
0,845
|
0,172
|
24,155
|
2,3
(1,6 -3,2)
|
<0,0001
|
|
Entérite fébrile
|
1,199
|
0,458
|
6,843
|
3,3
(1,4-8,2)
|
<0,01
|
Exclus : drépanocytose SS, sepsis, AVC, âge.
Il était démontré l'égalisation
(p>0,05) de la probabilité de survie au cours du temps total
d'hospitalisation entre les catégories des G.B. (Figure 21), du sepsis
(Figure 22) et de morbidité (drépanocytaire SS versus
témoins AA) (Figure 23).

Figure 21. Egalisation de survie entre les
patients avec globules blancs >15.000 elts/mm3 et les patients
avec globules blancs <15.000 elts/mm3.

Figure 22. Egalisation de survie
cumulée entre les cas de sepsis et de non sepsis au cours de
l`hospitalisation complète.

Figure 23. Egalisation de survie entre les
drépanocytaires SS et les témoins AA.
3.5. Définition des
valeurs usuelles des leucocytes (référence) chez les
drépanocytaires SS
3.5.1. Valeurs de
référence chez les drépanocytaires SS
Contrairement à la courbe de distribution normale des
globules des témoins AA entièrement incluse dans la distribution
normale des globules blancs de la population totale, celles des
drépanocytaires SS s'étale avec son sommet et une grande partie
en dehors de la distribution des globules blancs de la population totale
(Figure 24). Les courbes de distribution des globules blancs des
drépanocytaires SS et des témoins AA s'entrecroisaient au seuil
de 4000 GB/mm3 et de 15000 GB/mm3.
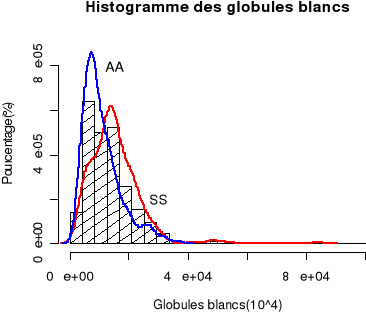
Figure 24. Comparaison des courbes de
distribution des courbes des globules blancs chez les témoins AA et les
drépanocytaires SS en rapport avec les histogrammes des globules blancs
de la population totale.
Ainsi, les variations physiopathologiques des leucocytes chez
les drépanocytaires SS étaient définies avec les valeurs
de référence suivantes :
- leucopénie : GB<4000/mm3 ;
- intervalle adéquat (physiologique)
4000/mm3 à 15000 GB/mm3 ;
- hyperleucocytose : GB > 15.000/mm3.
3.5.2. Extrême
variabilité des globules blancs chez les drépanocytaires
La variabilité interindividuelle des leucocytes
était discriminante entre les drépanocytaires SS et les
témoins AA (Figure 25) et entre les drépanocytaires SS en phase
intercritique montrant une variabilité des globules blancs similaire
à celles des témoins AA (Figure 26).

Figure 25. Variabilité des globules
blancs entre les drépanocytaires SS et les témoins AA.
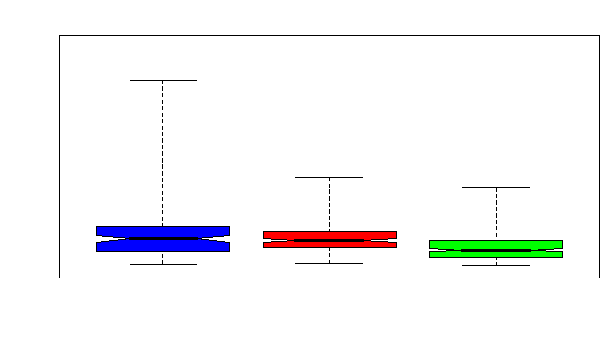
Figure 26. Variabilité des globules
blancs entre SS en phase critique, SS en phase intercritique et les
témoins AA.
L'extrême variabilité des leucocytes était
caractéristique de la présence du paludisme grave chez les
drépanocytaires SS (Figure 27) et non chez les témoins AA
(Figure 28).

Figure 27. Variabilité des globules
blancs chez les SS et paludisme grave.

Figure 28. Variabilité des globules
blancs entre les témoins AA et paludisme grave.
Les valeurs médianes des leucocytes ne
différaient pas entre les Quartiles des leucocytes chez les
drépanocytaires SS avec paludisme grave et extrême
variabilité des leucocytes au sein du Quartile IV des leucocytes (Figure
29). Contrairement à la discrète perte des valeurs
médianes des leucocytes à travers les quartiles des
drépanocytaires SS sans paludisme grave (Figure 30).

Figure 29. Variabilité très
marquée des globules blancs des patients SS avec hyperleucocytose et
paludisme grave.

Figure 30. Variabilité très
marquée des globules blancs des patients SS avec hyperleucocytose et
paludisme grave.
Mais tel n'était pas le cas chez les témoins AA
avec paludisme grave (Figure 31) et sans paludisme grave (figure 32) montrant
une évolution identique des valeurs médianes des leucocytes entre
les Quartiles des leucocytes.
Figure 31. Variabilité moins
prononcée des globules blancs les AA avec hyperleucocytose et paludisme
grave.


Figure 32. Absence de variabilité des
globules blancs des patients AA avec hyperleucocytose et paludisme grave.
Seuls les drépanocytaires SS
décédés présentaient une variabilité
extrême des leucocytes en comparaison avec les drépanocytaires SS
survivants, les témoins AA décédés et les
témoins AA survivants (Figure 33).
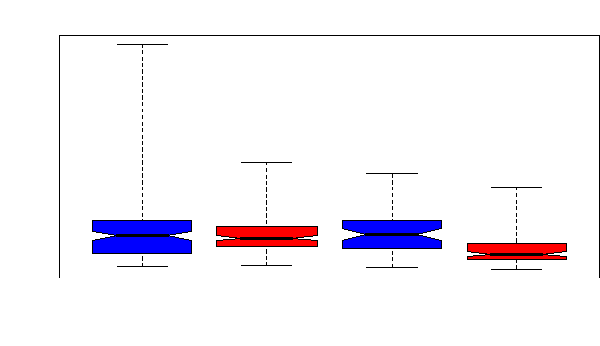
Figure 33. Variabilité des globules
blancs entre les SS (+) décédés, les SS (-) survivants,
les témoins AA (+) décédés et les témoins AA
(-) survivants
Contre toute attente, le sepsis n'était pas la base de
l'extrême variabilité des globules blancs chez les
drépanocytaires SS : les drépanocytaires SS
décédés sans sepsis présentaient une
variabilité plus importante des leucocytes que les
drépanocytaires SS décédés avec sepsis (Figure
34).
SS avec
sepsis
décédés
SS sans
sepsis décédés
SS avec
sepsis survivants
SS sans
sepsis survivants
AA avec
sepsis décédés
AA sans
sepsis décédés
AA avec sepsis survivants
AA sans
sepsis survivants
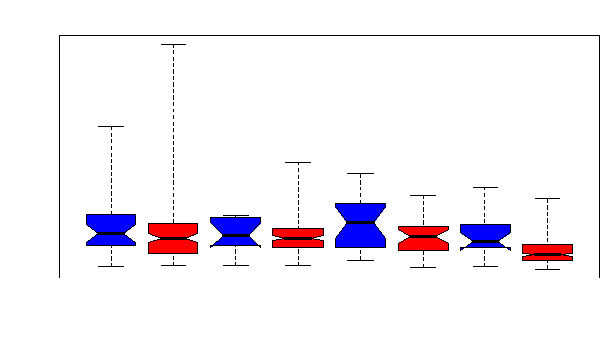
Figure 34. Variabilité des globules
blancs entre les catégories des patients selon l'issue vitale et le
sepsis.
3.5.3. Intervalle
adéquat (physiologiques) des globules blancs chez les
drépanocytaires SS
Il existait une relation curvilinéaire en forme de
lettre U entre la morbidité et les globules blancs chez les
drépanocytaires SS. En effet, comparé à la
leucopénie et à l'hyperleucocytose, l'intervalle adéquat
des leucocytes était plus protégé vis-à-vis du
paludisme grave, des crises vaso-occlusives et du sepsis (Figure 35).
Risque=Odds ratio
Globules blancs/mm3

Figure 35. Relation en forme de U entre le
paludisme grave ( ), les crises vaso-occlusives ( ), le
sepsis ( ) et les globules blancs chez les drépanocytaires
SS.
L'intervalle adéquat des leucocytes (OR= 0,438 IC95%
0,329-0,584 ; p<0,0001) et la leucopénie (OR=1,136 IC95%
0,221-0,456 ;p<0,001) conféraient respectivement une protection
très significative devant la létalité chez les
drépanocytaires SS. Mais le risque de létalité
était multiplié par 2 (OR= 2 IC95% 1,3 à 3,3; p<0,01)
en cas d'hyperleucocytose chez les drépanocytaires SS.
3.5.4. Variabilité de
l'hémoglobine chez les drépanocytaires SS
Contrairement à la variabilité de leurs
leucocytes, démontrée plus haut, les drépanocytaires SS ne
présentaient pas de variabilité importante de leur taux d'Hb
à l'opposé de l'extrême variabilité de l'Hb chez les
témoins AA (Figure 36).
AA
SS

Figure 36. Variabilité de
l'hémoglobine entre les drépanocytaires SS et les témoins
AA.
Cette variabilité de l'Hb était très
marquée chez les témoins AA survivants (Figure 37) et chez les
témoins AA décédés avec sepsis (Figure 38).
AA-
AA+
SS-
SS+
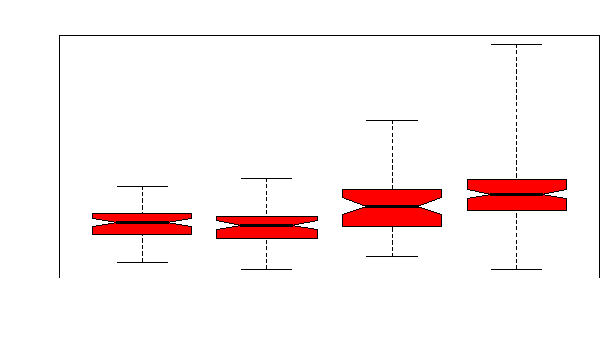
Figure 37. Variabilité de
l'hémoglobine entre les SS survivants, les SS
décédés et les témoins AA
décédés et survivants.
SS avec
se psis
décédés
SS sans
se psis décédés
SS avec
se psis survivants
SS sans
sepsis survivants
AA avec
sepsis décédés
AA sans
sepsis décédés
AA avec sepsis survivants
AA sans
sepsis survivants

Figure 38. Variabilité de
l'hémoglobine entre les catégories des patients selon l'issue
vitale et le sepsis.
CHAPITRE 4. DISCUSSION
La présente étude a analysé
l'augmentation et la diminution du nombre des leucocytes dans la
morbi-mortalité chez les drépanocytaires SS. Ce qui a permis de
comprendre le rôle péjoratif de l'hyperleucocytose dans la
susceptibilité aux infections graves et aux manifestations critiques
spécifiques chez les drépanocytaires SS. Mais cette
hyperleucocytose jouait plutôt un rôle protecteur vis-à-vis
du paludisme grave non encéphalique et au neuropaludisme chez le
drépanocytaire SS. C'est pourquoi, l'hyperleucocytose et la
leucopénie ont été respectivement définies selon
des seuils de référence propres aux témoins AA et aux
drépanocytaires SS. Un seuil létal dans l'hyperleucocytose du
drépanocytaire SS a ensuite était déterminé.
4.1. Phase critique et
globules blancs chez les drépanocytaires SS
La présente étude a démontré que
l'hyperleucocytose classique (traditionnelle) en général (valeurs
moyennes) était caractéristique chez le drépanocytaire SS
sans distinction de la sévérité de cette
hémoglobinopathie. En effet, le nombre de globules blancs moyens des
drépanocytaires SS en phase critique restait similaire à celui
observé chez ces mêmes patients en phase intercritique stable.
Mais tel n'était pas le cas chez les témoins AA avec la valeur
moyenne des leucocytes dans les limites normales des valeurs usuelles à
Kinshasa (57) et des valeurs de référence dans la
littérature (58).
En plus, l'hyperleucocytose était réellement un
facteur de gravité de la maladie drépanocytaire homozygote au
regard de la relation avec effet-dose dépendant entre les proportions
des drépanocytaires SS en phase critique (crises vaso-occlusives et
crises hyperhémolytiques) et les Quartiles des leucocytes. Mais une
relation inverse entre proportions des drépanocytaires SS en phase
intercritique et Quartiles des leucocytes était démontrée
dans la présente étude. Ces résultats suggèrent un
processus inflammatoire chronique dans l'histoire naturelle de la
drépanocytose homozygote (14,16-21,59-61). La mobilisation des
leucocytes (15,28,62-65), des plaquettes (66), la protéine C
réactive, l'á2 macroglobuline,la transferrine,l' IL-2,l'
IL-4,l'IL- 6 et l' IL- 8 (17) constituent les marqueurs de cette
réponse inflammatoire chez les SS(28).
La relation entre l'hyperleucocytose et la crise
vaso-occlusive démontrée dans la présente étude
s'explique par le rôle direct des leucocytes dans l'apparition de
l'obstruction microvasculaire (15, 16). Le substratum biologique de la relation
entre sévérité de la drépanocytaires SS et
augmentation des leucocytes, serait l'adhésion des leucocytes à
l'endothélium vasculaire médiée par l'action des
molécules d'adhésion (28).
C'est pourquoi, le schéma physiopathologique classique
centré sur la polymérisation de l'HbS puis la falciformation et
la déshydratation du GR s'avère aujourd'hui insuffisant pour
expliquer la sévérité et
l'hétérogénéité des manifestations cliniques
caractéristiques de la drépanocytose homozygote. En effet,
l'expression des molécules d'adhésion par des leucocytes
prédispose à l'apparition des crises chez les
drépanocytaires SS (28).
4.2. Infection et globules
blancs chez les drépanocytaires SS
La présente étude confirme la
susceptibilité aux infections des drépanocytaires SS en phase
critique, signe distinctif de l'évolution de la maladie
drépanocytaire (71). Il s'agissait de sepsis, de méningite
aiguë, d'entérite fébrile, ostéomyélite,
pneumonie et d'infection urinaire. Les germes les plus souvent mis en cause
tels que le Streptococcus pneumoniae, Haemophilus influenza ou des
Salmonella (7) n'ont pas été mis en évidence dans la
présente étude compte tenu des limites des ressources.
Mais les drépanocytaires SS en phase intercritique
avaient des taux des leucocytes similaires à ceux des témoins AA
(ne montraient aucune susceptibilité aux différentes maladies
infectieuses observées).
Et au regard de l'augmentation des globules blancs, la
susceptibilité aux infections était identique entre les
drépanocytaires SS en phase critique.
4.3. Paludisme et globules
blancs chez les drépanocytaires SS
Ce travail démontre la réalité non
anecdotique (72) et les particularités de la relation existant entre
paludisme grave et drépanocytaires SS.
Contrairement aux témoins AA présentant une
relation négative entre paludisme grave et taux des leucocytes, les
drépanocytaires SS toutes phases confondues étaient
caractérisés par une relation en U entre paludisme grave et taux
des leucocytes : les taux les plus élevés de
paludisme grave coïncidant avec les extrêmes des leucocytes.
Mais, les drépanocytaires SS en phase intercritique et
les drépanocytaires SS en phase critique montraient respectivement une
protection très significative non seulement pour le paludisme grave et
mais aussi pour le neuropaludisme par rapport aux témoins AA. Plusieurs
travaux rapportent la protection des drépanocytaires SS
vis-à-vis du paludisme sévère (72-78). Différentes
hypothèses ont été formulées pour expliquer
cette protection :
- le développement de l'hématozoaire serait
entravé par l'hémoglobine S qui ne lui offrirait pas des
conditions adéquates pour son développement d'où
limitation de la charge parasitaire ;
- la séquestration des globules rouges infectés
par le plasmodium falciforme par le biais des molécules
adhésives, situation qui favorise la polymérisation, la
gélification ainsi que la diminution de la solubilité de
l'hémoglobine S. Et le parasite est soit lysé
mécaniquement par les cristaux de l'hémoglobine S soit que le
parasite est privé de métabolite pour sa nutrition ;
- l'invasion et la croissance du plasmodium dans les globules
rouges sont compromises ;
- le stress oxydant produit par l'hématozoaire
accroîtrait la dénaturation de l'hémoglobine S produisant
la protoporphyrine IX toxique pour le parasite ;
- l'hème serait toxique aussi pour le parasite (72).
A côté de ces mécanismes, ce travail
montre que l'augmentation des taux des leucocytes renforçait la
protection des drépanocytaires SS en phase critique vis-à-vis du
paludisme grave. Et en considérant les valeurs usuelles des leucocytes
définies chez les drépanocytaires SS, les patients
drépanocytaires SS avec intervalle adéquat des leucocytes
(pseudonormalité des leucocytes) étaient moins vulnérables
au paludisme grave, aux crises vaso-occlusives et au sepsis en comparaison
avec les drépanocytaires SS avec leucopénie et hyperleucocytose.
Le paludisme entraîne donc des modifications significatives de la
fonction et du nombre des leucocytes au cours du paludisme grave des
drépanocytaires SS (77). En effet, l'hyperleucocytose plus que la
leucopénie est l'anomalie hématologique associée à
la présence du pigment malarique, à la
sévérité et au pronostic sombre du paludisme
(79-82).
Toutefois, dans certaines régions du Nigeria (83), du
Ghana (84) et de Côte-d'Ivoire (85), des taux très
élevés de paludisme sévère ont été
rapportés chez les drépanocytaires SS. Ce qui ferait
suggérer que certains génotypes sont moins concernés par
le Plasmodium falciparum que d'autres génotypes en fonction des souches
plasmodiales : l'allèle HLA classe I B53 et HLA classe II
DRB1* 1352 conférant une très grande résistance contre le
paludisme grave (76), un certain polymorphisme pourrait aussi modifier la
réponse de l'hôte vis-à-vis du paludisme : une
variante des molécules intercellulaires d'adhésion ICAM-1
(86,87).
4.4. Leucocytes,
hémoglobine et anémie chez les drépanocytaires SS
Le comportement de dispersion des leucocytes était
diamétralement opposé à celui de l'Hb selon la
présence du gène S, de la sévérité de la
maladie drépanocytaire et du paludisme, du sepsis et du
décès. En effet, les leucocytes montraient une variabilité
très marquée chez les drépanocytaires SS, les
drépanocytaires SS avec paludisme grave, chez les drépanocytaires
SS en phase critique et chez les drépanocytaires SS
décédés. Contrairement à l'hyperleucocytose
associée au paludisme grave qui accentuait la variabilité des
leucocytes chez les drépanocytaires SS, l'effet du sepsis était
indifférent sur cette variabilité des leucocytes. Par contre,
l'extrême variabilité de l'hémoglobine était
notée chez les témoins AA et non chez les drépanocytaires
SS. Cette variabilité de l'Hb était renforcée chez les
témoins AA décédés.
Les présentes données suggèrent une
physiopathologie des leucocytes indépendante de celle des globules
rouges chez les drépanocytaires SS. Mais tel n'était pas le cas
chez les témoins AA. En effet, la relation négative et
significative entre le taux des leucocytes et celui de l'Hb observée
chez les témoins AA, ne l'était plus chez les
drépanocytaires SS aussi bien en phase critique qu'en phase post
critique (intercritique). L'anémie hémolytique chronique chez les
drépanocytaires SS, entraînant à la longue l'augmentation
des indices érythrocytaires et la stimulation de
l'hématopoïèse (88), pourrait expliquer la tendance
à l'augmentation de l'Hb, du reste, très péjorative
(40). Ce qui serait à la base de l'absence de corrélation entre
les leucocytes et l'Hb chez les drépanocytaires SS. Ceci est d'autant
plus vrai que la valeur moyenne des leucocytes des anémiques
était identique à celle des non anémiques chez les
drépanocytaires SS en phase critique.
Les leucocytes ont certainement une importance
particulière dans l'environnement du GR drépanocytaire ;
là aussi on ignore si la variabilité des leucocytes aurait
particulièrement une base génétique. Une mutation, base
génétique à un moment précis de la physiopathologie
des leucocytes chez les drépanocytaires SS répondant au stimulus
externe devrait être identifié.
4.5. Mortalité
hospitalière et drépanocytose
En régression logistique, le drépanocytaire SS,
l'AVC, le sepsis, l'entérite fébrile, l'hyperleucocytose et
l'âge < 10 ans étaient les prédicteurs de la
mortalité hospitalière globale. Mais en considérant la
fonction temps, la drépanocytose SS, le sepsis, l'AVC et l'âge
n'étaient plus retenus comme prédicteurs significatifs de la
mortalité globale. Mais l'hyperleucocytose de la drépanocytose
était toujours prédicteur significatif de décès
endéans les 10 jours d'hospitalisation aux soins intensifs.
Le seuil > 15.000 leucocytes/mm3 a
été identifié comme le seuil discriminant les
drépanocytaires SS et les témoins AA décédés
des survivants. Et en considérant les valeurs usuelles des leucocytes
chez les drépanocytaires SS, seule l'hyperleucocytose des
drépanocytaires SS était identifiée comme
prédicteur de décès à l'opposé de la
leucopénie et de l'intervalle adéquat des leucocytes montrant une
protection statistique vis-à-vis du décès. Cette
étude confirme le rôle létal de l'hyperleucocytose
(27,89,90) et du taux de leucocyte >15.000
éléments/mm3 (27). L'hyperleucocytose, l'anémie
sévère et la dactylite sont bien connues comme facteurs de
mauvais pronostic chez les enfants noirs américains
drépanocytaires (4).
4.6. Implication de l'hyperleucocytose dans la prise en charge
des drépanocytaires SS
La présente étude démontre que
l'augmentation des leucocytes est plus péjorative que leur diminution
chez les drépanocytaires SS. Elle définit le taux des leucocytes
>15.000 éléments/mm3 à combattre par des
médicaments actuellement disponibles comme l'Hydroxyurée, les
glucocorticoïdes et l' Epo (14,22,28). L'Hydroxyurée, les
glucocorticoïdes et l' Epo diminuent l'expression des molécules
adhésives et corrigent la déficience membranaire des leucocytes
chez les drépanocytaires SS. Ce qui tend à diminuer
l'inflammation et l'agrégation cellulaire. Plusieurs études ont
démontré l'augmentation de l'Hémoglobine foetale, une
diminution des crises vaso-occlusives et la mortalité après
administration de l'Hydroxyurée (93-102).
La présente étude attire l'attention du
clinicien à ne pas basculer les drépanocytaires SS traités
par l'Hydroxyurée en état de leucopénie. Cette
leucopénie était aussi comme l'hyperleucocytose associée
à un taux élevé de paludisme grave chez les
drépanocytaires. Ainsi, la baisse des leucocytes (neutrophiles)
liée à l'Hydroxyurée viendrait fragiliser le
drépanocytaire SS devant le paludisme grave.
L'absence du rôle des infections sur l'hyperleucocytose
des drépanocytaires SS ne justifierait plus l'administration abusive
d'antibiotique aux drépanocytaires SS de Kinshasa ayant une leucocytose
entre 10.000 et 12.000 GB/mm3 : le taux le plus bas de paludisme
grave chez le drépanocytaire SS coïncidant avec un taux de
leucocyte égal à 7.400 - 12.000 GB/mm3 ;
l'infection intervenant au-delà de 17000 GB/mm3.
La prise en charge des drépanocytaires SS gagnerait en
maintenant les leucocytes des drépanocytaires SS dans le strict
intervalle adéquat et physiologique (4000-15000
éléments/mm3). Ce qui pourrait améliorer
l'espérance de vie et diminuer les infections
généralisées, les crises vaso-occlusives et le paludisme
grave.
CONCLUSION
Les variations physiopathologiques des leucocytes sont
particulières chez le drépanocytaire SS (grande
variabilité des leucocytes) : leucopénie, intervalle
adéquat des leucocytes et hyperleucocytose avec des implications
biocliniques.
L'hyperleucocytose détermine la
sévérité et le caractère létal de la
drépanocytose SS. Mais elle renforce la protection des
drépanocytaires SS vis-à-vis du paludisme grave et du
neuropaludisme. Le taux des leucocytes >15.000
éléments/mm3 est le seuil létal chez le
drépanocytaire SS.
En dépit du moindre risque de mortalité
liée à la leucopénie chez les drépanocytaires SS,
la leucopénie et l'hyperleucocytose constituent des facteurs de risque
de paludisme grave. Mais l'hyperleucocytose et l'intervalle adéquat des
leucocytes renforcent la protection des drépanocytaires SS
vis-à-vis du paludisme grave.
L'extrême variabilité des leucocytes est
significativement associée au paludisme grave et à la
mortalité hospitalière des drépanocytaires SS.
Ce travail ouvre des nouveaux horizons dans la prise en charge
des drépanocytaires SS.
RECOMMANDATIONS
Les recommandations suivantes sont formulées au terme
de ce travail :
· le Gouvernement de la République
Démocratique du Congo est invité à soutenir le Programme
National de Lutte contre la Drépanocytose et les Organisations Non
Gouvernementales à renforcer l'évaluation et le traitement de la
drépanocytose à la lumière du comportement biologique des
leucocytes ;
· les professionnels de santé en charge des
drépanocytaires devraient avoir à l'esprit les variations
physiopathologiques des leucocytes : un taux de leucocytes >
15.000/mm3 signifie l'hyperleucocytose, la
leucopénie pour un taux < 4000/mm3 et le taux
adéquat « normal » égal à 4001 -
14999/mm3. Ils ne devront donc plus prescrire abusivement
l'antibiothérapie devant l'hyperleucocytose sans infection chez les
drépanocytaires ;
· les chercheurs congolais sont invités à
identifier les conditions environnementales capables de maintenir les
leucocytes des drépanocytaires dans l'intervalle adéquat.
REFERENCES
1. OKPALA I.
Management Of pain in sickle cell disease.
Journal of the Royal Society of Medicine 2002; 95: 456-58.
2. GIROT R, BEGUE P, GALACTEROS F.
La drépanocytose. Paris : John Libbey 2003 .p1
3. DIALLO D , TCHERNIA G.
Sickle cell disease in Africa.
Curr Opin Hematol 2002; 9: 111-6.
4. MILLER ST, SLEEPER LA, PEGELOW CH , et al.
Prediction of adverse outcomes in children with sickle of
disease.
N Engl J Med 2000; 342: 83-9.
5. BUNN H F.
Pathogenesis and treatment of sickle all disease.
N Engl J Med 1997; 337 : 762-9.
6. TURHAM A, WEISS LA, MOHANDIS N , et al.
Primary role of adherent leukocytes in sickle cell vascular
occlusion ; a new paradigm.
PNAS 2002; 99 (5): 3047-57
7. LABIE D.
Le taux des polynucléaires neutrophiles est un facteur
déterminent de l'évolution de le maladie drépanocytaire.
Hématologie 2000 ; 6 : 1(Abstract).
8. BACHIR D, VIRAY R.
Traitement du priapisme dans la drépanocytose avec
l'étiléfrine. Développement et santé 1997 ;
132 : 3-6.
9. SERJEANT G R.
La drépanocytose. In :
Hémoglobinopathies.
Annales de Nestlé 1998 ; 56 : 55-66.
10. GDODOE AD , KAMPATIBE N, BAKONDE B, et al.
Attitudes thérapeutiques chez les
drépanocytaires en phase critique et intercritique au Togo.
Médecine d'Afrique noire 1998 ; 45
(3) : 154-60.
11. GINI EHUNGU K.
Influence du piracetam sur les facteurs
hémorhéologiques dans la drépanocytose.
Thèse d'agrégation. Faculté de
Médecine, KUL ;1985.
12. BERCHEL C, DIARA JP, LORET H, et al.
Histoire naturelle de la drépanocytose.
Rev Prat 1992 ;42:1885-91.
13. MEHTA S R , ANNAN A A, BYRNS PJ, et al.
Opportunities to improve outcomes in sickle cell disease.
Am Fam Phys 2006; 74:303-10.
14. OKPALA I.
Leukocyte adhesion and the pathophysiology of sickle cell
disease. Curr Opin Hematol 2006; 13: 40-3.
15. OKPALA I.
The intriguing contribution of white blood cells to sickle
cell disease a red cell disorder.
Blood 2004; 18:65-73.
16. Wunt T.
The role of inflammation and leukocytes in the pathogenesis of
sickle cell disease: haemoglobinopathy.
Hematology 2001; 5: 403-12.
17. HEBBEL R P, OSCAROGIA GBONE, KAWL D,et al.
The endothelium biology of sickle cell disease: inflammation
and chronic vasculopathy.
Microcirculation 2004; 11:124-8.
18. KAUL DK, LUIX D, CHAONG S, et al.
Antiinflammatory therapy ameliorates leukocyte adhesion and
microvascular flow abnormality in transgenic sickle mice.
Am J Physiol Heart Circ 2004; 287:293-301.
19. CHIES JA ,NARDI NB.
Sickle cell disease: a chronic inflammatory condition.
Medical Hypoth 2001 57: 46-50.
20. PLATT OS.
Sickle cell anemia as an inflammatory disease.
J Clin Invest 2000: 106;337-8.
21. WAGNER M C, ECKMAN JR , WICKS TM.
Histaminic increases sickle erythrocyte adherence to endothelium.
Br J Haematol 2005; 132: 512 -22.
22. STEINBERG M H.
Pathophysiological based drug treatment of sickle cell
disease.
Tr Phar Sc 2006; 27:204 - 10.
23. HEBBEL R P, BOOGAERTS MA, EATON J W, et al.
Erythrocyte adherence to endothelium in sickle cell anemia: a
possible determinant of disease severity.
N Engl J Med 1980; 302:992-3.
24. COLLER B S.
Leukocytes and ischemic vascular disease morbidity and
mortality. Is it time to intervene?
Artherioscler Thromb Vasc Biol 2005; 25: 658-70.
25. GLADWIN M T, RODGERS GP.
Pathogenesis and treatment of acute chest syndrome of sickle
cell anaemia.
Lancet 2000; 355:1476-8.
26. BALKARAN B, CHAR G, MORRIS JS , et al.
Stroke in cohort of patients with homozygous sickle cell disease.
J Pediatr 1992;120:360 - 6.
27. PLATT OS, BRAMBILA DJ, ROSSE WF , et al.
Mortality in sickle cell disease .Life expectancy and risk
factors for early death.
Blood 1994; 330:1639-44.
28. OKPALA I, DANIEL Y, HAYNES R , et al.
Relations between the clinical manifestations of sickle cell
disease and the expression of adhesion molecules on white blood cells.
Eur J Heamatol 2002; 69:135-44.
29. LONGO MBENZA B , NGIYULU R , KIZUNDA P, et al.
Gallblader disease in young Congolese with sickle cell anemia: An
ultrasound survey.
Journal of Tropical Pediatr 2004; 50:73-77.
30. NGIYULU R , LONGO-MBENZA B , LANKOKO NTOBE, et al.
Blood pressure and hypertension in African adults with sickle
cell disease.
Journal de l' IRSS 2005;1:1-6.
31. GALACTEROS F.
Drépanocytose. Encyclopédie Orphanet.
In :http://www.orpha.net/data/patho/fr-drepanocy.pdf,2000.
32. MIEKOUTIMA LE.
Les effets bénéfiques de l'Hydroxyurée dans
l'anémie falciforme de l'enfant. Thèse de Doctorat.
Faculté de Médecine, Université de Genève,2004.
33. LABIE D , ELION J.
Génétique et physiopathologie de la
drépanocytose .In : Girot R Bégue P, Galactéros
F . La drépanocytose
Paris : John Libbey 2003.p1-11.
34. GALACTEROS F.
La drépanocytose, physiopathologie, physiologie et
diagnostic.
Rev Prat 1995 ;45 :351-60.
35. WUN T PAGLIERONI T, RANGASWAMI A, et al.
Platelet activation in patients with sickle cell disease.
Br J Haematol 998 ; 100 :741-9.
36. CONSTANT I.
Drépanocytose et anesthésie. Conférence
d'actualisation
Paris : Elsevier ; 1997.
37. POILLON WN ,KIM BC, CASTRO O.
Intracellular hemoglobin S polymerization and clinical
severityofsicklecell anemia.
Blood 1998; 91:1777-83.
38. ELION J, LABIE D.
Bases physiopathologiques moléculaires et cellulaires du
traitement de la drépanocytose.
Hématologie 1996 ; 2 :499-510.
39. STEINBERG MN.
Treatment of sickle cell disease is based on pathophysiology Hbs
polymerization. In: Buchanan GR ,Debaun MR , Quinn CT, et al. Sickle cell
disease.
American Society of Heamatology USA 2004.p1-14
40. GALACTEROS F.
Bases physiopathologiques de la drépanocytose, prise en
charge et actualités thérapeutiques.
Bull Soc Pathol Exot 2001 ;94 :77-9.
41. LABIE D et ELION J.
L'endothélium vasculaire, composante majeure de la maladie
drépanocytaire : les cellules circulantes en sont le reflet.
M/S 1998 ; 14 :352-55.
42. LESPRIT E et LESPRIT P.
Infections bactériennes dans la drépanocytose.
Rev Prat 2004 ;54 :1574-7.
43. BLUNN A, YEGANEH S, VIGDER F, et al.
Endothelium function in patients with sickle cell anemia during
and after sickle cell crisis.
J Thromb Thro 2005; 19:83-86.
44. STEINBERG M H.
Management of sickle cell disease.
N Engl J Med 1999; 340:1021-30.
45. STEINBERG M H , RODGERS GP.
Pathophysiology of sickle cell disease: role of genetic and
cellular modifiers.
Semin Hematol 2001; 38:299-306.
46. LEPAGE PH , DRESS MF, FORGET P , et al.
Infections et prophylaxies anti infectieuses dans la
drépanocytose. Rev Méd de Liège 2004 ;
59 :145-148.
47. BEGUE P, HERBRETEAU VB.
Infections chez l'enfant drépanocytaire : aspects
cliniques et prévention. In :Girot R ,Bégue P,et
Galactéros F.La drépanocytose
Paris : John Libbey Paris 2003.
48. TAMOUZA R , NEONATO MG, BENSON M, et al.
Infections complications in sickle cell disease are influenced
by HLA class II alleles.
Hum Immune 2002; 63:194-9.
49. BELCHER JM, MARKER PM, WEBER JP, et al.
Activated monocytes in sickle cell disease: potential role in
the activation of vascular endothelium and vaso-occlusion.
Blood 2000; 96:2451-54.
50. LARD LR, MUL FP, DHAAS M, et al.
Neutrophil activation in sickle cell disease.
J Leukocyte Biol 1999; 66:411-5.
51. FRANCIS RB and HAYWOOD L.
Elevated immunoreactive tumor necrosis factor and
interleukin-1 in sickle cell disease.
J Natl Med Assoc 1992; 84: 611-615.
52. CROIZA TH.
Circulating cytokines in sickle cell patients during steady
state.
Br J Haematol 1994; 87:592-597.
53. FRENETTE PS.
Sickle cell vaso-occlusion: multistep and multi cellular
paradigm. Curr Opin Hematol 2002; 9:101-106.
54. OKPALA I , DANIEL Y R .
High expression of á Mâ2 integrin and
L-selectin by leukocytes of patients with severe manifestations of sickle cell
disease.
Blood 2002; 100 (suppl):11a.
55. WOLNEY AM , EVANDRO T M, DELMA MC.
Doppler echocardiography study in adolescents and young adults
with sickle cell anemia.
Arq Braq Cardiol 1999; 73: 469-74.
56. R. Development core team R:
A computing. R Fundation for statistical computing. R
Fundation for statistical computing Vienna, Austria 2005
57. WANE J.
Etude des valeurs de référence de quelques
paramètres biologiques de l'adulte jeune en milieu urbain
zaïrois.
Mémoire de spécialisation en Biologie Clinique,
Université de Kinshasa , 1985.
58. NGANGA M.
Valeurs de référence de quelques
paramètres hématologiques et biocliniques des enfants de 4
à 13 ans à Kinshasa
Mémoire de spécialisation en Biologie Clinique,
Université de Kinshasa , 2006.
59. WAGENER FA, EGGERT A, BOERMAN OC, et al.
Heme is a potent inducer of inflammation in mice and is
counteracted by heme oxygenase. Blood 2003: 98: 1802-11.
60. MAKIS AC, HATZIMICHAEL EC , et BOURANTAS IC.
The role of cytokines in sickle cell disease.
Annals of Haematol 2000; 79:407-13.
61. WALNET PS , ECKMAN JR.
Inflammatory mediators promote strong sickle cell adherence to
endothelium under venular flow conditions.
Am J Haematol 2003; 73:215-24.
62 .ANYAEGBU C C, OKPALA I , AKEN `OVA AY,et al.
Peripheral Blood neutrophil count and candidacidal activity
correlate with the clinical severity of sicke cell anaemia.
Eur J Haematol 1998 : 60 : 267 - 8
63. AWOGU AU.
Leukocyte counts in children with sickle cell anaemia :
Usefulness of stable state values during infections .
West Afr J Med 2000 ; 19 : 55 - 8.
64. KASSCHAU MR, BARABINO GA, BRIDGERS KR, et al.
Adhesion of sickle neutrophilis and erythrocytes to
fibrinonectin. Blood 1996; 87:771-80.
65. FALDON, VORDERMEIER S, PEARSON TC ,et al.
Blood polymorphonuclear leukocytes from the majority of sickle
cell patients in the crisis of disease show enhanced adhesion to vascular
endothelium and increased expression of CD64.
Blood 1998;91:266-74.
66. OKPALA IE.
Steady state platelet count and complications of sickle
disease. Haematol J 2002 ; 3 : 214 - 215.
67. VICHINSKY EP, NEUMAYR LM , EARLES AN, et al.
Causes and outcomes of the acute chest syndrome in sickle cell
disease . National acute chest syndrome study Group.
N Engl J Med 2002 : 342 : 1855 - 65.
68. PIETERS RC, ROGER RA, SALEH AW et al.
Molgramostin to treat SS - sickle cell leg ulcers.
Lancet 1995 : 343 ; 528.
69. FRENETTE PS.
Sickle cell vaso-occluscin : heterotypic, multicellular,
aggregations driven by leukocyte adhesion. Microcirculation 2004; 22: 167-
77.
70. OSAROGLAGBON UR, CHOOG S, BELCHER JD, et al.
Reperfusion injury Pathophysiology in sickle cell transgenic
mice. Blood 2000 ; 96 : 314 - 20.
71. OHENE F K , STEINBERG MH.
Clinical aspects of sickle cell anaemia in adults and
children. In: Steinberg MH, Forget BG, Thgys D, et al.
Disorders of haemoglobin. .
Cambrige : Cambridge University Press 2001.p 611 - 70.
72. MULUMBA M P.
Paludisme et drépanocytose : Problématique,
faits et hypothèses . Congo Médical 2003 ;3 : 1053 -
55.
73. GENDREL D, KOMBILA M, NORDOU M,et al.
Protection against plasmodium falciparum infection In children
with hemoglobin S.
Pediatr Inf Dis J 1991 ;10: 620 - 1 .
74. PRADA J, ALABI SA , BIENZLE U.
Bacterial strains isolated from blood cultures of Nigerian
Children with cerebral malaria.
Lancet 1993 ; 342 : 11-4.
75. CRAWLEY Y.
Seizures and status epilepticus in childhood cerebral malaria.
Q J Med 1996 ; 89 : 591 - 7.
76. MENDIS K, SINA BJ, MARCHESNI P, et al.
The neglected burden of Plasmodium vivax malaria.
Am J Trop Med Hyg 2001 ; 64 ( 1-2 suppl ) : 97 - 105.
77.WEATHERALL DJ ,MILLER LH, BARUCH DI ,et al.
Malaria and red cell.
American Society of Hematology USA2002.p35-57
78. ALUOCH JR.
Higher resistance to plasmodium infection patients with
homozygous sickle cell disease in Western Kenya.
Trop Med and Inter Health 1997; 2:568-71.
79 . PASCUAL CC , ROBERTS DJ
Clinical and haematological features of malaria. In: Malaria and
red cell. Weatherall DJ ,Miller LH, Baruch D I ,et al. Malaria and red cell.
American Society of Haematology USA 2002. p42-5.
80. English M
Deep Breathing in children with severe malaria : indicator
of metabolic acidosis and poor outcome .
Am J Trop Med Hyg 1996 ; 55 : 521 - 24.
81. HODDER AN.
The disulfide bond structure of plasmodium apical membrane
antigen - 1 .
J Biol Chem 1996 ; 271 : 29446 - 52.
82. TRIGLIA T.
Apical membrane antigen 1 plays a central role in erythrocyte
invasion by Plasmodium species.
Mol Microbiol 2000 : 38 : 706 - 18.
83. UZUEGWU P , ONWURAH AE
Prevalence of haemoglobinopathy and malaria diseases in the
population of old Aguata Division, Anambra state, Nigeria . Biokemistri
2003 ; 15 ( 2) : 57-66.
84. LANDGRAF B., KOLLARITSCH H., WIEDERMANN G ,et al.
Parasite density of plasmodium falciparum for influence of sex
hormone.
Trans Roy Sol Trop M & Hyg1994 ;1 :73 -4.
85. SANGARE A, SANAGO I, EBONGO E, et al.
Contribution à l'étude des relations entre la
drépanocytose et le paludisme.
Médecine d'Afrique noire 1990 ; 37 (51) : 268 -
73.
86. LUSE SA , MILLER LM.
Plasmodium falciparum malaria ultrastructure of parasitized
erythrocyes in candine vessels.
Am J Trop Med Hyg 1971 ; 20 : 655 -60.
87. LANGRETH SG , PETERSON E.
Pathogenicity, stability, and immunogenicity of a knoblers clone
of Plasmodium falciparum : separate roles of for ICAM-1, CD 36 and
thrombrospondim.
Br J Haemotol 1994 ; 87 : 167 - 70.
88. OMOTI CE.
Heamatological values in sickle cell anaemia in state state and
during vaso occlusive crisis in Benin city, Nigeria.
Annals of African Med 2005;4:62-7.
89. LEIKIN SL, GALLAGHER D , KINNEY TR, et al.
Mortality in children and adolescents with sickle cell
disease.
Pediatr 1989 ; 84 : 503-8.
90. POWARS DR.
Natural history of sickle cell disease : the first ten years
.
Semin Hemotol 1975 ; 12 : 267 - 285.
91. CHENE F.K., WEINER S.J., SLEEPER L.A.,et al.
Cerebrovascular accidents in sickle cell disease: rates and risk
factors.
Blood 1998;91:288-294.
92. PLATTOS, THORINGTON BD, BRAMBILA DJ, et al.
Pain in sickle cell disease: rates and risk factors.
N Engl j Med.325;11,191.
93. STEINBERG MH.
Foetal hemoglobin in sickle cell anemia . Determinants of
response to hydroxyurea.
Blood 1997 ; 89 : 1078 - 88.
94. BAKANARY SM .
Mortality in sickle cell patients in hydroxyurea
Blood 2004 : 105 ; 545 -7.
95. CHARACHE SM.
Design of the multicenter study of hydroxyurea in sickle cell
anemia Control. Clin. Trials 1995 : 16 : 432-46.
96. ZIMMERMAN SA.
Sustained Long terms hematologic efficacity of hydroxyurea at
maximum tolerated dose in children with sicke cell disease .
Blood 2004 : 103 : 2039 - 45.
97. FESTER A .
Five years of experience with hydroxyurea in children and young
adults with sickle cell disease.
Blood 2001 : 97 : 3028 - 32
98. KINNEY TR.
Safety of hydroxyurea in children with sickle cell anaemia:
results of the HUG-KIDS study, a phase I/II trial.
Blood 1998:94:1540-4.
99. WANG WC.
A two year pilot trial of hydroxyurea in very young children
with sickle cell anaemia.
J Pediatr 2001;139:790-6.
100. GULBIS B.
Hydroxyurea for sickle cell deisease in children and
prevention of cerebrovascular events: the Belgium experiency.
Blood 2005;105:2685-90.
101. STEINBERG MH.
Effects of hydroxyurea in mortality and morbity in adult
sickle cell anaemia :riks and benefits up to 9 years of treatment. J Am Med
Assoc 2003;89:1645-51.
102. WARE RE, ZIMMERMAN SA, SCHULTZ WH.
Hydroxyurea as an alternative to blood transfusion fort he
prevention of recurent stroke in children with sickle cell disease.Blood 1999;
3200-6.
TABLE DES MATIERES
DEDICACE
ii
REMERCIEMENTS
iii
ABREVIATIONS
vii
PLAN DU TRAVAIL
viii
INTRODUCTION
1
CHAPITRE 1. GENERALITES
6
1.1. La drépanocytose
6
1.1.1. Définition
6
1.1.2. Physiopathologie
6
1.1.3. Globules blancs et drépanocytose
9
CHAPITRE 2. MATERIEL ET METHODES
11
2.1. Nature et période de l'étude
11
2.2. Cadre d'étude
11
2.3. Population d'étude
11
2.3.1. Critères d'inclusion
11
2.3.2. Critères d'exclusion
12
2.4. Matériel
12
2.5. Approche méthodologique
13
2.5.1. Organisation et déroulement du travail
13
2.5.2. Collecte des données
13
2.6. Définitions opérationnelles
14
2.7. Analyses statistiques
15
CHAPITRE 3. RESULTATS
17
3.1. Morbidité
17
3.1.1. Données anamnestiques
17
3.1.2. Données hématologiques
18
3.1.3. Tableau clinique
19
3.1.4. Globules blancs et morbidité
19
3.1.4.1. Variations des globules blancs
19
3.1.4.1.1. Moyennes des globules blancs
19
3.1.4.1.2. Quartiles des globules blancs et types de
morbidité
20
3.1.4.1.3. Manifestations critiques et Quartiles des globules
blancs
22
3.1.4.2. Infections bactériennes et sepsis
24
3.1.4.3. Paludisme, sévérité de la
drépanocytose et globules blancs.
24
3.1.4.4. Infections, sepsis et globules blancs
29
3.1.4.4.1. Drépanocytaires en phase critique versus
drépanocytaires en phase post critique
29
3.1.4.4.3. Drépanocytaire SS en phase post critique versus
témoins AA
30
3.1.4.4.4. Influence des globules blancs sur la
susceptibilité aux sepsis ostéomyélite , pneumonie et
infection urinaire
30
3.1.4.5. Hémoglobine, anémies et globules blancs
32
3.2. Globules blancs et mortalité hospitalière
34
3.2.1. Globules blancs et issue vitale
34
3.2.2. Sepsis globules blancs et issue vitale
35
3.3. Déterminants indépendants de la
mortalité hospitalière après analyse multivariée au
sein de la population totale et sans considération du temps
36
3.4. Fonction de survie aux soins intensifs
37
3.5. Définition des valeurs usuelles des leucocytes
(référence) chez les drépanocytaires SS
41
3.5.1. Valeurs de référence chez les
drépanocytaires SS
41
3.5.2. Extrême variabilité des globules blancs chez
les drépanocytaires
42
3.5.3. Intervalle adéquat (physiologiques) des globules
blancs chez les drépanocytaires SS
48
3.5.4. Variabilité de l'hémoglobine chez les
drépanocytaires SS
49
CHAPITRE 4. DISCUSSION
52
4.1. Phase critique et globules blancs chez les
drépanocytaires SS
52
4.2. Infection et globules blancs chez les drépanocytaires
SS
54
4.3. Paludisme et globules blancs chez les drépanocytaires
SS
54
4.4. Leucocytes, hémoglobine et anémie chez les
drépanocytaires SS
57
4.5. Mortalité hospitalière et drépanocytose
58
CONCLUSION
61
RECOMMANDATIONS
62
REFERENCES
63
TABLE DES MATIERES
80
| 

