|
République Algérienne démocratique et
populaire
Ministère de l'enseignement supérieur et de la
recherche scientifique
UNIVERSITÉ DE
JIJEL
Faculté des sciences de
l'Ingénieur
Département de chimie industrielle
Mémoire
Réalisé par:
BOUZNIT YAZID
Pour obtenir le diplôme
«
Ingénieur d'état »
Spécialité: Chimie
Industrielle
Option : génie des
matériaux
Thème:
Synthèse et caractérisation de
pérovskites à base
de Lanthane
Encadreur : M me Amirouche Leila
Promotion 2007
Table des matières
|
Remerciements
|
|
|
Introduction générale
|
01
|
|
I Présentation générale des
pérovskites simples
|
02
|
|
I.1 Introduction
|
02
|
|
I.2 Description de la structure perovskite
|
02
|
|
I.3 conditions de stabilité d'une
structure pérovskite
|
..04
|
|
I.3. a Facteur de tolérance
|
..04
|
|
I.3. b Ionicité des liaisons
|
..04
|
|
I.4 Distorsions de la structure idéale
|
05
|
|
I.5 propriétés physiques des
pérovskites
|
.06
|
|
II Diffractométrie des rayons X
|
07
|
|
II.1 Introduction
|
..07
|
|
II.2 Production des rayons X
|
.07
|
|
II.2.1 Tube à cathode chaude
|
.....07
|
a) Dimension du foyer
|
08
|
b) Choix de l'anticathode
|
|
.08
|
|
|
c) Rendement d'émission des RX
|
08
|
|
II.3 Rayonnement monochromatique
|
....09
|
|
II.4. L'interaction des photons X avec la
matière
|
09
|
|
1I.4.1 Description microscopique des
interactions
|
..10
|
|
II.4.2 La loi d'atténuation du
rayonnement X
|
11
|
|
II.5 Diffraction des rayons X et
détermination d'une distance interatomique
|
12
|
|
II.5.1 condition de diffraction
|
13
|
|
II.5.2 intensité diffracté
|
13
|
a) Facteur de diffusion
|
....13
|
b) Facteur de structure
|
|
13
|
|
|
II. 6 Méthode de Debye-Scherrer
(méthode des poudres)
|
14
|
|
II.6.1 principe de la méthode
|
..14
|
|
II.6.2 appareillage de la méthode
|
15
|
|
II.6.3 source d'erreurs
|
.15
|
III méthodes de préparation des
pérovskites ....16
III.1 Introduction 16
III.2 Synthèse par Réaction
à l'état solide ...16
a) Matières premières 16
b) Mélange, Broyage .17
c) Calcination 17
d) Rebroyage 17
e) Problèmes rencontrés 17
III.3 Synthèse par voie humide (solution)
18
III.3.1 Procédé sol-gel 18
III.3.2 Co-précipitation 19
III.3.3 Hydrothermales ...20
|
IV Résultats expérimentaux
|
.....21
|
|
IV.1 Introduction
|
21
|
|
IV.2 Synthèse des échantillons
|
.... 21
|
a) Les produits de départ (les oxydes)
|
21
|
b) Les réactions mises en jeu
|
|
22
|
|
|
b.1 Exemple de calcul
|
22
|
|
IV.3 Diffraction des rayons X
|
22
|
|
IV.4 Etude des spectres
|
. 22
|
|
IV.5 Interprétation des spectres des
trois échantillons
|
.23
|
|
IV.5.1 L'échantillon E1
|
23
|
|
IV.5.2 L'échantillon E2
|
27
|
|
IV.5.2 L'échantillon E3
|
31
|
Conclusion générale 40
Annexe
Références bibliographiques
Reztree/bezteztree.rd?
Je voudrais exprimer ma profonde reconnaissance
à madame amirouche, qui a suivi ce travail
avec beaucoup d'intérêt et de
patience. Je la remercie pour les nombreuses discussions que
nous
avons mené sur ce thème. Sa gentillesse, sa
confiance et sa
disponibilité m'ont aussi été d'un grand
soutien.
De même, j'exprime mes remerciements les plus chaleureux
à
Monsieur Ben Abbas, professeur à l'université de
Jijel, pour les
conseils très précieux concernant les mesures de
diffraction des RX.
Aucun mot ne saurait témoigner de l'étendue des
sentiments que
j'épreuve à son égard.Je souhaite que dieu
le préserve une longue vie.
J'associe à ces remerciements tous les enseignants qui
ont
contribué, véridiquement
et sincèrement, en nous montrant les
vraies valeurs de la science et la technologie.
Je suis très reconnaissant à mes chers amis ;
Bouakacha Riad
et Hadadi Toufik pour l'intérêt qu'ils ont
apporté à ce travail.Je les
en remercie vivement d'être avec moi surtout en ce qui
concerne
l'outil informatique.
Je remercie infiniment tous ceux que je connais de prés
ou de
loin et que j'ai involontairement oubliés...
Introduction générale
Introduction générale
La matière dont est formé le monde qui nous
entoure est composée de particules discrètes, ayant une taille
submicroscopique, dont les lois de comportement sont décrites par les
théories atomiques. Les états d'organisation de la matière
sont très varies depuis le désordre complet des atomes ou des
molécules d'un gaz sous faible pression jusqu'à l'ordre quasi
parfait des atomes dans un monocristal.
Les propriétés des matériaux sont
définies par la nature des liaisons chimiques, l'arrangement atomique et
la microstructure, l'étude des relations entre l'organisation à
l'échelle atomique, la microstructure et les propriétés
des matériaux constituent le domaine de la science des
matériaux.
Les différentes études effectuées depuis
les années cinquante jusqu'à nos jours sur les matériaux
de type pérovskite ,démontrent la richesse de cette famille .En
effet ,il existe de multiple combinaisons possibles , notamment selon la
valence des cations utilisés,on peut avoir des combinaisons I-V comme
KNbO3, II-IV comme SrCoO3 ou bien III-III comme LaCoO3.d'autres combinaisons
sont encore possibles en remplaçant l'oxygène par un autre anion
comme S ou F.[1] Bon nombre de ces phases ABO3 acceptent des sous
stoechiométries plus ou moins importantes qui se traduisent par des
lacunes sur les deux sites cationiques mais aussi sur le site anionique. Cette
caractéristique unique permet d'atteindre des états de valence
mixtes qui confert aux phases pérovskites des propriétés
physiques ou chimiques remarquables.
Ainsi, ce travail a été axé sur la
synthèse des oxydes à base de Lanthane ayant la structure
pérovskite de formule générale LaMO3 ;(M= Fe,Mn,Ti) par la
méthode céramique et leur caractérisation par diffraction
des rayons X.
Notre travail comporte quatre chapitres :
Le premier chapitre fait une brève présentation
générale des pérovskites simples de type ABO3 du point de
vue de leurs structures cristalline ainsi que les propriétés
physiques.
Le deuxième chapitre présentera une approche
concernant les RX : de leur production jusqu'à leur utilisation en
radiocristallographie.
Le troisième chapitre présente une étude
visant les méthodes les plus utilisées pour la préparation
des oxydes à structure pérovskite.
Alors que le dernier chapitre englobe l'ensemble des
résultats expérimentaux, en donnant des remarques, tirant des
conclusions concernant notre travail.
En fin, en achèvant ce travail par une conclusion
générale qui rassemble le résumé des
résultats obtenus.
I.1 Introduction:
Les pérovskites forment une des principales familles
d'oxydes cristallins. Leur nom provient du minéral CaTiO3
qui présente une structure cristalline analogue. Ce
minéral fut décrit pour la première fois en 1830 par le
géologue Gustav Rose qui l'a nommé en l'honneur
d'un grand minéralogiste russe, le comte Lev Aleksevich von
Perovski.
La maille typique d'une pérovskite a une
symétrie cubique, mais un nombre important d'exceptions sont connues,
celles-ci présentent des structures voisines plus ou moins distordues.
La composition chimique d'un oxyde à structure pérovskite est le
plus souvent constitué d'un cation alcalino-terreux (A), un cation de
transition tétravalent (B) et des anions oxyde. Cette description
(AIIBIVO3) correspond à la composition de
référence CaTiO3 dont la structure est orthorhombique.
Cependant, des compositions AIIIBIIIO3
et AIBVO3 sont également connues depuis longtemps.
[1]
I.2 Description de la structure pérovskite
:
La pérovskite idéale ABO3 est décrite
dans le groupe d'espace Pm-3m. C'est une structure
tridimensionnelle dans laquelle le plus petit cation B, se trouve dans un
environnement octaédrique, les octaèdres étant
reliés entre eux par les sommets, le cation A, se trouvant au centre de
polyèdres de 12 oxygènes, reliés entre eux par des faces
carrées (Figure.I.1). En fonction du choix de l'origine, il y a deux
façons de décrire la structure. Dans la première A se
trouve à l'origine, dans la position 1a (0, 0, 0), B se
trouve au centre du cube, dans la position 1b (1/2, 1/2, 1/2),
et les oxygènes se trouvent au milieu de chaque face, dans la position
3d (0, 1/2,1/2) (Figure. I.2.a). Dans la deuxième
façon, l'origine est déplacée d'un vecteur (1/2, 1/2,
1/2), ce qui amène A à occuper la position 1b
(1/2, 1/2, 1/2), B la position 1a (0, 0, 0), les
oxygènes se trouvant au milieu de chaque arrête, dans la position
3c (1/2, 0, 0) (Figure. I.2.b). [2]
Le réseau pérovskite est un ensemble très
compact qui ne permet pas la formation des compositions interstitielles. En
revanche, de nombreuses substitutions sont possibles sur les sites A ou B ou
sur les anions oxygènes. Chaque composition ainsi obtenue peut
présenter une structure pérovskite distordue, en fonction de la
taille des ions occupant les sites A, B et O. [1]
On peut distinguer deux types de pérovskites suivant
l'occupation des sites A et B : -Les pérovskites simples dont les sites
A ou B sont occupés par un seul type d'atome : BaTiO3, KNbO3, NaTaO3,
PbTiO3....
-Les pérovskites complexes dont l'un des deux sites A ou B
est occupé par deux types d'atomes : PbMg1/3Nb2/3O3, PbSc1/2Ta1/2O3,
Na1/2Bi1/2TiO3,... [3].
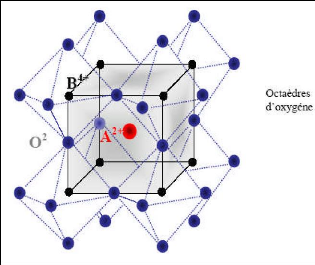
Fig.I.1 Arrangement des octaèdres dans
la maille idéale pérovskite (ABO3)
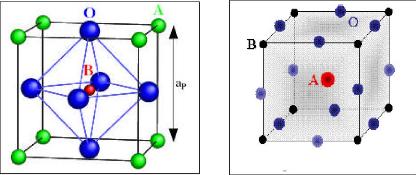
Fig.I.2.a maille élémentaire de la
pérovskite Fig.I.2.b maille élémentaire
de la pérovskite Simple cubique ABO3 (origine en A).
Simple cubique ABO3 (origine en B).
I.3 Conditions de stabilité d'une structure
pérovskite :
La stabilité de la structure pérovskite
dépend essentiellement de deux facteurs :
a) Facteur de tolérance t :
Goldschmidt a défini un critère
dimensionnel, appelé facteur de tolérance qui
tient compte de la taille des ions pour caractériser
les différentes structures dérivées de la structure
perovskite:
r ( ) ( )
A + O
r
t ? Ou
2 [ ( ) ( )]
r B + O
r
d ( )
A -- O
t ? (I.1)
2 ( )
d B -- O
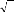
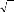
Où r(A), r(B) et r(O) sont respectivement les rayons
ioniques des cations A, B et de l'oxygène, d(A-O) et d(B-O) les
distances cation-oxygène.[2]
D'après ce critère, la structure cubique est
observée pour t très proche de 1, les limites de
stabilité de la phase pérovskite (plus ou moins distordue)
étant définies par t compris entre 0.75 et 1.06
. En fonction de la valeur du facteur de tolérance, on peut distinguer
plusieurs situations, schématisées dans le tableau suivant :
Tableau I.1 évolution des structures
cristallines en fonction de t. [1]
|
t<0.75 ilménite
|
0.75<t<1.06
pérovskite
|
t>1.06 hexagonal
|
|
0.75<t<0.95 Distorsion Orthorhombique
|
0.96<t<0.99
Distorsion
rhomboédrique
|
0.99<t<1.06
cubique
|
Remarques :
1) La structure idéale est rarement rencontrée. Le
plus souvent, on trouve des formes distordues, dans lesquelles la
symétrie est abaissée par la rotation des octaèdres BO6.
[2]
2) Pour la valeur idéale t 1, la structure perovskite
adopte une symétrie cubique. Un motif ABO3 par maille suffit alors pour
décrire l'arrangement structural. Le paramètre de maille,
noté a p, est proche de 4Å et correspond à
la distance B-O- B. Toutefois, beaucoup de pérovskites présentent
une symétrie moins élevée qui nécessite parfois le
choix d'une maille plus grande, multiple de la maille cubique idéale.
Ainsi, de nombreuses pérovskites présentent des transitions
structurales qui s'accompagnent d'un abaissement de symétrie. [4]
b) L'ionicité des liaisons anions-cations :
Le caractère ionique d'une composition ABO3 est
quantifié d'après l'échelle de Pauling
à partir de la différence
d'électronégativité :
AE = XA -- 0 + B -- 0
X
(I.2)
2
Où ÷ A-O et ÷ B-O sont
respectivement les différences d'électronégativité
entre A et O, B et O.
La structure pérovskite est d'autant plus stable que les
liaisons mises en jeu présentent un fort caractère ionique.
[5]
I.4 Distorsions de la structure idéale:
La structure idéale est rarement rencontrée. Le
plus souvent, on trouve des formes distordues, dans lesquelles la
symétrie est abaissée par la rotation des octaèdres BO6,
leurs mailles présentent alors de légères
déformations de type quadratique,
rhomboédrique ou orthorhombique
dûes à une très faible modification des
paramètres de la maille cubique .
Ces distorsions correspondent à une déformation
des octaèdres d'oxygène avec décentrage de l'ion B qui se
produit suivant certaines directions privilégiées par les
éléments de symétrie du nouveau système cristallin.
Ces directions sont les suivantes (Figure.I.3) :
-les 3 axes d'ordre 4 (A4 ) dans la phase quadratique.
-les 6 axes d'ordre 2 (A2 ) dans la phase orthorhombique.
-les 4 axes d'ordre 3 (A3 ) dans la phase
rhomboédrique.
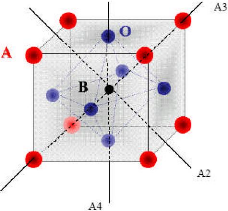
Figure.I.3 Directions de déformations
dûes au déplacement de l'ion B dans l'octaèdre.
Ces déplacements des ions B sont dûs
essentiellement à un problème de liaisons B-O dans
l'octaèdre des oxygènes. Par exemple, en prenant le même
ion A soit le Baryum on obtient BaTiO3 quadratique et ferroélectrique
parce que l'ion Ti4+ est assez petit, ce qui lui permet de se
décentrer dans l'octaèdre, tandis que BaSnO3 est cubique et
paraélectrique parce que l'ion Sn4+ plus gros est calé
au centre de l'octaèdre, il peut cependant y avoir des pivotements
d'octaèdres avec des symétries non cubiques et
paraélectriques ( CaTiO3, CaSnO3 etc.) [3]
I.5 Propriétés des pérovskites :
Les pérovskites, un véritable coffre au
trésor pour la science des matériaux. Ces matériaux
céramiques avec leur structure cristalline particulière
présentent une variété étonnante de
propriétés électroniques et magnétiques. Pour cela
on procède à la substitution de certains éléments
de leur grille cristalline, comme dans un jeu de construction, ce qui permet
d'obtenir des matériaux fonctionnels aux caractéristiques
«sur mesure» dans certaine mesures :
Les pérovskites jouent un rôle important dans
l'électronique moderne. Elles sont utilisées dans les
mémoires, les condensateurs, les appareils à micro-ondes, les
manomètres et l'électronique ultrarapide ; (train à
sustentation magnétique).
Elles sont supraconductrices à des températures
relativement élevées, elles transforment la pression
mécanique ou la chaleur en électricité
(piézoélectricité), accélèrent les
réactions chimiques (catalyseurs) et changent soudainement leur
résistance électrique lorsqu'elles sont placées dans un
champ magnétique (magnétorésistance).
Ces matériaux très prometteurs trouvent de plus
en plus des applications dans les céramiques transparentes, les
colorants non polluants, les cellules photovoltaïques ou les piles
à combustible. Les pérovskites ont des possibilités
d'utilisation quasi universelles car il est possible de faire varier dans des
limites très larges leurs propriétés. C'est aussi la
raison pour laquelle on les appelle aussi les caméléons
chimiques. [6]
Les exemples cités ci-dessus étaient
destinés à montrer combien les composés à structure
pérovskite présentent un intérêt dans les
applications industrielles. L'objectif de chimiste du solide consiste avant
tout à synthétiser de nouveaux matériaux et à
essayer de les bien caractériser, voir ressortir la potentialité
de ces matériaux à une application.
Chapitre II Diffractométrie des rayonx X
II.1 Introduction:
La diffractométrie de rayons X (DRX, on utilise aussi
souvent l'abréviation anglosaxone XRD pour X-ray diffraction) est une
technique d'analyse basée sur la diffraction des rayons X sur la
matière. La diffraction n'ayant lieu que sur la matière
cristalline, on parle aussi de radiocristallographie. Pour les matériaux
non cristallins, on parle de diffusion.
Les techniques de diffractions des rayonx X ont pris leur
essor à partir de 1912, date à laquelle Max von Laue
et ses collaborateurs Friedrich et Knipping
à Munich réussirent à obtenir le premier
diagramme de diffraction des rayonx X par un cristal, confirmation directe de
la structure périodique des milieux cristallisés.
L'appareil de mesure s'appelle un diffractomètre. Les
données collectées forment le diagramme de diffraction ou
diffractogramme. [7]
II.2 Production des rayonx X :
Les sources les plus courantes pour produire les rayons X sont
les tubes à rayons X dans lesquels le rayonnement est produit sous
l'effet du bombardement d'une cible métallique par des électrons
(Figure II.1).
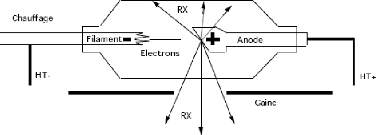
Fig.II.1 Schéma d'un tube à rayons
X. HT : haute tension d'accélération.
II.2.1 Tube à cathode chaude :
Le principe du tube à cathode chaude est le suivant :
La cible appelée anticathode joue également le
rôle d'anode est portée à un potentiel positif de quelques
dizaines de kilovolts par rapport à la cathode. Pour des raisons
pratiques,
particulièrement pour faciliter le refroidissement par
l'eau, l'anticathode est à la masse, la cathode se trouvant à une
haute tension négative produite par un système d'alimentation
classique comportant : transformateur, redresseurs, filtres . Pour les
longueurs d'ondes généralement utilisées en
cristallographie, les fenêtres de sorties sont en béryllium,
à absorption faible.
a) Dimension du foyer:
En raison de la forte absorption des électrons, les
rayons X proviennent d'une couche superficielle très mince de
l'anticathode. La zone source est appelée foyer. Pour de nombreuses
applications, il est souhaitable que la source des rayons X présentant
un faible diamètre apparent. Pour atteindre ce résultat, on
utilise une électrode de focalisation, appelée souvent
wehnelt. La surface métallique de cette
électrode est une équipotentielle, dont la forme est
étudiée pour focaliser les électrons sur la surface de
l'anticathode. On focalise généralement suivant un foyer
linéaire et on travail avec un faible angle de sortie de rayons X
(environ 6°). Par un effet de perspective, on obtient ainsi dans une
direction un foyer linéaire et dans la direction perpendiculaire un
foyer ponctuel.
b) Choix de l'anticathode :
Pour éviter la fusion de l'anticathode sous le
bombardement électronique, il faut un élément bon
conducteur de la chaleur et suffisamment réfractaire, ce qui limite
pratiquement le choix aux métaux. Pour avoir un spectre simple, on
utilise un élément pur sous forme d'une pastille rapportée
sur le corps de l'anticathode en cuivre.
Le spectre des rayonx X émis par bombardement
électronique est formé par la superposition de deux types
d'émission ; un fond continu et un spectre de
raies caractéristiques suivant les applications, on a besoin
d'un rayonnement polychromatique ou monochromatique.
c) Rendement d'émission des RX :
L'interaction des électrons rapides avec la
matière se traduit globalement par un ralentissement des
électrons, et l'énergie cinétique perdue se manifeste sous
différentes formes. Une fraction importante de cette énergie ( 99
%) est convertie en chaleur augmentant ainsi l'énergie interne de la
substance. Le reste (1%) est rayonné hors de la substance sous forme de
photons X.
II.3 Rayonnement monochromatique :
Pour la diffraction X, on s'intéresse principalement
aux raies Ká de la cible, pas au rayonnement de fond. En effet, la
direction de diffraction dépendant de la longueur d'onde (selon la loi
de Bragg), on cherche à avoir la plupart du temps une radiation
monochromatique (à l'exception des clichés de Laue). En fait, on
élimine en général la raie Kâ mais on conserve les
raies Ká1 et Ká2, ainsi que le rayonnement continu de freinage
qui contribuera au bruit de fond. Dans certains cas où le rapport signal
sur bruit est capital, on utilise un monochromateur, au prix d'une perte
importante d'intensité, on a alors une radiation «
réellement » monochromatique ; on peut aussi utiliser un
détecteur « solide » (diode de silicium dopé au lithium
ou diode de silicium à diffusion) ayant une très bonne
discrimination en énergie (principe de l'analyse dispersive en
énergie), ce qui permet de travailler en monochromatique tout en ayant
un signal intense.
On utilise typiquement des hautes tensions de 50 kV, et des
cibles de cuivre en général, parfois de molybdène, cobalt
ou de manganèse. En effet, la longueur d'onde des raies Ká1 du
cuivre (de l'ordre de 1,6 Å) permet d'observer le phénomène
de diffraction pour une grande plage de distances interréticulaires (d
allant de 0,9 à 9,2 Å sur une plage angulaire 2è de 10
à 120° (Loi de Bragg). Par contre, les raies du cuivre ont une
énergie suffisamment grande (8 keV pour la Ká1) pour exciter les
atomes de fer, la fluorescence induite sur les échantillons contenant
majoritairement du fer (comme les aciers et fontes) donne donc un bruit de fond
très élevé. L'utilisation d'un tube au cobalt ou au
manganèse permet de réduire ce bruit de fond parasite puisque les
énergies des photons sont insuffisantes pour exciter le fer (la raie
Ká1 du cobalt a une énergie de 6,9 keV, celle du manganèse
5,9 keV) ; une autre solution consiste à mettre un monochromateur
arrière (c'est-à-dire situé entre l'échantillon et
le détecteur) ou d'utiliser un détecteur filtrant de
manière précise les énergies des photons (détecteur
solide du type de ceux utilisés en analyse dispersive en énergie)
afin d'éliminer la composante fluorescente du fer.
II.4. L'interaction des photons X avec la matière
:
Lorsqu'un faisceau de rayons X pénètre dans un
milieu matériel, on constate une diminution progressive de son
intensité. Cette diminution du nombre de photons, l'atténuation
du faisceau, est dûe essentiellement à l'interaction des photons
avec les électrons. Dans un tel processus, l'énergie perdue se
retrouve sous deux formes: une partie EA est absorbée par le milieu, et
une partie ED est diffusée et sort de la matière dans une
direction différente de la
direction du faisceau initial. Les phénomènes
d'atténuation et d'absorption sont à l'origine des applications
et des effets des rayons X en radiodiagnostic et en radiothérapie.
[8]
1I.4.1 Description microscopique des interactions
Deux types d'interactions entre photon X et matière
sont envisageables : l'effet photoélectrique et l'effet Compton. L'effet
photoélectrique prédomine aux faibles énergies.
L'effet photoélectrique : le photon entre en collision
avec un électron des couches internes de l'atome. L'énergie E du
photon incident est transférée à l'électron qui est
éjecté de sa couche. Une partie de cette énergie est
utilisée pour «extraire» l'électron interne
(énergie de liaison W); l'excédent d'énergie se retrouve
sous forme d'énergie cinétique Ec de l'électron
éjecté. Par conséquent, E = W+Ec. L'effet
photoélectrique ne peut avoir lieu que si l'énergie du photon
incident est supérieure à l'énergie de liaison de
l'électron.
L'énergie cinétique du photo-électron est
finalement transférée au milieu lors d'ionisations
ultérieures. Le retour de l'atome à l'état fondamental
s'accompagne d'une émission d'énergie sous forme d'un photon de
fluorescence ou d'un électron Auger (fig.II.2).
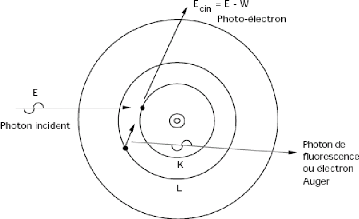
Fig.II.2 Effet photoélectrique.
Le photon de fluorescence est émis lorsqu'un
électron des couches supérieures prend la place laissée
vacante par l'électron éjecté. Parfois, pour des milieux
de Z petit, le photon de fluorescence produit un nouvel effet
photoélectrique avec émission d'un électron: c'est l'effet
Auger.
L'effet Compton : le photon entre en
collision avec un électron libre ou faiblement lié auquel il
cède une partie de son énergie. Un photon d'énergie plus
faible est diffusé dans une direction différente de la direction
initiale (fig.II.3). Pour les photons X étudiés ici, la majeure
partie de l'énergie est emportée par le photon diffusé.
[8]
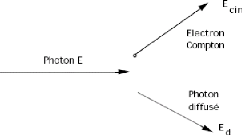
Figure II.3 Effet Compton : il s'agit de la
diffusion d'un photon par un électron.
II.4.2 La loi d'atténuation du rayonnement X :
Un faisceau unidirectionnel de photons
monoénergétiques traverse un écran matériel. Soit
I(x) l'intensité du faisceau (nombre de photons franchissant
l'unité de surface normale au faisceau par unité de temps)
à la position x. Appelons -dI la variation d'intensité sur une
épaisseur infiniment petite dx. L'expérience montre que -dI est
proportionnel à l'intensité incidente et à
l'épaisseur x :
-dI = m (E, M) I dx. (II.1)
Le coefficient de proportionnalité m(E, M),
appelé coefficient d'atténuation linéaire, dépend
de l'énergie E des photons incidents et du milieu M. Il a la dimension
de l'inverse d'une longueur. L'intégration de la relation (II.1) donne
la loi d'atténuation d'un faisceau parallèle
monoénergétique de rayonnement électromagnétique en
fonction de l'épaisseur x:
I(x) = I0 exp [-m (E, M) x] (II.2)
I(x) est l'intensité du faisceau après avoir
traversé une épaisseur x de matière et I0
l'intensité du faisceau incident [I0 = I(x = 0)].
L'intensité d'un rayonnement
électromagnétique décroît exponentiellement
en fonction de l'épaisseur de matière traversée.
Le coefficient d'atténuation varie fortement en fonction de la
matière et de l'énergie des photons. De manière
générale, il croît en fonction du numéro atomique du
milieu et décroît en fonction de l'énergie du
rayonnement.
La pénétration du rayonnement à travers
la matière est souvent caractérisée par l'épaisseur
de demi atténuation, épaisseur de matière telle que
l'intensité du faisceau incident est réduite de
moitié.[7]
II.5 Diffraction des rayons X et détermination d'une
distance interatomique :
La longueur d'onde des rayons X est du même ordre de
grandeur que la distance interatomique dans les cristaux. La structure
régulière d'un cristal diffractera donc un faisceau de rayons X
de même que les fentes équidistantes d'un réseau
diffractent la lumière. La diffraction par rayons X est un
procédé très puissant pour déterminer la structure
d'un cristal et la distance interatomique (cristallographie).
Les atomes ou les molécules d'un cristal appartiennent
à des familles de plans parallèles (plans réticulaires).
Chaque ensemble de plans parallèles se caractérise par une
distance d entre les plans. Un faisceau parallèle de rayons X
monochromatiques tombant sur le cristal sera diffracté dans toutes les
directions par chaque atome. Les ondes diffractées vont
interférer constructivement dans certaines directions
si elles sont en phase, c'est-à-dire si les différences des
chemins parcourus sont toutes égales à un nombre entier de
longueur d'onde.
On peut montrer que tout se passe comme si le faisceau de
rayons X était réfléchi partiellement par chaque plan
d'une famille (comme pour la réflexion de la lumière, l'angle de
réflexion est égal à l'angle d'incidence), avec la
condition supplémentaire que les faisceaux réfléchis par
les plans parallèles doivent tous être en phase. Ceci n'est
réalisé que dans certaines directions privilégiées
dépendant de la distance d et de la longueur d'onde
ë des rayons X.
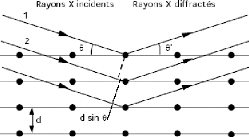
Figure II.4 diffraction des rayons X par une
structure cristalline.
Ainsi, on voit sur la figure (II.4) qu'il y aura
interférence constructive dans la direction
donnée par l'angle è si la différence
entre les parcours des rayons 1 et 2 est égale à
un nombre entier n de la longueur d'onde ë. Il ressort de la
figure (II.4) que ë = 2dsin è, d'où la loi de
Bragg:
2d (hkl) sin è = n ë n = 1, 2, 3...
(II.3)
Connaissant ë, il est possible de déterminer d par la
mesure de l'angle è.
La famille de plans atomiques séparés par la
distance d diffracte le faisceau dans la direction è si cet angle
satisfait la loi de Bragg. [8]
II.5.1 Condition de diffraction:
Les conditions de diffractions sont différentes suivant le
mode d'empilement. On peut montrer que pour une :
-Maille P : tous les plans diffractent.
-Maille I : seul les plans tels que (h+k+l) paire diffractent.
-Maille F : seul les plans tels que h, k, l soient de même
parité diffractent. [9]
II.5.2 Intensité diffracté :
a) Facteur de diffusion:
? sinkr
f = 4 j-j p r r dr
2
f ( ) 41'
Ou k = sin O (II.4)
kr ?
Le facteur de diffusion f d'un atome mesure sa capacité
à diffuser les rayons X.Si ce facteur est grand, cet atome contribue
beaucoup à la diffusion des rayonx X qui provoque la figure de
diffraction. Le facteur de diffusion est relié à la
densité électronique ñ et à l'angle de diffusion
è par:
0
Dans la direction du faisceau (è = 0),f est égal
au nombre d'électrons de l'atome.
b) Facteur de structure:
Le facteur de structure F hkl, est la somme des
facteurs de diffusion des atomes dans une maille cristallographique,
pondérée par un facteur de phase qui dépend de leur
position:
F hkl f i e [I
2 i ( hx i ky i lz
i )
? ?
=
(II.5)
i
L'intensité de la réflexion (hkl) est
proportionnelle à Fhkl 2. [10]
II. 6 Méthode de Debye-Scherrer (méthode des
poudres) :
L'idée d'utiliser la diffraction des rayons X pour
identifier une phase fût développée au début du
XXe siècle de manière indépendante par
Albert Hull en 1919 d'une part, et par Peter Debye
et Paul Scherrer d'autre part. En raison de la
guerre, la publication et la diffusion des journaux scientifiques était
difficile ; chronologiquement, c'est Hull qui publia le
premier ses travaux, mais la méthode porte le nom de Debye et
Scherrer.
II.6.1 Principe de la méthode :
Une poudre formée d'une phase cristalline
donnée, va toujours donner lieu à des pics de diffraction dans
les mêmes directions, avec des hauteurs relatives à peu
près constantes. Ce diagramme de diffraction forme ainsi une
véritable signature de la phase cristalline. Il est
donc possible de déterminer la nature de chaque phase cristalline au
sein d'un mélange (mélange de poudre ou échantillon massif
polyphasique), à condition d'avoir auparavant déterminé la
signature de chaque phase.
La détermination de cette signature peut se faire soit
de manière expérimentale (mesure d'un produit pur dans des
conditions idéales), soit par simulation numérique à
partir de la structure cristallographique connue -- structure ayant
elle-même pu être déterminée par diffraction X. Cette
signature est consignée dans une fiche sous la forme d'une liste de pics
; la position en 2è est convertie en distance interréticulaire d
par la loi de Bragg, afin d'avoir une valeur indépendante de la longueur
d'onde des rayons X utilisée. L'intensité I de chaque pic est
exprimée en pourcent %, 100 % étant la hauteur du pic le plus
intense. Cette liste de pics est souvent désignée par le terme
« liste de d--I ». On constitue ainsi des
bases de données, et le diagramme mesuré sur le produit inconnu
est comparé de manière informatique ou manuelle à toutes
les fiches de la base de données. La base de données la plus
complète à l'heure actuelle est la Powder diffraction file (PDF)
de l'ICDD (ex-JCPDS: Joint comittee for powder diffraction
standards, ex- comité E4 de l'ASTM), avec plus de 160 000
fiches.
L'intérêt de cette méthode est qu'elle
permet de distinguer les différentes formes de cristallisation d'un
même composé (par exemple pour la silice, distinguer le quartz de
la cristobalite). Cependant, elle ne peut généralement pas
permettre d'identifier des composés amorphes. Cette technique est donc
complémentaire de l'analyse élémentaire. La
procédure d'identification des phases se fait en deux étapes :
une étape de recherche dans une base
(search), puis une confrontation des fiches
probables avec ce qui est possible chimiquement (match) ; on
parle donc fréquemment de search/match pour
désigner cette procédure.
Il est important de noter qu'au final, c'est l'utilisateur qui
détermine si un produit est présent ou pas: en raison des
possibilités de confusion (plusieurs produits très
différents pouvant avoir des signatures très proches), un
algorithme automatisé ne peut pas prendre seul la décision. C'est
en dernier ressort la compétence de l'utilisateur, son habileté
et sa connaissance de l'échantillon qui interviennent. [7]
II.6.2 Appareillage de la méthode :
Du point de vue instrumental, on peut distinguer plusieurs
montages chambre DebyeScherrer, chambres à focalisation (Seeman-Bohlin
et Guinier) et diffractomètre de poudres en géométrie
Bragg-Brentano. L'avantage de ce dernier par rapport aux autres est que
l'enregistrement est fait à l'aide d'un goniomètre mobile muni
d'un détecteur au lieu d'un film photosensible.[1]
II.6.3 Sources d'erreurs :
Le fichier de sortie du calcul contient les déviations
standard estimées (sigmas) des paramètres
affinés (paramètres de maille, taux d'occupation, facteurs
thermiques, etc.), cellesci sont souvent utilisées comme indicateurs de
précision des affinements. Il faut tenir compte du fait que ces
déviations standard sont seulement issues de l'application du
modèle théorique sur un jeu de données, mais ne prennent
pas en compte les vraies erreurs expérimentales. Les sources d'erreurs
systématiques (expérimentales) qui ne sont pas prises en
considération dans les valeurs des sigmas sont :
orientation préférentielle des cristallites ,
ligne de base de diffractogramme, forme des raies(choix de la fonction de
profil), absorption de la radiation,transparence de
l'échantillon,déplacement de l'échantillon,erreur de
zéro(en degrés 2è),échantillon à gros
grains(très peu de cristallites qui diffractent ),instabilité du
faisceau incident,instabilité électrique ou mécanique du
diffractomètre.[1]
Chapitre III méthodes de préparation des
pérovskites
III.1 Introduction:
La littérature décrit plusieurs méthodes
pour la synthèse des oxydes mixtes à structure Pérovskite
comme la réaction à l'état solide, la
mécanosynthèse, la synthèse par combustion, la
méthode sol-gel, la synthèse par co-précipitation, la
méthode hydrothermale, etc... [2]
Nous nous intéressons aux plus courantes, en donnant
quelques exemples de techniques utilisées pour former des solides
(pérovskites) dont les propriétés sont
particulièrement intéressantes, on peut les classer en deux
types:
-Synthèse par voie sèche (méthode
céramique).
-Synthèse par voie humide.
III.2 Synthèse par Réaction à
l'état solide :
La synthèse des oxydes (pérovskites) par
réaction à l'état solide est l'une des méthodes les
plus utilisées dans la chimie du solide. A la base de cette
méthode se trouve la réaction par traitement thermique entre deux
ou plusieurs substances sous forme solide qui sont intimement
mélangées. Les réactifs, des oxydes et/ou des carbonates
sous forme des poudres, sont pesés en quantités
stoechiométriques et mélangés soigneusement par broyage
dans un mortier.
L'obtention d'un mélange homogène composé
des particules à faible taille facilitera ensuite la
cinétique de la réaction. La poudre est ensuite
soumise à des traitements thermiques successifs jusqu'à
l'obtention d'une seule
phase.la température retenue
avoisine, en général 1000°c.L'avantage de cette technique
est qu'elle assez facile et rapide à maître en oeuvre, car elle ne
nécessite aucune préparation antérieure des
précurseurs. La granulométrie des particules joue un rôle
très important sur la vitesse de la réaction et sur
l'homogénéité du composé obtenu, car la diffusion
à l'état solide est lente. On peut compenser ceci en faisant des
broyages intermédiaires. [2]
a) Matières premières:
Elles sont constituées d'oxydes, de carbonates, de
nitrates, etc. Une poudre idéale peut être décrite comme
étant formée de grains de petite taille (de l'ordre du 1 um), de
forme régulière, avec une répartition de taille
très étroite. La pureté ainsi que celle d'éventuels
ajouts sont contrôlés. Le problème principal concernant les
matières premières de base, qui sont sous forme de poudres, est
la difficulté d'évaluer les paramètre fondamentaux
traduisant la réactivité du matériau vis-à-vis des
autres avec lesquels il est amené à réagir, l'histoire
thermique du matériau joue ainsi un rôle très important.
b) Mélange, Broyage :
Il s'agit d'une des phases essentielles du cycle de
fabrication d'un solide à structure pérovskite. C'est
également au cours de cette opération que l'on obtient une
répartition uniforme des précurseurs. Les poudres sont
pesées suivant les quantités stoechiométriques
prévues par l'équation de réaction.
c) Calcination:
Dans ce but, les matériaux sont soumis à un
cycle thermique, éventuellement sous atmosphère
contrôlée, au cours duquel ils vont, par des
phénomènes de diffusion en phase solide,
réagir et former la phase recherchée. Au cours de cette
réaction il y a dégagement de dioxyde de carbone ou de dioxyde
d'oxygène et éventuellement d'un peu de vapeur d'eau.
d) Rebroyage :
Après le traitement thermique, la poudre est
rebroyée afin de réduire la taille des grains, de
l'homogénéiser et augmenter sa réactivité. La
poudre est alors soumise à un traitement thermique à haute
température, afin d'obtenir les phases recherchées. [3]
e) Problèmes rencontrés :
Un certain nombre de problèmes liés à cette
technique peuvent survenir, ils sont
énumérés dans le tableau suivant :
Tableau.III.1 problèmes et causes
liés à la méthode céramique. [3]
|
Problèmes possibles
|
causes
|
|
Défauts d'homogénéité
|
Mélange mal préparé, particules de trop
grande taille, mauvaise diffusion
|
|
Taille de grains trop élevée
|
Apparition d'une phase liquide (température trop
élevée), cristallisation des grains avec grossissement
|
|
Nombreuses phases parasites (impuretés)
|
Défaut de précision des pesées,
réaction incomplète (maintien en température trop bref ou
température trop basse)
|
|
Mauvaise distribution des constituants
|
Mauvaise homogénéité du mélange,
broyage inefficace.
|
|
Impuretés extrinsèques
|
Pollution par le broyeur où la nacelle, four pollué
par des oxydes volatils (Pb, Bi, Li) etc.) ou réaction avec
l'humidité atmosphérique.
|
III.3.Synthèse par voie humide (solution):
La méthode humide permet un contrôle précis
des propriétés physiques et chimiques des poudres et des
précurseurs avec, autre avantages, l'accentuation de :
-l'homogénéité.
-l'uniformité de la forme des particules.
La méthode humide inclut : co-décomposition,
processus sol-gel, co-précipitation, vaporisation de la glace,
pulvérisation et pyrolyse. [11]
III.3.1 Procédé sol-gel :
Parmi les différentes méthodes utilisées
pour la synthèse de matériaux, le procédé solgel
est particulièrement bien adapté à la fabrication de
matériaux homogènes, sous forme de poudres et de films. Lors
d'une synthèse par voie sol-gel, les précurseurs
moléculaires contenus dans la solution de départ (« le sol
») polymérisent suivant divers mécanismes et forment un
réseau d'oxydes (« le gel »). Une étape de
séchage suivie de traitements thermiques permet d'éliminer les
composés organiques pour former le matériau oxyde inorganique.
Cette technique présente de nombreux avantages. Parmi les plus
significatifs, citons la très grande pureté et
l'homogénéité des solutions liées au fait que les
différents constituants sont mélangés à
l'échelle moléculaire en solution, les contrôles de la
porosité des matériaux et de la taille des nanoparticules, les
traitements thermiques requis à basses températures ainsi que la
synthèse de matériaux inaccessibles par d'autres techniques.
Notons cependant que ce procédé génère un
rétrécissement du matériau lors du traitement thermique et
nécessite l'utilisation de certains précurseurs relativement
coûteux.
La solution élaborée par voie sol-gel permet de
réaliser des films par différentes méthodes (dip-coating,
spin coating, pulvérisation...). Des matériaux massifs peuvent
également être préparés par voie sol-gel lors de
l'évaporation rapide du solvant. Enfin, le sol peut aussi réagir
avec de l'eau dans un procédé d'émulsion et/ou de
séchage pour former des poudres. Il est possible d'obtenir des
matériaux hautement poreux dans lequel le réseau solide du gel
est maintenu après le séchage. Selon les conditions de mise en
oeuvre (en masse, dépôt de films, précipitation...) et de
traitement (chimique, physique, thermique...), des matériaux de formes
(matériaux massifs, couches minces, fibres, poudres) et de structures
(denses, mésoporeux, ultra poreux) très variées peuvent
être préparés.
La grande diversité de matériaux obtenus par
voie sol-gel fait que ces matériaux sont utilisés dans de
nombreuses applications. Un secteur particulièrement exploité est
celui de
l'optique, l'incorporation de colorants, de semi-conducteurs,
de particules métalliques, de terres rares permet de développer
des systèmes, tels des cellules solaires, des lasers à colorant,
des miroirs à conjugaison de phase, des luminophores, ...
Au cours du processus sol gel se produisent des
réactions constituant peu à peu les liens présents dans le
matériau final et selon leur degré d'avancement se
succèdent plusieurs états de la matière :
- Le sol qui est une suspension stable et transparente dans un
liquide d'identités moléculaires ou de particules plus ou moins
denses de taille comprise entre 1 et 100 nm.
- Le gel qui est un réseau tridimensionnel solide
continu dans un liquide. Le point de transition sol-gel est défini par
le moment ou un amas polymérique atteint la taille du récipient.
La viscosité du sol, qui augmentait avec la croissance de la taille des
particules, diverge alors et le solide formé acquiert un module
élastique.
- Le gel sec qui est un solide amorphe et poreux obtenu par
évaporation du liquide (xérogel ou aérogel selon les
conditions du séchage).
- Le matériau final, cristallisé, densifié
et débarrassé des résidus réactionnels par recuit
à plus hautes températures.
Les précurseurs utilisés dans le
procédé sol-gel sont les alcoxydes métalliques de formule
générale M(OR)n où M désigne un atome
métallique de valence n et R une chaîne alkyle (-CjH2j+1).[12]
III.3.2 Co-precipitation:
La co-précipitation permet l'obtention de produits de
précurseurs par précipitation simultanée de deux cations M
et M' (M étant un alcalin ou un alcalino-terreux et M'un métal de
transition).
Généralement après le mélange des
deux solutions contenant les cations métalliques, La mesure du pH est
nécessaire pour pouvoir suivre l'évolution de la
précipitation,après dissolution des masses adéquates
d'oxydes métalliques,les solutions sont mélangées
progressivement puis diluées.La précipitation a lieu à
froid ou à chaud à un pH donné.La chaleur active la
co-précipitation et le pH du milieu réactionnel une grande
importance pour la majorité des réactions de
co-précipitation puisqu'il détermine la nature et la
stoechiométrie du précipité.Cette dépendance du
mécanisme de la réaction avec le pH rend très difficile
l'obtention d'une stoechiométrie donnée et constitue un
inconvénient majeur lors du synthèse.
La méthode de co-précipitation a montré
quelques limites d'utilisation. La contrainte majeur que présente la
méthode consiste en la conservation de la stoechiométrie M'/M.
III.3.3 Hydrothermales :
La méthode hydrothermale consiste à chauffer des
réactifs en présence d'eau dans un récipient clos,un
autoclave.dans ces derniers ,la pression augmente et l'eau surchauffée
reste liquide au-dessus du point d'ébullition normal et la pression
dépasse la pression atmosphérique.Les conditions hydrothermales
se sont produites dans la nature et de nombreux minéraux,dont les
zéolithes naturelles,ont été formés ainsi.Les
émeraudes synthétiques sont obtenues sous les conditions
hydrothermales.L'utilisation de plus basses températures est l'un des
avantages de cette méthode.[11]
Chapitre IV Résultats expérimentaux
IV.1 Introduction:
Dans cette étude nous nous sommes intéressés
à la synthèse de trois phases à structure
pérovskite à base de lanthane qui sont :
LaFeO3, LaMnO3, LaTiO3 notées respectivement : P1, P2,
P3.Pour cela, nous avons préparé trois échantillons
notés E1, E2, E3 pour avoir, respectivement, les trois phases
citées ci-dessus.
Après la synthèse des échantillons vient
ensuite l'étape d'identification de ces derniers par DRX confirmant
ainsi la présence ou non des phases recherchées.
IV.2 Synthèse des échantillons :
Dans ce travail, la synthèse des échantillons a
été effectuée par réaction à l'état
solide à haute température en utilisant des oxydes sous forme de
poudres, Pour cela, des quantités stochiométriques ont
été pesées et broyées et mélangées
d'une manière intime. Obtenir un mélange homogène est
très important car cela facilitera la diffusion solide-solide et
augmentera d'avantage la cinétique des réactions.
Le traitement thermique est mené dans un four
électrique permettant de calciner à haute température
(>900°c) nos échantillons afin que la réaction entre
solides s'effectue. Les produits obtenus se présentent sous la forme de
cristallites de taille généralement proche de dizaine de microns
(voir paragraphe III.2).
La préparation des échantillons est l'un des
paramètres essentiels à l'obtention de résultats
reproductibles et de bonne qualité, car les quatre informations
principales obtenues à partir des données de diffraction sont
influencées par ce paramètre :
- la position des raies
- l'intensité des raies
- la forme des raies
- le fond continu
a) Les produits de départ (les oxydes)
:
Les produits de départ sont rassemblés dans le
tableau suivant :
Tableau IV.1 les oxydes (réactifs) de
départ.
|
Oxydes
|
nomenclature
|
|
La2O3
|
Oxyde de lanthane
|
|
TiO2
|
Oxyde de titane
|
|
Fe2O3
|
oxyde de fer (III)
|
|
MnO2
|
Oxyde de manganèse
|
b) Les réactions mises en jeu :
Pour des besoins concernant la diffraction des rayons X , nous
avons voulu préparer 1g pour chaque échantillon .Les
réactions chimiques mises en jeu sont :
4 E1

La2O3 + 2 FeO3 2 LaFeO3
4 E2
Y2 La2O3 + MnO2 LaMnO3 + 1/4 O2
4 E3
Y2 La2O3 + TiO2 LaTiO3 + 1/4 O2
b.1 Exemple de calcul (E1) :
1 mole de La2O3 + 1 mole de FeO3 donnent 2 moles de
LaFeO3
0,0020 mole 0,0020 mole n=m/M (LaFeO3) =1/242,75=0,0041 mole
0,6708 g 0,3288 g 1g
IV.3 Diffraction des rayons X :
Dans le cadre de ce travail nous avons utilisé un
diffractomètre de poudres, Un appareil D 8-ADVANCE de
BRUKER-AXS, qui utilise les rayonnements K á1 (1.54056
Å) et K á2 (1.54439 Å) du cuivre et la
géométrie Bragg-Brentano, équipé d'un
monochromateur arrière de graphite qui permet d'éliminer la
contribution de la fluorescence et du rayonnement Kâ. Le
générateur a été utilisé à 40 kV et
40 mA. Balayage entre 4 et 80° (2è) par pas de 0,04°, temps de
comptage 1 s par pas.
IV.4 Etude des spectres:
Les spectres sont sous format RAW (brut), une
conversion vers un format DAT a été nécessaires pour
adopter ce fichier au logiciel WINPLOTER.
Après avoir enregistré le diagramme de diffraction
X, notre méthodologie est résumée
ainsi :
1) Indexation du diagramme de diffraction (relation de Bragg),
cela consiste à faire une comparaison avec la base de données
(PCPDF) de ICDD (paragraphe II.6.1).
2) Affinement des paramètres de maille par la
méthode des moindres carres (logiciel CELREF, fourni avec le CDROM
NEXUS)
IV.5 Interprétation des spectres des trois
échantillons :
IV.5.1 L'échantillon E1 :
Les spectres RX de l'échantillon E1 qui
est traité à T=900°C et
T=1000°C pendant 24h sont représentés sur
les figures (IV.1.a) et (IV.1.b) :
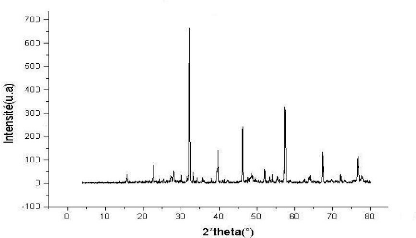
Fig.IV.1.a Spectre RX de l'échantillon
E1 traité à 900°C
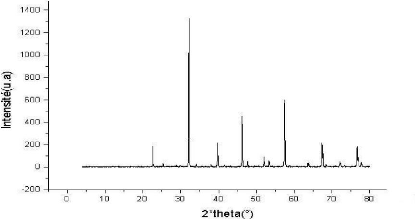
Fig.IV.1.b Spectre RX de l'échantillon
E1 traité à 1000°C
23
? L'indexation des pics est représentée sur les
tableaux IV.2.a (900°C) et IV.2.b (1000°C)
TableauIV.2.a : les phases de
l'échantillon E1 (900°C)
|
2è obs 2ècal
|
hkl
|
% Int
|
La phase
|
|
15.6960
|
|
|
5
|
NI
|
|
22.6065
|
22.6075
|
0 0 2
|
12
|
1
|
|
24.1957
|
|
|
3
|
NI
|
|
25.3037
|
25.3024
|
1 1 1
|
3
|
1
|
|
27.3157
|
|
|
5
|
NI
|
|
28.0109
|
|
|
8
|
NI
|
|
32.1855
|
32.1785
|
2 0 0
|
100
|
1
|
|
33.1850
|
|
|
7
|
NI
|
|
34.1590
|
34.1896
|
2 0 1
|
3
|
1
|
|
35.6885
|
|
|
3
|
NI
|
|
39.6834
|
39.6863
|
2 0 2
|
21
|
1
|
|
46.1503
|
46.1606
|
0 0 4
|
36
|
1
|
|
47.6397
|
47.6322
|
2 2 1
|
3
|
1
|
|
51.9892
|
51.9817
|
1 1 4
|
8
|
1
|
|
53.3070
|
53.3107
|
1 3 1
|
6
|
1
|
|
54.0851
|
|
|
5
|
NI
|
|
57.3796
|
57.3822
|
2 0 4
|
49
|
1
|
|
67.3297
|
67.3208
|
4 0 0
|
19
|
1
|
|
72.0183
|
72.0192
|
3 1 4
|
4
|
1
|
|
76.6079
|
76.6021
|
1 1 6
|
17
|
1
|
|
77.6419
|
77.6429
|
2 4 1
|
6
|
1
|
1: LaFeO3 ; NI: non identifiée
Tableau IV.2.b : les phases de
l'échantillon E1 (1000°C)
|
2è (obs)
|
2è (cal)
|
hkl
|
%Int
|
La phase
|
|
22.5919
|
22.5915
|
1 1 0
|
15
|
1
|
|
25.3011
|
25.3015
|
1 1 1
|
3
|
1
|
|
32.1787
|
32.1846
|
2 0 0
|
100
|
1
|
|
39.6856
|
39.6862
|
2 0 2
|
17
|
1
|
|
46.1435
|
46.1425
|
0 0 4
|
32
|
1
|
|
47.6358
|
47.6333
|
2 2 1
|
4
|
1
|
|
51.9775
|
51.9798
|
3 1 0
|
7
|
1
|
|
53.3017
|
53.3062
|
1 3 1
|
4
|
1
|
|
57.3744
|
57.3738
|
3 1 2
|
50
|
1
|
|
57.5600
|
|
|
21
|
|
|
63.6406
|
63.6424
|
1 3 3
|
3
|
1
|
|
67.3254
|
67.3349
|
4 0 0
|
17
|
1
|
|
67.5200
|
|
|
8
|
|
|
72.0274
|
72.0275
|
4 0 2
|
4
|
1
|
|
76.5945
|
76.5806
|
4 2 0
|
14
|
1
|
|
76.8200
|
|
|
7
|
|
T=900°C:
Les phases en presence:
LaFeO3 : cristallise dans le groupe d'espace
Pnma (62) du système orthorhombique avec les paramètres suivants
:
a=5.556 A°, b=5.565 A°, c=7.862 A° Après
l'affinement on obtient :
a=5.5590 A°
b=5.5647 A°
c=7.8598 A°
T=1000°C :
La seule phase présente est LaFeO3.
Après l'affinement on obtient :
a=5.5580 A° b=5.5652 A° c=7.8627 A°
Discussion des résultats :
D'une façon générale, lorsque on analyse
le spectre RX de l'échantillon E1 (900°C), on
remarque la présence de la phase P1 (LaFeO3).Cependant, il existe des
pics non identifiés, C'est-à-dire la présence des phases
parasites. Cela peut être expliqué par l'insuffisance de la
température de la cuisson.
Pour surpasser ce problème, on a encore effectué un
traitement thermique à 1000°C .Cette fois-ci, on a plus de phases
parasites.
La contribution du rayonnement Ká2 est apparue vers
(2è= 57.5600, 67.5200, 76.8200)
Lorsque on fait une comparaison entre les deux spectres, on
constate que : -les pics sont plus fins et plus intenses à 1000°C
qu'à 900°C.
-les pics non identifiés dans le spectre de RX à
900°C sont disparus à 1000°C.
Ainsi, la température-seuil adéquate
pour préparer de telles phases dans les mêmes conditions
avoisine 1000°C.
La structure détaillée de la maille
pérovskite LaFeO3 (réalisée à l'aide de logiciel
GRETEP 2) est représentée sur la figure ci-
dessous.
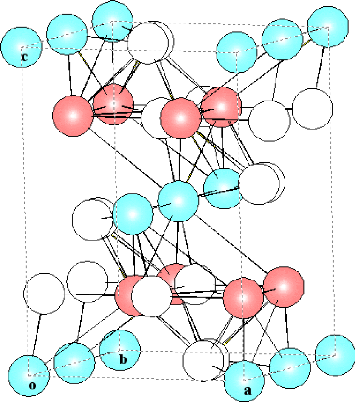
Fe
Figure IV.2 structure de la maille LaFeO3
IV.5.2 L'échantillon E2 :
Les spectres RX de l'échantillon E2 qui
est traité à T=900°C et
T=1000°C pendant 24h sont représentés sur
les figures (IV.3.a) et (IV.3.b) :
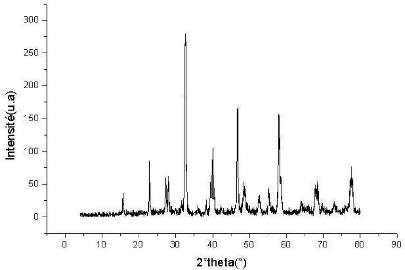
Fig.IV.3.a Spectre RX de l'échantillon
E2 traité à 900°C

Fig.IV.3.b Spectre RX de l'échantillon
E2 traité à 1000°C
? L'indexation des pics est représentée sur les
tableaux IV.3.a (900°C) et IV.3.b (1000°C)
Tableau IV.3.a
: les phases de l'échantillon E2 (900°C)
|
2è (obs)
|
2è (cal)
|
hkl
|
%INT
|
La phase
|
|
15.6670
|
|
|
13
|
NI
|
|
22.7367
|
22.3047
|
1 1 0
|
30
|
2
|
|
27.2970
|
|
|
20
|
NI
|
|
28.0051
|
|
|
20
|
NI
|
|
29.9500
|
|
|
5
|
NI
|
|
31.5361
|
|
|
7
|
NI
|
|
32.5073
|
32.4134
|
2 0 0
|
100
|
2
|
|
32.9400
|
36.0668
|
2 1 0
|
26
|
2
|
|
35.8669
|
|
|
6
|
NI
|
|
38.2755
|
|
|
9
|
NI
|
|
39.5256
|
|
|
22
|
NI
|
|
40.0353
|
40.1002
|
2 0 2
|
34
|
2
|
|
40.2804
|
|
|
10
|
NI
|
|
46.5880
|
46.9320
|
0 0 4
|
58
|
2
|
|
48.5172
|
|
|
19
|
NI
|
|
49.1661
|
|
|
5
|
NI
|
|
49.4230
|
|
|
5
|
NI
|
|
52.5428
|
52.5518
|
1 1 4
|
4
|
2
|
|
54.6715
|
|
|
6
|
NI
|
|
55.2166
|
|
|
4
|
NI
|
|
57.8179
|
57.7035
|
3 1 2
|
55
|
2
|
|
57.9733
|
|
|
14
|
NI
|
|
58.0800
|
58.1924
|
2 0 4
|
37
|
2
|
|
58.3252
|
58.1924
|
2 0 4
|
22
|
2
|
|
63.8060
|
63.7640
|
2 3 2
|
6
|
2
|
|
64.0299
|
64.2022
|
3 1 3
|
7
|
2
|
|
64.7707
|
64.5542
|
1 1 5
|
5
|
2
|
|
67.8068
|
67.8637
|
4 0 0
|
15
|
2
|
|
68.3478
|
68.4524
|
1 4 1
|
16
|
2
|
|
69.5210
|
69.7107
|
0 4 2
|
8
|
2
|
|
69.9709
|
69.9424
|
2 3 3
|
5
|
2
|
|
70.0708
|
|
|
7
|
NI
|
|
72.8807
|
72.7555
|
3 1 4
|
5
|
2
|
|
75.8770
|
75.6611
|
0 4 3
|
8
|
2
|
|
76.6573
|
76.5072
|
4 2 0
|
13
|
2
|
|
77.4414
|
|
|
16
|
NI
|
|
77.5365
|
77.6723
|
4 2 1
|
16
|
2
|
Tableau IV.3.b : les phases de
l'échantillon E2 (1000°C).
|
2è (obs)
|
2è (cal)
|
hkl
|
% Int
|
La phase
|
|
22.7478
|
22.9199
|
0 0 2
|
29.3
|
2
|
|
32.2843
|
32.3969
|
2 0 0
|
100
|
2
|
|
32.7166
|
|
|
97.9
|
|
|
38.2504
|
38.4223
|
1 0 3
|
4
|
2
|
|
40.0341
|
40.0568
|
2 0 2
|
31.6
|
2
|
|
40.4070
|
|
|
10.6
|
|
|
46.5658
|
46.8273
|
0 0 4
|
67.1
|
2
|
|
52.2885
|
52.1248
|
3 1 0
|
7.8
|
2
|
|
52.6792
|
52.4518
|
1 1 4
|
6.8
|
2
|
|
57.6304
|
57.6566
|
3 1 2
|
25.8
|
2
|
|
57.9327
|
|
|
53.2
|
|
|
58.4506
|
58.4475
|
2 2 3
|
20.8
|
2
|
|
67.6516
|
67.8256
|
4 0 0
|
15.5
|
2
|
|
68.3997
|
68.4329
|
1 4 1
|
18.3
|
2
|
|
77.2911
|
|
|
14.14
|
|
|
77.6943
|
77.6033
|
1 1 6
|
18
|
2
|
2 : LaMnO3
T=900°C :
Les phases en présence:
LaMnO3 : cristallise dans le groupe d'espace
Pbnm du système orthorhombique avec les paramètres suivants :
a=5.532 A° , b=5.722 A° , c=7.699 A°
Après affinement on obtient : a=5.5198 A° , b=5.7517
A° , c=7.7377 A°
T=1000°C :
LaMnO3 est la seule phase présente :
Après affinement on obtient : a=5.5226 A° , b=5.7527
A° , c=7.7541 A°
Discussion des résultats :
Les deux diagrammes de rayons X de l'échantillon E2
(900 et 1000°C) présentent des pics de diffraction correspondant
aux réflexions de la phase P2 (LaMnO3).cependant, à 900°C on
remarque l'existence d'un bruit de fond relativement intense et la
présence des phases parasites non indexées .la
quasi-totalité de ces dernières sont disparues à
1000°C ainsi que le bruit de fond.
La calcination à 900°C de l'échantillon E2
permet la formation quasi complète de la phase P2.mais la
présence des phases parasites montre bien que la formation totale de la
phase P2 s'effectuera à une température supérieure
à 900°C. En fin la calcination à 1000°C mène
à la formation de P2 sans phases secondaires.
Lorsqu'on fait une comparaison entre les deux spectres
(à 900°C et à 1000°C), on observe la diminution de
bruit de fond avec l'augmentation des intensités des pics
caractérisant la phase P2 et la disparition des pics non
identifiés.
La structure détaillée de la maille
pérovskite LaMnO3 est représentée sur la figure
cidessous.
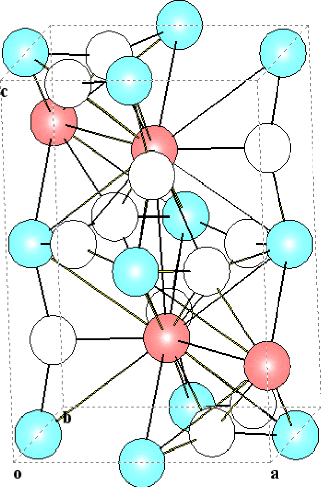
Figure IV.4 structure de la maille LaMnO3
IV.5.3 L'échantillon E3 :
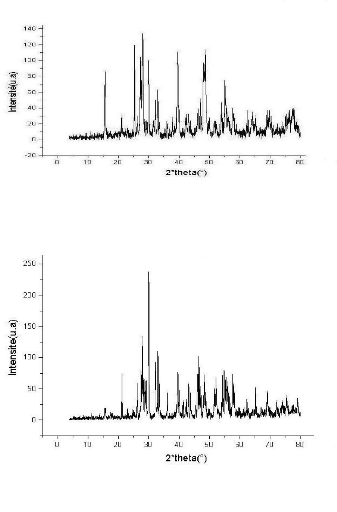
Fig.IV.5.a Spectre RX de l'échantillon E3
traité à 900°C
Fig.IV.5.b Spectre RX de l'échantillon E3
traité à 1000°C
33
Les spectres RX de l'échantillon E3 qui
est traité à T=900°C et
T=1000°C pendant 24h sont représentés sur
les figures (IV.5.a) et (IV.5.b) :
? L'indexation des pics est représentée sur les
tableaux IV.4 (1000°C) :
|
2è (obs)
|
2è (cal)
|
hkl
|
% Int
|
La phase
|
|
11.2573
|
|
|
4
|
NI
|
|
13.8275
|
13.7590
|
2 0 0
|
4
|
6
|
|
15.7539
|
|
|
8
|
NI
|
|
17.4933
|
17.4127
|
1 1 0
|
4
|
6
|
|
21.2188
|
21.1479
|
2 1 0
|
32
|
6
|
|
23.1136
|
23.1109
|
-2 1 1
|
6
|
6
|
|
24.8748
|
|
|
6
|
NI
|
|
25.0670
|
25.0851
|
2 1 1
|
5
|
6
|
|
26.2020
|
26.2615
|
3 1 0
|
25
|
6
|
|
27.5243
|
27.5222
|
1 1 0
|
32
|
4
|
|
27.5340
|
27.4522
|
-3 1 1
|
32
|
6
|
|
27.8327
|
|
|
58
|
NI
|
|
28.1952
|
28.1822
|
-1 1 2
|
29
|
6
|
|
28.7170
|
28.7436
|
-3 0 2
|
27
|
6
|
|
29.1486
|
|
|
22
|
NI
|
|
29.9259
|
29.9283
|
0 1 1
|
100
|
5
|
|
29.9809
|
29.9693
|
3 1 1
|
100
|
6
|
|
32.2966
|
32.2862
|
0 2 0
|
41
|
4
|
|
32.5320
|
32.3453
|
0 1 1
|
41
|
3
|
|
32.7942
|
32.7863
|
-4 1 1
|
9
|
6
|
|
33.0580
|
33.0570
|
-3 1 2
|
46
|
6
|
|
33.5228
|
33.5298
|
-4 0 2
|
24
|
6
|
|
33.8570
|
|
|
7
|
NI
|
|
34.1047
|
|
|
4
|
NI
|
|
34.4367
|
34.3658
|
0 2 1
|
3
|
6
|
|
36.1531
|
36.1448
|
1 1 1
|
18
|
4
|
|
37.3570
|
37.3498
|
-4 1 2
|
4
|
6
|
|
38.6073
|
38.5659
|
0 1 3
|
6
|
6
|
|
39.5815
|
39.5790
|
2 0 3
|
30
|
6
|
|
39.8120
|
39.8916
|
1 1 1
|
30
|
3
|
|
40.0583
|
40.0492
|
-1 2 2
|
21
|
6
|
|
41.2840
|
41.2861
|
1 2 2
|
13
|
6
|
|
42.1893
|
42.1198
|
-6 0 0
|
8
|
6
|
|
42.4538
43.1249
43.7581
|
42.4882 43.0624 43.7660
|
-5 1 2
4 2 0
-3 2 2
|
12
24
18
|
6
6
6
|
|
44.1400
|
|
|
6
|
NI
|
|
45.3428
|
45.3244
|
6 1 0
|
10
|
6
|
|
46.1500
|
46.0559
|
-5 0 3
|
31
|
6
|
|
46.3754
|
46.3953
|
0 0 2
|
31
|
3
|
|
46.5320
|
46.5389
|
-1 0 4
|
41
|
6
|
|
46.8588
|
|
|
15
|
NI
|
|
47.2043
|
47.2353
|
-4 2 2
|
21
|
6
|
|
48.2796
|
48.2763
|
-6 1 2
|
27
|
6
|
|
48.6583
|
48.7210
|
1 0 4
|
17
|
6
|
|
48.8658
|
48.8264
|
-2 2 3
|
12
|
6
|
|
50.0812
|
50.0770
|
-2 1 4
|
5
|
6
|
|
51.6388
|
51.6054
|
1 1 4
|
21
|
6
|
|
52.1920
|
|
|
29
|
NI
|
|
52.4121
|
52.2586
|
2 0 1
|
29
|
3
|
|
52.8950
|
52.8484
|
7 0 1
|
8
|
6
|
|
54.3891
|
54.4700
|
6 1 2
|
35
|
6
|
|
55.0687
|
55.0843
|
0 3 2
|
30
|
6
|
|
55.4909
|
55.5787
|
7 1 1
|
29
|
6
|
|
56.0488
|
56.0712
|
1 3 2
|
27
|
6
|
|
56.6660
|
56.6617
|
-6 2 2
|
16
|
6
|
|
57.1486
|
57.1195
|
-7 0 3
|
6
|
6
|
|
57.7098
|
57.7704
|
-5 1 4
|
32
|
6
|
|
57.9244
|
57.6888
|
2 1 1
|
32
|
3
|
|
58.0559
|
58.0682
|
-3 3 2
|
20
|
6
|
|
58.8393
|
58.8424
|
-8 0 2
|
5
|
6
|
|
59.8705
|
59.8526
|
8 1 0
|
7
|
6
|
|
60.4161
|
60.4044
|
7 2 0
|
4
|
6
|
|
61.3564
|
61.3967
|
-8 1 2
|
7
|
6
|
|
62.3364
|
62.2846
|
-2 3 3
|
9
|
4
|
|
62.8203
|
62.8415
|
0 0 2
|
7
|
6
|
|
62.8288
|
62.8015
|
-4 0 5
|
7
|
6
|
|
63.7053
|
|
|
4
|
NI
|
|
64.1094
|
64.0462
|
2 0 5
|
7
|
6
|
|
64.6749
|
64.6636
|
-7 0 4
|
5
|
6
|
|
65.2306
|
65.2333
|
-9 0 0
|
20
|
6
|
|
65.7766
|
65.7380
|
1 1 3
|
20
|
5
|
|
65.8244
|
65.7892
|
-5 0 5
|
4
|
6
|
|
66.9879
|
66.9218
|
-9 1 1
|
7
|
6
|
|
67.4620
|
67.4064
|
3 0 5
|
5
|
6
|
|
67.6657
|
67.7061
|
2 0 2
|
5
|
3
|
|
67.9852
|
68.0135
|
1 4 0
|
7
|
6
|
|
68.1219
|
68.1954
|
-5 1 5
|
5
|
6
|
|
68.6949
|
68.6649
|
-9 1 2
|
8
|
6
|
|
69.0624
|
69.0646
|
-1 2 5
|
18
|
6
|
|
69.2146
|
69.2154
|
3 0 1
|
18
|
4
|
|
69.2228
|
69.1879
|
4 2 4
|
18
|
6
|
|
69.8576
|
69.8566
|
6 2 3
|
8
|
6
|
|
70.2700
|
70.2332
|
8 2 1
|
5
|
6
|
|
70.4508
|
70.4621
|
-9 0 3
|
6
|
6
|
|
70.8575
|
70.8114
|
9 1 1
|
4
|
6
|
|
71.7877
|
71.8290
|
1 3 4
|
6
|
6
|
|
72.1147
|
72.0848
|
-3 4 1
|
11
|
6
|
|
72.3127
|
72.4357
|
1 2 2
|
11
|
3
|
|
72.62.78
|
72.6461
|
1 3 1
|
8
|
4
|
|
72.6359
|
72.6712
|
10 0 1
|
8
|
6
|
|
73.3067
|
73.3157
|
1 2 0
|
7
|
5
|
|
73.3525
|
73.3511
|
3 4 1
|
7
|
6
|
|
73.5542
|
73.5585
|
2 2 5
|
8
|
6
|
|
73.9923
|
73.9868
|
9 0 2
|
11
|
6
|
|
74.2391
|
74.2221
|
6 3 2
|
13
|
6
|
|
75.0117
|
75.0283
|
-3 4 2
|
8
|
6
|
|
75.3557
|
75.4155
|
-4 0 6
|
14
|
6
|
|
75.8212
|
75.8790
|
10 1 0
|
11
|
6
|
|
77.5072
|
77.5178
|
3 4 2
|
10
|
6
|
|
77.7216
|
77.7181
|
9 2 1
|
10
|
6
|
|
78.2756
|
78.3334
|
5 4 0
|
8
|
6
|
|
78.6794
|
78.7209
|
5 1 5
|
6
|
6
|
|
79.0052
|
79.0261
|
1 1 4
|
13
|
5
|
|
79.0492
|
79.1137
|
10 1 1
|
13
|
6
|
3 :LaTiO3 4 : TiO2 5 :La2O3 6 : La2Ti2O7
T=1000°C :
Les phases en présence sont :
LaTiO3 : cristallise dans le groupe d'espace
Pm-3m(221) du système cubique avec les paramètres suivants :
a=b=c=3.92 A°
Après affinement on obtient :
a=b=c=3,9085 A°
TiO2 : cristallise dans le groupe d'espace
P42/mmm (136) du système tetragonal avec les paramètres suivants
:
a=b= 4,58 A° c= 2,95 A°
Après l'affinement on obtient :
a=b= 4.5793 A° c= 2.9560 A°
La2O3 : cristallise dans le groupe d'espace
P-3m1(164) du système hexagonal avec les paramètres suivants :
a=b=3,933 A° c=6,129 A°
Après l'affinement on obtient
a= b= 3.9417 A° c=6.1372 A°
La2Ti2O7 : cristallise dans le groupe d'espace
P21/m (11) du système monoclinique avec les paramètres suivants
:
a=13,09 A° b= 5,54 A° c= 7,80 A° â=98,38
°
Après l'affinement on obtient
a=13.0031 A° b= 5.5410A° c= 7.7994A° â=98.46
°
Discussion des résultats :
Le spectre RX de l'échantillon E3 calciné
à 900°C pressente un bruit de fond intense ainsi que le
chevauchement des pics, ce qui rend difficile l'identification des phases et
par suit l'affinement des paramètres ,donc nous nous sommes
contenté d'étudier E3 à 1000°C seulement.
Le diffractogramme de rayons X de l'échantillon E3
traité à 1000°C (fig.IV.5.b) montre la présence de
trois phases secondaires (parasites) en l'occurrence de la phase
recherchée P3 (LaTiO3),ces phases sont :
La2O3 : sa présence indique que certaine
quantité de l'oxyde de Lanthane (réactif) n'a pas
réagit.
TiO2 : de même, une quantité de
l'oxyde de titane n'a pas réagit.
La2Ti2O7: c'est une phase parasite, sa
présence peut être expliqué par le fait que :
- L'atmosphère du four: il faut utiliser un four sous
atmosphère contrôlée(inerte ou à l'abri de
l'oxygène)
- une éventuelle erreur dans la pesée des oxydes de
départ (réactifs) : la stoechiométrie n'est pas
respectée.
Donc, l'échantillon E3 est polyphasique.
La structure détaillée de la maille
pérovskite LaTiO3 est représentée sur la figure qui
suit.
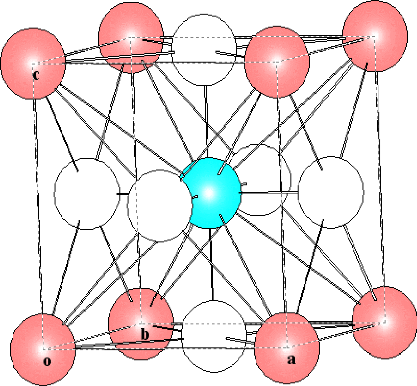
Figure IV.6 structure de la maille LaTiO3
Conclusion générale
Conclusion générale
L'ensemble des travaux présentés dans ce
mémoire a été axé sur la synthèse de trois
phases à structure pérovskite dont la formule
générale est LaMO3 (M=Fe, Mn, Ti) par voie solide (méthode
céramique), les différents échantillons ont
été traités à température 900°C puis
à 1000°C afin d'obtenir les phases recherchées, vient en
suit l'étape de caractérisation de ces échantillons par la
diffraction des rayons X. Les différents spectres obtenus ont
été comparés avec la base de données.
Les résultats de ce travail sont rassemblés comme
suit:
L'échantillon E1 :
La caractérisation par diffraction des RX nous a permis de
conclure que la poudre E1 est monophasique et après l'affinement par le
logiciel CELREF on trouve :
T=900°C :
LaFeO3 : SG : Pnma du système orthorhombique :
a=5.5590 A°, b=5.5647 A°, c=7.8598 A°
T=1000°C :
LaFeO3 : SG : Pnma du système orthorhombique :
a=5.5580 A° b=5.5652 A° c=7.8627 A°
L'échantillon E2 :
De même que l'échantillon E1 :
T=900°C :
LaMnO3 : SG : Pbnm du système orthorhombique :
a=5.5198 A° , b=5.7517 A° , c=7.7377 A°
T=1000°C:
LaMnO3 : SG : Pbnm du système orthorhombique :
a=5.5226 A° , b=5.7527 A° , c=7.7541 A°
L'échantillon E3:
T=1000°C:
LaTiO3 : SG : Pm-3m du système cubique
a=b=c=3,9085 A°
TiO2 : SG : P42/mmm du système
tetragonal
a=b= 4,58 A° c= 2,95 A°
La2O3 : SG : P-3m1du système hexagonal
a= b= 3.9417 A° c=6.1372 A°
La2Ti2O7 : SG : P21/m du système
monoclinique
a=13.0031 A° b= 5.5410A° c= 7.7994 A°
â=98.46 °
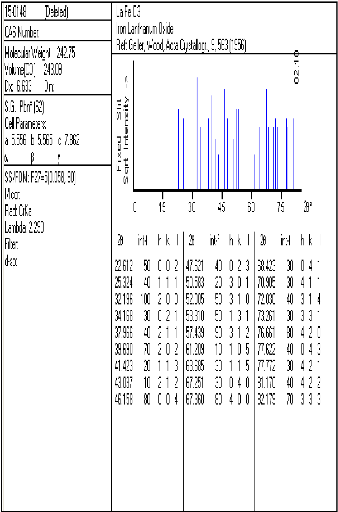
La phase LaFeO3 d'après la base de
données
CELREF VERSION 3
P1:LaFeO3
(T=900°C)
Initial values : (Refinement keys on 2nd line)
Zero Lambda a b c alpha beta gamma volume
|
0.000
0
H
|
1.54060
0
K L
|
5.5582 5.5645
1 1
2Th (obs)
|
7.8614 90.00
1 0
2Th_obs-shift
|
90.00 90.00 243.142
0 0
2Th (Calc) diff.
|
|
0
|
0
|
2
|
22.6065
|
22.6065
|
22.6028
|
0.0037
|
|
1
|
1
|
1
|
25.3037
|
25.3037
|
25.3033
|
0.0004
|
|
2
|
0
|
0
|
32.1855
|
32.1855
|
32.1835
|
0.0020
|
|
2
|
0
|
1
|
34.1590
|
34.1590
|
34.1935
|
-0.0345
|
|
2
|
0
|
2
|
39.6834
|
39.6834
|
39.6876
|
-0.0042
|
|
0
|
0
|
4
|
46.1503
|
46.1503
|
46.1506
|
-0.0003
|
|
2
|
2
|
1
|
47.6397
|
47.6397
|
47.6359
|
0.0038
|
|
1
|
1
|
4
|
51.9892
|
51.9892
|
51.9735
|
0.0157
|
|
1
|
3
|
1
|
53.3070
|
53.3070
|
53.3127
|
-0.0057
|
|
2
|
0
|
4
|
57.3796
|
57.3796
|
57.3768
|
0.0028
|
|
4
|
0
|
0
|
67.3297
|
67.3297
|
67.3322
|
-0.0025
|
|
3
|
1
|
4
|
72.0183
|
72.0183
|
72.0179
|
0.0004
|
|
1
|
1
|
6
|
76.6079
|
76.6079
|
76.5862
|
0.0217
|
|
2
|
4
|
1
|
77.6419
|
77.6419
|
77.6476
|
-0.0057
|
Final values : (Standard errors on 2nd line)
Zero Lambda a b c alpha beta gamma volume
|
0.000
0.0000
H
|
1.54060
0.00000
K L
|
5.5590 5.5647
0.0006 0.0007
2Th (obs)
|
7.8598 90.00
0.0007 0.000
2Th_obs-shift
|
90.00 90.00
0.000 0.000
2Th (Calc)
|
243.13
0.000
diff.
|
|
0
|
0
|
2
|
22.6065
|
22.6065
|
22.6075
|
-0.0010
|
|
1
|
1
|
1
|
25.3037
|
25.3037
|
25.3024
|
0.0013
|
|
2
|
0
|
0
|
32.1855
|
32.1855
|
32.1785
|
0.0070
|
|
2
|
0
|
1
|
34.1590
|
34.1590
|
34.1896
|
-0.0306
|
|
2
|
0
|
2
|
39.6834
|
39.6834
|
39.6863
|
-0.0029
|
|
0
|
0
|
4
|
46.1503
|
46.1503
|
46.1606
|
-0.0103
|
|
2
|
2
|
1
|
47.6397
|
47.6397
|
47.6322
|
0.0075
|
|
1
|
1
|
4
|
51.9892
|
51.9892
|
51.9817
|
0.0075
|
|
1
|
3
|
1
|
53.3070
|
53.3070
|
53.3107
|
-0.0037
|
|
2
|
0
|
4
|
57.3796
|
57.3796
|
57.3822
|
-0.0026
|
|
4
|
0
|
0
|
67.3297
|
67.3297
|
67.3208
|
0.0089
|
|
3
|
1
|
4
|
72.0183
|
72.0183
|
72.0192
|
-0.0009
|
|
1
|
1
|
6
|
76.6079
|
76.6079
|
76.6021
|
0.0058
|
|
2
|
4
|
1
|
77.6419
|
77.6419
|
77.6429
|
-0.0010
|
CELREF Version 3
P1:LaFeO3 (T=1000°C)
Initial values : (Refinement keys on 2nd line)
|
Zero
0.000
0
|
Lambda
1.54060
0
H K L
|
a b
5.5579 5.5646
1 1
2Th (obs)
|
c alpha
7.8646 90.00
1 0
2Th_obs-shift
|
beta gamma volume
90.00 90.00 243.232
0 0
2Th (Calc) diff.
|
|
1
|
1
|
0
|
22.5919
|
22.5919
|
22.5929
|
-0.0010
|
|
1
|
1
|
1
|
25.3011
|
25.3011
|
25.3015
|
-0.0004
|
|
2
|
0
|
0
|
32.1787
|
32.1787
|
32.1853
|
-0.0066
|
|
2
|
0
|
2
|
39.6856
|
39.6856
|
39.6834
|
0.0022
|
|
0
|
0
|
4
|
46.1435
|
46.1435
|
46.1307
|
0.0128
|
|
2
|
2
|
1
|
47.6358
|
47.6358
|
47.6356
|
0.0002
|
|
3
|
1
|
0
|
51.9775
|
51.9775
|
51.9813
|
-0.0038
|
|
1
|
3
|
1
|
53.3017
|
53.3017
|
53.3110
|
-0 .0093
|
|
3
|
1
|
2
|
57.3744
|
57.3744
|
57.3727
|
0.0017
|
|
1
|
3
|
3
|
63.6406
|
63.6406
|
63.6420
|
-0.0014
|
|
4
|
0
|
0
|
67.3254
|
67.3254
|
67.3363
|
-0.0109
|
|
4
|
0
|
2
|
72.0274
|
72.0274
|
72.0266
|
0.0008
|
|
4
|
2
|
0
|
76.5945
|
76.5945
|
76.5839
|
0.0106
|
Final values : (Standard errors on 2nd
line)
Zero Lambda a b c alpha beta gamma volume
|
0.000
0.0000
H
|
1.54060
0.00000
K L
|
5.5580 5.5652
0.0002 0.0005
2Th (obs)
|
7.8627 90.00
0.0009 0.000
2Th_obs-shift
|
90.00
0.000
2Th (Calc)
|
90.00 243.205
0.000 0.000
diff.
|
|
1
|
1
|
0
|
22.5919
|
22.5919
|
22.5915
|
0.0004
|
|
1
|
1
|
1
|
25.3011
|
25.3011
|
25.3015
|
-0.0004
|
|
2
|
0
|
0
|
32.1787
|
32.1787
|
32.1846
|
-0.0059
|
|
2
|
0
|
2
|
39.6856
|
39.6856
|
39.6862
|
-0.0006
|
|
0
|
0
|
4
|
46.1435
|
46.1435
|
46.1425
|
0.0010
|
|
2
|
2
|
1
|
47.6358
|
47.6358
|
47.6333
|
0.0025
|
|
3
|
1
|
0
|
51.9775
|
51.9775
|
51.9798
|
-0.0023
|
|
1
|
3
|
1
|
53.3017
|
53.3017
|
53.3062
|
-0.0045
|
|
3
|
1
|
2
|
57.3744
|
57.3744
|
57.3738
|
0.0006
|
|
1
|
3
|
3
|
63.6406
|
63.6406
|
63.6424
|
-0.0018
|
|
4
|
0
|
0
|
67.3254
|
67.3254
|
67.3349
|
-0.0095
|
|
4
|
0
|
2
|
72.0274
|
72.0274
|
72.0275
|
-0.0001
|
|
4
|
2
|
0
|
76.5945
|
76.5945
|
76.5806
|
0.0139
|
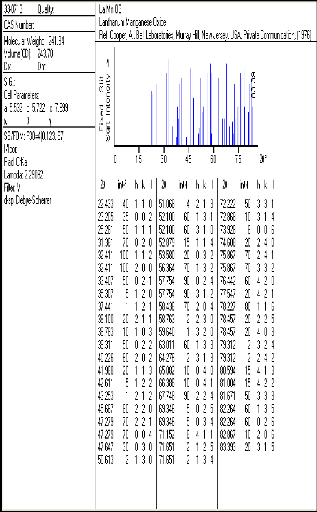
La phase LaMnO3 d'après la base de
données
CELREF version 3
P2:LaMnO3 (900°C)
Initial values : (Refinement keys on 2nd line)
Zero Lambda
a b c alpha beta gamma volume
|
0.000 0
H
|
K
|
1.54060 0
L
|
5.5341 5.7438
1 1
2Th (obs)
|
7.7235 90.00
1 0
2Th_obs-shift
|
90.00 90.00
0 0
2Th (Calc)
|
245.505
diff.
|
|
1
|
1
|
0
|
22.7367
|
22.7367
|
22.2893
|
0.4474
|
|
2
|
0
|
0
|
32.5073
|
32.5073
|
32.3275
|
0.1798
|
|
2
|
1
|
0
|
35.8669
|
35.8669
|
35.9983
|
-0.1314
|
|
2
|
0
|
2
|
40.2804
|
40.2804
|
40.0548
|
0.2256
|
|
0
|
0
|
4
|
46.5880
|
46.5880
|
47.0236
|
-0.4356
|
|
1
|
1
|
4
|
52.5428
|
52.5428
|
52.6288
|
-0.0860
|
|
3
|
1
|
2
|
57.8179
|
57.8179
|
57.6075
|
0.2104
|
|
2
|
0
|
4
|
57.9733
|
57.9733
|
58.2171
|
-0.2438
|
|
2
|
0
|
4
|
58.3252
|
58.3252
|
58.2171
|
0.1081
|
|
2
|
3
|
2
|
63.8060
|
63.8060
|
63.7881
|
0.0179
|
|
3
|
1
|
3
|
64.0299
|
64.0299
|
64.1354
|
-0.1055
|
|
1
|
1
|
5
|
64.7707
|
64.7707
|
64.6637
|
0.1070
|
|
4
|
0
|
0
|
67.8068
|
67.8068
|
67.6649
|
0.1419
|
|
1
|
4
|
1
|
68.3478
|
68.3478
|
68.5417
|
-0.1939
|
|
0
|
4
|
2
|
69.5210
|
69.5210
|
69.8249
|
-0.3039
|
|
2
|
3
|
3
|
69.9709
|
69.9709
|
69.9877
|
-0.0168
|
|
3
|
1
|
4
|
72.8807
|
72.8807
|
72.7232
|
0.1575
|
|
0
|
4
|
3
|
75.8770
|
75.8770
|
75.7933
|
0.0837
|
|
4
|
2
|
0
|
76.6573
|
76.6573
|
76.3412
|
0.3161
|
|
4
|
2
|
1
|
77.4414
|
77.4414
|
77.5114
|
-0.0700
|
Final values : (Standard errors on 2nd line)
Zero Lambda a b c alpha beta gamma volume
|
0.000
0.0000
H
|
K
|
1.54060
0.00000
L
|
5.5198
0.0074
2Th (obs)
|
5.7517 7.7377 90.00 90.00
0.0092 0.0139 0.000 0.000
2Th_obs-shift 2Th (Calc)
|
90.00 245.660
0.000 0.000
diff.
|
|
1
|
1
|
0
|
22.7367
|
22.7367
|
22.3047
|
0.4320
|
|
2
|
0
|
0
|
32.5073
|
32.5073
|
32.4134
|
0.0939
|
|
2
|
1
|
0
|
35.8669
|
35.8669
|
36.0668
|
-0.1999
|
|
2
|
0
|
2
|
40.2804
|
40.2804
|
40.1002
|
0.1802
|
|
0
|
0
|
4
|
46.5880
|
46.5880
|
46.9320
|
-0.3440
|
|
1
|
1
|
4
|
52.5428
|
52.5428
|
52.5518
|
-0.0090
|
|
3
|
1
|
2
|
57.8179
|
57.8179
|
57.7035
|
0.1144
|
|
2
|
0
|
4
|
57.9733
|
57.9733
|
58.1924
|
-0.2191
|
|
2
|
0
|
4
|
58.3252
|
58.3252
|
58.1924
|
0.1328
|
|
2
|
3
|
2
|
63.8060
|
63.8060
|
63.7640
|
0.0420
|
|
3
|
1
|
3
|
64.0299
|
64.0299
|
64.2022
|
-0.1723
|
|
1
|
1
|
5
|
64.7707
|
64.7707
|
64.5542
|
0.2165
|
|
4
|
0
|
0
|
67.8068
|
67.8068
|
67.8637
|
-0.0569
|
|
1
|
4
|
1
|
68.3478
|
68.3478
|
68.4524
|
-0.1046
|
0 4 2 69.5210 69.5210 69.7107 -0.1897
2 3 3 69.9709 69.9709 69.9424 0.0285
3 1 4 72.8807 72.8807 72.7555 0.1252
0 4 3 75.8770 75.8770 75.6611 0.2159
4 2 0 76.6573 76.6573 76.5072 0.1501
4 2 1 77.4414 77.4414 77.6723 -0.2309
CELREF version 3
P2: LaMnO3 (1000°C)
Initial values : (Refinement keys on 2nd line)
Zero Lambda a b c alpha beta gamma volume
0.000 1.54060 5.5308 5.7583 7.7447 90.00 90.00 90.00 246.653
0 0 1 1 1 0 0 0
H K L 2Th (obs) 2Th_obs-shift 2Th (Calc) diff.
0 0 2 22.7478 22.7478 22.9480 -0.2002
2 0 0 32.2843 32.2843 32.3473 -0.0630
1 0 3 38.2504 38.2504 38.4512 -0.2008
2 0 2 40.4070 40.4070 40.0325 0.3745
0 0 4 46.5658 46.5658 46.8872 -0.3214
3 1 0 52.2885 52.2885 52.0440 0.2445
1 1 4 52.6792 52.6792 52.4935 0.1857
3 1 2 57.6304 57.6304 57.5940 0.0364
2 2 3 58.4506 58.4506 58.4264 0.0242
4 0 0 67.6516 67.6516 67.7107 -0.0591
1 4 1 68.3997 68.3997 68.3596 0.0401
1 1 6 77.6943 77.6943 77.6931 0.0012
Final values : (Standard errors on 2nd line)
Zero Lambda a b c alpha beta gamma volume
0.000 1.54060 5.5226 5.7527 7.7541 90.00 90.00 90.00 246.342
0.0000 0.00000 0.0101 0.0163 0.0138 0.000 0.000
0.000 0.000
H K L 2Th (obs) 2Th_obs-shift 2Th (Calc) diff.
0 0 2 22.7478 22.7478 22.9199 -0.1721
2 0 0 32.2843 32.2843 32.3969 -0.1126
1 0 3 38.2504 38.2504 38.4223 -0.1719
2 0 2 40.4070 40.4070 40.0568 0.3502
0 0 4 46.5658 46.5658 46.8273 -0.2615
3 1 0 52.2885 52.2885 52.1248 0.1637
1 1 4 52.6792 52.6792 52.4518 0.2274
3 1 2 57.6304 57.6304 57.6566 -0.0262
2 2 3 58.4506 58.4506 58.4475 0.0031
4 0 0 67.6516 67.6516 67.8256 -0.1740
1 4 1 68.3997 68.3997 68.4329 -0.0332
1 1 6 77.6943 77.6943 77.6033 0.0910
CELREF Version 3
P3:LaTiO3 (1000°C)
Initial values : (Refinement keys on 2nd line)
Zero Lambda a b c alpha beta gamma volume
|
0.000
0
H
|
1.54060
0
K L
|
3.9111 3.9111
1 0
2Th (obs)
|
3.9111 90.00 90.00 90.00 59.827
0 0 0 0
2Th_obs-shift 2Th (Calc) diff.
|
|
0
|
1 1
|
32.5320
|
|
32.5320
|
32.3453
|
0.1867
|
|
1
|
1 1
|
39.8120
|
|
39.8120
|
39.8916
|
-0.0796
|
|
0
|
0 2
|
46.3754
|
|
46.3754
|
46.3953
|
-0.0199
|
|
2
|
0 1
|
52.4121
|
|
52.4121
|
52.2586
|
0.1535
|
|
2
|
1 1
|
57.9244
|
|
57.9244
|
57.6888
|
0.2356
|
|
2
|
0 2
|
67.6657
|
|
67.6657
|
67.7061
|
-0.0404
|
|
1
|
2 2
|
72.3127
|
|
72.3127
|
72.4357
|
-0.1230
|
|
|
Final values
|
: (Standard errors on 2nd line)
|
|
|
Zero
|
Lambda
|
a
|
b
|
c alpha
|
beta gamma
|
volume
|
|
0.000
|
1.54060
|
3.9085
|
3.9085
|
3.9085 90.00
|
90.00
|
90.00 59.706
|
|
0.0000
|
0.00000
|
0.0078
|
0.0000
|
0.0000 0.000
|
0.000
|
0.000 0.000
|
|
H
|
K L
|
2Th (obs)
|
2Th_obs-shift
|
2Th (Calc) diff.
|
|
0
|
1
|
1
|
32.5320
|
32.5320
|
32.3677
|
0.1643
|
|
1
|
1
|
1
|
39.8120
|
39.8120
|
39.9197
|
-0.1077
|
|
0
|
0
|
2
|
46.3754
|
46.3754
|
46.4285
|
-0.0531
|
|
2
|
0
|
1
|
52.4121
|
52.4121
|
52.2966
|
0.1155
|
|
2
|
1
|
1
|
57.9244
|
57.9244
|
57.7314
|
0.1930
|
|
2
|
0
|
2
|
67.6657
|
67.6657
|
67.7581
|
-0.0924
|
|
1
|
2
|
2
|
72.3127
|
72.3127
|
72.4924
|
-0.179
|
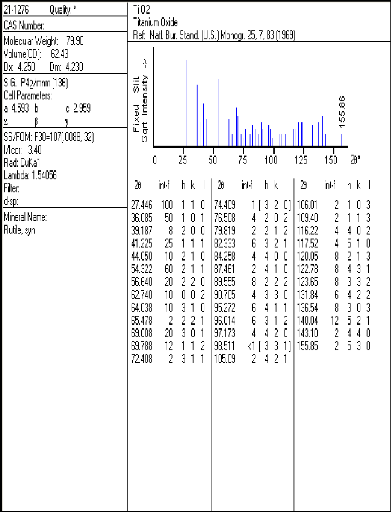
CELREF Version 3
P4:TiO2
Initial values : (Refinement keys on 2nd line)
Zero Lambda a b c alpha beta gamma volume
0.000 1.54060 4.5793 4.5793 2.9560 90.00 90.00 90.00 61.987
0 0 1 0 1 0 0 0
H K L 2Th (obs) 2Th_obs-shift 2Th (Calc) diff.
|
1
|
1
|
0
|
27.5243
|
27.5243
|
27.5241
|
0.0002
|
|
1
|
0
|
1
|
36.1531
|
36.1531
|
36.1383
|
0.0148
|
|
0
|
0
|
2
|
62.8203
|
62.8203
|
62.8225
|
-0.0022
|
|
3
|
0
|
1
|
69.2146
|
69.2146
|
69.2149
|
-0.0003
|
|
1
|
3
|
1
|
72.6278
|
72.6278
|
72.6462
|
-0.0184
|
Final values : (Standard errors on 2nd
line)
|
Zero
|
Lambda
|
a
|
b c
|
alpha beta gamma
|
volume
|
|
0.000
|
1.54060
|
4.5793
|
4.5793 2.9560
|
90.00 90.00
|
90.00
|
61.987
|
|
0.0000
|
0.00000
|
0.0010
|
0.0000 0.0003
|
0.000 0.000
|
0.000
|
0.000
|
|
H K
|
L
|
2Th (obs)
|
2Th_obs-shift
|
2Th (Calc)
|
diff.
|
|
|
1
|
1
|
0
|
27.5243
|
27.5243
|
27.5243
|
0.0000
|
|
1
|
0
|
1
|
36.1531
|
36.1531
|
36.1380
|
0.0151
|
|
0
|
0
|
2
|
62.8203
|
62.8203
|
62.8215
|
-0.0012
|
|
3
|
0
|
1
|
69.2146
|
69.2146
|
69.2152
|
-0.0006
|
|
1
|
3
|
1
|
72.6278
|
72.6278
|
72.6465
|
-0.0187
|
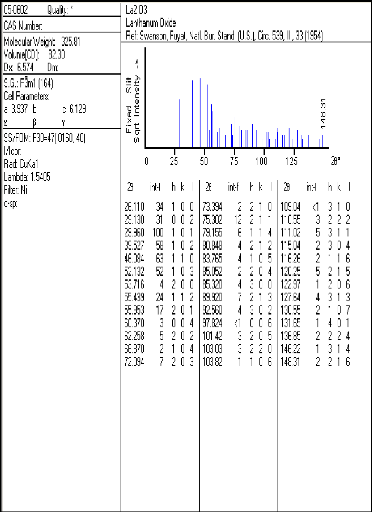
CELREF Version 3
P 5:La2O3
Initial values : (Refinement keys on 2nd
line)
Zero Lambda a b c alpha beta gamma volume
0.000 1.54060 3.9415 3.9415 6.1373 90.00 90.00 120.00 82.572
0 0 1 0 1 0 0 0
H K L 2Th (obs) 2Th_obs-shift 2Th (Calc) diff.
0 1 1 29.9259 29.9259 29.9292 -0.0033
1 1 3 65.7766 65.7766 65.7391 0.0375
1 2 0 73.3067 73.3067 73.3190 -0.0123
1 1 4 79.0052 79.0052 79.0269 -0.0217
Final values : (Standard errors on 2nd line)
Zero Lambda a b c alpha beta gamma volume
0.000 1.54060 3.9417 3.9417 6.1372 90.00 90.00 120.00 82.577
0.0000 0.00000 0.0049 0.0000 0.0015
0.000 0.000 0.000 0.001
H K L 2Th (obs) 2Th_obs-shift 2Th (Calc) diff.
0 1 1 29.9259 29.9259 29.9283 -0.0024
1 1 3 65.7766 65.7766 65.7380 0.0386
1 2 0 73.3067 73.3067 73.3157 -0.0090
1 1 4 79.0052 79.0052 79.0261 -0.0209
CELREF Version 3
P6:La2Ti2O7
Initial values : (Refinement keys on 2nd line)
Zero Lambda a b c alpha beta gamma volume
0.000 1.54060 13.0031 5.5410 7.7994 90.00 98.46 90.00 555.833
0 0 1 1 1 0 1 0
H K L 2Th (obs) 2Th_obs-shift 2Th (Calc) diff.
|
2
|
0
|
0
|
13.8275
|
13.8275
|
13.7591
|
0.0684
|
|
1
|
1
|
0
|
17.4933
|
17.4933
|
17.4127
|
0.0806
|
|
2
|
1
|
0
|
21.2188
|
21.2188
|
21.1479
|
0.0709
|
|
-2
|
1
|
1
|
23.1136
|
23.1136
|
23.1104
|
0.0032
|
|
2
|
1
|
1
|
25.0670
|
25.0670
|
25.0859
|
-0.0189
|
|
3
|
1
|
0
|
26.2020
|
26.2020
|
26.2617
|
-0.0597
|
|
-3
|
1
|
1
|
27.5340
|
27.5340
|
27.4517
|
0.0823
|
|
-1
|
1
|
2
|
28.1952
|
28.1952
|
28.1821
|
0.0131
|
|
-3
|
0
|
2
|
28.7170
|
28.7170
|
28.7426
|
-0.0256
|
|
3
|
1
|
1
|
29.9809
|
29.9809
|
29.9703
|
0.0106
|
|
0
|
2
|
0
|
32.2966
|
32.2966
|
32.2861
|
0.0105
|
|
-4
|
1
|
1
|
32.7942
|
32.7942
|
32.7858
|
0.0084
|
|
-3
|
1
|
2
|
33.0580
|
33.0580
|
33.0562
|
0.0018
|
|
-4
|
0
|
2
|
33.5228
|
33.5228
|
33.5287
|
-0.0059
|
|
0
|
2
|
1
|
34.4367
|
34.4367
|
34.3657
|
0.0710
|
|
-4
|
1
|
2
|
37.3570
|
37.3570
|
37.3488
|
0.0082
|
|
0
|
1
|
3
|
38.6073
|
38.6073
|
38.5664
|
0.0409
|
|
2
|
0
|
3
|
39.5815
|
39.5815
|
39.5808
|
0.0007
|
|
-1
|
2
|
2
|
40.0583
|
40.0583
|
40.0490
|
0.0093
|
|
1
|
2
|
2
|
41.2840
|
41.2840
|
41.2866
|
-0.0026
|
|
-6
|
0
|
0
|
42.1893
|
42.1893
|
42.1204
|
0.0689
|
|
-5
|
1
|
2
|
42.4538
|
42.4538
|
42.4871
|
-0.0333
|
|
4
|
2
|
0
|
43.1249
|
43.1249
|
43.0626
|
0.0623
|
|
-3
|
2
|
2
|
43.7581
|
43.7581
|
43.7652
|
-0.0071
|
|
6
|
1
|
0
|
45.3428
|
45.3428
|
45.3249
|
0.0179
|
|
-5
|
0
|
3
|
46.1500
|
46.1500
|
46.0543
|
0.0957
|
|
-1
|
0
|
4
|
46.5320
|
46.5320
|
46.5391
|
-0.0071
|
|
-4
|
2
|
2
|
47.2043
|
47.2043
|
47.2344
|
-0.0301
|
|
-6
|
1
|
2
|
48.2796
|
48.2796
|
48.2751
|
0.0045
|
|
1
|
0
|
4
|
48.6583
|
48.6583
|
48.7226
|
-0.0643
|
|
-2
|
2
|
3
|
48.8658
|
48.8658
|
48.8260
|
0.0398
|
|
-2
|
1
|
4
|
50.0812
|
50.0812
|
50.0767
|
0.0045
|
|
1
|
1
|
4
|
51.6388
|
51.6388
|
51.6069
|
0.0319
|
|
7
|
0
|
1
|
52.8950
|
52.8950
|
52.8502
|
0.0448
|
|
6
|
1
|
2
|
54.3891
|
54.3891
|
54.4725
|
-0.0834
|
|
0
|
3
|
2
|
55.0687
|
55.0687
|
55.0844
|
-0.0157
|
|
7
|
1
|
1
|
55.4909
|
55.4909
|
55.5804
|
-0.0895
|
|
|
|
|
|
|
Annexe
|
|
1
|
3
|
2
|
56.0488
|
56.0488
|
56.0715
|
-0.0227
|
|
-6
|
2
|
2
|
56.6660
|
56.6660
|
56.6605
|
0.0055
|
|
-7
|
0
|
3
|
57.1486
|
57.1486
|
57.1176
|
0.0310
|
|
-5
|
1
|
4
|
57.7098
|
57.7098
|
57.7687
|
-0.0589
|
|
-3
|
3
|
2
|
58.0559
|
58.0559
|
58.0675
|
-0.0116
|
|
-8
|
0
|
2
|
58.8393
|
58.8393
|
58.8411
|
-0.0018
|
|
8
|
1
|
0
|
59.8705
|
59.8705
|
59.8534
|
0.0171
|
|
7
|
2
|
0
|
60.4161
|
60.4161
|
60.4049
|
0.0112
|
|
-8
|
1
|
2
|
61.3564
|
61.3564
|
61.3955
|
-0.0391
|
|
-2
|
3
|
3
|
62.3364
|
62.3364
|
62.2841
|
0.0523
|
|
-4
|
0
|
5
|
62.8288
|
62.8288
|
62.8002
|
0.0286
|
|
2
|
0
|
5
|
64.1094
|
64.1094
|
64.0488
|
0.0606
|
|
-7
|
0
|
4
|
64.6749
|
64.6749
|
64.6612
|
0.0137
|
|
-9
|
0
|
0
|
65.2306
|
65.2306
|
65.2343
|
-0.0037
|
|
-5
|
0
|
5
|
65.8244
|
65.8244
|
65.7873
|
0.0371
|
|
-9
|
1
|
1
|
66.9879
|
66.9879
|
66.9217
|
0.0662
|
|
3
|
0
|
5
|
67.4620
|
67.4620
|
67.4096
|
0.0524
|
|
1
|
4
|
0
|
67.9852
|
67.9852
|
68.0133
|
-0.0281
|
|
-5
|
1
|
5
|
68.1219
|
68.1219
|
68.1935
|
-0.0716
|
|
-9
|
1
|
2
|
68.6949
|
68.6949
|
68.6637
|
0.0312
|
|
-1
|
2
|
5
|
69.0624
|
69.0624
|
69.0651
|
-0.0027
|
|
4
|
2
|
4
|
69.2228
|
69.2228
|
69.1908
|
0.0320
|
|
6
|
2
|
3
|
69.8576
|
69.8576
|
69.8596
|
-0.0020
|
|
8
|
2
|
1
|
70.2700
|
70.2700
|
70.2349
|
0.0351
|
|
-9
|
0
|
3
|
70.4508
|
70.4508
|
70.4600
|
-0.0092
|
|
9
|
1
|
1
|
70.8575
|
70.8575
|
70.8135
|
0.0440
|
|
1
|
3
|
4
|
71.7877
|
71.7877
|
71.8301
|
-0.0424
|
|
-3
|
4
|
1
|
72.1147
|
72.1147
|
72.0843
|
0.0304
|
|
-10
|
0
|
1
|
72.6359
|
72.6359
|
72.6712
|
-0.0353
|
|
3
|
4
|
1
|
73.3525
|
73.3525
|
73.3513
|
0.0012
|
|
2
|
2
|
5
|
73.5542
|
73.5542
|
73.5608
|
-0.0066
|
|
9
|
0
|
2
|
73.9923
|
73.9923
|
73.9901
|
0.0022
|
|
6
|
3
|
2
|
74.2391
|
74.2391
|
74.2240
|
0.0151
|
|
-3
|
4
|
2
|
75.0117
|
75.0117
|
75.0276
|
-0.0159
|
|
-4
|
0
|
6
|
75.3557
|
75.3557
|
75.4143
|
-0.0586
|
|
10
|
1
|
0
|
75.8212
|
75.8212
|
75.8801
|
-0.0589
|
|
3
|
4
|
2
|
77.5072
|
77.5072
|
77.5185
|
-0.0113
|
|
9
|
2
|
1
|
77.7216
|
77.7216
|
77.7201
|
0.0015
|
|
5
|
4
|
0
|
78.2756
|
78.2756
|
78.3334
|
-0.0578
|
|
5
|
1
|
5
|
78.6794
|
78.6794
|
78.7253
|
-0.0459
|
|
10
|
1
|
1
|
79.0492
|
79.0492
|
79.1161
|
-0.0669
|
Final values : (Standard errors on 2nd line)
Zero Lambda a b c alpha beta gamma volume
|
0.000
|
1.54060
|
13.0031
|
5.5410
|
7.7994
|
90.00
|
98.46
|
90.00
|
555.842
|
|
0.0000
|
0.00000
|
0.0023
|
0.0011
|
0.0017
|
0.000
|
0.013
|
0.000
|
0.126
|
H K L 2Th (obs) 2Th_obs-shift 2Th (Calc) diff.
|
2
|
0
|
0
|
13.8275
|
13.8275
|
13.7590
|
0.0685
|
|
1
|
1
|
0
|
17.4933
|
17.4933
|
17.4127
|
0.0806
|
|
2
|
1
|
0
|
21.2188
|
21.2188
|
21.1479
|
0.0709
|
|
-2
|
1
|
1
|
23.1136
|
23.1136
|
23.1109
|
0.0027
|
|
2
|
1
|
1
|
25.0670
|
25.0670
|
25.0851
|
-0.0181
|
|
3
|
1
|
0
|
26.2020
|
26.2020
|
26.2615
|
-0.0595
|
|
-3
|
1
|
1
|
27.5340
|
27.5340
|
27.4522
|
0.0818
|
|
-1
|
1
|
2
|
28.1952
|
28.1952
|
28.1822
|
0.0130
|
|
-3
|
0
|
2
|
28.7170
|
28.7170
|
28.7436
|
-0.0266
|
|
3
|
1
|
1
|
29.9809
|
29.9809
|
29.9693
|
0.0116
|
|
0
|
2
|
0
|
32.2966
|
32.2966
|
32.2862
|
0.0104
|
|
-4
|
1
|
1
|
32.7942
|
32.7942
|
32.7863
|
0.0079
|
|
-3
|
1
|
2
|
33.0580
|
33.0580
|
33.0570
|
0.0010
|
|
-4
|
0
|
2
|
33.5228
|
33.5228
|
33.5298
|
-0.0070
|
|
0
|
2
|
1
|
34.4367
|
34.4367
|
34.3658
|
0.0709
|
|
-4
|
1
|
2
|
37.3570
|
37.3570
|
37.3498
|
0.0072
|
|
0
|
1
|
3
|
38.6073
|
38.6073
|
38.5659
|
0.0414
|
|
2
|
0
|
3
|
39.5815
|
39.5815
|
39.5790
|
0.0025
|
|
-1
|
2
|
2
|
40.0583
|
40.0583
|
40.0492
|
0.0091
|
|
1
|
2
|
2
|
41.2840
|
41.2840
|
41.2861
|
-0.0021
|
|
-6
|
0
|
0
|
42.1893
|
42.1893
|
42.1198
|
0.0695
|
|
-5
|
1
|
2
|
42.4538
|
42.4538
|
42.4882
|
-0.0344
|
|
4
|
2
|
0
|
43.1249
|
43.1249
|
43.0624
|
0.0625
|
|
-3
|
2
|
2
|
43.7581
|
43.7581
|
43.7660
|
-0.0079
|
|
6
|
1
|
0
|
45.3428
|
45.3428
|
45.3244
|
0.0184
|
|
-5
|
0
|
3
|
46.1500
|
46.1500
|
46.0559
|
0.0941
|
|
-1
|
0
|
4
|
46.5320
|
46.5320
|
46.5389
|
-0.0069
|
|
-4
|
2
|
2
|
47.2043
|
47.2043
|
47.2353
|
-0.0310
|
|
-6
|
1
|
2
|
48.2796
|
48.2796
|
48.2763
|
0.0033
|
|
1
|
0
|
4
|
48.6583
|
48.6583
|
48.7210
|
-0.0627
|
|
-2
|
2
|
3
|
48.8658
|
48.8658
|
48.8264
|
0.0394
|
|
-2
|
1
|
4
|
50.0812
|
50.0812
|
50.0770
|
0.0042
|
|
1
|
1
|
4
|
51.6388
|
51.6388
|
51.6054
|
0.0334
|
|
7
|
0
|
1
|
52.8950
|
52.8950
|
52.8484
|
0.0466
|
|
6
|
1
|
2
|
54.3891
|
54.3891
|
54.4700
|
-0.0809
|
|
0
|
3
|
2
|
55.0687
|
55.0687
|
55.0843
|
-0.0156
|
|
7
|
1
|
1
|
55.4909
|
55.4909
|
55.5787
|
-0.0878
|
|
1
|
3
|
2
|
56.0488
|
56.0488
|
56.0712
|
-0.0224
|
|
-6
|
2
|
2
|
56.6660
|
56.6660
|
56.6617
|
0.0043
|
|
-7
|
0
|
3
|
57.1486
|
57.1486
|
57.1195
|
0.0291
|
|
-5
|
1
|
4
|
57.7098
|
57.7098
|
57.7704
|
-0.0606
|
|
-3
|
3
|
2
|
58.0559
|
58.0559
|
58.0682
|
-0.0123
|
|
-8
|
0
|
2
|
58.8393
|
58.8393
|
58.8424
|
-0.0031
|
|
8
|
1
|
0
|
59.8705
|
59.8705
|
59.8526
|
0.0179
|
|
7
|
2
|
0
|
60.4161
|
60.4161
|
60.4044
|
0.0117
|
|
-8
|
1
|
2
|
61.3564
|
61.3564
|
61.3967
|
-0.0403
|
|
-2
|
3
|
3
|
62.3364
|
62.3364
|
62.2846
|
0.0518
|
|
-4
|
0
|
5
|
62.8288
|
62.8288
|
62.8015
|
0.0273
|
|
2
|
0
|
5
|
64.1094
|
64.1094
|
64.0462
|
0.0632
|
|
-7
|
0
|
4
|
64.6749
|
64.6749
|
64.6636
|
0.0113
|
|
-9
|
0
|
0
|
65.2306
|
65.2306
|
65.2333
|
-0.0027
|
|
-5
|
0
|
5
|
65.8244
|
65.8244
|
65.7892
|
0.0352
|
|
-9
|
1
|
1
|
66.9879
|
66.9879
|
66.9218
|
0.0661
|
|
3
|
0
|
5
|
67.4620
|
67.4620
|
67.4064
|
0.0556
|
|
1
|
4
|
0
|
67.9852
|
67.9852
|
68.0135
|
-0.0283
|
|
-5
|
1
|
5
|
68.1219
|
68.1219
|
68.1954
|
-0.0735
|
|
-9
|
1
|
2
|
68.6949
|
68.6949
|
68.6649
|
0.0300
|
|
-1
|
2
|
5
|
69.0624
|
69.0624
|
69.0646
|
-0.0022
|
|
4
|
2
|
4
|
69.2228
|
69.2228
|
69.1879
|
0.0349
|
|
6
|
2
|
3
|
69.8576
|
69.8576
|
69.8566
|
0.0010
|
|
8
|
2
|
1
|
70.2700
|
70.2700
|
70.2332
|
0.0368
|
|
-9
|
0
|
3
|
70.4508
|
70.4508
|
70.4621
|
-0.0113
|
|
9
|
1
|
1
|
70.8575
|
70.8575
|
70.8114
|
0.0461
|
|
1
|
3
|
4
|
71.7877
|
71.7877
|
71.8290
|
-0.0413
|
|
-3
|
4
|
1
|
72.1147
|
72.1147
|
72.0848
|
0.0299
|
|
-10
|
0
|
1
|
72.6359
|
72.6359
|
72.6712
|
-0.0353
|
|
3
|
4
|
1
|
73.3525
|
73.3525
|
73.3511
|
0.0014
|
|
2
|
2
|
5
|
73.5542
|
73.5542
|
73.5585
|
-0.0043
|
|
9
|
0
|
2
|
73.9923
|
73.9923
|
73.9868
|
0.0055
|
|
6
|
3
|
2
|
74.2391
|
74.2391
|
74.2221
|
0.0170
|
|
-3
|
4
|
2
|
75.0117
|
75.0117
|
75.0283
|
-0.0166
|
|
-4
|
0
|
6
|
75.3557
|
75.3557
|
75.4155
|
-0.0598
|
|
10
|
1
|
0
|
75.8212
|
75.8212
|
75.8790
|
-0.0578
|
|
3
|
4
|
2
|
77.5072
|
77.5072
|
77.5178
|
-0.0106
|
|
9
|
2
|
1
|
77.7216
|
77.7216
|
77.7181
|
0.0035
|
|
5
|
4
|
0
|
78.2756
|
78.2756
|
78.3334
|
-0.0578
|
|
5
|
1
|
5
|
78.6794
|
78.6794
|
78.7209
|
-0.0415
|
|
10
|
1
|
1
|
79.0492
|
79.0492
|
79.1137
|
-0.0645
|
Références bibliographiques
Références bibliographiques
[1] : Ciprian Bogdan JURCA <<
Synthèse et caractérisation de pérovskites double
Magnétorésistives dérivées de Sr2FeMoO6 >>
thèse doctorat, Université Paris XI (2004).
[2] : Cristian PERCA << Structure
cristalline et magnétique de pérovskites RBaMn2O6-ä
(où ä = 0 et 0,5) >> thèse doctorat,
Université Paris XI (2005)
[3] : Abdelhadi Aydi << Elaboration et
caractérisation diélectriques de céramiques
ferroélectriques et/ou relaxeur de formule MSnO3-NaNbO3 (M= Ba, Ca)
>> thèse doctorat, Université Bordeaux 1. (2005)
[4] : Sébastien Courjault <<
Physico-chimie de cobaltites à monocouche de thallium de type 1222
>> thèse doctorat, Université Bordeaux 1(2002).
[5] : Abdelhadi Ben Ayad <<
Matériaux monocristallins à forte activité
piézoélectrique : élaboration, caracterisation et
application >> thèse doctorat, Institut national des science
appliquées de Lyon (2005).
[6] : Site internet <<
http://
www.Empa-pérovskite >>
[7] : Site internet <<
http:// www.wikipedia.org
>>
[8] :Jean-Pierre Eberhart <<Analyse
structurale et chimique des matériaux >>.DUNOD, Paris, 1997.
[9] : Florence Daumarie << La chimie au
capes >> HERMANN, Paris, 2002.
[10] : Peter W.Atkins << Les concepts de
chimie physique >> DUNOD, Paris, 1998.
[11] : Boukechira. D et Boufroua. N <<
Synthèse et caractérisation d'oxyde mixte à base de Nickel
de type : Pérovskite >> Mémoire de fin d'étude,
Université de Jijel (2005)
[12] : Maricela Villanuva-Ibanez « HfO2 et
SrHfO3 dopés terres rares : propriétés otiques et
potentialités en scintillation >> thèse doctorat,
université de Claude Bernard-Lyon 1 (2005)


| 

