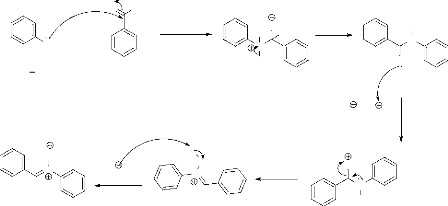
Synthèse des isoxazolidines par réaction de cycloaddition dipolaire-1,3 entre nitrones et olefines( Télécharger le fichier original )par Abdelmalek KHORIEF NACEREDDINE Université Badji-Mokhtar Annaba - Magister 2007 |

2. Synthèse de l'a, N-diphénylnitrone 8 à partir de la N-phénylhydroxylamine 2 et du benzaldéhyde.La a,N-diphénylnitrone 8 a été synthétisée en utilisant la condensation du benzaldehyde16 avec l'hydroxylamine 2. Cette dernière a été préparée au niveau de notre laboratoire à partir du benzène comme produit de départ, selon l'enchaînement réactionnel décrit dans le schéma 7. La réaction de substitution électrophile, sur le noyau benzénique, a été réalisée en prenant de multiples précautions du fait de la toxicité du benzène et de l'emploi des acides forts. Le nitrobenzène obtenu a été ensuite réduit en N-phénylhydroxylamine 2. Le rendement obtenu après recristallisation est de 67 %. 2 8 - Schéma 7 -
HNO3 H2SO4 NO2 Zn / NH4Cl H2O NHOH O EtOH,TA,24h
N O La condensation de l'hydroxylamine 2 avec le benzaldéhyde conduit finalement à la formation de la nitrone 8 avec un rendement satisfaisant (76%). Le mécanisme réactionnel de la condensation est détaillé dans le schéma suivant :
O H EtOH OH N OH NHOH 2 EtO , H H O O N EtO N O N H -H2O OH2 N OH 8 - Schéma 8 - La spectroscopie IR a permis de justifier la présence des bandes caractéristiques de la nitrone, à savoir 1570 cm-1 qui est la bande de vibration de valence de C=N et 1210 celle de N-O. Le spectre RMN 1H de la nitrone 8 montre que le pic du proton, de la fonction imine, apparaît sous forme d'un singulier à 7,90 ppm, ce qui confirme bien la structure de notre produit. En spectroscopie de masse ce produit est caractérisé par l'apparition du pic d'ion moléculaire à m/z =197. 3. Synthèse de a-phényl-N-(4-hydroxy)-phényl-nitrone 9 par réaction entre le benzaldéhyde et le para-hydroxynitrobenzène :Cette réaction nous a permis d'obtenir des structures très complexes de nitrones39 à partir de produits commerciaux simples. Nous avons pu réaliser la synthèse de la a-phényl-N-4- hydroxyphénylnitrone 9 en utilisant le benzaldéhyde et le 4-hydroxynitrobenzène. Cette nitrone possède un substituant hydroxyle situé en position para dans le noyau benzénique porté par l'atome d'azote. C'est par conséquent une réaction clé pour la préparation de ce genre de composés, qui est la condensation entre les nitroarènes et les composés carbonylés39, donc c'est la raison pour laquelle nous avons employé cette méthode pour l'obtention de la nitrone 9. La 4- hydroxyphénylhydroxylamine est préparée, à partir de la réduction du 4-hydroxynitrobenzène, in situ, que nous faisons,ensuite, réagir avec le benzaldéhyde pour aboutir à la nitrone attendue 9 (Schéma 9).
NH4Cl, EtOH N H2O,Zn,16h, 0 - 25C° O OH + O H NO2 OH 9 - Schéma 9 - Le produit 9 a été identifié par spectroscopie infrarouge. L'apparition de la bande d'absorption caractéristique de OH à 3300 cm-1, et les autres bandes caractéristiques de la fonction nitrone (1680 cm-1 et 1100 cm-1) confirment d'une manière claire la structure du produit 9.
CONCLUSION La condensation entre les aldéhydes aliphatiques et aromatiques para substitués, en l'occurrence le benzaldéhyde, le paranitrobenzaldéhyde, anisaldéhyde et propanaldéhyde, nous fournit les nitrones correspondantes avec des rendements variés en fonction de la structure de ces derniers et aussi de par la nature des groupements portés par le noyau benzénique de l'aldéhyde. Les nitrones préparées à partir des aldéhydes aliphatiques sont instables et se décomposent en leurs produits de départ, pendant la purification ou au contact avec des solvants polaires comme l'éthanol. Tandis que les nitrones aromatiques sont généralement plus stables et cristallisent dans l'éthanol ou l'éther diéthylique. L'introduction des substituants donneurs d'électrons tel que le groupement méthoxy, joue un rôle important dans la formation des nitrones correspondantes, où l'on obtient de bons rendements (de l'ordre de 80%). Par contre les substituants attracteurs d'électrons, comme le groupement nitro, défavorisent la formation de nitrones, et l'on constate l'obtention de rendements faibles (environ 12%). Les nitrones aromatiques porteurs du groupement aryles sur l'atome d'azote sont plus stables et ceci est dû à la forte conjugaison qui existe dans la molécule.
TECHNIQUES GENERALES Sauf indication contraire, les analyses ont été conduites dans les conditions générales indiquées cidessous. Les spectres IR ont été enregistrés sur le spectrophotomètre infrarouge SHIMADZU- FTIR 8400S, avec des cellules en chlorure de sodium à épaisseur fixe pour les solutions liquides en film. Pour les solides, les pastilles sont préparées (2-4mg) de l'échantillon dans 200-225mg de KBr. La position des bandes caractéristiques est donnée en cm-1, les lettres F, m, et f placés après ces derniers signifient respectivement forte, moyenne, faible. Les spectres de masse ont été enregistrés avec un spectromètre de masse couplé à un appareil de chromatographie en phase gazeuse équipé d'une colonne capillaire de polarité moyenne de type (25m FS-OV-1701-CB-0.25 CS-25292-82). On utilise l'ionisation par impact électronique (70ev), les intensités relatives sont indiquées entre parenthèses, le chiffre 100 est attribué au pic de base. Les spectres de Résonance Magnétique Nucléaire du proton et de carbone 13 (RMN 1H et RMN 13C) ont été enregistrés sur un appareil BRUKER AC 200 à 250 MHz. Les spectres ont été effectués dans le CDCl3. Les déplacements chimiques (ö) sont exprimés en partie par million (ppm) par rapport au tétraméthylsilane (TMS) pris comme référence interne. Pour la description des signaux, nous utiliserons les abréviations suivantes: S : singulet, d : doublet, t : triplet, q : quadruplet, m : multiplet, M: massif, dd : doublet dédoublé. Les points de fusion (Pf) ont été déterminés en tube capillaire au moyen de l'appareil BUCHI electrothermal 9100. Chromatographie : Les chromatographies sur couche mince (ccm) ont été réalisées sur des plaques de gel de silice sur aluminium 60 F 254 SDS, les ccm sont observées en lumière ultraviolette à 254 nm ou trempées dans un révélateur d'iode (SiO2 +I2). Les chromatographies sur colonne ont été effectuées sur gel de silice SDS. Solvants et réactifs : Le benzène et le toluène sont distillés et stockés sur tamis moléculaire 4 Å. Les réactifs ont généralement été utilisés sans purification supplémentaire. I. Synthèse de nitrones par réaction de
condensation entre un composé carbonylé et
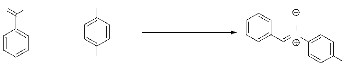
Me N O C8H9NO La réaction est réalisée sur 1.06 g (0.01mol) de benzaldéhyde dissous dans 20ml de toluène et 0.835 g (0.01mol) de 1 en solution dans 20ml du toluène et de 1.4 ml (0.01 mol) de triéthylamine. Temps de réaction 2 heures à reflux du solvant. La purification par recristallisation permet d'isoler 900 mg (0.0066mol, 67%) de 5. Rdt =67% Aspect : cristaux (recristallise dans l'éther diéthylique) Rf: 0.27 (Et2O/ CH2Cl2) Pf : 80 - 81°C tr: 10.90 min (80°C +10°C/min) IR (KBr) : 3055 (m) VC-H, 2993 (m) VC-H, 1589 (f) VC=N, 1558 (f) v C=C, 1080 (m) v N-C, 1296 (m) VNO. SM: [M.] +=135 (70), 134 (100), 118(20), 107(25), 89 (30), 77(50), 65(30), 44(73). RMN 1H (CDCl3): ö = 3.75 (s, 3H, CH3); 7.28-7.38 (m, 5H,C6H5), 8.15 (q, 1H, J=3.22 Hz, CH=N).
a-(4-nitro)-phényl-N-méthyl-nitrone 6 Me N O O2N C8H8N2O3 M =180 g/mole La réaction est effectuée à partir de 1.51 g (0.01 mol) de 4-nitrobenzaldéhyde en solution dans 20 ml de toluène et de 0.835 g (0.01 mol) de 1 dissous dans 20 ml de toluène et de 1.4 ml de triéthylamine en solution dans 20 ml de toluène. Après purification par chromatographie sur colonne de silice (10 g), nous avons isolé 200 mg (0.0011 mol, 11 %) de 6. Rd =11 % Aspect : poudre rouge brique Rf : 0.48 (Et2O/ CH2Cl2 (1/1)). Pf : 199 -200 °C IR (KBr) 2900 (m) VC-H ; 1680 (f) VC=N ; 1600 (m) VC=C ; 1350 (f) vC-N ; 1250 (m) VN-O 3400 (L) due à la présence d'une quantité d'eau dans la pastille de KBr.
a-(4-méthoxy)-phényl-N-méthylnitrone 7 Me N O MeO C9H11NO2 M =165 g/mole 2.27 g (0.02 mol ) d'anisaldéhyde (4-méthoxybenzaldéhyde) est dissous dans 30 ml du benzène, on additionne 1.67 g (0.02 mol) de 1 en solution de 30 ml de benzène, puis on ajoute, goutte à goutte, 2.8 ml (0.02 mol) de triéthylamine dans 20 ml du même solvant. Après purification du brut par chromatographie sur colonne on obtient 2.40 g (0.0145 mol, 73%) de la nitrone 7. Rd =73 % Aspect: huile jaune Rf: 0.20 (Et2O/ CH2Cl2). Tr: 9.71min (80C° +10C°/min) IR (CH2Cl2): 2900 (m) VC-H ; 1750 (m) VC=N ; 1600 (f) VC=C ; 1420 (m) VC-N ; 1280 (m) VN-O . a,N-diphénylnitrone 8 C13H11NO M =197 g/mole
N O Mode opératoire : Dans un bicol de 250ml, on introduit 0,33 g (0,30 mmol) de la N-phénylhydroxylamine 2 que l'on dissous dans 30 ml d'éthanol absolue, à une température comprise entre 40 - 60 °C, puis on ajoute 0,30 ml (2,94 mmol) de benzaldéhyde anhydre. On agite à température ambiante pendant 24 heures. Après concentration du mélange, la recristallisation dans l'éthanol donne 0.45 g (0.22 mmol, 76%) de 8. Rd =76 % Aspect : cristaux (recristallise dans l'éthanol) Rf: 0.30 (Et2O/ CH2Cl2, 8/2). tr: 27min (80C°+10C°/min) Pf : 112-113C° IR (KBr) : 2990 (f) VC-Harom ; 1570 (m) VC=N ; 1550 (m) VC=C ; 1470 (m) VC-N ; 1400 (m) VN-O . SM: [M.] + =197 (20); 182(5); 169(20); 141(15); 115(10); 105(20); 91(25); 77(100); 65(30); 51(40); 41(05). RMN 1H(CDCl3) : 7.38-7.68 (m, 4H) ; 7.70-7.77 (m, 2H); 7.90(s, 1H); 8.20 (d, 1H, J=2.9Hz); 8.33 (d, 1H, J=2.38) ; 8.37-8.50 (m, 2H). Mode opératoire de la réaction de nitration du benzène : Dans un bicol de 100 ml équipé d'un barreau magnétique, d'une ampoule à brome et d'un réfrigérant, on introduit 20 ml d'acide nitrique concentré et 10 ml d'acide sulfurique concentré. On laisse refroidir à l'aide d'un bain de glace fondante et on fait tomber goutte à goutte dans l'ampoule à brome, 20ml de benzène anhydre. Il faut régler le goutte à goutte de telle sorte que la couleur brune, crée par une goutte, disparaisse avant d'ajouter la goutte suivante. Lorsque l'addition est complète, on agite encore pendant 10 minutes avant de transférer le mélange réactionnel dans une ampoule à décanter et récupérer la phase organique. On la reprend dans 20ml d'éther et on sèche sur du sulfate de magnésium. L'évaporation du solvant sous pression réduite donne 15 ml de nitrobenzène. Mode opératoire de la réduction du nitrobenzène en N-phénylhydroxylamine : Dans un bicol de 250 ml on introduit 2,32 g (0,043 mol) de chlorure d'ammonium, 50ml d'eau distillée et 47,24 g (39,31ml, 0,31 mol) de nitrobenzène. Après agitation du mélange on additionne lentement 5,46 g (0,083 mol) de zinc en solution dans 20 ml d'eau distillée, pendant 15 minutes. Après augmentation de la température jusqu'à 60 - 65 °C, on agite encore le mélange jusqu'à l'abaissement de la température qui signale la fin de la réduction. On filtre le mélange réactionnel pour enlever l'oxyde de zinc (patte blanche), puis on lave le filtrat avec 50 ml d'eau distillée. On transvase le filtrat dans un Becher de 200 ml qu'on sature par du sel (NaCl). Le refroidissement dans un bain de glace pendant une heure du temps donne le maximum de recristallisation de l'hydroxylamine attendue. Cette dernière contient un petite quantité de sel, la recristallisation dans un solvant organique tel que l'éther donne 0.67 g (0,61 mol, 51%) d'hydroxylamine 2. Synthèse de a-phényl-N-(4-hydroxy) phénylnitrone 9. C13H11NO2 M=213 g/mole
N O OH Rd = 83% Aspect : cristaux (recristallise après chromatographie) Rf: 0.46 (Et2O/CH2Cl2 1/1) Pf: 170 °C IR (KBr): 3350(L) vO-H ; 3000 (f) VC-Harom ; 1680 (m) VC=N ; 1580 (m) VC=C ; 1320 (m) vC-N ; 1100 (m) VN-O Mode opératoire : Dans un bicol de 100ml on introduit 190 mg (1.36 mmole) de para-hydroxynitrobenzène dissous dans 2.6 ml d'éthanol, puis on ajoute 90.4 mg de NH4Cl dissous dans 3 ml d'eau distillée. Après agitation, on additionne 138 mg (1.30 mmol) de benzaldéhyde. Le mélange est refroidi jusqu'à 0°C, puis on ajoute goutte à goutte 170 mg (0.21 mmol) de Zn en solution dans 100 ml d'éthanol. On agite pendant 12 heures. Après filtration sous vide, le filtrat est lavé par deux fois 20 ml de dichlorométhane. La phase organique est ensuite séchée sur sulfate de sodium (Na2SO4). Après avoir filtré, la solution est alors évaporée sous vide. Le produit brut obtenu est purifié par chromatographie sur colonne de silice (10 g). On recueille 242 mg (1.136 mmol, 83%) de produit 9 sous forme de cristaux.
g tt-evai 2 .221 I H2 2048_0 2044.01 040.39 2136..0 2.1134 .24 2 0V2. 23 213.68 1847.ti6 160.06 103'9.4 ----..., 14131.16 183b.S3 1833,Ba 1033.2b li30.5 le3C.A0 1423.0 W..5-,a 3821.7E
s-6-3 PS g».24 1 964 934.24
co 1,11 ...1210.1.99 2104,35 2101.86 _7-'49#3.146 1949,95 1944.09 1141.24 1939.02 111111 -----1991.06 1912.99 1971.21 1870.68 1965,03 1961.24 1965.70 1E64.54 1E62.31 1659.98 1923.2?
L 27.73 10.15 10.64 10.26 9,91 5.54 1.70 .6.01 4.36 41,2,7QEa1WW22Z FARM-2MR2NtilgMFAVW Agq pRk i Y8 P, - Et2:::!2.414 r aiXi xi o 2 g AiWit UU tv1N I I I Iliiillii 1 'I i . . .. . . , II 6 oP XI 0.1-PocTs TIT I rrin PI r...) r ji \Ufs $ i 01 1 1.4 boat osu -1)46, 11 I it if r (1\ u 1,.. .. . ,..: ,1 ,
|
|