Chapitre 1
Méthodes de calcul de structure
électronique
1.1 Introduction
Dans ce chapitre, nous présentons les méthodes
de calcul de structure qui sont implémentées dans la plupart des
codes de calcul utilisés dans la détermination des grandeurs
caractéristiques des molécules. De nos jours, plusieurs
méthodes sont disponibles à cet effet et ne peuvent donner les
mêmes résultats. Certaines ne servent qu'à des fins
qualitatives car elles ne prennent pas en compte la corrélation
électronique. Par contre pour obtenir des résultats plus
précis, il faut inclure cette corrélation dans le traitement.
Dans ce chapitre, nous présentons en particulier les méthodes
RHF, CASSCF, et MRCI qui sont utilisées dans ce travail.
1.2 Équation de Schrödinger.
Toute l'information que l'on peut obtenir sur un
système moléculaire est contenue dans la fonction d'onde
ø(R, r), solution de l'équation de Schrödinger
indépendant du temps[13, 14, 15]
Hø(R, r) = Eø(R, r) (1.1)
où R et r représentent
respectivement l'ensemble des coordonnées des noyaux et des
électrons par rapport au centre de masse de la molécule, H
est le hamiltonien du système et E son énergie.
CHAPITRE 1. MÉTHODES DE CALCUL DE STRUCTURE
ÉLECTRONIQUE 4
Le hamiltonien totale H non relativiste
représentant un système à n électrons et
N noyaux peut s'écrire :
H(R, r) = TN(R) + Te(r) +
VNN(R) + VNe(R, r) + Vee(r)
(1.2)
Dans le système d'unité atomique
(me = e = ú = 4ðåo
= 1). Les différents termes constituant le hamiltonien
ont la signification et la forme définie comme suit :
>N
1
TN(R) = -
2 á=1
qui est l'énergie cinétique des noyaux,
1 V2 á = V2 (1.3)
N
Má
|
Xn
1
Te(r) = -2
i=1
qui est l'énergie cinétique des
électrons,
|
V2 i = V2 (1.4)
e
|
|
VNN(R) =
|
Xn á=1
|
XN â>á
|
ZáZâ (1.5)
Ráâ
|
qui est le terme d'interaction noyau-noyau,
VNe(R,r) = -
Zá (1.6)
Rái
XN á=1
Xn i=1
qui est le terme d'interaction électron-noyau,
Vee(r) =
1 (1.7)
rij
Xn i=1
Xn j<i
qui est le terme d'interaction
électron-électron. Avec Má la masse du noyau
á; Ráâ la distance entre le noyau
á et le noyau â de charges respectivement
Zá et Zâ ; Rái la distance entre le
noyau á et l'électron i; rij
la distance entre l'électron i et j ; les
indices N et e sont liés respectivement aux noyaux et
aux électrons.
En général, l'équation de
Schrödinger n'a de solution analytique que pour des systèmes
moléculaires ayant un ou deux électrons au plus. Dans les autres
cas, il faut recourir aux techniques numériques et faire appel à
des approximations à l'effet de simplifier la technique de
résolution. Parmi les plus couramment utilisées figure
l'approximation de Born-Oppenheimer qui est d'ailleurs fondamentale dans les
méthodes de calcul ab initio.
CHAPITRE 1. MÉTHODES DE CALCUL DE STRUCTURE
ÉLECTRONIQUE 5
Le potentiel d'interaction électron-électron
rend impossible la résolution de l'équation de Schrödinger
pour un nombre d'électron supérieur à un. Pour contourner
cette difficulté,
1.3 Approximation de Born-Oppenheimer
L'une des approximations que nous pouvons effectuer afin de
simplifier l'équation de Schrödinger (1.1) est celle
proposée en 1927 par Max Born et Robert Oppenheimer connue sous le nom
de l'approximation de Born-Oppenheimer (B-O) [15, 16]. Elle consiste à
séparer les mouvements des noyaux et des électrons. Elle est
basée sur le fait que la masse des noyaux d'un système
moléculaire est beaucoup plus grande que celle des électrons
(Mp 1836 me) ce qui suppose
que les noyaux se déplacent plus lentement et peuvent par
conséquent être considérés comme fixes.
L'énergie cinétique TN est donc négligée
en comparaison avec Te et le potentiel d'interaction
noyau-noyau (V NN) ne dépend plus que de la position
des noyaux et joue le rôle de paramètre dans cette
équation. Le hamiltonien total du système peut donc
s'écrire sous la forme d'une somme d'un hamiltonien électronique
He et d'un opérateur énergie cinétique
du noyau TN :
H = He + TN (1.8)
Avec :
He = Te(r) +
Vee(r) + VNe(R, r) + VNN(R)
(1.9)
La fonction d'onde totale peut s'écrire sous la forme
d'un développement en série sur une base complète de
fonctions électroniques.
ø(R, r) = X8
øNi(R)øi(R, r) (1.10)
i=1
telle que :
He(R)øi(R,r) =
Ei(R)øi(R,r) (1.11)
où les coefficients øNi(R)
dépendent des coordonnées des noyaux.
L'approximation de B-O est une très bonne
approximation, car dans la plupart des systèmes moléculaires,
elle introduit une erreur très négligeable. Toutefois, cette
approximation cesse d'être valable lorsque la molécule est dans un
état électronique, vibrationnel et rota-tionnel trop
excité car les noyaux sont rapides et leurs vitesses ne sont pas
négligeables.
1.4 Approximation de Hartree-Fock
CHAPITRE 1. MÉTHODES DE CALCUL DE STRUCTURE
ÉLECTRONIQUE 6
Hartree [17] puis Fock [18] ont proposé de
résoudre cette équation de Schrödinger électronique
en décrivant la fonction d'onde électronique sous la forme d'un
déterminant de Slater [19] composé de spin-orbitales
mono-électriques de type á ou â. Pour un système
à n électrons, ce déterminant s'écrit :
|
ö1(1) ö1(2)
1
~~~~~~~~~~~~~
Öe
|
ö2(1) ...
ö2(2) ...
... ...
... ...
ö2(N) ...
|
... öN(1)
... öN(2)
... ...
... ...
... öN(N)
|
|
,(1.12)
|
|
= ...
CAN!
... ö1(N)
|
où
v1N!est le facteur de
normalisation, une colonne représente une spin orbitale et une ligne
représente les coordonnées des électrons suivants la base
d'ordre N. La fonction d'onde décrite sous cette forme est
antisymétrique, conséquence directe du principe d'exclusion de
Pauli. L'énergie électronique Ee s'écrit sous
la forme :
Ee = (öe|
He |öe) (1.13)
où
h(i) +
1
(1.14)
He = Xn
i=1
rij
Xn i=1
n
E
j>i
L'hamiltonien électronique avec
XN
1
h(i) = -2?2i
-
Zá
(1.15)
Rái
á=1
un opérateur mono-électronique et,
|
gij =
|
1
=
rij
|
1 (1.16)
| ri-rj |
|
|
Ee =
|
Xn i=1
|
h(i) +
|
Xn i=1
|
n
E
j>i
|
(Jij - Kij)(1.17)
|
où
l'opérateur a deux électrons associé
à la répulsion Coulombienne entre un électron i et un
électron j.
L'énergie HF totale du système va donc
s'écrire :
CHAPITRE 1. MÉTHODES DE CALCUL DE STRUCTURE
ÉLECTRONIQUE 7
Jij =
höi(i)öj(j)| gij
|öi(i)öj(j)i (1.18)
qui est l'intégrale de Coulomb et
Kij =
höi(i)öj(j)| gij
|öj(i)öi(j)i (1.19)
qui est l'intégrale d'échange. Pour
obtenir l'énergie HF, on minimise l'énergie calculée en
utilisant la technique de variation à l'aide des multiplicateurs de
Lagrange. Ainsi la variation de l'énergie au premier ordre pour un
système à couches fermées s'écrit :
äEe = 2 XN
häöi| hi + XN (Jj -
Kj) |öii (1.20)
i=1 j=1
On définit l'opérateur de Fock pour un
électron par :
|
Fi = hi +
|
Xn j
|
(Ji - Kj) (1.21)
|
où les Jj et Kj sont respectivement
l'opérateur de coulomb et d'échange qui se définissent par
leur action sur les spinorbitales de la manière suivante :
Ji |öj(j)i =
höi(i)| gij
|öi(i)öj(j)i , (1.22)
Ki |öj(j)i =
höi(i)| gij
|öj(i)öi(j)i . (1.23)
Les équations de Hartree-Fock prennent la forme :
|
Fi |öii =
|
XN j
|
Àij |öji . (1.24)
|
Roothaan et Hall ont démontrés de manière
indépendante qu'en introduisant un jeu de fonction spatiales connues,
les équations intégro-différantielles peuvent être
transformées en
Les équations de HF forment un ensemble
d'équations intégrodifférentielles couplées et
peuvent être résolues par la méthode du champ
auto-cohérent (SCF) qui est une méthode itérative. La
résolution numérique de l'équation (1.24) de HF s'effectue
en utilisant l'approxi-mation LCAO proposée par Roothaan et Hall
[20].
1.5 Méthode de Roothaan-Hall : Approximation
LCAO
CHAPITRE 1. MÉTHODES DE CALCUL DE STRUCTURE
ÉLECTRONIQUE 8
un système d'équations algébriques et
être résolu en utilisant la méthode des matrices. Dans
cette approximation, les OMsøi sont exprimées comme
une combinaison linéaire de M fonctions atomiques appelées
»orbitales atomiques»
öi = XM cpi÷p. (1.25)
p
où M est la dimension de la base et les facteurs
cpi sont les coefficients des OMs développées sur les
fonctions de base ÷p.
En combinant les équations (1.24) et (1.25) et en
multipliant de chaque côté par ÷q puis en
intégrant, on obtient les équations de HAll-Roothaan :
FC = SCå (1.26)
Ici F est la matrice de Fock dont les éléments sont
:
Fpq = h÷p| F |÷qi,
(1.27)
S est la matrice de recouvrement avec
Spq = h÷p |÷qi.
(1.28)
C est la matrice carrée des coefficients du
développement et å est le vecteur des énergies
d'orbitales.
Les équations de Roothan-Hall (1.26) ne sont valables
que pour des systèmes à couches fermées avec un nombre
pair d'électrons. Les électrons sont appariés et l'un
d'entre eux a un spin á et l'autre un spin â. La fonction d'onde
correspondante est du type Restricted Hartree-Fock (RHF). La technique
correspondante est illustrée dans la référence [14], ainsi
que d'autres détails.
1.6 Méthodes Post Hartree-Fock
L'approximation de Hartree-Fock donne la meilleure
représentation d'une fonction d'onde décrite par un seul
déterminant de Slater. Cependant, les électrons interagissent
relativement à un champ moyen. Par conséquent, l'énergie
calculée n'est pas très précise et ne représente
qu'environ ~ 99 % de l'énergie totale. La différence
(c'est-à-dire ~ 1 % restant) entre l'énergie exacte et
celle de Hartree-Fock (HF) porte le nom d'énergie de corrélation
[21] :
CHAPITRE 1. MÉTHODES DE CALCUL DE STRUCTURE
ÉLECTRONIQUE 9
Ecorr = Eexacte - EHF. (1.29)
C'est pour récupérer cette énergie de
corrélation qu'on a recours aux méthodes dites Post-HF. Il existe
deux types méthodes de corrélation électronique :
- Les méthodes variationnelles telles que l'Interaction
de configuration (CI), l'Interaction de configuration
multiréférence (MRCI), et la méthode du champ
autocohérent multiconfi-gurationnelle (MCSCF),
- Les méthodes de perturbation telles que la
Théorie des perturbations multicorps (MBPT) et la méthode des
Clusters couplés (CC).
Dans le cadre de ce travail, nous allons utiliser la
première classe de ces méthodes qui offre l'avantage de pouvoir
traiter les états excités. Il est important de rappeler que le
point de départ de toutes ces méthodes de corrélation est
par principe la fonction d'onde de HF. Par conséquent, pour tout
traitement de la corrélation, la fonction d'onde s'écrira :
>2 W = a0ÖHF +
ajÖj, (1.30)
j=1
avec a0 ~ 1. Les méthodes de
corrélation diffèrent dans la manière de calculer les
coefficients aj. Les déterminants additionnels Öj
sont obtenus par excitation des électrons des orbitales
occupées de la fonction d'onde HF vers les orbitales virtuelles suivant
le diagramme de la figure 1.1. Les Öj s'appellent encore
déterminants de Slater excités
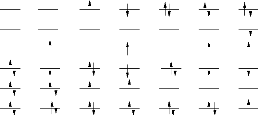
HF S-type S-type D-type D-type T-type Q-type
FIGURE 1.1: Diagramme des déterminants de Slater
excité généré à partir de la
référence HF.
CHAPITRE 1. MÉTHODES DE CALCUL DE STRUCTURE
ÉLECTRONIQUE 10
Les différentes méthodes Post Hartree-Fock sont
détaillées dans les références [14]. Nous
insisterons sur la méthode MRCI, qui est celle que nous avons
utilisée dans ce mémoire et qui offre l'avantage de pouvoir
traiter les états excités.
1.6.1 La méthode MRCI
La méthode MRCI est celle que nous utilisons dans ce
travail, c'est une méthode d'inter-action de configuration qui utilise
comme référence, la fonction d'onde MCSCF [22] à partir de
laquelle on n'autorise que les simples et doubles excitation. La fonction
d'onde ØMRCI est décrite par :
|
EØMRCI = ØMCSCF +
S
|
a
|
Eca SÖa S +
D
|
E
ab
|
cab D Öab D . (1.31)
|
öo = E CRöR (1.33)
R
où a et b constituent les orbitales de l'espace
externe, S et D sont les excitations simple et double des configurations de
référence, øi correspondant aux configurations internes (
incluant la fonction d'onde de référence et les configurations
obtenues par excitation d'un ou deux électrons à partir des
couches fermées dans les couches internes)
øas et øabD sont
associés respectivement aux configurations issues d'excitation simples
et doubles des orbitales internes vers les orbitales externes.
Dans l'espace de référence, il y a trois types
d'orbitales : les orbitales de coeur doublement occupées et non
corrélées, les orbitales de valence qui sont occupées par
0, 1 ou 2 électrons et corrélées au niveau CI et les
orbitales externes qui sont non occupées dans la fonction d'onde de
référence. Cette méthode de calcul engendre un très
grand nombre de configurations à traiter et demande beaucoup de temps de
calcul. Pour y remédier, une technique a été
implémentée dans le code MOLPRO par Werner et Knowles,
nommée « internally contracted ». Il s'agit de faire des
combinaisons linéaires de configurations qui ont la même partie
externe, mais des parties internes différentes. La contraction des
configurations est obtenue par des combinaisons linéaires entre elles
avec des coefficients fixes qui ne seront pas optimisés. Les
configurations contractées avec des excitations doubles dans l'espace
externe sont définies par :
ab 1
øijp = 2(Eai,bj +pEaj,bi)öo (1.32)
où p correspond au facteur de spin (il prend la valeur
+1 pour un couplage singulet entre les orbitales externes a et b et la valeur
-1 pour un couplage triplet). Eai,bj sont des opérateurs
biélectroniques et öo est la fonction d'onde de
référence MCSCF :
CHAPITRE 1. MÉTHODES DE CALCUL DE STRUCTURE
ÉLECTRONIQUE 11
En remplaçant öo par son expression,
l'équation (1.32) devient
|
X
øab
ijp =
R
|
XCR(Eai,bj + pEaj,bi)öR =
R
|
CRöab (1.34)
ijp,R
|
Après contraction, la fonction d'onde MRCI s'écrit
donc:
|
XøMRCI =
i
|
>
Ciøi +
s
|
X
a
|
XCa s øa s +
i=j
|
X
b
|
X
ab
|
Cijp
ab øab (1.35)
ijp
|
où lesøab
ijp sont les configurations contractées obtenues
après orthonormalisation et C les coefficients obtenus par un calcul CI
direct.
Ceci permet de réduire d'un facteur 30 les CSFs
à considérer avec très peu d'effet sur la qualité
des résultats. Cependant, malgré ces contractions, le nombre de
configurations à prendre en compte reste toujours trop
élevé. Afin de réduire encore ce nombre, il ne faut
considérer que les configurations dont les coefficients C
dépassent un certain seuil de référence. Cette
méthode CI tronquée n'est pas extensive en taille.
L'énergie de corrélation est donc mal décrite lorsque le
nombre de particules augmente. On peut la corriger grâce à
l'approximation de Davidson [23] où la contribution de l'énergie
de corrélation des configurations quadri-excitées est
estimée à partir des configurations doublement excitées.
La correction de l'énergie se calcule de la manière suivante :
ÄEQ = ÄESD(1 - X
C2 R) (1.36)
R
où ÄSD représente la
contribution à l'énergie de corrélation des excitations
simples et doubles prisent en compte dans la fonction de
référence MCSCF. Grâce à cette correction, cette
méthode est plus souvent privilégiée par rapport au calcul
Full CI pour la même base.
1.7 Le choix des bases d'orbitale
Une base est une description mathématique des orbitales
au sein d'un système [24]. Son choix est fonction de la précision
recherchée et du temps de calcul. En effet, une base contenant un nombre
infini d'orbitales moléculaires permettrait d'obtenir l'énergie
la plus précise par rapport à une méthode donnée.
Mais ceci rendrait les calculs impossibles compte tenu de la taille de la
matrice d'IC. Pour cela, elle doit obéir aux deux critères
essentiels suivants :
- La base doit être telle que l'erreur de superposition
soit le plus faible possible,
- La base doit reproduire avec une bonne précision les
grandeurs caractéristiques de la molécule, telles que les
constantes spectroscopiques.
CHAPITRE 1. MÉTHODES DE CALCUL DE STRUCTURE
ÉLECTRONIQUE 12
Il est à noter, qu'on utilise principalement deux types de
fonctions de base pour les calculs
numériques :
- les orbitales de type Slater (STOs)
- les orbitales de type gaussienne (GTOs)
Pour réaliser ce présent travail, nous avons
utilisé le logiciel MOLPRO [25].
1.8 Logiciel de calcul MOLPRO
Molpro[25] est un système complet de programme ab
initio pour le calcul de structure électronique des
molécules. Il a été conçu par H.-J. Werner et P. J.
Knowles et contient aussi les contributions de beaucoup d'autres auteurs. Il se
distingue des autres programmes utilisés en chimie quantique parcequ'il
donne avec la plus grande exactitude les résultats concernant le calcul
sur des petites molécules.
1.9 Conclusion
Les méthodes Post H-F ont pour point de départ,
la méthode H-F. Nous constatons que, malgré leurs
différences aucune d'entre elles n'est parfaite. Car toutes ces
méthodes présentent des limites et, au fur et à mesure
qu'on corrige ces défauts, on obtient des résultats plus
meilleurs.
13
| 

