Chapitre I
Méthodes Théoriques utilisées pour le
calcul de l'énergie d'interaction
La nature et l'importance des interactions
intermoléculaires dans les agrégats ioniques
OH-(H2O)n ont été étudiées
par les méthodes de la supermolécule HartreeFock et post
Hartree-Fock, Möller-Plesset, théorie de la perturbation (MPPT),
à l'ordre deux (MP2), trois (MP3) et quatre avec excitations simple,
double et quadruple (M P4SDQ), clusters couplés, simple double
excitations [CCSD(T)] avec une inclusion approchée de la triple
excitation, par la méthode de la perturbation, symmetry-adapted
perturbation theory (SAPT) et la théorie de la fonctionnelle de la
densité (DFT).
Les différentes méthodes utilisées pour
le calcul de l'énergie d'interaction donnent des informations
complémentaires sur les contributions énergétiques non
additives.
I.A/ Le Modèle de la supermolécule au niveau
SCF : Calculs ab intio:
Dans l'approche supermolécule (le système total
étant considéré comme une supermolécule),
l'énergie d'interaction est calculée comme dans l'équation
(1), utilisant n'importe quelle méthode fiable du calcul de
l'énergie électronique. Dans le cadre de la supermolécule,
des calculs ab initio ont été faits aux niveaux SCF, MP2, M P3, M
P4SDQ et CCSD(T).
Dans le modèle de la supermolécule, les
énergies d'interaction, à deux et trois corps non additive, sont
données respectivement, par :
+(1)
M (C
- (3)
st t (
= ---+++(2)
Où l'exposant SM est l'abréviation pour la
méthode de la supermolécule. SM est remplacé par HF pour
la méthode H artree- Fock, MPn pour la théorie de la perturbation
Möller-Plesset à l'ordre n et par CCSD(T) pour les calculs
coupled-cluster simple, double excitations et la triple excitation
approchée.
Nous avons calculé les énergies de la
première itération pour tous les systèmes AB,
AC, BC et
ABC, ce qui nous permet d'obtenir l'énergie d'interaction à trois
corps à la
première itération : . La différence
entre l'énergie à trois corps HF non
s it a
additive, obtenue à la convergence, et est notée
par [22].
F ? n ? st t a HF ? na ?
Cette quantité communément appelée
«l'énergie de déformation Hartree-Fock[23,24]
» ou énergie de délocalisation, nous donne une idée
sur la manière dont se déforme la distribution de charge des
monomères isolés (délocalisée sur toute la
supermolécule) pendant le processus SCF[25].
I.B/ La Méthode Hartree Fock [28]:
L'équation de Schr~dinger non relativiste et
indépendante du temps pour des systèmes polyélectroniques
est résolue de façon approchée via les approximations
suivantes :
L'approximation de Born-Oppenheimer considère les
noyaux figés par rapport aux mouvements des électrons
[9] . Nous pouvons négliger l'énergie cinétique
des noyaux et la répulsion entre noyaux est considéré
comme une constante. Dans ce cas l'opérateur hamiltonien de
l'énergie électronique d'un système à n
électrons et N noyaux, est :

Soit et E la fonction d'onde et l'énergie décrivant
ce système. L'équation de
Schr~dinger s'écrit :
Par la suite, on omettera de mettre l'indice e pour
électronique, dans toutes les équations développées
cette approximation est prise en compte.
La plus importante approximation, est celle de hartree-Fock, qui
consiste à ramener le
problème à n
éléctrons, comliqué, à un problème à
un életrcon où la
répulsion
électron-électron est traîté
comme une moyenne. La fonction d'onde approchée
donnée par
Hartree-Fock en 1928 [32] où la fonction d'onde du
système
est représentée par le produit des fonctions
d'onde monoélectroniques
( )
r
qui ne dépendent chacune que des coordonnées d'un
seul électron est :
r
(4)
Ø ( ) ( r
Dans la théorie de Pauli [33], pour mieux
décrire l'électron, il est nécessaire de
spécifier
son spin. Ceci est possible en introduisant deux fonctions
de spin ,
s e s
correspondant respectivement au spin haut et spin bas. La
fonction d'onde monoélectronqiue, dite fonction spinorbitale, est alors
écrite comme le produit de deux fonctions indépendantes : la
fonction d'espace et la fonction de spin :
)
(5)
? ( r = Ö ( x )
où ri désigne les variables d'espace et de spin, x
la variable d'espace et s la variable
de spin. ne peut prendre que deux
vaeurs . Ces deux
?
J ? ? J 1
e ?
fonctions de spin sont orthonormales :
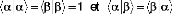
| 

