|

Mémoire présenté en vue de l'obtention du
diplôme d'Ingénieur de Conception de Génie Informatique
Développement d'un modèle
d'évolution de l'architecture
des gènes
PAR: Esaie KUITCHE KAMELA
Sous la direction de AïDA OUANGRAOUA,
Professeur
MEMBRES DU JURY:
Président: CREPIN TIMOLEON KOFANE,
Professeur Rapporteur: THOMAS BOUETOU BOUETOU, Professeur
Examinateur: GEORGES KOUAMOU, Docteur
Date de soutenance : 26 septembre 2016
ii
DÉDICACE
Je dédie ce travail :
-- À ma très chère et tendre maman, Mme
KAMELA née MAFOKAM Maria et à mon papa KAMELA Martin, qui
grâce à leur concours ont toujours su me soutenir au prix de
sacrifices incommensurables.
iii
REMERCIEMENTS
La réalisation de ce mémoire a été
possible grâce au concours de plusieurs personnes à qui je
voudrais témoigner toute ma reconnaissance. Il s'agit notamment :
-- de mes encadreurs, les professeurs Aïda
OUANGRAOUA et Thomas BOUETOU BOUETOU, pour leurs
disponibilité, indications, supports et surtout leurs judicieux
conseils, qui ont contribué à alimenter de manière
soutenue ma réflexion.
-- du professeur Crepin TIMOLEON KOFANE et
docteur Georges Edouard
KOUAMOU, pour avoir accepté
de faire partie de notre jury d'évaluation.
-- des enseignants de l'ENSPY en particulier ceux du
Génie Informatique pour m'avoir fourni les outils nécessaires
à la réussite de mes études d'ingénierie.
-- également de mes collègues du laboratoire
CoBIUS : Safa JAMMALI, Jean-David AGUILAR, Sarah BELHAMITI, Nilson
COIMBRA, et Michael LUCE pour nos discussions enrichissantes et
constructives qui ont permis à l'amélioration de la
qualité de mon travail.
-- de tous mes frères et soeur dont Livane
MAMOUBÉ KAMELA, Thierry NGATEU KAMELA, Armand FOTSING KAMELA, Valery
Martial TANKOU KAMELA ainsi qu'à mes parents papa
Martin KA-MELA et maman Maria MAFOKAM.
-- de toute la communauté estudiantine de l'ENSPY, en
particulier ceux de la Promotion 2016 du Génie Informatique dont leur
dynamisme et humour indubitables ont été pour moi une source
continue de courage.
-- de mes amis Floriane MEPOUBONG, Jean BIEMEWOU,
Pascal
DONGMO, Alain TSAYO, vous tous qui ne cessez
de me motiver. -- de la communauté camerounaise du Canada et
spécialement celle de Sher-
brooke pour leur accueil et hospitalité.
-- de tous(tes) ceux et (celles) qui de près ou de
loin, d'une manière comme d'une autre, pensent toujours à moi et
que j'ai oublié de citer.
iv
RÉSUMÉ
Les analyses génomiques récentes ont
révélé la capacité des gènes eucaryotes de
produire plusieurs ARN et protéines. Ce mécanisme joue un
rôle majeur dans la diversification fonctionnelle des gènes [1].
Cependant, les modèles de reconstruction actuelles de la
phylogénie de famille de gènes sont basés sur une seule
protéine de référence par gène, négligeant
ainsi toutes les autres protéines alternatives produites par les
gènes [31]. Ensuite, la réconciliation de l'arbre de gènes
dans l'arbre d'espèces est utilisée pour déduire
l'histoire évolutive des familles de gènes [12].
Le problème de la reconstruction de l'évolution
de l'expression du gène a été introduit dans [2],
où un modèle et un algorithme pour la reconstruction de la
phy-logénie des transcipts, ayant pour entrée l'arbre de
gènes et les structures des gènes. Ici, nous explorons une
approche différente en utilisant la réconciliation. Nous
proposons une extension de la réconciliation afin de reconstruire
à la fois l'arbre des gènes et l'histoire de l'évolution
de toutes les protéines produites par les gènes d'une famille de
gènes, compte tenu de l'arbre d'espèces. Nous proposons un
modèle d'évo-lution des protéines impliquant deux nouveaux
types d'événements évolutif appelé "création
de protéines" et "perte de protéines", en plus des
événements classiques de spéciation, duplication et de
perte de gènes.
Ce mémoire présente également les limites
des méthodes actuelles et propose une méthode heuristique pour la
reconstruction des arbres de gènes et d'arbres de protéines.
Cette méthode est résumée dans un processus à sept
étapes et vise à minimiser le coût de la
réconciliation. Aussi, nous avons implémenté et
appliqué ce modèle pour les familles de gènes de la base
de données Ensembl, montrant que notre modèle permet de
réduire le coût de la réconciliation des arbres de
gènes reconstruits avec des arbres d'espèces, par rapport aux
arbres de gènes Ensembl correspondants.
v
ABSTRACT
Recent genome analyses have revealed the ability of eukaryotic
genes to produce multiple transcripts and proteins. This mechanism plays a
major role in the functional diversification of genes [1]. Still, current
reconstructions of gene family phylogenies are based on a single reference
protein per gene, thus neglecting all other alternative products of genes
[31].
The problem of reconstructing gene product evolution was first
introduced in [2], where a model and an algorithm for transcript phylogeny
reconstruction, given the gene tree and the gene exon structures, were
introduced. Here, we explore a different approach using reconciliation. Gene
trees reconciliation with species trees is used to infer the evolutionary
history of gene families [12]. We propose an extension of the framework of
reconciliation in order to reconstruct both the gene tree and the evolutionary
history of all the proteins produced by the genes of a gene family, given the
species tree. We propose a model of protein evolution involving two types of
evolutionary event called "protein creation" and "protein loss", in addition to
the classical speciation, gene duplication and gene loss events.
In this report, we introduce new reconciliation problems
derived from the protein evolutionary model. We present some preliminary
algorithmic results and a heuristic method for the joint reconstruction of gene
trees and proteins trees. We applied this algorithm to gene families of the
Ensembl database, showing that our framework allows to lower the reconciliation
cost of the reconstructed gene trees with species trees, as compared to the
corresponding Ensembl gene trees.
vi
Table des figures
2.1 Structure de la cellule chez les Eucaryotes 5
2.2 Illustration d'une molécule d'ADN d'une cellule
d'eucaryote 6
2.3 Illustration des deux brins de l'ADN et de la relation de
complémen-
tarité des différentes bases 7
2.4 Chaque chromosome contient plusieurs gènes. 7
2.5 Épissage alternatif d'un gène chez les
eucaryotes 8
2.6 Code génétique 9
2.7 Exemple de deux séquences de nucléotides
à aligner 11
2.8 Résultat d'alignement possible des deux
séquences 11
2.9 Exemple d'alignement multiple de neuf séquences
protéiques. Les colonnes d'acides aminés conservés dans
l'alignement sont surlignées en
vert et bleu. Crédit : wikipedia. 11
2.10 Arbre
d'espèce enraciné, montrant les trois domaines de vivant :
bactéries, archées et eucaryotes, reliant les trois branches
d'organismes
au dernier ancêtre universel (le tronc noir en bas de
l'arbre) 12
2.11 Arbre d'espèces 13
2.12 Arbre de gènes non étiqueté 14
2.13 Résultat de la réconciliation de l'arbre de
gène 2.14 avec l'arbre d'es-
pèce 2.11 14
2.14 Arbre de gènes
étiquetés extrait de la réconciliation de l'arbre de
gène
dans l'arbre d'espèce 15
3.1 Application de UPGMA 18
3.2 Matrice et graphe additif 19
3.3 Enraciner un arbre à l'aide d'un outgroup. 19
3.4 Méthode de Ensembl-Compara pour la construction des
arbres de gènes 22
3.5 Produit de l'épissage alternatif 24
vii
TABLE DES FIGURES
4.1 Arbre de protéines étiqueté avec les
événements de spéciation, dupli-
cation, création et perte 26
4.2 Exemple de réconciliation d'arbres de protéines
avec arbre de gènes 28
4.3 Arbre de gènes étiqueté 29
4.4 arbre d'espèces 29
4.5 Processus de construction d'arbre de gènes à
sept étapes 31
4.6 Extrait d'arbre de protéines 34
4.7 Les trois principales catégories d'algorithmes
utilisées pour le regrou-
pement des séquences de protéines 35
4.8
Illustration présentant la différence entre le regroupement avec
che-
vauchement et le regroupement sans chevauchement 36
4.9
Exemple d'application de l'algorithme de regroupement avec chevauchement sur
sept protéines de cinq gènes. Les cinq gènes sont p1, p2
et p4 ayant chacun une protéine, et p3 et p5 ayant chacun deux
protéines. 37
4.10 Application de l'algorithme
glouton de fusion de 6 arbres. Les arbres sont greffés à l'arbre
de référence par des créations matérialisées
sur
la figure par des triangles au fond noir. 42
5.1 Arbre de gènes de la famille MAG obtenu avec notre
méthode . . . 44
5.2 Arbre de gène de la famille MAG obtenu de Ensembl
45
5.3 Arbre de gène de la famille FAM86 obtenu de notre
modèle 46
5.4 Arbre de gène de la famille FAM86 obtenu de Ensembl
47
viii
Table des Matières
DÉDICACE ii
REMERCIEMENTS iii
RÉSUMÉ iv
ABSTRACT v
LISTE DES FIGURES vii
GLOSSAIRE xi
|
1
|
INTRODUCTION
|
1
|
|
1.1
|
Problématique
|
2
|
|
1.2
|
Hypothèses
|
2
|
|
1.3
|
Objectif général
|
2
|
|
1.4
|
Objectifs spécifiques
|
2
|
|
1.5
|
Résumé des contributions
|
3
|
|
1.6
|
Plan du mémoire
|
3
|
|
2
|
GÉNOMIQUE
|
4
|
|
2.1
|
Bio-informatique et biologie computationnelle
|
4
|
|
|
2.1.1 Cellule
|
4
|
|
|
2.1.2 Chromosome
|
5
|
|
|
2.1.3 Acide désoxyribonucléique (ADN)
|
5
|
|
|
2.1.4 Gène
|
6
|
|
|
2.1.5 Acide ribonucléique (ARN)
|
6
|
|
|
2.1.6 Protéine
|
7
|
|
2.2
|
Bases de données
|
8
|
ix
TABLE DES MATIÈRES
|
2.3
2.4
|
2.2.1 Bases de données existantes
2.2.2 Accès aux bases de données
Évolution des séquences biologiques
2.3.1 Recherche de similarités entre séquences
2.3.2 Construction d'arbres phylogénétiques
Conclusion sur la génomique
|
8
9
10 10 10 14
|
|
3
|
ÉTAT DE L' ART
|
16
|
|
3.1
|
Méthodes de construction d'arbres
phylogénétiques
|
16
|
|
|
3.1.1 Énumération des arbres
|
16
|
|
|
3.1.2 Construction d'arbres à partir des distances entre
feuilles . . .
|
17
|
|
|
3.1.2.1 UPGMA
|
17
|
|
|
3.1.2.2 Neighbour Joining
|
17
|
|
|
3.1.3 Les méthodes de parcimonie
|
20
|
|
|
3.1.4 Méthodes de maximum de vraisemblance
|
20
|
|
3.2
|
Construction des arbres de gènes
|
21
|
|
|
3.2.1 Exemple de construction d'arbres de gènes -
EnsemblCompara
|
21
|
|
|
3.2.2 Limites des arbres de gènes actuels
|
23
|
|
3.3
|
Conclusion sur l'état de l'art
|
23
|
|
4
|
MÉTHODOLOGIE ET IMPLÉMENTATION
|
25
|
|
4.1
|
Modèle d'évolution des protéines et
problème de réconciliation . . . .
|
25
|
|
|
4.1.1 Idée de base
|
25
|
|
|
4.1.2 Définitions formelles
|
26
|
|
4.2
|
Méthodologie : Processus à sept étapes
|
30
|
|
|
4.2.1 Étape 1 : Définition du jeu de données
|
30
|
|
|
4.2.2 Étape 2 : Alignement multiple de toutes les
séquences codantes
|
32
|
|
|
4.2.3 Étape 3 : Calcul de la matrice de similarité
|
32
|
|
|
4.2.4 Étape 4 : Regroupement avec chevauchement des
séquences
|
|
|
|
codantes
|
32
|
|
|
4.2.4.1 Définition du regroupement
|
33
|
|
|
4.2.4.2 Problème de regroupement dans le cas
présent . . . .
|
33
|
|
|
4.2.4.3 Regroupement avec chevauchement hiérarchique . .
.
|
35
|
|
|
4.2.5 Construction d'un groupe de référence
|
36
|
|
|
4.2.6 Étape 6 : Création des arbres de
protéines pour les groupes . .
|
40
|
|
|
4.2.7 Étape 7 : Construction de l'arbre de
protéines et de l'arbre
|
|
|
|
des gènes
|
41
|
|
4.3
|
Implémentation
|
41
|
X
TABLE DES MATIÈRES
|
5
|
ÉTUDE DE CAS
|
43
|
|
5.1
|
Étude de cas de la famille The Myelin-Associated
Glycoprotein (MAG) 43
|
|
5.2
|
Étude de cas de la famille FAM86 46
|
|
6
|
CONCLUSION GÉNÉRALE
|
48
|
xi
Glossaire
ADN Acide Désoxyribonucléique.
API Application Programming Interface. ARN Acide
Ribonucléique.
BLAST Basic Local Alignment Search Tool.
CoBIUS Complexité Biologique &
Informatique de l'Université de Sherbrooke. FTP File Transfer
Protocol.
MACSE Multiple Alignment of Coding SEquences. MUSCLE Multiple
Sequence Alignment by log-Expectation.
NJ Neighbour joining.
TreeBest Tree Building guided by Species
Tree.
UPGMA Unweighted Pair Group Method with Arithmetic Mean.
1
CHAPITRE UN
INTRODUCTION
L'aboutissement du projet d'annotation du génome humain
en 2003 a permis aux scientifiques de mieux comprendre les processus
biologiques qui se déroulent dans les cellules des êtres vivants
au travers de l'Acide Désoxyribonucléique (ADN). Cet ADN est
l'entité qui contient toutes les informations régissant le
développement, le fonctionnement et la reproduction des êtres
vivants. Ces nouvelles connaissances ont eu le mérite de susciter un
regain d'attention de la part du public et des chercheurs.
L'ADN est composé de gènes, supports de
l'information, qui sont la base de la structure et du fonctionnement des
génomes. Il est donc naturel que nous nous intéressions tout
particulièrement à ce programme génétique qui
détermine notre fonctionnement.
Une bonne caractérisation et compréhension du
fonctionnement des gènes sera d'un apport considérable dans des
domaines tels que l'industrie pharmaceutique ou encore des traitements contre
les cancers. Les recherches dans ces domaines constituent une tâche
collective faisant appel à des collaborations multidisciplinaires.
De nombreux travaux en bio-informatique ont été
réalisés dans le but de proposer des modèles
d'évolution des gènes, tenant de diverses caractéristiques
séquentielles ou structurales des gènes. Ce mémoire se
focalise principalement à l'analyse des limites de ces modèles et
propose un nouveau modèle d'évolution basé sur
l'expression des gènes, suivi d'un algorithme pour la reconstruction de
l'histoire évolutive des gènes.
Ce mémoire qui décrit des approches
computationnelles pour résoudre une problématique issue des
sciences de la vie aura la particularité de marquer des temps
d'arrêt pour introduire les notions biologiques sous-jacentes.
2
CHAPITRE 1. INTRODUCTION
1.1 Problématique
Chez les eucaryotes1, il est maintenant reconnu que
chaque gène de l'ADN d'une espèce peut produire plusieurs
protéines et que les gènes des organismes eucaryotes peuvent
être classés en familles de gènes homologues 2.
La problématique soulevée par ce mémoire est de
définir un modèle d'évolution et un algorithme
associé pour reconstruire l'histoire évolutive d'une famille de
gènes (un ensemble de gènes homologues) en prenant en compte
toutes les protéines issues de chacun de ces gènes.
Cette approche diffère de celle des méthodes actuelles qui ne
considèrent qu'une seule protéine par gène, conduisant
ainsi souvent à des arbres (phylogénies) de gènes
erronés.
1.2 Hypothèses
Nous admettons dans nos travaux:
~ L'hypothèse de la théorie de
l'évolution [5], qui stipule que tous les êtres vivants sont issus
d'un ancêtre commun;
~ Les arbres d'espèces utilisés dans les
applications de nos algorithmes sont corrects.
1.3 Objectif général
L'objectif général est de proposer un
modèle d'évolution, un algorithme associé, ainsi qu'une
implémentation de l'algorithme pour la reconstruction de
l'évolution d'une famille de gènes en tenant compte de toutes les
protéines de chaque gène.
1.4 Objectifs spécifiques
~ Modéliser le mécanisme de production de
plusieurs protéines par un même gène;
~ Modéliser l'évolution de l'ensemble des
protéines au sein d'une famille de gènes;
1. les eucaryotes sont un domaine du vivant regroupant toutes
les espèces, unicellulaires ou pluricellulaires, qui se
caractérisent par la présence d'un noyau dans leurs cellules. Il
s'oppose aux domaines des procaryotes.
2. les gènes homologues sont des gènes qui sont
issus d'un gène ancêtre commun qui a divergé par la suite.
Ces gènes peuvent appartenir à la même espèce ou
non.
3
CHAPITRE 1. INTRODUCTION
-- Proposer une modélisation globale suivie d'un
algorithme de reconstruction de l'évolution;
-- Étudier un cas et comparer les résultats
obtenus avec ceux issus de méthodes existantes.
1.5 Résumé des contributions
Nous avons proposer un modèle d'évolution des
protéines dans les arbres de gènes avec nouveaux
événements (création et pertes de protéines), puis
définit du problème d'optimisation (Entrée : sous-arbres
de protéines (groupes); arbre d'espèces; Sortie: arbre global des
protéines, et arbre des gènes), par la suite nous avons proposer
une heuristique pour résoudre le problème et enfin nous avons
réaliser une implémentation et effectuer une étude de
cas.
1.6 Plan du mémoire
Le mémoire est structuré autour de six
chapitres. Le premier chapitre introduit le problème de construction des
arbres de gènes et présentera les objectifs à atteindre.
Le chapitre 2 décrit les notions biologiques et bio-informatiques
nécessaires à la compréhension du sujet. Le chapitre 3 est
l'état de l'art, et présente les différents
modèles, méthodes existantes pour la construction des arbres
phylogénétiques. Le chapitre 4 quant à lui présente
les modèles et méthodes que nous avons développés,
ainsi que l'implémentation que nous avons réalisée. Le
chapitre 5 est dédié à l'étude de cas et le
chapitre 6 conclut le mémoire et pose des axes d'amélioration.
4
CHAPITRE DEUX
GÉNOMIQUE
Ce chapitre a pour but de fournir les notions biologiques et
bio-informatiques de base nécessaires pour mieux appréhender le
problème d'optimisation que nous considérons dans ce
mémoire. On commercera par définir la bio-informatique et la
biologie computationnelle. Ensuite on définira, par ordre d'inclusion,
les notions de cellules, chromosomes, ADN, gènes et protéines. On
finira en présentant quelques usages fréquents de ces types
d'objets biologiques en biologie computationnelle.
2.1 Bio-informatique et biologie computationnelle
La biologie est la science du vivant
s'intéressant en particulier à l'origine, la croissance, la
reproduction, la structure et le comportement des êtres vivants.
La bio-informatique se définit comme
l'application de l'informatique à la biologie. Notamment, il s'agit
d'utiliser des notions et ressources informatiques pour faciliter la
compréhension et la manipulation de données biologiques.
La biologie computationnelle quant à
elle vise le développement de modèles et algorithmes s'inspirant
des processus biologiques afin de répondre à des questions issues
de la biologie.
2.1.1 Cellule
La cellule est l'unité structurale, fonctionnelle et
reproductive constituant toute ou partie des êtres vivants, à
l'exception des virus. On distingue deux types d'orga-nisme. D'une part, il y a
les organismes unicellulaires qui sont constitués d'une seule cellule.
C'est le cas des bactéries ou des levures. D'autre part, il y a les
organismes multicellulaires composés de plusieurs cellules tels que
l'humain. Tout être vivant
5
CHAPITRE 2. GÉNOMIQUE
est composé de cellules dont la structure fondamentale
est commune [22, 26]. La figure 2.1 illustre une cellule d'eucaryote.
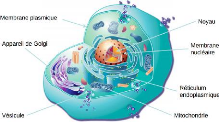
Crédit
:fr.bio.wikia.com/wiki/Cellule_Eucaryote
FIGURE 2.1 - Structure de la cellule chez les Eucaryotes
2.1.2 Chromosome
Le chromosome est une structure cellulaire microscopique
représentant le support physique du matériel
génétique. Tous les êtres vivants disposent des chromosomes
en nombre variable propre à chaque espèce. Par exemple, chaque
cellule humaine possède 22 paires de chromosomes homologues
numérotés de 1 à 22, et une paire de chromosomes sexuels,
soit un total de 23 paires. La figure 2.2 présente de manière
explicite la relation entre cellule, chromosome, ADN et gène, chez les
eucaryotes. [22, 26]
2.1.3 Acide désoxyribonucléique (ADN)
L'ADN est une macromolécule biologique constituant les
chromosomes des cellules. L'ADN contient toute l'information
génétique, appelée génome, permettant le
développement, le fonctionnement et la reproduction des êtres
vivants.
Les molécules d'ADN sont formées de deux brins
antiparallèles enroulés l'un autour de l'autre pour former une
double hélice comme présentée sur la figure 2.2. Chacun de
ces brins est un polymère constitué d'une succession d'acides
nucléiques. Chaque nucléotide est caractérisé par
une base azotée, adénine (A), cytosine (C), guanine (G) ou
thymine (T). L'ADN est représenté sous la forme d'une
séquence sur un alphabet de quatre lettres correspondant à l'un
de ses brins. Exemple : ATTCCGATCGGAAATTGC.
6
CHAPITRE 2. GÉNOMIQUE
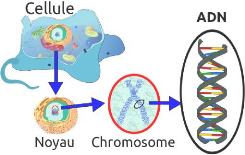
Crédit
:fr.wikipedia.org/wiki/Acide_desoxyribonucleique
FIGURE 2.2 - Illustration d'une molécule d'ADN d'une
cellule d'eucaryote
2.1.4 Gène
La définition de gène a évolué au
cours des dernières années. La définition
générale repose sur les trois aspects ci-dessous [14] :
1. Le gène est une portion de séquence d'ADN
générant des produits fonctionnels (ARNs ou protéines).
2. Le gène est la principale composante utile de
l'ADN.
3. Les gènes sont le support de l'évolution,
transmis aux générations suivantes avec l'ensemble de mutations
qu'ils subissent.
2.1.5 Acide ribonucléique (ARN)
L'acide ribonucléique (ARN) est une copie d'un
gène composée d'un seul brin d'acides nucléiques. Il
existe deux principaux types d'ARN. Les ARNs codants destinés à
être lus par des ribosomes pour permettre la synthèse de
protéines. Les ARNs non-codants sont directement impliqués dans
des fonctions au sein de la cellule.
Chez les eucaryotes, l'épissage [3,
15] est un processus par lequel, les ARNs transcrits à partir des
gènes subissent des étapes de coupure et ligature qui conduisent
à l'élimination de certains segments pour aboutir à un ARN
final dit mature (ARNm). Les segments d'ARN conservés s'appellent des
exons et ceux qui sont éliminés s'ap-pellent des introns. Un
gène eucaryote peut donc être redéfini comme une suite
d'exons et d'introns alternés. L'épissage alternatif
est un mécanisme qui permet à un gène de produire
plusieurs ARNm en générant différentes combinaisons des
exons [3, 15, 4].
7
CHAPITRE 2. GÉNOMIQUE
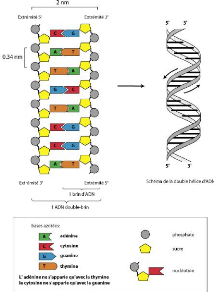
Crédit :https : //
www.bioutils.ch/protocoles/9extractiondadn
FIGURE 2.3 - Illustration des deux brins de l'ADN et de la
relation de complémentarité des différentes bases.
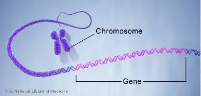
FIGURE 2.4 - Chaque chromosome contient plusieurs
gènes.
Crédit : U.S. National Library of Medicine
2.1.6 Protéine
Un des enjeux majeurs de l'ère postgénomique
1 est la description et la caractérisation
fonctionnelle de l'ensemble des protéines exprimées par les
gènes d'un
1. Le projet d'annotation du génome humain
débute en 1993 et voit la participation de milliers de chercheurs de par
le monde, dix ans après il est arrivé au terme et l'ère
postgénomique débute.
8
CHAPITRE 2. GÉNOMIQUE
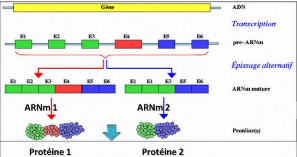
Crédit :https : //
fr.wikipedia.org/wiki/Epissage
FIGURE 2.5 - Épissage alternatif d'un gène chez
les eucaryotes
organisme.
Une protéine est un polymère constitué
d'une succession d'acides aminés. La traduction [16]
est le processus biologique permettant de traduire une séquence d'ARNm
en séquence d'acides aminés suivant le code
génétique présenté à la figure 2.6. Ce code
universel (identique pour tous les êtres vivants) spécifie 20
acides aminés, et permet d'associer à tout triplet de
nucléotides, un acide aminé. Une protéine est
représentée sous la forme d'une séquence sur un alphabet
de 20 lettres correspondant aux acides aminés du code
génétique. Par exemple, la séquence d'ARNm ci-après
est traduite en la séquence d'acides aminés ci-dessous.
>ARNm
ATGTTCCTGCCGCTGTTCTTGGCCATGCTGTGGGGCGGGTCGCAGGCTCTGGACTCATTC
>Protéines
MFLPLFLAMLWGGSQALDSF
2.2 Bases de données
2.2.1 Bases de données existantes
Des bases de données biologiques ont été
développées pour faciliter le stockage et l'accès aux
données biologiques de taille volumineuse. Ces bases de données
sont de deux grands types, les bases de données primaires qui
contiennent les données provenant des résultats
expérimentaux et les bases de données secondaires qui
contiennent
9
CHAPITRE 2. GÉNOMIQUE
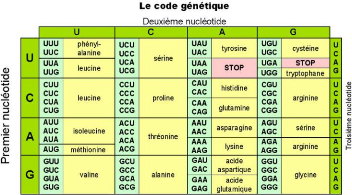
Crédit :http : //
raymond.rodriguez1.free.fr/Textes/1s13.htm
FIGURE 2.6 - Code génétique
les résultats de l'analyse des bases de données
primaires. On distingue trois principales bases de données primaires
à avoir :
1. DNA Data Bank of Japan (National Institute of Genetics)
Site web : http://www.ddbj.nig.ac.jp/
2. EMBL (European Bioinformatics Institute) Site web :
http://www.ebi.ac.uk/
3. GenBank (National Center for Biotechnology Information)
Site web :
http://www.ncbi.nlm.nih.gov/genbank/
Il existe une panoplie de bases de données secondaires
qui diffèrent suivant l'usage auquel elles sont destinées. Pour
nos travaux, nous avons utilisé Ensembl, qui est l'une
des base de données génomique secondaire la mieux annotée
pour les eucaryotes.
2.2.2 Accès aux bases de données
Afin de faciliter la récupération et la
manipulation des données répertoriées dans les bases de
données, différentes méthodes d'accès sont
généralement proposées :
1. via des interfaces web (navigateurs)
10
CHAPITRE 2. GÉNOMIQUE
2. via des Datamart2
3. via des accès directs aux bases de données en
lecture
4. via FTP
5. via des API spécialisées
La section suivante présente différents
modèles, algorithmes et notions de biologie computationnelle,
liées aux objets biologiques précédemment
décrits.
2.3 Évolution des séquences
biologiques
Après acquisition des séquences biologiques
(gènes, ADN, ARN, protéines), ces données biologiques sont
ensuite traitées pour en extraire des informations et des
connaissances.
2.3.1 Recherche de similarités entre
séquences
Une opération fréquente en bio-informatique est
la recherche de similarité entre séquences dans le but de
déterminer les régions conservées entre ces
séquences. Plusieurs techniques de recherche de similarité
existent parmi lesquelles on peut citer l'alignement des séquences qui
est une façon d'agencer les séquences d'acides nucléiques
ou d'acides aminés afin d'identifier des régions de
similarité qui peuvent être une conséquence de relations
fonctionnelles, structurales ou d'évolution entre les séquences.
Il s'agit d'un problème d'optimisation consistant à trouver la
meilleure combinaison des nucléotides ou des acides aminés de
chacune des séquences qui va maximiser la similarité entre elles.
Les figures 2.7 et 2.8 montrent un exemple d'ali-gnement de deux
séquences nucléiques. La figure 2.9 montre un exemple
d'aligne-ment multiple de neuf séquences protéiques.
2.3.2 Construction d'arbres
phylogénétiques
Phylogénie. Charles Darwin a
posé les bases de la théorie de l'évolution [5]. La
phylogénie se définit comme l'étude de l'histoire de
l'évolution d'un groupe de gènes ou d'espèces. L'analyse
phylogénétique est fréquemment réalisée pour
découvrir comment des groupes d'organismes ont évolué au
fil du temps depuis un ancêtre commun. De nombreux modèles ont
été proposés pour reconstruire l'histoire évolutive
des êtres vivants [25]. On peut citer entre autres :
2. Un datamart est un sous-ensemble d'un data warehouse
destiné à fournir des données aux utilisateurs, et souvent
spécialisé vers un groupe ou un type d'affaires.
11
CHAPITRE 2. GÉNOMIQUE

FIGURE 2.7 - Exemple de deux séquences de
nucléotides à aligner
FIGURE 2.8 - Résultat d'alignement possible des deux
séquences

FIGURE 2.9 - Exemple d'alignement multiple de neuf
séquences protéiques. Les colonnes d'acides aminés
conservés dans l'alignement sont surlignées en vert et bleu.
Crédit : wikipedia.
-- Les modèles basés sur les réseaux
phylogénétiques [24, 18, 19] dans lesquels on suppose que
l'évolution peut-être, représentée par un graphe;
-- Les modèles basés sur les arbres
phylogénétiques qui s'appuient sur le concept de base de la
descendance avec modification [11, 17, 28]. Cette représentation est la
plus répandue et est celle considérée dans ce
mémoire. Un arbre phylogénétique est un arbre
enraciné, étiqueté tel que :
-- ses feuilles représentent les espèces ou
gènes étudiés;
-- ses noeuds internes représentent les ancêtres
virtuels ayant divergé;
-- ses arêtes représentent les liens de
descendance entre les espèces (ou gènes).
Un arbre phylogénétique représente les
relations évolutives d'un ensemble d'espèces ou de gènes.
Il permet, de représenter comment à partir d'un ancêtre
12
CHAPITRE 2. GÉNOMIQUE
commun qui est la racine de l'arbre, on a obtenu toutes les
espèces (ou gènes) qui sont aux feuilles de l'arbre.
Les arbres d'espèces. L'arbre
d'espèces est un arbre phylogénétique qui présente
les relations évolutives d'un ensemble d'espèces.
L'évolution des espèces depuis une espèce ancestrale
commune se fait par un événement appelé la
spéciation 3. La figure 2.10 présente
l'arbre d'espèces, communément accepté, des trois grandes
familles du vivant.
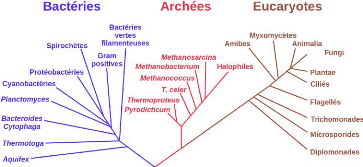
Crédit :https : //
fr.wikipedia.org/wiki/Arbrephylogenetique
FIGURE 2.10 - Arbre d'espèce enraciné, montrant
les trois domaines de vivant : bactéries, archées et eucaryotes,
reliant les trois branches d'organismes au dernier ancêtre universel (le
tronc noir en bas de l'arbre)
Les arbres de gènes. L'arbre de
gènes est un arbre phylogénétique qui décrit
comment un gène particulier appartenant à une espèce
ancestrale commune a évolué au fil du temps. L'évolution
d'un gène se fait notamment à travers trois
événements: la duplication de gènes
4, la spéciation de gènes qui
résulte de la spéciation d'espèces et la perte de
gènes.
3. La spéciation est le processus évolutif par
lequel deux nouvelles espèces vivantes apparaissent à partir
d'une espèce ancestrale.
4. La duplication est une mutation génétique
aboutissant au dédoublement d'un gène dans le génome d'une
espèce.
13
CHAPITRE 2. GÉNOMIQUE
Réconciliation d'arbre de gènes avec
arbre d'espèces. La réconciliation d'un arbre de
gènes avec un arbre d'espèces est une imbrication de l'arbre des
gènes dans l'arbre d'espèces qui permet de représenter
l'histoire évolutive d'une famille de gènes à
l'intérieur de l'arbre d'espèces. La réconciliation permet
d'étiqueter les noeuds de l'arbre de gènes comme duplication ou
spéciation. Les gènes d'une famille de gènes sont tous
homologues (descendants d'un même gène ancestral). Deux
gènes d'une famille sont soit paralogues5 ou orthologues. Par
définition, deux gènes orthologues appartiennent toujours
à deux espèces différentes.
Le problème de réconciliation est un
problème d'optimisation qui consiste, à trouver une
réconciliation de l'arbre des gènes dans l'arbre d'espèces
qui minimisent les différences entre les deux arbres, étant
donnés un arbre de gènes et un arbre d'espèces. Ces
différences sont évaluées comme le nombre de duplications
et de pertes de gènes induits par la réconciliation dans l'arbre
des gènes, les spéciations étant communes aux deux
arbres.
Une réconciliation optimale de l'arbre de gènes
décrit à la figure 2.12 avec l'arbre d'espèces
décrit à la figure 2.11 est présentée à la
figure 2.13. Après cette réconciliation on obtient l'arbre de
gènes étiqueté présenté à la figure
2.14.
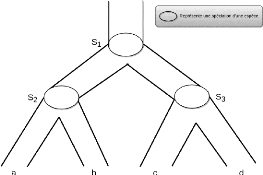
FIGURE 2.11 - Arbre d'espèces
5. Deux gènes sont paralogues (resp. orthologues)
lorsque leur plus proche ancêtre commun dans l'arbre des gènes a
subi une duplication (resp. spéciation).
14
CHAPITRE 2. GÉNOMIQUE
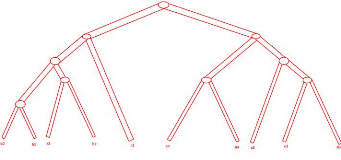
FIGURE 2.12 - Arbre de gènes non
étiqueté
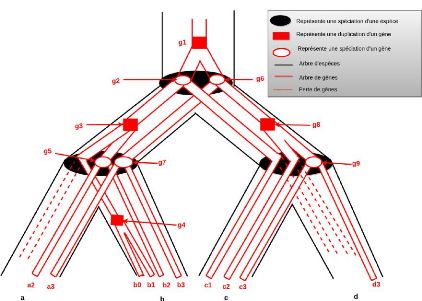
FIGURE 2.13 - Résultat de la réconciliation de
l'arbre de gène 2.14 avec l'arbre d'espèce 2.11
2.4 Conclusion sur la génomique
Ce chapitre a défini la bio-informatique et la biologie
computationnelle, puis a poursuivi en introduisant les notions de biologie et
biologie computationnelle né-
15
CHAPITRE 2. GÉNOMIQUE
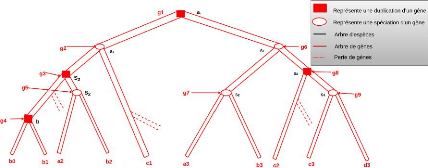
FIGURE 2.14 - Arbre de gènes étiquetés
extrait de la réconciliation de l'arbre de gène dans l'arbre
d'espèce
cessaires pour la compréhension du sujet. La
dernière partie du chapitre a présenté deux grands types
de traitement informatique sur les objets biologiques qui sont la recherche de
similarité entre séquences et l'évolution des
séquences biologiques.
Les concepts de base étant présentés, le
chapitre suivant va s'atteler à présenter un état de l'art
des méthodes computationnelles actuelles pour la construction des arbres
phylogénétiques, en présentant les limites de ces
approches pour lesquelles nous allons proposer de nouvelles solutions.
16
CHAPITRE TROIS
ÉTAT DE L' ART
Les différents processus biologiques sur lesquels
s'appuie la phylogénie ont été introduits au chapitre
précédent. Ce présent chapitre a pour objectif de faire un
état des lieux de l'art des solutions qui sont actuellement
utilisées pour reconstruire la phylogénie des
gènes.
Nous commencerons par présenter les approches
générales de construction d'arbres phylogénétiques
puis, plus spécifiquement, celles de construction des arbres de
gènes. Nous relèverons les limites des méthodes actuelles
qui seront corrigées par la suite.
Il faut cependant noter que dans ce chapitre, tous les arbres
phylogénétiques considérés seront binaires,
c'est-à-dire que tous les noeuds internes d'un arbre possèdent
deux enfants. Cette propriété ne constitue pas une limitation,
car, tout arbre non binaire pourra être approximé par un arbre
binaire.
3.1 Méthodes de construction d'arbres
phylogénétiques
Il existe plusieurs techniques de construction d'arbres
phylogénétiques. Ces méthodes basées sur des
problèmes d'optimisation reposent sur plusieurs critères à
savoir : (i) la distance entre les séquences, (ii) la parcimonie et
(iii) vraisemblance. Nous commencerons par présenter la méthode
naïve consistant à énumérer toutes les topologies
d'arbre possible, puis nous décrirons les trois méthodes
basées sur les critères ci-dessus.
3.1.1 Énumération des arbres
Cette méthode consiste à générer
toutes les topologies d'arbre possibles étant donné un ensemble
de gènes qui sont les feuilles de l'arbre, pour ensuite choisir parmi
ces topologies, un arbre minimisant un critère donné. Par
récurrence, on peut
17
CHAPITRE 3. ÉTAT DE L' ART
démontrer que pour n gènes, il existe
C(n) = (2n-3)!
22n-2(n-2)!
topologies d'arbre binaire
possibles.
Cette méthode a le mérite de
générer tous les arbres possibles pour un ensemble de
gènes donné. Néanmoins, son utilisation demeure
limitée à cause de sa complexité qui croit à un
rythme exponentiel en fonction du nombre de feuilles
(C(30)estsuprieur1038). Conséquemment, la
recherche exhaustive ne peut se faire sur des données de grandes
tailles.
3.1.2 Construction d'arbres à partir des distances
entre feuilles
Cette approche prend en entrée une matrice de distance
donnant les distances entre les gènes deux à deux, puis construit
progressivement l'arbre phylogénétique des feuilles de l'arbre
vers la racine. Nous décrivons ci-après deux méthodes
basées sur cette approche [11] : la méthode Unweighted Pair Group
Method with Arithmetic Mean (UPGMA) et la méthode Neighbour Joining
(NJ).
3.1.2.1 UPGMA
UPGMA est un algorithme permettant, à partir d'une
matrice de distance de construire un arbre enraciné en un sommet qui est
le plus distant des feuilles. Cette méthode est simple et intuitive.
Les principales étapes de UPGMA
sont:
1. Initialiser l'ensemble des noeuds (groupes
1) de l'arbre à l'ensemble des
gènes
2. De façon itérative, jusqu'à ce qu'il ne
reste plus qu'un groupe :
~ Créer un nouveau groupe regroupant deux groupes les
plus proches et retirer ces deux groupes de la liste des groupes.
~ Ajouter dans l'arbre un nouveau noeud correspondant au
nouveau groupe, comme parent des deux noeuds correspondant aux groupes
retirés.
L'arbre est construit des feuilles vers la racine, en ajoutant un
noeud à chaque itération. À chaque étape, on
définit la distance entre deux groupes Ci et Cj comme
>
suit : dij = 1
pECi,qECj dpq. Si Ck
= Ci ? Cj la distance entre Ck et un autre
|Ci||Cj|
groupe Cl est donnée par la formule récursive
suivante : dklj =
dil|Ci|+djl|Cj|
|Ci|+|Cj|
.
La figure 3.1 montre un exemple d'application de UPGMA
3.1.2.2 Neighbour Joining
Neighbour Joining est un autre algorithme dédié
à la construction des d'arbres phylogénétiques tenant
compte des différences de vitesse d'évolution sur les branches
1. Un groupe est un ensemble de données partageant des
caractéristiques communes.
CHAPITRE 3. ÉTAT DE L' ART
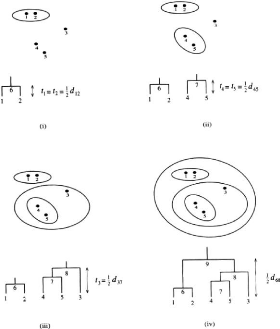
18
FIGURE 3.1 - Application de UPGMA
de l'arbre. La prise en compte des vitesses d'évolution
utilise la notion d'arbre additif.
Un arbre phylogénétique non-enraciné sur
n gènes est additif pour une matrice de distance D
symétrique entre les n gènes si les arcs de l'arbre
sont étiquetés avec des distances de sorte que pour chaque paire
de feuilles (i, j) dans l'arbre, la somme des distances des
arêtes du chemin de i à j est égale
à D(i, j).
La méthode NJ se base sur l'hypothèse qu'il
existe un arbre additif pour la matrice de distance donnée comme
entrée, et produit un tel arbre non enraciné.
Les principales étapes de NJ sont :
1. Initialiser l'ensemble des noeuds (groupes) de l'arbre
à l'ensemble des gènes
2. De façon itérative, jusqu'à ce qu'il ne
reste plus qu'un groupe : ~ Créer un nouveau groupe Ck
regroupant deux groupes Ci et Cj minimisant
19
CHAPITRE 3. ÉTAT DE L' ART
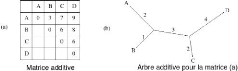
FIGURE 3.2 - Matrice et graphe additif
1 P
la formule d(i,j) - (ri +rj) avec ri =
k?L dik, et retirer ces deux
|L| - 2
groupes de la liste des groupes.
~ Ajouter dans l'arbre un nouveau noeud correspondant au
nouveau groupe Ck, comme parent des deux noeuds correspondant aux
groupes retirés, de sorte que les nouvelles arêtes de l'arbre sont
étiquetés dik = 1 2(dij +ri -rj)
et djk = dij - dik.
~ Pour chaque autre groupe Cm, recalculer
la distance entre Cm et Ck suivant la formule:
dkm = 1 2(dim + djm - dij)
La méthode NJ construit un arbre non-enraciné.
Pour enraciner cet arbre, il suffit d'ajouter un gène très
éloigné des autres gènes considérés
(outgroup). La position du branchement de ce gène sur l'arbre indique la
position de la racine de l'arbre.
FIGURE 3.3 - Enraciner un arbre à l'aide d'un
outgroup.
Une autre stratégie d'enracinement d'arbre est de
considérer comme racine le milieu d'un plus long chemin dans l'arbre
(hypothèse de l'horloge moléculaire).
20
CHAPITRE 3. ÉTAT DE L' ART
3.1.3 Les méthodes de parcimonie
L'approche par parcimonie consiste à rechercher un
arbre qui minimise le nombre de modifications évolutives (mutations,
délétions, ou insertions) pour passer d'une séquence
à l'autre sur les branches de l'arbre. Les deux principaux algorithmes
permettant de calculer le nombre de modifications évolutives induites
par un arbre donné sont celui de Fitch [13] et Sankoff [6].
Le principe de l'algorithme classique de Fitch est le
suivant :
1. Initialiser le nombre de modifications C à
0.
2. Pour chaque noeud k de l'arbre, en allant des
feuilles vers la racine (parcours
postfixe des noeuds)
~ Si k est une feuille, poser Rk =
{étiquette de k}
~ Si k n'est pas une feuille,
~ Calculer Inters = Ri n R ,
où i, j sont des enfants de k ;
~ Si Inters == Ø, poser Rk = Ri
U R et incrémenter C de 1
~ Sinon, poser Rk = Inters
L'algorithme de parcimonie pondérée de Sankoff
est plus général que celui de Fitch. Il ne calcule pas juste le
nombre de modifications, mais considère un poids S(a;
b) pour la substitution d'une lettre a en b. Il
étiquette les noeuds internes de l'arbre de sorte à minimiser le
poids total de l'arbre. L'étiquetage des noeuds est déduit par
récurrence, en calculant l'étiquette d'un noeud à partir
des étiquettes de ses noeuds enfants.
Les principales étapes des méthodes de
parcimonie sont :
1. Calculer un alignement multiple des séquences
(gènes)
2. Pour chaque colonne de l'alignement, trouver un arbre
minimisant le nombre de modifications évolutives pour cette colonne sur
les branches de l'arbre. La recherche de l'arbre se fait par
énumération des arbres, ou en utilisant des heuristiques
d'exploration de l'espace de recherche qui évite
d'énumérer tous les arbres.
3. À partir de l'ensemble des arbres obtenus, trouver
un super-arbre qui minimise la somme totale des nombres de modifications
évolutives pour toutes les colonnes de l'alignement.
3.1.4 Méthodes de maximum de vraisemblance
Le maximum de vraisemblance est une méthode
probabiliste permettant de trouver la séquence de noeuds internes la
plus probable. Les hypothèses de cette méthode sont : Le
processus de substitution d'une séquence en une autre suit un
modèle
21
CHAPITRE 3. ÉTAT DE L' ART
probabiliste dont on connaît l'expression
mathématique. Les sites évoluent indépendamment les uns
des autres; Les sites évoluent selon la même loi. Les taux de
substitution ne changent pas au cours du temps le long d'une branche. Ils
peuvent varier entre branches.
3.2 Construction des arbres de gènes
Les arbres de gènes présentés dans la
littérature sont tous des approximations de la réalité car
la modélisation qui conduit à leur construction ne prend pas en
compte toute la structure des gènes. En particulier, les méthodes
de construction d'arbres de gènes actuelles ne prennent en compte qu'une
seule des protéines (généralement la plus longue) de
chaque gène. Une partie des informations contenues dans les gènes
est ainsi négligée.
3.2.1 Exemple de construction d'arbres de gènes -
Ensembl-Compara
Les arbres de gènes de la base de données
Ensembl sont construits suivant une méthode décrite dans [31]. La
figure 3.4 décrit cette méthode composée de sept
étapes pour la construction des arbres de gènes.
Nous détaillons ci-après le principe des sept
étapes de la méthode de Ensembl-Compara pour la construction des
arbres de gènes.
1. Définition des données de
protéines : Pour chaque gène codant dans une
espèce, considérer la plus longue protéine produite par le
gène.
2. Basic Local Alignment Search Tool Protein
(BLASTP)2 : Chaque protéine est interrogée
à l'aide de WU BLASTP contre la base de données de
protéines de chaque espèce, y compris celle de l'espèce
à laquelle la protéine appartient.
3. Construction du graphe : La relation
entre deux protéines est conservée si elle satisfait soit
à un meilleur partenaire réciproque par BLASTP ou à un
score de BLASTP supérieur à 0.33. On construit alors un graphe
dans lesquels, les noeuds correspondent aux protéines et les
arêtes aux relations conservées.
4. Regroupement : On extrait à partir
du graphe, les composantes connexes. Chaque composante connexe
représente une famille de gènes. Si une famille
2. BLASTP est un programme d'alignement de protéines
permettant, étant donnée une séquence protéique,
d'autres séquences similaires à elle dans une base de
données de séquences protéiques.
22
CHAPITRE 3. ÉTAT DE L' ART
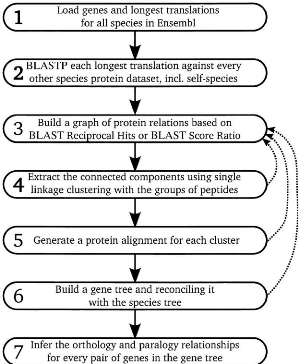
FIGURE 3.4 - Méthode de Ensembl-Compara pour la
construction des arbres de gènes
23
CHAPITRE 3. ÉTAT DE L' ART
de gènes a plus de 750 membres, les étapes 3 et
4 sont répétées en augmentant le seuil de conservation de
relation entre deux protéines.
5. Alignements multiples : Les
protéines d'une même famille de gènes sont alignées
à l'aide du programme "Multiple Sequence Comparison by Log-Expectation"
(MUSCLE) [8, 9] pour obtenir un alignement multiple.
6. Arbre de gène et réconciliation
: L'alignement multiple des protéines d'une famille de
gènes et l'arbre des espèces sont donnés en entrée
au programme de construction d'arbres "Tree Building guided by Species Tree"
(TreeBeST) 4, pour construire l'arbre des gènes de la
famille. L'arbre de gènes est alors réconcilié avec
l'arbre d'espèces afin d'étiqueter comme duplication ou
spéciation.
7. Inférence des orthologues et paralogues
: Les arbres de gènes obtenus sont finalement aplatis en des
tables d'orthologues et de paralogues décrivant les relations
d'homologie entre paires de gènes.
De manière générale, tous les arbres de
gènes disponibles dans les bases de données et dans la
littérature sont construits sur ce modèle.
3.2.2 Limites des arbres de gènes actuels
Dans cette section, nous illustrons les limites des arbres de
gènes construits par la méthode d'Ensembl Compara. Suivant cette
méthode, seule la protéine 3 du gène décrit dans la
figure 3.5 devrait être considérée pour la construction de
l'arbre des gènes de la famille à laquelle le gène
appartient. En effet, cette protéine est la plus longue. Elle
possède quatre exons sur les six que compte le gène. Cependant,
bien qu'étant la plus longue, elle ne couvre pas les exons E2 et E3 qui
ne seront jamais considérés par la suite, alors qu'ils sont
présents dans les protéines 1 et 2 qui sont produites par le
même gène.
3.3 Conclusion sur l'état de l'art
En résumé, nous avons passé en revue les
méthodes les plus utilisées pour la construction d'arbres
phylogénétiques. En particulier, pour les arbres de gènes,
nous
3. MUSCLE est un programme d'alignement multiple de
séquences. MUSCLE peut aligner des centaines de séquences en
quelques secondes.
http://www.drive5.com/muscle/
4. TreeBeST est un programme polyvalent qui construit,
manipule et affiche des arbres phylogénétiques. Il est
particulièrement destiné à la construction d'arbres de
gènes avec un arbre d'espèce connu.
CHAPITRE 3. ÉTAT DE L' ART
24
FIGURE 3.5 - Produit de l'épissage alternatif
avons expliqué les limites des méthodes de
construction actuelles qui font abstraction d'informations importantes portant
sur la structure des gènes. Dans le chapitre suivant, nous allons
proposer une approche qui permet d'adresser ce problème.
25
CHAPITRE QUATRE
MÉTHODOLOGIE ET
IMPLÉMENTATION
Ce chapitre présente la méthodologie que nous
proposons pour la construction des arbres de gènes en tenant compte de
toutes les protéines issues des gènes. D'abord, nous introduisons
formellement un nouveau modèle d'évolution de protéines
à l'intérieur d'un arbre de gènes. Ensuite, nous
étendons la notion de réconciliation afin de définir un
problème d'optimisation qui consiste à reconstruire un arbre de
protéines et un arbre de gène optimal, étant donné
l'arbre des espèces. Nous terminons, en proposant une heuristique pour
la résolution de ce problème, et une implémentation de
cette heuristique.
4.1 Modèle d'évolution des
protéines et problème de réconciliation
Dans un premier temps, nous commençons par introduire
l'idée de base du nouveau modèle d'évolution de
protéines que nous proposons. Puis, dans un second temps, nous
décrirons formellement le modèle et les problèmes
d'optimisation associés.
4.1.1 Idée de base
Le modèle d'évolution de protéines que
nous proposons s'appuie sur l'hypothèse selon laquelle toutes les
protéines sont issues d'un ancêtre commun et ont
évolué au fil du temps au travers de quatre types
d'événements qui sont : duplication 1,
1. Duplication: une protéine a donné naissance
à deux protéines par le biais d'une duplication de
gènes
26
CHAPITRE 4. MÉTHODOLOGIE ET
IMPLÉMENTATION
spéciation 2, création 3,
et perte 4 de protéines. La figure 4.1 montre un exemple
d'arbre d'évolution d'un ensemble de protéines. On observe qu'il
y a deux créations et trois pertes de protéines dans cet
arbre.
FIGURE 4.1 - Arbre de protéines étiqueté
avec les événements de spéciation, duplication,
création et perte
4.1.2 Définitions formelles
Dans cette section, 8 représente, l'ensemble des
espèces, g représente l'ensemble des gènes d'une famille
de gènes et P représente l'ensemble de protéines produites
par les gènes d'une famille. On définit sur ces ensembles des
fonctions de correspondance : s : g - 8 qui à tout gène
fait correspondre l'espèce à laquelle il appartient et g :
P - g qui à toute protéine fait correspondre le gène
qui l'a produit. Les trois ensembles 8, g et P sont tels que 8 =
{s(x) : x E g} et g = {g(x) : x E P}, c'est
à dire que les fonctions s et g sont surjectives.
Arbres phylogénétiques : Tous
les arbres phylogénétiques considérés sont
enracinés et binaires. Un arbre T pour un ensemble L
est un arbre binaire enraciné dont l'ensemble des feuilles est
L. L'ensemble des feuilles d'un arbre T est notée
G(T)
2. Spéciation : une protéine a donné
naissance à deux protéines par le biais d'une spéciation
d'espèce.
3. Création : une protéine est apparue à
un certain moment dans un gène
4. Perte : un gène a perdu la fonctionnalité de
produire une protéine à un certain moment
27
CHAPITRE 4. MÉTHODOLOGIE ET
IMPLÉMENTATION
et l'ensemble des sommets de T est noté
V(T). Étant donné un noeud x E V(T), le
sous-arbre complet5 de T enracinée en x est
notée T[x]. Le plus proche ancêtre commun (en
anglais, least common ancestor (lca)) d'un sous-ensemble L'
de £(T) dans T, notée
lcaT(L'), est l'ancêtre commun à toutes les
feuilles L' qui est le plus éloigné de la
racine de T. Étant donné un noeud interne x de
T, les deux enfants de x sont arbitrairement
désignés par xl et xr (pour left et
right, en anglais).
Arbres de protéines, gènes, et espèces :
Nous notons S un arbre des espèces pour l'ensemble S,
G est un arbre des gènes pour l'ensemble G, et P
un arbre des protéines pour l'ensemble P.
La fonction de correspondance s est étendue
pour devenir une fonction de V(G) E G vers V(S) comme suit :
si x est un noeud interne de G, alors s(x) =
lcaS({s(x') : x' E £(G[x])}), c'est à dire que
l'image d'un noeud x E V(G) dans V(S) est le plus proche
ancêtre commun, dans l'arbre S, de toutes les images des
feuilles du sous-arbre G[x].
De même, nous étendons la fonction de
correspondance g pour qu'elle devienne une fonction de V(P)
vers V(G) : si x est un noeud interne de P,
alors g(x) = lcaG({g(x') : x' E
£(P[x])}).
Réconciliation par LCA : Chaque noeud interne de
l'arbre G représente un gène ancestral au moment d'un
événement de spéciation (Spec) ou de duplication (Dup) de
gènes. La réconciliation par LCA de G avec
S est une fonction d'étiquetage lG : V(G) - £(G) ?
{Spec, Dup} tels que l'étiquette d'un noeud interne x de
G est lG(x) = Spec si s(x) =6 s(xl) et s(x) =6
s(xr), et sinon lG(x) = Dup.
Nous étendons la notion de réconciliation aux
arbres de protéines comme suit : chaque noeud interne de l'arbre P
représente une protéine ancestrale au moment d'un
événement de spéciation (Spec), de duplication (Dup) ou de
création (Creat) de protéines. La réconciliation par
LCA de P avec G est une fonction d'étiquetage
lP : V(P) - £(P) ? {Spec, Dup, Creat} tels que l'étiquette
d'un noeud interne x de P est lP (x) = Spec si
g(x) =6 g(xl) et g(x) =6 g(xr) et lG(g(x)) =
Spec, sinon lP (x) = Dup si g(x) =6 g(xl) et g(x) =6
g(xr) et lG(g(x)) = Dup, et sinon lG(x) = Creat
.
La figure 4.2 illustre la réconciliation de l'arbre des
protéines de la figure 4.1 (à considérer comme
non-étiqueté) avec l'arbre de gènes de la figure 4.3, ce
dernier ayant été réconcilié avec l'arbre
d'espèces illustré à la figure 4.4. Sur la figure 4.1, on
peut noter que, d'une part les créations de protéines
correspondent uniquement aux noeuds placés sur les branches de l'arbre
de gènes et d'autre part il n'y a que
5. Un sous-arbre enraciné en un noeud x d'un arbre T
est complet si il contient toutes les feuilles descendantes de x dans T.
28
CHAPITRE 4. MÉTHODOLOGIE ET
IMPLÉMENTATION
les arbres enracinés en un noeud de création qui
peuvent contenir deux protéines du même gène.
FIGURE 4.2 - Exemple de réconciliation d'arbres de
protéines avec arbre de gènes
Coût de réconciliation : Le
coût de réconciliation par LCA de l'arbre G avec l'arbre
S est le nombre de noeuds de duplication et d'événements de perte
de gènes dans G résultant de la réconciliation par LCA de
G avec S.
Par extension, le coût de réconciliation par
LCA de l'arbre P avec l'arbre G est le nombre de noeuds de création
et d'événements de perte de protéines dans P
résultant de la réconciliation par LCA de P avec G.
Le coût de double réconciliation de G
avec P et S est la somme du coût de réconciliation par LCA de G
avec S et du coût de réconciliation par LCA de P avec G.
Les définitions précédentes nous
permettent maintenant d'introduire le problème d'optimisation dont
l'objectif est de reconstruire un arbre de gènes optimal G, étant
donné l'arbre des protéines P et l'arbre d'espèces S.
Problème: MINDRGT (POUR "MINIMUM DOUBLE
RECONCILIATION GENETREE" EN ANGLAIS) :
Entrées : Un arbre d'espèce S pour
un ensemble d'espèces S ; un arbre de protéines P pour un
ensemble de protéines P
Sortie : Un arbre de gènes G pour G
= {g(x) : x E L(P)} tel que le
coût de double
29
CHAPITRE 4. MÉTHODOLOGIE ET
IMPLÉMENTATION
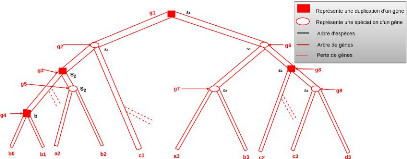
FIGURE 4.3 - Arbre de gènes étiqueté
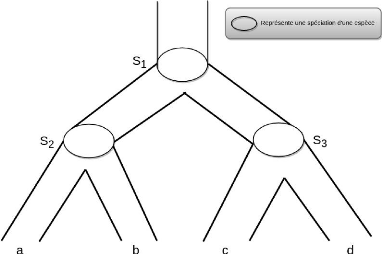
FIGURE 4.4 - arbre d'espèces
30
CHAPITRE 4. MÉTHODOLOGIE ET
IMPLÉMENTATION
réconciliation de P vers G et S
est minimum.
Le problème d'optimisation MinDRGT suppose que l'arbre
des protéines P est connu. Dans le cas où l'arbre des
protéines n'est pas entièrement connu, mais un ensemble de
sous-arbres partiels P1. . . P, couvrant tout l'ensemble P
est connu, le problème devient :
Problème: MINDRPGT (POUR "MINIMUM
DOUBLE RECONCILIATION PROTEIN AND GENE TREES" EN ANGLAIS) :
Entrées : Un arbre d'espèce
S pour un ensemble d'espèces S ; un ensemble de sous-arbres de
protéines P1 . . . P, couvrant l'ensemble des
protéines P
Sortie : Un arbre de protéines P
tel que chaque Pi, 1 = i = k, est un sous-arbre
de P, et un arbre de gènes G pour G =
{g(x) : x E L(P)} tel que le coût de double
réconciliation de P vers G et S est
minimum.
La méthodologie de construction d'arbre de gènes
présentée dans la section suivante consiste à construire
un ensemble de sous-arbres P1 . . . P, couvrant l'ensemble
des protéines P, puis à utiliser une solution heuristique6
du problème MinDRPGT pour reconstruire l'arbre de protéines
P et l'arbre de gènes G.
4.2 Méthodologie : Processus à sept
étapes
L'approche méthodologique utilisée pour la
construction des arbres de gènes en tenant compte de toutes les
protéines de chaque gène est basée sur une solution
heuristique gloutonne pour résoudre le problème MinDRPGT.
L'approche méthodologique est résumée dans un processus
à sept étapes illustrées à la figure 4.5.
4.2.1 Étape 1 : Définition du jeu de
données
La première étape consiste à
définir l'ensemble des protéines de la famille de gènes
dont on veut reconstruire l'arbre. Par définition, une famille de
gène est composée d'un gène donné, tous ses
orthologues, ses paralogues intra-spécifiques (au sein de la même
espèce) et ses paralogues inter-spécifiques (dans des
espèces différentes). Puis, dans la famille de gène, nous
retenons uniquement les gènes produisant au moins une protéine.
Pour chaque protéine produite par un gène de la famille, nous
6. L'analyse de la complexité des problèmes
MinDRPT et MinDRPGT, et la conception de solution algorithmique exacte, le cas
échéant, seront approfondie dans le cadre d'une thèse de
doctorat.
3
4
CHAPITRE 4. MÉTHODOLOGIE ET
IMPLÉMENTATION
|
Définition du jeu de donnée (génes
et séquences codantes)
|
NJ'
2 r Alignement multiple de toutes les séquences
codantes A l'aide de MACSE
|
Calcul d'une matrice de similarité â l'aide
de FsePA
|
|
Regroupement avec chevauchement des
--)
séquences codantes
|
C'oustructiou du groupe de
référence
|
Création des sous-arbres A l'aide de
TreeBest
|
|
Construction de l'arbre de gènes et de
protéines
|
31
FIGURE 4.5 -- Processus de construction d'arbre de gènes
à sept étapes
32
CHAPITRE 4. MÉTHODOLOGIE ET
IMPLÉMENTATION
retenons sa séquence codante, c'est-à-dire la
séquence d'ARNm qui a été traduite pour obtenir la
protéine.
4.2.2 Étape 2 : Alignement multiple de toutes les
séquences codantes
Afin de pouvoir avoir une mesure de similarité entre
les séquences codantes, nous procédons à un alignement
multiple des séquences. Pour ce faire, nous utilisons le programme MACSE
(Multiple Alignment of Coding SEquences Accounting for Frameshifts and Stop
Codons) [29], qui est actuellement l'unique programme d'alignement multiple de
séquences codantes prenant en compte les décalages de cadre de
lecture dans la traduction7 en protéine. Contrairement
à l'approche des méthodes actuelles de construction d'arbre de
gènes, nous avons choisi d'aligner les séquences codantes
plutôt que les protéines elles-mêmes, afin de tenir compte
des phénomènes de décalage de cadre de lecture qui
induisent des alignements erronés des séquences
protéiques.
4.2.3 Étape 3 : Calcul de la matrice de
similarité
Une fois les alignements effectués, nous calculons le
score de similarité de chaque paire de séquences. On obtient
comme résultat une matrice de similarité. Pour le calcul du score
de similarité, nous utilisons un schéma de score tenant compte
simultanément de l'échelle des nucléotides et celle des
protéines définies dans [20]. Le but est de tenir compte à
la fois des décalages de cadre de lecture, et des longueurs des
différences, entre les protéines, issues de ces décalages.
Les scores de similarités sont normalisés en les divisant par les
longueurs des alignements.
4.2.4 Étape 4 : Regroupement avec chevauchement des
séquences codantes
NB : il existe une bijection entre l'ensemble des
protéines et l'ensemble des séquences codantes. Par la suite on
utilisera le terme protéine par souci de simplification.
7. Les séquences codantes sont traduites en
protéines en considérant un cadre de lecture des triplets de
nucléotides consécutifs (codons) dans la séquence pour les
traduire en acides aminés. Le décalage du cadre de lecture
résulte d'un changement du positionnement du cadre de lecture d'un
nombre de nucléotides non multiple de 3 conduisant à un
changement de traduction des codons, et à des séquences
protéiques différentes.
33
CHAPITRE 4. MÉTHODOLOGIE ET
IMPLÉMENTATION
4.2.4.1 Définition du regroupement
Le regroupement appelé en anglais clustering [21] est
une méthode de classification non supervisée, l'objectif
étant de découvrir les regroupements naturels d'un ensemble de
motifs, de points ou d'objets quelconques, ou dans notre cas des
protéines. En pratique, la plupart des scientifiques décrivent un
groupe en tenant compte de son homogénéité interne et de
son hétérogénéité externe. En d'autres
termes, les motifs au sein du même groupe devraient être
similaires, tandis que les motifs dans différents groupes ne le
devraient pas. Le regroupement est utilisé pour grouper les
séquences de protéines en sous-familles en fonction de leurs
similarités, ce qui fournira par ailleurs des indices importants sur les
caractéristiques générales des familles de
protéines. Cette approche de regroupement est également utile
pour inférer la fonction biologique des protéines en
déterminant leur appartenance à des familles de protéines
bien connues et annotées.
4.2.4.2 Problème de regroupement dans le cas
présent
Le modèle de regroupement que nous proposons s'appuie
sur le modèle d'évolu-tion de protéines
présenté dans la section précédente. Nous
construisons un ensemble de groupes de protéines, tel que pour tout
groupe C, le plus proche ancêtre commun de deux protéines du
groupe ne doit jamais correspondre à une création dans l'arbre
des protéines étiqueté.
La figure 4.6 illustre un exemple d'arbre de protéines
étiqueté dans lequel le noeud 6 est une création de
protéines qui conduit à deux protéines (1 et 2)
appartenant au même gène A. À l'inverse, la création
au niveau du noeud 9 va produire des protéines qui appartiennent dans un
premier temps au même gène ancestral, mais par la suite de
l'évolution, à des gènes différents à cause
de la spéciation et de la duplication qui ont mené à trois
gènes différents. Les protéines 1, 2, 4 appartiennent donc
au même gène A, la protéine 3 au gène B, et les
protéines 5, 11 au gène C.
L'ensemble des groupes de taille maximum en termes d'inclusion
que l'on voudrait obtenir est {(1, 3, 11), (2, 3, 11), (4, 5)} dans lequel
chaque groupe contient au plus une protéine par gène.
Cependant, dans notre cas, l'arbre des protéines et son
étiquetage ne sont pas connus et nous cherchons à les
reconstruire, en même temps que l'arbre des gènes. Par
conséquent, nous simplifions le problème de regroupement des
protéines au problème qui consiste à former un
ensemble de groupes dans lesquels chaque groupe contient une seule
protéine par gène, tout en permettant qu'une protéine
puisse se retrouver dans plusieurs groupes. Ceci nous amène
donc à définir une méthode de regroupement avec
chevauchement 8 pour créer des groupes
8. le regroupement avec chevauchement est une technique de
regroupement qui permet qu'un
34
CHAPITRE 4. MÉTHODOLOGIE ET
IMPLÉMENTATION
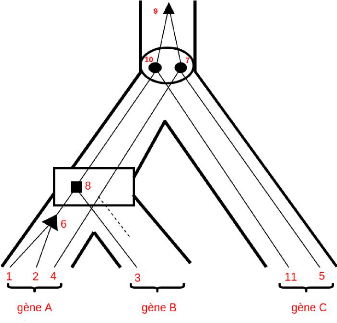
FIGURE 4.6 - Extrait d'arbre de protéines
qui respectent nos contraintes.
Les principales méthodes de regroupement de
protéines présentes dans la littérature sont
décrites à la figure 4.7. Elles sont basées sur les
méthodes par partition-nement [23], les méthodes
hiérarchiques [7] et les méthodes utilisant des graphes [30].
Toutes ces méthodes approchent le problème posé en
4.2.4.2, mais présentent des limites majeures qui nous ont conduits
à développer notre propre méthode de regroupement.
D'abord, les méthodes de regroupement
hiérarchique, bien qu'étant les plus utilisées en
bio-informatique du fait de leur complexité quadratique, conduisent
automatiquement à des regroupements sans chevauchement. Ensuite, les
approches par partitionnement quant à elles sont très efficaces,
car chaque élément à classer est mis dans la meilleure
classe préexistante. Mais leur principale limite est qu'elles exigent
élément puisse appartenir à plus d'un
groupe, par opposition au regroupement sans chevauchement qui ne le permet
pas
35
CHAPITRE 4. MÉTHODOLOGIE ET
IMPLÉMENTATION
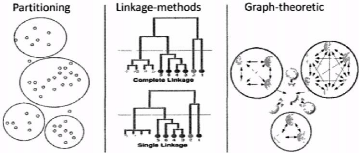
FIGURE 4.7 - Les trois principales catégories
d'algorithmes utilisées pour le regroupement des séquences de
protéines
de connaître au préalable le nombre de groupes,
alors que dans notre cas ce nombre doit être découvert. Enfin, les
approches basées sur la théorie de graphe conduisent
également à des regroupements sans chevauchement. Bien qu'il
existe des cas rares où l'on peut obtenir des regroupements avec
chevauchement.
4.2.4.3 Regroupement avec chevauchement
hiérarchique
Idée de base. L'idée de base de
notre méthode de regroupement avec chevauchement est de proposer une
version modifiée du regroupement hiérarchique (qui est à
la base du regroupement sans chevauchement) pour créer des regroupements
avec chevauchement. Tel que chaque groupe contient au plus une protéine
par gène tout en permettant qu'une protéine puisse appartenir
à plus d'un groupe. La figure 4.8 illustre graphiquement la
différence majeure entre le regroupement sans chevauchement et le
regroupement avec chevauchement que nous choisissons comme approche.
Présentation globale du modèle
Soit n le nombre de protéines et á un
seuil de similarité. Initialement, chaque protéine forme un
groupe. La méthode va, de manière récursive, choisir
à chaque étape les deux groupes les plus proches, les fusionner
afin d'en former un nouveau. On choisit deux groupes à fusionner si et
seulement si le score de similarité entre eux est inférieur au
seuil á. Le score de similarité pour deux groupes A
et B est calculé ainsi comme suit (D étant
la matrice de similarité calculé à l'étape 3) :
A' ? A - B
36
CHAPITRE 4. MÉTHODOLOGIE ET
IMPLÉMENTATION
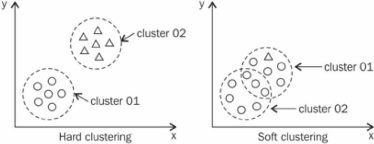
FIGURE 4.8 - Illustration présentant la
différence entre le regroupement avec chevauchement et le regroupement
sans chevauchement
B' B - A
Si Card(A') == 0 or Card(B')
== 0 Alors
D(A',B') oc
Sinon >I >I
1
D(A', B') yEB
D(x, y)
Card(A').Card(B')
xEA
La figure 4.9 présente un exemple d'application de
l'algorithme de regroupement avec chevauchement sur sept protéines de
cinq gènes.
Algorithme détaillé de regroupement avec
chevauchement hiérarchique Définition des
entrées
DistanceMatrix D :
est une matrice de similarité qui contient les mesures de
similarité entre chaque paire de séquences. C'est donc une
matrice symétrique. Les valeurs de cette matrice sont normalisées
entre 0 et 1, avec 1 représentant la plus grande similarité;
SimilarityLevel á :
est une valeur de similarité comprise entre 0 et 1;
Header h : est un
tableau associatif, qui associe à chaque gène la liste de ses
protéines. Il est utile pour satisfaire la contrainte que chaque groupe
doit contenir au plus une protéine de chaque gène.
L'algorithme 4.1 décrit formellement la méthode.
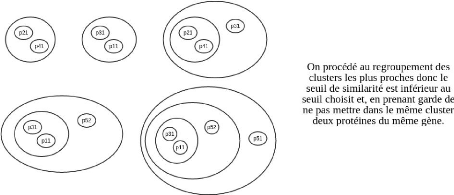
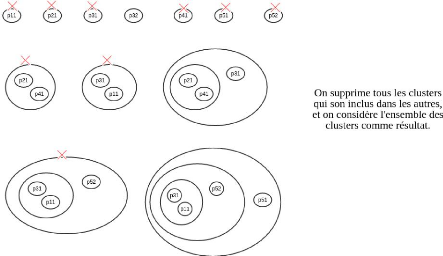
4.2.5 Construction d'un groupe de
référence
Une fois les groupes obtenus, on choisit un groupe de
référence de cardinalité maximale. Si la
cardinalité de ce cluster est strictement inférieure au nombre
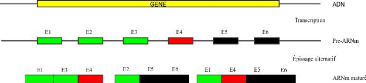
de
CHAPITRE 4. MÉTHODOLOGIE ET
IMPLÉMENTATION
37
FIGURE 4.9 - Exemple d'application de l'algorithme de
regroupement avec chevauchement sur sept protéines de cinq gènes.
Les cinq gènes sont p1, p2 et p4 ayant chacun une protéine, et p3
et p5 ayant chacun deux protéines.
38
CHAPITRE 4. MÉTHODOLOGIE ET
IMPLÉMENTATION
Algorithm 4.1 Soft Clustering
1: procedure CLuTTERING(DistanceMatrix
D, SimilarityLevel á, ListeDesClus-ters listeCluster
h)
2: groupeBanis ? []
3: loop :
4: i, j, min ? ChoisirPlusPetitEltV alide(D,
groupeBanis, listeCluster, á)
5: if (i == -1 or j ==
-1 or min == +8) then
6: ConsiliderGroupe.
7: else
8: listeCluster ? listeCluster ?
[listeCluster(i), listeCluster(j)]
9: groupeBanis ? groupeBanis ?
[i,j]
10: Ajouter une ligne et une colonne à la matrice
D
11: n ? nombreLinge(D) + 1
12: miseAJourValeur(i, j, n).
13: goto loop.
14: end if
15: ChoisirPlusPetitEltValide(D, groupeBanis, listeCluster,
á) :
16: for i = 1 to nombreDeLigne(D)
do
17: for j = i + 1 to
nombreDeColonne(D) do
18: if (listerCluster[i] ?
listeCluster[j] ?/ groupeBanis) then
19: if ((? p1 ? listeCluster[i] and ?
p2
? listeCluster[j]) tel que gene(p1) =6 gene(p2))
and V aleurDeSimilarite(listeCluster[i], listeCluster[2],
á)) then
20: Return (i,j,D[i,j])
21: else
22: Continue
23: end if
24: else
25: Continue
26: end if
27: end for
28: end for
29: Return (-1,
-1, +8)
39
CHAPITRE 4. MÉTHODOLOGIE ET
IMPLÉMENTATION
30: ValeurDeSimilarite(clusterI, clusterJ, á)
:
31: clusterI' ? clusterI -
ClusterJ
32: clusterJ' ? clusterJ -
ClusterI
33: flag ? TRUE
34: for i = 1 to
longeur(clusterI') do
35: for j = i + 1 to
longeur(clusterJ') do
36: if (D[i,j] >= á)
then
37: flag = FALSE
38: end if
39: end for
40: end for
41: Return flag
42: miseAJourValeur(i,j, n) :
43: B ? sequence(n) //
44: for i = 1 to n
do
45: A ? sequence(i)
46: A' ? A - B
47: B' ? B - A
48: if (Card(A) == 0 or Card(B) ==
0) then
49: D(A', B') ? 8
50: else >I >I
51: D((A', B') ?
1 y?B
D(x, y)
Card(A).Card(B)
x?A
52: end if
53: end for
54: Return D
55: ConsiliderGroupe :
56: for i = 1 to
longeur(listeCluster) do
57: if (?k ? [1,
longeur(listeCluster)] avec k =6 i) and
listeCluster[i] ? listeCluster[k] then
58: listeCluster ? listeCluster -
listeCluster[i]
59: end if
60: end for
61: end procedure
CHAPITRE 4. MÉTHODOLOGIE ET
IMPLÉMENTATION
gènes, on le complète en lui ajoutant une
protéine de chaque gène non représenté dans ce
groupe. La protéine d'un gène non représenté dans
le groupe est choisie de sorte à minimiser le coût moyen de
similarité entre cette protéine et les protéines
présentes dans le groupe. L'algorithme 4.2 décrit le processus de
construction du groupe de référence le cas
échéant.
Algorithm 4.2 AugmenterGroupe
1: procedure
AUGMENTERGROUPE(Cref, G)
2: Entrées : Cref groupe de
référence, G = {G1, ..., Gl}
Collection des ensembles de protéines des l gènes non
représentés dans Cref
3: if (E = Ø)
then
4: Terminer
5: else
P =
argmax{D(Cref
,P )}
6: P ??i?{1,...,l}
Gi . D étant la
distance entre p et le groupe Cref
7: Gp = gène auquel p
appartient
8: Cref = Cref U {P}
9:
G = G \ {GP }
1: AugmenterGroupe(Cref, G)
2: end if
3: end procedure
40
À la fin de cette procédure, on obtient une
liste de groupes, dans laquelle un groupe, le groupe de
référence, contient une protéine de chaque gène et
chaque protéine du groupe de référence n'appartient
à aucun autre groupe, et les groupes sont tous deux à deux
disjoints. L'étape suivante consiste à créer un arbre de
protéines par groupe et de fusionner tous les arbres en un seul
super-arbre de protéines.
4.2.6 Étape 6 : Création des arbres de
protéines pour les groupes
Rendu à cette étape, on dispose d'un ensemble de
groupes, il s'agit de créer un arbre par groupe. Pour ce faire, on
utilise le programme TreeBest[10]. L'entrée de ce programme doit
être un ensemble de séquences alignées. Pour ce faire, on
utilise encore MACSE pour aligner les séquences codantes du groupe avant
de passer l'alignement multiple obtenu à TreeBest. La sortie du
programme est un arbre au format Newick9
9. Newick est le nom d'un format de fichier utilisé en
bio-informatique pour décrire un arbre phylogénétique.
41
CHAPITRE 4. MÉTHODOLOGIE ET
IMPLÉMENTATION
4.2.7 Étape 7 : Construction de l'arbre de
protéines et de l'arbre des gènes
Disposant d'une forêt de l arbres pour l
groupes telle que l'un des arbres est considéré comme
l'arbre de référence (celui contenant une protéine pour
chacun des gènes), l'objectif de cette étape est de fusionner les
arbres en un seul arbre.
Les principales phases de l'algorithme de cette
étape sont :
1. Définir l'arbre de gènes G comme
égal à l'arbre de référence (contenant une
protéine pour chacun des gènes)
2. Initialiser le super-arbre de protéines P
comme égal à l'arbre de référence
3. De façon itérative jusqu'à fusionner
tous les arbres
~ Choisir l'arbre Tk avec k E [1,
l] tel que .C(Tk) n .C(P) est maximal.
~ Construire l'arbre induit T =
Tk|L(Tk)\L(P
~ Calculer le noeud de P, x =
lcaP{P1 E .C(P) | ?P2 E
(.C(Tk)\.C(P)),g(p1) =
g(p2)}
La figure 4.10 montre un exemple d'application de l'algorithme
glouton de concaténation d'une forêt d'arbres. L'arbre de
référence étant l'arbre ayant pour racine
étiqueté par 1.
4.3 Implémentation
Afin d'implémenter le modèle
précédent, le langage python à
été choisi pour deux raisons. D'une part, c'est un excellent
langage de script et d'autre part, c'est l'un des langages les plus
utilisés en bio-informatique, pour lequel de nombreuses
bibliothèques sont disponibles. Chacune des sept étapes a
été implémentée séparément, mais de
telle sorte que chaque étape rende des résultats sous format
réutilisable pour les étapes suivantes.
Les données ont été
récupérées de la base de données de EnsemblCompara,
version 84 et de NCBI, nous avons utilisé PyCongent, une API Python pour
la récupération de données de la base de données
Ensembl. Nous avons utilisé la bibliothèque numpy pour la
manipulation des structures de données optimisées pour les
tableaux et matrices.
42
CHAPITRE 4. MÉTHODOLOGIE ET
IMPLÉMENTATION
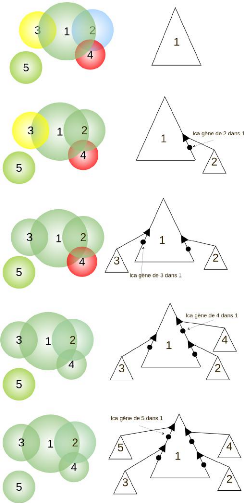
FIGURE 4.10 - Application de l'algorithme glouton de fusion de
6 arbres. Les arbres sont greffés à l'arbre de
référence par des créations matérialisées
sur la figure par des triangles au fond noir.
43
CHAPITRE CINQ
ÉTUDE DE CAS
Afin de tester le modèle, les méthodes et les
implémentations décrits précédemment, nous avons
fait des études de cas. Pour nos tests, nous avons retenu les
espèces suivantes : la vache, l'humain, la souris, le fugu, le zebrafich
et le poulet, représentatives de l'ensemble des espèces
vertébrés.
5.1 Étude de cas de la famille The
Myelin-Associated Glycoprotein (MAG)
Le premier cas d'étude est réalisé sur la
famille de gène MAG [27, 3]. Elle a été largement
étudiée dans la littérature [3]. En raison du fait que les
gènes de cette famille subissent l'épissage alternatif, ce cas
constitue un candidat parfait pour tester notre algorithme. Le tableau 5.1
récapitule les informations sur cette famille MAG.
|
Nom de la famille
|
Source de données
|
Nombre de gènes
|
Nombre de protéines
|
|
1
|
MAG
|
Ensembl
|
28
|
70
|
TABLE 5.1 - Données utilisées pour l'étude
de cas de la famille MAG
La figure 5.1 présente l'arbre de
référence (arbre de gènes) sur lequel viendront se greffer
les arbres provenant des groupes restants. Notre arbre de gène partage
18 protéines sur les 28 au total avec celui de Ensembl et a un plus
petit coût de réconciliation plus faible. Après
réconciliation avec l'arbre d'espèce, l'arbre de gène
obtenu comporte 5 pertes de gènes et 14 duplications. La figure 5.2
présente l'arbre de gènes construit par Ensembl-Compara, qui
compte 12 pertes et 20 duplications. Ce cas permet de montrer que notre
méthode permet la construction d'arbres de gènes de coût de
réconciliation optimaux
CHAPITRE 5. ÉTUDE DE CAS
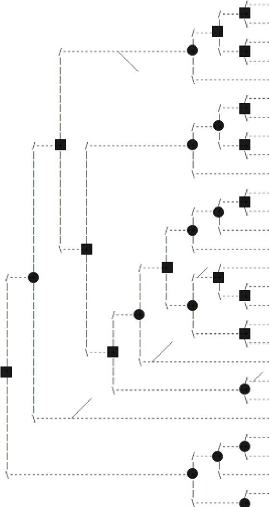
--.._ ~.,.,. _--..---
- i
1-
-------
-
L
I I I
I I k
I
Jr- --
I
11.
f
k-
EN5F 9668$321977 ENSFVG4UCW rZU r3 ENSP6E069323329
EN5PMEMA13961 FNSMJSFIE3E0I37i87 ENSPUd5I1d55I7 ENSPO6659A161Z6
ENSMJSPOINEBUDA720 ENSMJSPOME4130O32 ENSETAF60005.196'51 EN5F 96690345243 EN5P!
!EW1Z3a1 ENSMUSNO0E0505592 ENSETPF900E6304195 ENSIIR8Ad354A, kriSPOEUUDA15ZEU
ENSP46$50A7323.S EN5B7APOIMEi 6424 ENSEARMIE+3E4a.54Z3 ENSBTANO.5 0522603
EN51115P500 6113245 FN5RTPPAEt7F.0013174F1 ENSLAAF'9d9E412aà12
EN5P26699073245 EN5MJ5PR M1399$1 ENSR1AP!Eb64I238a EN56ARP600E01196615
EN5MLP999E6823556
44
FIGURE 5.1 -- Arbre de gènes de la
famille MAG obtenu avec notre méthode
45
CHAPITRE 5. ÉTUDE DE CAS
4 -ENSDARP00000126612
· ENSTRUPO0000006444
- ENSBTAP00000011656
·
--
·E N S BTA P00000049B61
GEN SP00000401502
.' ENSP00000262262
~
·E N S BTAP00000055070
· ENSMUSP00000113245 -ENSMUSP00000058875
.#
·ENSMUSP00000004728
· EN SBTAP00000031748
/
·ENSMUSP00000032667
_.--ENSPOO000413861
· ENSP00000323328
· ENSP00000291707 \
·EN5P00000321077
· ENSBTAP00000022603
· ENSP00000354090
EN SP00000470259 1ENSP00000455510
o
·E N S B TAP0000002 6431
· -ENSBTAPODO00055422
· ENSBTAP00000026429
JENSP00000345243 s
ENSP00000412361
· ENSMUSP00000005592 ENSBTAPOO000004195
· ENSDARP00000136924 o
·ENSTR
UP00000023555
4 SP00000376048
.
·E N SBTAP00000052 366
· ENSMUSP00000139564
FIGURE 5.2 -- Arbre de gène de la famille MAG obtenu de
Ensembl
CHAPITRE 5. ÉTUDE DE CAS
5.2 Étude de cas de la famille FAM86
Le second cas d'étude concerne la famille FAM86. Les
gènes de cette famille subissent également l'épissage
alternatif. Il s'agit ainsi d'un bon candidat pour notre étude. Le
tableau 5.2 récapitule les informations sur cette famille FAM86.
|
Nom de la famille
|
Source de données
|
Nombre de gènes
|
Nombre de protéines
|
|
3
|
FAM86
|
Ensembl
|
8
|
16
|
TABLE 5.2 - Données utilisées pour l'étude
de cas de la famille FAM86
La figure 5.3 présente l'arbre de
référence (arbre de gènes) sur lequel viendront se greffer
les arbres provenant des groupes restant. L'arbre de gène obtenu
comporte 0 pertes de gènes et 3 duplications contre 3 pertes et 4
duplications Notre arbre de gène partage 6 protéines sur 8 avec
celui de Ensembl et a un plus petit coût de réconciliation.
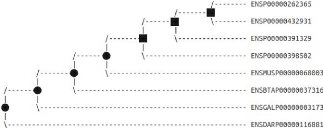
46
FIGURE 5.3 - Arbre de gène de la famille FAM86 obtenu de
notre modèle
47
CHAPITRE 5. ÉTUDE DE CAS
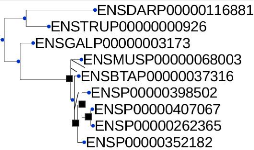
FIGURE 5.4 - Arbre de gène de la famille FAM86 obtenu de
Ensembl
48
CHAPITRE SEPT
CONCLUSION GÉNÉRALE
Parvenu au terme de ce mémoire, dont le but
était de proposer un modèle d'évo-lution de l'architecture
des gènes en tenant compte de toutes les séquences codantes, nous
avons dans un premier temps posé le problème et montré les
limites des solutions actuelles. Puis nous avons proposé un
modèle d'évolution de protéines et introduit des
problèmes d'optimisations dont le but est de reconstruire des arbres de
protéines et de gènes, étant donné les arbres
d'espèces. Nous avons également proposé une méthode
heuristique gloutonne résumée dans un processus à sept
étapes pour résoudre le problème posé. Nous avons
terminé avec des applications sur des familles de gènes de la
base de données Ensembl.
Bien qu'ayant eu des résultats ayant des coûts de
réconciliation mouilleurs que ceux d'Ensembl avec cette approche
heuristique, il demeure qu'une solution algorithmique exact qui
considère simultanément tous les arbres de chaque groupe de
l'étape 4 du processus permettra de reconstruire une solution plus
précise.
Le problème étant formellement posé, il
sera question pour les prochains travaux sur la même problématique
d'affiner.
49
Références
[1] Pea Carninci, T Kasukawa, S Katayama, J Gough, MC Frith,
N Maeda, R Oyama, T Ravasi, B Lenhard, C Wells, et al. The transcriptional
landscape of the mammalian genome. Science, 309(5740) :1559-1563,
2005.
[2] Yann Christinat and Bernard ME Moret. Inferring
transcript phylogenies. BMC bioinformatics, 13(9) :1, 2012.
[3] Yann Christinat and Bernard ME Moret. Inferring
transcript phylogenies. BMC bioinformatics, 13(9) :1, 2012.
[4] Yann Christinat and Bernard ME Moret. A transcript
perspective on evolution. IEEE/ACM Transactions on Computational Biology
and Bioinformatics (TCBB), 10(6) :1403-1411, 2013.
[5] Charles Darwin. R.(1859) : On the origin of species by
means of natural selection. Murray. London, 1871.
[6] William HE Day, David S Johnson, and David Sankoff. The
computational complexity of inferring rooted phylogenies by parsimony.
Mathematical bios-ciences, 81(1) :33-42, 1986.
[7] I Dondoshansky and Y Wolf. Blastclust (ncbi software
development toolkit). NCBI, Bethesda, Md, 2002.
[8] Robert C Edgar. Muscle : a multiple sequence alignment
method with reduced time and space complexity. BMC bioinformatics,
5(1) :1, 2004.
[9] Robert C Edgar. Muscle : multiple sequence alignment with
high accuracy and high throughput. Nucleic acids research, 32(5)
:1792-1797, 2004.
[10] Heng Li et Al. Incorporating species phylogeny in the
reconstruction of gene trees.
[11] R. Durbin et Al. Biological Sequence Analysis.
Cambridge university press, 1998.
50
RÉFÉRENCES
[12] Oliver Eulenstein, Snehalata Huzurbazar, and David A
Liberles. Reconciling phylogenetic trees. Evolution after gene duplication,
pages 185-206, 2010.
[13] Walter M Fitch. Toward finding the tree of maximum
parsimony. In Proceedings of the 8th International Conference on Numerical
Taxonomy, pages 189-230. Freeman, 1975.
[14] Mark B Gerstein, Can Bruce, Joel S Rozowsky, Deyou
Zheng, Jiang Du, Jan O Korbel, Olof Emanuelsson, Zhengdong D Zhang, Sherman
Weissman, and Michael Snyder. What is a gene, post-encode? history and updated
definition. Genome research, 17(6) :669-681, 2007.
[15] Brenton R Graveley. Alternative splicing : increasing
diversity in the proteomic world. TRENDS in Genetics, 17(2) :100-107,
2001.
[16] P. H. The origin of the genetic code. 38 :367-379,
1968.
[17] John P. Huelsenbeck, Fredrik Ronquist, et al. Mrbayes :
Bayesian inference of phylogenetic trees. Bioinformatics, 17(8)
:754-755, 2001.
[18] Daniel H Huson and David Bryant. Application of
phylogenetic networks in evolutionary studies. Molecular biology and
evolution, 23(2) :254-267, 2006.
[19] Daniel H Huson, Regula Rupp, and Celine Scornavacca.
Phylogenetic networks: concepts, algorithms and applications.
Cambridge University Press, 2010.
[20] Safa Jammali, Esaie Kuitche, Ayoub Rachati,
Bélanger François, and Aïda Ouangraoua. Aligning coding
sequences with frameshift extension penalties. 2016.
[21] Abdellali Kelil. Contribution à l'analyse des
séquences de protéines Similarité, Clustering et
Alignement. Université de Sherbrooke, 2011.
[22] Christina Kyriakopoulou. From fundamental genomics
to systems biology : understanding the book of life, volume 23132.
European Communities, 2008.
[23] Weizhong Li and Adam Godzik. Cd-hit : a fast program for
clustering and comparing large sets of protein or nucleotide sequences.
Bioinformatics, 22(13) :1658-1659, 2006.
[24] David A Morrison. Networks in phylogenetic analysis :
new tools for population biology. International journal for parasitology,
35(5) :567-582, 2005.
[25] Luay Nakhleh. Computational approaches to species
phylogeny inference and gene tree reconciliation. Trends in ecology &
evolution, 28(12) :719-728, 2013.
[26] SB Primrose and RM Twyman. Basic biology of plasmid and
phage vectors. Principles of Gene Manipulation and Genomics, 2006.
[27] Richard H Quarles. Myelin-associated glycoprotein (mag)
: past, present and beyond. Journal of neurochemistry, 100(6)
:1431-1448, 2007.
51
RÉFÉRENCES
[28] Vincent Ranwez, Sébastien Harispe,
Frédéric Delsuc, and Emmanuel JP Dou-zery. Macse : Multiple
alignment of coding sequences accounting for frameshifts and stop codons.
PLoS One, 6(9) :22594, 2011.
[29] Vincent Ranwez, Sébastien Harispe,
Frédéric Delsuc, and Emmanuel JP Dou-zery. Macse : Multiple
alignment of coding sequences accounting for frameshifts and stop codons.
PLoS One, 6(9) :e22594, 2011.
[30] Stijn Van Dongen. A cluster algorithm for graphs.
Report-Information systems, (10) :1-40, 2000.
[31] Albert J Vilella, Jessica Severin, Abel Ureta-Vidal, Li
Heng, Richard Durbin, and Ewan Birney. Ensemblcompara genetrees : Complete,
duplication-aware phylogenetic trees in vertebrates. Genome research,
19(2) :327-335, 2009.
| 

