CHAPITRE 4. MÉTHODOLOGIE ET
IMPLÉMENTATION
37
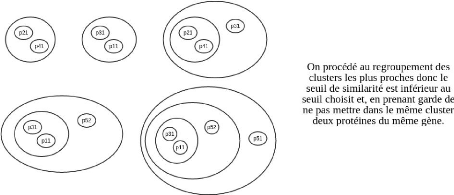
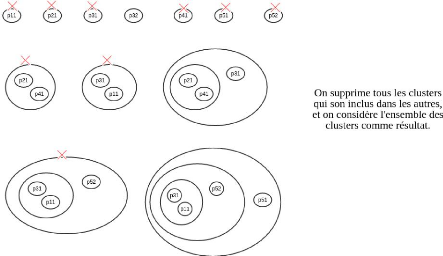
FIGURE 4.9 - Exemple d'application de l'algorithme de
regroupement avec chevauchement sur sept protéines de cinq gènes.
Les cinq gènes sont p1, p2 et p4 ayant chacun une protéine, et p3
et p5 ayant chacun deux protéines.
38
CHAPITRE 4. MÉTHODOLOGIE ET
IMPLÉMENTATION
Algorithm 4.1 Soft Clustering
1: procedure CLuTTERING(DistanceMatrix
D, SimilarityLevel á, ListeDesClus-ters listeCluster
h)
2: groupeBanis ? []
3: loop :
4: i, j, min ? ChoisirPlusPetitEltV alide(D,
groupeBanis, listeCluster, á)
5: if (i == -1 or j ==
-1 or min == +8) then
6: ConsiliderGroupe.
7: else
8: listeCluster ? listeCluster ?
[listeCluster(i), listeCluster(j)]
9: groupeBanis ? groupeBanis ?
[i,j]
10: Ajouter une ligne et une colonne à la matrice
D
11: n ? nombreLinge(D) + 1
12: miseAJourValeur(i, j, n).
13: goto loop.
14: end if
15: ChoisirPlusPetitEltValide(D, groupeBanis, listeCluster,
á) :
16: for i = 1 to nombreDeLigne(D)
do
17: for j = i + 1 to
nombreDeColonne(D) do
18: if (listerCluster[i] ?
listeCluster[j] ?/ groupeBanis) then
19: if ((? p1 ? listeCluster[i] and ?
p2
? listeCluster[j]) tel que gene(p1) =6 gene(p2))
and V aleurDeSimilarite(listeCluster[i], listeCluster[2],
á)) then
20: Return (i,j,D[i,j])
21: else
22: Continue
23: end if
24: else
25: Continue
26: end if
27: end for
28: end for
29: Return (-1,
-1, +8)
39
CHAPITRE 4. MÉTHODOLOGIE ET
IMPLÉMENTATION
30: ValeurDeSimilarite(clusterI, clusterJ, á)
:
31: clusterI' ? clusterI -
ClusterJ
32: clusterJ' ? clusterJ -
ClusterI
33: flag ? TRUE
34: for i = 1 to
longeur(clusterI') do
35: for j = i + 1 to
longeur(clusterJ') do
36: if (D[i,j] >= á)
then
37: flag = FALSE
38: end if
39: end for
40: end for
41: Return flag
42: miseAJourValeur(i,j, n) :
43: B ? sequence(n) //
44: for i = 1 to n
do
45: A ? sequence(i)
46: A' ? A - B
47: B' ? B - A
48: if (Card(A) == 0 or Card(B) ==
0) then
49: D(A', B') ? 8
50: else >I >I
51: D((A', B') ?
1 y?B
D(x, y)
Card(A).Card(B)
x?A
52: end if
53: end for
54: Return D
55: ConsiliderGroupe :
56: for i = 1 to
longeur(listeCluster) do
57: if (?k ? [1,
longeur(listeCluster)] avec k =6 i) and
listeCluster[i] ? listeCluster[k] then
58: listeCluster ? listeCluster -
listeCluster[i]
59: end if
60: end for
61: end procedure
CHAPITRE 4. MÉTHODOLOGIE ET
IMPLÉMENTATION
gènes, on le complète en lui ajoutant une
protéine de chaque gène non représenté dans ce
groupe. La protéine d'un gène non représenté dans
le groupe est choisie de sorte à minimiser le coût moyen de
similarité entre cette protéine et les protéines
présentes dans le groupe. L'algorithme 4.2 décrit le processus de
construction du groupe de référence le cas
échéant.
Algorithm 4.2 AugmenterGroupe
1: procedure
AUGMENTERGROUPE(Cref, G)
2: Entrées : Cref groupe de
référence, G = {G1, ..., Gl}
Collection des ensembles de protéines des l gènes non
représentés dans Cref
3: if (E = Ø)
then
4: Terminer
5: else
P =
argmax{D(Cref
,P )}
6: P ??i?{1,...,l}
Gi . D étant la
distance entre p et le groupe Cref
7: Gp = gène auquel p
appartient
8: Cref = Cref U {P}
9:
G = G \ {GP }
1: AugmenterGroupe(Cref, G)
2: end if
3: end procedure
40
À la fin de cette procédure, on obtient une
liste de groupes, dans laquelle un groupe, le groupe de
référence, contient une protéine de chaque gène et
chaque protéine du groupe de référence n'appartient
à aucun autre groupe, et les groupes sont tous deux à deux
disjoints. L'étape suivante consiste à créer un arbre de
protéines par groupe et de fusionner tous les arbres en un seul
super-arbre de protéines.
4.2.6 Étape 6 : Création des arbres de
protéines pour les groupes
Rendu à cette étape, on dispose d'un ensemble de
groupes, il s'agit de créer un arbre par groupe. Pour ce faire, on
utilise le programme TreeBest[10]. L'entrée de ce programme doit
être un ensemble de séquences alignées. Pour ce faire, on
utilise encore MACSE pour aligner les séquences codantes du groupe avant
de passer l'alignement multiple obtenu à TreeBest. La sortie du
programme est un arbre au format Newick9
9. Newick est le nom d'un format de fichier utilisé en
bio-informatique pour décrire un arbre phylogénétique.
41
| 

