3.6 Résultats du la dynamique moléculaire
:
3.6.1 Détail du calcul :
La simulation de la dynamique moléculaire du dioxyde de
titane anatase a été réalisée par le code CASTEP
intégré dans Materials Studio MS. Ce choix de calcul nous a
permis d'acquérir d'avantages d'informations par rapport aux autres
modules.
Dans ce cadre, notre objectif est de prédire les
paramétres des mailles d'une part et d'une autre part pour
déterminer les structures des bandes et les densités
d'états électroniques et de les comparer avec les autres
résultats.
Comme d'habitude nous avons commencé le calcul par
l'optimisation de la géométrie. L'optimisation est
effectué par l'approximation GGA-PBE avec une énergie de coupure
450 eV.
Pour décrire les interactions d'électrons de
valence, nous avons utilisé le potentiel ultrasoft avec 3×3×3
k-points.
Nous avons obtenu la convergence après trois
itérations pour une énergie de - 9.92631981E+003 eV.
Par la suite, nous avons lancé le calcul de la
dynamique moléculaire. Nous avons utilisé l'approximation de
GGA-PBE pour décrire l'interaction d'échange -
corrélation.
Le plus important ici est que le calcul ne marche pas que si
la structure doit être dans le groupe d'espace P1 pour garantir une
meilleur relaxation structurale de tous les paramétres
cristallographiques.
Pour tenir compte des variations de la pression et de la
température, les équations du mouvement des ions et de la cellule
sont intégrées par l'algorithme de saute-mouton « Verlet
Leap-Frog »
La configuration initiale correspond à la
température minimale, choisie pour être identique à celle
de la structure expérimentale. Le systéme est alors
équilibré à la pression et la température de
l'atmosphère (T= 300K).
Nous avons adopté aussi l'algorithme de Nosé -
Anderson - Barostat de l'ensemble thermodynamique NPT.
Notre systéme est intégré de 100 pas
d'intégration avec un pas Lt = 1 ???? .
La figure (3.16) présente la structure anatase
optimisé au cours de calcul de la dynamique moléculaire.

Chapitre III Résultats et
discussions
57 | Page
FIGURE 3.16 - La structure
anatase en cours de calcul dynamique
Au cours de calcul, cette structure de la figure (3.16), nous
avons marqué des fluctuations et des vibrations de cette structure. Ce
calcul a duré plusieurs heures il y a environ 9 heures. Nous avons
contrôlé notre structure et nous avons constaté qu'elle
commence à déformer légèrement. La figure (3.17)
ci-dessous, montre la variation de la constante du mouvement en fonction du
temps de la simulation. On remarque que cette constante décroit
progressivement de -9756.94 jusqu'à -9756.99, puis une
légère augmentation et enfin un chute brisque de -9756.96
jusqu'à elle s'annule.
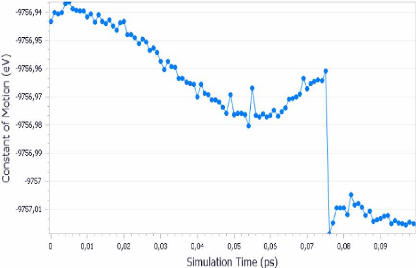
FIGURE 3.17 - Evolution de la
constante du mouvement en fonction du temps de simulation
Chapitre III Résultats et
discussions
La figure suivante (3.18) montre l'évolution de la
pression au cours de la simulation en fonction du temps de calcul. Cette figure
indique une augmentation continue de la pression jusqu'à elle atteint 36
GPa.
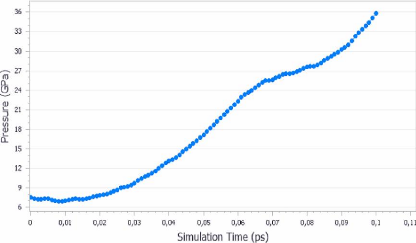
FIGURE 3.18 - Evolution de la
pression en fonction du temps de simulation
La figure (3.19) montre aussi l'évolution de la
température en fonction du temps de la simulation.

FIGURE 3.19 - Evolution de la
température en fonction du temps de simulation
58 | P a g e
Chapitre III Résultats et
discussions

200 GPa
59 | P a g e
La température varie entre 0 et 450 K et le temps de la
simulation varie entre 0 et 0.1 pc. Pendant ces intervalles la
température initiale est 300 K comme que déjà la fixer,
puis elle commence à varier toute l'intervalle. On constate que la
température al fin de la simulation est presque celle de la
température initiale 300 K, cela nous indique que le systéme a
subit la relaxation à la fin de cycle et donc un retour à
l'état d'équilibre de systéme.
| 

