évaluation des paramètres pharmacotechniques et chimiques des comprimés d'isoniazide distribués à Lubumbashi.par Eric LAU SALION Université de Lubumbashi - Grade de pharmacien en sciences pharmaceutiques 2020 |

masse moyenne La masse individuelle de 2 au plus des 20 comprimés peut s'écarter de la masse moyenne d'un pourcentage plus élevé que celui qui est indiqué, mais la masse d'aucune unité ne peut s'écarter de plus du double de ce pourcentage. Cette approche est recommandée par la pharmacopée Européenne, est avantageuse dans le cas où elle permet de lire ou de voir rapidement les différents écarts des poids des différentes unités par rapport à la masse moyenne. Tableau I : écarts des limites acceptables pour l'uniformité des masses
6
La deuxième concerne la lecture des deux limites ; inférieure et supérieure, la lecture de la limite inferieur se fait en prenant le poids le plus bas de toutes les unités qu'on soustrait à la masse moyenne divisé par la masse moyenne et le tout multiplié par cent. Psup-Pmoyen Limite sup = Pmoyen x 100 La lecture de la limite supérieure se réalise en prenant le poids le plus bas de toutes les unités qu'on soustrait à la masse moyenne divisé par la masse moyenne et le tout multiplié par cent.
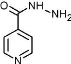
Dans le cadre de notre travail, nous allons interpréter nos résultats en suivant la première approche qui est recommandée par la pharmacopée est qui est fiable et claire à illustrer I.2. Test de friabilitéI.2.1. Intérêt et principeLe test de friabilité permet de s'assurer que les comprimés non enrobés présentent une résistance mécanique suffisante, pour que leurs surfaces ne soient pas endommagées ou ne présentent pas des signes d'abrasion ou de rupture, sous l'effet de toutes les manipulations (chocs mécaniques, frottements, attrition) qu'ils vont subir jusqu'au moment de leur utilisation. (Phr Eur, 2004). Le test de friabilité appliqué à un certain nombre de comprimé non enrobés, consiste à apprécier la perte de masse de ces comprimés, sous l'effet des frottements et des chutes qui leurs ont été imposés dans certaines conditions. (Phr Eur, 2007). I.2.2. AppareillageL'appareil utilisé pour le test de friabilité des comprimés non enrobé est décrit dans la la pharmacopée européenne 2004. Il s'agit d'un tambour rotatif d'un diamètre intérieur de 283 mm à 291 mm et d'une hauteur de 36 mm à 40 mm, constitué d'un polymère synthétique transparent 7 à surfaces intérieures polies ne produisant pas d'électricité statique. L'une des faces du tambour est amovible. A chaque rotation, les comprimés sont projetés du centre du tambour vers la paroi extérieure, selon une trajectoire curviligne de rayon intérieur compris entre 75,5 mm et 85,5 mm. Le tambour est monté sur l'axe horizontal d'un dispositif d'entraînement dont la vitesse de rotation est de 25 #177; 1 tours/minute. Par conséquent, à chaque rotation, les comprimés roulent ou glissent et tombent sur la paroi ou les uns sur les autres (Phr Eur. 2004). I.2.3. Critères d'évaluationLa friabilité est exprimée en termes de perte de masse, et calculée en pourcentage de la masse initiale des comprimés non enrobés prélevés. La perte de masse maximale considérée comme acceptable, pour la plupart des produits, est de 1 pour cent de la masse des comprimés soumis à l'essai (Phr Eur, 2004). I.3. Test de désagrégationI.3.1. Intérêt et principeLe test de désagrégation des comprimés et les gélules permet de s'assurer, que leur vitesse de désagrégation ne constitue pas le facteur limitant de la dissolution du principe actif qu'ils contiennent (Le Hir, 2001). Le test de désagrégation appliqué aux comprimés non enrobés, est destiné à déterminer leur plus ou moins grande aptitude à se désagréger, en milieu liquide, dans un temps prescrit et dans des conditions expérimentales bien définies (Phr Eur, 200). Le test de désagrégation des comprimés non enrobés fait partie des essais pour contrôler la « disponibilité in vitro » du principe actif qu'ils contiennent (Le Hir, 2001). I.3.2. AppareillageEn fonction des dimensions des comprimés à analyser, deux types d'appareils sont utilisés pour le test de désagrégation : Pour les comprimés non enrobés de dimensions normales (inférieures à 18 mm), on utilise l'appareil A, pour le test de désagrégation. La partie principale de ce l'appareil est constituée par un assemblage rigide supportant 6 tubes cylindriques transparents. Chaque tube a une longueur de 77,5 #177; 2,5mm et un diamètre intérieur de 21,5mm. Chacun de ces tubes est pourvu d'un disque cylindrique (diamètre 20,7 #177; 0,15 mm et épaisseur 9,5 #177; 0,15 mm) en 8 matière plastique transparente. Les tubes sont maintenus verticaux par 2 plaques séparées, superposées, en matière plastique rigide et percées chacune de 6 trous. Sous la plaque inférieure est fixée une toile métallique en fils d'acier inoxydable. La plaque supérieure porte fixée en son centre, une tige métallique qui permet de relier l'assemblage à un dispositif mécanique destiné à assurer un mouvement vertical, alternatif et régulier dont l'amplitude est de 50mm à 60mm ; le nombre de déplacements complets (montée-descente) est de 29 à 32 par minute. L'appareil est plongé dans un liquide de délitement (eau distillée) contenu dans un vase cylindrique de 1 litre ou dans tout autre récipient convenable. Le volume de liquide à verser dans le récipient est tel que, lorsque l'assemblage est dans la position la plus élevée, le tamis métallique est au moins à 15 mm en dessous de la surface du liquide et, lorsque l'assemblage est dans sa position la plus basse, le tamis est au moins à 25 mm du fond, les extrémités supérieures des tubes ouverts demeurant au-dessus de la surface du liquide. Un dispositif adéquat maintient la température de l'ensemble à 37 #177; 2 °C. (Phr Eur, 2004). Pour les comprimés non enrobés de grandes dimensions (supérieures à 18 mm), on utilise pour le test de désagrégation, l'appareil B. Ce dernier est semblable à l'appareil A, mais ne comporte que 3 tubes cylindriques transparents de diamètre intérieur 33,0 #177; 0,5 mm. Le disque cylindrique de l'appareil B a un diamètre de 31,4 #177; 0,13 mm et une épaisseur de 15,3 #177; 0,15 mm (Phr Eur, 2004). I.3.3. Critères d'évaluationPour les comprimés non enrobés de dimensions inférieures à 18 mm, le test de désagrégation est satisfaisant si tous les 6 comprimés sont désagrégés au bout de 15 minutes. On dira que le comprimé s'est désagrégé s'il n'y a plus de résidu sur la grille de l'appareil de désagrégation, ou s'il subsiste un résidu, ce dernier est constitué seulement par une masse molle ne comportant pas de noyau palpable et non imprégné. Si sur les 6 comprimés soumis à l'essai, 1 ou 2 ne sont pas désagrégés, répétez l'essai sur 12 comprimés supplémentaires. Dans ce dernier cas, les exigences de l'essai sont satisfaites si au moins 16 comprimés sur 18 soumis à l'essai sont désagrégés. Pour les comprimés non enrobés de dimensions supérieures à 18 mm, le test de désagrégation est satisfaisant si tous les 6 comprimés prélevés au total sont désagrégés au bout de 15 minutes. Pour les comprimés non enrobés de dimensions normales ou de grandes dimensions, lorsque les 6 comprimés ne satisfont pas à l'essai de désagrégation en raison de leur 9 adhérence au disque, répétez l'essai sur 6 autres comprimés en omettant les disques (Phr Eur, 2004). I.4. Test de dissolutionLes essaies de dissolution sont utilisés tout au long de processus de mise au point du médicament et constitue une exigence pour toutes les formes galéniques orales en phase solide dans le cadre des essaies de libération et de stabilité du produit. Il s'agit d'un test analytique essentiel utilisé pour détecter les changements physiques dans un ingrédient pharmaceutique actif. Une fois que le médicament est formé, ses propriétés de dissolution ont un effet direct sur son absorption par l'organisme. Par conséquent la vitesse de dissolution d'un comprimé ou d'une capsule par exemple est essentielle à ce processus. C'est un processus physico-chimique dans lequel une substance ou un médicament est plongé dans un solvant pour former ensuite un mélange homogène appelé solution. Les tests de dissolution font partis des tests de stabilité nécessaire au processus de développement d'un médicament ou pour des tests de contrôle-qualité. Ces tests permettent de : d'évaluer les mécanismes et les propriétés de dissolution du médicament pouvant avoir un effet direct sur son absorption dans l'organisme, de définir la biodisponibilité du principe actif (Phr Eur, 2004). I.4.1. Intérêt et principeLe test de dissolution in vitro appliqué aux comprimés, permet de s'assurer, qu'une fois administrés, ces derniers libèreront le principe actif qu'ils contiennent, pour le mettre à la disposition de l'organisme, et ceci dans les limites de concentration et de vitesse déterminées, afin de garantir l'effet thérapeutique désiré. Utilisé en contrôle de routine de la production, le test de dissolution permet aussi de démontrer la reproductibilité du procédé de production, la conformité du produit fini avec les lots précédents (reproductibilité inter lot) et d'apprécier la variabilité à l'intérieur d'un même lot de fabrication (reproductibilité intra lot). L'essai de dissolution in vitro appliqué aux comprimés est destiné à déterminer leur plus ou moins grande aptitude à laisser passer en solution dans un milieu déterminé, le ou les principes actifs qu'ils contiennent. Le passage en solution est apprécié par dosage du principe actif dans des échantillons prélevés dans le milieu de dissolution à intervalles de temps 10 différents. Le test de dissolution des comprimés est le principal essai réalisé pour contrôler la « disponibilité in vitro » du principe actif qu'ils contiennent. Ainsi, lorsqu'un essai de dissolution est prescrit, un essai de désagrégation peut ne pas être exigé. I.4.2. AppareillageLes pharmacopées préconisent 4 types d'appareils pour réaliser le test de dissolution in vitro des formes pharmaceutiques orales solides : l'appareil à palette tournante ; l'appareil à panier tournant ; l'appareil à cylindres réciproques ; l'appareil à flux continu. L'appareil à cylindres réciproques a été particulièrement développé pour étudier la dissolution des formes multi particulaires à libération prolongée. L'appareil à flux continu est plus particulièrement destiné à étudier les formes à libération modifiée et les formes multi particulaires. L'appareil à palette tournante et l'appareil à panier tournant sont quant à eux plus adaptés pour réaliser le test de dissolution in vitro des formes pharmaceutiques orales solides à libération immédiate que sont les comprimés. L'appareil à palette tournante est constitué de : un récipient cylindrique à fond hémisphérique, d'une capacité de 1000 millilitres, en verre borosilicaté ou en un autre matériau transparent, approprié. Le récipient est muni d'un couvercle évitant l'évaporation et comportant un orifice central destiné au passage de la tige de l'agitateur ainsi que de plusieurs autres orifices permettant l'introduction d'un thermomètre et celle des dispositifs de prélèvement du liquide. C'est au fond du récipient que sont placés les comprimés à contrôler ; un agitateur constitué d'une tige verticale à la partie inférieure de laquelle est fixée une palette dont la forme correspond à celle de la portion d'un cercle délimitée par deux plans parallèles. Un bain d'eau thermo statée qui permet de maintenir la température du milieu de dissolution à 37 #177; 0,5 °C pendant l'essai. Concernant ce travail, nous n'avons pas pu effectuer le test de dissolution vu que l'appareil de dissolution de la faculté des sciences pharmaceutiques n'était pas prêt à utiliser. 11 I.5. Test de résistance à la ruptureI.5.1. Intérêt et principeLe test de dureté permet de s'assurer que les comprimés non enrobés présentent une résistance mécanique suffisante pour ne pas se briser lors de leurs manipulations ou d'étapes de production ultérieures (Phr Brit, 2007). La détermination de la dureté d'un comprimé non enrobé se fait en mesurant l'intensité de la force qui lui est diamétralement appliquée pour provoquer sa rupture par écrasement (Phr Eur, 2004). I.5.2. AppareillageL'appareil de mesure de la dureté des comprimés non enrobé est décrit dans la PE-04 et dans la PB-07. Il est constitué de 2 mâchoires se faisant face, l'une se déplaçant vers l'autre. La surface plane des mâchoires est perpendiculaire au sens du déplacement. La surface d'écrasement des mâchoires est plane et plus grande que la zone de contact avec le comprimé. L'appareil est étalonné à l'aide d'un système précis à 1 newton près (Phr Eur, 2004). I.5.3. Critères d'évaluationExprimez les résultats en donnant la valeur moyenne, les valeurs minimales et maximales des forces mesurées, toutes exprimées en newtons (N). Aucunes des pharmacopées consultées pour notre étude ne présentent de normes de dureté pour les comprimés non enrobés. En effet, la dureté d'un comprimé est fonction de sa formulation qui peut varier d'une spécialité pharmaceutique à une autre. Les normes de dureté de comprimés sont donc fixées pour chaque spécialité pharmaceutique dans le dossier de l'autorisation de mise sur le marché (Phr Eur, 2004). Ici il n'y a aucune pharmacopée qui donne les règles spécifiques concernant le test de résistance à la rupture, les analyses sont faites sur base des données décrites dans le dossier de l'autorisation de mise sur le marché de chaque spécialité. 12 I.6. Test de variation des masses I.6.1. Intérêt et principeCe test ressemble un tout petit peu au test d'uniformité des masses, sauf qu'ici il est effectué sur dix comprimés au lieu de vingt comprimés. L'essai de variation de masse des comprimés permet de s'assurer qu'au cours de la fabrication, la répartition du mélange initial de poudre ou de granulés, en unités de prises (chaque comprimé), a été suffisamment précise et uniforme pour garantir une même masse et donc une même teneur en principe actif pour l'ensemble des comprimés d'un même lot et que les variations de masses ne se diffère pas ou ne s'écarte pas au de la recommandation de la pharmacopée britannique. I.6.2. Critères d'évaluationSi AV est supérieur à 15,0 %, recommencer l'essai avec 20 autres comprimés ou gélules et recalculer l'AV comme indiqué ci-dessus. L'AV final de 30 comprimés ou gélules doit être inférieur ou égal à 15 (USP, 2018). Le test de variation des masses prend en compte la teneur en principe actif et la dispersion de ce dernier dans le milieu physiologique, il est plus intéressant par rapport au test d'uniformité des masses qui ne tient compte que des masses de chaque unité. 13 CHAPITRE II. PHARMACOLOGIE DES ANTITUBERCULEUXLes antituberculeux constituent du point de vue de leur structure, une classe hétérogène de médicaments caractérisée par la diversité de leur origine pharmaco chimique. Aussi, leur classification a été envisagée peu après leur introduction en thérapeutique infectieuse par l'OMS en termes de « médicaments de première ligne ou première intention » et « médicaments de seconde ligne » associés dans les régimes thérapeutiques en considération des critères suivants : efficacité et résistance, toxicité, tolérance, volume et prix (OMS, 2013). Les médicaments antituberculeux sont de deux types, majeurs et mineurs. Les majeurs sont à quatre à savoir l'Isoniazide (INH), la Rifampicine (RIF), le Pyrazinamide (PZA) et l'Éthambutol (EMB), ils sont utilisés en première intention sauf en pédiatrie (Lucht et al., 2019). Et les mineurs sont Les antibiotiques appartenant à la famille des aminosides Lorsqu'on soupçonne fortement ou que l'on a prouvé la résistance à la Streptomycine, il est possible d'utiliser l'effet bactéricide d'un autre aminoside contre les bactéries actives en multiplication en l'occurrence la Kanamycine et l'Amikacine qui sont aussi actives et mieux tolérée que la Kanamycine (Collazos et al., 2012). - Les antibiotiques appartenant à la famille des polypeptides. - La Capréomycine peut être utilisé à la Kanamycine et à l'Amikacine, l'Ethionamide et le Prothionamide appartenant à la famille des thioamides. A noter que le Prothionamide pourrait être mieux toléré que l'Éthionamide dans certaines populations. - Les quinolones antibactériennes, elles sont dotées d'une bonne action bactéricide, elles sont utiles en association avec d'autres médicaments. Ce sont la Moxifloxacine et la Lévofloxacine. - La D-Cyclosérine est un antituberculeux bactériostatique utilisé en association pour prévenir la résistance. Cependant, sa forte toxicité limite son emploi. L'acide Para-Amino-Salicylique était utile dans le passé pour prévenir la résistance à l'Isoniazide et à la Streptomycine. Il continue de jouer le même rôle aujourd'hui pour d'autres molécules bactéricides. 14 La Thioacétazone est un bactériostatique, utilisé dans le traitement de la tuberculose des patients infectés par Mycobacterium tuberculosis multi résistants dans certains pays en voie de développement (Lucht et al ; 2019). II.1. Les antituberculeux essentielsCes antituberculeux dits essentiels sont : la rifampicine, le pyrazinamide, l'éthambutol et l'isoniazide II.1.1. Mécanisme d'action des antituberculeux essentielsLes deux antituberculeux bactéricides les plus efficaces sont l'isoniazide (IT) et la rifampicine (R) qui agissent sur les bacilles en multiplication rapide et continue, mais aussi sur ceux qui sont en multiplication ralentie sur les macrophages. Un avantage supplémentaire de la rifampicine est de détruire plus rapidement les bacilles en multiplication ralentie, car elle agit à une étape très précoce de la multiplication bacillaire (Lucht et al ; 2019). II.1.1.1. La rifampicineLa rifampicine déploie son activité antibactérienne en se liant de façon spécifique à l'ARN polymérase bactérienne, inhibant ainsi la transcription des ARN messagers. Il s'agit d'un antituberculeux majeur bactéricide sur les bacilles en multiplication, intra et extracellulaires et sur les bacilles dormants, grâce à sa puissante activité stérilisante. Son spectre d'action est large et comprend les bactéries à Gram positif, à Gram négatif et intracellulaires (Mycobacterium leprae, Mycobacterium tuberculosis). Elle n'est cependant pas active sur les mycobactéries atypiques sauf sur Mycobacterium kansasii (Durand, 2014). II.1.1.2. L'EthambutolL'Ethambutol agirait sur les bactéries en croissance au niveau de la paroi mycobactérienne. Le mécanisme d'action évoqué est que la cible de l'Éthambutol serait une enzyme, l'arabinosyltransférase, intervenant dans la liaison arabinose-galactane. Son inhibition par l'Ethambutol, en effet conduirait à l'inhibition de la synthèse de l'arabinane, de l'arabinogalactane et du lipoarabinogalactane de la paroi cellulaire (Mitchison at al., 2012). De ce fait, les acides mycoliques ne peuvent pas se lier sur le motif arabinogalactane (constituant de la paroi). Ceci entraine une désorganisation de la paroi cellulaire et la mort de la bactérie soumise au stress du milieu extérieur (Mitchison et al., 2012). 15 II.1.1.3. PyrazinamideLe Pyrazinamide nécessite un PH acide et la présence d'une pyrazinamidase qui permet de transformer le pyrazinamide en acide pyrazoique, le compose actif, qui agirait en inhibant la synthèse des acides gras à chaines courtes du bacille (Mitchison et al., 2012). II.1.2. Propriétés pharmacocinétiques des antituberculeux essentielsPour qu'un antibiotique soit efficace ; il faut qu'il arrive en quantité suffisante sur le lieu de l'infection afin d'atteindre la concentration minimale inhibitrice capable d'inhiber toute culture de bactéries et que le germe soit sensible à cet antibiotique (Pilly E., 2012). II.1.2.1. RifampicineL'absorption orale est bonne mais diminuée par la prise d'aliments. Elle doit donc être prise à jeun, le pic plasmatique est atteint 2 heures après la prise, la diffusion tissulaire est bonne dans de nombreux tissus : os, poumons autres tissus du fait de sa liposolubilité. Elle franchit peu la barrière hématoméningée ; on obtient toutefois des concentrations efficaces dans le liquide cephalorachidien lors d'inflammation des méninges.la fixation aux protéines plasmatiques est de 80 %, la métabolisation se fait via une désacétylation au niveau hépatique sous forme de métabolite actif. C'est un inducteur enzymatique puissant à l'origine de nombreuses interactions médicamenteuses. Elle accélère son propre métabolisme. La demi-vie d'élimination est de 2 à 5 heures, et l'élimination se passe à 70 à 80 % par voie biliaire (cycle entérohépatique) et 10 à 30 % par voie rénale (Pilly E., 2012). II.1.2.2. EthambutolL'absorption par voie orale est assez bonne (environ 80 %), non modifiée par la prise concomitante d'aliments, la demi-vie d'élimination bi phasique est de 2 à 4 heures pour la première phase et 12 à 14 heures pour la seconde. La fixation protéique est faible, de 20 à 40 %, il a une faible métabolisation au niveau hépatique et l'élimination est principalement rénale à 80 % sous forme active (OMS 2015). II.1.2.3. Pyrazinamide (PZA)L'absorption est retardée par la prise concomitante d'aliments, il doit donc être pris à jeun, sa demi-vie est d'environ 9 heures, la fixation protéique est faible, le métabolisme hépatique conduit a un métabolite principal qui est l'acide pyrazinoïque qui inhibe la sécrétion 16 tubulaire de l'acide urique, provoquant ainsi des hyper uricémies. La distribution est large dans l'organisme y compris dans le système nerveux central. Et l'élimination est rénale à 70 % (OMS 2015). II.1.3. Propriétés pharmacologiques des antituberculeuxLes antituberculeux sont soit bactériostatiques (la bacteriostase est le ralentissement de la croissance des bactéries, pouvant aller jusqu'à l'arrêt de cette croissance, lorsque la bacteriostase est maximale), soit bactéricides (ils provoquent la mort directe des bactéries) (OMS 2015). Les bacilles dont la multiplication est lente situés à l'intérieur des macrophages. Leur multiplication est ralentie par le manque d'oxygène et le pH acide du cytoplasme macrophagique. L'action des médicaments est variable selon leur activité bactéricide et/ou stérilisante sur ces populations bacillaires. Les deux antituberculeux bactéricides les plus efficaces sont l'isoniazide (II) et la rifampicine (R) qui agissent sur les bacilles en multiplication rapide et continue, mais aussi sur ceux qui sont en multiplication ralentie sur les macrophages. Un avantage supplémentaire de la rifampicine est de détruire plus rapidement les bacilles en multiplication ralentie, car elle agit à une étape très précoce de la multiplication bacillaire (F. Lucht, 2019). Deux autres antituberculeux bactéricides d'efficacité moindre et d'action complémentaire sont le pyrazinamide (Z), qui est actif en milieu acide et détruit ainsi les bacilles intracellulaires et la streptomycine (S) qui n'est active que sur les bacilles se multipliant activement dans le milieu extracellulaire parce qu'elle est incapable de traverser la membrane cellulaire. Deux antituberculeux sont bactériostatiques et donc beaucoup moins efficaces, l'éthambutol et le thioacétazone. Ils sont utilisés en association avec des bactéricides puissants pour éviter l'apparition de bacilles résistants. D'autres médicaments sont d'efficacité mineure et ne sont pas utilisés dans la chimiothérapie de courte durée. Ce sont l'éthionamide, la Kanamycine et la capréomycine, les quinolones, la cyclosérine. Les seuls médicaments qui détruisent les bacilles persistants et qui ont une action stérilisante sont la rifampicine et le pyrazinamide. Ces médicaments sont toujours employés dans la chimiothérapie de courte durée. 17 Point pratique : La chimiothérapie de courte durée est possible grâce à l'utilisation combinée des trois médicaments les plus actifs : l'isoniazide, la rifampicine et le pyrazinamide (F. Lucht, 2019). II.1.4. Mécanisme de résistance aux antituberculeuxLa résistance aux médicaments antituberculeux est due à une mutation chromosomique spontanée. La proportion de mutants résistants de type sauvage dans une population de M. tuberculosis non traitée est habituellement très faible. Le traitement par les médicaments antituberculeux exerce une pression sélective sur la population de M. tuberculosis, entraînant une diminution des bacilles sensibles, une multiplication des mutants pharmaco-résistants et l'émergence d'une pharmaco-résistance : il s'agit d'une résistance acquise. La résistance primaire concerne les patients infectés par M. tuberculosis qui sont résistants aux antituberculeux dès le début, avant tout traitement. Des schémas thérapeutiques inappropriés, une posologie inférieure à celle recommandée, une qualité douteuse des médicaments, une mauvaise observance du traitement sont souvent associés à l'émergence d'une pharmaco-résistance (Albert, 2018). L'isoniazide (hydrazide d'acide isonicotinique) est un pro-médicament qui nécessite une activation par l'enzyme catalase-peroxydase codée par le gène katG. Le principe actif (radical acyle isonicotinique ou anion) réagit avec le nicotinamide adénine dinucléotide (NAD), formant le produit d'addition INH-NAD, qui inhibe l'InhA provoquant une inhibition de la synthèse de l'acide mycolique de la paroi cellulaire. L'INH n'est actif que contre les formes dynamiques de M. tuberculosis, mais pas contre les bacilles dormants. La tolérance à l'INH dans les mycobactéries dormantes peut être causée par la protéine 1 de liaison à l'ADN mycobactérienne (MDP1), une protéine analogue aux histones, qui régule la transcription de katG et peut conduire à une tolérance à l'INH. La résistance à l'INH est un processus complexe. Les mutations dans plusieurs gènes, y compris katG, ahpC, inhA, kasA et ndh ont toutes été associées à une résistance à l'INH. Les mutations dans katG sont le principal mécanisme de résistance à l'INH. La mutation katG S315 est la mutation la plus fréquente dans les souches résistantes à l'INH, représentant 50 à 95 % des isolats cliniques résistants. Contrairement aux mutations katG, qui provoquent habituellement une résistance élevée, les mutations du gène inhA ou dans la région de son promoteur sont habituellement associées à une résistance de bas niveau avec des Le programme national de la lutte contre la tuberculose de la république démocratique du Congo exige un schéma thérapeutique basé sur l'utilisation de l'association isoniazide, 18 concentrations minimales inhibitrices de 0,2-1 g/L et sont moins fréquentes que les mutations katG. Les mutations dans inhA ne causent pas seulement la résistance à l'INH, mais elles confèrent également une résistance croisée à l'éthionamide éthylique (ETH), structurellement lié (Clin et al., 2003). II.1.5. Schéma thérapeutique de la prise en charge de la tuberculoseLe schéma thérapeutique se sur deux phases ; la première est la phase intensive et la deuxième étant la phase d'entretien. Les recommandations de l'OMS distinguent les traitements de première ligne et ceux de deuxième ligne réservés aux patients porteurs de bacilles multi résistants. En première ligne, les molécules utilisables sont l'isoniazide, la rifampicine, l'éthambutol, le pyrazinamide, la streptomycine. En deuxième ligne, ce sont les aminoglycosides (Amikacine, Kanamycine), les polypeptides (capréomycine), les fluoroquinolones (moxifloxacine, lévofloxacine), les thioamides (éthionamide, Prothionamide), la cyclosérine, l'acide AminoSalicyclique, la Thiacétazone, la clofazimine, l'amoxicilline-acide clavulanique, la clarithromycine, le linézolide (OMS ; 2010). Le programme national de lutte contre la tuberculose de la Tunisie préconise un s thérapeutique sur l'utilisation de l'association isoniazide, Pyrazinamide, Rifampicine et Ethambutol en phase d'attaque pendant deux mois et l'association de l'Isoniazide+rifampicine en phase d'entretient pendant quatre mois (PNLT Tunisie, 2015). Le programme national de lutte contre la tuberculose du Mali utilise un schéma préconisant l'association de l'association isoniazide, Pyrazinamide, Rifampicine et Ethambutol en phase d'attaque durant deux mois et l'association de l'Isoniazide+rifampicine phase d'entretient pendant une période de quatre mois (PNLT Mali, 2017). Le programme national de lutte contre la tuberculose du Sénégal utilise un schéma préconisant l'association de l'association isoniazide, Pyrazinamide, Rifampicine et Ethambutol en phase d'attaque pendent deux mois et l'association de l'Isoniazide+rifampicine en phase d'entretient six mois. Mais l'apparition des résistances a fait en sorte que ce schéma soit ramené à six mois (PNLT Sénégal, 2014). 19 Pyrazinamide, Rifampicine et Ethambutol en phase d'attaque pendent deux mois et l'association de l'Isoniazide+rifampicine en phase d'entretient quatre mois (PNLT RDC 2010). Le succès du traitement de la tuberculose repose d'abord sur l'application de régimes de chimiothérapie standardisés choisis par les programmes nationaux et conformes aux régimes recommandés par l'OMS. Il repose aussi sur des mesures organisationnelles appropriées visant notamment à assurer la compliance du malade au traitement et la supervision directement observée de la prise des médicaments durant au moins la phase initiale (OMS, 2015). Vue les propositions des différents programmes nationaux consultés, nous disons que le schéma proposé par l'OMS est suivi à la lettre par tous les programmes nationaux, vue que ce schéma répond aux attentes thérapeutiques. Des études cliniques contrôlées ont montré que le Traitement Prophylactique à l'Isoniazide réduit le risque d'évolution vers la tuberculose active chez les personnes co-infectées par le VIH et M. tuberculosis (WHO, 2019). 20 II.2. Spécificités pharmacologiques de l'isoniazideL'isoniazide est synthétisé au début du vingtième siècle, mais son activité contre la tuberculose n'est signalée pour la première fois que dans les années 1950. Elle fait partie de la liste des médicaments essentiels de l'organisation mondiale de la santé (HANSCH, et al. ; 1995) Structure
Formule brute C6H7N30Masse molaire 137.13 g/mol Pka 1.82 LogP -0.7 (Phr Eur 2014). II.2.1. Propriétés physico-chimiquesL'isoniazide se présente sous forme de cristaux blancs ou incolores ou de poudre cristalline blanche. Il est inodore et s'altère lentement lorsqu'il est exposé à l'air et à la lumière. Il est très soluble dans l'eau, peu soluble dans l'alcool, légèrement soluble dans le chloroforme et très légèrement soluble dans l'éther. Son point de fusion se situe entre 170 et 173 °C. En solution (1/10), son pH s'établit entre 6,0 et 7,5 (Phr Eur 2014). II.2.2. Spectre d'activité et mécanisme d'actionL'isoniazide est efficace contre les mycobactéries, en pratique le Mycobacterium Tuberculosis, mais également contre des mycobactéries atypiques comme Mycobacterium Kansasii et Mycobacterium xenopi (OMS 2015). 21 L'isoniazide agit en bloquant la synthèse de l'acide mycoliques, molécule indispensable des parois cellulaires des mycobactéries, entrainant la mort des mycobactéries intracellulaires ou extracellulaires. L'isoniazide est un antituberculeux bactéricide qui inhibe la synthèse de la paroi bactérienne. Deux mécanismes d'action ont été décrits : le premier concerne l'action de l'isoniazide sur l'enzyme catalase-peroxydase codée par le gène katG. L'oxydation de l'isoniazide par cet enzyme abouti à la formation d'un métabolite actif entraînant la mort cellulaire. Le deuxième mécanisme concerne l'inhibition de la synthèse de la protéine InhA codée par le gène InhA. Cette protéine joue un rôle important dans la synthèse des acides mycoliques, composants majeurs de la paroi des mycobactéries. La prise de cet antibiotique peut entrainer un antibiotique peut entrainer une inflammation du foie mais aussi parfois une inflammation des nerfs (OMS 2015). II.2.3. Indications thérapeutiquesL'isoniazide est utilisé dans le traitement des infections tuberculeuses latentes actives. Dans le traitement de la tuberculose active et afin de limiter le développement de résistance et pour raccourcir la durée du traitement, l'isoniazide est administré pour un patient adulte, quotidiennement en poly thérapie avec d'autres antibiotiques, la pyrazinamide, l'éthambutol et la rifampicine pendant 2nmois puis en association avec la rifampicine pendant 4 autres mois. Pour un patient enfant, la polytherapie est composée de rifampicine, de pyrazinamide et d'isoniazide pendant deux mois, puis de rifampicine et d'isoniazide les quatre mois suivants (OMS 2015). L'isoniazide est utilisé comme traitement prophylactique pour les populations suivantes : Les personnes infectées par le VIH et présentant une induration d'au moins 5 mm au test Mantoux ; Les personnes au contact de malade de la tuberculose et qui présentent une induration d'au moins 5 mm ; Les personnes qui, après un test Mantoux négatif, voient le test devenir positif dans les deux ans et présentant une induration d'au moins 10mm pour les personnes de plus de 35 ans et d'au moins 15 mm pour les patients de moins de 35 ans ; Les personnes atteintes de lésions pulmonaires visibles sur leur radiographie pulmonaire susceptibles d'être dues à la tuberculose, guéries et présentant une induration d'au moins 10 mm, Il fait partie de tous les schémas thérapeutiques antituberculeux actuellement recommandés par l'OMS. 22 L'isoniazide est parfois utilisé seul pour
éviter : la transmission aux contacts proches à haut risque ; La
progression de l'infection vers le complexe primaire chez le sujet
récemment infecté et encore asymptomatique Le plus souvent le traitement préventif ou la chimio prophylaxie par l'isoniazide est un traitement de la primo-infection visant à stériliser les lésions et empêcher le développement d'une tuberculose évolutive. Il s'agit d'un traitement que d'une prophylaxie au sens strict. Le traitement préventif consiste en l'administration d'isoniazide pendant 6 à 9 mois (Borthamley 2001). II.2.4. Propriétés pharmacocinétiquesL'isoniazide est rapidement absorbé et diffusé facilement dans tous les liquides et tissus organiques. Sa demi-vie plasmatique, déterminée au niveau génétique, se situe entre moins d'1 heure et plus de 3 heures selon la vitesse d'acétylation. Il est excrété en grande partie dans les urines au bout de 24 heures, principalement sous forme de métabolites inactifs. L'absorption de l'isoniazide est rapide avec une biodisponibilité proche de 100%, le temps maximal après une prise unique à raison de 5mg/Kg/jour est de 1 à 2 heures. La concentration maximale pour la même dose est de 1-2.5 mg/L à la 3eme heure. Il se distribue rapidement dans tous les liquides organiques (liquide céphalorachidien, liquide pleural et liquide ascitique), dans les tissus, dans les organes et dans les excréta (salive, expectorations et fèces). L'isoniazide ne se lie pas dans une mesure importante aux protéines plasmatiques. Il traverse la barrière placentaire et passe dans le lait maternel, où il se retrouve à une concentration semblable à la concentration plasmatique (Ellard et al. ; 1976). Sa demi-vie plasmatique chez les personnes présentant des fonctions rénale et hépatique normales varie entre 1 et 4 heures, selon la vitesse du métabolisme et son élimination est essentiellement urinaire forme de métabolite appelé Acetylhydrazine. L'isoniazide est métabolisé dans le foie, surtout par acétylation et par déshydrazination. La vitesse d'acétylation est déterminée par des facteurs génétiques. Environ 50 % des personnes de race noire et des personnes de race blanche sont des inactivateurs lents, et les autres sont des inactivateurs rapides ; la plupart des Orientaux sont des inactivateurs rapides. La vitesse 23 d'acétylation n'influe pas de façonnable sur l'efficacité de l'isoniazide ; cependant, une acétylation lente peut entraîner un accroissement des concentrations du médicament dans le sang et, par conséquent, une augmentation des effets toxiques. Une carence en pyridoxine (vitamine B6) peut être observée chez les adultes qui prennent des doses élevées d'isoniazide ; cette carence est probablement due à une compétition entre l'isoniazide et le phosphate de pyridoxal pour l'enzyme apotryptophanas (OMS 2015). II.2.5. Réactions indésirablesLes réactions indésirables les plus fréquentes causées par l'isoniazide sont celles qui intéressent le système nerveux et le foie, troubles sanguins et lymphatiques, troubles gastro-intestinaux, nausées, vomissements, douleurs épigastriques et pancréatite, troubles hépatobiliaires, (élévation de la concentration sérique des transaminases, troubles immunitaires (réactions d'hypersensibilité), troubles métaboliques et nutritionnels (carence en pyridoxine, pellagre, hyperglycémie et acidose métabolique), troubles de l'appareil locomoteur et du tissu conjonctif, troubles de l'appareil reproducteur et des seins (Gynécomastie), troubles de la peau et des tissus sous-cutanés ; Acné, syndrome de Stevens-Johnson, pemphigus, rash, dermatite cutanées (morbilliformes, maculopapuleuses, purpuriques ou exfoliatrices),troubles vasculaires (OMS 2015). 24 DEUXIEME PARTIE : PARTIE EXPERIMENTAL25 CHAPITRE III. PRESENTATION DU MILIEUX DE
RECHERCHE,
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
OCC Laboratoire d'analyse des médicaments de la faculté des sciences pharmaceutiques |
- Test de délitement - Identification du principe actif - Dosage du principe actif - Uniformité des masses - Test de friabilité - Test de dureté - Test de désagrégation |
Ce présent tableau présente les deux laboratoires qui ont servis de cadre expérimental pour l'analyse de nos échantillons et il présente en même temps les activités réalisées par laboratoire.
Une fois collectés, les échantillons ont été acheminés dans un local spécialement aménagé où le contrôle de la température a été réalisé à l'aide d'un thermomètre et le taux d'humidité par un hygromètre.
Les réactifs et solvants utilisés dans ce travail ont été les suivants :
Eau distillée ; NH2 ou ammoniac ; AgNO3 (nitrate d'argent) Les appareils ci-après ont été utilisés dans notre étude :
- Spectrophotomètre UV-Vis de marque HACH, série DR 5000
- Appareil de délitement, marque DISKET, série ez tab 300
- Appareil de friabilité, marque DISKET, série ez tab 300
- Appareil de dureté ou durometre, marque DISKET, série ez tab 300
- Agitateur automatique de marque de marque MS-H80
27
La verrerie et petit matériel utilisés aux laboratoires sont les suivants :
Tube à essai ; Becher ; Pipette pasteur ; Verre de montre ; Pissette ; Ballon jaugé à fond plat de 100 mL
Comme nous l'avons mentionné dans le point IV.1.1., les échantillons ont été collectés dans 12 structures sélectionnées sur base des informations obtenues à la Division Provinciale de la Santé du Haut-Katanga. En effet, la politique nationale en RDC veut que les antituberculeux soient distribués gratuitement aux malades. Sur ce, il existe des structures spécialisées pour assurer leur distribution. Nous avons donc visité toutes ces structures suivant les informations recueillies par la Division Provinciale de la Santé. Sur terrain, nous avons commencé par voir l'administration de la structure sanitaire en vue d'obtenir l'autorisation de mener notre étude. Nous avons été ensuite conduit auprès de services chargés de la prise en charge de la tuberculose.
Les échantillons ont été acquis soit gratuitement soit par achat. En effet, aux hôpitaux SNCC et Jason Sendwe, les échantillons ont été acquis gratuitement, mais dans les dix structures restantes, les échantillons ont été acquis par achat.
Concernant le choix de la molécule antituberculeuse, nous avons sélectionné l'isoniazide parce que la détermination de la teneur en principe actif devrait se faire en utilisant la méthode
non séparative (spectrophotométrie UV visible).
Seule isoniazide était disponible en
monothérapie et
utilisé dans le traitement préventif de la tuberculose.
Il convient de souligner qu'avant de solliciter les échantillons de l'isoniazide, nous avons questionné les personnels chargés de la distribution des antituberculeux sur l'utilisation et la disponibilité de l'isoniazide seul ou en association. Le modèle de ce questionnaire est repris en annexe I de ce travail. Ceci nous a permis notamment de prendre connaissance de la forme et le dosage de l'isoniazide qui est couramment distribué aux patients atteints de la tuberculose.
Pour être pris en compte, les échantillons devraient répondre aux critères d'inclusions qui ont été clairement définis :
- La date de péremption qui devrait être en cours de validité,
28
- La forme de l'isoniazide qui devrait être sous forme de comprimés non enrobé et non associé pour être analyser.
Au moins 40 comprimés étaient collectés pour chaque échantillon. Les échantillons devraient être collectés avec leur emballage primaire et secondaire tout en se rassurant qu'il y a suffisamment des comprimés disponibles afin d'éviter ainsi une rupture de stock du médicament dans la structure sanitaire.
Les échantillons d'isoniazide ayant un même : numéro de lot, date de fabrication et de péremption ont été considérés comme un seul et même échantillon et ce, même s'ils étaient collectés dans de sites différents.
Les échantillons collectés ont été soumis à une inspection visuelle attentive de l'emballage primaire, secondaire et du produit lui-même. Cette démarche a consisté à vérifier notamment le numéro de lot, l'emplacement des mentions sur l'emballage primaire et secondaire, la couleur de l'emballage et de la forme galénique, le logo du fabricant, l'adresse du fabricant, le pays de provenance, l'hologramme, la notice etc.
Pour effectuer ce test, nous avons suivi la procédure ci-après :
- A l'aide d'une balance analytique, peser individuellement 20 comprimés, prélevés
au hasard et déterminer la masse moyenne
- Calculer les écarts limites de la moyenne en % et comparer aux exigences (tableau
III) (Phr Eur, 2020).
Les résultats sont exprimés en moyenne, les valeurs minimales et maximales des forces mesurées, toutes exprimées en newtons (N). Aucunes des pharmacopées consultées pour
29
Tableau III : Ecarts limites en pourcentage de la moyenne admise pour mesurer l'uniformité de masse des comprimés et gélules
Forme pharmaceutique Masse moyenne (??) Ecarts limites de la
moyenne admis (%)

Comprimés non enrobés ?? ~ 80 mg 10
80 mg > ?? <250 mg 7,5
250 mg ~ ?? 5
Capsules ?? < 300 mg 10
?? ~ 300 mg 7,5
Spécifications (Pharmacopée Européenne, 2020) :
Au plus 2 unités peuvent s'écarter de la masse moyenne d'un pourcentage indiqué.
La masse d'aucune unité ne peut s'écarter du double de ce pourcentage. Si le premier essai ne satisfait pas aux spécifications, il faut reprendre le test avec 10 autres unités.
Il permet de s'assurer que les comprimés présentent une résistance mécanique suffisante pour ne pas se briser lors de leurs manipulations ou d'étapes de production ultérieures (Phr Eur, 2004).
Mode opératoire
- Placez le comprimé entre les mâchoires en tenant compte, le cas échéant, de sa
forme, de la barre de cassure et de la gravure ; pour chaque détermination,
- Orienter le comprime de la même façon par
rapport à la direction d'application de
la force.
- Effectuez la mesure sur 10 comprimé, en prenant soin
d'éliminer tout débris de
comprimé avant chaque
détermination. Indiquez le type d'appareil et, le cas
échéant, l'orientation des comprimés. Ce
procédé ne s'applique pas lorsqu'un appareil entièrement
automatisé est utilisé (Phr Eur, 2004).
30
notre étude ne présentent de normes de dureté pour les comprimés non enrobés. En effet, la dureté d'un comprimé est fonction de sa formulation qui peut varier d'une spécialité pharmaceutique à une autre. Les normes de dureté de Comprimés sont donc fixées pour chaque spécialité pharmaceutique dans le dossier d'AMM (Phr Eur,, 2004).
Le protocole ci-après a été suivi
- Prélever 10 comprimés entiers soigneusement dépoussiérés avant l'essai
- Peser exactement l'échantillon et placer les comprimés dans le tambour
- Procéder à 100 rotations (25 rotations par minutes pendant quatre minutes), puis
sortir les comprimés du tambour, éliminer les poussières libres comme précédemment et peser à nouveau exactement.
Si, au terme du cycle de rotations, l'échantillon comporte des comprimés visiblement fêlés, fissurés ou cassés, il ne satisfait pas à l'essai. Si les résultats sont difficiles à interpréter ou si la perte de masse est supérieure à la valeur cible, répéter l'essai à deux reprises et calculer la moyenne des trois résultats. Pour la plupart des produits, la perte de masse maximale (résultant d'un seul essai ou de la moyenne de 3 essais) considérée comme acceptable est de 1,0 pour cent (Pharmacopée Européenne, 2016).
-
-
Pour effectuer ce test, nous avons défini en premier lieu les conditions expérimentales :
Température : 37°C, Volume : 700 ml,
- Durée de l'opération : 30 minutes,
- Tour par minute : 30 tours/minutes
Après que ces conditions soient définies, la procédure ci-après a été suivie :
- Placer le liquide d'immersion dans les deux vases cylindriques ;
- Lancer l'analyse et attendre la fin de l'étape du réchauffement de liquide
d'immersion.
- Une fois la température fixée (37°C)
atteinte, placer une unité de l'échantillon à
examiner
dans chacun des six tubes cylindriques transparents et démarrer
l'analyse ;
31
- Au temps indiqué, remonter le porte-tube hors du liquide d'immersion et
examiner l'état des unités soumises à l'essai.
Toutes les unités sont complètement désagrégées. Si un ou deux d'entre elles ne sont pas désagrégées, répéter l'essai sur 12 unités supplémentaires. Les exigences de l'essai sont satisfaites si au moins 16 des 18 unités soumises à l'essai sont désagrégées (Phr Eur, 2020).
Le tableau IV reprend les spécifications relatives au temps de désagrégation des différentes formes galéniques.
Tableau IV : Spécifications relatives au temps de désintégration des formes galéniques
Formes galéniques Milieu Spécifications
Comprimés conventionnels Eau à 37°C < 15 minutes
|
Comprimés recouverts d'un enrobage ordinaire |
Eau à 37°C < 60 minutes |
Gélules Eau à 37°C < 30 minutes
Comprimés disperssibles Eau à 15-25 °C < 3 minutes
|
Comprimés et capsules entéro-solubles |
HCl > 120 minutes |
Nous avons déterminé la valeur d'acceptation de la manière suivante :
- Peser individuellement 10 comprimés et calculer la teneur estimée individuelle de 10
X wi
comprimés à partir de l'équation : xi= A
??
Où :
- Xi= teneur estimée de chaque comprimé ou gélule
- A= résultat du dosage de principe actif réalisé sur le mélange de 10 comprimes ou gélules
- W= masse moyenne de 10 comprimés ou gélules
- wi= masse de chaque comprimé ou capsule
- Xi= teneur estimée de chaque comprimé ou gélule
- A= résultat du dosage de principe actif réalisé sur le mélange de 10 comprimes ou gélules
Dissoudre une masse de 0.05g de la poudre dans 1mL d'eau distillée et ajouter respectivement 1mL de AgNO3 (nitrate d'argent) et d'ammoniac (HNH2), en présence de
32
- W= masse moyenne de 10 comprimés ou gélules - wi= masse de chaque comprimé ou capsule
N.B : Pour les gélules peser individuellement 10 capsules pleines, ouvrir les 10 capsules et les vider. Peser individuellement, les 10 capsules vides et calculer la masse du contenu par différence.
Déterminer la valeur d'acceptation (AV : Acceptante Value) de la manière ci-après :
- 1er cas : si 98,5% ~ x ~ 101,5%, alors AV= ks
- 2ème cas : si x ~ 98,5% alors AV= 98,5- x + ks
- 3ème cas : si x > 101.5%, alors AV= x -101,5 + ks
Où :
x = Moyenne de la valeur estimée individuelle de 10 comprimés ou gélules exprimé en %de la teneur déclarée ;
K= Constante d'acceptabilité (k= 2,4 pour n= 10 et k= 2,0 pour n= 30) S= Ecart type
Limites d'acceptation : AV = 15,0 %
Si AV est supérieur à 15,0 %, recommencer l'essai avec 20 autres comprimés ou gélules et recalculer l'AV comme indiqué ci-dessus. L'AV final de 30 comprimés ou gélules doit être inférieur ou égal à 15 (USP, 2018).
Ce test a permis de s'assurer que les échantillons à base de l'isoniazide contenaient bien la substance active telle déclarée par le fabricant. Dans le cadre de ce travail, nous avons utilisé les méthodes colorimétriques pour pouvoir confirmer la présence de l'isoniazide dans nos échantillons. Ainsi donc, la procédure ci-après a été suivie (Phr Eur, 2004).
Mode opératoire
33
l'isoniazide il y aura apparition des boules d'azotes qui se dégagent et de l'argent métallique qui se dépose sous forme de précipités brun-noir et l'apparition d'un miroir métallique brillant (OMS,2007).
La détermination de la quantité de l'isoniazide dans nos échantillons collectés a été effectuée à l'aide de la méthode spectrophotométrique UV visible. La procédure ayant servie à doser nos échantillons est la suivante (Phr Eur, 2004 et al). :
- Pesé individuellement 20 comprimés
- Déterminer la moyenne de ces 20 comprimés et calculer la prise d'essai (100 mg)
en partant de la règle de trois simple sur base de la masse moyenne de chaque échantillon
- A l'aide d'un mortier et d'un pilon broyer les 20
comprimés afin d'obtenir une
poudre
- Peser la prise d'essai obtenue et la transférer dans
un ballon jaugé de 100 mL,
répéter l'opération
deux fois avec la même prise d'essai
- Dissoudre la prise d'essai avec de l'eau distillée
puis porter au volume avec le
même solvant (solution mère),
- Prélever 1 mL de la solution mère et le
transférer dans un ballon jaugé de 100mL,
puis porter au
volume avec de l'eau distillée
- Mesurer les absorbances de trois solutions au
spectrophotomètre UV visible de
marque HACH DR 5000 à 263nm
N.B : le coefficient d'extinction molaire de l'isoniazide est de .307 à 263nm dans une solution d'isoniazide dissoute à une concentration de 1g/100 mL.
34
Le tableau V présente les informations relatives aux échantillons collectés.
Tableau V: Médicaments sélectionnés pour l'analyse au laboratoire
|
DCI |
Fabricants |
Date de fabrication |
Numéro du lot |
Date d'expiration |
Sites de collecte |
|
Isoniazid |
Macleos |
Le |
Le |
Hôpital général |
|
|
Pharmaceuticals |
04/2018 |
NIB8150A |
03/2022 |
SENDWE, KENYA, RUASH, Cliniques |
|
|
Universitaires |
|||||
|
Isoniazid |
Mylan |
Le |
Le |
RUASHI, |
|
|
Labooratories |
09/2018 |
NIB8149A |
12/2022 |
TSHAMILEMBA |
|
|
Limited |
|||||
|
Isoniazid |
Macleos |
Le |
Le |
KAMALONDO, |
|
|
Pharmaceuticals |
O1/2019 |
NIB914A |
12/2022 |
KATUBA, SNCC |
|
|
Isoniazid |
Cadila Pharmaceuticals |
10/2019 |
039E9020 |
09/2020 |
GECAMINES SUD, KISANGA, |
CINQUANTENAIRE
Comme le montre le tableau V seulement quatre échantillons ont été collectés. Ceci ce se justifierait par le petit nombre de fabricants (n =3) identifiés sur terrain. En effet, plus le nombre de fabricantes est élevé plus les lots sont importants.
L'enquête menée sur l'utilisation de l'isoniazide dans les structures chargées de distribuer les antituberculeux a montré que l'isoniazide dosé à 300mg était le plus utilisé (figure I). En revanche l'isoniazide dosé à 100 mg et 150 mg était moins disponible et leur quantité était tellement faible que leur acquisition n'était pas autorisée de peur qu'il n'y ait pas de rupture de stock.
35

Ces images illustrent les quelques échantillons collectés.
FREQUENCE D'UTILISATION
100MG 150MG 300MG
80%
15%
5%
Figure 1 : Fréquence d'utilisation d'Isoniazide selon les doses
L'inspection visuelle attentive réalisée sur les échantillons collectés n'a pas révélé une
anomalie au niveau du produit et de l'emballage. Le tableau ci-dessous présente les échantillons collectés et la description de leur emballage et forme galénique.
Aucun échantillon n'a présenté de fissures ou de cassures au niveau de la forme galénique ; tous les comprimés présentaient une forme ronde de couleur blanche et bien ferme, concernant les emballages primaires, le numéro de lot, les dates de fabrication et de péremption étaient bien présentes et lisibles.
36
Tableau VI: Paramètres organoleptiques
|
code |
Forme galénique |
caractéristiques |
Date de fabrication et péremption |
|
INH1 |
comprimés |
Couleur blanche, forme ronde, non sécable, sans fissure |
10/2019 09/2020 |
|
INH2 |
Comprimés |
Couleur blanche, forme ronde, non sécable, sans fissure |
O1/2019 12/2022 |
|
INH3 |
Comprimés |
Couleur blanche, forme ronde, non sécable, sans fissure |
04/2018 03/2022 |
|
INH4 |
comprimés |
Couleur blanche, forme ronde, non secable, sans fissure, présence d'une lettre S sur le |
09/2018 12/2022 |
comprimé
37
Tableau VII: Résultats de l'uniformité de masse
comprimé INH 1 % en INH 2 % en INH 3 % en INH 4 % en (poids rapport (poids rapport (poids rapport (poids rapport
en g) avec la en g) avec la en g) avec la en g) avec la
masse masse masse masse
|
moyenne |
moyenne |
moyenne |
moyenne |
|||||
|
C1 |
0.5624g |
100.02 |
0.5605 |
99.9 |
0.6013g |
100.2 |
0.4495g |
100.5 |
|
0.5607g |
100.09 |
0.5593 |
99.7 |
0.5973g |
99.5 |
0.4454g |
99.6 |
|
|
C3 |
0.5688g |
101.5 |
0.5576 |
99.4 |
0.5993g |
99.8 |
0.4486g |
100.3 |
|
C4 |
0.5611g |
100.1 |
0.5592 |
99.7 |
0.6051g |
100.8 |
0.4482g |
100.2 |
|
C5 |
0.5571g |
99.4 |
0.5572 |
99.3 |
0.5959g |
99.3 |
0.4472g |
100.0 |
|
C6 |
0.5622g |
100.3 |
0.5662 |
100.9 |
0.6042g |
100.6 |
0.4466g |
99.8 |
|
C7 |
0.5562g |
99.2 |
0.5568 |
99.3 |
0.6053g |
100.8 |
0.4473g |
100.04 |
|
C8 |
0.5576g |
99.5 |
0.5645 |
100.6 |
0.6017g |
100.2 |
0.4438g |
99.2 |
|
C9 |
0.5552g |
99.1 |
0.5614 |
100.1 |
0.6109g |
101.8 |
0.4499g |
100.6 |
|
C10 |
0.5612g |
100.1 |
0.5617 |
100.1 |
0.6045g |
100.7 |
0.4495g |
100.5 |
|
C11 |
0.5527g |
98.6 |
0.5642 |
100.6 |
0.5922g |
99.6 |
0.4451g |
99.5 |
|
C12 |
0.5621g |
100.3 |
0.5593 |
0.997 |
0.5925g |
98.7 |
0.4431g |
99.1 |
|
C13 |
0.5634g |
100.5 |
0.5541 |
98.8 |
0.5946g |
99.1 |
0.4495g |
100.5 |
|
C14 |
0.5587g |
99.74 |
0.5584 |
99.6 |
0.6012g |
100.2 |
0.4492g |
100.5 |
|
C15 |
0.5621g |
100.3 |
0.5580 |
99.5 |
0.5994g |
99.8 |
0.4462g |
99.7 |
|
C16 |
O.5681g |
99.6 |
0.5657 |
100.9 |
0.6017g |
100.2 |
0.4492g |
100.4 |
|
C17 |
0.5639g |
100.6 |
0.5615 |
100.1 |
0.5963g |
99.3 |
0.4433g |
99.1 |
|
C18 |
0.5612g |
100.1 |
0.5599 |
98.8 |
0.6026g |
100.4 |
0.4476g |
99.8 |
|
C19 |
0.5527g |
99.5 |
0.5625 |
100.3 |
0.5995g |
99.9 |
0.4466g |
99.9 |
|
0 |
0.5609g |
100.1 |
0.5645 |
100.6 |
0.5961g |
99.33 |
0.4473g |
100.04 |
Le tableau VII met en évidence le poids de chaque unité pesée individuellement et leur pourcentage calculé en rapport avec la masse moyenne en vue de voir les différentes variations.
38
Les observations faites sur les résultats présentés dans le tableau V montrent qu'aucun échantillon n'a présenté une unité qui s'est écarté de limites d'acceptation qui est de 95 - 105 % dans notre cas (figure 2) telles que décrite dans la pharmacopée européenne. En effet, aucune unité ne peut s'écarter de cet intervalle.
uniformité des masses
Série1 Série2

98.67
101.54
100.99
98.84
98.69
101.8
100.62
99.1
ECH1 ECH2 ECH3 ECH4
105.00 103.00 101.00 99.00 97.00 95.00
Figure2 : Limites inférieure et supérieure relative au test de l'uniformité de masse
Tous nos échantillons sont donc conformes au test d'uniformité de masse. Les résultats obtenus dans ce travail sont différents de ceux trouvés en Guinée par Zamble (2004) (n= ) et Ewoudou (2014) (n= ???) qui avaient respectivement trouvé 77,5 % et 77,7 % des échantillons n'ayant pas satisfait au test d'uniformité de masse. En revanche, Tous (n=101) les échantillons analysés par Kouonang (2004) au Mali avaient satisfait au test d'uniformité de masse.
39
Nous présentons dans le tableau VIII les résultats de friabilité.
Tableau VIII: Résultat de friabilité
|
ECH |
Poids avant (g) |
Poids après (g) |
Taux de friabilité |
Conclusion |
|
(%) |
||||
|
INH1 |
5.579 |
5.678 |
0.101 |
Conforme |
|
INH2 |
5.598 |
5.583 |
0.015 |
Conforme |
|
INH3 |
5.991 |
5.980 |
0.011 |
Conforme |
|
INH4 |
4.444 |
4.431 |
0.013 |
Conforme |
Concernant la friabilité, tous les lots étaient conformes et ont présenté un taux de friabilité inférieur à 1% conformément aux spécifications de la Pharmacopée Européenne. Dans son étude au Cameroun, Mnanga et al (2016) ont trouvé que sur 29 échantillons soumis au test de friabilité, près de 7 % étaient non conformes. De leur part, Musa et al (2011) avaient aussi obtenu 100 % de conformité en rapport avec le test de friabilité.
Le tableau IX présente les résultats de test de résistance à la rupture de comprimés.
Tableau IX: Résultat de la résistance à la rupture
|
Code |
Epaisseur |
Diametre |
Dureté |
|
Ech1 |
12.233#177;0.102 |
12.226#177;0.104 |
6.16#177;8.355 |
|
Ech2 |
12.288#177;0.104 |
12.286#177;0.103 |
6.41#177;5.326 |
|
Ech3 |
11.534#177;0.154 |
11.532#177;0.291 |
9.42#177;29.84 |
|
Ech4 |
11.086#177;0.692 |
10.987#177;2.867 |
6.519#177;45.142 |
Le test de la résistance à la rupture a donné des valeurs qui nous ont permis de faire la moyenne. Le résultat de la moyenne de chaque lot ne dépasse pas le 10% de différence inter-lots. L'interprétation devrait se faire en se basant sur le document de l'autorisation de mise sur le marché de l'isoniazide, mais fort malheureusement nous n'avons pas pu accéder au dossier de
40
demande d'autorisation de mise sur le marché de fabricant pour interpréter le résultat de la dureté.
Les échantillons soumis à ce test ont tous présenté (figure 3) un temps de désagrégation satisfaisant par rapport aux spécifications de la Pharmacopée Européenne.
Temps de délitement
|
Ech4 Ech3 Ech2 Ech1 |
||||
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15
Figure 3: résultats du test de désagrégation Légende :
- Ech1 = échantillon 1 ou INH1
- Ech2 = échantillon 2 ou INH2
- Ech3 = échantillon 3 ou INH3
- Ech4 = échantillon 4 ou INH4
Sur 101 échantillons soumis au test de désagrégation par Kouonang, (2004) au Mali, 4.9% des échantillons avaient présenté un temps de désagrégation supérieur aux normes prescrites. Oumarou (2003) au Cameroun (n=12), avait trouvé 2.7% de non-conformité relative avec le test de désagrégation. Et ils ont travaillés au MALI en 2003 sur le contrôle qualité de certains antiparasitaires.
41
Le test de variation de masse effectué sur nos échantillons collectés a montré que tous les échantillons ont présenté une valeur d'acceptation inférieur à 15 % comme l'illustre la figure 4.
ACCEPTANCE VALUE
2.41%
2.39%
2.40%
2.37%
INH1 INH2 INH3 INH4
Figure 4 : Acceptances values
Nos échantillons analysés étaient donc conformes aux spécifications de la pharmacopée Américaine qui recommande que la valeur d'acceptation ne doit pas être supérieure à 15 % Dans son étude, Mwamba (2017) avait trouvé que sur 41 lots analysés, 32 % avaient présenté une valeur d'acceptation supérieure à 15 % et ont donc été déclarés non conformes.
Nos quatre échantillons ont donné un résultat positif et conformes à l'identification. C'est-à-dire il y a eu apparition de précipités brun noir et l'apparition d'un miroir métallique brillant (annexe II).
42
Les résultats de dosage des échantillons d'isoniazide dosés à 300 mg sont présentés dans le tableau XI.
Tableau X: Résultat du dosage
|
Echantillon Masse moyenne (n=20) |
Masse pesée (n=3) |
Absorbance (n=3) |
Ecart type relatif (%) |
Teneur en % |
INH 1 0.295 0,295 3,01 96.09
INH2 0.301 0.307 0,325 1,99 98.05
INH3 0.305 0.307 0,303 2,22 99.35
INH4 0.303 0.307 0,302 1,32 98.69
Au regard des résultats présentés dans le tableau XI, la détermination de la teneur en principe actif des échantillons à base d'isoniazide a montré que tous les échantillons étaient conformes aux spécification de la Pharmacopée Britannique (90 - 112 %). La détermination de la teneur de l'isoniazide dans les préparations pharmaceutiques par Shah et al (1995) avait montré que sur trois échantillons analysés par chromatographie Liquide à Haute Performance, tous les échantillons contenaient une quantité d'isoniazide conforme aux spécifications.
43
Notre étude a porté sur l'évaluation des caractéristiques pharmacotechniques et chimiques des comprimés d'isoniazide utilisés à Lubumbashi.
Une enquête a été menée dans différentes structures qui prennent en charge la tuberculose à Lubumbashi en vue d'obtenir des informations concernant l'utilisation de l'isoniazide et y collecter les échantillons à analyser. Des paramètres pharmacotechniques effectués ont été : uniformité de masses, les tests de friabilité, de résistance à la rupture, de désagrégation et de variations des masses. La détermination de la teneur en principe actif a été réalisée par spectrophotométrie UV visible et l'identification par les méthodes colorimétriques.
Les résultats de tests pharmacotechniques réalisés ont montré que tous les échantillons (n=4) ont satisfait aux tests de l'uniformité de masse, de friabilité, de dureté, de désagrégation ainsi que de variations de masse. Le test d'identification a permis de confirmer la présence de l'isoniazide dans les échantillons d'isoniazide qui ont été collectés. En ce qui concerne la détermination de la teneur, tous les échantillons ont présenté une quantité en isoniazide conforme aux spécifications de la Pharmacopée Britannique (90 - 112 %).
Ce travail a donc montré que les comprimés d'isoniazide distribués dans les structures sanitaires dans la ville de Lubumbashi sont de bonne qualité au regard des résultats qui ont été obtenus dans notre étude. A l'issue de cette étude, nous pouvons envisager de mener des études pour déterminer les impuretés susceptibles d'accompagner l'isoniazide lors de sa synthèse, d'effectuer le contrôle qualité des autres antituberculeux majeurs (rifampicine, pyrazinamide et éthambutol) et de réaliser des études de biodisponibilité et de bioéquivalence des médicaments à base des antituberculeux majeurs consommés dans la ville de Lubumbashi.
Carbonelle B., Dailloux M., Lebrun L., Maugein J. et Pernot C. (2003). Cahier de formation biologie médicale. Mycobactéries et mycobactérioses. Bioforma 29 : 157
44
Acocella, G., R. Conti, M. Luisetti, E. Pozzi et C. Orassi. (1985). Pharmacokinetic Studies on Antituberculosis Regimens in Humans 1. Absorption and Metabolism of the Compounds Used in the Initial Intensive Phase of the Short Course Regimens : Single Administration Study. Am Rev Respir Dis. 132 : 510-515
Agence de la santé publique du Canada (2015). La tuberculose: La résistance aux antituberculeux au Canada. Ottawa (Ontario 2017): Ministre de Travaux publics et Services gouvernementaux Canada. Accessible à l'adresse: https://www.canada.ca/fr/sante-publique/services/publications/maladieset-affections/tuberculose-resistance-aux-antituberculeux-canada-2015.html, Consulté le 13/07/2020
Ahlburg D. (2000). The economic impacts of tuberculosis. Genève, Organisation mondiale de la Santé, http://www.stoptb.org/conference/ahlburg.pdf), consulté 13/07/2020
Alberto P. (2018), Union Internationale Contre la Tuberculose et les Maladies respiratoires, guide pratique pour la prise en charge de la tuberculose résistante. L'union, 979 : 11-42
Armand V., (2000). Mycobacteriology Unit, Institute of Tropical Medicine, Nationalestraat, Antwerp, Belgique, 54: 16-21
Anderson RJ., Groundwater PW., Todd A. et Worsley AJ. (2012). Antibacterial Agents: Chemistry, Mode of Action, Mechanisms of Resistance and Clinical Applications: Agents Targeting Protein Synthesis: Aminoglycoside antibiotics. Durham UNIVERSITY; First edition. 435: 150-172
Bartmann, K. (1990). National Committee for Clinical Laboratory Standards. Antimycobacterial susceptibility testing (Proposed Standard). Antituberculosis Drugs. Dans Handbook of Experimental Pharmacology, 84, 3è Éd.; 23
Bhowruth V, Dover LG et Besra GS. (2007). Tuberculosis Chemotherapy: Recent Developments and Future Perspectives. Progress in Medicinal Chemistry. 45: 169
Enarson D., Rieder H., Arnadottir T. et Trébucq A. (1996). Guide de la tuberculose pour les pays à faibles revenus. 4ème édition. Paris : UICTMR ; Organisation mondiale de la santé.
45
CMIT, tuberculose. In E. PILLY. (2008). Vivactis Plus Ed, Alinéa plus, le collège des universités, 26 : 412-9.
Collazos J., Mayo J. et Martinez E. (1995). The chemotherapy of tuberculosis from the past to the future. Respiratory Medicine. 89: 463-69
Crofton J., Chaulet P., Maher D., et coll. (1997). Principes pour la prise en charge de la tuberculose à bacilles résistants. WHO/TB/96.210 Genève : OMS. 1 : 18
CSHPF. (2006). Enquête autour d'un cas de tuberculose - Recommandations pratiques. www.santépublique.com, le 13/08/2020
Daniel T., Ellner J. et Bloom R. (2005). Tuberculosis, pathogens, protection and control. Elsevier copyright 100: 75-102.
Djim M. (2008). Contrôle qualité des médicaments: cas des antipaludéens au Burkina Faso. Th D, Pharmacie. Université de Bamako. 105.
Danzabe F. (2014) Evaluation de la qualité de l'amoxicilline 500 mg comprimé générique du circuit formel et informel de la ville de Yaoundé [Thèse de pharmacie]. Douala; 2015. 108: 1595
Ellard, G.A. (1976). Gammon. Pharmacology of Isoniazid Metabolism in Man. Pharmacokinet Biopharm; 4 : 83-113
Ewoudou M. (2014) Evaluation pharmacotechnique des molécules antipaludiques du marché légal et illicite dans la ville de Yaoundé: cas de l'artemether + lumefantrine 20/120 comprimé [thèse de pharmacie]. Douala; 2014. 9.
Gonberine B. (2006). phase biopharmaceutique des médicaments anti hypertenseurs et antibiotique. 35-40
46
Huchon G. (1994). Tuberculose. Science en marche. Paris (France) : Editions Scientifiques, Techniques et Médicale. 5-119 ,ESTEM ; Université franconphone ;
HANSCH. (1995). Explorer QSAR-Constantes hydrophobes, éléctroniques et stériques. Washington, DC : American Chemical Society. 21
IDSA Guidelines. (2016). Clinical Infectious Diseases.; 63:147-195. www.idsociety.org; le 2/09/2020
Interpretation of Negative Epidemiological Evidence for Carcinogenicity : Isonizid. Publication de l'IARC no 65. (1985). htpp:// www.tuberculosepid.ca/isoniazid.com, le 13/08/2020
Jonathan I. (2019). taux de succes du traitement chez ls patients atteints de la tuberculose en Afrique; these de doctorat en pharmacie
Kouonang K.S. (2004). Contrôle de la qualité de trois antipaludiques dérivés de l'Artémisinine : Artésunate - Artéméther - Dihydroartémisinine; FMPOS/ Université de Bamako; 76.
Lee A., Teo A. et Wong SY. (2001). Novel mutations in ndh in isoniazid-resistant Mycobacterium tuberculosis isolates. Antimicrob Agents Chemother. 45:9-2157
Marigot D.et Perrone C. (2009). Les nouveaux antituberculeux. Reanimation. 18 : 334-42.
Maher D., Chaulet P., Spinaci S. et Harries A. (1997). Le traitement de la tuberculose : principes à l'intention des programmes nationaux. (Traduit de l'original en anglais WHO/TB/97.220).
Mbadinga C.G. (2004). Contrôle de qualité de l'Amodiaquine et de la Quinine au LNS du Mali; FMPOS/ Université de Bamako; 95. These.
Mitchison D. et Davies G. (2012). The chemotherapy of tuberculosis: past, present and future. International Journal of Tuberculosis and Lung Disease. London (England): The Union. 16: 724- 32
N'Guessan K., Kouassi Y. et Bouzid S. (2001). Intérêt et limites de la microscopie des exsudats au cours de l'infection cutanée à Mycobacterium ulcerans en Côte d'Ivoire. Bulletin de la Société de pathologie exotique. 94 : 9-10
47
OMS, 19th WHO Model List of Essential Medecines (Avril 2015)., «Drug treatment for tuberculosis during pregnancy; safety, 24, 553-565. www.OMS.com
OMS (1974). Comité OMS d'experts de la tuberculose. Rapport n°9.Genève (Suisse) :
Organisation Mondiale de la Santé ; 1974. Accessible
à :
http://whqlibdoc.who.int/trs/WHO_TRS_552_fre.pdf
; consulté le 20/10/2020
OMS (2013) ; Le traitement de la tuberculose : principes a l'intention des programmes nationaux Troisième édition Organisation mondiale de la Santé. 13
Oumarou M. (2003). Contrôle de qualité de certains antiparasitaires (Métronidazole, Mébendazole, Niclosamide, Praziquantel) au Laboratoire National de la Santé [thèse de pharmacie]. Bamako ;77.
P. BURI, et LESNE.(2015). Traité biopharmacie et pharmacocinétique deuxième édition
Pilly E. (2012). Maladies infectieuses et tropicales. Collège des universitaires de maladies infectieuses et tropicales (CMIT), 23e éd.
Pharmacopée américaine (2007). (USP30-NF25) -Version électronique. 3è éd. 147
Pharmacopée britannique (2009). Ed IV, 346-790
Pharmacopée européenne 2014.Tome II ; 8è éd. 2729
Pouillot R., Bilong C., Boisier P., Ciss M., Moumouni A. et Amani I.(2007) Le circuit informel des médicaments à Yaoundé et à Niamey : étude de la population des vendeurs et de la qualité des médicaments distribués. Santé publique. 101:113 - 118.
Pr F. Lucht, Pr E. Botelho-Nevers ; le traitement médical de la tuberculose. 2019
Programme national de lutte contre la tuberculose. (2015). Tunisie
Programme national de lutte contre la tuberculose. (2010). République démocratique du Congo
Programme national de lutte contre la tuberculose. (2017). Mali
Programme national de lutte contre la tuberculose. (2014). Sénégal
Rapport sur la lutte contre la tuberculose dans le monde. OMS 2017 ( http://www.who.int/tb/publications/global_report/fr/), The End TB Strategy Global strategy and
48
targets for tuberculosis prevention, care and control after 2015. WHA65/2012/REC/3 ; le 19/09/2020
Rapport sur la lutte contre la tuberculose dans le monde. OMS 2017 ( http://www.who.int/tb/publications/global_report/fr/), The End TB Strategy Global strategy and targets for tuberculosis prevention, care and control after 2015. WHA65/2012/REC/3 14/09/2020
Réseau Médicaments et Développement (REMED). Marché pharmaceutique parallèle, vente illicite et santé publique, 1994.
Revue francophone des laboratoires
· n° 496
· novembre
2017. www.em-consulte.com
Consulté le 10/10/2020
Stratégie de Coopération de l'OMS avec le pays république démocratique du Congo (2017).
Singapore Tuberculosis Service/British Medical Research Council. Assessment of a Daily Combined Preparation of Isoniazid, Rifampicin and Pyrazinamide in a Controlled Trial of Three 6-month Regimens for Smear-Positive Pulmonary Tuberculosis. (1991) Am Rev Respir Dis; 143 : 707-712
Shea K.M., Kammerer J.S., Winston C.A., Navin T.R., et Horsburgh C. (2014). Estimated rate of reactivation of latent tuberculosis infection in the United States, overall and by population subgroup. 179: 216-25
Silva MS., Senna SG. Et Ribeiro MO. (2009). Mutations in katG, inhA, and ahpC genes of Brazilian isoniazid-resistant isolates of Mycobacterium tuberculosis. J Clin Microbiol. 41: 4-447
Singapore Tuberculosis Service/British Medical Research Council. Assessment of a Daily Combined Preparation of Isoniazid, Rifampicin and Pyrazinamide in a Controlled Trial of Three 6-month Regimens for Smear-Positive Pulmonary Tuberculosis. Am Rev Respir Dis 1991 ; 143 : 707-712
Treatment of Tuberculosis: Guidelines, 4th edition Geneva: World Health Organization; 2010. Treatment of tuberculosis, GUIDELINES: Fourth Edition WHO (2010). OMS.com
49
World Health Organisation. Global Tuberculosis Control. WHO Report 2011.
World Health Organisation. Anti-tuberculosis drug resistance in the world. Fourth Global Report, 2008
Villemagne B., Crauste C., Flipo M., Baulard A.R., Deprez B., et Willand N. (2012). Tuberculosis : The drug development pipeline at a glance. Eur J Med Chem; 51 : 1-16.
a

BALLONS A FOND PLAT SUR L'AGITATEUR AUTOMATIQUE

ECTROPHOTOMETRE UV VIS
Aneexe 4
b


BALANCE ANALYTIQUE


DUROMETRE
Appareil de délitement
C
FRIABILIMETRE