Cristallographie de deux enzymes : mutant de l'uridine monophosphate kinase d' E. coli insensible à la régulation allostérique et la cytokinine oxydase( Télécharger le fichier original )par Ahmed Meksem Institut National Agronomique Paris Grignon - Master en Nutrition Santé 2006 |

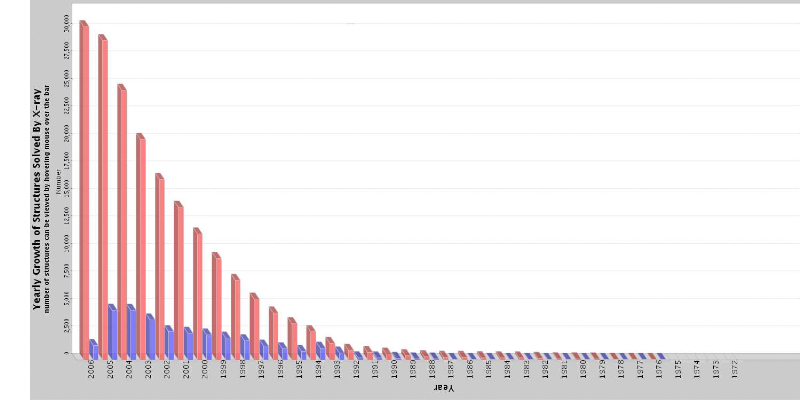
A- IntroductionLa génomique décrypte des séquences protéiques en quantité exponentielle (plus de 2 600 000 séquences connues actuellement : ftp://ftp.ncbi.nih.gov/refseq/release/, 05 Mai 2006). Mais ces séquences ne renseignent pas plus sur la biologie des systèmes considérés qu'un annuaire téléphonique de Paris sur l'organisation et les merveilles de cette ville. En effet, les fonctions biologiques des protéines dépendent étroitement de leur structure tridimensionnelle (3D). Celle-ci ne peut être prédite correctement d'après la séquence et doit être résolue expérimentalement, avec trois méthodes possibles : la microscopie électronique, la résonance magnétique nucléaire (RMN) et la cristallographie. La microscopie électronique a une faible résolution : de l'ordre de 20 ? ; la RMN est surtout efficace pour des molécules d'une masse moléculaire (MM) inférieure à 30 kDa. La cristallographie est la seule méthode qui permette la détermination de la structure 3D à résolution atomique : de 1 à 2 ?, et l'étude des macromolécules sans limitation de la MM. La cristallographie constitue une méthode fondamentale, en biologie structurale, pour l'étude et l'analyse des structures 3D des acides nucléiques et complexes : protéine-acides nucléiques, protéine- protéine et protéine-ligand, dans leur conformation active (Duée et Dideberg, 1997 ; Deleu et al, 1998). Cette technique permet d'obtenir une description 3D détaillée des molécules, et elle apporte des informations concernant les interactions protéine-ligand. Elle réclame un équipement lourd et très onéreux : environ 300 000 € pour le seul ensemble générateur de rayons X / détecteur. Son principal inconvénient est la nécessité d'obtenir des cristaux de la protéine étudiée. Ses premiers succès datent des années 60 avec la résolution de la structure 3D d'une protéine monomérique (myoglobine, J.C. Kendrew, 1960), d'une protéine oligomérique (hémoglobine, M.F. Perutz, 1962) et d'une enzyme (lysozyme, D.C. Philips, 1965). Les changements fondamentaux intervenus dans les techniques propres à la cristallographie et à la biologie (séquençage des génomes, biologie moléculaire, purification de protéines étiquetées, sources intense de rayons X avec les synchrotrons, détecteurs, informatique...) ont entraîné une véritable explosion de la cristallographie des protéines. Le nombre de structures résolues ne cesse d'augmenter : 30000 structures protéiques (soit 86% du total des structures résolues) sont disponibles en mai 2006 dans la Protein Data Bank1 contre 11000 en 2000 (Figure 1 & Tableau 1). Mais ce chiffre ne correspond en fait qu'à 1,3 % des 2 millions de séquences protéiques connues (ftp://ftp.ncbi.nih.gov/refseq/release/). Cet aspect quantitatif est doublé d'un aspect qualitatif : des structures des protéines de plus en plus complexes et importantes sont résolues : virus, ribosome 70S bactérien (52 protéines, 3 ARN, au total 2500 kDa)...(Figure 2). Même un nombre non négligeable de protéines membranaires, malgré leur insolubilité, ont été cristallisées et leur structure 3D résolue.
Données actualisées le 28 Mai 2006 Figure 1 : Nombre de structures 3D résolues par cristallographie, déposées et accessibles dans la PDB. En bleu : le nombre de structures déposées dans l'année, en rose : le nombre total de structure. Le 28 Mai 2006, le nombre est exactement de 31 111 Structures (http://www.rcsb.org/pdb/Welcome.do) Tableau 1 : Structures 3D résolues par les différentes techniques analytiques déposées dans la PDB / Le 28 Mai 2006
Etant pluridisciplinaire, la cristallographie est une méthode qui nécessite de bonnes compétences en biologie, en chimie, en physique et en informatique. Cette méthode comporte une étape limitante : la cristallisation, obtention de cristaux stables à partir des macromolécules pures en solution, de taille suffisante (environ 0,1 mm) et qui diffractent suffisamment les rayons X. La connaissance de la physico-chimie des macromolécules n'est pas suffisante pour établir une méthode rationnelle de cristallogenèse, qui reste pour une grande part très empirique. Cette étape délicate et assez longue ne réussit que pour, environ, un quart des cas. La résolution de la structure 3D est longue, elle peut prendre plus d'une année. En revanche, dans les cas faciles et avec les technologies actuelles, les résultats peuvent être obtenus en quelques mois (Figure 3) (Stelter, 2003). Les données déduites de l'étude structurale, par cristallographie, des macromolécules en général et des protéines en particulier ont un fort impact sur l'élaboration des concepts fondamentaux de la biologie (recherche fondamentale). Elles permettent aussi de comprendre le mécanisme d'action des médicaments [antibiotiques, anti-viraux, anti-cancéreux, antihypertenseurs ...] et des produits phytosanitaires, et d'en concevoir de nouveaux (« drug design »). Dans ce même contexte, les principales caractéristiques des enzymes qui intéressent les industries agro-alimentaires (spécificité et thermosensibilité) sont comprises et on peut ainsi les modifier et/ou les optimiser selon les exigences de l'industrie en question (recherche appliquée). 1 La Protein Data Bank / PDB (Brookhaven, Etats-Unis) : est la banque de données créée en 1977 pour archiver les structures 3D des macromolécules, obtenues par diffraction X ou RMN... A
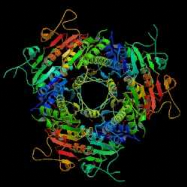
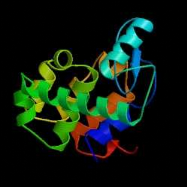
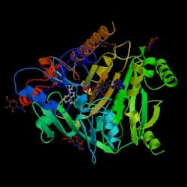
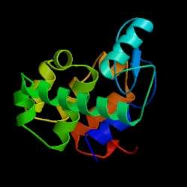
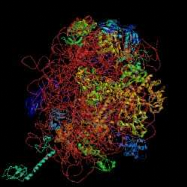
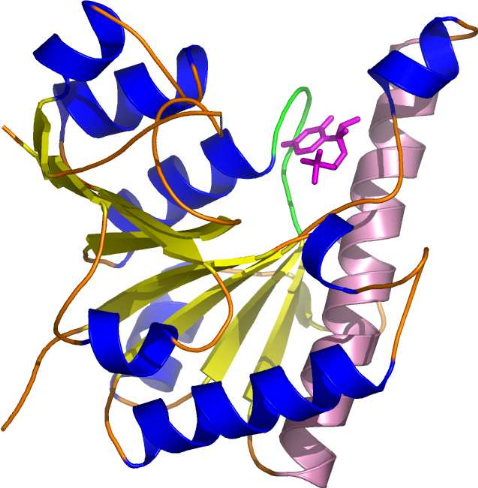
B C D Figure 2 : Evolution de la cristallographie des protéines Les premières protéines étudiées par cristallographie étaient classiques : des molécules compactes et globulaires qui cristallisent facilement (lysozymes, A). Par le suite, de nouvelles protéines de plus grande taille et plus délicats à cristalliser (comme celles faisant l'objet de notre stage) voient leur structure 3D résolue : Cytokinine oxydase du maïs, B ; Uridile monophosphate kinase d'E. coli, C. Récemment, les structures de très grosses molécules ont été résolues : sous unité 50 S du ribosome bactérien, D : Structures résolues de plus en plus complexes. Les étapes de détermination de la structure 3D d'une protéine par cristallographie Protéine pure (97 à 99 %) /Stable ou fraîche / Concentration min = 5 mg/ml / Soluble / Monodisperse et repliée Cristaux protéiques très fragiles / sensibles aux irradiations I S O L E M E I N II Montage sur des cryoboucles T Surexpression et purification Environ 1 protéine sur 4 cristallise I- Cristallisation Taches de diffraction (de Braggs) des rayons X : direction hkl/amplitude et phase
Structure 3D détaillée : vision atomique détaillée de la protéine Comprendre : Fonction / Spécificité / Interactions dynamiques entre molécules / Régulation / Ingénierie des protéines (mutagenèse dirigée, drug design...) / évolution moléculaire (relations Séquence-Evolution-Fonction) / modélisations moléculaires II- Enregistrement des données de diffraction : cliché de diffraction IV- Construction et affinement III Carte de densité électronique VI Figure 3 : De la protéine à sa structure 3D. Toute étude structurale des protéines par cristallographie débute par sa purification, se poursuit par la préparation de cristaux, enregistrement des données de diffraction des rayons X et l'exploitation des clichés pour obtenir la structure 3D. Malgré les progrès considérables réalisés aux différents niveaux de la cristallographie, l'écueil principal reste la cristallogenèse. III- Phasage |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||