I.2. Pharmacocinétique d'un médicament :
I.2.1. Définition :
C'est la partie de la pharmacologie qui étudie, en
fonction du temps, le devenir d'une substance après son introduction
dans un organisme vivant. Cette destinée comporte essentiellement quatre
phases : l'absorption ou résorption, la distribution, les
biotransformations ou métabolismes, et l'élimination, qui peuvent
être, en partie ou en totalité, fonction de la voie
d'administration et de la forme sous laquelle elle est administrée. Ces
facteurs conditionnent la quantité du produit pouvant atteindre les
cibles biologiques et produire une modification, à l'origine de l'effet
thérapeutique (Anonyme 3).
I.2.2. Devenir du médicament dans l'organisme
:
Après avoir pénétrer dans l'organisme, le
PA traverse un nombre variable de cellules pour aboutir dans la circulation
sanguine. Au niveau du tube digestif, et en particulier de l'intestin, la
résorption est facilitée par la très grande surface de
contact avec le contenu intestinal. La voie orale est donc la plus
utilisée, meme si elle n'est pas adaptée à toutes les
situations (Anonyme 3).
I.2.2.1. Absorption :
A) Les niveaux d'absorption :
L'absorption est le transfert d'un PA de son site
d'administration jusqu'à la circulation sanguine. Le taux et
l'efficacité d'absorption dépendent de la voie
d'administration (Champe et al., 2000).
> Absorption à partir des points
d'administration :
Elle est proportionnelle à la solubilité dans l'eau
du liquide extracellulaire. Les complexes peu solubles dans l'eau seront
absorbés lentement.
> Absorption au niveau de l'estomac
:
Elle peut avoir lieu à partir de la paroi stomacale
pour les petites molécules (alcool, eau) et pour les molécules
non ionisées au pH de l'estomac (ex. Aspirine). Mais l'absorption
stomacale est limitée dans le temps en cas de vidange rapide de
l'estomac. Cette motricité gastrique déterminera la vitesse de
délivrance à l'intestin, et sera ralentie par la présence
d'aliments.
> Absorption au niveau de l'intestin
:
La plus grande partie de l'absorption se fera au niveau de
l'intestin, qui présente une grande surface (Helali,
1994).
B) Mécanismes d'absorption :
> Transport passif :
Il peut se faire par :
- Diffusion simple le long du gradient de concentration pour les
substances liposolubles.
- Diffusion facilitée : dans ce cas la substance se
combine avec une molécule transporteuse de la membrane, qui agira sans
dépense d'énergie.
- Filtration sous un gradient de pression (cas de la filtration
glomérulaire).
> Transport actif :
Il est comparable à la diffusion facilitée, mais il
peut agir contre un gradient de concentration avec dépense
d'énergie. Le transport actif est sélectif et saturable
(Schmitt, 1980).

C) Facteurs influençant l'absorption
:
1. pH :
La plupart des médicaments sont des acides ou des bases
faibles (Champe et al., 2000). > Au
niveau de l'estomac :
Au niveau de l'estomac les acides faibles sont absorbés,
et les bases faibles sont excrétées.
> Au niveau de l'intestin :
Le pH intestinal étant alcalin, les pores aqueux rendent
possible l'absorption de molécules hydrosolubles (Schmitt,
1980).
La figure (03), illustre les formes ionisées du
médicament en fonction du pH du milieu d'absorption.
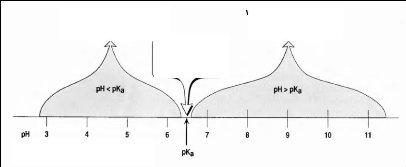
pK est le pH auquel 50 % de la
substance est
ionisée
Si : pH < pKa
HA et
BH+
prédominantes
Si : pH = pKa
HA = A-
BH+ = B
Si : pH > pKa
A- et
B
prédominantes
Figure (03) : Distribution d'un
médicament entre les deux formes : ionisée et non
ionisée
(Pour cette illustration : le médicament a un pKa =
6.5) (Champe et al, 2000).
2. Flux sanguin :
Le flux sanguin au niveau de l'intestin est plus grand qu'au
niveau de l'estomac, ce qui justifie l'importance d'absorption à ce
niveau.
3. Etendue de la surface absorbante
:
L'intestin présente une surface 1000 fois large que
l'estomac donc l'absorption intestinal est plus efficace.
4. Temps de contact avec la surface absorbante
:
Si le médicament se déplace rapidement tout le long
de tractus gastro-intestinal (cas d'une diarrhée sévère),
Il sera mal absorbé (Schmitt, 1980).
Il est à signaler que :
La biodisponibilité se
définit comme étant la fraction de la dose de médicament
administré qui atteint la circulation générale et la
vitesse à laquelle elle l'atteint (Lechat, 2007).
I.2.2.2. Distribution :
L'étape de distribution du PA, lorsqu'il atteint la
circulation générale, peut se diviser en deux phases :
A) Fixation d'un médicament aux
protéines plasmatiques (phase plasmatique) :
Les protéines plasmatiques ont un rôle très
important dans la distribution des

médicaments dont vont être transportés,
leurs fixations sur ces protéines se fait par des liaisons
réversibles parmi ces protéines l'albumine qui est la
plus importante sur le plan quantitatif (Touitou, 1995).
La molécule d'albumine est chargée, et
peut se lier de nombreuses substances. La partie liée aux
protéines par rapport à la quantité totale plasmatique est
variable : 90 % pour la pénicilline, moins de 10 % pour la
caféine (Helali, 1994).
La partie liée aux protéines n'a pas d'action
pharmacologique. Elle sert de réserve car le médicament se
défixe des protéines en fonction les besoins. C'est cette
fonction non liée aux protéines qui sera responsable de
l'activité thérapeutique (Touitou, 1995).
La liaison d'un médicament aux protéines
plasmatiques se caractérise par le pourcentage de médicament
fixé mais aussi par la force de la liaison (constante d'affinité
et le nombre de sites de fixation qui sont plus ou moins importants et pourront
être plus ou moins saturés).
B) Pénétration tissulaire (phase
tissulaire) :
La fraction libre du PA, véhiculé par le sang, va
pouvoir pénétrer dans les tissus de différents organes. La
pénétration tissulaire dépend de plusieurs facteurs :
- La taille de la molécule du PA ;
- L'hydrosolubilité et la liposolubilité du
médicament ;
- La fixation aux protéines plasmatiques et aux
protéines tissulaires : il existe un équilibre entre les formes
libres et les formes liées aux différentes protéines
plasmatiques et tissulaires ;
- La vascularisation : plus l'organe est vascularisé,
plus la pénétration sera importante et rapide. La liaison d'un
médicament sur ses récepteurs tissulaires et le plus souvent
réversible (Jolliet et al., 2000).
Les étapes de distribution sont résumées
dans la figure (04).
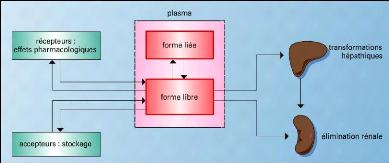
Figure (04) : Schéma
général de la distribution d'un médicament
(Anonyme 3).
Il est à signaler que :
Le volume de distribution : exprime
la quantité de médicament dans l'organisme divisée par la
concentration plasmatique du médicament. Ce volume est un volume
théorique qui serait atteint en cas de répartition
homogène de la molécule dans le volume, C'est à dire que
la concentration du médicament serait partout identique à celle
du plasma.
Vd =Quantité administrée /
Concentration plasmatique (Moulin et Coqurel, 2002).
I.2.2.3. Biotransformation (métabolisme)
:
Le terme métabolisme fait référencer
à la transformation par une réaction enzymatique d'un
médicament, en un ou plusieurs autres composés actifs ou inactifs
au plan pharmacologique.
De nombreux tissus peuvent réaliser cette
transformation (peau, poumon, rein, intestin...). Néanmoins le principal
site de biotransformation se situe au niveau hépatique (Moulin
et Coqurel, 2002) (fig.05).
MEDICAMENT
PHASE 1
METABOLITES
PHASE 2



Métabolites libres
Métabolites conjugués
Médicament
conjugué
Médicament
inchangé



Figure (05) : Phases de transformations
des médicaments (Anonyme 2).
Les réactions de biotransformation sont divisées
en deux phases, phase 1 et phase 2, mais il faut signaler que
le passage d'une phase à autre et l'ordre dépend de la structure
du médicament lui-même et la capacité des enzymes
hépatiques (Anthony, 2002).
A) Réactions de la phase 1 :
Au cours de la phase 1, des réactions biochimiques
transforment la substance initiale en métabolites.
- Le métabolite formé peut être
pharmacologiquement actif. C'est un processus d'activation.
- Le métabolite formé peut être dangereux
pour l'organisme qui le fabrique. On parle de «métabolite
réactif », Il s'agit surtout de radicaux libres.
- Les métabolites formées peuvent être
inactifs (inactivation) ou moins actifs (désactivation) que la
molécule initiale.
Parmi les réactions de la phase 1 on trouve :
- Oxydation ;
- Réduction ;
- Hydrolyse (Anonyme 2).
B) Réactions de la phase 2 (conjugaison)
:
Les conjugaisons réalisent l'union des
médicaments ou de leurs métabolites avec un agent conjuguant
provenant du métabolisme physiologique. Le produit formé,
appelée conjugué, est inactif et facilement
éliminé. Le siège des conjugaisons est essentiellement
hépatique.
La glycurono-conjugaison est la conjugaison la plus
fréquente chez l'homme (Anonyme 2).
C) Facteurs influençant la biotransformation
:
- Les gènes : l'activité des enzymes du
métabolisme peut varier d'un individu à l'autre ; - L'age : sujet
âgé, enfant ;
- L'induction/inhibition : certains médicaments ont la
propriété de stimuler les systèmes enzymatiques
responsables du métabolisme ou de les inhiber ;
- L'insuffisance hépatique ;
- La grossesse (Jolliet et al., 2000).
I.2.2.4. Elimination (excrétion)
:
A) Voies d'élimination :
L'élimination correspond à la disparition du
médicament sous forme active. Cette élimination peut se faire
sous une forme inchangée dans l'urine et les selles ou bien après
une biotransformation en métabolites inactifs le plus souvent
(Saint-Maurice et al., 2004). L'excrétion d'un PA peut
s'effectuer par différentes voies :
> Excrétion par voie rénale
:
La voie rénale est la voie d'excrétion
principale, le rein joue son rôle physiologique d'excrétion. Le PA
passe de la circulation sanguine dans l'urine par deux processus : la
filtration glomérulaire, et la sécrétion tubulaire
(Jolliet et al., 2000).
> Excrétion par voie hépatique
:
Le foie participe à l'excrétion des
médicaments hors de l'organisme par le biais du système biliaire.
Après excrétion dans la bile, le médicament se retrouve
dans la lumière intestinale où il peut être
réabsorbé.
> Autres voies
d'excrétion:
Les autres voies (salivaires, pulmonaire...) sont usuellement
négligeables par rapport aux voies rénale et hépatique.
Néanmoins on soulignera l'importance de la voie lactée pouvant
donner des risques d'intoxications du nourrisson lors de l'allaitement
(Lechat, 2007).
Il est à signaler que :
La clairance : c'est la
capacité globale de l'organisme à éliminer une
molécule, elle correspond au volume de plasma totalement
épuré par unité de temps ; elle est ainsi habituellement
exprimée comme un débit en ml/min.
La clairance totale est égale à la somme des
clairances de chaque organe susceptible d'intervenir dans l'élimination
du médicament: clairance rénale, hépatique, intestinale,
pulmonaire ...etc (Lechat, 2007).
Le T (1/2) : c'est le temps
nécessaire pour que la concentration plasmatique d'un
médicament tombe par la moitie. Le T
(1/2) est déterminé par le volume de
distribution et la clairance ou bien l'élimination (Yang et al.,
2004).
I.3. Pharmacodynamique d'un médicament
:
I.3.1. Définition :
Le terme pharmacodynamique réfère à
l'action d'un médicament au niveau cellulaire, ce terme englobe la
fixation d'un médicament sur son récepteur, ou site du fixation,
et la relation entre la dose et la réponse physiologique
(Anthony, 2002).
I.3.2. Mécanismes d'action des médicaments
:
La plupart des médicaments agissent sur l'organisme grace
à leur affinité avec les
récepteurs de nos cellules. La nature des liaisons entre
le récepteur et le médicament conditionne fortement
l'activité thérapeutique (Anonyme 3).
I.3.2.1. Mécanisme général
:
> Action par fixation spécifique
:
Les médicaments agissent en général par
fixation sur des récepteurs, cette fixation est spécifique du
médicament et de son effet. Elle dépend étroitement de sa
structure et de ses propriétés chimiques.
> Sans fixation dans l'organisme
:
Ces médicaments agissent grâce à leurs
propriétés physiques (volume, pouvoir couvrant, etc.) ou en
modifiant celles du milieu extra cellulaire (pouvoir osmotique,
équilibre acidobasique, équilibre électrolytique, etc.).
Les structures chimiques peuvent être très différentes pour
un même effet.
> Action sur des organismes étrangers
:
Certains médicaments agissent sur des organismes
pathogènes (bactéries, virus, parasites, champignons). Les
mécanismes d'action sont semblables à ceux énumères
ci-dessus (Anonyme 2).
I.3.2.2. Mécanisme moléculaire
:
A) Définition d'un récepteur
:
Les récepteurs peuvent être définis comme
les éléments sensibles dans le système de communication
chimique qui coordonne les fonctions des différentes cellules de
l'organisme, les messagers chimiques étant des hormones, des
neurotransmetteurs ou des facteurs de croissance.
Les récepteurs sont classés en récepteurs
intra-cellulaires, principalement nucléaires et en récepteurs
membranaires (Jolliet et al., 2000).
B) Interaction entre un médicament et un
récepteur:
L'association chimique du PA avec le récepteur provoque
l'action thérapeutique, cette association se fait selon la
réaction réversible suivante :
1 2

M + Re MRe effet pharmacologique
Cette réaction est caractérisée par :
- Efficacité ;
- Effet pharmacologique (Helali, 1994).
C) Notion relation dose-effet :
La relation qui existe entre le logarithme de dose ou
concentration d'un médicament et la réponse biologique obtenue
après l'action de celui-ci, est une courbe sigmoïde approche la
réponse 0 % à faibles doses, puis la réponse maximale 100
% à hautes doses (Helali, 1994).
Il est à signaler que :
La DE 50 : est la dose (ou
concentration) de médicament produisant une réponse qui est
égale à la moitie (ou 50 %) de l'effet maximal obtenue chez
l'animal. La DE50 est utilisé pour la comparaison entre deux
médicaments: si deux médicaments ont la même
activité intrinsèque celui qui a la plus forte affinité
pour le récepteur a une représentation graphique
concentrationeffet déplacée vers la gauche et sa DE50 est plus
faible mais la hauteur des plateaux (l'effet maximum) est identique
(Helali , 1994).
La DL50 : est la dose (ou
concentration) de médicament qui tue 50 % des animaux au cours d'une
expérience. Les animaux les plus utilisés sont la souris, le
rat.
Les médicaments qui possèdent une DL50 plus
élevée, sont les médicaments qui offrent une
sécurité élevée et le contraire est juste
(Helali, 1994).
| 

