Développement d'un portail web pour le criblage virtuel sur la grille de calcul( Télécharger le fichier original )par Farida LOUACHENI Institut de la Francophonie pour l'Informatique - Master 2 Informatique 2013 |

2.4 AutoDockAutoDock [4] est un programme flexible, utiliser pour le docking protéine-ligand. Il s'agit d'un ensemble de procédures, dont le but de prédire l'interaction de petites molécules, telles que des médicaments candidats »ligand» ou des substrats à un récepteur dont la structure 3D est connue. AutoDock fonctionne essentiellement comme une procédure en deux étapes : le calcul de la carte d'interaction du site de liaison du récepteur qui est réaliséavec autogrid, et la position de ligand sur la carte d'interaction, qui est effectuée avec autodock. Le programme AutoGrid est chargéde calculer les cartes d'interaction des grilles afin de maximiser l'étape d'évaluation des différentes configurations du ligand. Pour cela une grille entoure la protéine réceptrice et un atome sonde est placéà chaque intersection. L'énergie d'interaction de cet atome avec la protéine est calculée et attribuée à l'empla-cement de l'atome sonde sur la grille. Une grille d'affinitéest calculépour chaque type d'atome du ligand. Le temps de calcul de l'énergie en utilisant les grilles est proportionnel au nombre d'atomes du ligand uniquement, il est indépendant du nombre d'atomes du récepteur. Le programme AutoDock effectue la partie de recherche et d'évaluation des différentes configurations du ligand. Il est possible d'utiliser plusieurs techniques pour obtenir les configurations (par recuit simulé, algorithme génétique ou par algorithme génétique La-marckien). Pour la méthode Monte Carlo, à chaque pas un déplacement au hasard de tous les degrés de libertéest effectué(translation, rotation, torsion). Les énergies de la nouvelle et de l'ancienne configuration sont comparées. Si la nouvelle est plus basse elle est gardée, sinon elle est conservée ou rejetée. La version actuelle du programme est la version 4.2, qui fournit de nouvelles fonctionnalités importantes pour le docking comme la flexibilitédes résidus de protéines et des fonctions de score de haute qualité. Afin de pouvoir réaliser le criblage virtuel avec AutoDock, un ensemble d'outils nécessaires doivent être mis en place :
15 2.4.1 Docking avec AutoDockAutoDock a besoin de connaître les types, les charges et la liste de liaison de chaque atome, afin de pouvoir effectuer la procédure de docking. Tout d'abord, il faut chercher dans la base de donnée PDB (Protein Data Bank) dans le site (http: // www. pdb. org,http: // www. rcsb. org ), les fichiers pdb pour la protéine et le ligand.
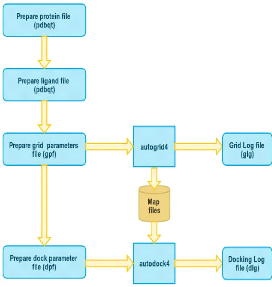
FIGURE 7 - Procédures de docking avec AutoDock La procédure de docking avec AutoDock se décompose en plusieurs étapes : 1. Préparer le fichier d'entrée de protéine. Dans cette étape un fichier PDBQT(Protein Data Bank, Partial Charge (Q), & Atom Type (T)) sera créé, qui contient les atomes et les charges partielles. > input protein.pdb > output protein.pdbqt L'utilisateur possède 2 choix pour préparer son protéine, soit il utilise l'outil »ADT», soit via la commande suivante : > /usr/local/MGLTools-1.5.6/bin/pythonsh /usr/local/MGLTools-1.5.6 /MGLToolsPckgs/AutoDockTools/Utilities24/prepare receptor4.py -r protein.pdb
> input ligand.pdbqt & protein.pdbqt > output ligand protein.dpf > /usr/local/MGLTools-1.5.6/bin/pythonsh /usr/local/ MGLTools-1.5.6/MGLToolsPckgs/ AutoDockTools/ Utilities24/prepare dpf4.py -l ligand.pdbqt -r protein.pdbqt 17 On peut préparer les fichiers de paramètres pour la grille et pour le docking sans utiliser l'outil ADT, en utilisant un script shell (voir annexe) pour préparer ces fichiers. Le résultat de ce script sont respectivement les fichiers : dpf »docking parameter file» et gpf »grid parameter file».
|
|