2.1.5- Indice de fixation FIS
Cet indice est la mesure de l'écart entre la population
d'individus trouvés à l'état hétérozygote
(HO) et l'hétérozygote attendu (HE). Les trois populations
présentent des indices de fixation (FIS) positifs plus ou
moins élevés compris entre (FIS = 0,083) pour Tataouine et (FIS =
0,153) pour Kebili, alors que Médenine (FIS = 0,114) prend une valeur
intermédiaire entre les deux. Ces indices positifs traduisent aussi un
déficit en hétérozygote dont le loci CVRL07 et CVRL01 sont
en partie responsables, car les dits loci présentent des FIS
sont souvent supérieurs à 0,35 dans toutes les populations
analysées. Les coefficient de consanguinité sont respectivement
15,3%, 11,4% et 8,3% pour Kebili, Médenine et Tataouine. Mis à
part le hasard, trois principaux facteurs pourraient expliquer ce
déséquilibre observé. Il s'agit de facteurs
génétiques, l'existence d'allèles nuls et l'effet de
Wahlund (Jordana et al., 2003). En ce qui concerne les causes
génétiques, il est bien connu que la consanguinité
(accouplement entre un individu et ses ascendants, ses collatéraux et/ou
ses descendants) modifie les fréquences génotypiques. La
conséquence en est une perte de variabilité
génétique au cours des générations. Le second
facteur pourrait être inhérent à l'existence
d'allèles nuls,
allèles ne donnant lieu par PCR à aucune
amplification. Une délétion au niveau des amorces ou une mutation
dans les séquences flanquantes du microsatellite pourraient
entraîner la présence d'allèles nuls (Laliberté,
1998). Enfin, le dernier facteur fait référence à la
présence de sous populations à l'intérieur de chaque
population (région) pouvant induire l'effet de Wahlund. A
l'échelle de la population totale, les loci affichent un excès
d'homozygote assez important avec FIS = 0,33 pour CVRL01 et FIS=0,78 pour
CVRL07. L'excès en homozygotie ou hétérozygotie
observée par rapport à l'homozygotie ou
l'hétérozygotie attendue sous l'hypothèse de
Hardy-Weinberg a été testé pour chaque locus et
population. Ainsi, il apparaît que certaines populations vis-à-vis
certains loci présentent des excès d'homozygotes significatifs
par rapport aux proportions de l'équilibre de Hardy Weinberg. D'autres
populations ont des excès en hétérozygotes
significativement différents des proportions de l'équilibre de
Hardy-Weinberg et bien sûr, quelles populations ont montré
l'équilibre panmictique au niveau de certains loci. Les valeurs moyennes
du FIS : 0,153, 0,114 et 0,083 pour Kebili, Médenine et Tataouine,
respectivement sont toutes positives suggérant ainsi un déficit
en hétérozygotes chez toutes les populations camelines
étudiées. L'indice moyen pour la population globale est de 0,071
indiquant un déficit d'hétérozygote relativement
modéré.
2.2- Diversité génétique inter
population
2.2.1- Indice de fixation ou F statistiques
Les indices de fixation ont été calculés
pour tous les loci. Les valeurs de FIS varie de -0,84 pour VOLP32
à 0,78 pour CVRL07 et celle du FIT varient de 0,364 au locus
CVRL06 à 0,816 pour YWLL02. La valeur moyenne de FIS=0,071 indique un
déficit d'hétérozygotes moins important au niveau des
sous-populations que dans la population totale (FIT=0,150). La valeur de
FIT indique un déficit global d'hétérozygotes
de 15% en tenant compte des trois populations étudiées. La
différentiation génétique FST est relativement
élevée pour YWLL02 et VOLP32. En revanche FST affiche
des valeurs faibles aux loci CVRL02 et CVRL06. La différenciation
moyenne entre les populations est de FST=0,083, ce qui peut être
considéré comme une valeur globalement modérée,
indiquant l'origine de la variation génétique totale dans
l'espèce. Rappelons que, la diversité génétique
totale est la somme de la diversité génétique intra
population et de la diversité génétique inter population.
La valeur FST=0,083 montre qu'une grande part (91,7%) de la variabilité
génétique totale est expliquée par la variation intra
population et que le reste de cette variabilité (8,3%) est
attribuée aux différences entre populations. Ce niveau de
différenciation génétique reste, globalement,
semblable à ceux reportés dans d'autres
études pour d'autres espèces domestiques : 8,2 % pour les races
camelines indiennes (Vijh et al., 2007), 8 % pour les
équidés en Espagne (Canon et al., 2000) et 10 % entre
les populations bovines européennes (MacHugh et al., 1998). Le
coefficient FST a été calculé entre les
populations, le paramètre de différentiation
génétique (FST=0,031) entre Médenine et Tataouine traduit
l'absence de structuration géographique et génétique entre
ces deux populations qui apparaissent homogènes. Ce net rapprochement
suppose des échanges d'animaux entre ces deux régions. Cependant,
on retrouve en revanche une distinction entre la population de Kebili et les
deux autres régions, qui peut s'expliquer par l'isolement de cette
région (tableau 16).
Tableau 16 : Les FST entre les paires
des populations
Kebili Médenine Tataouine
Kebili
Médenine 0,131
Tataouine 0,108 0,031
2.2.2 - Distance génétique et construction
des dendrogrammes
La matrice des distances estimées entre les individus
de la population totale, a servi pour la constriction des dendrogrammes. Ces
distances varient de 0 à 0,9, ce qui montre une large variabilité
génétique au sein de la population cameline
étudiée. La distance nulle entre deux individus suggère
une similarité vis-à-vis les loci étudiés. Par
contre une distance élevée traduit l'éloignement
génétique d'un individu par apport à un autre.
Rappelons que la construction des différents arbres
phylogénétiques ont été réalisé
à l'aide du logiciel Darwin version 5.0.155 (Perrier et
Jacquemoud-Collet, 2006) selon la procédure Neighbor Joining. L'examen
du dendrogramme de la population totale (figure 11), permet de distinguer trois
groupes principaux et qui à leur tour présentent des sous
groupes. L'analyse de l'arbre, montre que le regroupement des individus se fait
indépendamment de l'origine géographique et le nom ethnique de
l'écotype. Cette répartition sur l'arbre phylogénique,
peut être expliquée par l'existence d'une large base
génétique commune entre les individus de différentes
populations.
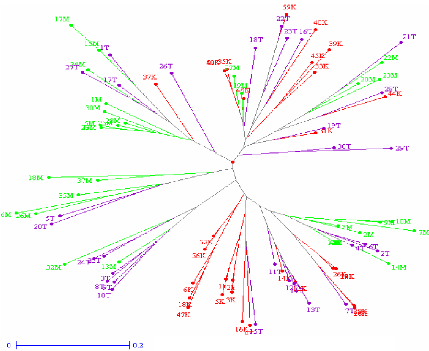
· K : Kebili
· M :
Medenine
· T : Tataouine
Figure 11: Arbre phylogénétique
entre les individus de la population totale
Pour étudier la structuration de la diversité
génétique au sein des populations, la distance de Nei (1978) a
été estimée entre les paires de populations. La matrice
des distances génétiques entre les populations indique une
variation de 0,104 entre Médenine et Tataouine à 0,29 entre
Kebili et Tataouine (Tableau 17). Ces valeurs relativement peu
élevées indiquent que les populations sont
génétiquement comparables et probablement appartiennent à
un même groupe génétique.
Tableau 17 : Distance génétique
entre les paires des populations
Kebili Médenine Tataouine
Kebili
Médenine 0,280
Tataouine 0,290 0,104
Le dendrogramme des populations (Figure 12) montre qu'en
général les regroupements des populations sont en relation avec
des proximités géographiques. Le rameau de
Nefzawa (Kebili) constitue un groupe isolé.
Cependant, le rameau de l'Aaradh (Médenine
et
Tataouine) forme un groupe divisé en deux classes. Cette
classification génétique confirme bien la classification
géographique avancée dans le chapitre précédent.
+ Kebeli
--2
! + Medenine
+ 1
|
+
|
|
Tataouine
|
|
|
Figure 12 : Relations phylogénique entre
les trios populations
|
|
L'analyse de dendrogramme de chaque population (figures 13, 14
et 15) montre bien que les subdivisions de chaque population ne correspondent
pas aux écotypes et que le regroupement se fait indépendamment de
ces écotypes. Ce qui mène à l'hypothèse que les
écotypes identifiés et nommés par les éleveurs ne
constituent pas des entités génétiques bien
individualisées. Cette hypothèse peut être
consolidée par les résultats du chapitre précèdent
tel que les pratiques de croisements incontrôlés, le choix et
l'utilisation des mâles reproducteurs (85% des géniteurs sont
choisis du troupeau lui-même et utilisés pendant une durée
moyenne de 7 ans) et la consanguinité. Tous ces facteurs limitent
significativement la différentiation.
| 

