CHAPITRE 5. ÉTUDE DE CAS
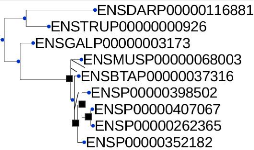
FIGURE 5.4 - Arbre de gène de la famille FAM86 obtenu de
Ensembl
48
CHAPITRE SEPT
CONCLUSION GÉNÉRALE
Parvenu au terme de ce mémoire, dont le but
était de proposer un modèle d'évo-lution de l'architecture
des gènes en tenant compte de toutes les séquences codantes, nous
avons dans un premier temps posé le problème et montré les
limites des solutions actuelles. Puis nous avons proposé un
modèle d'évolution de protéines et introduit des
problèmes d'optimisations dont le but est de reconstruire des arbres de
protéines et de gènes, étant donné les arbres
d'espèces. Nous avons également proposé une méthode
heuristique gloutonne résumée dans un processus à sept
étapes pour résoudre le problème posé. Nous avons
terminé avec des applications sur des familles de gènes de la
base de données Ensembl.
Bien qu'ayant eu des résultats ayant des coûts de
réconciliation mouilleurs que ceux d'Ensembl avec cette approche
heuristique, il demeure qu'une solution algorithmique exact qui
considère simultanément tous les arbres de chaque groupe de
l'étape 4 du processus permettra de reconstruire une solution plus
précise.
Le problème étant formellement posé, il
sera question pour les prochains travaux sur la même problématique
d'affiner.
49
Références
[1] Pea Carninci, T Kasukawa, S Katayama, J Gough, MC Frith,
N Maeda, R Oyama, T Ravasi, B Lenhard, C Wells, et al. The transcriptional
landscape of the mammalian genome. Science, 309(5740) :1559-1563,
2005.
[2] Yann Christinat and Bernard ME Moret. Inferring
transcript phylogenies. BMC bioinformatics, 13(9) :1, 2012.
[3] Yann Christinat and Bernard ME Moret. Inferring
transcript phylogenies. BMC bioinformatics, 13(9) :1, 2012.
[4] Yann Christinat and Bernard ME Moret. A transcript
perspective on evolution. IEEE/ACM Transactions on Computational Biology
and Bioinformatics (TCBB), 10(6) :1403-1411, 2013.
[5] Charles Darwin. R.(1859) : On the origin of species by
means of natural selection. Murray. London, 1871.
[6] William HE Day, David S Johnson, and David Sankoff. The
computational complexity of inferring rooted phylogenies by parsimony.
Mathematical bios-ciences, 81(1) :33-42, 1986.
[7] I Dondoshansky and Y Wolf. Blastclust (ncbi software
development toolkit). NCBI, Bethesda, Md, 2002.
[8] Robert C Edgar. Muscle : a multiple sequence alignment
method with reduced time and space complexity. BMC bioinformatics,
5(1) :1, 2004.
[9] Robert C Edgar. Muscle : multiple sequence alignment with
high accuracy and high throughput. Nucleic acids research, 32(5)
:1792-1797, 2004.
[10] Heng Li et Al. Incorporating species phylogeny in the
reconstruction of gene trees.
[11] R. Durbin et Al. Biological Sequence Analysis.
Cambridge university press, 1998.
50
RÉFÉRENCES
[12] Oliver Eulenstein, Snehalata Huzurbazar, and David A
Liberles. Reconciling phylogenetic trees. Evolution after gene duplication,
pages 185-206, 2010.
[13] Walter M Fitch. Toward finding the tree of maximum
parsimony. In Proceedings of the 8th International Conference on Numerical
Taxonomy, pages 189-230. Freeman, 1975.
[14] Mark B Gerstein, Can Bruce, Joel S Rozowsky, Deyou
Zheng, Jiang Du, Jan O Korbel, Olof Emanuelsson, Zhengdong D Zhang, Sherman
Weissman, and Michael Snyder. What is a gene, post-encode? history and updated
definition. Genome research, 17(6) :669-681, 2007.
[15] Brenton R Graveley. Alternative splicing : increasing
diversity in the proteomic world. TRENDS in Genetics, 17(2) :100-107,
2001.
[16] P. H. The origin of the genetic code. 38 :367-379,
1968.
[17] John P. Huelsenbeck, Fredrik Ronquist, et al. Mrbayes :
Bayesian inference of phylogenetic trees. Bioinformatics, 17(8)
:754-755, 2001.
[18] Daniel H Huson and David Bryant. Application of
phylogenetic networks in evolutionary studies. Molecular biology and
evolution, 23(2) :254-267, 2006.
[19] Daniel H Huson, Regula Rupp, and Celine Scornavacca.
Phylogenetic networks: concepts, algorithms and applications.
Cambridge University Press, 2010.
[20] Safa Jammali, Esaie Kuitche, Ayoub Rachati,
Bélanger François, and Aïda Ouangraoua. Aligning coding
sequences with frameshift extension penalties. 2016.
[21] Abdellali Kelil. Contribution à l'analyse des
séquences de protéines Similarité, Clustering et
Alignement. Université de Sherbrooke, 2011.
[22] Christina Kyriakopoulou. From fundamental genomics
to systems biology : understanding the book of life, volume 23132.
European Communities, 2008.
[23] Weizhong Li and Adam Godzik. Cd-hit : a fast program for
clustering and comparing large sets of protein or nucleotide sequences.
Bioinformatics, 22(13) :1658-1659, 2006.
[24] David A Morrison. Networks in phylogenetic analysis :
new tools for population biology. International journal for parasitology,
35(5) :567-582, 2005.
[25] Luay Nakhleh. Computational approaches to species
phylogeny inference and gene tree reconciliation. Trends in ecology &
evolution, 28(12) :719-728, 2013.
[26] SB Primrose and RM Twyman. Basic biology of plasmid and
phage vectors. Principles of Gene Manipulation and Genomics, 2006.
[27] Richard H Quarles. Myelin-associated glycoprotein (mag)
: past, present and beyond. Journal of neurochemistry, 100(6)
:1431-1448, 2007.
51
RÉFÉRENCES
[28] Vincent Ranwez, Sébastien Harispe,
Frédéric Delsuc, and Emmanuel JP Dou-zery. Macse : Multiple
alignment of coding sequences accounting for frameshifts and stop codons.
PLoS One, 6(9) :22594, 2011.
[29] Vincent Ranwez, Sébastien Harispe,
Frédéric Delsuc, and Emmanuel JP Dou-zery. Macse : Multiple
alignment of coding sequences accounting for frameshifts and stop codons.
PLoS One, 6(9) :e22594, 2011.
[30] Stijn Van Dongen. A cluster algorithm for graphs.
Report-Information systems, (10) :1-40, 2000.
[31] Albert J Vilella, Jessica Severin, Abel Ureta-Vidal, Li
Heng, Richard Durbin, and Ewan Birney. Ensemblcompara genetrees : Complete,
duplication-aware phylogenetic trees in vertebrates. Genome research,
19(2) :327-335, 2009.
| 

